
1
Istotą procesów dyfuzyjno-cieplnych jest wymiana masy
i ciepła między stykającymi się fazami.
Intensywność tej wymiany zależy m. in. od stopnia
odchylenia stanu okłady od tzw. stanu równowagi
międzyfazowej.
Najczęściej przyjmuje się, że układ znajduje się w stanie
równowagi międzyfazowej wówczas, gdy efektywność wymiany
ciepła i masy między stykającymi się fazami jest równa zero.
Zgodnie z definicją termodynamiczną stan równowagi
osiągany jest wówczas, gdy potencjał termodynamiczny układu
jest minimalny.
2
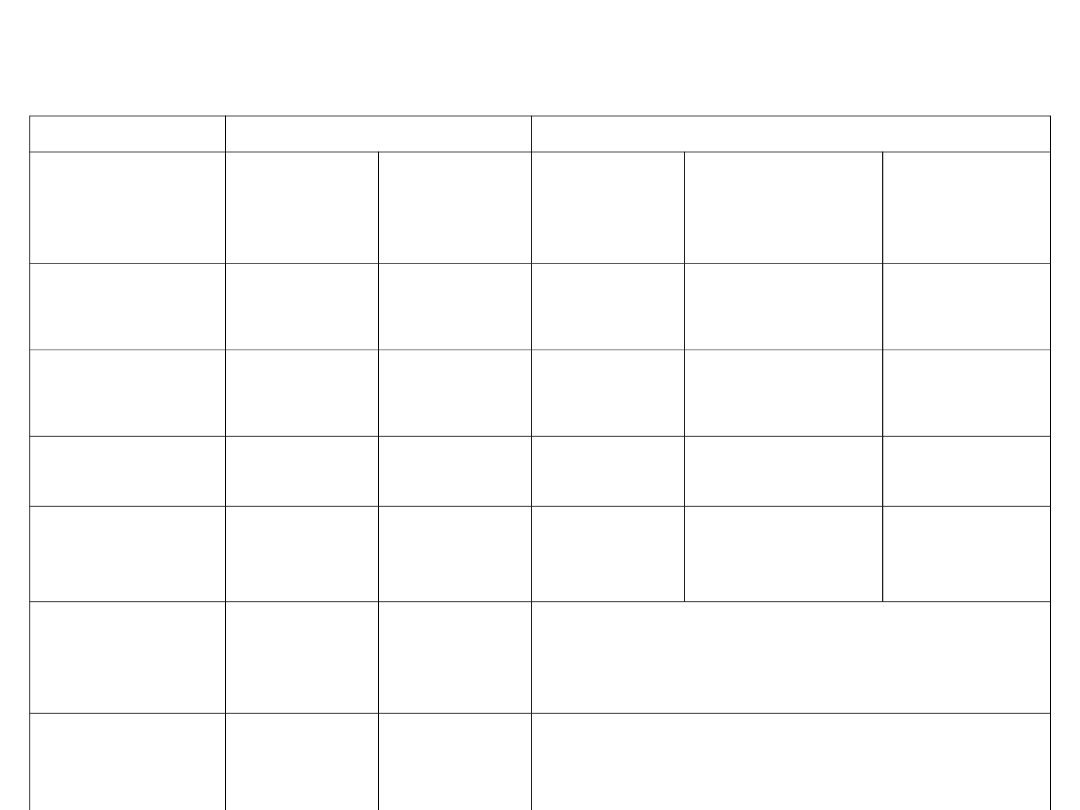
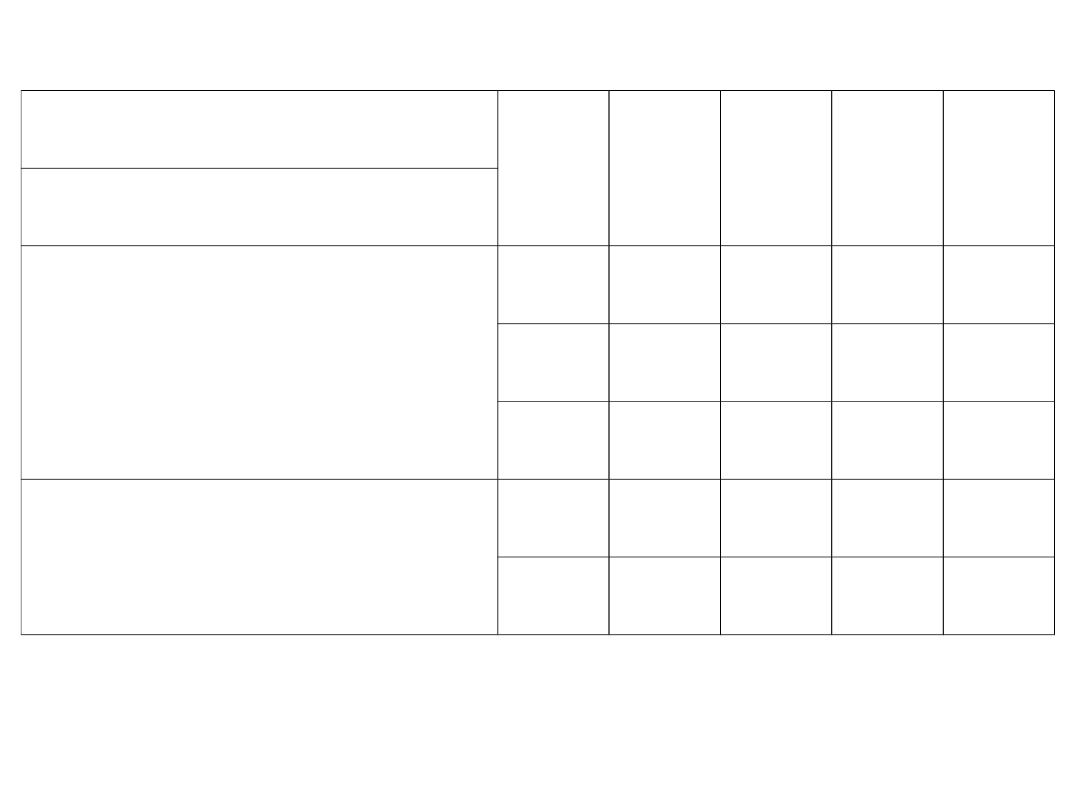
Tab. 1. Definicje stężeń
Stężenia uogólnienie: Z – faza gazowa, S – faza ciekła
Nazwa
Oznaczenie
Definicja
układ dwu-
składniko
wy
układ
wielo-
składniko
wy
układ
dwu-
składniko
wy
układ wielo-
składnikow
y
Ułamek
(udział)
masowy
x
y
x
A
y
A
m
A
/
(m
A
+m
B
)
[kg A /
kg miesz.]
m
A
/
i=A
k
m
i
Ułamek
(udział)
molowy
x
mol
y
mol
x
molA
y
molA
n
A
/
(n
A
+n
B
)
[kmol A /
kmol miesz.]
n
A
/
i=A
k
n
i
Stosunek
masowy
X
Y
X
A
Y
A
m
A
/
m
B
[kg A /
kg nie A]
m
A
/
i=B
k
m
i
, i ≠ A
Stosunek
molowy
X
mol
Y
mol
X
molA
Y
molA
n
A
/
n
B
[kmol A /
kmol nie A]
n
A
/
i=B
k
n
i
,
i ≠ A
Stężenie
masowe
(koncentracja
masowa)
c
c
A
m
A
/ V [kg A/m
3
miesz.]
Stężenie
molowe
(koncentracja
molowa)
C
C
A
n
A
/ V [kg A/m
3
miesz.]

3
! UWAGA !
OZNACZENIA NA WYKŁADACH
wykłady
ćwiczenia
x
A
x
molA
y
A
y
molA
! ułamki molowe!
4
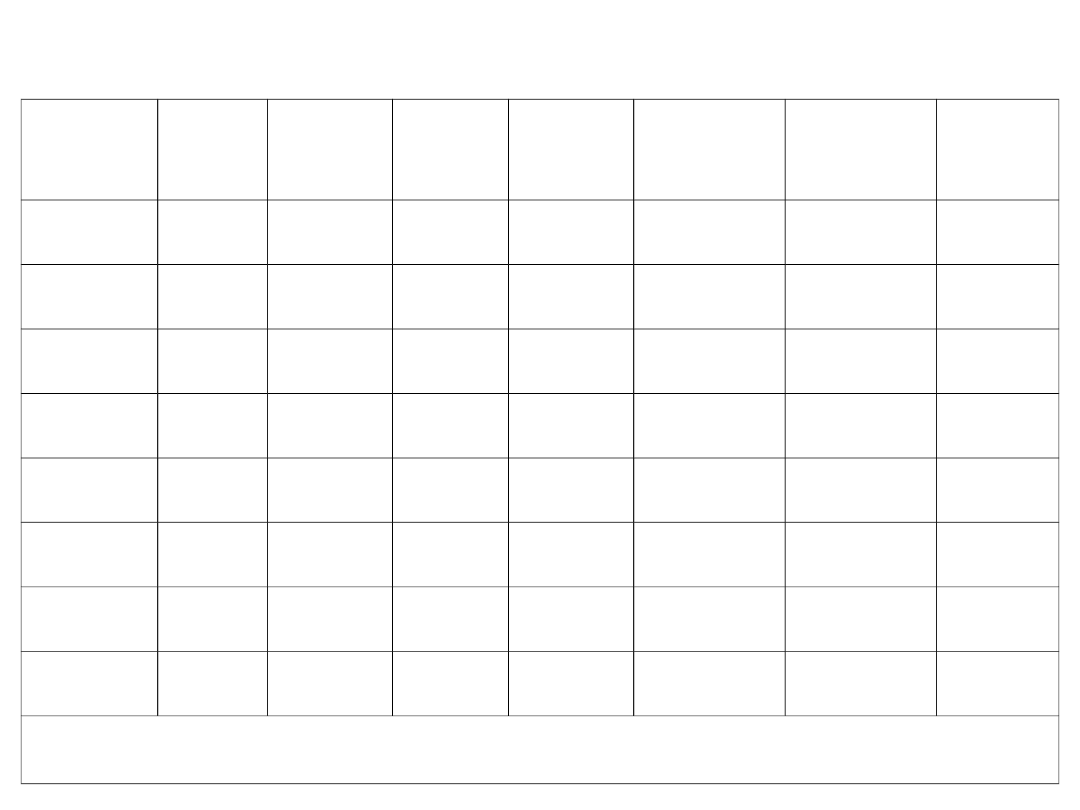
Tab. 2. Wzory do przeliczania stężeń w mieszaninach
Dane:
Szukane:
Udział
masowy
x`
A
kg/kg
Udział
molowy
x
A
kmol/kmol
Stosunek
masowy
X`
A
kg/kg
Stosunek
molowy X
A
kmol/kmol
Koncentracja
masowa
c`
A
kg/m
3
Koncentracja
molowa
c
A
kg/m
3
Ciśnienie
cząstkowe
p
A
Pa
x`
A
=m
A
/m=
(x
A
M
A
) /
i
k
(x
j
M
j
)
X`
A
/
i
k
X`
j
(X
A
M
A
) /
i
k
(X
j
M
j
)
c`
A
/
i
k
c
j
(c
A
M
A
) /
i
k
(c
j
M
j
)
(p
A
M
A
) / *
i
k
(p
j
M
j
)
x
A
=n
A
/n=
(x`
A
/M
A
) /
i
k
(x`
j
/M
j
)
(X`
A
/M
A
) /
i
k
(X`
j
/M
j
)
X
A
/
i
k
X
j
(c`
j
/M
j
) /
i
k
(c`
j
/M
j
)
c
A
/
i
k
c
j
p
A
/ *
p
X`
A
=m
A
/m
t
=
x`
A
/
x`
t
x
A
M
A
/
x
t
M
t
X
A
M
A
/
M
t
c`
A
/
c
t
c
A
M
A
/
c
t
M
t
p
A
M
A
/ *
p
t
M
t
X
A
=n
A
/n
t
=
x`
A
M
t
/
x`
t
M
A
x
A
/
x
t
X`
A
M
t
/
M
A
c`
A
M
t
/
c`
t
M
A
c
A
/
c
t
p
A
/ *
p
t
ć
A
=m
A
/V=
x`
A
/ *
i
k
(x`
j
/
j
)
(x
A
M
A
) / *
i
k
(x
j
M
j
/
j
)
X`
A
/ *
i
k
(X`
j
/
j
)
(X
A
M
A
) / *
i
k
(X
j
M
j
/
j
)
c
A
M
A
p
A
M
A
/ *
RT
c
A
=n
A
/V=
(x`
A
/M
A
) / *
i
k
(x`
j
/
j
)
x
A
/ *
i
k
(x
j
M
j
/
j
)
(X`
A
/M
A
) / *
i
k
(X`
j
/
j
)
X
A
/ *
i
k
(X
j
M
j
/
j
)
c`
A
/
M
A
p
A
/ *
RT
c
A
=n
A
m
/m=
**
(x`
A
m
) / **
M
A
(x
A
m
)
/ **
i
k
(x
j
M
j
)
(X`
A
m
) / **
M
A
i
k
X`
j
(X
A
m
) / **
i
k
X
j
M
j
p
A
=n
A
RT/V=
*
[(y`
A
RT)/M
A
]
/
i
k
(y`
j
/
j
)
y
A
p *
[(Y`
A
RT)/M
A
]
/
i
k
(Y`
j
/
j
) *
Y
A
RT / *
i
k
(Y
j
M
j
/
j
)
c`
A
RT
.
/ *
M
A
c
A
RT *
Oznaczenia: A – składnik, j – składniki bieżące, r – składnik inertny (rozpuszczalnik).
Uwagi: Wzory „*” dotyczą mieszanin idealnych; „**” dotyczą mieszanin o znanej gęstości
m
.
Dla fazy gazowej symbole x, x`, X, X` należy zastąpić symbolami y, y`, Y, Y`.
5
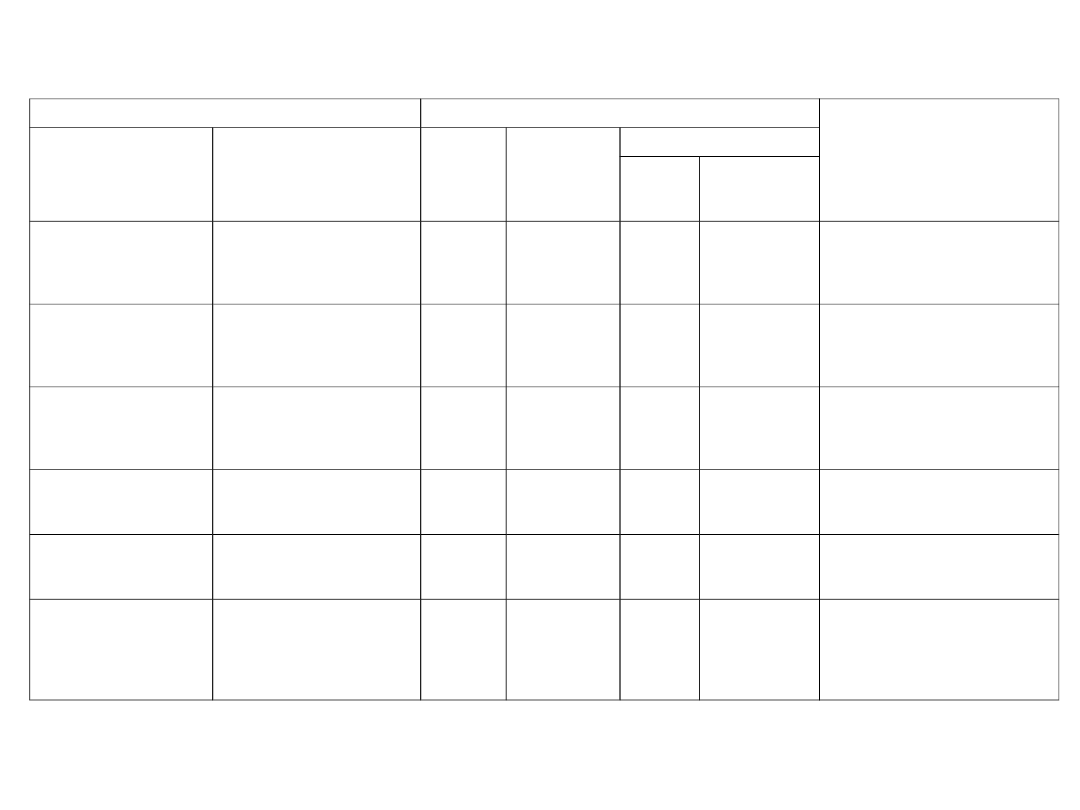
Tab. 3. Zdolność przepuszczania granicy rozdziału układów
Granica rozdziału
Możliwy przepływ
Rodzaj wymiennika w
jakim występuje
Rodzaj
Stosowane nazwy
substa
ncji
niektórych
składnikó
w
energii
wewn
ętrzne
j
ciepła
z fazy stałej zwartej
(np. metal, grafit,
szkło, ceramika)
przepona, ściana,
przegroda, wypełnienie
(w wymienniku
regeneracyjnym)
nie
nie
nie
tak
wymiennik ciepła
(powszechnie)
z fazy stałej
makroporowatej (np.
drobne otwory, albo
materiały spiekane)
ściana porowata
tak
nie
tak,
wraz z
substa
ncją
tak
możliwa w wymienniku
ciepła z odsysaniem
z fazy stałej
mikroporowatej
sorbenty, węgle
aktywowane, sita
molekularne, jonity,
anionity i kationity
nie
tak
tak, ze
składni
kami
możliwy
adsorber, wymiennik jonowy
błona
półprzepuszczalna
membrany z polimerów,
membrany o
porowatości włoskowatej
nie
tak
tak, ze
składni
kami
możliwy, ale
nie istotny
wymiennik masy, dializer
powierzchnia
międzyfazowa (np.
ciecz-gaz)
-
nie
tak
tak, ze
składni
kami
nie
wymiennik masy: absorber,
desorber, ekstraktor
powierzchnia
międzyfazowa (np.
ciecz-gaz)
-
nie
tak
tak, ze
składni
kami
tak
jednoczesny wymiennik
ciepła i masy: skruber,
kondensator par
wieloskładnikowych,
chłodnia kominowa
6
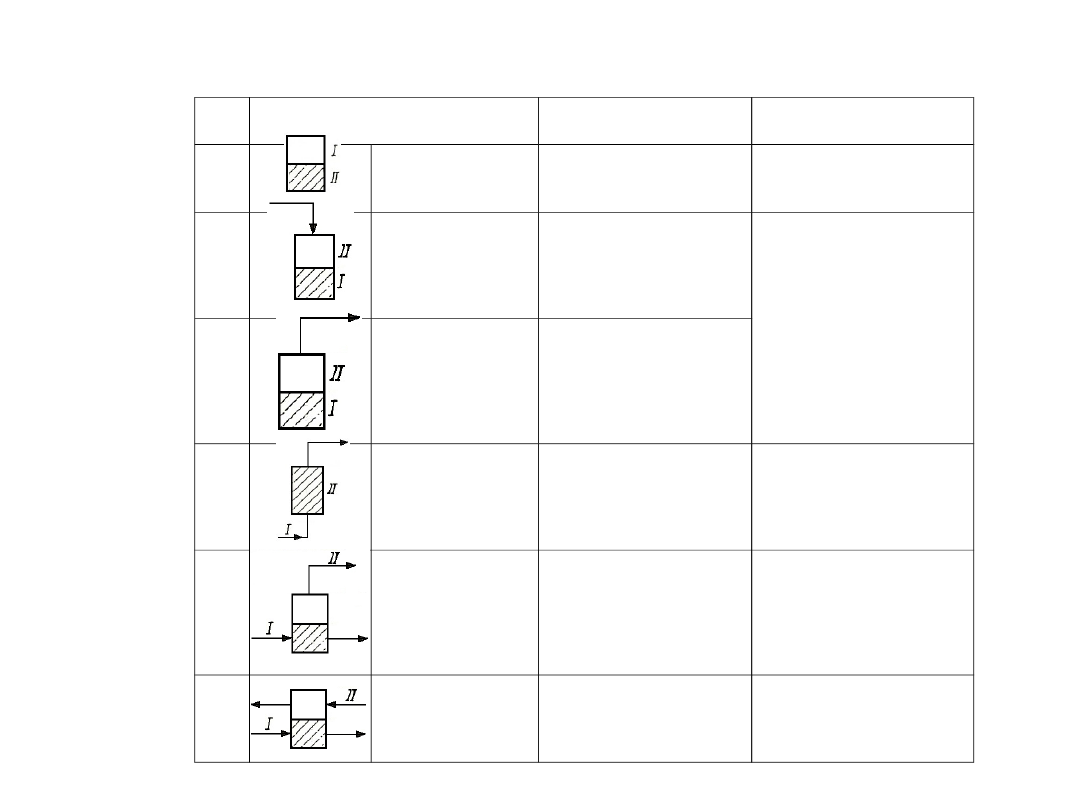
Tab. 4. Sposoby prowadzenia procesów wymiany masy
L.p.
Sposób prowadzenia procesu
Krótka
charakterystyka
Przykłady zastosowań
1
proces okresowy
obie fazy umieszczone w
wymienniku masy
ekstrakcja okresowa
ługowanie okresowe
adsorpcja okresowa
2a
proces z ciągłym
doprowadzeniem
jednej fazy
jedna z faz umieszczona
w wymienniku masy,
druga faza
doprowadzona do
wymiennika masy
destylacja okresowa
desorpcja okresowa
2b
proces z ciągłym
odprowadzeniem
jednej fazy
jedna z faz umieszczona
w wymienniku masy,
druga faza
odprowadzana w sposób
ciągły z wymiennika
masy
3
proces z ciągłym
przepływem jednej
fazy przez
wymiennik masy
jedna z faz umieszczona
w wymienniku masy,
druga faza w sposób
ciągły przepływa przez
wymiennik masy
suszenie
adsorpcja
absorpcja
desorpcja
ługowanie
4
proces przepływu
jednej fazy z
równoczesnym
powstaniem
drugiej fazy
jedna z faz w sposób
ciągły przepływa przez
wymiennik masy,
powstająca druga faza
jest usuwana z
wymiennika masy
destylacja okresowa
kondensacja
krystalizacja
5
proces ciągły
obie fazy przepływają w
sposób ciągły przez
wymiennik masy
rektyfikacja
suszenie ciągłe
absorpcja ciągła
nawilżanie powietrza
7
Tab. 5. Modelowa przepuszczalność granicy rozdziału utworzonej z
powierzchni międzyfazowej gaz-ciecz
Modelowa
przepuszczalność
Przepływ przez
granicę rozdziału
Warunki
temperaturowe w
obszarze
membrany
Nazwa
występującej
wymiany w
wyniku
określonej
przepuszczalnośc
i granicy
rozdziału
Przykładowy
wymiennik, w
którym
realizowany jest
proces
przepuszcza jeden
składnik w jedną
stronę, drugi w
drugą (oba
składniki
rozpuszczają się w
obu układach)
dwóch składników
przy równych i
przeciwnie
skierowanych
strumieniach
molowych
izotermiczne
wymiana masy w
warunkach dyfuzji
równomolowej
wymiennik masy,
kolumna
rektyfikacyjna
przepuszcza tylko
jeden składnik, ten
jeden składnik
rozpuszcza się w
obu układach
tylko jednego
składnika
izotermiczne
wymiana masy w
warunkach dyfuzji
przez gaz obojętny
wymiennik masy:
absorber, desorber,
adsorber
przepuszcza jeden
składnik, ze
składnikiem płynie
strumień energii
wewnętrznej, ciepło
płynie w tym
samym kierunku lub
przeciwnym
tylko jednego
składnika oraz
ciepła
nieizotermiczne
jednoczesna
wymiana ciepła i
masy
jednoczesny
wymiennik ciepła i
masy; skraplacz
mieszanin par,
chłodnia kominowa,
skruber do
chłodzenia gazu

8
{ilość substancji składnika j-ego w
układzie}
{stężenie składnika j-ego w układzie} =
------------------------------------------------------------
{ilość substancji w układzie}
{dowolnie wyrażona ilość przepływającej wielkości}
{strumień} = ------------------------------------------------------------------------
{przedział czasu}
{siła napędowa} U
{strumień przepływający przez powierzchnię A} = ----------------------- = ------ =
---- = I
{opór} R R

9
WYMIENNIKI MASY
Sposób prowadzenia procesu:
1. ciągły;
2. okresowy.
1. W procesie ciągłym istnieje stały przepływ faz przez wymiennik
parametry procesu są tylko funkcją miejsca.
2. W procesie periodycznym aparat pracuje szarżami, a parametry
procesu są funkcjami nie tylko miejsca, ale i czasu.
W zależności od czasu trwania rozróżnia się procesy:
- krótko trwające (t
m
< 30s);
- średni trwające (30s < t
m
< 2h);
- długo trwające (2h < t
m
< 1 doba).
10
Pod względem konstrukcyjnym aparaty do wymiany masy
powinny umożliwiać maksymalny kontakt faz oraz gwarantować
mechaniczny rozdział tych faz tak, aby możliwe było odprowadzenie ich z
aparatu w osobnych strumieniach.
Procesy wymiany masy prowadzi się bądź w jednym aparacie,
bądź też w kilku aparatach podobnego typu ustawionych obok siebie lub
jeden nad drugim. Często aparaty ustawione jeden nad drugim są
połączone jednym płaszczem i noszą nazwę KOLUMNY.
m*
p
Dopływ
m*
k
Odpły
w
Przestrzeń
bilansowa
m
Akumulacja
Dopływ = Odpływ + Akumulacja
m*
p
= m*
k
+ m
m*
jp
= m*
jk
+ m
j
(dla j = A, B, C, …, k)
m*
p
= m*
k
m*
jp
= m*
jk
(dla j = A, B, C, …, k)

Bilans energetyczny
(uproszczony)
Dla procesów ustalonych
Q*
p
± Q* = Q*
k
Q*
p
strumień ciepła zawartego w substancjach
wchodzących do przestrzeni bilansowej;
Q*
k
strumień ciepła zawartego w substancjach
wychodzących do przestrzeni bilansowej;
Q* strumień ciepła dostarczony (+) lub
odprowadzony (-) z
przestrzeni bilansowej
niezależnie od substancji
wchodzących lub
wychodzących. Najczęściej są to tzw. straty ciepła
oraz efekty cieplne reakcji.

Równowaga międzyfazowa
Reguła faz Gibbsa
s = k – f +2
W układzie zawierającym czynnik jednorodny (np. wodę i parę
wodną)
s = 1 – 2 + 2 = 1
Jeden stopień swobody oznacza, że dowolnie można odebrać
temperaturę lub ciśnienie, wówczas drugi parametr jest jednoznacznie
wyznaczony za pomocą pewnej zależności funkcyjnej: p
An
= f(T) lub T
n
=f(p).
W układzie zawierającym składniki A, B, C, …, k w pełni
rozpuszczalne w fazie ciekłej i gazowej (przypadek występujący w
rektyfikacji)
s = k – 2 + 2 = k
Liczba stopni swobody równa liczbie składników oznacza, że
jeżeli proces prowadzi się przy obranym p = const, to pozostaje jeszcze
(k – 1) stopni swobody, które służą do określania składu mieszaniny.
Dowolny dobór pozostałych parametrów, np. temperatury, jest już
niemożliwy, gdyż wynika ona z założonego ciśnienia i składu.
Równowaga między fazą
ciekłą i gazową (parową)
Równowaga faz para-ciecz dla czynnika
jednorodnego
Układ dwufazowy ciecz-gaz jest bardzo ważny, gdyż
występuje często m. in. w procesach destylacji, rektyfikacji, adsorpcji i
desorpcji.
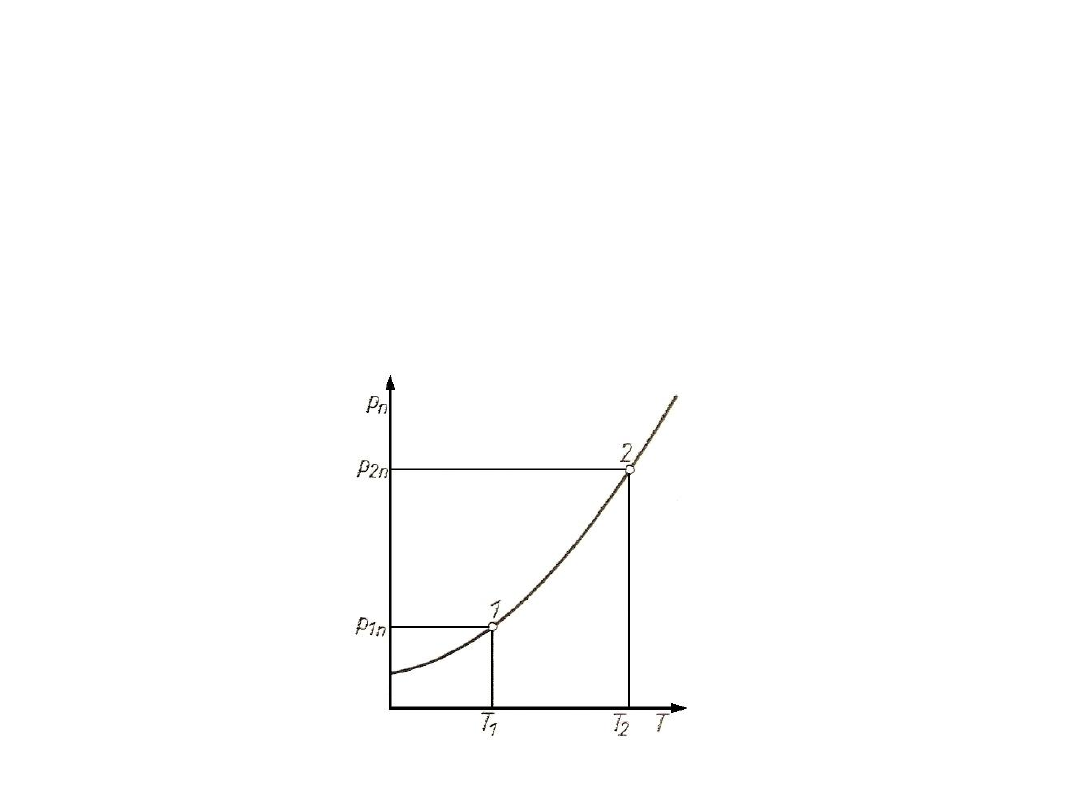
Rys. 1. Krzywa nasycenia dla czynnika jednorodnego

14
Zależność p
n
= f(T) jest opisana przez prawo Clausiusa-
Clapeyrona:
dp
n
/ dT = Ĥ / [T (`` - `)]
gdzie:
Ĥ - zmiana entalpii 1 kmola substancji przy przejściu z fazy ciekłej
do
gazowej
(ciepło parowania pod stałym ciśnieniem)
J/kmol;
``, ` -
objętość 1 kmola substancji w stanie gazowym i
ciekłym m
3
/kmol.
W praktyce bardzo popularne jest empiryczne uogólnienie
nazywane równaniem Antoine`a:
ln(p
n
) = A – [B / (T + C)]
Znajomość zależności p
n
= f(T) dla konkretnej substancji
pozwala na wyznaczenie temperatury wrzenia pod dowolnym
ciśnieniem.

Równowaga między fazą
ciekłą i gazową w mieszaninach
dwu- i wielo-składnikowych
Układy, w których znajduje się więcej niż jeden związek chemiczny
nazywane są układami wieloskładnikowymi lub mieszaninami. Jeżeli dany
układ wieloskładnikowy składa się z dwu faz – ciekłej i gazowej, to na ogół
składy tych faz są różne.
W stanie równowagi przy ustalonym ciśnieniu i ustalonej
temperaturze skład jednej z faz jest wyznaczony jednoznacznie przez skład
drugiej fazy. Można to zapisać następująco:
Z*
j
= f(S
j
)
j = A, B, …, k
lub
S*
j
= f(Z
j
)
j = A, B, …, k
gdzie: Z i S – stężenia uogólnione, gwiazdka oznacza stan równowagi.
Przebieg funkcji dla konkretnego rodzaju stężenia w ustalonej
temperaturze i pod ustalonym ciśnieniem, np. y*
j
= f(x
j
), nazywamy krzywą
równowagi. Zależnie od postaci użytych stężeń przebieg tej linii będzie różny,
ale charakter jej będzie podobny.

16
Tradycyjnie przyjęło się, że stosunek stężenia
równowagowego w fazie gazowej do odpowiadającego mu stężenia w
fazie ciekłej jest nazywany stałą równowagi i oznaczany przez K:
Z*
j
/ S
j
= K
j
j = A, B, …, k
W dalszych rozważaniach stężeń równowagowych jako
składnik przykładowy będzie brany pod uwagę składnik A.
Wartość K zależy od sposobu wyrażania stężeń w
poszczególnych fazach:
K
y,x
= y*
A
/ x
A
, K
Y,X
= Y*
A
/ X
A
, K
pc
= p*
A
/ c
Ac
itp.
Stałą równowagi w dowolnym punkcie wykresu Z*
j
= f(S
j
) da
się przedstawić następująco:
tg = Z*
A
/ S
A
= K
17
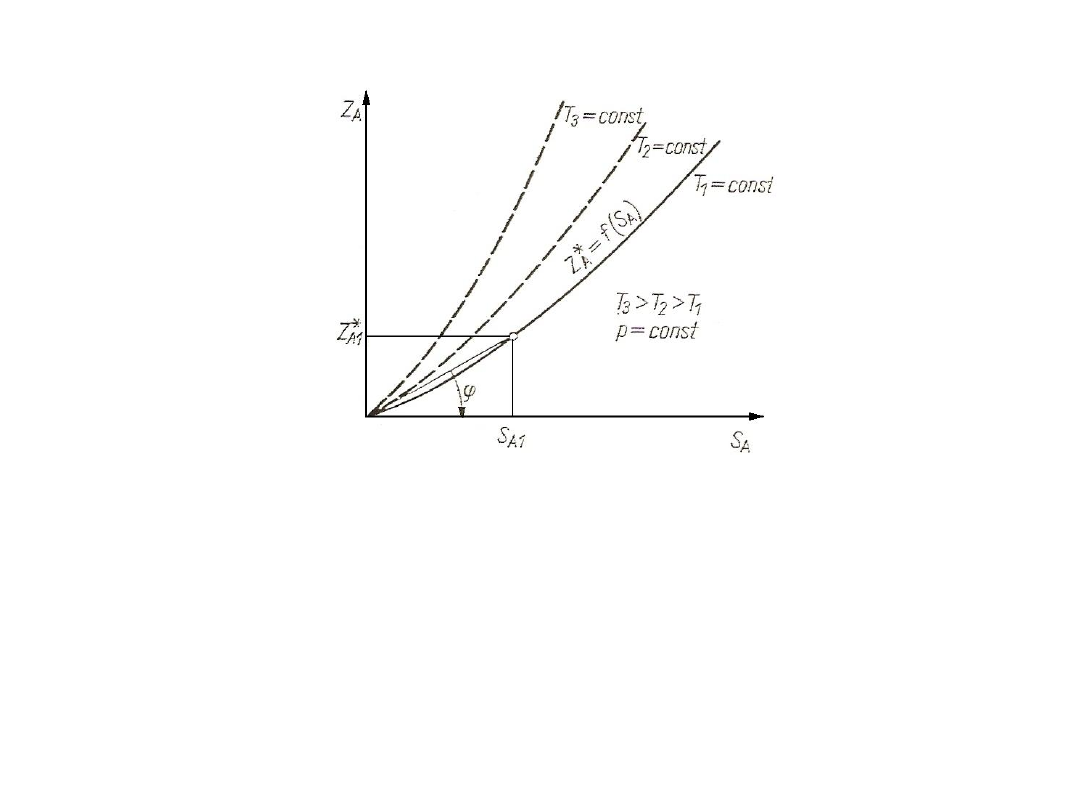
Gdy K = const, linia równowagi jest linią prostą. Linie równowagi dla
wyższych temperatur leżą wyżej.
Rys. 2. Linie równowagi Z*
A
= f(S
A
) dla różnych temperatur
Najogólniej można powiedzieć, że K = f(T, p, S
A
). Określenie stała
równowagi jest nieco mylące, gdyż wielkość ta jest stała tylko w bardzo
nielicznych przypadkach. Jeżeli wartość K nie zależy od stężenia, to dla
stałego ciśnienia można zapisać:
(K)
p
= f(T)
lub
(K)
p,T
= const
W 1803 r. Henry odkrył prawo, które rządzi rozpuszczalnością gazów
w cieczach i które dla T = const ma następującą postać:
p*
A
= H x
A

18
Stała Henry`ego H ma wymiar ciśnienia, a jej wartość można znaleźć w
różnych poradnikach technicznych. Jest ona szczególną postacią stałej K:
(K
p,x
)
T
= (H)
T
Prawo Henry`ego dotyczy tylko małych stężeń, niezbyt niskiej
temperatury oraz umiarkowanego ciśnienia. Jeżeli w równowadze z
cieczą znajduje się nie jeden gaz, ale mieszanina gazów, to prawo
Henry`ego obowiązuje dla każdego ze składników oddzielnie.
W ogólnym przypadku stała K ≠ const, więc linia równowagi nie
jest linią prostą, ale krzywą o zmiennym nachyleniu. Nachylenie to jest
ważnym parametrem we wzorach określających przenikanie masy. W
przypadku, gdy znane jest równanie:
Z*
A
= f(S
A
) nachylenie linii równowagi można obliczyć z definicji:
m = dZ*
A
/ dS
A
= df(S
A
) / dS
A
W przypadku, gdy linia równowagi jest dana w postaci wykresu,
dwa niezbyt odległe punkty krzywej umożliwiają określenie przybliżonej
wartości nachylenia linii równowagi w pewnym zakresie:
m = dZ*
A
/ dS
A
= Z*
A
/ S
A
= (Z*
A2
- Z*
A1
) / (S
A2
– S
A1
)
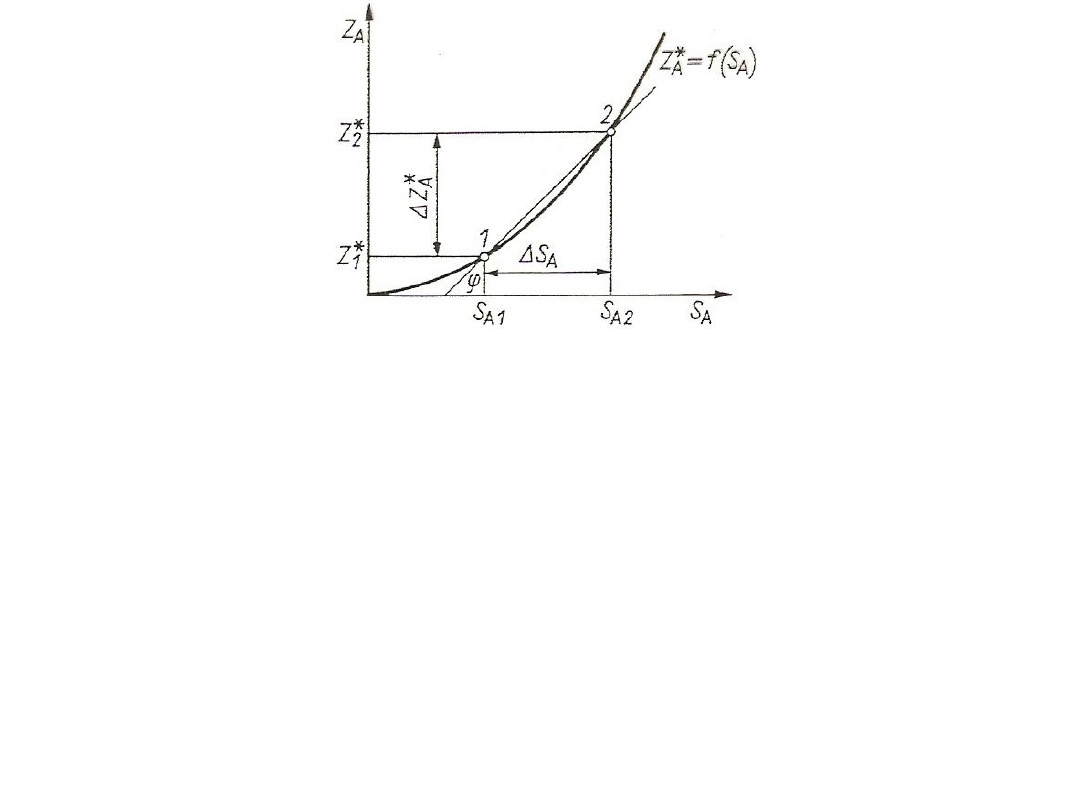
19
Rys. 3. Wyznaczanie nachylenia linii równowagi
W zakresie S
A1
≤ S
A
≤ S
A2
można krzywą równowagi aproksymować w
pewnym przybliżeniu za pomocą linii prostej:
Z*
A
= Z*
A1
+ m(S
A
– S
A1
)
W obliczeniach praktycznych jest potrzebna liczbowa wartość m tylko
w pewnych określonych punktach krzywej równowagi, np. u wlotu albo wylotu
wymiennika masy. Wyznaczenie jej graficznie lub z przyrostów skończonych
nie zawsze jest dokładne. Przybliża się wtedy krzywą równowagi funkcją
potęgową (S do potęgi r)
Z*
A
= B S
a
Po zlogarytmowaniu otrzymuje się linię prostą:
lgZ*
A
= lgB + r lgS
A
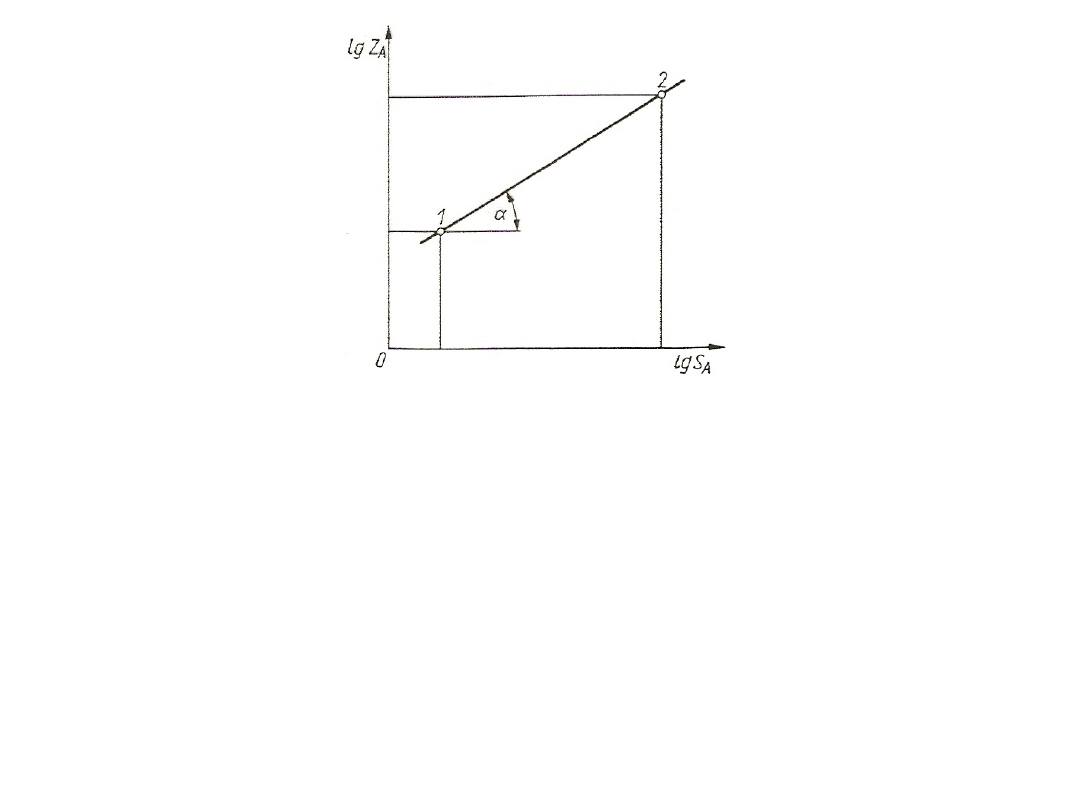
20
Rys. 4. Wyznaczanie wykładnika r krzywej równowagi
Nachylenie tej prostej określa wykładnik r:
r = tg = d(lgZ*
A
) / d(lgS
A
) = d(lnZ*
A
) / d(lnS
A
) = (dZ*
A
/ Z*
A
) (S
A
/ dS
A
) =
m / K
Tym sposobem można więc znaleźć wykładnik r na podstawie
znajomości dwu punktów dość dalekich od siebie, np. stanu na wlocie i
wylocie z wymiennika masy.
W punkcie 1: m
1
= r K
1
, a w punkcie 2: m
2
= r K
2
. Dla linii
równowagi odbiegających od funkcji potęgowych podane związki są
tylko przybliżeniem. Gdy układ stosuje się do prawa Henry`ego, wtedy
dla stężeń prężność cząstkowa – ułamek molowy r = 1 i m = K
px
= H

21
Równowaga w roztworach
(mieszaninach) idealnych
Jeżeli ciśnienie w układzie jest umiarkowane (dużo mniejsze od
ciśnień krytycznych wszystkich składników), a budowa strukturalna
składników jest podobna, to równowagę w układzie opisuje prawo
Raoulta będące szczególnym przypadkiem prawa Henry`ego.
Zgodnie z prawem Raoulta stała Henry`ego składnika A w
równaniu przyjmuje wartość prężności pary nasyconej tego składnika w
temperaturze roztworu tzn. H = p
An
, a równanie przybiera postać:
p*
A
= p
An
x
A
(forma I)
Po podzieleniu tego równania przez wartość ciśnienia w
wymienniku masy otrzymuje się:
p*
A
/ p = (p
An
/ p) x
A
i dalej:
y*
A
= K
yx
x
A
(forma II)
przy czym: - K
yx
stała równowagi w równaniu Raoulta.

22
Prawo Raoulta jest więc szczególnym przypadkiem prawa
Henry`ego dla roztworów idealnych, gdy składniki w fazie gazowej
mogą się wykraplać w temperaturze roztworu. W zasadzie zarówno
prawo Henry`ego, jak też prawo Raoulta są prawami graficznymi.
Równanie Raoulta przedstawia asymptotę do rzeczywistego
przebiegu krzywej ciśnień cząstkowych p*
A
= f(x
A
) dla x
A
1.
Równanie Henry`ego opisuje natomiast graniczną styczną dla tej
samej krzywej, gdy x
A
0, a więc dla gazów o bardzo małym
stężeniu składnika absorbowanego. Jak stwierdzono powyżej prawo
Raoulta obowiązuje dla mieszanin idealnych. Są to takie mieszaniny,
w których siły występujące między cząsteczkami różnych
składników i między cząsteczkami jednego (tego samego) składnika
są jednakowo wielkie. Po zmieszaniu takich składników, gdy p*
A
=
const, nie ma zmiany objętości, ani też nie powstaje dodatkowe
ciepło mieszania.
Jeżeli układ stosuje się do prawa Raoulta, to przebieg linii
równowagi można opisać równaniem analitycznym, wychodząc z
prawa Raoulta dla składników A i B:
y*
A
= (p
An
/ p) x
A
y*
B
= (p
Bn
/ p) x
B
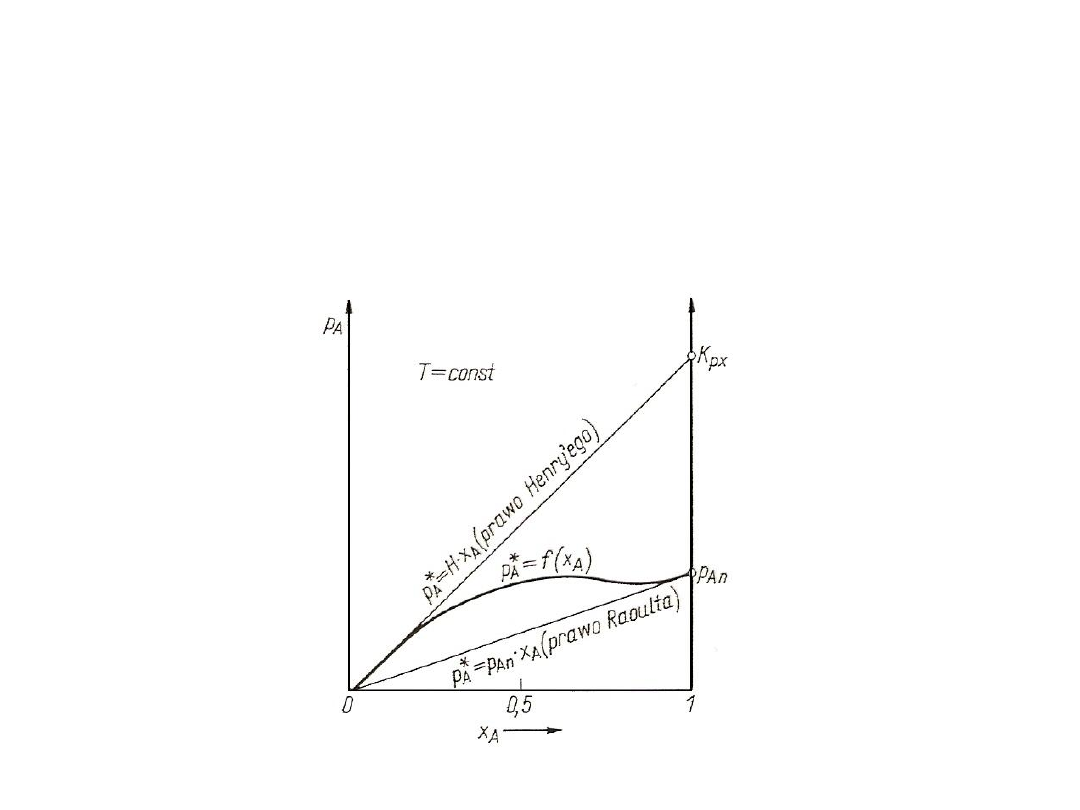
23
Rys. 5. Graficzna interpretacja prawa Raoulta i prawa Henry`ego
Przez podzielenie tych dwóch równań stronami i podstawienie:
x
B
= 1 – x
A
oraz y*
B
= 1 - y*
A
,
po przekształceniach otrzymuje się równanie linii równowagi
Fenskego:
y*
A
= ( x
A
) / [1 + x
A
( - 1)]
gdzie: = p
An
/ p
Bn
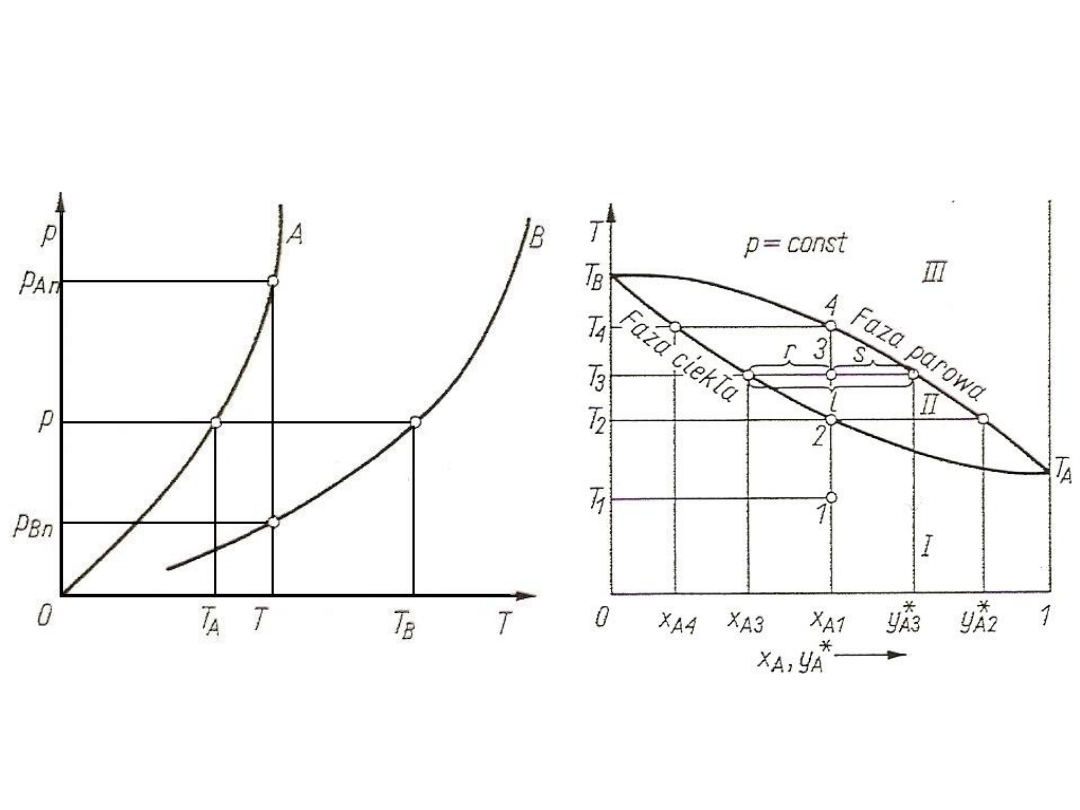
24
Rys. 7. Krzywa składu w układzie
dwuskładnikowym
Do pełnej interpretacji procesu destylacji i warunków
równowagowych w układach dwuskładnikowych są stosowane wykresy
pomocnicze:
1. Krzywa składu, którą można wyznaczyć na podstawie krzywych
prężności pary nasyconej poszczególnych składników.
Rys. 6. Krzywa prężności pary
nasyconej składników A i B
mieszaniny dwuskładnikowej
25
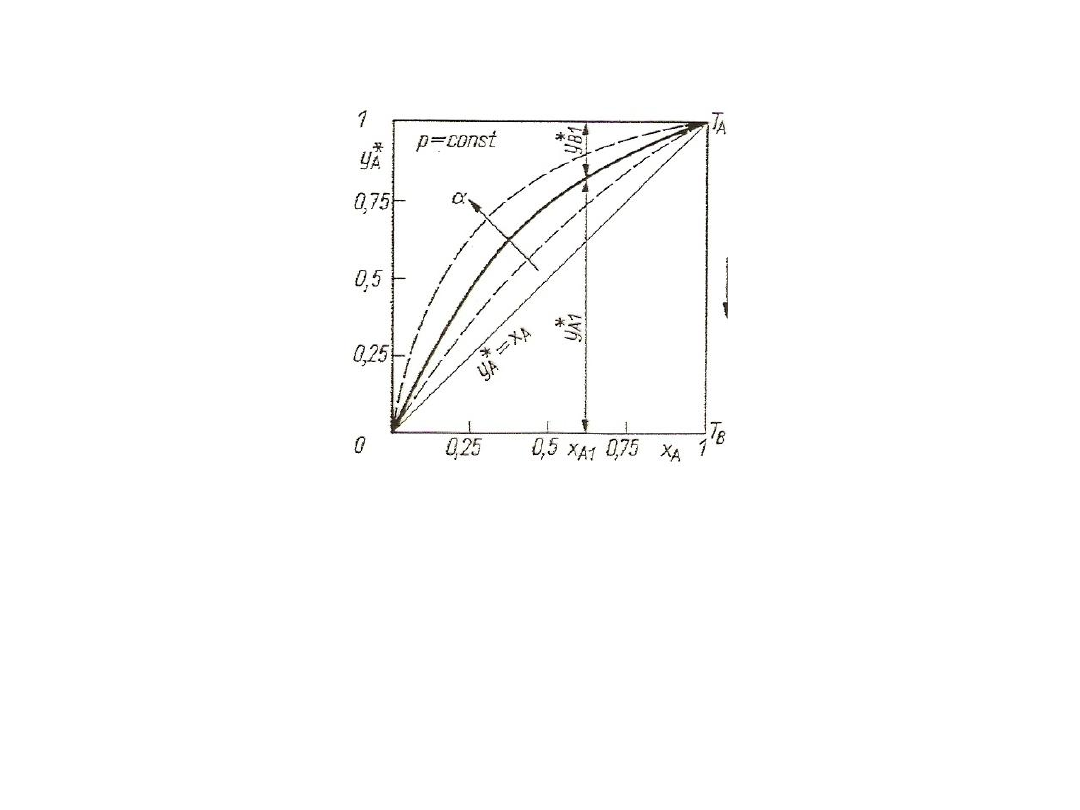
2. Wykresy krzywej równowagi można zbudować z wykresu składu lub z
obliczeń na podstawie równania: y*
A
= (p
An
/ p) x
A
.
Rys. 8. Krzywa równowagi w układzie dwuskładnikowym
Współczynnik względnej lotności α dla wielu mieszanin
dwuskładnikowych zależy bardzo nieznacznie od temperatury. W praktyce
oblicza się go bardzo często jako średnią geometryczną dla skrajnych
wartości temperatury w układzie. Dla bardzo małych stężeń x
A
wyrażenie
( - 1) x
A
jest dużo mniejsze od jedności, więc y*
A
= ( x
A
) . Z tego też
powodu w pobliżu x
A
≈ 0 linia równowagi jest prawie linią prostą. Z kolei
dla małych stężeń składnika B tzn. dla wartości x
A
zbliżonych do jedności,
zachodzi zależność graniczna y*
A
(1 / ) x
A
+ ( - 1) , czyli linia
równowagi również jest prawie linią prostą.
26
Współczynnik względnej lotności jest miarą trudności rozdziału
danej mieszaniny. Współczynnik ten rośnie, gdy ciśnienie układu maleje,
a linia równowagi ma wtedy przebieg bardziej wypukły, tzn. że różnice w
składach fazy parowej i ciekłe się powiększają. Gdy = 1, y*
A
= x
A
, znika
więc wzbogacanie fazy parowej przez odparowanie.
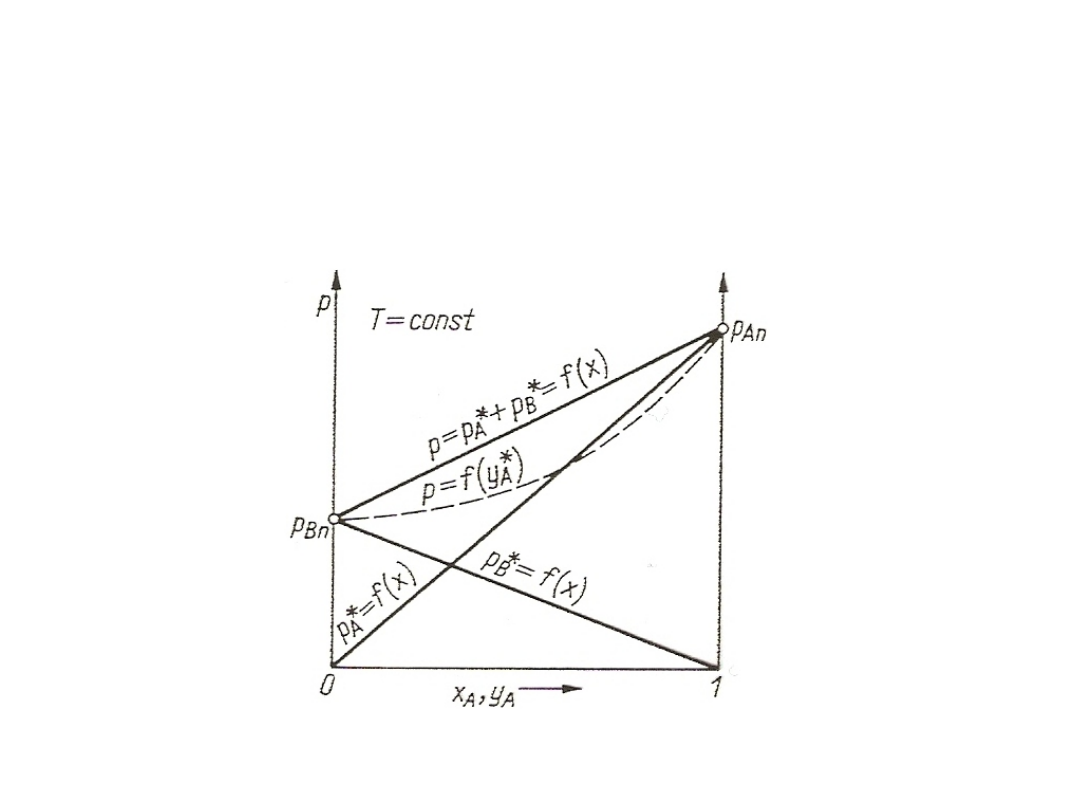
3. Wykres ciśnień cząstkowych.
Rys. 9. Zależność prężności par dla składu cieczy w stałej temperaturze dla
układu dwuskładnikowego stosującego się do prawa Raoulta

27
Zgodnie z prawem Daltona dla mieszaniny idealnej w
stałej temperaturze suma prężności cząstkowych par
składników jest równa ciśnieniu całkowitemu układu:
p = p*
A
+ p*
B
Na rysunku pokazano przebieg zmian ciśnień cząstkowych i
ciśnienia całkowitego w zależności od udziałów molowych w
fazie ciekłej w stałej temperaturze. Dodatkowo zaznaczony jest
przebieg zmian ciśnienia całkowitego p od stężenia y*
A
w fazie
gazowej.

28
Układy dwufazowe (równowaga w roztworach rzeczywistych)
składające się z fazy gazowej i ciekłej można podzielić na 4 grupy:
1. Zarówno gaz, jak i ciecz można traktować jako mieszaniny idealne.
W układach takich spełnione jest prawo Raoulta i Daltona. Sytuacja
taka dotyczy mieszaniny związków o podobnej budowie pod niskim
ciśnieniem.
2. Fazę gazową można uznać za mieszaninę idealną, natomiast gaz jest
mieszaniną rzeczywistą. Spełnione jest prawo Raoulta, prawo
Daltona nie obowiązuje. Dotyczy to mieszanin związków o podobnej
budowie pod zwiększonym ciśnieniem. Rzeczywiste zachowanie się
mieszanin gazowych tym bardziej odróżnia się od zachowania gazów
idealnych, im większe są wartości ciśnienia zredukowanego i im
niższa jest temperatura zredukowana.
3. Mieszanina gazowa jest idealna (spełnia prawo Daltona) natomiast
mieszanina ciekła jest rzeczywista – prawo Raoulta nie jest spełnione
mieszaniny zarówno o różnej budowie pod niskim ciśnieniem.
4. Ani faza gazowa, ani ciekła nie są idealne. Związki (mieszaniny
związków) o różnej budowie pod wysokim ciśnieniem. Tu
modyfikacji prawa Raoulta:
p*
A
=
A
p
A
x
A
gdzie:
A
- współczynnik aktywności.

29
Równowaga w roztworach
(mieszaninach) rzeczywistych
p*
A
=
A
p
A
x
A
A
- współczynnik aktywności
A
= f(p
A
,x
A
)
Siły przyciągania między cząsteczkami różnych składników A i
B w cieczy:
F
AB
= (F
AA
F
BB
)
1/2
F
AA
F
BB
– siły przyciągania między cząstkami tego samego związku
(
A
> 1)
F
AB
< (F
AA
F
BB
)
1/2
dodatnie odchylenie
(b)
(
A
< 1)
F
AB
> (F
AA
F
BB
)
1/2
ujemne odchylenie
(a)
Mieszaniny o składzie odpowiadającym punktom ekstremalnym
azeotropy.
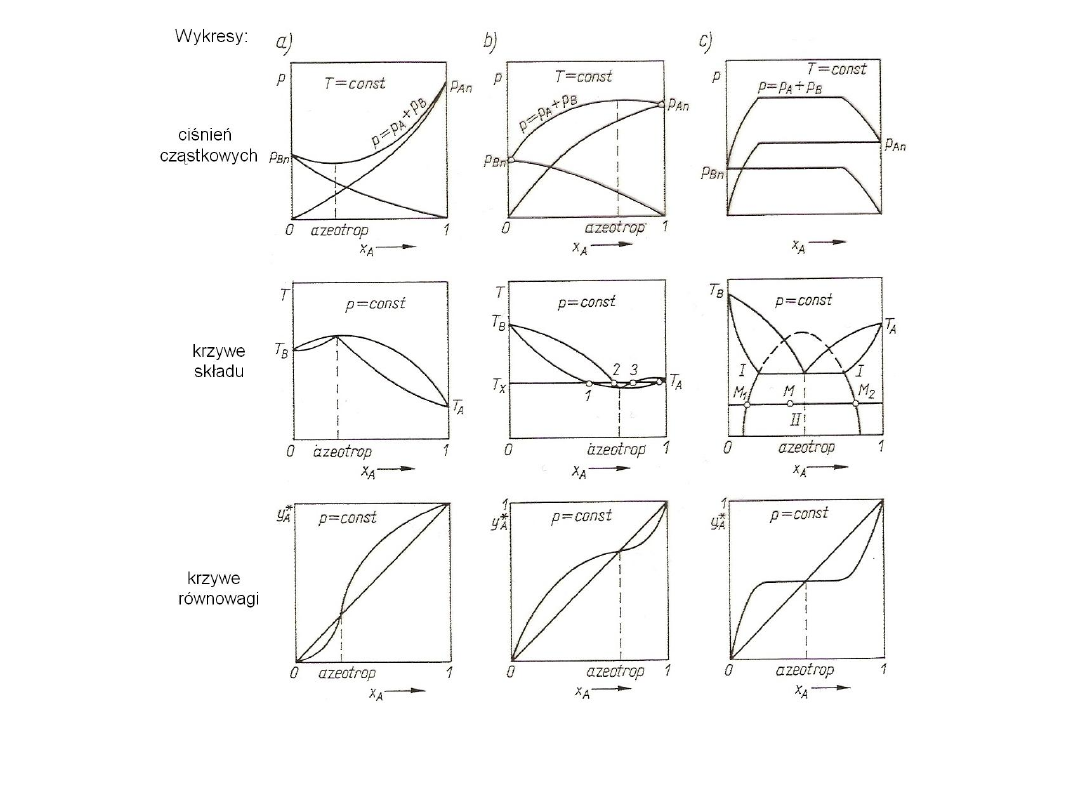
30
Rys. 10. Mieszaniny dwuskładnikowe odchylające się od prawa Raoulta:
a)
A
<1; b)
A
>1; c) mieszaniny o częściowej rozpuszczalności w fazie ciekłej

31
Równowaga międzyfazowa w
układzie ciecz-ciecz
Zagadnienie równowagi w układzie ciecz-ciecz występuje w
procesie ekstrakcji składnika C z cieczy A za pomocą ciekłego
rozpuszczalnika B. Strumień rozpuszczalnika B o dużej zawartości
składnika C jest nazwany ekstraktem, strumień A (zubożały w składnik
C) – rafinatem, a mieszanina składników A + C = F stanowi tzw.
surówkę ekstrakcyjną. W procesie ekstrakcji ekstrakt i rafinat tworzą
dwie fazy ciekłe, które można rozdzielić metodami mechanicznymi.
Składnik C zawarty jest w obydwu fazach. Podobnie jak dla przypadku
układu gaz-ciecz, stężenia składnika C w ekstrakcie i w rafinacie w
stanie równowagi są powiązane zależnością:
X*
CE
= f(X
CR
)
gdzie: X
CR
– stosunek molowy składnika C w rafinacie;
X*
CE
- stosunek molowy składnika C w ekstrakcie (w stanie
równowagi).
Tradycyjnie wprowadza się tutaj wielkość K zwaną
współczynnikiem podziału Nersta:
K = X*
CE
/ X
CR
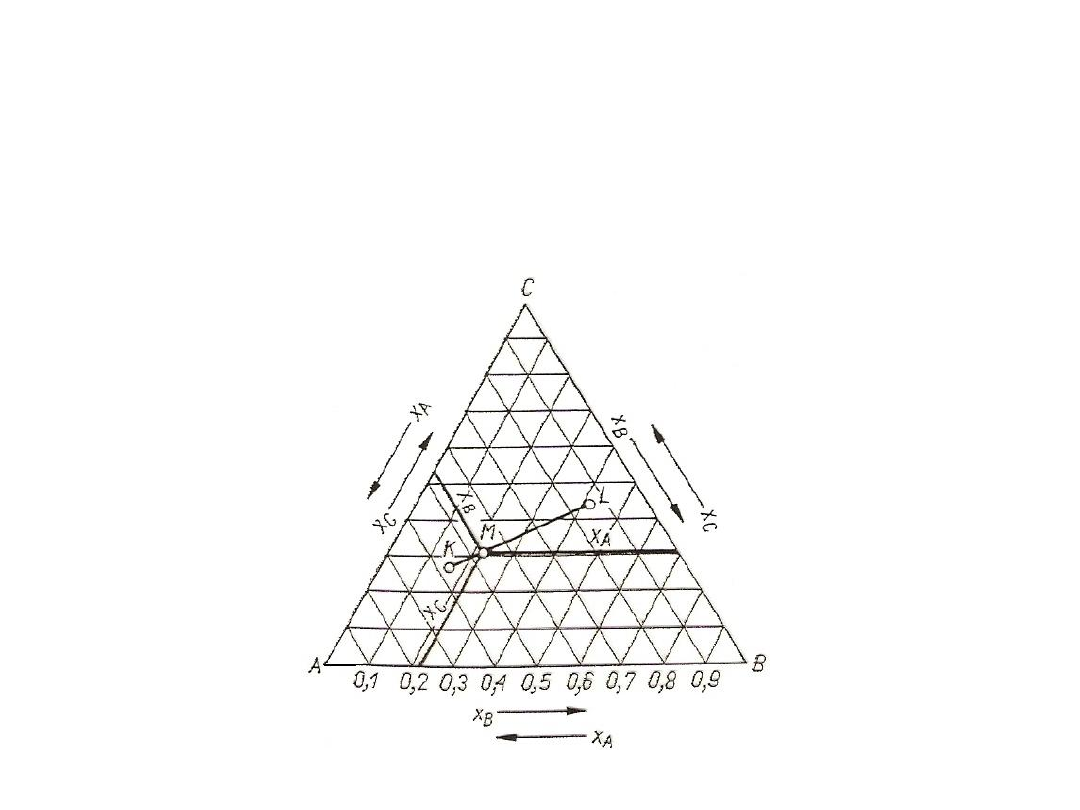
32
Wartość współczynnika K zależy od temperatury, ciśnienia oraz
składu faz pozostających w równowadze. W przypadku, gdy składniki A i
B są absolutnie w sobie nierozpuszczalne, a stężenia składnika C w
ekstrakcie i w rafinacie są niewielkie, współczynnik K jest wielkością
stałą, a zależność ta nosi nazwę prawa podziału Nersta. Jednak w
większości przypadków składniki A i B są częściowo w sobie
rozpuszczalne i sytuacje należy rozważać na wykresie trójkątnym
nazywanym też trójkątem Gibbsa.
Rys. 11. Wykres trójkątny Gibbsa dla mieszanin trójskładnikowych

33
Ilościowe parametry transportu
masy
Podstawową jednostką masy jest kilogram. Bardzo często
używaną jednostką jest kilomol. Masę wyrażoną w kilogramach oznacza
się literą m, a w kilomolach literą n i czasami nazywa się ją ilością
substancji.
Podstawową wielkością kinetyczną charakteryzującą każdy ruch
jest jego prędkość. Prędkość ruchu masy określa się przez strumień
masy lub gęstość strumienia masy.
Strumieniem przepływającej masy (substancji) składnika i,
nazywa się ilość masy (substancji) składnika i, przepływającej w
jednostce czasu przez wybraną w rozważanej przestrzeni powierzchnię
A:
m`
i
= dm
i
/ d n`
i
= dn
i
/ d
Gęstością strumienia nazywa się strumień przepływający przez
jednostkę powierzchni. Strumień jest wielkością charakterystyczną dla
wybranej powierzchni, a gęstość strumienia jest charakterystyczna dla
pewnego punktu leżącego na tej powierzchni:
J
i
= lim
A0
n`
i
/ A=dn`
i
/ dA
gdzie: n`
i
- strumień przepływający przez powierzchnię A.

34
Gęstość strumienia jest w ogólnym przypadku wielkością
wektorową, gdyż zależy od ustawienia powierzchni względem kierunku
ruchu masy. Całkowity strumień składnika i przepływający przez
wybraną powierzchnie A można wyznaczyć z zależności:
n`
i
= ∫
A
J
i
dA
Jeżeli gęstość strumienia J
i
nie zależy od położenia, a powierzchnia A
jest prostopadła do kierunku przepływu substancji, to powyższa
zależność przybiera znacznie prostszą postać:
n`
i
= J
i
A
gdzie: A – powierzchnia, przez którą przepływa substancja.
Transport masy może być ustalony lub nieustalony. Transport
ustalony jest wówczas, gdy wielkość J
i
nie zależy od czasu.
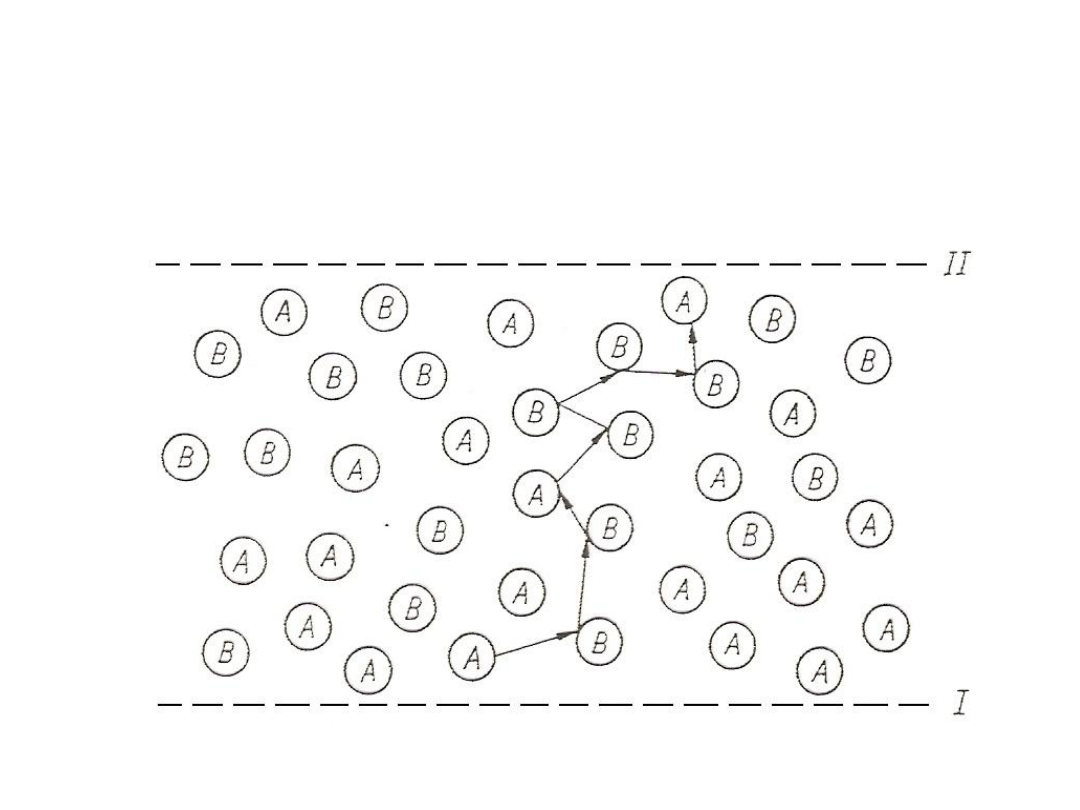
Podstawowe prawa dyfuzji
Wstępny opis dyfuzji
czącząsteczkowej
Rys. 12. Dyfuzja cząsteczkowa

36
I prawo Ficka
Dla rozważanego przypadku układu dwuskładnikowego (A, B)
prawo to ma postać:
J
A
= - D
AB
dc
A
/ dz
gdzie: D
AB
– kinematyczny współczynnik dyfuzji, m
2
/s;
z – odległość mierzona w kierunku ruchu masy, m;
„-” –
znak minus wynika z przepływu masy w
kierunku spadku
stężenia.
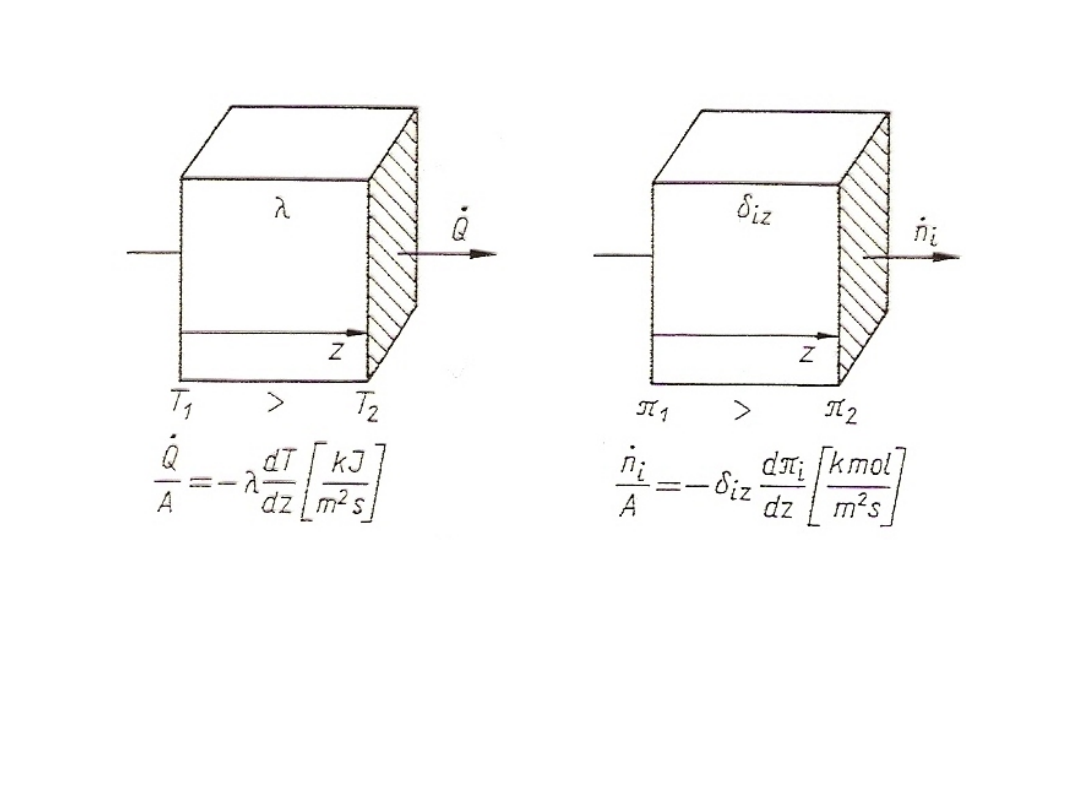
37
Rys. 13. Porównanie dyfuzji substancji z przewodzeniem ciepła
Zjawisko dyfuzji jest analogiczne do zjawiska przewodzenia
ciepła, co przedstawiono poglądowo na rysunku powyżej. Podobieństwo
praw opisujących te zjawiska wynika z tego, że są one wywołane tym
samym mechanizmem, a mianowicie chaotycznym ruchem cząstek.
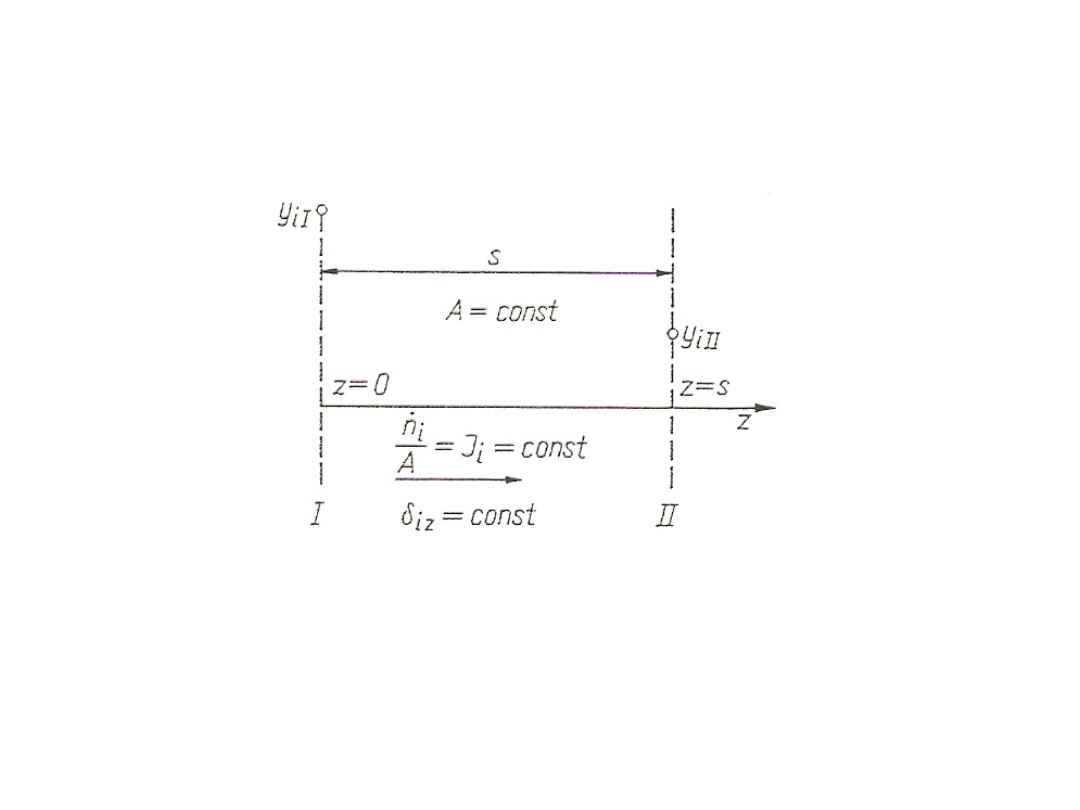
Dyfuzja ustalona
Rozważania ogólne
Rys. 14. Dyfuzja ustalona przez nieruchomą warstwę
Jeżeli zastępczy współczynnik dyfuzji i jest stały od przekroju
I do przekroju II, to:
J
i
=
iz
/ s ∫
yiII
yiI
dy
i
/ y
fi
gdzie: s – odległość przekrojów I i II.

39
Uwzględniając definicje y
fi
:
∫
yiII
yiI
dy
i
/ y
fi
= ∫
yiII
yiI
y
iI
/ (1 +
i
y
i
) = (1 /
i
) ln[(1 +
i
y
iI
) / (1 +
i
y
iII
)]
Ostatecznie otrzymuje się wzór na gęstość strumienia składnika i
dla ustalonej dyfuzji między przekrojami I i II:
J
i
= [
iz
/ (s
i
)] ln(y
fI
/ y
fII
)
gdzie: y
fI
i y
fII
– przeciwstężenia składnika i odpowiednio w przekrojach I i
II.
Znacznie wygodniej jest stosować inną postać wzoru po
wprowadzeniu średniej logarytmicznej:
(y
fi
)
m
= (y
fI
- y
fII
) / ln(y
fI
/ y
fII
)
stąd:
ln(y
fI
/ y
fII
) = (y
fI
- y
fII
) / (y
fi
)
m
ale:
y
fI
- y
fII
= 1 +
i
y
iI
- 1 -
i
y
iII
=
i
(y
iI
- y
iII
)
Po podstawieniu:
J
i
= (
i
/ s) [(y
fI
- y
fII
) / (y
fi
)
m
]
Wzór ten jest najbardziej ogólnym wzorem określającym gęstość
strumienia dyfundującego składnika i w stanie ustalonym.

40
Podstawowe prawa dyfuzji
w fazie gazowej
Na podstawie teorii kinetycznej można dla fazy gazowej
wyprowadzić bardziej ogólne prawo dyfuzji, którego szczególnym
przypadkiem jest prawo Fica. Opierając się na tzw. założeniach
Maxwella, otrzymuje się dla składnika i w układzie k-składnikowym
następujący wzór określający gęstość strumienia masy:
J
i
= -
iz
(d
i
/ dz)
gdzie:
iz
- zastępczy dynamiczny współczynnik dyfuzji składnika i w
układzie
wieloskładnikowym, kmol/(ms);
iz
= (1 +
i
y
i
) / [
(j=1, j≠i)
k
(y
i
/
ij
) - y
i
(j=1, j≠i)
k
(y
ji
/
ij
)]
gdzie:
ij
– binarny dynamiczny współczynnik dyfuzji w układzie
dwuskładnikowym i-j;
ij
= D
ij
c
m
c
m
=
j=1
k
c
j
=
m
/ M
m
gdzie: D
ij
- binarny kinematyczny współczynnik dyfuzji w układzie
dwuskładnikowym i-j, m
2
/s;

41
i
- parametr określający rodzaj dyfuzji:
i
= -
j=1
k
ji
ji
= J
j
/ J
i
(j = 1, 2, 3, …i, …k)
d
i
- różniczkowy moduł napędowy dyfuzji określony za pomocą wzoru:
d
i
= dy
i
/ y
fi
y
fi
- przeciwstężenie składnika i:
y
fi
= 1 +
i
y
i
Według Hoblera wzory te będą słuszne również dla fazy ciekłej.

42
Szczególne przypadki
dyfuzji
Wyprowadzone uprzednio wzory są prawdziwe dla każdego
przypadku dyfuzji. W praktyce bardzo często występują znaczne
uproszczenia.
Dyfuzja ekwimolarna przeciwkierunkowa
Jest to przypadek dyfuzji gdy:
j=1
k
J
j
= 0
Warunek ten oznacza, że dyfuzyjny ruch pewnych składników
w jedną stronę musi być stowarzyszony z dyfuzyjnym ruchem
pozostałych składników w stronę przeciwną. Dla układu
dwuskładnikowego A-B oznacza to, że:
J
A
= - J
B
i
=
j=1
k
J
j
/ J
i
= (1 / J
i
) J
j
= 0
y
fi
= 1
J
i
= (
i
/ s) (y
iI
- y
iII
)
Dla układu dwuskładnikowego A-B, otrzymujemy wyrażenie na
zastępczy współczynnik dyfuzji:
Az
= 1 / {(y
B
/
AB
) – [y
A
( -1 /
AB
)]} =
AB

43
Po podstawieniu otrzymanych wyników do podstawowego
równania różniczkowego dyfuzji otrzymujemy:
J
A
= -
AB
(dy
A
/ dz)
lub:
J
A
= - D
AB
(dc
A
/ dz)
Jest to klasyczne prawo Ficka. Zatem dla dyfuzji ekwimolarnej
przeciwkierunkowej prawo Ficka jest spełnione w sposób ścisły.
Dyfuzja składnika przez jeden lub kilka składników
inertnych
Z definicji przypadek ten występuje wówczas, gdy w układzie
dyfunduje tylko składnik i, pozostałe natomiast nie ulegają dyfuzji i
dlatego nazywa się je składnikami inertnymi (obojętnymi), zatem:
J
i
≠ 0; J
j
= 0 dla
j ≠ i
Po uwzględnieniu tego warunku w definicji parametru
i
otrzymuje się zależność:
i
= - (J
i
/ J
i
) = - 1
y
fi
= 1 - y
i
Wyrażenie określające zastępczy współczynnik dyfuzji przybiera
postać:
iz
= (1 - y
i
) / [
(j=1, j≠i)
k
(y
i
/
ij
)]

44
Dla układu dwuskłanikowego A-B (dyfuzja składnika A przez
inert B):
Az
= (1 – y
A
) / (y
B
/
AB
) =
AB
Wyrażenie określające gęstość strumienia składnika i dla
dyfuzji przez warstwę grubości s przybiera postać:
J
i
= (
iz
/ s) ln[(1 - y
iII
) / (1 – y
iI
)]
lub
J
i
= (
iz
/ s) [(y
iI
- y
iII
) / (1 - y
i
)
m
]
lub
J
i
= (
iz
/ s) [(Y
iI
- Y
iII
) / (1 - Y
i
)
m
]
Wyrażenia (1 - y
i
)
m
i (1 - Y
i
)
m
oznaczają średnie logarytmiczne
odpowiednich wartości w przekrojach I i II.

45
Dyfuzja nieustalona
Bardzo często w rzeczywistych układach strumień
dyfundującej masy zmienia się w czasie. Taki przypadek nazywamy
dyfuzja nieustaloną, Dyfuzję nieustaloną często omawia się przy okazji
różnego rodzaju rozważań teoretycznych. Podstawowym prawem,
opisującym dyfuzje nieustaloną, jest drugie prawo Ficka, które jest
ściśle słuszne dla dyfuzji ekwimolarnej, a w przybliżeniu dla małych
stężeń składnika i. W kartezjańskim układzie współrzędnych prawo to
ma postać równania:
c
i
/ = D
iz
[(
2
c
i
/ x
2
) + (
2
c
i
/ y
2
) + (
2
c
i
/ z
2
)] = D
iz
2
c
i
W bardzo wielu przypadkach dyfuzja zachodzi tylko w jednym
kierunku. Jeżeli tym kierunkiem jest oś z, to drugie prawo Ficka ma
postać najczęściej stosowaną:
c
i
/ = D
iz
(
2
c
i
/ z
2
)
Są to równania różniczkowe cząstkowe II rzędu (typu
parabolicznego).

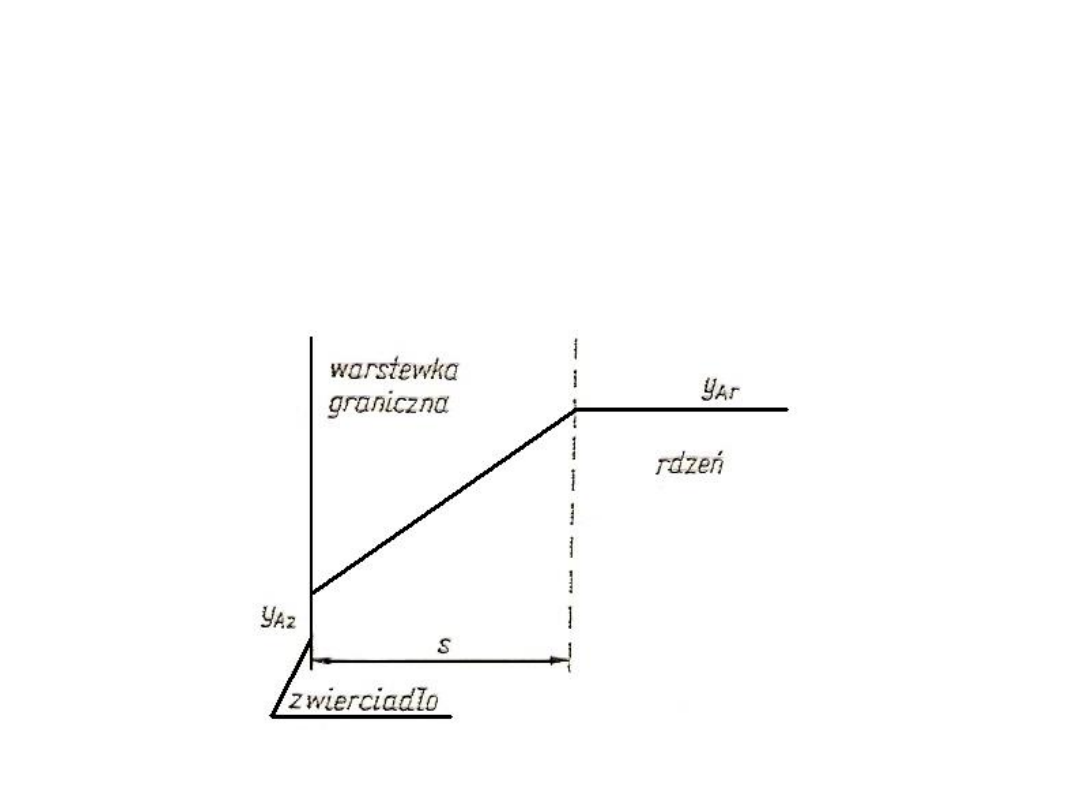
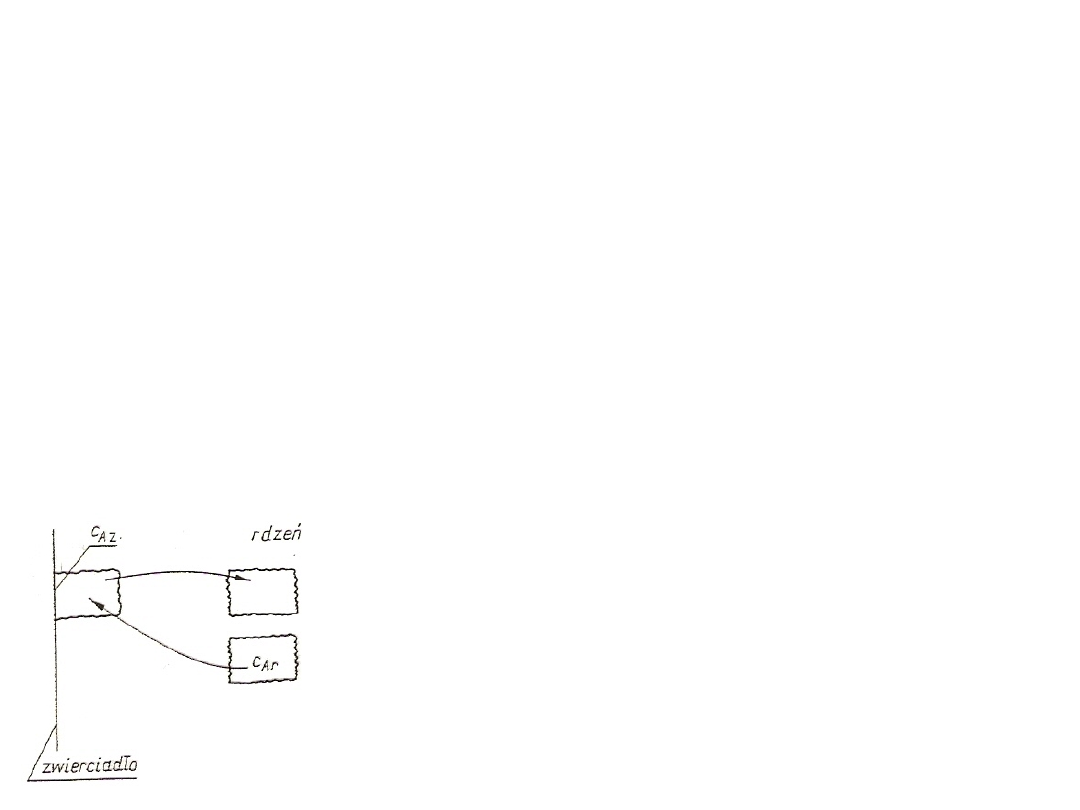
Transport masy przez wnikanie
Ogólna charakterystyka i podział wnikania
masy
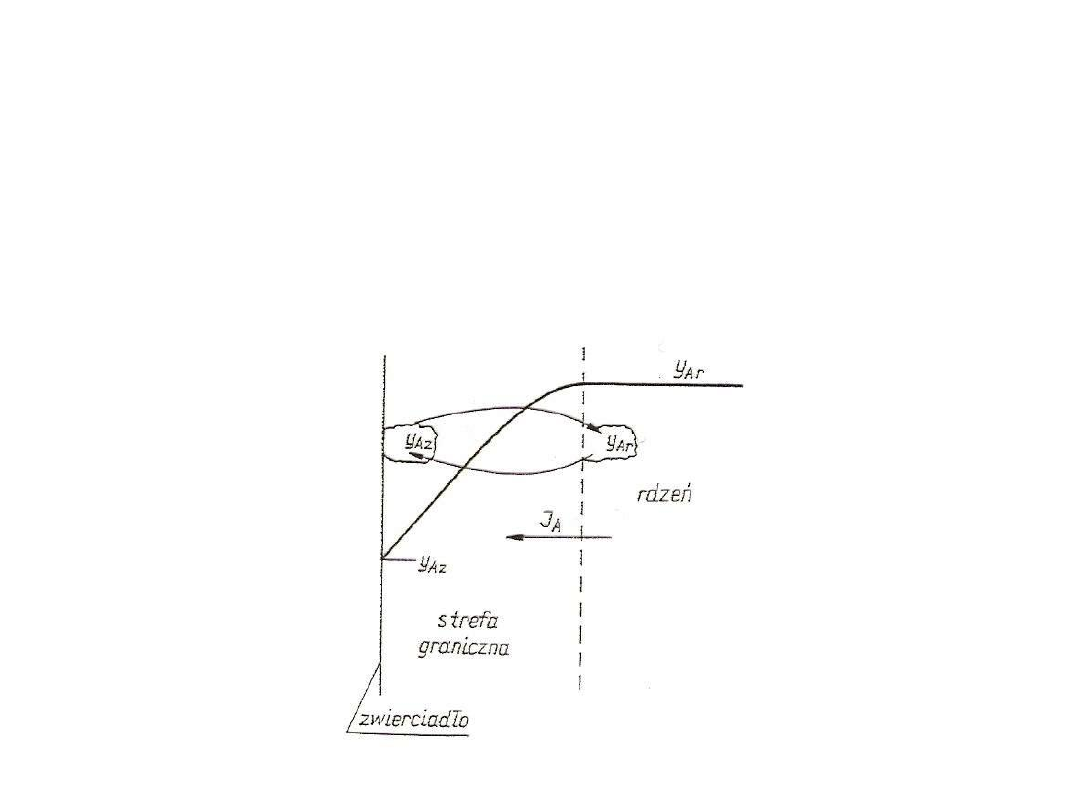
W punkcie tym omówiono ruch jednego wyróżnionego składnika
A w układach jednofazowych wieloskładnikowych. Na wstępie zakłada
się, że w rozważanym układzie można wydzielić rdzeń, w którym
stężenia wszystkich składników są wyrównane, oraz pewną strefę
graniczną zawierającą brzeg układu. Tę część brzegu układu, która
styka się z inną fazą nazywa się powierzchnią międzyfazową lub
zwierciadłem i oznacza indeksem z.
Proces przenoszenia składnika A z rdzenia do zwierciadła lub ze
zwierciadła do rdzenia nazywa się wnikaniem masy. Składa się ono z
dwóch różnych mechanizmów – dyfuzji i konwekcji. Aby proces dyfuzji
był efektywny, konieczna jest różnica stężeń składnika A przy
zwierciadle i rdzeniu układu, czyli c
Az
≠ c
Ar
. Jeżeli c
Az
> c
Ar
, to składnik
jest przenoszony od zwierciadła do rdzenia, a jeżeli c
Az
< c
Ar
, to składnik
jest przenoszony od rdzenia do zwierciadła.
Konwekcja, czyli mechaniczne przenoszenie składnika A wraz z
innymi składnikami bądź do zwierciadła, bądź do rdzenia może
zachodzić nawet przy c
Az
= c
Ar
, jednak wówczas nie będzie ona
efektywna. Konwekcję dzieli się na wymuszoną (odbywa się na wskutek
wymuszonego ruchu w obrębie danej fazy) lub naturalną (ruch w
obrębie fazy wytwarza się samorzutnie, np. wskutek różnic gęstości w
różnych miejscach układu).
Wnikanie masy w fazach gazowej i ciekłej może się odbywać
zarówno poprzez dyfuzję, jak i konwekcję. W fazie stałej natomiast jest
możliwa tylko dyfuzja.
Rys. 15. Schemat procesu wnikania masy

48
Ilościowe określenie wnikania
masy
Ilościowo wnikanie masy składnika A określa się za pomocą
równania definicyjnego:
J
A
=
A
(
A
)
gdzie:
A
-
współczynnik wnikania masy, kmol/(m
2
s);
A
-
moduł napędowy wnikania masy.
Wartość współczynnika wnikania masy określa gęstość
strumienia wnikającej masy składnika A przy module napędowym (
A
)
= 1.
Moduł napędowy wnikania masy jest określony za pomocą
wzoru:
A
= (y
Ar
- y
Az
) / (y
fA
)
m
gdzie: (y
fA
)
m
-
średnia logarytmiczna z przeciwstężeń
składnika A w rdzeniu i
przy zwierciadle.
Dla dyfuzji ekwimolarnej lub dla małych stężeń składnika A
obowiązuje znacznie prostszy wzór:
A
= y
Ar
- y
Az
Dla innych przypadków dyfuzji można znaleźć odpowiednie
wyrażenia w literaturze specjalistycznej.

49
Współczynnik wnikania masy określony za pomocą wzoru
odnosi się do rzeczywistej powierzchni międzyfazowej, którą często jest
rudno wyznaczyć. Dlatego wnikanie masy jest odnoszone bądź do
jakiejś umownej powierzchni A
p
, np. do powierzchni przekroju aparatu,
bądź do jakiejś umownej objętości V
p
, np. do objętości aparatu. W
takich przypadkach rozważa się od razu cały strumień:
n`
A
=
Ap
A
p
A
lub:
n`
A
=
Vp
V
p
A
Współczynniki
Ap
lub
Vp
są ważne tylko dla aparatów
charakteryzujących się podobnymi parametrami geometrycznymi.
Bardzo często, zwłaszcza w ujęciach tradycyjnych, spotyka się
opis, w którym zamiast modułu napędowego
A
używa się różnicy
stężeń. Ponieważ sposobów wyrażania stężeń jest dosyć dużo, więc jest
wiele możliwych kombinacji. Stosunkowo najczęściej używa się do tego
celu koncentracji molowej c
A
. Gęstość strumienia wnikającej masy w
takim ujęciu wyraża się wzorem:
J
A
=
c
c
A
Indeks przy współczynniku oznacza rodzaj stężenia użytego
do określenia siły napędowej (wyrażenia użytego zamiast modułu
napędowego
A
).
50
Wzory określające gęstość strumienia wnikającej masy,
wyrażone za pomocą ułamków molowych lub stosunków molowych:
J
A
=
y
y
A
J
A
=
Y
Y
A
Tab. 6. (skrypt) k =
Faza gazowa
Faza ciekła
Ozn.
dyfuzja
p-kierun.
dyfuzja
jednokierun.
wzór uprosz.
wymiar
Ozn.
dyfuzja
p-kierun.
dyfuzja
jednokierun.
wzór
uprosz.
k
CG
k
G
(k
G
C
G
) /
(C
BG
m)
k
G
kmol A /
[m
2
s (kmol A
/
m
3
)] = m/s
k
CL
k
L
(k
L
C
L
) /
(C
BL
m)
k
L
k
ymol
k
G
C
G
(k
G
C
G
) /
(1-y
mol
)
m
k
G
C
G
kmol A /
[m
2
s (kmol A
/
kmol A+B)]
k
xmol
k
L
C
L
(k
L
C
L
) /
(1-x
mol
)
m
k
L
C
L
k
p
k
G
/
RT
(k
G
C
G
) /
(p
B
m)
k
G
/
RT
kmol A /
(m
2
s Pa)
-
-
-
-
k
Ymol
(k
G
C
G
) /
[(1+Y
mol
)
(1+Y
mol z
)]
(k
G
C
G
) /
(1+Y
mol
)
m
k
G
C
G
kmol A /
[m
2
s (kmol A
/
kmol B)]
k
Xmol
(k
L
C
L
) /
[(1+X
mol
)
(1+X
mol z
)]
(k
L
C
L
) /
(1+X
mol
)
m
k
L
C
L
k
Y
(k
G
C
G
m*) /
[(m*+Y)
(m*+Y
z
)]
(k
G
C
G
) /
(m*+Y)
m
(k
G
C
G
) /
m*
kmol A /
[m
2
s (kg A /
kg B)]
k
X
(k
L
C
L
m*) /
[(m*+X)
(m*+X
z
)]
(k
l
C
L
) /
(m*+X)
m
(k
L
C
L
) /
m*

51
Aby dokładnie scharakteryzować proces wnikania masy,
należy przede wszystkim zbadać zależności, jakim podlega
współczynnik wnikania masy .
- zależy od właściwości dyfuzyjnych układu, jak też od
warunków
hydrodynamicznych panujących w układzie.
Istnieje kilka teoretycznych modeli próbujących
matematycznie opisać proces wnikania masy.
We wszystkich rozważaniach zakłada się dyfuzję ekwimolarną
przeciwkierunkową lub małe stężenie składnika wnikającego oraz, że
słuszne jest zarówno pierwsze, jak i drugie prawo Ficka.
Należy zauważyć, że w tym ujęciu współczynnik wnikania
zależy od rodzaju użytego stężenia i od rodzaju dyfuzji. Obydwie te
zależności nie będą występować jeżeli użyje się modułu napędowego
A
. Oto zależność między współczynnikami
A
i
c
:
A
= (y
f
)
m
c
m
c
52
Teoretyczne modele wnikania
masy
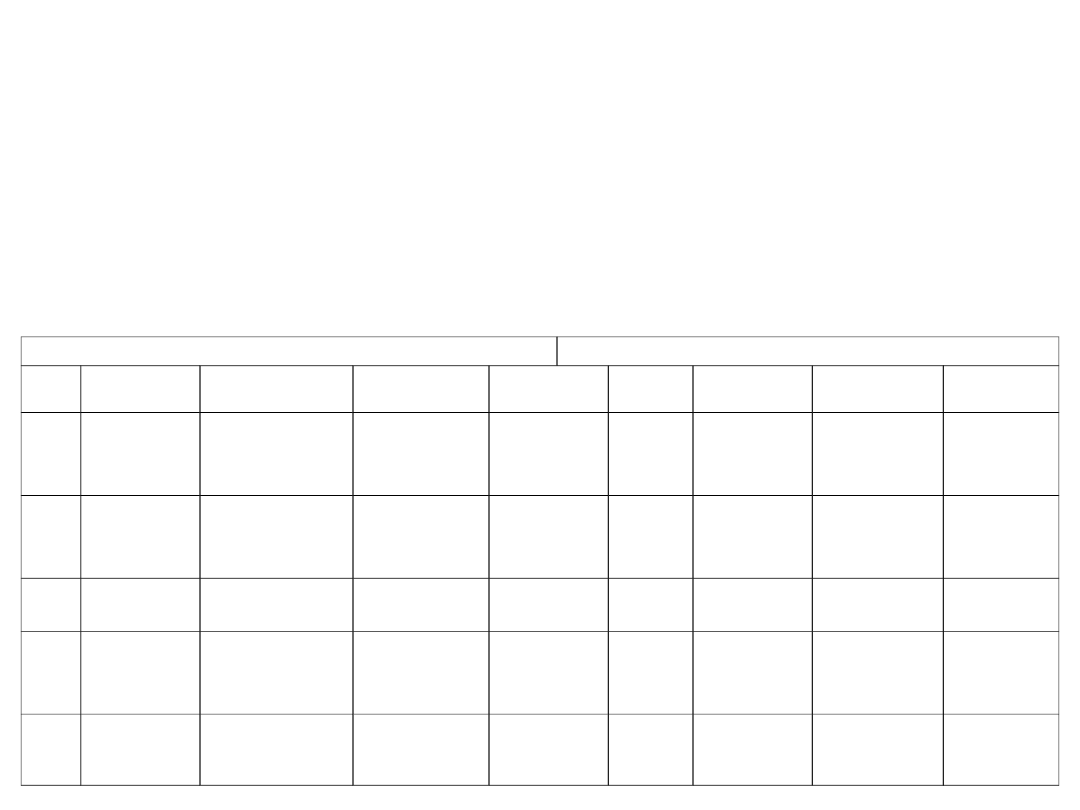
Model warstewki granicznej Lewisa-Whitmana
Model ten zakłada, że rozważany układ składa się z rdzenia, w
którym stężenia wszystkich składników są wyrównane oraz z
nieruchomej warstewki granicznej. W rdzeniu zachodzi czysta
konwekcja, a w warstewce granicznej czysta dyfuzja.
Rys. 16. Rozkład stężeń składnika A wg modelu warstwy granicznej

53
Rozkład stężeń wnikającego składnika A przedstawiono na rys.
16. Ponieważ w rdzeniu stężenia są wyrównane, efektywny ruch masy
zachodzi tylko w warstewce granicznej. Gęstość strumienia wnikającej
masy:
J
A
= (
Az
/ s) ( y
Ar
– y
Az
)
Wartość J
A
można również wyrazić wzorem określającym wnikanie
masy:
J
A
=
A
( y
Ar
– y
Az
)
Z porównania tych wzorów wynika:
A
= (
Az
/ s) = (D
A
c
m
) / s
Do obliczenia współczynnika
A
konieczna jest znajomość
grubości warstewki granicznej s.
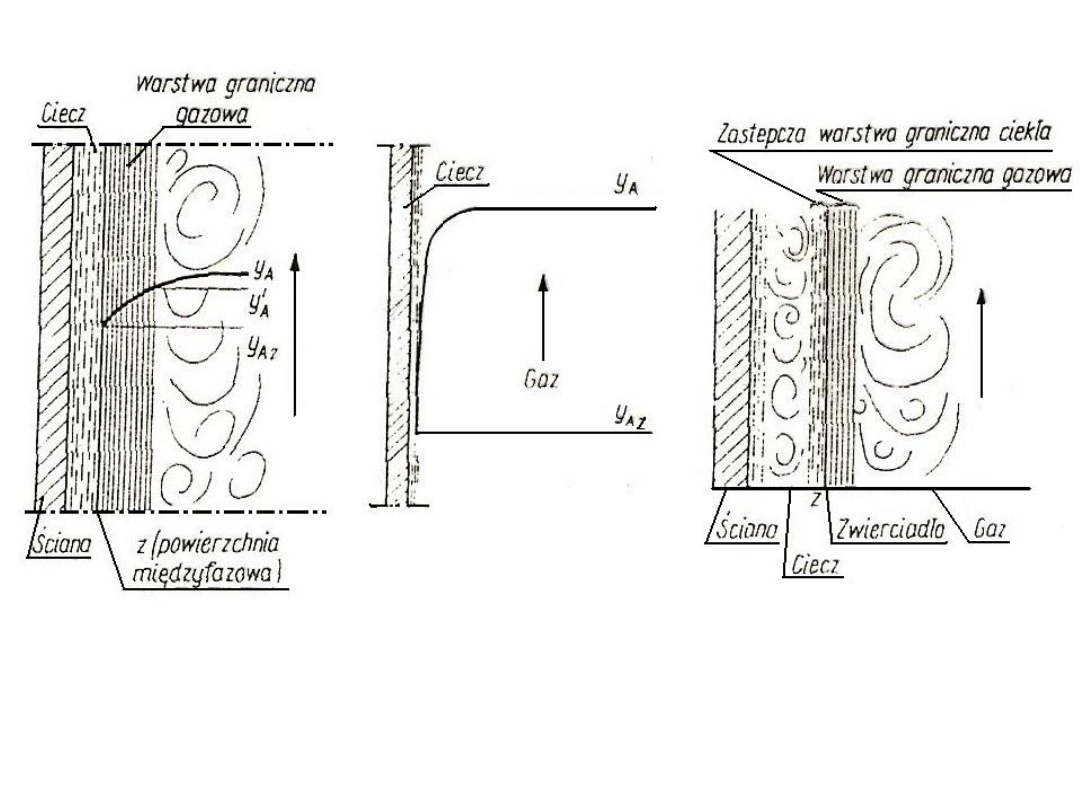
54
Rys. 17. Przykładowy
model wnikania masy
w fazie gazowej,
oznaczony przy
pomocy stężeń y
A
Rys. 18. Rozkład
stężeń przy wnikaniu
masy w fazie gazowej
Rys. 19. model ilustrujący
koncepcję dwóch warstw
granicznych
55
Model penetracji Hibgiego
Model ten został w zasadzie opracowany dla fazy ciekłej, jednakże
można za jego pomocą opisywać również wnikanie w fazie gazowej.
Założenia są następujące:
1. Stężenie składnika A na powierzchni międzyfazowej jest stałe i
wynosi c
Az
.
2. Stężenie składnika A w rdzeniu fazy jest wyrównane i wynosi c
Ar
.
3. Na powierzchni międzyfazowej znajdują się nieruchome elementy tej
fazy. Elementy te przebywają na powierzchni przez czas , a
następnie wskutek ruchu burzliwego zostają przenoszone do rdzenia.
Na powstałe miejsce przedostaje się inny element z rdzenia.
4. Długość elementu jest nieskończona. Stężenie składnika A w dużej
odległości od zwierciadła jest równe jego stężeniu w rdzeniu, czyli:
lim
z ∞
c
A
= c
Ar
Rys. 20. Schemat penetracyjnego wnikania
masy

56
Podczas przebywania elementu przy powierzchni odbywa się
nieustalona dyfuzja składnika A. Strumień wnikającego składnika zmienia
się w czasie. Średnia wartość tego strumienia jest równa średniej
całkowej z zawartości chwilowych względem czasu.
Na podstawie powyższych założeń przez rozwiązanie równania
dyfuzji nieustalonej uzyskuje się wzór teoretyczny na obliczanie
współczynnika wnikania masy przy znanej wartości czasu przebywania
elementów na powierzchni :
A
= 2 [D
Az
/ ()]
1/2
c
m
Model odnawiania powierzchni Danckwertsa
Jest to pewna modyfikacja modelu Higbiego. Wszystkie założenia
są w nim takie same, jak w modelu penetracji. Jedyna różnicą jest
założenie, że czas przebywania elementów na powierzchni jest losowy
(przypadkowy), natomiast stały jest parametr rozkładu czasów
przebywania p
s
zwany szybkością odnawiania powierzchni. Wynikiem
końcowym modelu jest wzór:
A
= [D
Az
p
s
]
1/2
c
m
Praktyczne zastosowanie wzoru wymaga znajomości parametru
p
s
.
Badania doświadczalne wykazują, że w układach o przepływie
zbliżonym do laminarnego wnikanie masy jest lepiej opisane przez teorię
warstewkową. W układach o większej burzliwości przepływu, bardziej
adekwatne są modele penetracyjne.

57
Sposoby wyznaczania
współczynników wnikania masy
Znajomość współczynników wnikania masy jest nieodzowna w
obliczeniach wymienników masy. Współczynniki wnikania masy mogą
być wyznaczone w sposób teoretyczny, półempiryczny i czysto
doświadczalny. Końcowe wzory określające współczynniki wnikania masy
są najczęściej przedstawiane za pomocą liczb bezwymiarowych.
Przedstawianie takie daje pewne uogólnienie wynikające z teorii
podobieństwa. najczęsciej stosuje się następujące liczby bezwymiarowe:
Liczba Sherwooda – Sh:
Sh = (
A
L) /
Az
≈ (
C
L) / D
Az
„L” oznacza tutaj wymiar liniowy. W każdym przypadku wymiar ten
powinien być ściśle zdefiniowany.
Liczba Reynoldsa – Re:
Re = (w L ) /
Zarówno prędkość w, jak i wymiar liniowy L są wielkościami
umownymi i powinny być ściśle zdefiniowane w każdym przypadku.
Liczba Reynoldsa charakteryzuje w pewnym sensie warunki
hydrodynamiczne panujące w układzie. Można przyjąć, że większej
wartości Re odpowiada większa burzliwość.

58
Liczba Schmidta – Sc:
Sc = v / D
Az
= / (M
m
Az
)
Liczba Schmidta jest zależna tylko od właściwości
fizykochemicznych układu. może być zależna od temperatury poprzez
lepkość i współczynnik dyfuzji.
Liczba Fouriera – Fo:
Fo = (D
A
) / L
2
Wielkość oznacza tutaj czas trwania procesu. Liczba Fouriera
charakteryzuje niestacjonarność procesu.
Liczba Pecleta – Pe:
Pe = (w L) / D
A
= Re · Sc
Wzory określające wnikanie masy bardzo często doprowadza się do
postaci:
Sh = f(Re, Sc, Fo, Pe, …)
Funkcja f najczęściej ma postać potęgową. Szczególnie postacie
wzorów dla różnych procesów i aparatów można znaleźć w literaturze
fachowej i naukowej.

59
Współczynnik dyfuzji
W poprzednich rozważaniach wprowadzono dwa współczynniki
dyfuzji: kinematyczny D
ij
[m
2
/s] i dynamiczny
ij
[kmol/(m s)].
Współczynniki te są związane równaniem:
ij
= D
ij
c
m
dla fazy ciekłej:
c
m
=
m
/ M
m
w którym c
m
oznacza całkowitą koncentrację.
Wielkość tę dla fazy gazowej pod umiarkowanym ciśnieniem
można obliczyć ze wzoru:
c
m
= p / (R T)
Współczynniki D
ij
i D
ji
oraz
ij
i
ji
nazywa się binarnymi
współczynnikami dyfuzji. Wychodząc z założenia Maxwella można
wykazać, że dla układów dwuskładnikowych:
D
AB
= D
BA
Binarne współczynniki dyfuzji są zatem sobie równe. Zależność
ta jest ścisła dla dwuskładnikowych układów gazowych.

60
Tab. 7.

61
Jeżeli całkowanie podstawowego równania różniczkowego
przeprowadzi się w granicach od y
iI
do pewnego y
i
, to otrzyma się
rozkład stężeń składnika i wzdłuż drogi dla tego przypadku:
y
i
= 1 – (1 – y
iII
)
z/s
(1 – y
iI
)
1-(z/s)
Przypadek dyfuzji jednego składnika przez inerty występuje
często w praktyce w procesach absorpcji lub desorpcji. Pewne
zastrzeżenie może tutaj budzić założenie, że inertne składniki nie
dyfundują. Przecież również dla nich występują gradienty stężeń w
kierunku przeciwnym, a więc powinny one dyfundować w tym kierunku.
W rzeczywistych układach absorpcyjnych i desorbcyjnych dyfuzja
składników inertnych jednak nie występuje. Dzieje się tak dlatego, że
opór w drugiej fazie jest tak duży dla tych składników, że nie mogą one
przepływać z jednej fazy do drugiej. A zatem można przyjąć, że w
warunkach ustalonych nie przepływają one (nie dyfundują) również w
obrębie jednej fazy.
W celu otrzymania rozkładu stężenia składnika i na drodze
dyfuzji całkuje się równanie w granicach od y
iI
do pewnego y
i
:
y
i
= y
iI
– (z / s) (y
iI
– y
iII
)
Jest to rozkład liniowy, przedstawiony graficznie na rys. 21.
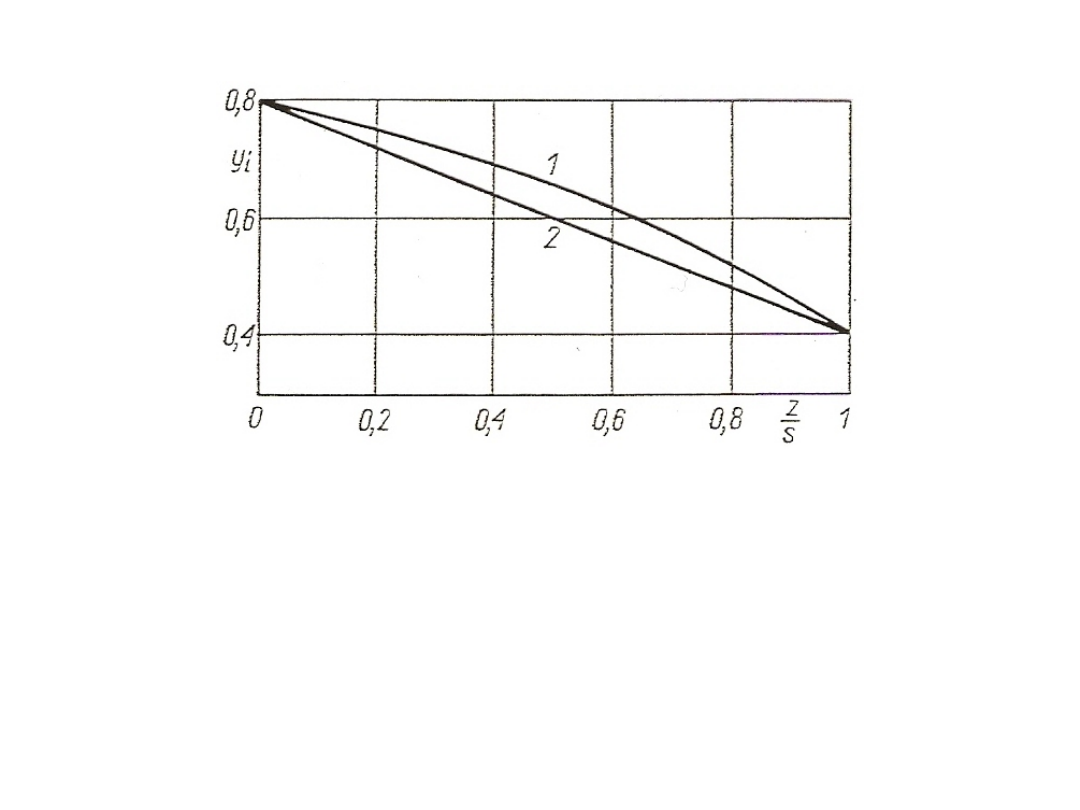
62
Rys. 21. Rozkład stężeń: 1 – podczas dyfuzji przez inert;
2 – podczas dyfuzji ekwimolarnej przeciwkierunkowej
Przypadek dyfuzji ekwimolarnej występuje przede wszystkim
w procesach rektyfikacji. Otrzymane wzory można stosować również
w innych przypadkach, gdy stężenie rozważanego składnika i jest
małe. Wtedy y
i
<< 1 y
fi
≈ 1.

63
Przenikanie masy (dwie fazy) k-składników założenia:
- rozważać będziemy ruch wyróżnionego składnika A. Pozostałe
składniki mogą być obojętne lub również mogą się poruszać;
- każda faza składa się z rdzenia o wyrównanych stężeniach wszystkich
składników oraz ze strefy granicznej;
- strefy graniczne obydwu faz stykają się ze sobą wzdłuż powierzchni
międzyfazowej, którą nazywa się zwierciadłem.
Proces polegający na przenoszeniu składnika A z rdzenia jednej
fazy do rdzeni drugiej fazy nazywa się przenikaniem masy składnika
A:
Proces przenikania masy można podzielić na:
1. Proces wnikania masy od rdzenia fazy I do zwierciadła.
2. Proces przechodzenia masy przez zwierciadło.
3. Proces wnikania masy od zwierciadła do fazy II.
x – ułamki molowe w fazie I;
y - ułamki molowe w fazie II;
Ilościowo przenikanie masy jest dla danego układu określone
przez strumień składnika A przechodzący z fazy do fazy.
Podstawowym zagadnieniem w opisie przenikania masy jest więc
określenie gęstości strumienia J
A
. Rozważamy proces ustalony.

Gęstość strumienia przenikającej masy.
Współczynnik przenikania masy
W celu wyprowadzenia ogólnego wzoru określającego
przenikanie masy trzeba przyjąć następujące założenia:
1. Znane są współczynniki wnikania masy w obu fazach
AI
i
AII
.
2. Znana jest linia równowagi y*
A
= f(x
A
).
3. Na powierzchni międzyfazowej panuje równowaga międzyfazowa
składnika A:
y
Az
= y*(x
Az
)
Założenie to jest jednoznaczne ze stwierdzeniem, że powierzchnia
międzyfazowa nie stawia żadnego oporu dla ruchu składnika A.
4. Rodzaj dyfuzji w obu fazach jest taki sam, tzn. współczynnik ε
charakteryzujący dyfuzję jest jednakowy w obydwu fazach. Założenie
to jest słuszne dla ustalonego wnikania masy bez przebiegu reakcji
chemicznej. w przypadku wystąpienia reakcji chemicznej założenie
to może nie być słuszne.
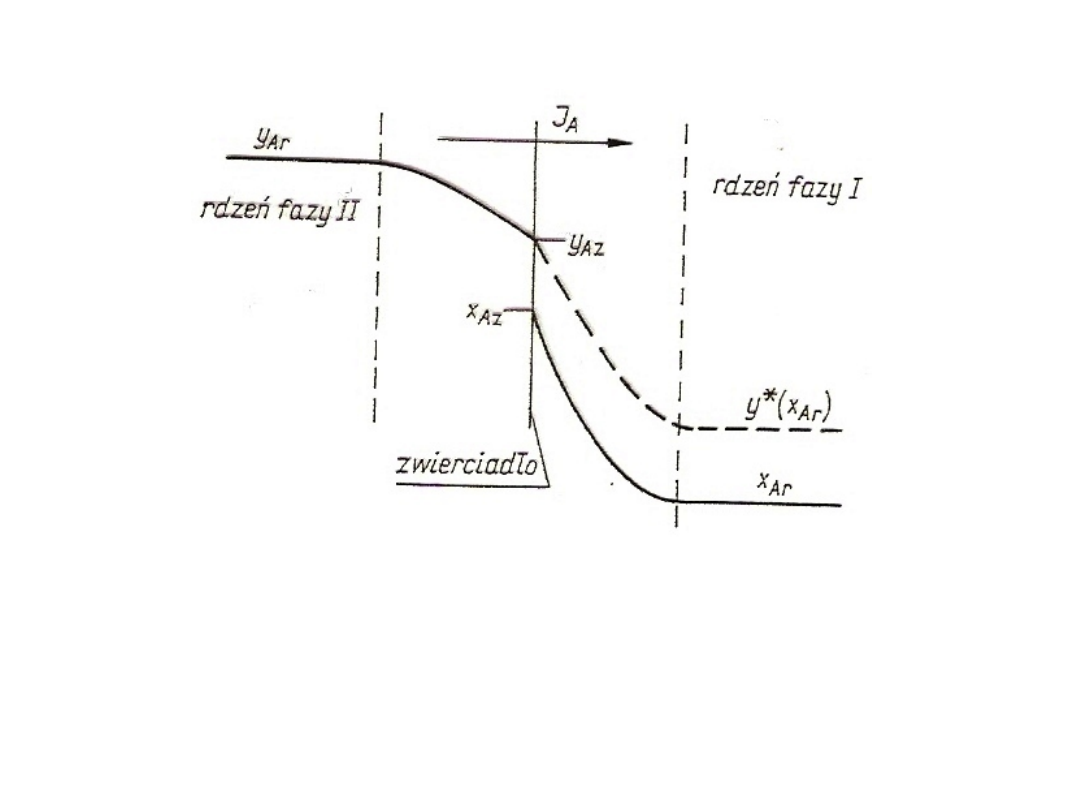
65
Rys. 22. Przykład rozkładu stężeń składnika A podczas przenikania masy
Ponieważ przenikanie jest ustalone, całkowita gęstość
strumienia masy przenikającej J
A
musi być równa gęstości strumienia
masy składnika A wnikającej od rdzenia fazy II do zwierciadła, jak
również gęstość strumienia masy składnika A wnikającej od
zwierciadła do rdzenia fazy I.

66
Gęstość strumienia masy przy wnikaniu można opisać
podstawowymi równaniami wnikania masy:
J
A
=
AII
(
A
)
II
oraz:
J
A
=
AI
(
A
)
I
gdzie: (
A
)
II
- moduł napędowy wnikania masy w fazie II;
(
A
)
I
-
moduł napędowy wnikania masy w fazie I;
Wielkości te można rozpisać:
(
A
)
I
= (y
Ar
– y
Az
) / (y
f
)
m
= (1 /
A
) ln[(1 +
A
y
Ar
) / (1 +
A
y
Az
)]
(
A
)
II
=(x
Ar
– x
Az
) / (x
f
)
m
= (1 /
A
) ln[(1 +
A
x
Ar
) / (1 +
A
x
Az
)]
zatem:
J
A
= (
AII
/
A
) ln[(1 +
A
y
Ar
) / (1 +
A
y
Az
)]
J
A
= (
AI
/
A
) ln[(1 +
A
x
Ar
) / (1 +
A
x
Az
)]
Następnie wzory te dzieli się stronami odpowiednio przez
AII
i
AI
:
J
A
/
AII
= (1 /
A
) ln[(1 +
A
y
Ar
) / (1 +
A
y
Az
)] = (1 /
A
) [ln(1 +
A
y
Ar
) - ln(1 +
A
y
Az
)]
J
A
/
AI
= (1 /
A
) ln[(1 +
A
x
Ar
) / (1 +
A
x
Az
)] = (1 /
A
) [ln(1 +
A
x
Ar
) - ln(1 +
A
x
Az
)]
J
A
=
AII
(
A
)
II
=
AII
(y
Ar
– y
Az
)
J
A
=
AI
(
A
)
I
=
AI
(x
Ar
– x
Az
)

67
Po podzieleniu powyższych równań stronami oraz odpowiednim
przekształceniu otrzymuje się równanie:
y
Az
= - (
AI
/
AII
) (x
Az
– x
Ar
) + y
Ar
Ze względu na x
Az
i y
Az
jet to równanie prostej przechodzącej
przez punkt (x
Ar
, y
Ar
) i mającej nachylenie (-
AI
,
AII
).
Na powierzchni międzyfazowej panuje równowaga, co oznacza,
że:
y
Az
= y*(x
Az
)
Zatem:
y*(x
Az
) = (-
AI
/
AII
) (x
Az
– x
Ar
) + y
Ar
Punkt, którego współrzędne określają stężenia x
Az
i y
Az
, leży
zatem na przecięciu prostej z linią równowagi.
Można też pokazać graficzną interpretacje zamiennika n dla
przenikania z dyfuzją ekwimolarną przeciwkierunkową lub przy małych
stężeniach składnika A. Ze wzoru definicyjnego można otrzymać:
n = [y*(x
Az
) - y*(x
Ar
)] / (x
Az
– x
Ar
)
Jak wynika z powyższego wzoru, zamiennik n dla tego
przypadku jest równy nachyleniu linii prostej przechodzącej przez
punkty B i C.

68
Następnie w celu wyeliminowania stężeń przy zwierciadle
wprowadza się wielkość n określoną wzorem:
n = {ln[1 +
A
y*(x
Az
)] – ln[1 +
A
y*(x
Ar
)]} / [ln(1 +
A
x
Az
) – ln(1 +
A
x
Ar
)]
gdzie: - równowagowe stężenie składnika A do jego stężenia w rdzeniu
fazy I.
Obie strony równania mnoży się przez n:
(n J
A
) /
AJ
= (1 /
A
) {ln(1 +
A
y
Ar
) - ln[1 +
A
y*(x
Az
)]}
Następnie dodaje się stronami równania:
J
A
[(1 /
AII
) + (n /
AI
)] = (1 /
A
) {ln(1 +
A
y
Ar
) - ln[1 +
A
y*(x
Az
)]}
Prawą stronę powyższej równości oznacza się przez (
A
)
0II
i
nazywa ogólnym modułem napędowym przenikania masy:
(
A
)
0II
=(1 /
A
) {ln(1 +
A
y
Ar
) - ln[1 +
A
y*(x
Az
)]}
Wyrażenie określające można przedstawić w prostszej postaci,
wprowadzając średnią logarytmiczną. Można bardzo łatwo wykazać, że:
(
A
)
0II
= [y
Ar
- y*(x
Az
)] / (y
f
)
m
gdzie: (y
f
)
m
- średnia logarytmiczna z wyrażeń (1 +
A
y
Ar
) i [1 +
A
y*(x
Ar
)].
Moduł napędowy (
A
)
0II
można obliczyć znając stężenie
składnika A w rdzeniu obu faz.
J
A
= (
A
)
0II
/ [(1 /
AII
) + (n /
AI
)]

69
Bezpośrednie korzystanie ze wzoru definicyjnego, jest niemożliwe z
powodu nieznajomości wartości stężeń przy powierzchni międzyfazowej.
Dlatego trzeba go tak przekształcać, aby otrzymać wyrażenie nadające się do
bezpośredniego zastosowania:
n = {ln[1 +
A
y*(x
Ar
)]} / ln(1 +
A
y
Ar
)
Jeżeli występujące w tym wzorze przyrosty skończone zastąpi się
przyrostami różniczkowymi, to popełniony błąd nie będzie duży. Zatem:
n = {dln[1 +
A
y*(x
Ar
)]} / dln(1 +
A
y
Ar
) = { (1 +
A
x
Ar
) / [1 +
A
y*(x
Ar
) ] } (dy*
/ dx)
x=xAr
Wyrażenie (dy* / dx) określa nachylenie linii równowagi w układzie x-
y, które oznacza się symbolem m
y
:
(dy* / dx)
x=xAr
= m
y
Wyrażenie określające zamiennik n otrzymuje zatem postać:
n = m
y
{(1 +
A
x
Ar
) / [1 +
A
y*(x
Ar
)]}
Znając wartość m
y
,
A
, x
Ar
i y*(x
Ar
) można obliczyć wartość zamiennika
n, a następnie wartość współczynnika przenikania masy k
A
.
Po wprowadzeniu współczynnika przenikania masy k
AII
otrzymuje się
podstawowe równanie przenikania masy:
J
A
= k
AII
(
A
)
0II
Równanie to jest prawdziwe w zastosowaniu do ustalonego przenikania, dla
różnych rodzajów dyfuzji, pod warunkiem, że w obydwu fazach zachodzi ten
sam rodzaj dyfuzji.

70
Jeżeli w obydwu fazach występuje dyfuzja ekwimolarna
przeciwkierunkowa lub stężenia składnika A są małe, powyższe wzory
znacznie się upraszczają. Ponieważ
A
= 0 i y
f
= 1, więc:
(
A
)
0II
= y
Ar
- y*(x
Ar
)
oraz:
n = m
y
Przypadki dyfuzji ekwimolarnej przeciwkierunkowej oraz dyfuzji w
małych stężeniach są bardzo częste, dlatego dokładnie rozważono
przenikanie masy w tych warunkach.
J
A
= (
A
)
0II
/( 1 /
AII
) + (n /
AI
)
Wzór ten jest nieco podobny do wzoru Ohma znanego z elektroniki.
Gęstość strumienia odpowiada natężeniu prądu, a moduł napędowy
odpowiada napięciu. Wobec tego wyrażenie w mianowniku powinno
odpowiadać oporowi przewodnika. Zgodnie z tą analogią mianownik
wyrażenia nazywa się oporem przenikania masy, a odwrotność oporu
przenikania nazywa się współczynnikiem przenikania masy i oznacza literką
k
A
. Zgodnie z tym:
1 / k
AII
= (1 /
AII
) + (n /
AI
)
Indeks II przy współczynniku k
A
oraz przy ogólnym module
napędowym (
A
)
0
oznacza, że za punkt odniesienia jest przyjęta faza II.
Moduł napędowy jest wyrażony za pomocą stężeń właściwych dla tej fazy.
W rozważanym przypadku stężeniami tymi są ułamki molowe y
A
.
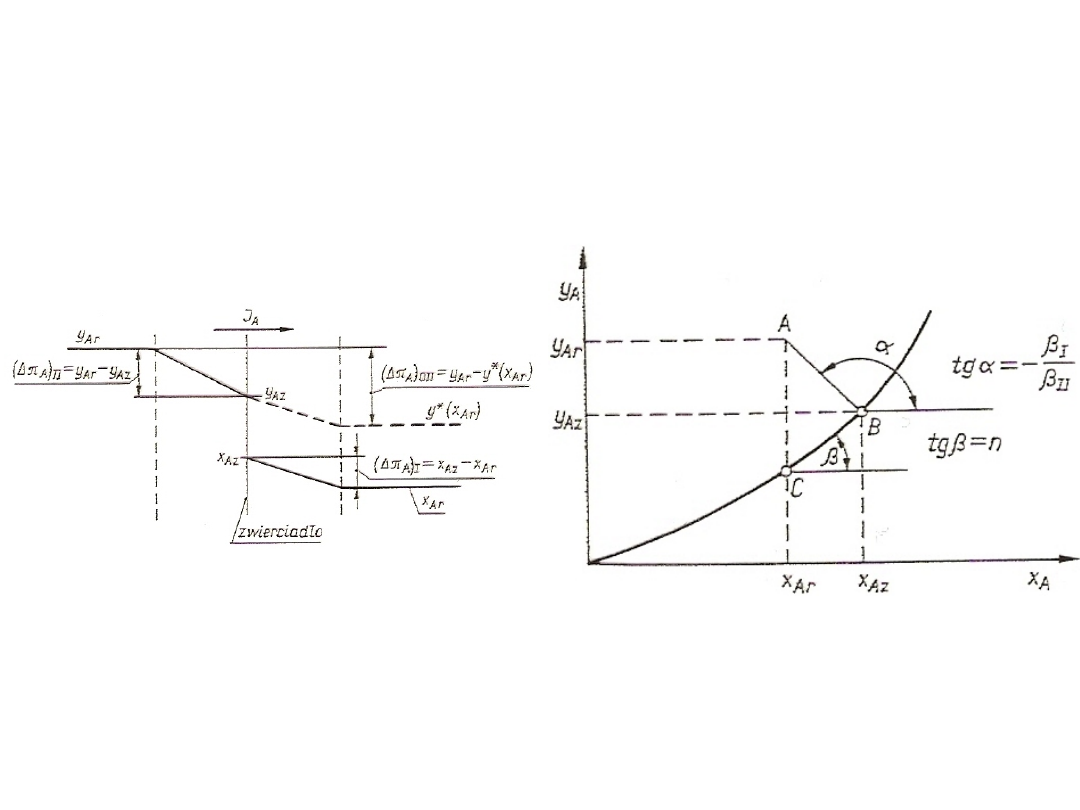
71
Do obliczenia wartości współczynnika przenikania masy,
potrzebne są wartości współczynników wnikania
AII
i
AI
oraz wielkość
n, która jest nazywana zamiennikiem stężeń.
Rys. 23. Rozkład stężeń przy
przenikaniu składnika A w przypadku
dyfuzji ekwimolarnej
przeciwprądowej
Rys. 24. Wykreślna ilustracja
przenikania masy w układzie stężeń
x
A
-y
A
w przypadku dyfuzji
ekwimolarnej przeciwprądowej

72
Analogie
Wartości współczynników wnikania masy potrzebne do obliczeń
projektowych otrzymuje się najczęściej z badań eksperymentalnych.
Dzięki możliwości porównywania różnych układów wyniki uzyskane dla
jednego układu można przenieść na inny i w ten sposób częściowo
zaoszczędzić czas i koszty eksperymentu.
Jest to możliwe wówczas, gdy zachodzi analogia
MATEMATYCZNA różne pod względem fizycznym procesy stosują się
do tych samych praw matematycznych.
Analogia taka zachodzi w przypadku transportu pędu, ciepła i
masy. Dzięki niej, ze znanych np. współczynników wnikania ciepła,
można wnioskować o wartości współczynników wnikania masy i
odwrotnie.
Należy jednak pamiętać, że tylko w niektórych przypadkach ta
analogia jest ścisła, jest to w większości przypadków przybliżenie.
które umożliwia właściwe ukierunkowanie badań teoretycznych lub
eksperymentalnych w celu znalezienia właściwych zależności.

73
Analogie między transportem
pędu, ciepła i masy
Podstawą analogi transportu pędu, ciepła i masy jest
podobieństwo podstawowych praw opisujących te procesy, tzn. prawa
Newtona, Fouriera i Ficka, które dla jednego kierunku z mają postać:
F
S
/ (A ) = - (dw / dz) (prawo Newtona)
Q* / (A c
P
) = - a (dT / dz)
(prawo Fouriera)
n`
A
/ A = - D
Az
(dc
A
/ dz) (prawo Ficka)
gdzie: , a i D
Az
- podstawowe współczynniki kinetyczne, m
2
/s;
Q*
- strumień ciepła, W;
n`
A
- strumień masy, kmol/s;
F
s
- siła naprężeń statycznych odpowiadająca
strumieniowi pędu, N.

74
W rozważaniach analogi często wykorzystuje się analizę
wymiarową, w której operuje się liczbami bezwymiarowymi zwanymi
liczbami podobieństwa. Definicje podstawowych liczb bezwymiarowych
wynikają ze stosunków współczynników , a i D
Az
w powyższych
wzorach – są to odpowiednio liczby Prandtla – Pr, Schmidta – Sc i
Lewisa – Le:
Pr = / a = (c
P
) / = (c
P
) /
Sc = / D
Az
= ( c
m
) /
Az
= / (M
m
Az
)
Le = a / D
Az
= / (c
P
Az
M
m
) = Sc / Pr
W teorii analogi transportu masy i pędu często stosuje się
Liczbę Stantona St:
St = Sh / (Re Sc)
gdzie: Sh i Re – liczby Sherwooda i Reynoldsa zdefiniowane wzorami.
Analogia transportu masy i pędu daje wyniki w przypadku
przepływu płynu przez rurociągi. Przy założeniu odpowiedniego
rozkładu prędkości w rurociągu otrzymuje się wzory nazywane
odpowiednio analogami Reynoldsa, Taylora, Karmana i Colburna.
Analogia Reynoldsa:
St = / 8
W analogi tej zakłada się w całym przekroju ruch burzliwy i równość Sc
= 1.

75
Analogia Taylora
St = ( / 8) / [1 + 5 ( / 8)
1/2
(Sc – 1)]
Do wyprowadzenia powyższego wzoru wykorzystuje się uniwersalny
rozkład prędkości.
Analogia Karmana
W analogi tej zakłada się istnienie w rurociągu trzech stref: przepływu
laminarnego, przepływu burzliwego i przejściowej:
St = ( / 8) / {1 + 5 ( / 8)
1/2
[Sc – 1 + ln((5 Sc + 1)/6)]}
Jeżeli Sc = 1, to zarówno analogia Taylora jak i Karmana sprowadzają się
do prostej analogi Reynoldsa.
Analogia Colburna
St = ( / 8) / Sc
2/3
Po rozwinięciu liczby Stantona otrzymuje się analogię Colburna w
postaci:
Sh / (Re Sc
1/3
) = ( / 8)
Doświadczenia wykazały, że analogia Colburna daje bardzo dobre
wyniki.
Na podstawie powyższych wzorów można obliczyć współczynnik
wnikania masy korzystając ze znajomości łatwego do wyznaczenia
współczynnika oporu występującego we wzorze Darcy`ego-
Weissbacha.

76
Dzięki analogi w procesach transportu ciepła i masy można
wyznaczyć współczynnik wnikania masy, korzystając ze wzorów
wyprowadzonych na podstawie badań wnikania ciepła.
Współczynnik przewodzenia temperatury (współczynnik
wyrównania temperatury, dyfuzji ciepła) a:
a = / (c
P
)
[m
2
/s]
Kinematyczny współczynnik lepkości [m
2
/s].
=
Zastępczy wymiar liniowy
z
:
z
= [
2
/ (g
2
)]
1/3
(zastępczy wymiar poprzeczny dla przepływu nieustalonego).
77
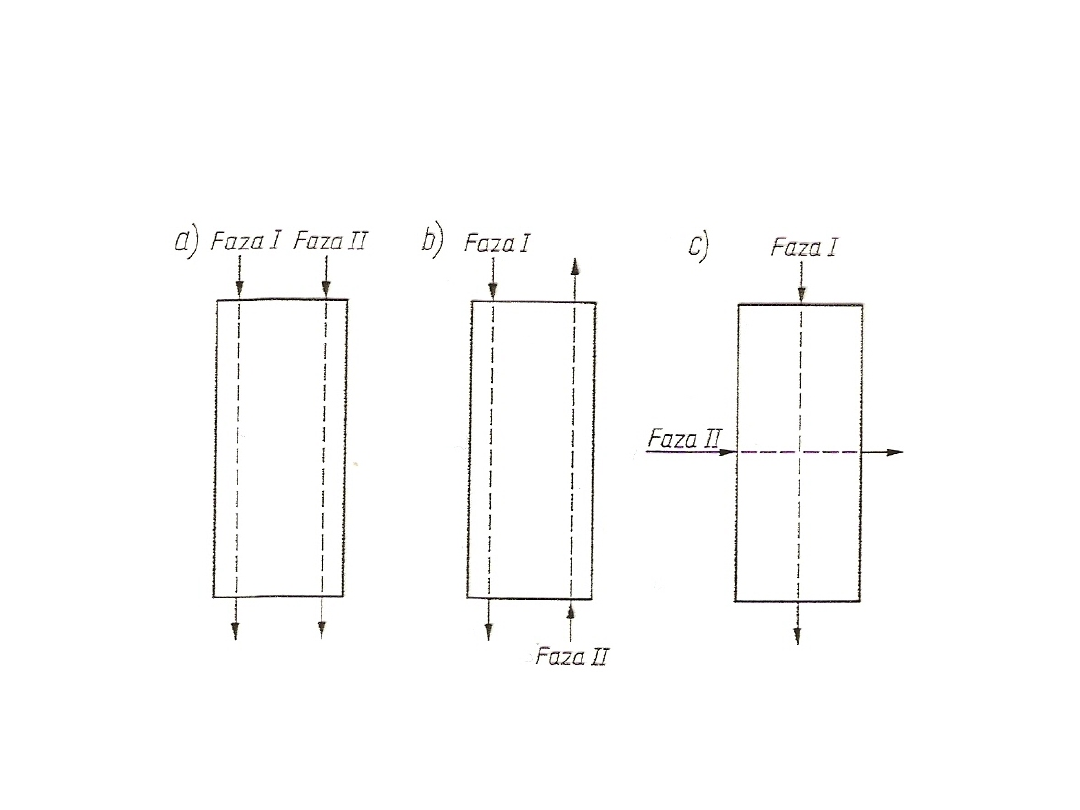
Ogólne zasady obliczania
wymienników masy
Rys. 25. Sposoby prowadzenia faz w wymiennikach masy:
a) współprąd, b) przeciwprąd, c) prąd krzyżowy

Bilanse materiałowe
ciągłych wymienników
masy
Rozważania ogólne
W obliczeniach wymienników masy bardzo ważną rolę
odgrywają bilanse materiałowe, a zwłaszcza bilans wymienianego
składnika, np. A. Zakłada się, że w wymienniku kontaktują się fazy I i II
oraz, że jest wymieniany składnika A. Aby rozważania uogólnić stosuje
się tzw. uogólnione strumienie
I
,
II
i
A
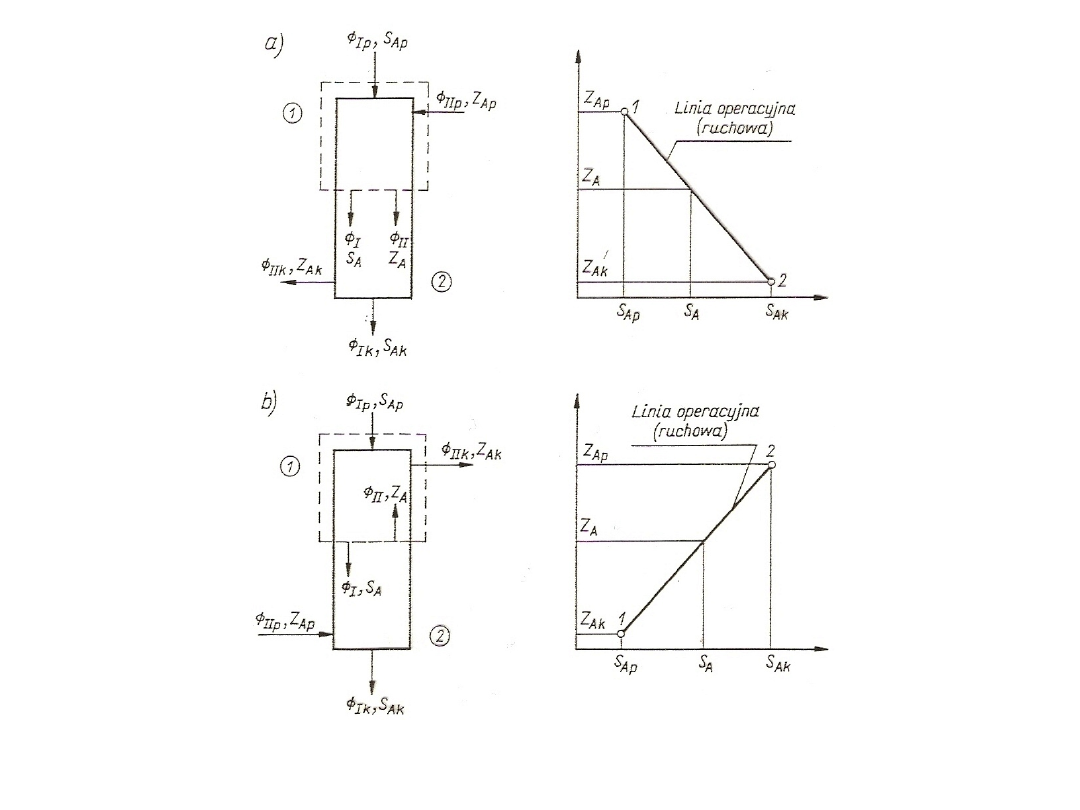
oraz uogólnione stężenia S
A
i
Z
A
. Szczegółowe znaczenie tych symboli dla konkretnych procesów
zestawiono w tabeli poniżej. Indeksem r oznaczono tam strumienie
składników inertnych, czyli rozpuszczalników. Na kolejnym rysunku
przedstawiono graficzną ilustracje bilansu materiałowego w układzie
stężeń uogólnionych dla współprądu oraz przeciwprądu. Bilans ogólny
nie zależy od sposobu prowadzenia faz i zapisać można go w postaci
równania:
Ip
S
Ap
+
IIp
Z
Ap
=
Ik
S
Ak
+
IIk
Z
Ak
Strumień wymienianego składnika A można wyrazić za pomocą
równań:
A
=
IIp
Z
Ap
-
IIk
Z
Ak
=
Ik
S
Ak
-
Ip
S
Ap
79
Tab. 8. Zestawienie strumieni i stężeń uogólnionych
Na ogół strumienie przepływających faz zmieniają się wzdłuż
aparatu (kolumny). Często jednak można tak dobrać stężenia S
A
i Z
A
,
aby odpowiednie strumienie
I
i
II
były stałe wzdłuż całego aparatu.
Oznaczenie:
I
I
A
S
A
Z
A
Proces:
Absorpcja, desorpcja
- dyfuzja przez inert
n`
cr
n`
gr
n`
A
X
A
Y
A
m`
cr
m`
gr
m`
A
X`
A
Y`
A
V`
c
V`
g
n`
A
c
cA
c
gA
Destylacja, rektyfikacja
- dyfuzja ekwimolarna
przeciwkierunkowa
n`
c
n`
g
n`
A
x
A
y
A
m`
c
m`
g
m`
A
x`
A
y`
A
80
Rys. 26. Ilustracja bilansu materiałowego składnika A na wykresie stężeń
uogólnionych: a) współprąd, b) przeciwprąd

81
W dalszych rozważaniach zakłada się, że strumienie
I
i
II
są
stałe tzn.:
I
=
Ip
=
Ik
II
=
IIp
=
IIk
Równani na
A
otrzymuje postać:
A
=
II
(Z
Ap
- Z
Ak
) =
I
(S
Ak
- S
Ap
)
Częściowe bilansowanie wymiennika (w przestrzeni oznaczonej
linią kreskowaną na rysunku) umożliwia uzależnienie od siebie stężeń
składników A w obydwu fazach w dowolnym przekroju aparatu.
Bilans wymiennika współprądowego
Bilans częściowy wymiennika współprądowego ma postać
równania:
Ip
S
Ap
+
IIp
Z
Ap
=
I
S
A
+
II
Z
A
Po przekształceniu;
Z
A
= - (
I
/
II
) S
A
+ (
Ip
/
II
) S
Ap
+ (
IIp
/
II
) Z
Ap
Jest to zależność w układzie Z
A
-S
A
w określonym przekroju
wymiennika. Graficzna ilustracja tej zależności nosi nazwę linii
operacyjnej (ruchowej).
Z założenia o stałości strumieni:
Z
A
= - (
I
/
II
) S
A
+ (
I
/
II
) S
Ap
+ Z
Ap

82
Bilans wymiennika przeciwprądowego
Bilans przestrzeni zakreślonej na ostatnim rysunku:
Ip
S
Ap
+
II
Z
A
=
I
S
A
+
IIk
Z
Ak
stąd
Z
A
= (
I
/
II
) S
A
+ (
IIk
/
II
) Z
Ak
- (
Ip
/
II
) S
Ap
Równanie to jest ogólną postacią równania linii operacyjnej
przeciwprądowego wymiennika masy. Z założenia o stałości strumieni:
Z
A
= (
I
/
II
) S
A
+ Z
Ak
- (
I
/
II
) S
Ap
W procesach absorpcji i desorpcji używa się stosunków
masowych i molowych i zakłada się, że stałe są strumienie inertów w
fazie ciekłej i gazowej (n`
gr
= const, n`
cr
= const).
W procesie rektyfikacji, przy założeniu ekwimolarnej wymiany
masy stałe są molowe strumienie czynników (n`
g
= const, n`
c
= const).

83
Gabaryty
Najczęściej spotykanym zagadnieniem wymienników masy jest
wyznaczenie wymaganej wielkości (wysokości) aparatu
zapewniającego założone stężenie wylotowe.
Wielkość ta zależy przede wszystkim od intensywności
wymiany masy, który z kolei określa szybkość przenikania masy.
Przenikanie masy jest możliwe wówczas, gdy stężenie
składnika A w określonym przekroju jest różne od stężenia
równowagowego. Warunki równowagowe określa się za pomocą linii
równowagi, jeśli na wykresie Z
A
-S
A
leży ona niżej niż linia operacyjna,
to w całym aparacie Z
A
> Z*
A
, co oznacza, że składnik A przenika od
fazy II do fazy I (absorpcja).
Gdy linia równowagi leży wyżej niż linia operacyjna, wówczas
Z
A
< Z*
A
i składnik A przechodzi od fazy I do fazy II (desorpcja).
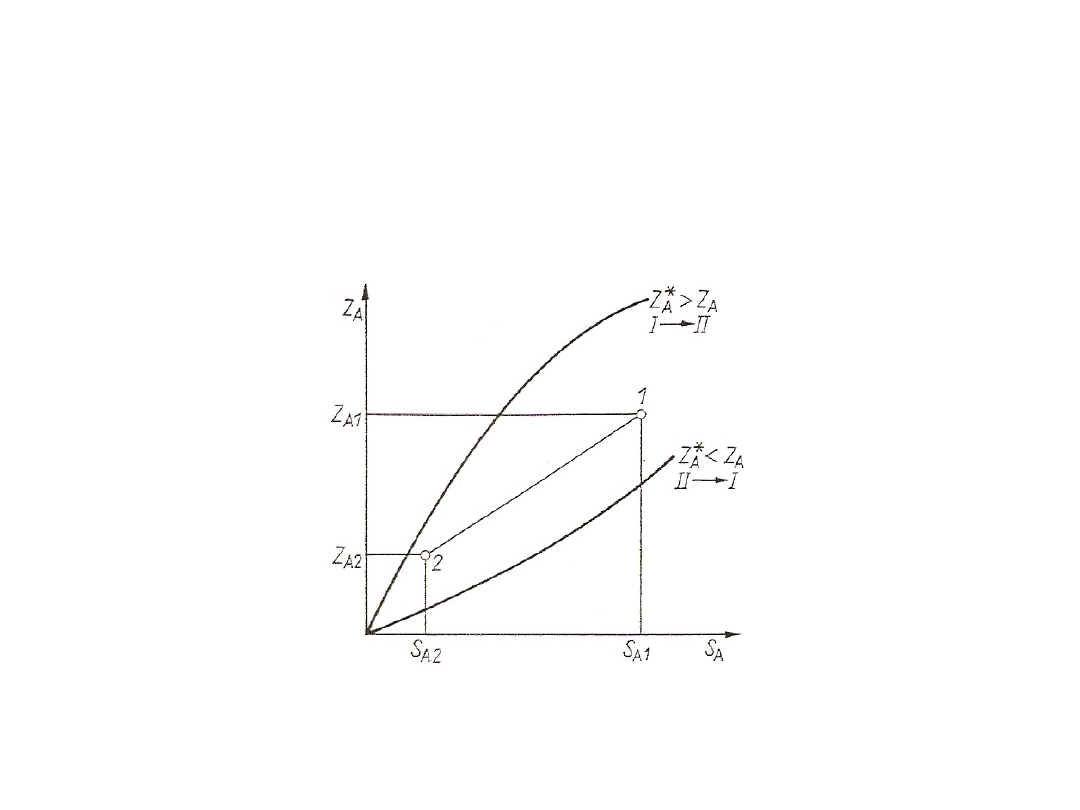
Metody obliczania
gabarytów wymienników
masy
Rozważania ogólne
Rys. 27. Wpływ wzajemnego usytuowania linii równowagi
i operacyjnej na kierunek transportu składnika A
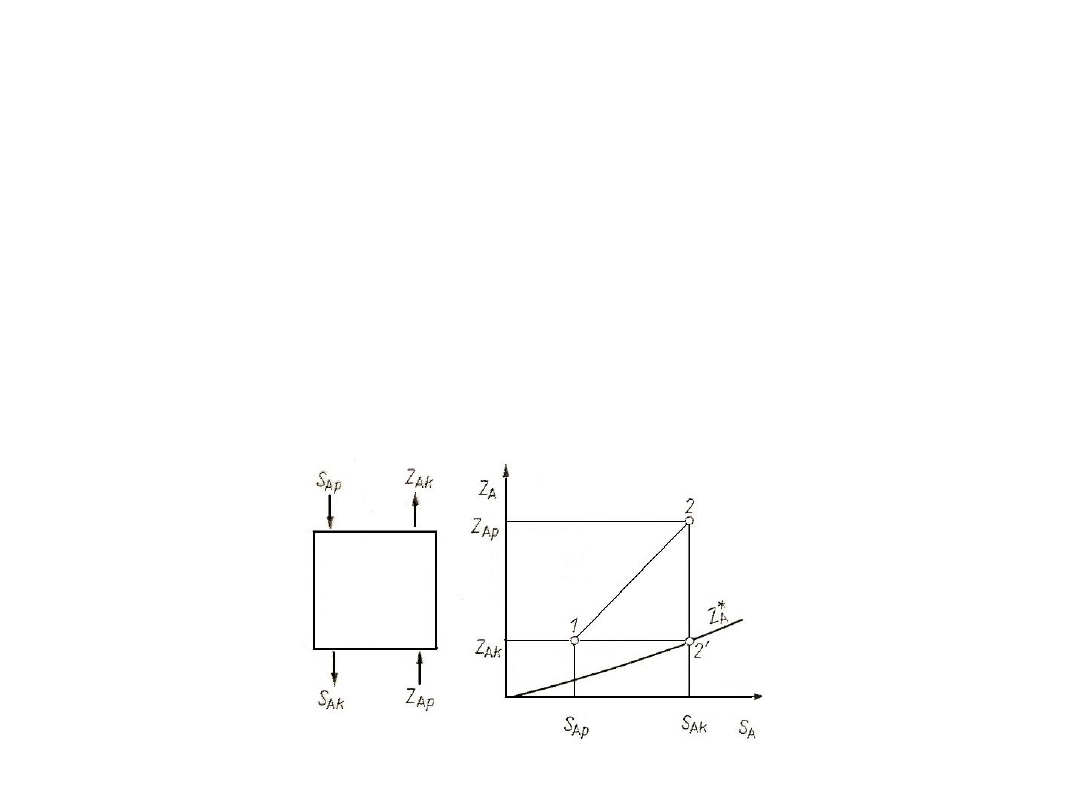
85
W praktyce najczęściej stosuje się 3 metody obliczania
wielkości wymienników masy:
- metodę stopni teoretycznych;
- metodę kinetyczną;
- metodę Colburna.
Metoda stopni teoretycznych
Na rysunku 28 przedstawiono wymiennik z zaznaczonymi
stężeniami wlotowymi i wylotowymi. Zakłada się, że dla punktu 2`
jest spełniona zależność:
Z
Ak
= Z*(S
Ak
)
Rys. 28. Metoda stopnia teoretycznego
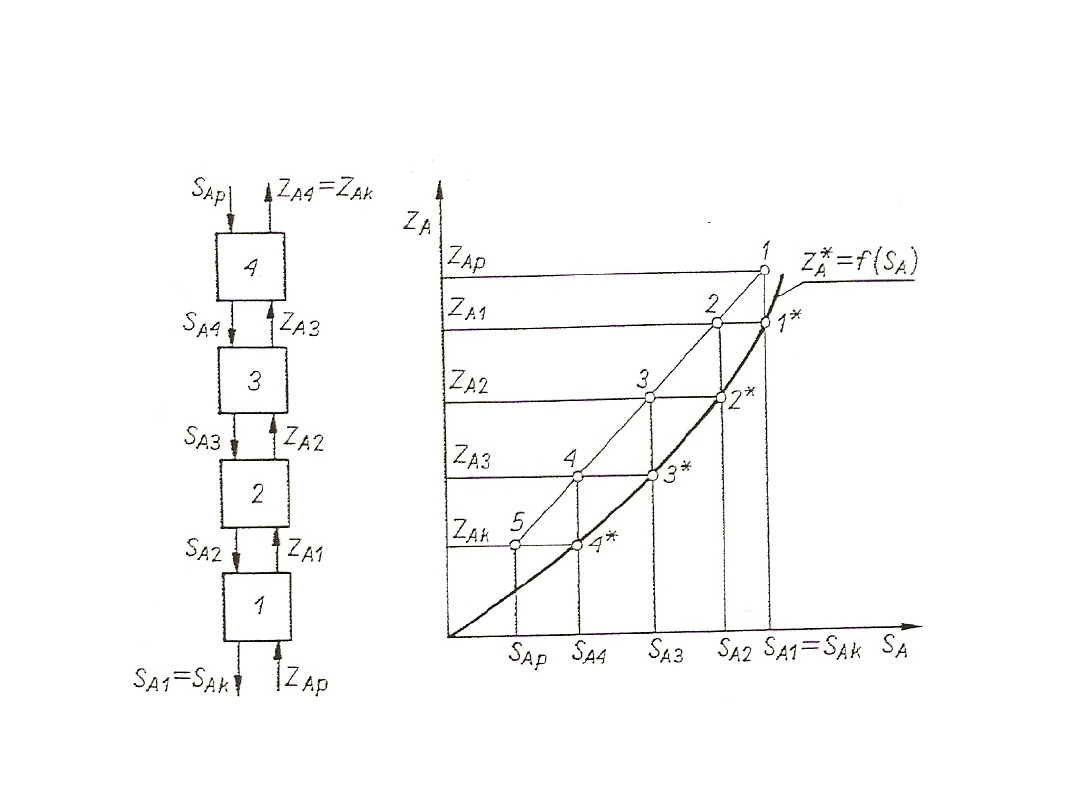
86
Z
Aj
= Z*(S
Aj
)
Rys. 29. Praca przeciwprądowej kaskady wymienników (stopni teoretycznych)
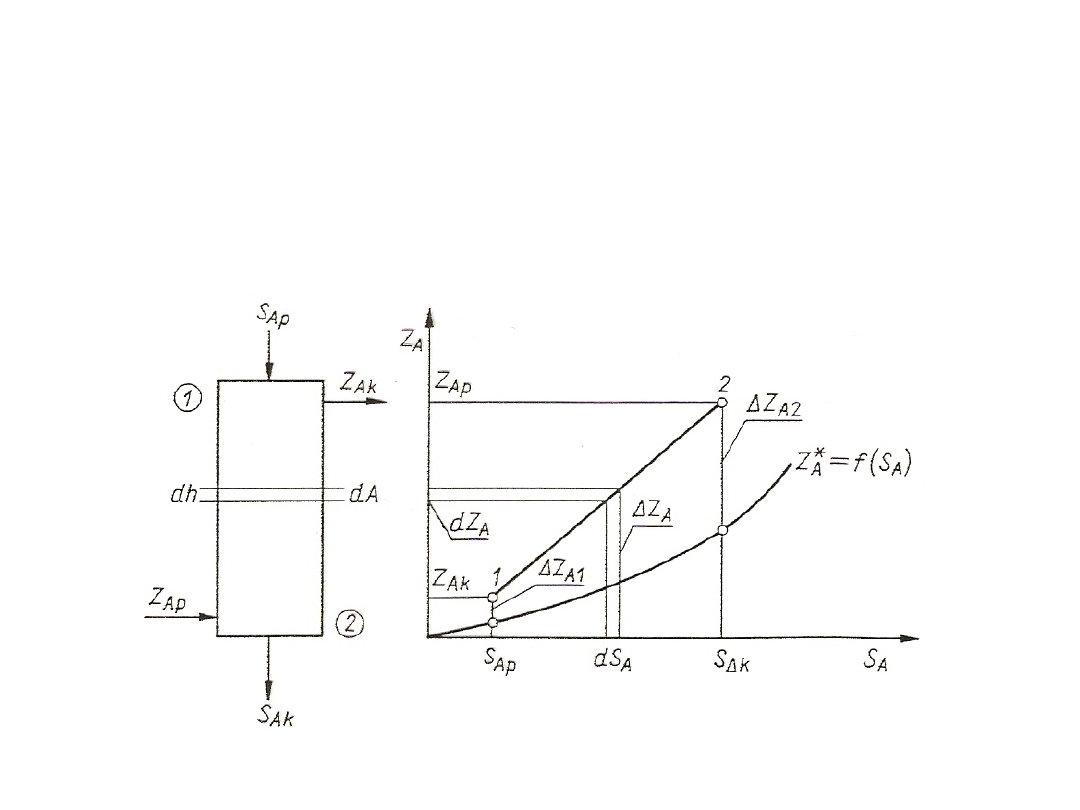
87
Metoda kinetyczna
W metodzie kinetycznej stosuje się wzór określający szybkość
przenikania masy składnika A. Podstawową jej ideą jest założenie. że
całkowity strumień wymienianego składnika A można zapisać za
pomocą wzoru:
A
= k
A
A (
A
)
m
gdzie: (
A
)
m
- średni moduł napędowy przenikania asy składnika A.
Rys. 30. Wykres pomocniczy do kinetycznej metody obliczania wymienników
masy

88
W różniczkowej warstwie wymiennika o powierzchni wymiany dA
następują przyrosty stężeń dS
A
i dZ
A
. W rozważanej warstwie zachodzi
przenikanie masy, dla którego:
J
A
= d
A
/ dA = k
AII
(
A
)
0II
Jednocześnie z bilansu warstwy:
d
A
=
II
dZ
A
=
I
dS
A
Połączenie tych dwóch wzorów prowadzi do zależności:
dA = (
II
dZ
A
) / [k
AII
(
A
)
0II
]
Po scałkowaniu w granicach od punktu 1 do 2 otrzymuje się:
A = ∫
1
2
(
II
dZ
A
) / [k
AII
(
A
)
0II
]
(
A
)
m
= [
II
(Z
Ap
- Z
Ak
)] / {k
A
∫
1
2
(
II
dZ
A
) / [k
AII
(
A
)
0II
]}
Przy założeniu, że w całym wymienniku
II
= const oraz k
A
=
const = k
AII
otrzymuje się wyrażenie określające średni moduł napędowy
napędowy wymiennika masy:
(
A
)
m
= (Z
Ap
- Z
Ak
) / ∫
1
2
dZ
A
/ (
A
)
0II

89
Obliczanie całki w mianowniku w istotny sposób zależy od sposobu
wyrażania modułu napędowego przenikania masy oraz od
zależności((
A
) = f(Z
A
). Jeżeli założy się zależność liniową, to średni
moduł napędowy będzie średnią logarytmiczną z modułów na końcach
wymiennika, tzn.:
(
A
)
m
= (
A1
–
A2
) / ln
A2
/
A1
Metoda Colburna
Jest to pewna modyfikacja metody kinetycznej. W literaturze
znana jest również pod nazwą metody wysokości jednostkowej
wymiany masy HTU [Hight of Transfer Unit].
Podstawowy wzór tej metody jest następujący:
H = (h
og
) (N
og
)
lub:
H = (h
oc
) (N
oc
)
gdzie: H
- wysokość wymiennika masy, m;
h
og
i h
oc
- jednostkowa wysokość wymiany masy odniesiona do
fazy
gazowej
lub ciekłej, m;
N
og
i N
oc
- liczba jednostek wymiany masy w fazie gazowej lub
ciekłej.

90
Wysokość różniczkowej warstwy wymiennika można związać z
różniczkową powierzchnią wymiany masy dA za pomocą zależności:
dh = dA /a dA
k
gdzie: a
– tzw. jednostkowa powierzchnia wymiany masy, tzn.
powierzchnia odniesiona do objętości, m
2
/m
3
;
A
k
– przekrój poprzeczny aparatu, m
2
.
dh = (
II
dz
A
) / [a A
k
k
AII
(
A
)
0II
]
Po scałkowaniu w granicach od punktu 1 do 2:
H = ∫
ZAk
ZAp
{(
II
dz
A
) / [a A
k
k
AII
(
A
)
0II
]}
Po założeniu, że wielkość
II
/ (a A
k
k
AII
) = const, można napisać:
H = [
II
/ (a A
k
k
AII
)] ∫
ZAk
ZAp
[ dz
A
/ (
A
)
0II
] = (h
og
) (N
og
)
gdzie:
h
og
=
II
/ (a A
k
k
AII
)
N
og
= ∫
ZAk
ZAp
[ dz
A
/ (
A
)
0II
]

91
Rektyfikacja
Destylacja prosta umożliwia jedynie ograniczony rozdział
mieszaniny na składniki, przy czym stopień czystości produktów
(destylatu i wyczerpanej cieczy) zależy od tego, jak dalece linia
równowagi odbiega od przekątnej kwadratu stężeń x-y. Aby osiągnąć
lepszy rozdział mieszaniny, a w granicznym przypadku zupełnie
czyste składniki, należy stosować bardziej skomplikowaną metodę
rozdziału – rektyfikację. Proces ten prowadzi się najczęściej w
aparatach nazywanych kolumnami rektyfikacyjnymi.
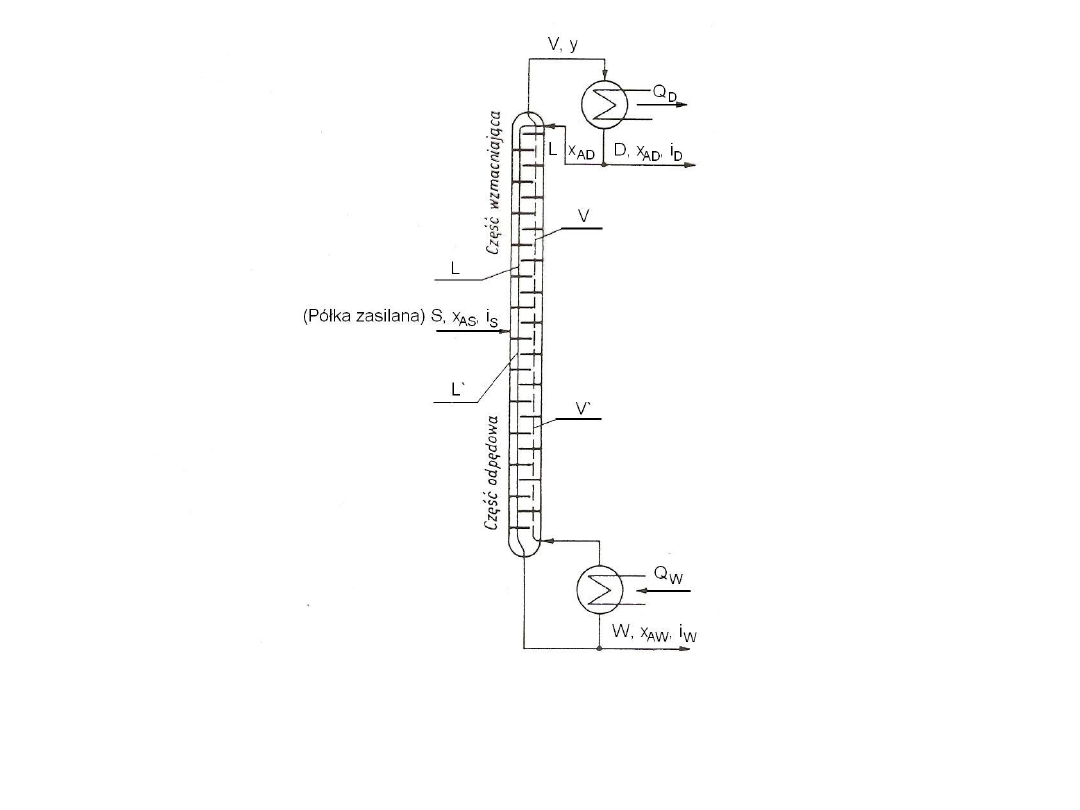
92
Rys. 31. Schemat kolumny rektyfikacyjnej do rozdzielania mieszaniny
dwuskładnikowej
CZĘŚĆ WZMACNIAJĄCA ≡ REKTYFIKACJA
CZĘŚĆ ODPĘDOWA ≡ ODPAROWUJĄCA ≡ WYCZERPUJĄCA
93
Rys. 32. Zasada numeracji półek w kolumnie rektyfikacyjnej

94
W praktyce najczęściej jest dany strumień surowca S oraz jego
skład wyrażony przez stężenie x
AS
. Celem rektyfikacji jest rozdział
surowca na destylat o stężeniu składnika D x
AD
i ciecz wyczerpaną o
stężeniu x
AW
. W dalszym ciągu rozważań indeks D będzie oznaczał
wielkość charakterystyczną dla destylatu, natomiast indeks W wielkość
charakterystyczną dla cieczy wyczerpanej (wypływającej z kotła
destylacyjnego).
Pierwszym zadaniem jest wyznaczenie otrzymanych w danej
instalacji strumieni destylatu i cieczy wyczerpanej. W tym celu
wykonuje się bilans molowy całej kolumny.
Bilans masowy kolumny
S = D + W
S x
AS
= D x
AD
+ W x
AW
Stąd np.:
D = (x
AS
- x
AW
) / (x
AD
- x
AW
) S
W = (x
AD
- x
AS
) / (x
AD
- x
AW
) S
Bilans cieplny kolumny
Q
W
+ S i
S
= D i
D
+ W i
W
+ Q
D
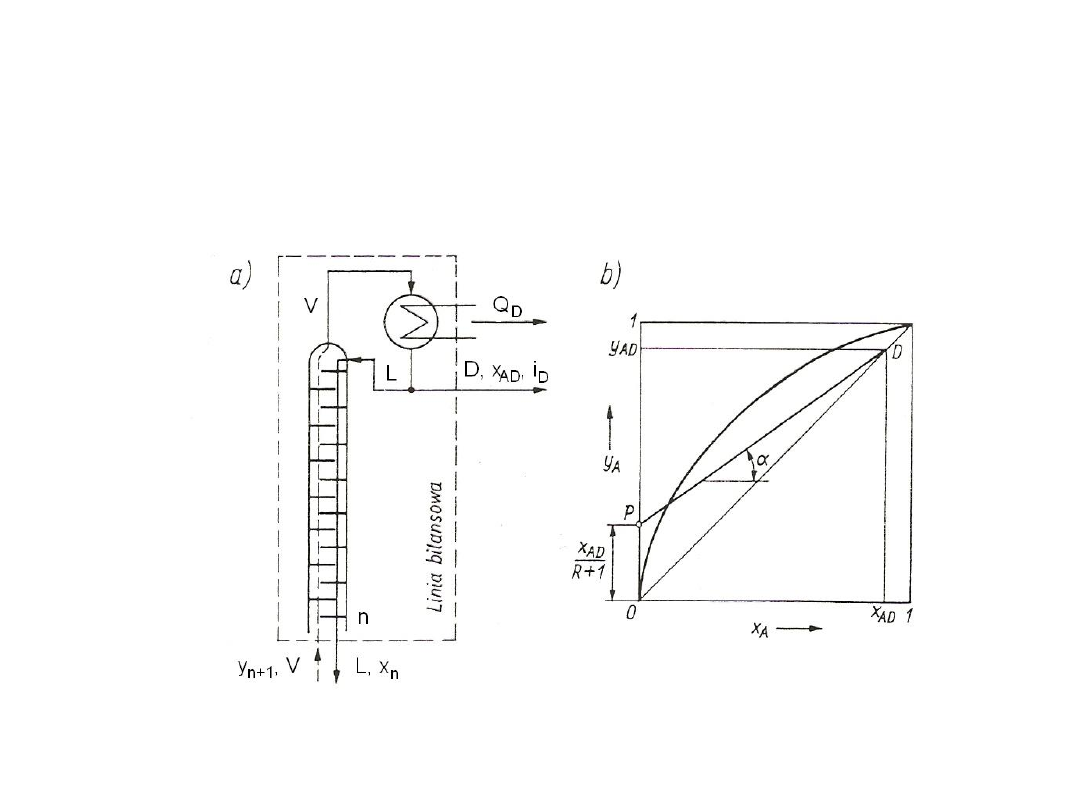
95
Bilanse masowe i linie operacyjne
dla pełnego zestawu
rektyfikacyjnego
Część wzmacniająca (górna) kolumny rektyfikacyjnej
Linią bilansową (kreskowaną) wyodrębnia się część wzmacniającą
kolumny.
Rys. 33. Część wzmacniająca kolumny rektyfikacyjnej:
a) schemat bilansowania, b) linia operacyjna

96
Bilans skraplacza
V = D + L
Założenia początkowe: kolumna pracuje z pełnym zawracaniem
destylatu D = 0
V = L
V y
n+1
= L x
n
Równanie linii bilansowej ma więc postać:
y
A
= x
A
linia bilansowa pokrywa się z podstawą kwadratu stężeń x-y –
część b rysunku.

97
Jak zmieniają się parametry strumienia ciekłego i parowego
wzdłuż kolumny, gdy odbiera się część destylatu D > 0.
Założenia upraszczające:
- mamy do czynienia z układem dwuskładnikowym który
charakteryzuje się jednakowymi kilomolowymi entalpiami wrzenia
czystych składników A i B oraz, że entalpia wrzącej mieszaniny cieczy
złożonej ze składników A i B nie zależy od składu i równa się entalpii 1
kilomola składnika A albo B:
i`
n+1
≈ i`
n
≈ i`
n-1
≈ … ≈ i`
- zmiany ciśnienia wzdłuż wysokości kolumny są bardzo małe
i = const
- ciepło parowania 1 kmol składnia w Ai B oraz i kmol mieszaniny
wrzącej cieczy złożonej ze składników A i B gdzie stałe:
r
n+1
≈ r
n
≈ r
n-1
≈ … ≈ r
- kolumna pracuje adiabatycznie ciepło wyzwolone podczas
skraplania 1 kmol mieszaniny ze strumienia pary do strumienia cieczy
powoduje odparowanie 1 kmol cieczy i powstanie 1 kmol pary bogatej
w składnik bardziej lotny Strumienie molowe cieczy i pary podczas
przepływu przez daną część kolumny są stałe.

98
V = L + D
dla n-tej półki i składnika A
V y
n+1
= L x
n
+ D x
D
y
n+1
= (L / V) x
n
+ (D / V) x
D
y
n+1
= [L / (L + D)] x
n
+ [D / (L + D)] x
D
Wielkości x
n
, y
n+1
możemy traktować jako zmienne bieżące
(punkt o tych współrzędnych leży na linii operacyjnej), a równanie
ostatnie można napisać wtedy w postaci ogólnej:
y = [L / (L + D)] x + [D / (L + D)] x
D
Jest to równanie GLO.
Wprowadzając pojęcie powrotu (orosienia):
R = L / D
otrzymujemy:
y = [R / (R + 1)] x + [I / (R + 1)] x
D
[UWAGA: to są stężenia molowe]
Jest to najczęściej spotykane równanie linii operacyjnej (bilansowej)
GLO. Uwaga: linia operacyjna jest linią prostą tylko przy spełnieniu
wszystkich założeń upraszczających.
99
GLO przecina przekątną kwadratu stężeń w punkcie yAD =xAD
(D) jej nachylenie wynosi:
tg = R / (R + 1) ≤ 1
tg = 1 występuje gdy L = V, a więc dla całkowitego powrotu (R ∞)
wówczas linia operacyjna pokrywa się z przekątną kwadratu stężeń.
Współrzędne punktu P [0, x
D
/(R+1)].
Rys. 34. Schemat bilansowania półki zasilającej
100
Część odpędowa (dolna) kolumny rektyfikacyjnej
Rys. 35. Część odpędowa kolumny rektyfikacyjnej:
a) schemat bilansowania, b) linia operacyjna

101
Bilans:
L` = V` + W
L` x
m
= V’ y
m+1
+ W x
W
y
m+1
= (L` / V’) x
m
– (W / V’) x
W
lub:
y
m+1
= [L` / (L` - W)] x
m
– [W / (L` - W)] x
W
ogólnie:
y = [L` / (L` - W)] x
– [W / (L` - W)] x
W
RDLO
Uwzględniając molowe strumienie cieczy i pary dopływające do
półki zasilanej i odpływające z niej układamy bilans materiałowy
półki zasilanej:
L + V` + S = L` + V
Należy znaleźć związek miedzy liczbą moli cieczy płynącej w
górnej części kolumny L, a liczbą moli płynącej w dolnej części kolumny
L`.
Podobnie należy ustalić związek między molowym natężeniem
przepływu pary w górnej V i dolnej V` części kolumny.
Rozwiązanie tego zagadnienia staje się możliwe po
uwzględnieniu wpływu stanu cieplnego surówki na strumienie cieczy i
pary w obu częściach kolumny.

102
Wprowadzamy pomocniczą wielkość e, która charakteryzuje
stan cieplny surówki.
Linia operacyjna (DLO) przecina przekątną kwadratu stężeń
x-y w punkcie x
W
=y
W
=x
w
i jej nachylenie wynosi:
tg = L` / V` ≥ 1
gdy: V` = L` to tg = 1 i linia operacyjna pokrywa się z przekątną
kwadratu stężeń.

103
Definicja e
Wielkość e oznacza stosunek ilości ciepła, jaką należy dostarczyć 1
kmol surówki, aby zamienić ją w nasyconą parę suchą do kilomolowego
ciepła parowania:
e = (i`` - i
S
) / r
gdzie: i
S
- entalpia 1 kmol surówki, J/kmol;
i``
- entalpia 1 kmol pary suchej w temperaturze wrzenia
surówki, J/kmol;
r
- molowe ciepło parowania, J/kmol.
Uwzględniając definicje wielkości e mamy:
L` = L + e S
L + V` + S = L + e S + V
V` = V + S (e - 1)
Jeśli uwzględnimy te zależności w równaniach linii operacyjnych
DLO:
y = [L` / (L` - W)] x
– [W / (L` - W)] x
W
= [(L + eS) / (L + eS - W)] x
– [W / (L +
eS - W)] x
W
przebieg DLO zależy m. in. od stanu cieplnego surówki e.

104
Oznacza to również, że współrzędne punktu przecięcia obu linii
operacyjnych dla górnej i dolnej części kolumny zależą od wartości e.
Zauważmy, że nachylenie DLO wyniesie:
tg` = (L + eS) / (L + eS - W) = f(e)
Wyznaczenie współrzędnych punktu przecięcia obu linii
operacyjnych sprowadza się do wyznaczenia miejsca geometrycznego
punktów przecięć tych linii dla różnych stanów cieplnych surówki.
Matematycznie oznacza to rozwiązanie układu dwóch równań
linii operacyjnych dla górnej i dolnej części kolumny przy jednoczesnym
wyeliminowaniu wielkości L:
y = [L / (L + D)] x + [D / (L + D)] x
D
y = [(L + eS) / (L + eS - W)] x
– [W / (L + eS - W)] x
W
po przekształceniach mamy:
y = [e / (e -1)] x – [x
S
/ (e – 1)]
Równanie linii surowca (równanie linii e).
105
Rys. 36. Wykreślanie linii operacyjnych i linii surowca
Na linii surowca przecinają się obydwie linie operacyjne (punkt
S na rysunku). Dla y
A
= 0 , x
A
= x
AS
/ e jest to punkt przecięcia się
prostej surowca z osią odciętych (punkt E).
Dla x
A
= y
A
= x
AS
/ e mamy punkt przecięcia linii surowca z
przekątną kwadratu stężeń (punkt F). Znajomość położenia punktu E
umożliwia wykreślenie linii surowca jako przedłużenie odcinka EF.
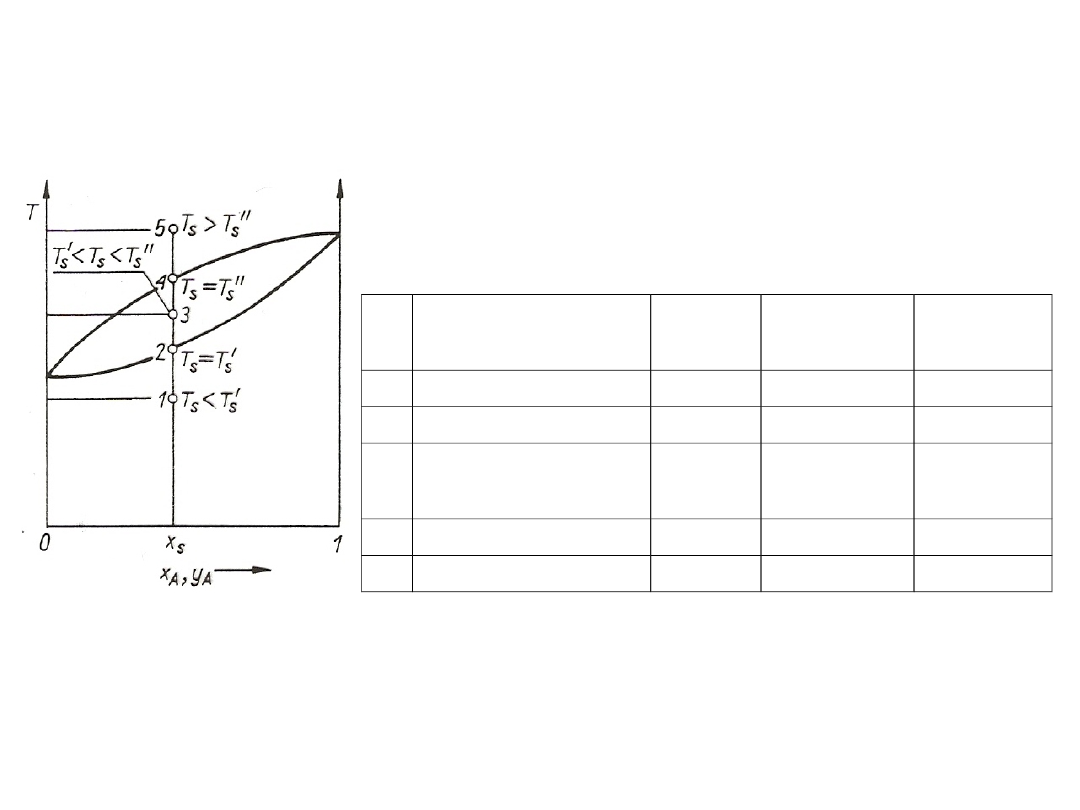
106
Różna możliwe stany surowca przedstawia rysunek poniżej. W
tablicy przedstawiono wpływ stanu surowca na parametry linii operacyjnej
surowca.
Rys. 37. Możliwe stany
surowca w rektyfikacji
mieszaniny
dwuskładnikowej
Tab. 9. Wpływ surówki na parametry linii
surowca
tg = e / (e – 1)
Lp
.
Stan surówki
Temp.
Współczynni
k kaloryczny
Nachylenie
linii
surowca
1
Ciecz przechłodzona
T
S
< T`
S
e > 1
tg > 1
2
Ciecz w stanie wrzenia
T
S
= T`
S
e =1
tg = ∞
3
Mieszanina cieczy
wrzącej
i pary nasyconej
T
S
> T`
S
T
S
< T``
S
o < e < 1
tg < 1
4
Para nasycona
T
S
= T``
S
e = 0
tg = 0
5
Para przegrzana
T
S
> T``
S
e < 0
0 < tg < 1

107
Minimalna i optymalna ilość
powrotu (orosienia) w kolumnie
Kolumna rektyfikacyjna może być rozpatrywana jak wymiennik
masy, w którym składnik bardziej lotny A przechodzi z fazy ciekłej do
fazy gazowej. Intensywność wymiany masy jest proporcjonalna do
modułu napędowego, tj. różnicy między równowagowym ułamkiem
molowym y*
A
a rzeczywistą wartością y
A
reprezentowaną przez linie
operacyjne. Intensywność wymiany masy rośnie zatem, gdy linie
operacyjne oddalają się od linii równowagi i maleje, gdy zbliżają się one
do tej linii. Z punktu widzenia wymiany masy korzystne jest więc, gdy
linie operacyjne oddalają się od linii równowagi, Położeniem granicznym
jest przekątna kwadratu stężeń x-y odpowiadająca wartości n`
D
=0, czyli
R=∞. Moduł napędowy wymiany masy jest wtedy największy. W
praktyce zwiększenie przepustowości kolumny wymaga zmniejszenia
wartości R, co powoduje zbliżenie punktu S do linii równowagi.
Graniczną wartością R jest taka wartość, dla której punkt przecięcia się
linii operacyjnych leży na krzywej równowagi – położenie S`. W punkcie
S` moduł napędowy osiąga wartość zerową, a to oznacza, że transport
masy ustaje. Wartość współczynnika orosienia osiąga wartość
minimalną R=R
min.
Wyznaczenie wartości R
min
jest ważnym etapem projektowania
kolumn rektyfikacyjnych.

108
(tg)
min.
= R
min.
/ (R
min.
+ 1) = [x
AD
– (y
AS
)
min.
] / [x
AD
– (x
AS
)
min.
]
Stąd:
R
min.
= [x
AD
– (y
AS
)
min.
] / [x
AD
– (x
AS
)
min.
]
gdzie: (x
AS
)
min
, (y
AS
)
min
- współrzędne punktu S``. Współrzędne te można
wyznaczyć
dla x=const rozwiązując układ równań:
(y
AS
)
min
= y* (x
AS
)
min
=
[
A
(x
AS
)
min
] / [1 + (x
AS
)
min
(
A
- 1)]
(y
AS
)
min
= [e / (e-1)] (x
AS
)
min
– [x
AS
/ (e-1)]
Jeżeli surowiec jest podawany w postaci cieczy wrzącej, e=1, to
oraz .
R
min.
= [1 / (-1)] { (x
AD
/ x
AS
) – [(1 - x
AD
) / (1 - x
AS
)] }
Rzeczywista wartość stosunku orosienia R musi być większa od
wartości minimalnej. Najczęściej w praktyce stosuje się:
R = k R
min.
gdzie: k – liczba większa od jedności (najczęściej zawarta w granicach od
1,3 do 2).
Przy doborze wartości R należy kierować się względami
ekonomicznymi. Dysponując danymi dotyczącymi kosztów inwestycyjnych
(budowy kolumny, aparatów cieplnych, pomp, fundamentów, budynków
itp.) oraz kosztów eksploatacyjnych (przetłaczanie surowca, pary grzejnej,
wody chodzącej, obsługi itp.) można określić minimum kosztów
sumarycznych, przy których obrany powrót R będzie optymalny.
109
Rys. 38. Sposób wyboru rzeczywistego powrotu w kolumnie rektyfikacyjnej na
podstawie kosztów
Zmniejszenie wartości R powoduje zwiększenie kosztów
inwestycyjnych, gdyż zmniejszający się moduł napędowy wymiany masy
musi być zrekompensowany wielkością aparatu (wysokością kolumny).
Zwiększenie wartości R powoduje natomiast wzrost kosztów
eksploatacyjnych.
110
Rys. 39. Szacunkowy wybór rzeczywistego powrotu w kolumnie
rektyfikacyjnej
W przypadku, gdy dane ekonomiczne są niedostępne problem
przyjęcia wartości R można rozwiązać graficznie. Dla kilku dowolnie
przyjętych wartości R (w granicach nadmiaru 1 … 10) określa się
równania linii operacyjnych i wyznacza odpowiadającą danemu R
wysokość kolumny H lub konstruuje się wykres. Na wykresie szuka się
punktu M, od którego dalsze zmniejszanie wartości R powoduje
gwałtowny wzrost liczby n
t
. Tak otrzymana wartość R, odpowiadająca
punktowi M jest przyjmowana do dalszych obliczeń.

111
Metody wyznaczania wysokości
kolumn rektyfikacyjnych
Tradycyjnie stosuje się metodę stopni (półek) teoretycznych.
Graficzna wersja dla tej metody dla rektyfikacji dwuskładnikowej została
zaproponowana przez McCabe`a i Thielego i polega na zastosowaniu
tzw. konstrukcji schodkowej między linią równowagową a operacyjną.
Schodkowanie można wykonać osobno dla każdej części kolumny albo
też dl obydwu części łącznie.
Na rysunku pokazano przykładową konstrukcje McCabe`a-
Thielego dla dwuskładnikowej mieszaniny i surowca w postaci cieczy
przechłodzonej ( > 1). W graficznej metodzie wyznaczania liczby półek
teoretycznych należy uwzględnić to, że wyparka zastępuje jedną półkę
teoretyczną oznaczoną na rysunku punktem 7.
Jeżeli założy się, że współczynnik względnej lotności jest stały,
to liczbę półek teoretycznych można wyznaczyć analitycznie metodą
Smokera, odpowiednie wzory dostępne są w literaturze specjalistycznej.
112
Rys. 40. Wyznaczanie liczby półek teoretycznych w kolumnie
rektyfikacyjnej metodą McCabe`a-Thielego
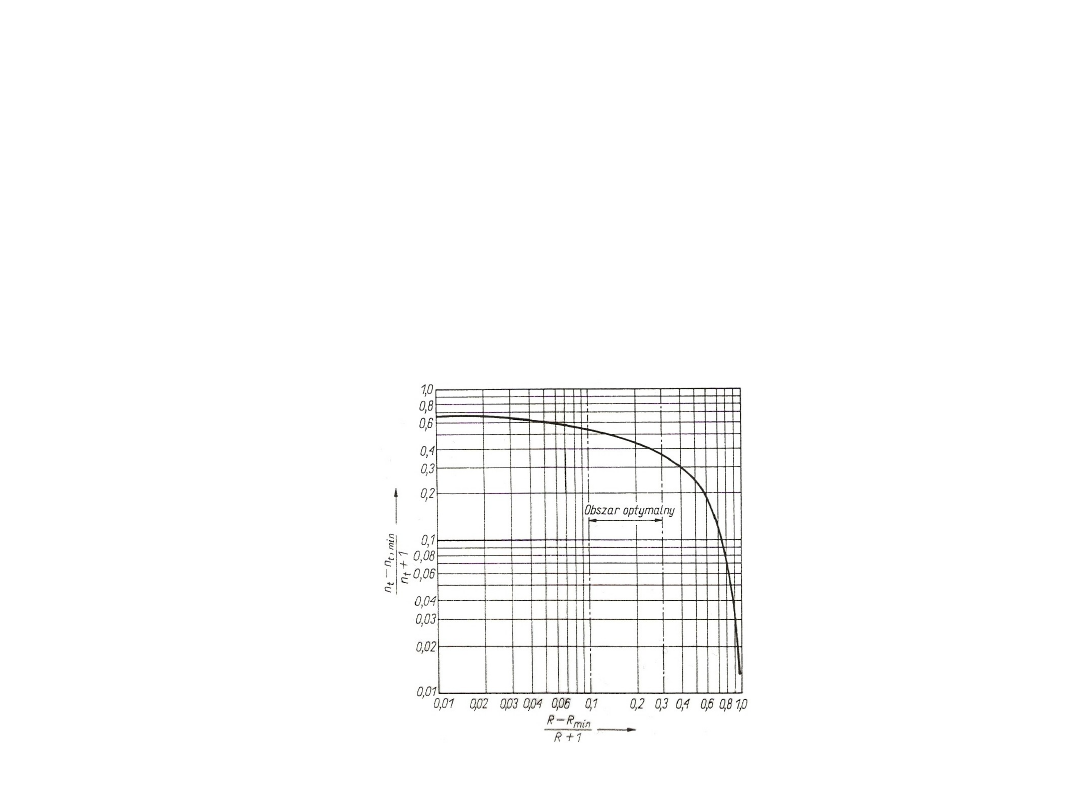
113
Do szybkiego oszacowania liczby półek teoretycznych można
zastosować metodę Gillilanda, która na podstawie rozważań Fenskiego
i Underwooda opracował zależność empiryczną między teoretyczną
liczbą półek i powrotem (orosieniem) R. Zależność tę można stosować
również dla mieszanin nieidealnych i wieloskładnikowych. W celu
wyznaczenia liczby półek teoretycznych n
t
, należy wcześniej wyznaczyć
wartość R
min.
oraz n
t, min
dla R=∞. Tę ostatnią wartość wyznacza się
albo metodą McCabe`a-Thielego przez schodkowanie między linią
równowagi, a przekątną kwadratu stężeń x-y, albo też dla A=const za
wzoru Fenskiego:
n
t, min
= {ln { [x
AD
(1 - x
AK
)] / [x
AK
(1 - x
AD
)] } / ln
A
} - 1
Rys. 41. Wykres Gillialanda do określania liczby półek teoretycznych

114
Jak wspomniano wcześniej sposób prowadzenia procesu
wymiany masy może być: ciągły lub okresowy (periodyczny).
W procesie ciągłym istnieje stały przepływ faz przez wymiennik
masy, więc parametry procesu są funkcją tylko miejsca , a nie czasu.
W procesie periodycznym aparat pracuje szarżami, a parametry
procesu są funkcją miejsca i czasu.
Niekiedy z aparatu odbiera się w sposób ciągły tylko jedną fazę,
podczas gdy druga pozostaje w nim aż do czasu przerobienia danej
szarży.
Dla określenia pracy aparatu ważnym parametrem jest czas
potrzebny do rozdziału mieszaniny, który powinien być równy
efektywnemu czasowi przebywania substancji w aparacie.
Natomiast dla eksploatacji ciągłej definiuje się średni czas
przebywania substancji w aparacie:
m
= V / V*
gdzie: V – objętość zajęta przez mieszaninę, m
3
;
V* -
objętościowy strumień przepływu mieszaniny
m
3
/s.

115
Metoda stopni teoretycznych polega na zastosowaniu
wymiennika masy hipotetyczną kaskadą n stopni teoretycznych.
Liczbę n dla zadanych położeń linii operacyjnej i równowagowej
wyznacza się za pomocą konstrukcji schodkowej, polegającej na
przemiennym kreśleniu odcinków pionowych i poziomych, aż do
osiągnięcia wymaganych stężeń. Konstrukcje tą można wykonać
graficznie lub za pomocą obliczeń komputerowych. Numeryczna
wersja tej konstrukcji metodą „z półki na półkę” sprawność.

116
Sprawność ogólna kolumny
półkowej
Bardzo ważnym pojęciem określającym intensywność wymiany
masy na półkach jest pojęcie sprawności kolumny lub półki. Pojęcie
sprawności umożliwia przejście z liczby półek teoretycznych na liczbę
półek rzeczywistych, niezbędnych do zapewnienia założonych składów
produktów. Termin sprawność jest niejednoznaczny. W praktyce używa
się kilku rodzajów sprawności inaczej definiowanych i mających inne
wartości. Oto najważniejsze definicje i wzory mające zastosowanie w
praktyce.
Sprawność ogólna kolumny (sekcji) E
k
– wyraża najstarszą, a
jednocześnie najprostszą metodę wyznaczania liczby półek
rzeczywistych w kolumnie. Zgodnie z definicją:
E
k
= n
t
/ n
gdzie: n – liczba półek rzeczywistych;
n
t
- liczba półek teoretycznych.
Znajomość wartości E
k
umożliwia bezpośrednie obliczenie
liczby półek teoretycznych n:
n = n
t
/ E
k

117
W literaturze podaje się wzory lub wykresy do oszacowania
wartości E
k
. Są to zazwyczaj zależności czysto empiryczne, ważne tylko
dla określonego typu półek i określonego procesu. Na przykład dla
destylacyjnych kolumn kołpakowych sprawność ogólną kolumny można
obliczyć z korelacji O`Connela:
E
k
= 0,0906 (
c
)
-0,245
gdzie: – współczynnik względnej lotności.
Po założeniu, że linie równowagowa i operacyjna są proste,
sprawność ogólną można obliczyć za pomocą wzoru Lewisa:
dla D ≠ 1
E
k
= { ln [1 + E
Mg
(D - 1)] } / lnD
dla D = 1
E
k
= E
Mg
gdzie:
D = m (
g
/
c
)
E
Mg
- sprawność umowna półki;
g
i
c
-
uogólnione strumienie gazu i cieczy;
m -
nachylenie linii równowagi w uogólnionym
układzie stężeń.

118
Strumienie gazu i cieczy opuszczające półkę teoretyczną są ze
sobą w równowadze, ale na półce rzeczywistej tek nie jest. Stopień
oddalenia wartości stężeń rzeczywistych od wartości równowagowych
można wyrazić za pomocą sprawności umownie zdefiniowanej przez
Murphree`go. Definicję można sformułować używając albo stężeń w
fazie gazowej, albo w fazie ciekłej.
Jeżeli zastosuje się stężenia uogólnione S i Z, i założy, że
rozważany składnik A przechodzi od fazy ciekłej do fazy gazowej;
definicja jest następująca:
E
Mg
= [(Z
n
)
śr
– (Z
n-1
)
śr
] / [Z*
n
- (Z
n-1
)
śr
]
gdzie: Z*
n
– równowagowe stężenie składnika A do stężenia w cieczy
opuszcającej półkę, czyli:
Z*
n
= K S
n
W przypadkach, gdy wskutek silnej zmiany stężenia cieczy
wzdłuż drogi na półce, może zajść nierówność Z*
n
< (Z
n
)
śr
, wówczas E
Mg
przybiera wartości większe od jedności. Świadczy to o umownym
charakterze tej definicji i dlatego niektórzy autorzy unikają nawy
sprawność, np. w literaturze niemieckiej wielkość ta określana jest jako
stopień wzmocnienia.
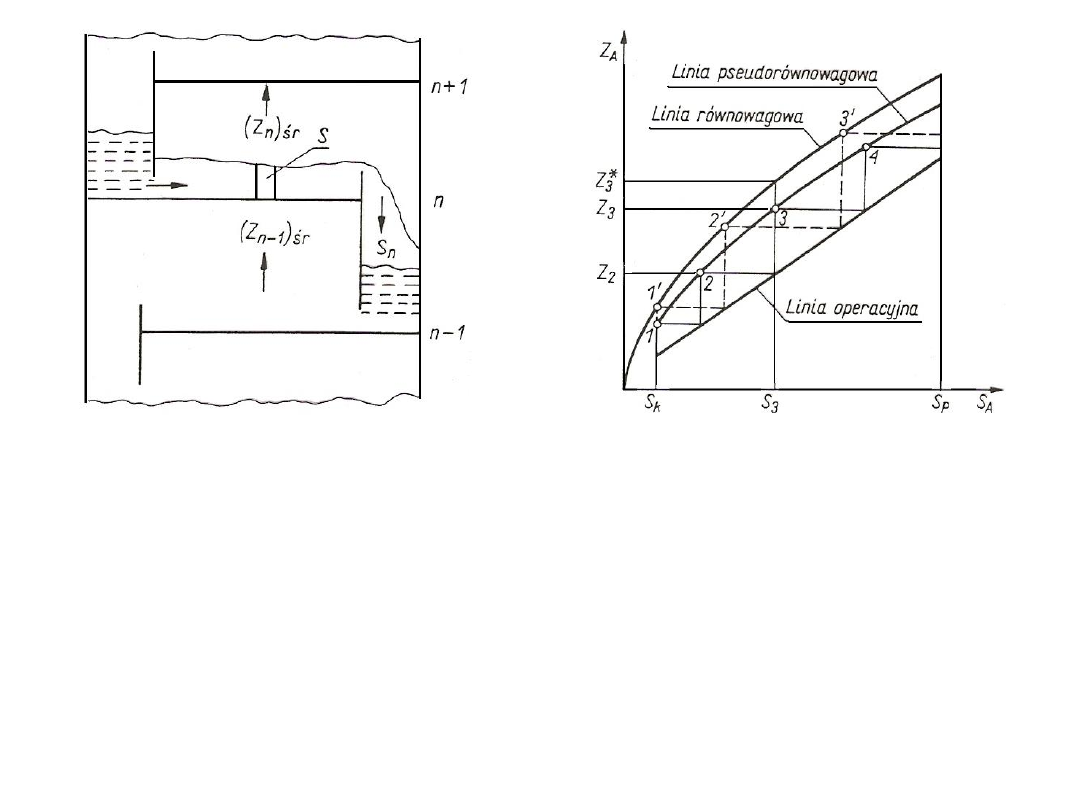
119
Rys. 42. Oznaczenia do definicji
umownej sprawności półki
Rys. 43. Zmodyfikowana metoda
McCabe-Thielego wyznaczania liczby
półek rzeczywistych
Wartość sprawności E
Mg
może być wykorzystana do wyznaczania
liczby półek rzeczywistych za pomocą zmodyfikowanej metody
graficznej McCabe-Thielego. Modyfikacja polega na tym, że pionowe
odcinki między linią operacyjną i równowagową mnoży się przez E
Mg
.
Otrzymane punkty tworzą tzw. linię pseudorównowagową. Konstrukcja
schodkowania między linią operacyjną a pseudorównowagową daje
rzeczywistą liczbę półek. Na rysunku przedstawiono przykład takiej
konstrukcji. Zaznaczono tam też stężenia faz dla półki 3.
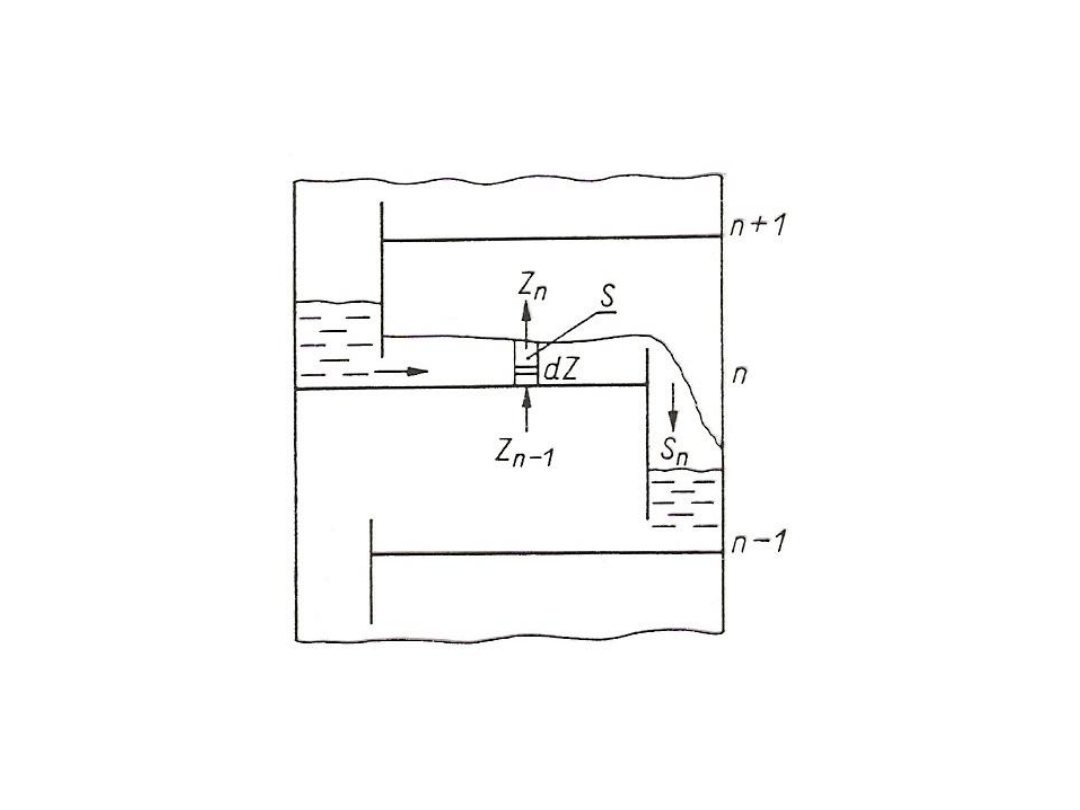
120
Sprawność lokalna półki
Rys. 44. Oznaczenia do definicji sprawności lokalnej półki

121
Pojęcie sprawności lokalnej pozwala na powiązanie
zagadnienia sprawności z teorią wymiany masy. Sprawność lokalna
jest wielkością charakterystyczną dla wybranego punktu na półce –
stąd nazwa. Oznaczenia potrzebne do definicji przedstawiono na
rysunku.
Sprawność lokalna (wyrażona przez stężenia w fazie gazowej) E
g
dla
wybranego punktu na półce wynosi:
E
g
= (Z
n
– Z
n-1
) / (Z*
n
– Z
n-1
)
gdzie: Z*
n
- równowagowe stężenie składnika A do stężenia w cieczy
w danym punkcie, w tym przypadku:
Z*
n
= K S
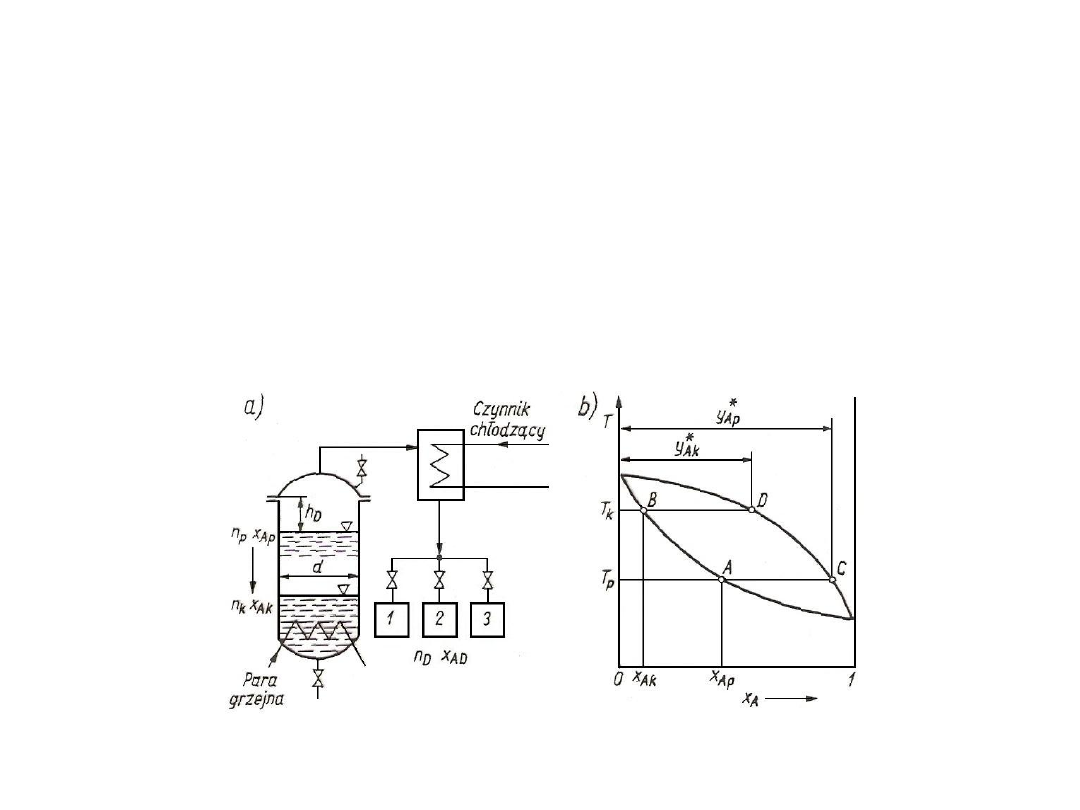
122
Destylacja prosta różniczkowa
(kotłowa)
Na rysunku przedstawiono schemat destylacji różniczkowej
periodycznej. Produkt górny (destylat) jest odbierany kolejno do oddzielnych
zbiorniczków. Otrzymuje się w ten sposób frakcje o różnym składzie składnika
lżejszego (bardziej lotnego). Przebieg takiej destylacji przedstawiono na
wykresie składu. Temperatura w kotle do prowadzenia mieszaniny do wrzenia
zmienia się od T
p
do T
k
, skład cieczy w kotle - od A do B, a skład fazy parowej
(destylatu) – od C do D. Przy założonych stężeniach na początku i na końcu
destylacji można z bilansu obliczyć ilość odparowanej cieczy.
Rys. 45. Schemat prostej destylacji różniczkowej (kotłowej):
a) schemat instalacji, b) wykres składu
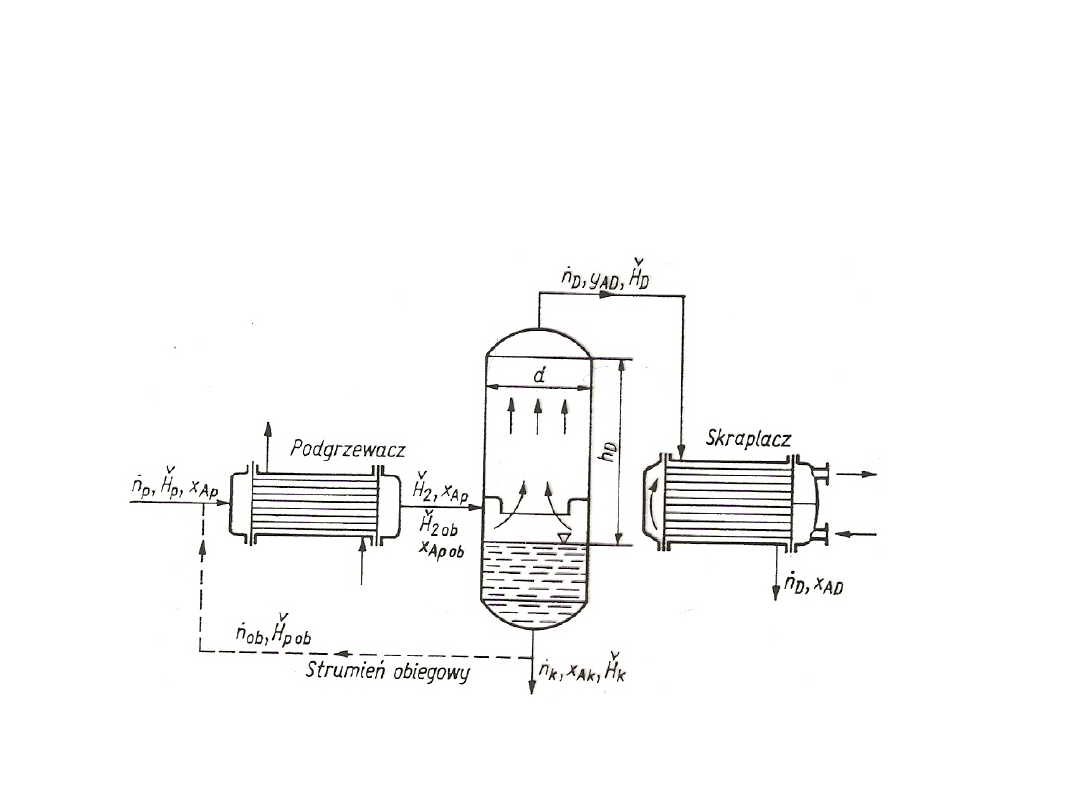
123
Rys. 46. Schemat instalacji do prostej destylacji równowagowej (rzutowej)
Destylację prostą równowagową najczęściej prowadzi się w
sposób ciągły. Jej zasada wynika ze schematu pokazanego na rysunku.
Mieszanina cieczy i pary może pochodzić ze stopnia reakcyjnego lub z
podgrzewacza, w których ciśnienie jest wyższe niż w rozdzielaczu. Taka
metoda destylacji nadaje się szczególnie do mieszanin, których krzywe
składu są od siebie bardzo odsunięte. Wtedy efekt rozdziału przy
jednokrotnym odparowaniu jest dostatecznie duży.
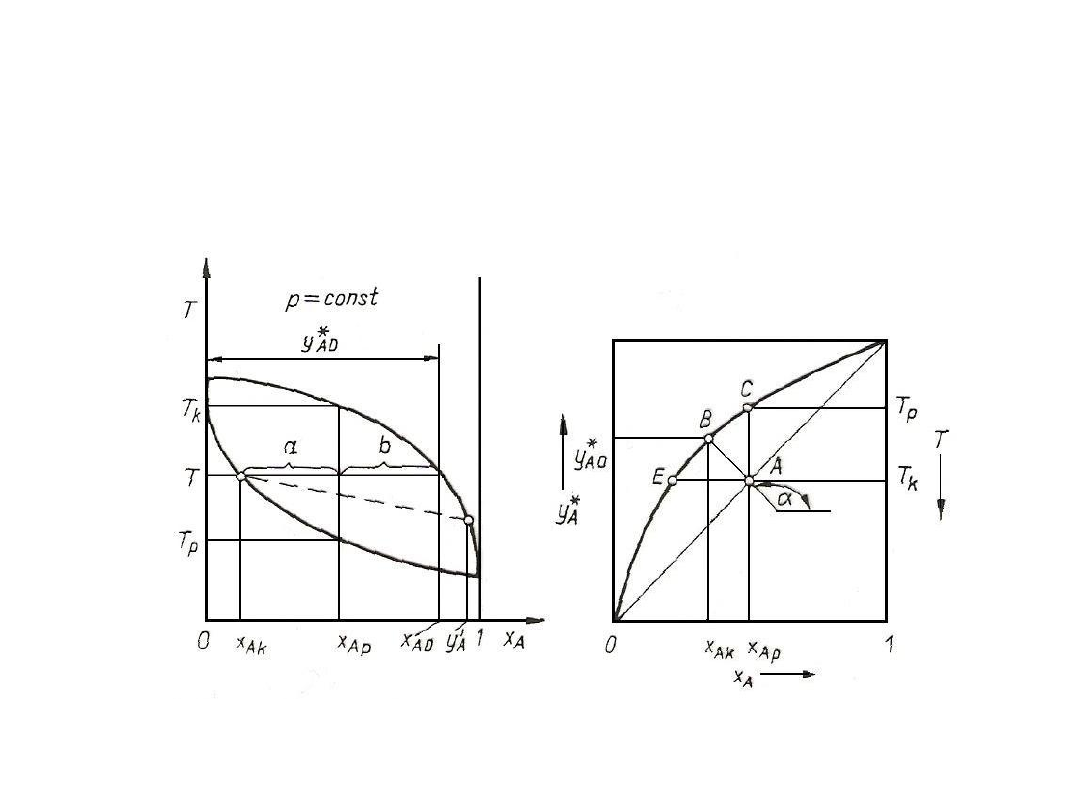
124
Rys. 47. Krzywa składu i krzywa równowagi wyjaśniające zasadę
destylacji równowagowej (rzutowej)
Jeżeli wymieszanie obu faz podczas przepływu przez instalację
nie jest pełne, to ostateczne składy obydwu faz mogą się różnić od
składów wynikających ze stanu równowagi (linia kreskowana na
rysunku). W takiej sytuacji, przy ograniczonym czasie kontaktu obu faz,
separator opuszcza para o stężeniu y`
A
> y*
AD
i rozdział jest lepszy,
ponieważ stężenia nie zdążą się wyrównać.

125
Destylacja z parą wodną
Destylacja z parą wodną służy przede wszystkim do usuwania
różnych zanieczyszczeń z mieszanin o wysokiej temperaturze wrzenia
za pomocą czynnika nie rozpuszczającego lub słabo rozpuszczającego w
fazie ciekłej. Czynnikiem tym jest najczęściej para wodna (niekiedy
stosuje się inny składnik inertny nie mieszający się z cieczą) stąd nazwa
– destylacja z parą wodną. Proces prowadzi się tak, że do cieczy w kotle
doprowadza się nasyconą lub przegrzaną parę wodną. Powoduje to
odparowanie składnika zanieczyszczającego w znacznie niższej
temperaturze. Para wodna spełnia ponadto rolę nośnika ciepła do
podgrzewania cieczy w kotle do temperatury wrzenia i dostarcza ciepła
destylacji.
Temperatury wrzenia mieszaniny można wyznaczyć graficznie.
Ciśnienie par nad mieszaniną np. dwóch składników A i B zupełnie w
sobie nierozpuszczalnych jest równe sumie ciśnień par czystych
składników:
p
An
+ p
Bn
= p
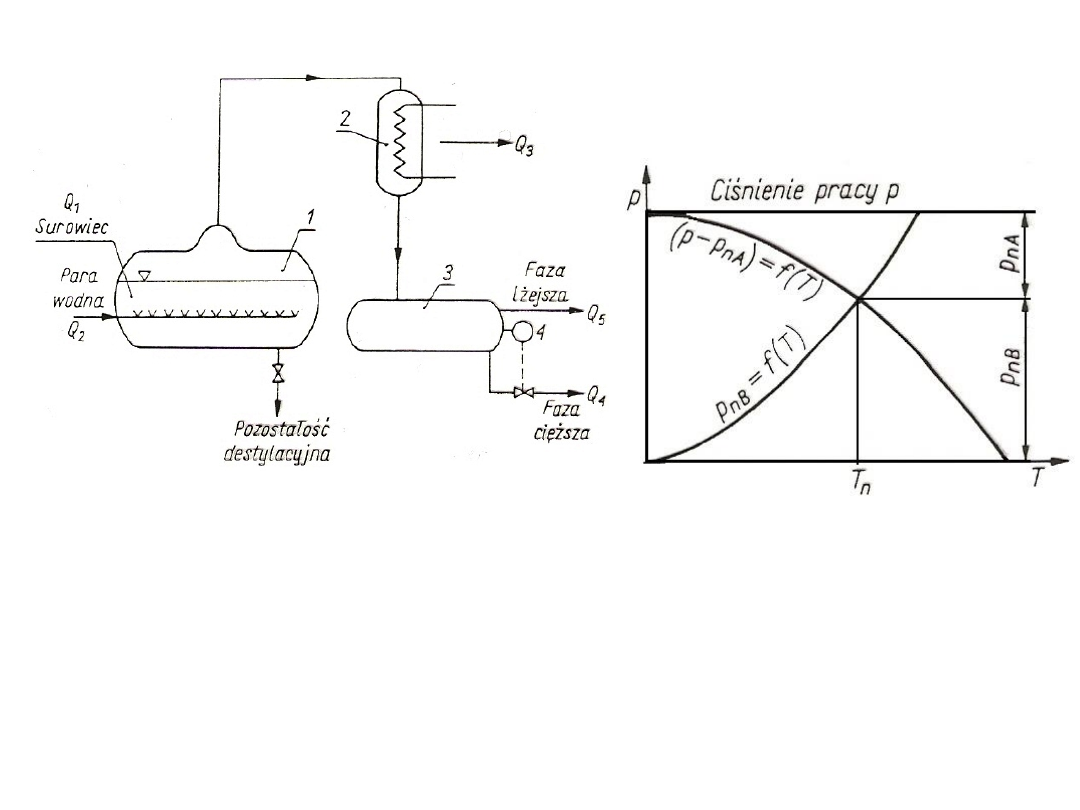
126
Rys. 50. Schemat instalacji periodycznej
do destylacji z parą wodną:
1 – kocioł, 2 –skraplacz,
3 – rozdzielacz faz, 4 – regulator poziomu
Rys. 51. Wykres wyjaśniający
destylację z parą wodną
(sposób wyznaczania
temperatury wrzenia)

127
W celu przeanalizowania procesu sporządza się bilans molowy:
ń
p
= ń
k
+ ń
D
ń
p
x
Ap
= ń
k
x
Ak
+ ń
D
x
AD
Na podstawie tych dwóch równań można napisać:
ń
k
/ ń
D
= (ń
p
– ń
D
) / ń
D
= (x
AD
– x
Ap
) / (x
Ap
– x
Ak
) = b / a = (1 – ) /
gdzie: = ń
D
/ ń
p
–
oznacza stopień odparowania; wielkość
odcinków a oraz b
pokazano na rysunku.
Przebieg procesu destylacji równowagowej mieszaniny
dwuskładnikowej można przedstawić na wykresie krzywej równowagi. Z
punktu A kreśli się linię prostą pod kątem , gdzie tg() = - ń
k
/ń
D
i
otrzymuje się punkt B (y*
AD
, x
Ak
) na linii równowagi.
Linia AB jest linią operacyjną charakteryzującą przebieg procesu:
- gdy = 0 (linia AC), nie ma odparowania, jest tylko podgrzewanie
cieczy do temperatury T
p
;
- gdy = 1 (linia AE), występuje pełne odparowanie odpowiadającego
temperaturze T
k
.

128
W dowolnym punkcie trwania procesu możemy napisać bilans
molowy składnika A:
n x
A
= (n – dn)(x
A
– dx
A
) + dn y*
A
Po wymnożeniu otrzymuje się:
n x
A
= n x
A
– dn x
A
– n (dx
A
) + dn (dx
A
) + dn y*
A
Po uproszczeniu i pominięciu iloczynu dn dx
A
:
dn (y*
A
- x
A
) = n dx
A
albo:
dn / n = dx
A
/ (y*
A
- x
A
)
Równanie całkuje się w granicach od n
p
n
k
i x
Ap
x
Ak
∫
np
nk
dn / n = ∫
xAk
xAp
dx
A
/ (y*
A
- x
A
) = - Ra(całka Rayleigha)
Lewą stronę równania można z łatwością scałkować otrzymując:
[ln(n)]│
np
nk
= ln(n
k
) - ln(n
p
) = - Ra (n
k
/ n
p
) = e
- Ra
lub:
n
k
= n
p
e
- Ra
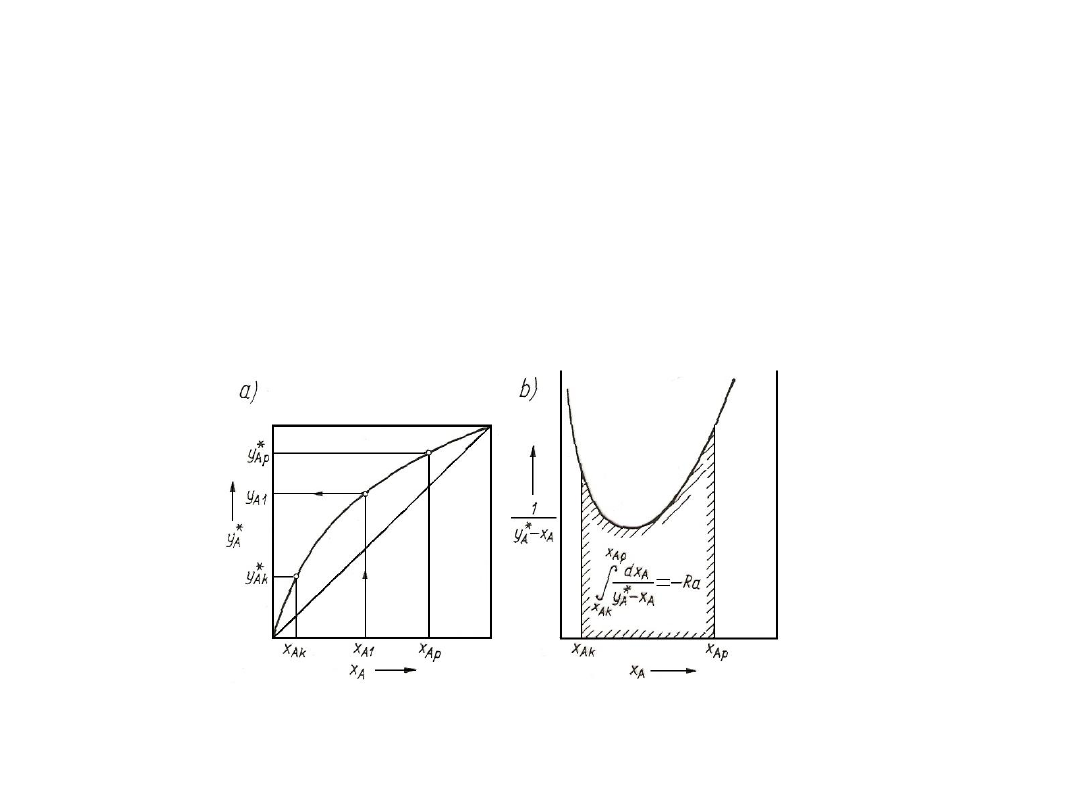
129
Ostateczne zagadnienie sprowadza się do wyznaczenia całki
Rayleigha, którą zwykle otrzymuje się graficznie przez planimetrowanie
powierzchni pod krzywą:
1 / (y*
A
– x
A
) = f(x
A
)
w zaznaczonych granicach.
W przypadku szczególnym, gdy współczynnik względnej
lotności jest prawie wielkością stałą można do całki wstawić y*
A
z
równania Fenskiego:
y*
A
= { ( x
A
) / [1 + x
A
( – 1)] }
Rys. 52. Graficzne wyznaczenie całki Rayleigha:
a) krzywa równowagi, b) wykres pomocniczy
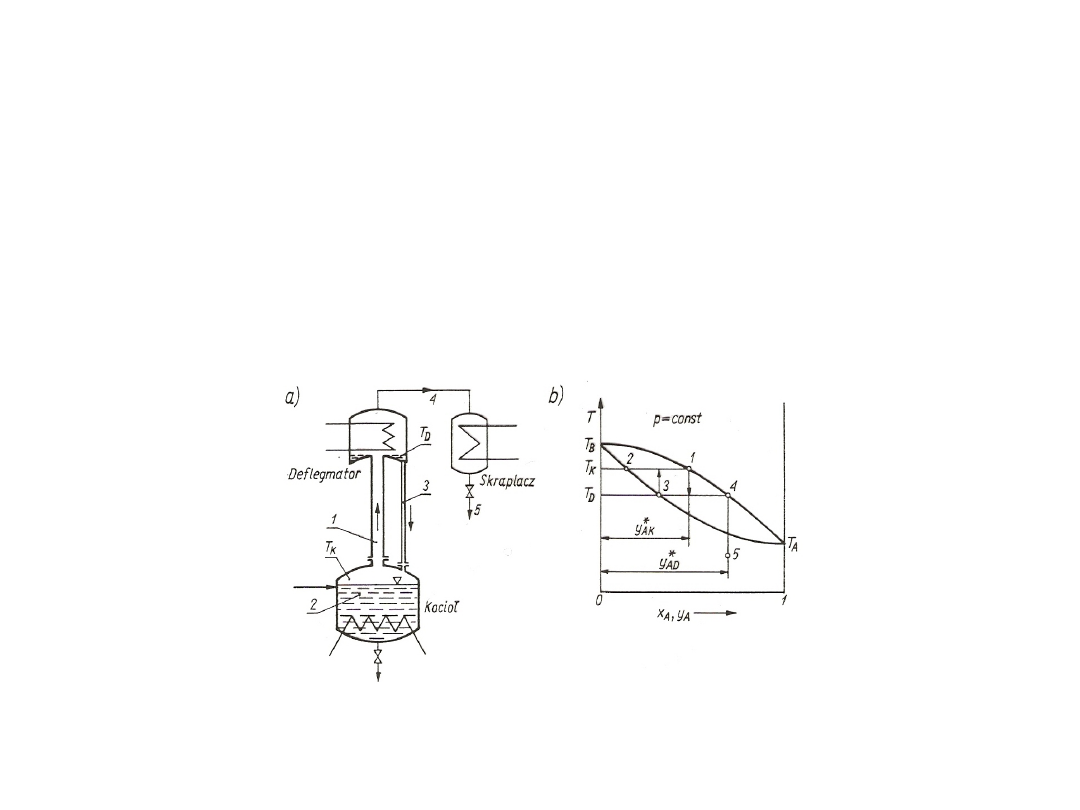
130
Rektyfikacja mieszanin
dwuskładnikowych
Destylacja z deflegmatorem – zakłada się, że poddawana
rektyfikacji mieszanina składa się z dwu składników A i B, przy czym
składnik A jest bardziej lotny.
Pośrednią instalacją między kolumną rektyfikacyjną a destylacją
prostą jest destylacja z deflegmatorem to jest z częściowym
skraplaczem.
Rys. 53. Destylacja z deflegmatorem: a) schemat,
b) wyjaśnienie działania na wykresie składu

131
Jeżeli w kotle i w deflegmatorze przy dwóch różnych temperaturach
T
K
i T
D
ustalą się stany równowagi, to stężenie składnika bardziej lotnego w
oparach z deflegmatora będzie znacznie większe niż w oparach z kotła y
AD
>y
AK
. Skropliny z deflegmatora zawraca się do kotła, ich skład odpowiada
punktowi 3 na wykresie składu, natomiast opary z kotła mają stężenie
odpowiadające punktowi 1. Pozostałość w kotle, tzn. produkt dolny ma
stężenie odpowiadające punktowi 2.
Kolumna rektyfikacyjna z częścią odpędową i wzmacniającą
Jednoczesne otrzymywanie w sposób ciągły obydwu produktów o
odpowiednim stopniu czystości można uzyskać w kolumnie zawierającej
część odpędową (dolną) i część wzmacniającą (górną) – jak to pokazana na
rysunku. Miejsce doprowadzenia surówki dzieli kolumnę na wymienione dwie
części. Jeżeli surówkę doprowadza się w postaci cieczy, dołącza się ona do
fazy ciekłej spływającej z części wzmacniającej, której skład powinien być w
przybliżeniu równy składowi surowca i wspólnie spływają przez część
odpędową wzbogacając się w składnik mniej lotny. Na dole jest odbierana
ciecz wyczerpana (produkt dolny), a górą rektyfikat (produkt górny).
Tego rodzaju instalacje rektyfikacyjne mogą mieć również wbudowany
deflegmator (częściowy skraplacz), w którym przez regulację ilości wody
chłodzącej tylko część par ulega skraplaniu, a resztę stanowi destylat parowy
przepływający do kondensatora. kondensator może być też ustawiony w dole
kolumny, co wymaga pompowania skroplin, ale oszczędza tłoczenia wody
chłodzącej. Zamknięcie hydrauliczne między kolumną deflegmatorem
uniemożliwia ucieczkę pary z pominięciem deflegmatora. Element grzejny
(wyparka) może znajdować się w korpusie kolumny u dołu, albo na zewnątrz
w postaci recyrkulatora.

132
Rektyfikacja adiabatyczna ciągła z częścią wzmacniającą
W dążeniu do jeszcze lepszego rozdziału mieszaniny wlotowej na
składniki o bardzo dużej czystości należy stosować wiele stopni rozdziału,
tzn. kolumnę rektyfikacyjną.
Zasadę działania kolumny rektyfikacyjnej wyjaśniono na wykresie
składu. Założono ustalenie się temperatury na każdym stopniu aparatu
kolumnowego i tym samym osiągnięcie kolejnych stanów równowagi. Od
stopnia do stopnia można prześledzić wzbogacenie się oparów w składnik
lżejszy A i coraz większe zubożenie w ten składnik spływającej w dół
kolumny fazy ciekłej, która jednocześnie wzbogaca się w składnik B.
Jeżeli kolumna zawiera dużo takich stopni, to istnieje możliwość
otrzymania bardzo czystych składników. Elementy zabudowy kolumny
(półki lub wypełnienia) maja za zadanie wytworzenie dużej powierzchni
styku obydwu faz i dużej turbulencji przepływu, co przyczynia się do
intensywnej wymiany ciepła i masy między strumieniami cieczy i pary.
Ze skraplacza jedynie część cieczy odbiera się jako destylat
(produkt górny), część natomiast misi być zawrócona na szczyt kolumny
jako powrót (refluks, orosienie). Ze względu na przepustowość instalacji
odbiór destylatu jako produktu górnego powinien być jak największy, a
kolumna powinna zawracać tylko minimalną jego część. O wielkości
zawracania destylatu jest mowa w dalszym toku analizy pracy kolumny.
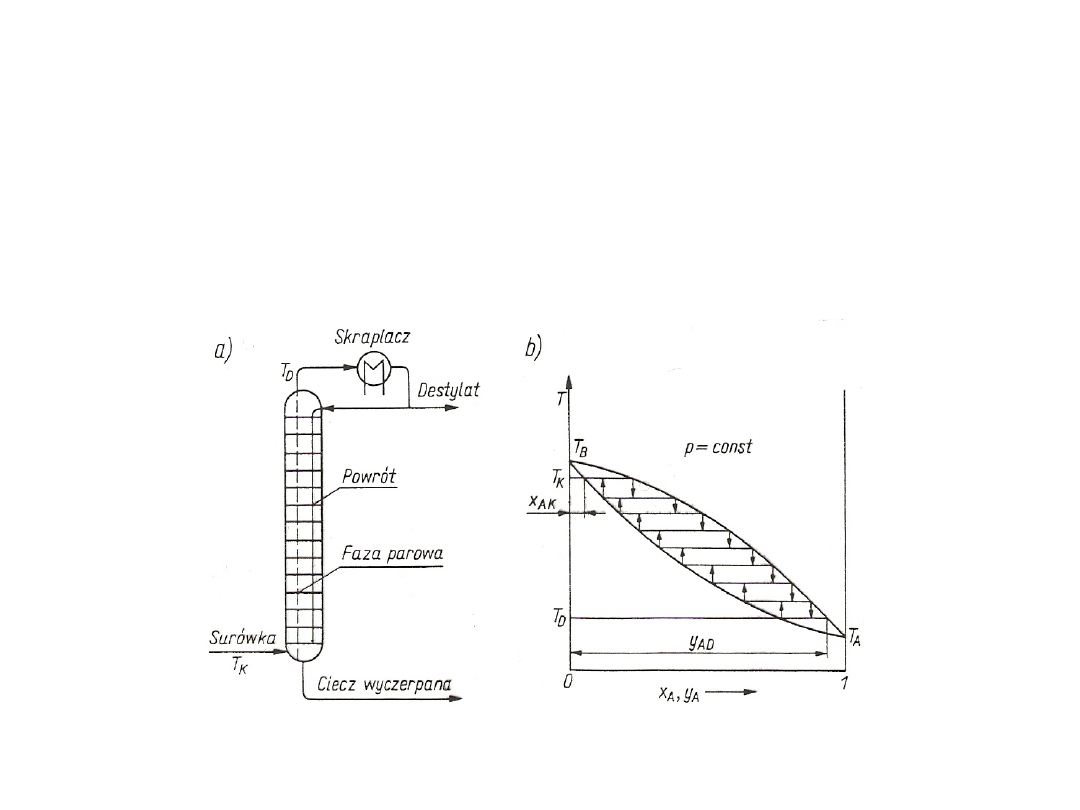
133
Przedstawiona na rysunku instalacja rektyfikacji zawiera jedynie
część wzmacniającą, leżącą powyżej miejsca doprowadzenia surówki.
Tego rodzaju kolumny są stosowane bardzo często do pracy okresowej.
Kocioł (nie pokazany na schemacie) napełnia się surowcem i prowadzi
rektyfikację, otrzymując u góry kolumny składnik bardziej lotny, a u dołu
w kotle – jako pozostałość – składnik cięższy (mniej lotny). Kolumna taka,
pracująca w sposób ciągły, dostarcza jedynie produkt górny o
wymaganej czystości, natomiast u dołu jest odprowadzany produkt
dolny – wzbogacony wprawdzie w składnik mniej lotny, ale o
niewystarczającym stopniu czystości.
Rys. 54. Rektyfikacja ciągła tylko z częścią wzmacniającą:
a) schemat, b) wyjaśnienie działania na wykresie składu

Zasady bilansowe w procesach
absorpcji i desorpcji
Rozważania ogólne
Jeżeli w danym procesie rozpuszczalnik jest obojętny
(inertny), tzn. nie bierze udziału w transporcie masy, to jego ilość w
poszczególnych fazach jest stała. Fakt ten ułatwia bilansowanie
procesu, pod warunkiem odpowiedniego doboru rodzaju strumieni i
stężeń. W przypadku procesów absorpcyjno-desorpcyjnych najczęściej
przyjmuje się strumienie molowe rozpuszczalnika tzn. ń
cr
i ń
gr
oraz
stosunki molowe składnika wymienianego tzn. X
A
i Y
A
.
UWAGA: w skrypcie masowe natężenie przepływu:
G
A
ilość absorbowanego składnika A, kg/s;
G
i
natężenie przepływu inertu gazowego, kg/s;
L
i
natężenie przepływu inetru ciekłego, kg/s;
X, Y stosunki masowe składnika A w cieczy i w gazie.
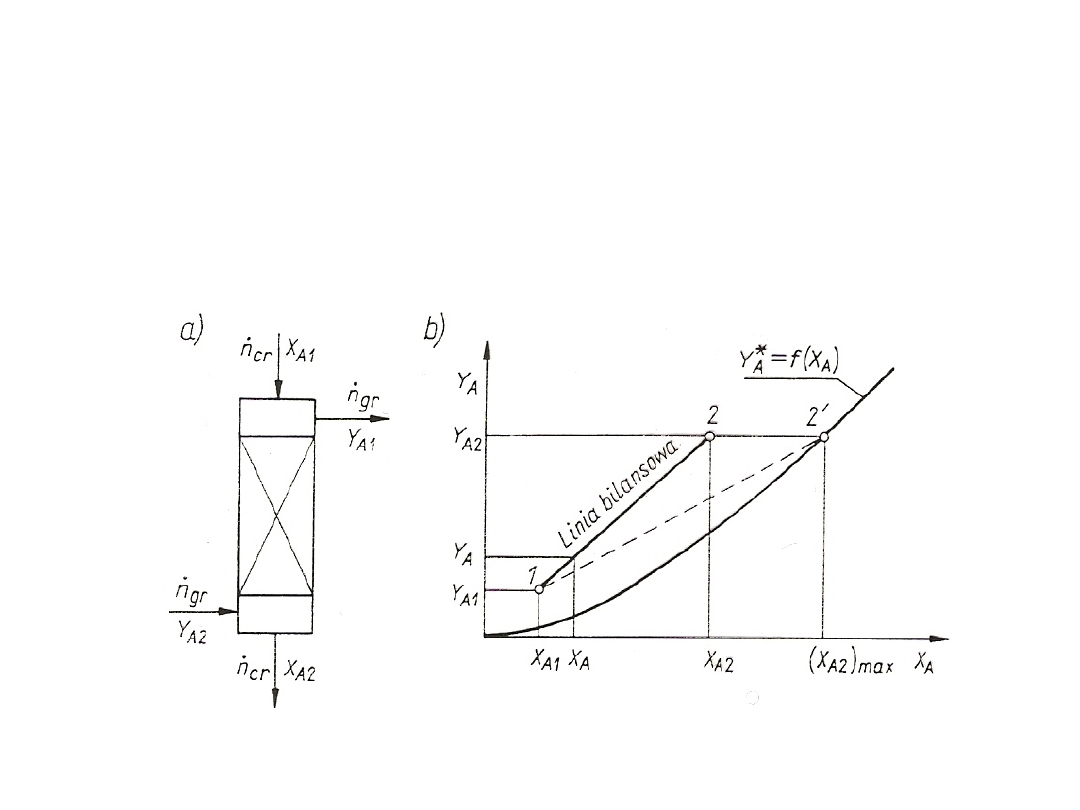
135
Bilans procesu absorpcji
W procesie absorpcji, którego celem jest usunięcie składnika A z
fazy gazowej najczęściej zadane z góry są stężenia tego składnika na
wlocie i wylocie absorbera np. Y
A2
– wlot i Y
A1
– wylot. Zazwyczaj znane
jest też stężenie składnika A w świeżej cieczy chłonnej doprowadzanej do
aparatu np. X
A1
. Przypadek ten przedstawiono na rysunku:
Rys. 55. Schemat absorpcji składnika A z fazy gazowej za pomocą cieczy
chłonnej:
a) oznaczenia strumieni i stężeń, b) wykres stężeń

136
Jeżeli znane są strumienie ń
cr
i ń
gr
, to powinno być spełnione
równanie bilansowe:
ń
A
= ń
cr
(X
A2
– X
A1
) = ń
gr
(Y
A2
– Y
A1
)
Równanie to umożliwia obliczenie strumienia absorbowanego składnika, a
także stężenia składnika A w cieczy opuszczającej absorber X
A2
. Jeżeli
podobne równanie bilansowe napisze się dla górnej części absorbera
ograniczonej przekrojem, w którym stosunki molowe w cieczy i gazie
wynoszą X
A
i Y
A
, to otrzyma się równaie linii bilansowej w postaci:
Y
A
= (ń
cr
/ ń
gr
) X
A
+ [Y
A1
- (ń
cr
/ ń
gr
) X
A1
]
Gdy wartość (ń
cr
/ ń
gr
) jest stała, wówczas jest to równanie linii prostej. Na
rysunku jest to linia prosta przechodząca przez punkty 1 i 2. Nachylenie
linii bilansowej jest określone stosunkiem (ń
cr
/ ń
gr
) . Gdy stosunek ten
maleje, wówczas przy stałym położeniu punktu 1 linia bilansowa przesuwa
się w kierunku linii równowagi. W skrajnym przypadku linia bilansowa
przecina się z linią równowagi w punkcie 2`. Wartość parametru w tym
przypadku nosi nazwę minimalnego stosunku orosienia, dla którego
stężenie składnika A w cieczy osiąga wartość maksymalną (X
A2
)
max
.
Ponieważ punkt 2` leży na linii równowagi, wartość (X
A2
)
max
odpowiada
równowagowemu stężeniu składnika A w cieczy do wartości Y
A2
, czyli:
(X
A2
)
max
= X*
A
(Y
A2
) = Y
A2
/ K
XY
Znajomość stężenia (X
A2
)
max
umożliwia obliczenie minimalnego stosunku
orosienia:
R
min.
= (ń
cr
/ ń
gr
)
min.
= (Y
A2
- Y
A1
) / [(X
A2
)
max
- X
A1
]

137
Minimalnemu stosunkowi orosienia odpowiada przerywana linia
bilansowa 1-2` na rysunku.
Stosując minimalną ilość cieczy chłonnej, podobnie jak w
procesie destylacji, można osiągnąć założony rozdział mieszaniny
jedynie przy nieskończenie dużej liczbie stopni rozdziału. Rzeczywista
wartość R musi być większa i dobiera się ją zwykle na podstawie
doświadczeń (praktyki przemysłowej) za pomocą zależności:
R = k R
min.
gdzie: k – liczna większa od jedności (najczęściej zawarta w granicach
od 1,3 do 1,6)
W procesie desorpcji najczęściej z góry zadane są stężenia
składnika A w cieczy na wlocie X
A2
, na wylocie X
A1
oraz stężenie A w
świeżym gazie Y
A1
. Bilans ogólny procesu jest identyczny jak bilans
procesu absorpcji.
Wielkością analogiczną do stosunku orosienia jest stosunek strumieni
fazy gazowej i ciekłej. Stosunek ten oznaczymy przez R
g
:
R
g
= ń
gr
/ ń
cr
Po zbilansowaniu dolnej części desorbera do pewnego przekroju
otrzymuje się równanie linii bilansowej w postaci:
Y
A
= (1 / R
g
) X
A
+ [Y
A1
– (X
A1
/ R
g
)]

138
Jeżeli R
g
=const to równanie jest równaniem linii prostej. Na rysunku jest
to prosta łącząca punkty 1 i 2. Do zrealizowania założonego rozkładu
stężeń konieczne jest, aby stosunek R
g
był większy od pewnej wartości
minimalnej (R
g
)
min
. Ponieważ linia równowagi w układach absorpcyjno-
desorpcyjnych zazwyczaj jest krzywą wklęsłą, więc przy wyznaczaniu
wartości (R
g
)
min
mogą zajść dwa przypadki. W przypadku gdy linia
równowagi jest słabo wklęsła lub wypukła, punkt 2` odpowiadający
minimalnej wartości R
g
jest punktem przecięcia linii bilansowej z linią
równowagi. Wylotowe stężenie składnika A w gazie wylotowym osiąga
wartość maksymalną (Y
A2
)
max
. Minimalną wartość (R
g
)
min
można obliczyć
ze wzoru:
(R
g
)
min
= (X
A2
– X
A1
) / [(Y
A2
)
max
– Y
A1
]
Jeżeli krzywa równowagi jest bardziej wklęsła (np. w zakresie małych
stężeń w układzie SO
2
-H
2
O) to punkt 2` odpowiadający minimalnej
wartości R
g
jest punktem styczności linii równowagowej i bilansowej.
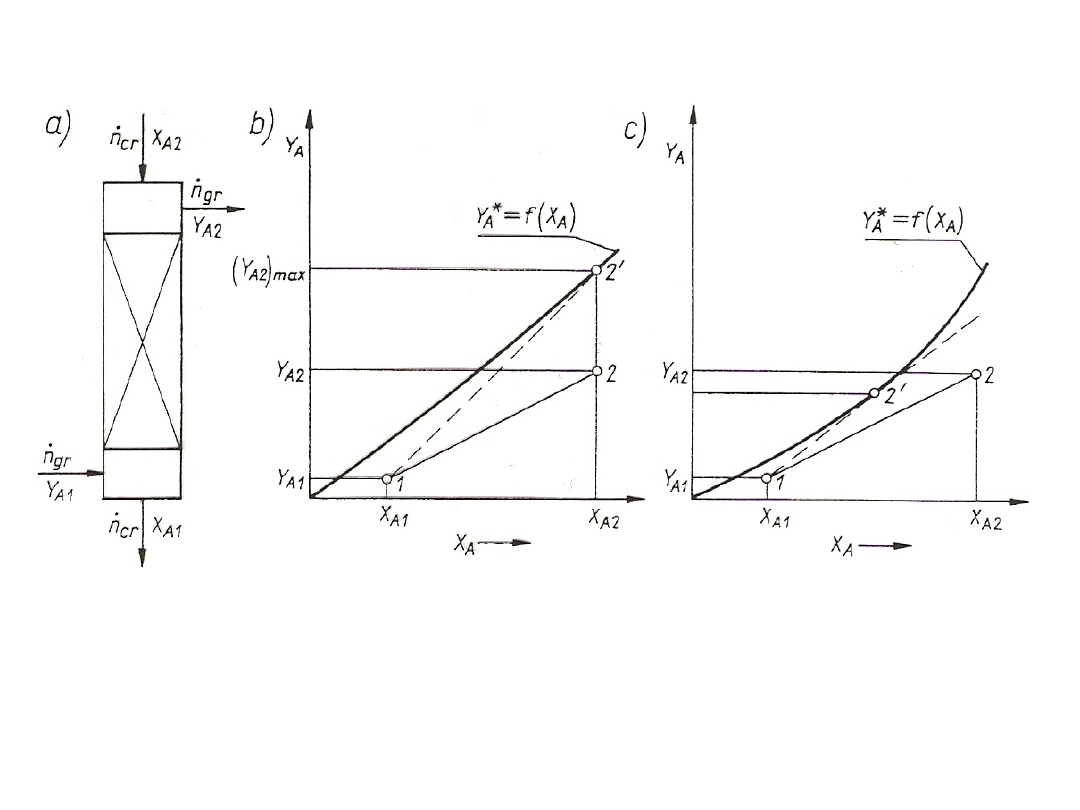
139
Rys. 56. Schemat desorpcji składnika A z cieczy za pomocą gazu:
a) oznaczenia strumieni i stężeń,
b) krzywa równowagi słabo wklęsła lub wypukła,
c) krzywa równowagi silnie wklęsła

140
Cyrkulacja cieczy chłonnej w
procesie absorpcji
Jeżeli ilość cieczy chłonnej, wynikają ca z bilansu molowego, jest
stosunkowo niewielka i nie gwarantuje właściwego zwilżania
powierzchni wypełnienia absorbera albo w czasie procesu absorpcji
wydzielają się bardzo duże ilości ciepła, to stosuje się cyrkulację cieczy
z chłodzeniem. Bilans materiałowy takiego układu jest następujący:
ń
gr
(Y
Ap
- Y
Ak
) = z ń
cr
(X
Ak
- X
Ap
)
gdzie: z – krotność cyrkulacji (stosunek ilości cieczy zraszającej do ilości
cieczy świeżej).
Ten sam bilans dla przypadku bez cyrkulacji ma postać:
ń
gr
(Y
Ap
- Y
Ak
) = ń
cr
(X
Ak
- X
Ap
)
Z porównania równań otrzymuje się krotność cyrkulacji
wyrażoną stężeniami:
z = (X
Ak
- X
Ap
) / (X
Ak
– X`
Ap
) > 1
Wymieszanie się strumieni cieczy o stężeniu i daje pewne
stężenia pośrednie:
X`
Ap
= [X
Ap
+ (z – 1) X
Ak
] / z
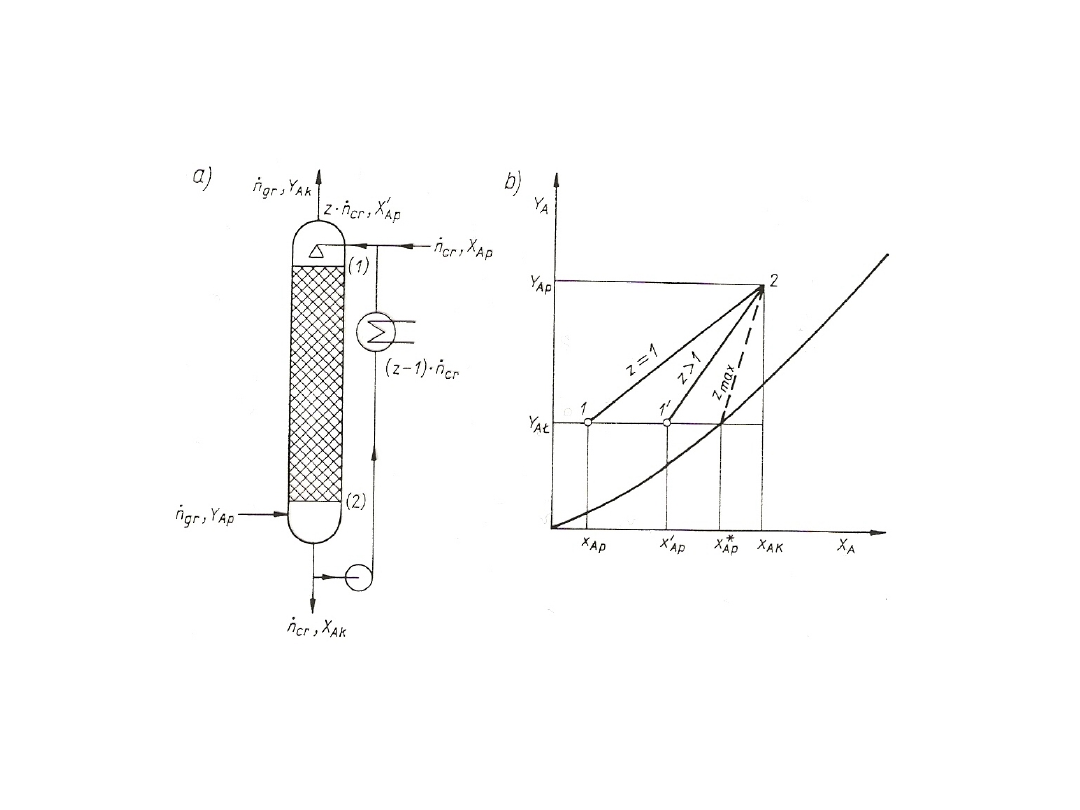
141
Rys. 57. Zależność położenia linii operacyjnej od krotności cyrkulacji:
a) schemat, b) wykres operacyjny

142
Zwiększenie strumienia cieczy cyrkulacyjnej powoduje bardziej
stromy przebieg linii bilansowej, a tym samym zmniejsza średni moduł
napędowy transportu masy. Jest to duża wada tego rozwiązania.
W przypadku gdy:
z = z
max
= (X
Ak
- X
Ap
) / (X
Ak
– X*
Ap
)
wówczas średni moduł napędowy maleje do zera, co uniemożliwia
stosowanie takiej cyrkulacji w praktyce.
Rozłożenie procesu absorpcji na kilka stopni zmniejsza ujemny
wpływ cyrkulacji na moduł napędowy. Rezultat jest tym lepszy, im więcej
stopni jest zastosowanych w procesie.
Jak widać z wykresu na rysunku przy zastosowaniu cyrkulacji
jednostopniowej linia bilansowa I przecięłaby linię równowagi, co
uniemożliwiłoby przeprowadzenie procesu. Na podstawie teoretycznych
rozważań Hoblera można stwierdzić, że zastosowanie cyrkulacji cieczy
daje duże korzyści tam, gdzie jest mało cieczy chłonnej i gdzie moduły
napędowe transportu masy są dość duże.
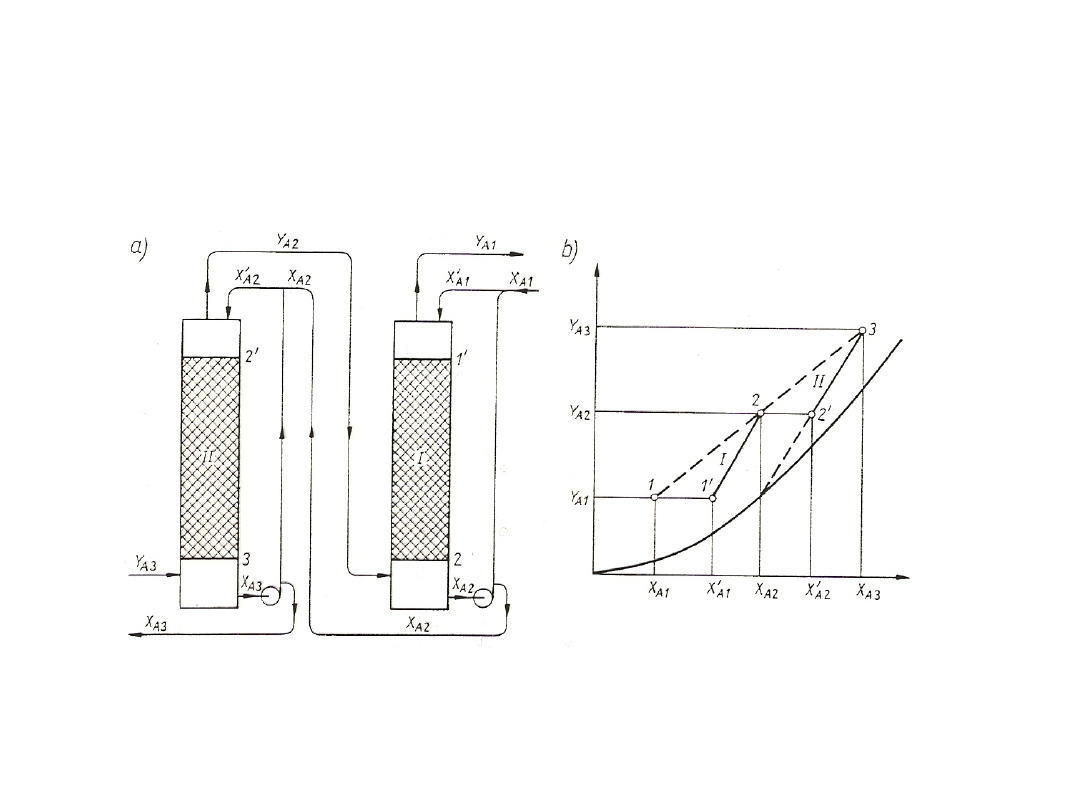
143
Rys. 58. Dwustopniowy proces absorpcji z cyrkulacją cieczy:
a) schemat, b) wykres operacyjny.
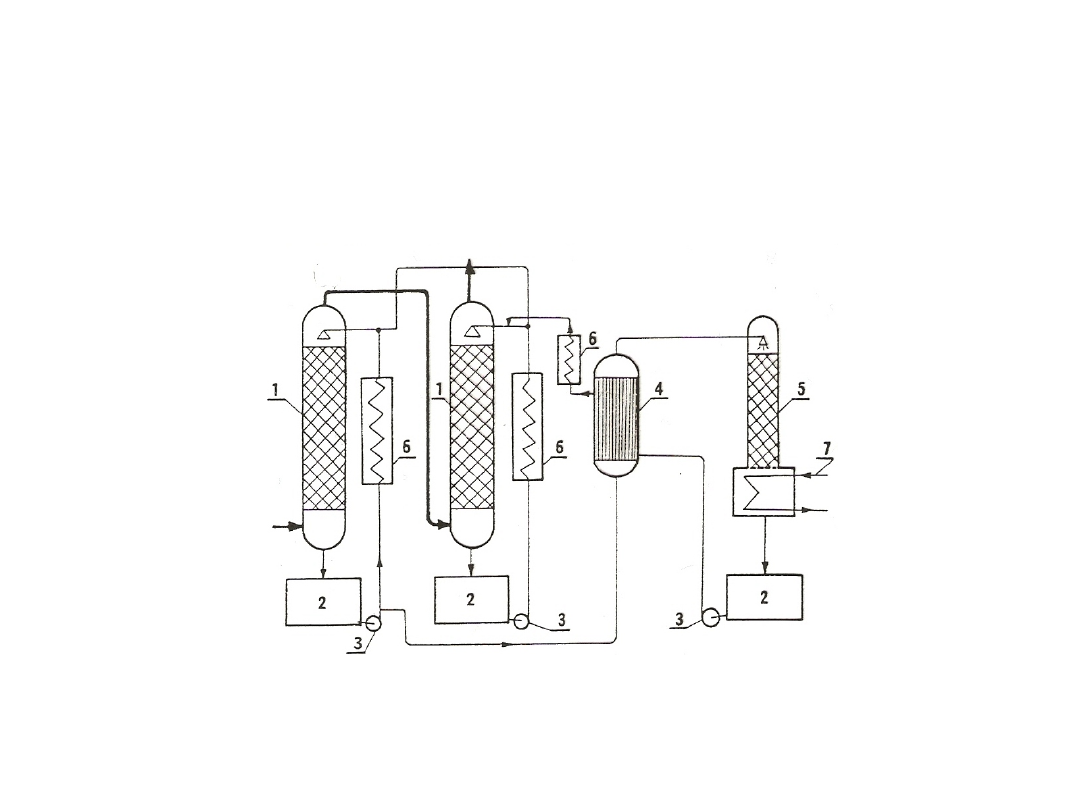
144
Korzystne warunki dla procesu absorpcji (wysokie ciśnienie i
niska temperatura) są przeciwieństwem warunków korzystnych dla
procesu desorpcji. Kierunek przebiegu procesu transportu masy zależy
każdorazowo od stanów równowagi w danych warunkach.
Na rysunku przedstawiono typowy schemat instalacji
absorpcyjno-desorpcyjnej z cyrkulacją cieczy chłonnej.
Rys. 59. Schemat instalacji absorpcyjno-desorpcyjnej z cyrkulacją cieczy:
1 – absorbery; 2 – zbiorniki, 3 – pompy, 4 – wymiennik ciepła,
5 – kolumna desorpcyjna; 6- chłodnice, 7 – para grzejna

145
Ciecz absorpcyjna (chłonna) powinna charakteryzować się:
- dużą rozpuszczalnością wymaganego składnika (korzystnym
przebiegiem linii równowagi);
- odpowiednią selektywnością, np. podczas absorpcji fizycznej
rozpuszczalność innych składników nie powinna przekraczać ilości 1/5
składnika absorbowanego;
- łatwością regeneracji (nie powinna tworzyć azeotropu z
absorbowanym składnikiem, powinna wykazywać dużą różnicę
temperatur wrzenia w stosunku do absorbowanego składnika oraz mieć
korzystny przebieg linii równowagi desorpcyjnej);
- niskim ciśnieniem par w temperaturze procesu absorpcji w celu
zmniejszenia strat cieczy chłonnej przez parowanie;
- małą lepkość;
- brak własności korodujących;
- brakiem toksyczności;
- niską ceną zakupu;
- brakiem ujemnego wpływu ewentualnego zanieczyszczenia
przerabianej mieszaniny cieczą chłonną na ilość produktów.
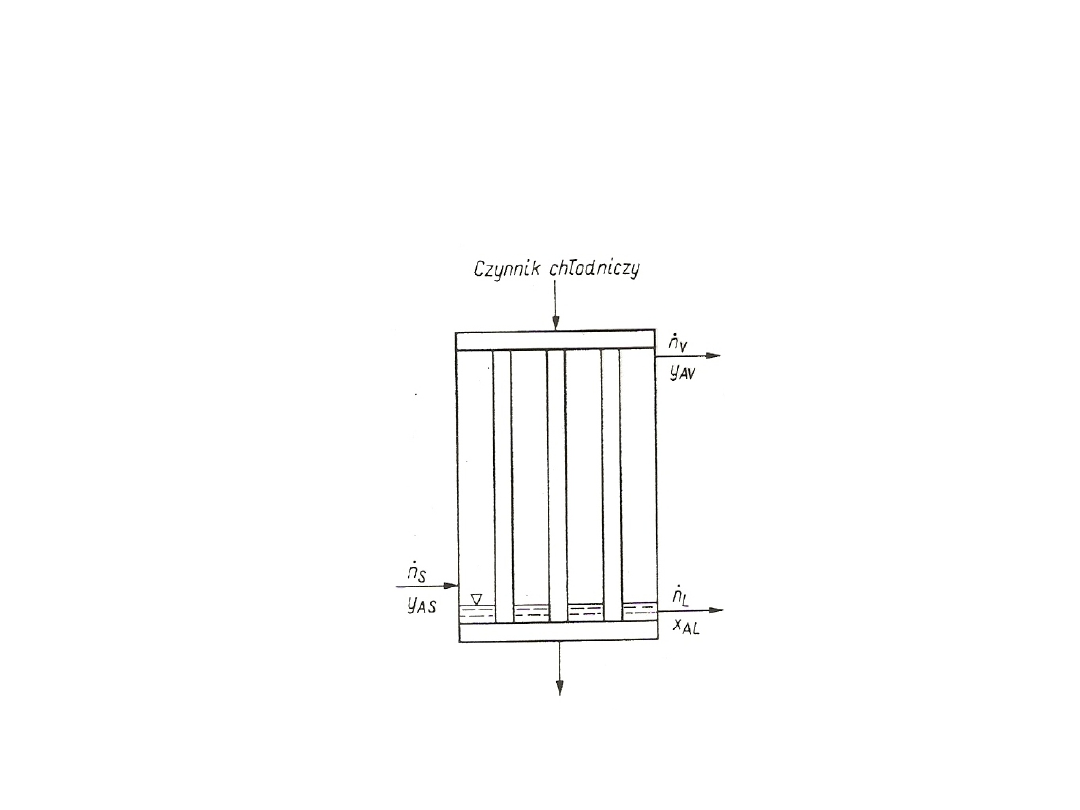
146
Kondensacja częściowa
Procesem analogicznym do destylacji różniczkowej jest
kondensacja częściowa. odbywa się ona w aparatach nazywanych
deflegmatorami. Schematycznie proces ten przedstawiono na rysunku.
Rys. 60. Schemat kondensacji częściowej w deflegmatorze

147
Deflegmator jest typowym wymiennikiem ciepła, najczęściej
rurkowym, ustawionym pionowo. Jako surowiec do deflegmatora dopływa
mieszanina par różnych składników w ilości ń
S
. Ułamek molowy bardziej
lotnego składnika A w surowcu wynosi y
AS
. Przepływająca w górę para
kondensuje się częściowo na powierzchni chłodzonej, wzbogacając się
jednocześnie w składnik A. Nie skroplona część par w ilości ń
V
jest
odbierana z górnej części aparatu. Ułamek molowy składnika A w tym
produkcie wynosi y
AV
. Kondensat spływa po powierzchni chłodzonej i
odpływa dołem aparatu w ilości ń
AL
zubożony w składnik A, którego ułamek
molowy wynosi x
AL
. Istotnym parametrem w procesie jest stosunek
kondensacji r zdefiniowany wzorem:
r = ń
L
/ ń
V
= (ń
S
– ń
V
) / ń
V
Równania bilansu molowego procesu mają postać:
ń
S
= ń
L
+ ń
V
ń
AS
y
AS
= ń
L
x
AL
+ ń
V
y
AV
x
AL
= y
AS
– [ (y
AV
– y
AS
) / r ]
Stosunek kondensacji r zależy od intensywności chłodzenia. Dla
mieszaniny dwuskładnikowej, przy założeniu że w każdym przekroju
skraplacza ciecz jest w równowadze z parą, można wyprowadzić zależność
między wartością r a ułamkami molowymi y
AS
i y
AV
:
ln (r + 1) = ∫
yAS
yAV
[ dy
A
/ (y
A
– x*
A
)]
gdzie: x*
A
- wartość równowagowa do ułamka y
A
na styku faz w dowolnym
przekroju deflegmatora

148
Zakładając, że w rozważanym zakresie stężeń współczynnik
względnej lotności jest stały:
x*
A
= y
A
/ [ – y
A
( -1)]
r = (y
AV
/ y
AS
)
/( -1)
[(1 - y
AS
) / (1- y
AV
)]
1/( -1)
- 1
W przypadku szczególnym, gdy współczynnik względnej
lotności w zaznaczonym zakresie jest wielkością prawie stałą:
ln (n
k
/ n
p
) = - ∫
xAk
xAp
{ dx
A
/ [ ( ( x
A
) / (1 + x
A
( -1)) ) – x
A
] }
Po scałkowaniu i przyjęciu, że n
D
= n
p
– n
k
otrzymuje się:
n
D
= n
p
{ 1 – [ 1 / ( (x
Ap
/x
Ak
)
1/(-1)
( (1-x
Ak
)/(1-x
Ap
) )
1/(-1)
) ] }

149
Ogólne wskazania przy projektowaniu
instalacji do prowadzenia procesów
dyfuzyjno-cieplnych
Podczas projektowania instalacji do procesów dyfuzyjno-
cieplnych należy uwzględnić podane niżej czynności w takiej lub innej
kolejności w zależności od indywidualnych warunków:
1. Wybór najodpowiedniejszego procesu dla zadanego problemu
rozdziału mieszaniny. Oprócz właściwości rozdzielanej mieszaniny
należy uwzględnić również ekonomikę procesu, jego wpływ na
środowisko i niezawodność eksploatacji.
2. Zebranie danych fizyczno-chemicznych obydwu faz (stany
równowag).
3. Badanie procesowe mające na celu dopasowanie procesu do danego
przypadku rozdziału mieszaniny, tzn. właściwy dobór zabudowy
wnętrza aparatu i jego kształtu. Badania mogą być prowadzone w
skali laboratoryjnej, a jeśli to konieczne, to w instalacji pilotowej. Przy
przeniesieniu wyników należy uwzględnić problemy powiększania
skali.
4. Ustalenie sposobu eksploatacji aparatu i jego warunków ruchu.
5. Wykonanie obliczeń procesowych aparatu:
a) wstępne oszacowanie długości lub wysokości aparatu na
podstawie skróconych obliczeń rachunkowych lub graficznych;
b) iteracyjne powtórne przeliczenie;

150
c) uwzględnienie sprawności stopni rozdziału na podstawie badań
półtechnicznych (najlepiej) lub danych z przemysłu;
d) rozplanowanie wybranej zabudowy wnętrza aparatu zgodnie z
wynikami przeprowadzonych badań i sprawdzonych danych z
literatury.
6. Zaprojektowanie lub dobranie zgodnie z zasadami techniki
procesowej wszystkich elementów peryferyjnych, np. pomp,
wymienników ciepła, zbiorników itp..
7. Przeprowadzenie próby optymalizacji instalacji do rozdziału
mieszaniny, np. ze względu na minimum kosztów ogólnych.
8. Opracowanie schematu urządzeń kontrolno-pomiarowych i
regulujących dla całej instalacji.
9. Przedstawienie rozwiniętego schematu całej projektowanej
instalacji.
10. Przeliczenie i przedstawienie schematu pełnej sieci rurociągów
danej instalacji.
11. Przeliczenia wytrzymałościowe i konstrukcja elementów
projektowanej instalacji.
12. Ustalenie przestrzennego rozmieszczenia elementów instalacji
(plan fundamentów, izometryczny plan sieci rurociągów,
ewentualnie model instalacji w odpowiednio dobranej skali).
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
- Slide 51
- Slide 52
- Slide 53
- Slide 54
- Slide 55
- Slide 56
- Slide 57
- Slide 58
- Slide 59
- Slide 60
- Slide 61
- Slide 62
- Slide 63
- Slide 64
- Slide 65
- Slide 66
- Slide 67
- Slide 68
- Slide 69
- Slide 70
- Slide 71
- Slide 72
- Slide 73
- Slide 74
- Slide 75
- Slide 76
- Slide 77
- Slide 78
- Slide 79
- Slide 80
- Slide 81
- Slide 82
- Slide 83
- Slide 84
- Slide 85
- Slide 86
- Slide 87
- Slide 88
- Slide 89
- Slide 90
- Slide 91
- Slide 92
- Slide 93
- Slide 94
- Slide 95
- Slide 96
- Slide 97
- Slide 98
- Slide 99
- Slide 100
- Slide 101
- Slide 102
- Slide 103
- Slide 104
- Slide 105
- Slide 106
- Slide 107
- Slide 108
- Slide 109
- Slide 110
- Slide 111
- Slide 112
- Slide 113
- Slide 114
- Slide 115
- Slide 116
- Slide 117
- Slide 118
- Slide 119
- Slide 120
- Slide 121
- Slide 122
- Slide 123
- Slide 124
- Slide 125
- Slide 126
- Slide 127
- Slide 128
- Slide 129
- Slide 130
- Slide 131
- Slide 132
- Slide 133
- Slide 134
- Slide 135
- Slide 136
- Slide 137
- Slide 138
- Slide 139
- Slide 140
- Slide 141
- Slide 142
- Slide 143
- Slide 144
- Slide 145
- Slide 146
- Slide 147
- Slide 148
- Slide 149
- Slide 150
Wyszukiwarka
Podobne podstrony:
Dyfuzja prezentacja
DYFUZJONIZM prezentacja
prezentacja finanse ludnosci
prezentacja mikro Kubska 2
Religia Mezopotamii prezentacja
Prezentacja konsument ostateczna
Strategie marketingowe prezentacje wykład
motumbo www prezentacje org
lab5 prezentacja
Prezentacja 18
Materialy pomocnicze prezentacja maturalna
Prezentacja na seminarium
Lato prezentacja 3
Prezentacja1
więcej podobnych podstron