
HUMAN MUTATION Mutation in Brief #815 (2005) Online
MUTATION IN BRIEF
© 2005 WILEY-LISS, INC.
Received 13 November 2004; accepted revised manuscript 21 March 2005.
Two ATM Variants and Breast Cancer Risk
Deborah Thompson
1/8
, Antonis C. Antoniou
1
, Mark Jenkins
2
, Anna Marsh
3
, Xiaoqing Chen
3
,
Tierney Wayne
4
, Andrea Tesoriero
5
, Roger Milne
2
, Amanda Spurdle
3
, Yvonne Thorstenson
4
,
Melissa Southey
5
, Graham G. Giles
6
, kConFab Investigators
7
, Kum Kum Khanna
3
,
Joseph Sambrook
7
, Peter Oefner
4
, David Goldgar
8
, John L. Hopper
2
, Doug Easton
1
and
Georgia Chenevix-Trench
3
1
Cancer Research United Kingdom Genetic Epidemiology Unit, University of Cambridge, Cambridge, United
Kingdom;
2
Centre for Genetic Epidemiology, The University of Melbourne, Melbourne, Australia;
3
Queensland
Institute of Medical Research, Brisbane, Australia;
4
Stanford Genome Technology Center, Palo Alto, California;
5
Genetic Epidemiology Laboratory, Pathology Department, University of Melbourne, Melbourne, Australia;
6
Cancer Epidemiology Centre, The Cancer Council Victoria, Melbourne, Australia;
7
Peter MacCallum Cancer
Institute, Melbourne, Australia;
8
Unit of Genetic Epidemiology, International Agency for Research on Cancer,
Lyon, France
*Correspondence to: Dr Georgia Chenevix-Trench, Queensland Institute of Medical Research, c/o Royal Brisbane
Hospital Post Office, Herston, Queensland 4029 Australia; E-mail: georgiaT@qimr.edu.au
Grant sponsor: National Health and Medical Research Council, National Breast Cancer Foundation
Communicated by Dvorah Abeliovich
The ATM gene is mutated in ataxia-telangiectasia (AT). Heterozygote female relatives of AT
cases have a 2-7fold increased risk of breast cancer. We previously reported high risks of
breast cancer associated with certain ATM variants. To estimate the risks more precisely, we
have examined two ATM variants, c.1066-6T>G (IVS10-6T>G) and c.4258C>T
(p.Leu1420Phe), in additional cases and controls from the same Australian cohorts
previously used to estimate the risk of breast cancer associated with c.1066-6T>G. A total of
775 and 84 population-based controls were genotyped for the c.1066-6T>G and c.4258C>T
ATM variants respectively, as were index cases from 378 and 373 non-BRCA1/2 breast
cancer families. Penetrance was estimated by Bayes factor analysis. The allele frequencies of
ATM c.1066-6T>G and c.4258C>T estimated from controls were 0.005 (95% CI = 0.002 to
0.009) and 0.012 (95% CI = 0.001 to 0.042), respectively. We identified three new breast
cancer families with c.1066-6T>G, and seven families with c.4258C>T. Combining with the
two c.1066-6T>G families previously reported, the estimated penetrance to age 70 of c.1066-
6T>G was 17.2% (95% CI = 4.7% to 37.5%). For c.4258C>T, the estimated average
penetrance was 4.8% (95% CI 1.7% to 10.1%). In conclusion, we found no evidence that the
ATM c.4258C>T variant increases breast cancer risk, and little evidence that c.1066-6T>G
confers an elevated risk. Analysis of additional families will be necessary to define more
precisely the risk, if any, associated with c.1066-6T>G.
KEY WORDS: AT; ATM; breast cancer; ataxia telangiectasia mutated; risk; variant
INTRODUCTION
Ataxia telangiectasia (AT; MIM# 208900) is a rare autosomal recessive disorder caused by mutations in the
ATM gene (MIM# 607585) and characterised by progressive neuronal degeneration, immunological deficiency,
radiosensitivity, and increased cancer risk. At the cellular level, AT is characterized by hypersensitivity to ionizing
radiation (IR) and other agents that cause double strand breaks in DNA, by defects in G1/S and G2/M checkpoint
responses, and by defective activation of p53 in response to IR. An association between AT and breast cancer was
first suggested by Swift et al (Swift et al., 1990) when they reported an excess of breast
cancers in the female
DOI: 10.1002/humu.9344

2 Thompson et al.
relatives of AT patients. Subsequent studies of ATM carriers have found a moderate (two- to sevenfold) increased
risk of breast cancer (Athma et al., 1996; Geoffroy-Perez et al., 2001; Inskip et al., 1999; Janin et al., 1999; Olsen
et al., 2001). Based on an estimated ATM carrier frequency of 1% and a three-fold increased risk of breast cancer,
ATM heterozygosity may account for 5% of all breast cancer (Easton, 1994), compared with the 1 to 2%
attributable to mutations in BRCA1 and BRCA2. Other studies have compared the frequency of germline ATM
variants in breast cancer cases, unselected for family history, with that in controls, but all have been too small to
detect modest increases in risk (Bishop and Hopper, 1997; Chen et al., 1998; FitzGerald et al., 1997; Shayeghi et
al., 1998; Vorechovsky et al., 1996). More recently, it has been suggested that the elevated risk of breast cancer
may be confined to missense mutations of ATM (Gatti et al., 1999). Most studies of germline ATM mutations in
breast cancer cases have used methods that are biased towards detecting protein-truncating mutations but recent
studies that used denaturing high performance liquid chromatography (DHPLC) or single strand conformation
polymorphism analysis for mutation scanning have found an overall increased frequency of missense variants of
ATM in breast cancer cases compared with controls (Maillet et al, 2002, Dork et al, 2001, Sommer et al, 2002,
Sommer et al, 2003, Thorstenson et al, 2003, Teraoka et al., 2001)]. Nevertheless, case-control studies evaluating
specific, rare, missense ATM variants, have not shown an association with increased risk of breast cancer
(Bernstein et al., 2003, Bretsky et al., 2003, Spurdle et al, 2002).
Similarly, linkage analysis of breast cancer families has not provided evidence of a major role for ATM in
breast cancer predisposition (Cortessis et al., 1993; Wooster et al., 1993). However, such analyses lacked power to
detect a moderate effect, particularly in the presence of genetic heterogeneity. If some specific ATM mutations
confer a high risk of breast cancer, they would occur in rare multiple-case breast cancer families more frequently
than in unselected breast cancer cases. Stankovic et al (Stankovic et al., 1998) identified a missense variant in
ATM (c.7271T>G; p.Val2424Gly), in two AT families, that was associated with a 13-fold (95% CI 4 to 46)
increased risk of breast cancer in both heterozygotes and homozygotes. More recently, Thorstenson et al (2003)
identified the ATM missense variant, c.4258C>T (p. Leu1420Phe), in eight of 202 multiple-case breast-ovarian
cancer families without BRCA1 or BRCA2 mutations, and in none of 421 controls. The cumulative risk of breast
cancer to age 70 was estimated to be almost complete (99%; 95% CI = 25% to 100%). A study of 82 Dutch early
onset breast cancer cases, who had been exposed to low-dose IR at an early age and had survived their cancer for
more than five years, found seven germline ATM variants including the splice site variant, c.1066-6T>G, in three
cases. c.1066-6T>G was estimated to confer an approximate 9-fold elevation in breast cancer risk (Broeks et al.,
2000). The c.1066-6T>G variant has also been identified in the homozygous state in a German patient of Turkish
descent with classical AT, and has been shown to result in 90% skipping of exon 11 in homozygotes (resulting in
premature truncation of the protein) and 40-60% skipping in heterozygotes (as opposed to 15% in wild type
individuals) (Dork et al., 2001).
We have previously reported the identification of a single multiple-case breast cancer family with an ATM
7271T>G variant, and two families with c.1066-6T>G variant, from among 76 non-BRCA1/2 breast cancer
families (Chenevix-Trench et al., 2002). The estimated average penetrances to age 70 for ATM 7271T>G and
c.1066-6T>G were 55% (95% CI = 26% to 88%) and 78% (95% CI = 36% to 99%) respectively. The c.1066-
6T>G variant was not found in any of 262 breast cancer cases unselected for family history, nor in 68 controls
from an Australian population-based case-control series of breast cancer families. Despite this apparent rarity of
the c.1066-6T>G variant in Australia, other studies in Holland, Germany and the Czech Republic have observed a
carrier frequency of 0.6%-1.1% in unselected controls, with unknown personal and/or family histories of breast
cancer (Broeks et al., 2000; Dork et al., 2001; Lei et al., 2002). These carrier frequencies in controls are not
consistent with a highly penetrant mutation. In addition, subsequent studies in the United States, France and
Australia found that the c.1066-6T>G variant occurred in 1.3% unilateral breast cancer cases, but in only 0.2% of
bilateral cases (Bernstein et al., 2003) and at similar frequencies in breast cancer families as in controls (Lindeman
et al., 2004, Szabo et al., 2004).
In order to improve the precision of our estimation of breast cancer risk associated with ATM c.1066-6T>G,
we evaluated its frequency in additional Australian population-based controls and multiple-case breast cancer
families from the sources previously described (Chenevix-Trench et al., 2002). We hypothesized that the
penetrance of c.1066-6T>G might vary depending on the haplotype on which it occurred and so we determined the
haplotypes, and carried out functional evaluation of the variant in lymphoblastoid cell lines (LCLs) derived from
both cases and controls heterozygous for the variant, as well as in a single prophylactic mastectomy specimen from
a carrier. We re-estimated the penetrance of c.1066-6T>G for breast cancer using Bayes factor analysis. Given the
recent report that c.4258C>T confers a high risk of breast cancer, we also screened members of the same
population-based control and multiple-case family cohorts for c.4258C>T variant, and estimated its penetrance.

Two ATM Variants and Breast Cancer Risk
3
MATERIALS AND METHODS
Ascertainment
Controls genotyped for the c.1066-6T>G and c.4258C>T (p.Leu1420Phe) ATM (ACCESSION U82828.1,
GI:2304970
;
ACCESSION
AAB65827.1) variants were from the Australian Breast Cancer Family
Study/Australian Breast Cancer Family Registry (ABCFS/ABCFR), a population-based, case-control-family study
which recruited subjects between 1992-1999 from metropolitan Melbourne and Sydney, Australia via population-
based breast cancer registries (Hopper et al., 1994; Hopper et al., 1999). Voting is compulsory in Australia and
female controls, with no personal history of breast cancer, were ascertained through the electoral roll. In addition
to the 68 controls from the ABCFS/ABCFR tested in our previous study (Chenevix-Trench et al., 2002), we
genotyped the c.1066-6T>G variant in the remaining 707 controls from the ABCFS/ABCFR (775 controls in total).
The c.4258C>T variant was genotyped in 84 ABCFS/ABCFR controls, randomly selected from the 775 available.
The c.1066-6T>G and c.4258C>T variants were also genotyped in a total of 378 (302 from this study, 76
from the previous study (Chenevix-Trench et al., 2002) and 373 index cases respectively from multiple-case
families with breast cancer ascertained throughout Australia and New Zealand by The Kathleen Cuningham
Foundation Consortium for Research into Familial Breast Cancer (kConFab; http://www.kconfab.org, most of
whom were not available at the time of the previous study. Five index cases were genotyped only for c.1066-
6T>G.
These multiple-case families, sequentially ascertained through Familial Cancer Clinics, had four or more
members with breast or ovarian cancer in the same or adjacent generations, or two individuals with breast cancer
of whom one had high-risk features (onset before age 40, bilateral breast cancer, male breast cancer or breast plus
ovarian cancer in the same woman). These criteria are slightly less restrictive than those used in our earlier study in
which we excluded all families with male breast cancer, reflecting increased confidence in the sensitivity of our
continued BRCA1 and BRCA2 mutation testing. ATM variant status was not part of the ascertainment criteria. None
of the kConFab families were found to carry a mutation in BRCA1 or BRCA2 as determined by a variety of testing
strategies, and were thus defined as non-BRCA1/2 families. The index case in each family was the woman with the
youngest age at diagnosis of breast cancer in the family from whom blood was available. DNA from blood was
used for all germline genotyping except in two relatives of an index case where DNA was extracted from an
archival paraffin block of gall bladder or normal breast (adjacent to a tumor) because the patients were deceased.
In the ABCFS/ABCFR, 87.6% of participants reported only Caucasian ancestry, as did 96.9% kConFab
participants. An additional 2.9% ABCFS/ABCFR and 1.7% kConFab participants reported some Caucasian
ancestry. All subjects gave informed consent for the study that was approved by the ethics committees of the
Queensland Institute of Medical Research, the Peter MacCallum Cancer Institute, the University of Melbourne,
The Cancer Council Victoria and the New South Wales Cancer Council.
Genotyping
Genotyping of the variants was performed by DHPLC. For c.1066-6T>G a 350bp product was amplified in
a 20
µl reaction with 16pmol of each primer (forward 5’-GATCGTGCTGTTCCACTCC-3’ and reverse 5’-
GATGAAAGGATTCCACTGAAAG-3’) using the PCR conditions previously described (Chenevix-Trench et al.,
2002). Cycling conditions were 94
°C for 12 minutes, followed by four sets of 4 cycles of 94°C for 30 seconds,
63
°C-57°C for 45 seconds and 72°C for 30 seconds, with the annealing temperature dropping 2°C after each set of
4 cycles, followed by 30 cycles of 94
°C for 30 seconds, 55°C for 45 seconds and 72°C for 30 seconds, and a final
extension of 72
°C for 7 minutes. PCR products were then denatured at 95
o
C for five minutes and cooled to 60
o
C
over 30 minutes (1
o
C/min), and injected onto the Varian Helix System (Varian, Walnut Creek, CA). DHPLC was
carried out at 54
°C using the Medium method for fragments of 200-400 bp (Varian). A positive control with the
relevant variant was included in all DHPLC analysis. All PCR products showing aberrant shifts on DHPLC were
re-amplified and sequenced with both forward and reverse primers using ABI Prism Big Dye Terminator cycle
Sequencing Ready reaction kit (PE Applied Biosystem), and analyzed on an ABI 377 sequencer. For c.4258C>T
genotyping, a 475bp product was amplified in a 20
µl reaction with 16 pmol of each primer (forward 5’-
CTGAAAATTAAATAAATTGGCAAT-3’ and reverse 5’-AAAACAGGAAGAACAGGATAGAAA-3’). All
remaining reagents, cycling conditions, denaturing and slow reannealing were the same as for c.1066-6T>G. The
heteroduplex was observed at 53
°C using the Medium method as above. All relatives of index cases identified as
having the c.4258C>T or c.1066-6T>G variant, and for whom germline DNA as available, were genotyped for the
relevant variant.

4 Thompson et al.
Haplotype and loss of heterozygosity analyses
Three polymorphic markers at the ATM locus (IVS4+36insAA, IVS25-15delA and D11S2179) were used
for haplotype and loss of heterozygosity (LOH) analysis, using the PCR primers described by Broeks et al (Broeks
et al., 2003). PCR products were analysed on an ABI 377 with Genescan software (PE Biosystems, Foster City,
CA) and loss of heterozygosity was defined as a decrease of one allele by more than 50% (normal:tumor allelic
ratios of < 0.5 or > 2.0) (Wang et al., 2001) if the flanking markers were informative. DNA from the c.1066-6T>G
homozygote was used as a control in the haplotype analysis.
Analysis of splicing and protein truncation in c.1066-6T>G carriers
RNA was obtained from LCLs of a c.1066-6T>G homozygous AT patient (HA141), five heterozygous
carriers of c.1066-6T>G and four wildtype individuals using TRI REAGENT (Sigma). In addition, RNA from
prophylactic mastectomy material was made available by kConFab from one heterozygous carrier of c.1066-6T>G
who had prophylactic surgery because of her strong family history of breast cancer. Approximately 1
µg total
RNA was reverse transcribed using random hexamer primers and Superscript III Rnase H
-
reverse transcriptase
(Invitrogen). The resulting cDNA was used as template in a 20
µl PCR reaction with 8 pmol of each primer as
described by Dork et al (Dork et al., 2001). Cycling conditions were 94
°C for 12 minutes, followed by four sets of
4 cycles of 94
°C for 30 seconds, 65°C-59°C for 45 seconds and 72°C for 30 seconds, with the annealing
temperature dropping 2
°C after each set of 4 cycles, followed by 30 cycles of 94°C for 30 seconds, 57°C for 45
seconds and 72
°C for 30 seconds, and a final extension of 72°C for 7 minutes. Two fragments were produced, a
659bp product containing exon 11, and a 489bp fragment lacking exon 11, and these were quantified on an ABI
377 sequencer.
For the Protein Truncation Test, LCLs were treated for 4 hours with cycloheximide (100ug/mL) and RNA
was extracted using the TriPure Isolation Reagent (Roche). A 1315bp RT-PCR product was amplified using the
one step Titan
TM
One Tube RT PCR-system (Roche) with forward
primer5’GGATCCTAATACGACTCACTATAGGGAGACCACCATGAGTCTAGTACTTAATGATCTGCTT
(exon 4) and reverse primer 5’GATAGTATCATCAGTAATGGAGACAGCTC (exon 12). In vitro translation was
achieved using the Promega TNT
®
Coupled Reticulocyte Lysate System incorporating radio labeled S
35
methionine (NEN). Products were separated on a 14% SDS-PAGE gel and protein products visualized via
overnight autoradiography.
Penetrance analysis
The penetrance was estimated using a maximum likelihood approach. The analysis was based on the
likelihood of the observed genotypes for the variant in the family (V), conditional on the proband’s genotype (V
p
),
the family’s disease phenotype (D). The likelihood is maximised over possible values of the hazard ratio (HR),
ψ
for breast cancer associated with the variant:
(
)
(
)
Pr
,
|
( )
Pr( |
,
, )
Pr
,
|
P
P
V D
L
V D V
V D
ψ
ψ
ψ
ψ
=
=
This approach corrects for the ascertainment of families with respect to disease phenotypes. It is
equivalent to maximising the Bayes’ factor for causality of the variant, as described by Thompson et al. (2003).
The analyses were performed using the program MENDEL (Lange et al., 1987). Briefly, the numerator
likelihood was computed by dividing the likelihood for a full version of the pedigree (all genotypes and
phenotypes) by the likelihood for a version of the same pedigree where all phenotypes are given, but only the
proband’s genotype. The denominator likelihood was computed by dividing the likelihood for a version of the
pedigree containing all genotypes but no phenotypes by the likelihood of the proband’s genotype alone. We used a
penetrance model with liability classes based on six age groups; 0 – 29 years, 30 – 39 years, 40 – 49 years, 50 – 59
years, 60 – 69 years and 70+ years. All males and all females for whom phenotypic information was not available
were coded as being unaffected at age 30 years. The incidence rates used for non-carriers were the population
rates given for Australia (1988-1992) in Cancer in Five Continents Volume VII (Parkin et al., 1999) (cumulative
risk of 5.9% by age 70). Incidence rates in carriers assumed a dominant mode of inheritance and a single hazard
ratio for all ages. The overall age-specific incidences in carriers and non-carriers, weighted according to their
relative frequencies within the population, were constrained to equal the population incidence rate, for each age-
group (Antoniou et al., 2000). Allele frequencies for each variant were those estimated from the control series.

Two ATM Variants and Breast Cancer Risk
5
The allowed parameter space for the HR was restricted to the range of HRs compatible with a variant accounting
for no more than 50% of the total breast cancer familial RR. Assuming a familial RR of 1.9 (Dite et al., 2003) and
using the allele frequencies observed for each variant in the population-based controls (see Results) this is
equivalent to maximising the likelihood over all HRs
≤8 for c.4258C>T, or ≤11 for c.1066-6G>T. A likelihood
ratio test was used to evaluate whether the HR differed significantly from 1. 95% likelihood-based confidence
limits for the HR were also computed:
{
}
: 2 ln ( ) 2 ln( ( )
3.84
L
L
ψ
ψ
ψ
−
≤
%
where
ψ
%
i
s the maximum likelihood estimate of the HR. The penetrance estimates were obtained by applying the
best-fitting HR to the population incidence rates in the standard way. The analysis was performed separately for
the c.1066-6T>G variant and the c.4258C>T variant.
RESULTS
We found four ATM c.1066-6T>G carriers among
index cases from multiple-case kConFab families, in
addition to the two previously reported in families B and C (Chenevix-Trench et al., 2002). Although at the onset
of the study all the families were defined as non-BRCA1/2 following mutation testing by a variety of methods, one
of the four c.1066-6T>G carriers was subsequently shown to carry a pathogenic BRCA1 truncating mutation during
the course of this study. ATM c.1066-6T>G did not co-segregate with the BRCA1 mutation in this family, nor with
affected status. The co-occurrence of ATM c.1066-6T>G with a pathogenic BRCA1 mutation in the same
individual does not influence the evidence for it being a high-risk mutation, because pathogenic mutations in
BRCA1 and BRCA2 have previously been reported in the same individual (Friedman et al., 1998; Ramus et al.,
1997). Nevertheless, this BRCA1 family was excluded in order to estimate the penetrance due to ATM c.1066-
6T>G alone. All the index cases of the remaining five c.1066-6T>G families, including families B and C, have
been found to be BRCA1/2-negative by full sequence analysis. All available DNA samples (n=19) from the
remaining three families were then genotyped for c.1066-6T>G, as well as one newly ascertained paternal first
cousin of the index case of family B (Chenevix-Trench et al., 2002) who was recently diagnosed with bilateral
breast cancer at age 48. Seven additional carriers were thus identified, including the new family B cousin. We also
identified seven ATM c.4258C>T carriers among 373 index cases from multiple-case families that we screened.
DNA was available from 68 family members of these index cases and further genotyping identified a total of 20
c.4258C>T carriers.
In order to obtain a precise estimate of the frequency of ATM c.1066-6T>G, we screened 707 additional
controls from the ABCFS/ABCFR and found seven c.1066-6T>G carriers, none of whom had a family history of
breast cancer. This relatively high frequency of c.1066-6T>G is different, but not significantly so, from our
previous finding of no carriers among 262 cases and 68 controls from the ABCFS/ABCFR. The combined data
from 775 population-based controls from Australia gave an allele frequency estimate of 0.005 (95% CI = 0.002 to
0.009). Fewer controls were genotyped for c.4258C>T than for c.1066-6T>G, because two carriers were found
among the first 84 controls genotyped (neither of whom had a family history of breast cancer), suggesting that the
variant is not disease-causing (estimated allele frequency = 0.012, 95% CI 0.001 to 0.042).
We found that every c.1066-6T>G variant identified in the kConFab families, and the ABCFS/ABCFR
controls, occurred on the same haplotype (insAA at IVS4+36insAA, delA at IVS25-15delA, and allele 140 at
D11S2179). Analysis of three tumors from c.1066-6T>G carriers showed that one did not undergo LOH (as we
reported previously in a fourth tumour from family B (Chenevix-Trench et al., 2002)), one showed loss of the
wildtype allele and one showed loss of the variant allele. We generated LCLs from three kConFab cases carrying
the c.1066-6T>G variant, and from two of the control carriers, as well as four wildtype controls. RT-PCR analysis,
using PCR primers to exons 10 and 12, showed that in all these LCLs approximately equal amounts of each
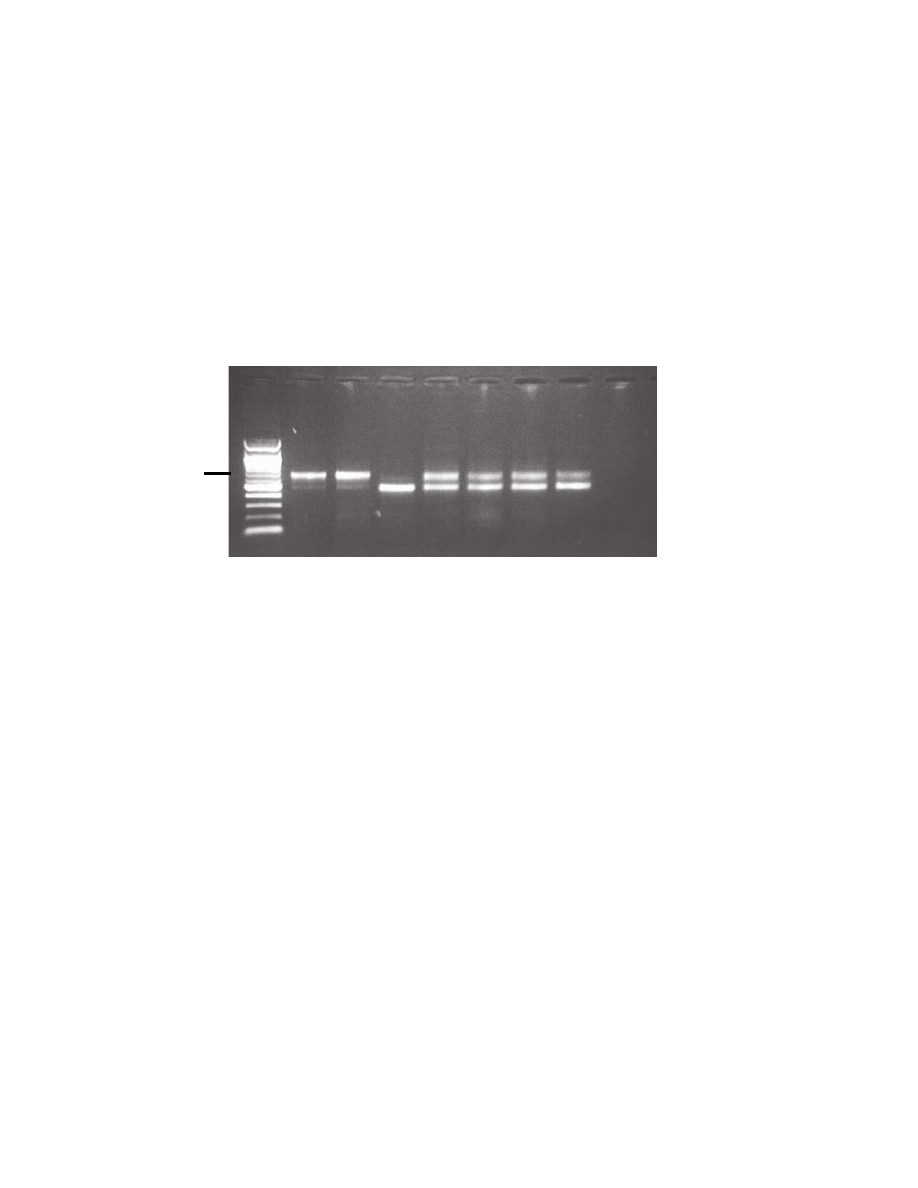
mRNA species (with and without exon 11) were expressed (Fig. 1). A similar result was found in the single
prophylactic mastectomy specimen examined. Wildtype LCLs showed predominance (approximately 95%) of the
exon 11-containing mRNA species but the LCL from a c.1066-6T>G homozygous individual showed expression
of mainly (approximately 95%) the exon 11-deleted mRNA.
We confirmed the relative proportions of products with and without exon 11 in individuals of different
genotypes at the protein level using the Protein Truncation Test. DNA from ATM c.1066-6T>G heterozygous
LCLs derived from six kConFab cases and two ABCFS/ABCFR controls were compared with wildtype individuals
by PTT. In all heterozygous individuals, two bands were observed in approximately equal amounts but analysis of
wild type individuals showed a predominance of the larger, wildtype band and the homozygous mutant showed
mainly the smaller band (data not shown). These results indicate that the ATM c.1066-6T>G variant behaves in a
6 Thompson et al.
similar manner in LCLs derived from women with and without breast cancer, and regardless of their family history
of breast cancer.
hom LC
L (HA141)
het
LC
L - cont
rol
N
egative control
het LCL - case
het LCL - case
wt LCL
wt LCL
Marke
r
het
PM
500bp
Figure 1. Results of ATM exon 11 RT-PCR showing both 659 bp and 489 bp (exon 11 deleted) bands in all lanes except the
homozygote c.1066-6T>G carrier. wt – wild type, het – c.1066-6T>G heterozygote; hom – c.1066-6T>G homozygote LCL –
lymphoblastoid cell line; PM – prophylactic mastectomy.
The likelihood ratio test provided no evidence that the c.4258C>T variant is associated with breast cancer
risk (p=0.6). The best fitting HR for this variant was 0.8 (95% CI = 0.3 to 1.8), equivalent to a penetrance of 4.8%
by age 70 years (95% CI = 1.7% to 10.1%). Estimates were robust to misspecification of the allele frequency (the
lower and upper bounds of the 95% CI gave best fitting HRs of 0.8 and 0.7 respectively). The best fitting HR for
the five c.1066-6T>G families was 3.4 (95% CI = 0.80 to 11.0, p = 0.1), equivalent to a penetrance of 17.2% (95%
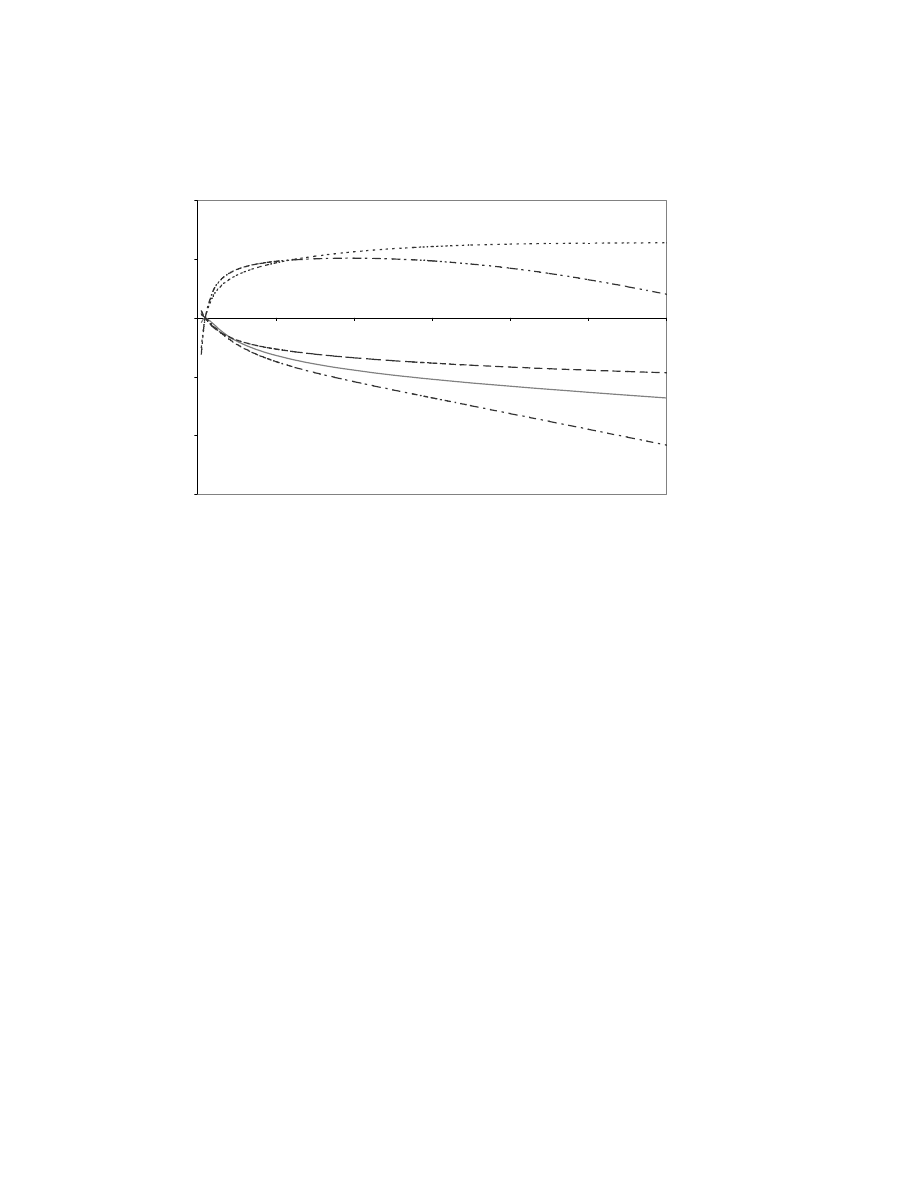
CI = 4.7% to 37.5%) to age 70 i.e. no significant increase in risk. There was substantial heterogeneity between
families (p = 0.009) (Fig. 2); the best-fitting HR for the three families reported for the first time here was just 0.51
(95% CI = 0.03 to 3.5, p = 0.5), whereas the best-fitting HR for the two families including in the original study was
at the upper boundary of the possible HRs (HR = 11.0, p = 0.003).
Since the overall penetrance estimate was considerably lower than previously reported (Chenevix-Trench et
al., 2002), we reanalyzed the original dataset from the first two families identified with the c.1066-6T>G variant to
determine whether these differences were due to the new methodology for estimating causality, or the inclusion of
additional families and family members that had subsequently been genotyped. The best fitting HR for the
original dataset and the analysis method used in the current study (without restricting the HR, for equivalence with
the previous analysis) gave a penetrance to age 70 of 55.4% (95% CI = 11.0% to 61.1%, p = 0.01). Although this
is lower than the estimate of 78% obtained from the segregation analysis in the original study (Chenevix-Trench et
al., 2002), it is still considerably higher than the estimate of 17.2% obtained in the present study, suggesting that
the change in effect size between the two studies is chiefly attributable to changes in the available data, rather than
in the statistical methodology.
Two ATM Variants and Breast Cancer Risk
7
0.001
0.01
0.1
1
10
100
0
10
20
30
40
50
Hazard Ratio
Bayes F
a
ctor
ped 3
ped 5
ped 2
ped 1
ped 4
60
Figure 2. Bayes factors for the ATM IVS10-6T>G variant at different hazard ratios, showing substantial heterogeneity between
families, with only the two families included in the original study (pedigrees 3 and 5 are the updated versions of the previously
published families B and C) providing evidence in favor of causality for this variant.
DISCUSSION
In genotyping index patients from non-BRCA1/2 families for two ATM variants, c.1066-6T>G and
c.4258C>T, we identified five index cases with the c.1066-6T>G variant (including two reported previously
(Chenevix-Trench et al., 2002)), and seven with 1420L>F. We also genotyped a large number of population-based
controls with no personal history of breast cancer, and found that c.1066-6T>G occurred at a frequency of 0.005
(95% CI = 0.002 to 0.009), and c.4258C>T at 0.012 (95% CI = 0.001 to 0.042). These high control frequencies
were inconsistent with previous high penetrance estimates of breast cancer (penetrance estimates to age 70 of 78%
(95% CI = 36% to 99%) for c.1066-6T>G (Chenevix-Trench et al., 2002) and 99% (95% CI = 25% to 100%) for
c.4258C>T (Thorstenson et al., 2003). To explain the inconsistency between the previous estimates of the
penetrance of c.1066-6T>G (Chenevix-Trench et al., 2002) and its high allele frequency we originally
hypothesised that the variant occurred on different haplotypes and that the haplotype background influenced the
effect of the variant on splicing. Haplotype analysis showed that for all individuals, whether from non-BRCA1/2
families or controls, the ATM c.1066-6T>G variant occurred on the same M5 haplotype found in the Dutch breast
cancer cases (Broeks et al., 2003), and the homozygote AT patient with this variant (Dork et al., 2001). Consistent
with the common haplotype background of all the carriers, we found no differences in the splicing phenotype
between LCLs derived from different carriers, nor in their behavior in the Protein Truncating Test.
Using the data from all five families with ATM c.1066-6T>G, the excess risk of breast cancer associated
with the variant was not statistically significant. The new penetrance estimate for ATM c.1066-6T>G (17.2% to age
70, 95% CI = 4.7% to 37.5%) was substantially lower than our original estimate of 78% (95% CI = 36% to 99%)
(Chenevix-Trench et al., 2002). A re-analysis of the original data, using the method used throughout the present
analysis, confirmed that the previous high estimate of penetrance was broadly appropriate for the first two families
analyzed, in which ATM IVS10-6T >G segregated well with breast cancer. We conclude that the lower penetrance
estimate in the current analysis of all five families is mainly due to the larger dataset, rather than to the method of
analysis. Nevertheless, the confidence intervals on the penetrance estimates are still wide, and more multiple-case
breast cancer families with the c.1066-6T>G variant, and multiple available blood samples, will need to be

8 Thompson et al.
identified to refine the penetrance estimates, and narrow the confidence intervals. We also note that the method of
analysis makes no allowance for the possible effects of other causes of familial aggregation. While this does not
invalidate the test statistics, it may imply that the penetrance estimates are larger than those which would apply to
population-based series.
The pathogenicity of the c.1066-6T>G variant is also unclear with respect to AT. The aberrant splicing
observed in lymphocytes and LCLs is consistent with a pathogenic mutation, and this is supported by its
occurrence in a single homozygote patient with classical AT who appears to have no other ATM mutations and
shows aberrant splicing and less than 5% normal ATM protein (Dork et al., 2001). However, c.1066-6T>G
appears to be too frequent in controls to be a pathogenic AT mutation (the allele frequency estimate here, 0.005, is
twice some estimates of the combined frequency of all AT-pathogenic ATM mutations, e.g. Olsen et al., 2001) and
has never been reported in a compound heterozygote AT patient. The rarity of this variant in AT cases is also
difficult to explain if c.1066-6T>G is a benign polymorphism since almost 1% of AT patients would be expected
to carry the variant by chance, and c.1066-6T>G can be detected by PTT and other standard diagnostic methods.
However, it is possible that the variant is not always recorded in the PTT assay because normal splicing of exon 11
is ‘leaky’ resulting in a quantitative change in the assay, rather than a qualitative change.
We found no evidence that the ATM c.4258C>T variant increased breast cancer risk (HR = 0.8, 95% CI =
0.3 – 1.8), which is consistent with published analyses of the functional consequences of this variant (Gutierrez-
Enriquez et al., 2004). Therefore, in conclusion, there is currently little evidence that c.1066-6T>G confers an
elevated breast cancer risk, and even less for an increased risk associated with c.4258C>T. The loss of the c.1066-
6T>G allele in some of the tumours, as we and others have reported (Broeks et al., 2000), is consistent with the
conclusions from our penetrance analysis. Analysis of additional families, in which multiple individuals can be
genotyped, will be necessary to more precisely define the risk, if any, associated with c.1066-6T>G and to
determine whether this ATM variant modifies the penetrance of BRCA1/2 mutations. Therefore, findings of
increased risk in previous studies should not be used clinically to make decisions about risk reduction strategies.
ACKNOWLEDGMENTS
We wish to thank Thilo Dork for the gift of the HA141 LCL from the c.1066-6T>G homozygous patient,
Margaret Cummings for review of archival pathology blocks, Chris Clarke and Lucy Webster for RNA from the
prophylactic mastectomy specimen, Dale Nyholt for useful discussions, and Heather Thorne, Sandra Picken,
Eveline Niedermayr and all the kConFab research nurses and staff for their contributions to this resource. kConFab
is supported by grants from the National Breast Cancer Foundation, the National Health and Medical Research
Council (NHMRC) and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria,
Tasmania and South Australia, and the Cancer Foundation of Western Australia. GCT, JH, and KK are NHMRC
Research Fellows, and ABS is funded by an NHMRC Career Development Award. This work is supported by
grants from the NHMRC and the Susan G. Komen Foundation. DT, AA, and DFE are supported by Cancer
Research U.K. DT was supported by an IARC postdoctoral fellowship.
REFERENCES
Antoniou, A.C., Gayther, S.A., Stratton, J.F., Ponder, B.A., Easton, D.F. 2000. Risk models for familial ovarian and breast
cancer. Genet Epidemiol. Feb;18:173-90.
Athma, P., Rappaport, R. and Swift, M.,1996. Molecular genotyping shows that ataxia-telangiectasia heterozygotes are
predisposed to breast cancer. Cancer Genet Cytogenet, 92(2): 130-4.
Bernstein, J.L. et al., 2003. ATM variants 7271T>G and IVS10-6T>G among women with unilateral and bilateral breast cancer.
Br J Cancer, 89(8): 1513-6.
Bishop, D.T. and Hopper, J., 1997. AT-tributable risks? Nat Genet, 15(3): 226.
Bretsky, P. et al., 2003. The relationship between twenty missense ATM variants and breast cancer risk: the Multiethnic Cohort.
Cancer Epidemiol Biomarkers Prev, 12(8): 733-8.
Broeks, A. et al., 2003. IVS10-6T>G, an ancient ATM germline mutation linked with breast cancer. Hum Mutat, 21(5): 521-8.
Broeks, A. et al., 2000. ATM-heterozygous germline mutations contribute to breast cancer- susceptibility. Am J Hum Genet,
66(2): 494-500.

Two ATM Variants and Breast Cancer Risk
9
Chen, J., Birkholtz, G.G., Lindblom, P., Rubio, C. and Lindblom, A., 1998. The role of ataxia-telangiectasia heterozygotes in
familial breast cancer. Cancer Res, 58(7): 1376-9.
Chenevix-Trench, G. et al., 2002. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst, 94(3): 205-
15.
Cortessis, V. et al., 1993. Linkage analysis of DRD2, a marker linked to the ataxia-telangiectasia gene, in 64 families with
premenopausal bilateral breast cancer. Cancer Res, 53(21): 5083-6.
Dite, G.S., Jenkins, M.A., Southey, M.C., Hocking, J.S., Giles, G.G., McCredie, M.R., Venter, D.J., Hopper, J.L. Familial
risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. 2003. J Natl Cancer Inst. 95(6):448-57.
Dork, T. et al., 2001. Spectrum of ATM gene mutations in a hospital-based series of unselected breast cancer patients. Cancer
Res, 61(20): 7608-15.
Easton, D.F., 1994. Cancer risks in AT heterozygotes. Int J Radiat Biol, 66(6 Suppl): S177-82.
FitzGerald, M.G. et al., 1997. Heterozygous ATM mutations do not contribute to early onset of breast cancer. Nat Genet, 15(3):
307-10.
Friedman, E. et al., 1998. Double heterozygotes for the Ashkenazi founder mutations in BRCA1 and BRCA2 genes. Am J Hum
Genet, 63(4): 1224-7.
Gatti, R.A., Tward, A. and Concannon, P., 1999. Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic
differences between missense and truncating mutations. Mol Genet Metab, 68(4): 419-23.
Geoffroy-Perez, B. et al., 2001. Cancer risk in heterozygotes for ataxia-telangiectasia. Int J Cancer, 93(2): 288-93.
Gutierrez-Enriquez S., Fernet M., Dork T., Bremer M., Lauge A., Stoppa-Lyonnet D., Moullan N., Angele S., Hall J.
Functional consequences of ATM sequence variants for chromosomal radiosensitivity. Genes Chromosomes Cancer. 2004
Jun;40(2):109-19.
Hopper, J.L. et al., 1999. Design and analysis issues in a population-based, case-control-family study of the genetic
epidemiology of breast cancer and the Co-operative Family Registry for Breast Cancer Studies (CFRBCS). J Natl Cancer
Inst Monogr, 26: 95-100.
Hopper, J.L., Giles, G.G., McCredie, M.R.E. and Boyle, P., 1994. Background, rationale and protocol for a case-control-family
study of breast cancer. Breast, 3: 79-86.
Inskip, H.M., Kinlen, L.J., Taylor, A.M., Woods, C.G. and Arlett, C.F., 1999. Risk of breast cancer and other cancers in
heterozygotes for ataxia- telangiectasia. Br J Cancer, 79(7-8): 1304-7.
Janin, N. et al., 1999. Breast cancer risk in ataxia telangiectasia (AT) heterozygotes: haplotype study in French AT families. Br
J Cancer, 80(7): 1042-5.
Lange, K., Weeks, D. and Boehnke, M., 1988. Programs for Pedigree Analysis: MENDEL, FISHER, and dGENE. Genet
Epidemiol, 5(6): 471-2.
Lei, H., Pospisilova, D., Lindblom, A. and Vorechovsky, I., 2002. Re: Dominant negative ATM mutations in breast cancer
families. J Natl Cancer Inst, 94(12): 951-2; author reply 952.
Lindeman G.J., Hiew M., Visvader J.E., Leary J., Field M., Gaff C.L., Gardner R.J., Trainor K., Cheetham G., Suthers G., Kirk
J. Frequency of the ATM IVS10-6T-->G variant in Australian multiple-case breast cancer families. Breast Cancer Res.
2004;6(4):R401-7. Epub 2004 Jun 02.
Maillet, P. et al., 2002. Constitutional alterations of the ATM gene in early onset sporadic breast cancer. J Med Genet, 39(10):
751-3.
Olsen, J.H. et al., 2001. Cancer in Patients With Ataxia-Telangiectasia and in Their Relatives in the Nordic Countries. J Natl
Cancer Inst, 93(2): 121-127.
Parkin, D.M., Pisani, P. and Ferlay, J., 1999. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer,
80: 827-841.

10 Thompson et al.
Petersen, G.M., Parmigiani, G. and Thomas, D., 1998. Missense mutations in disease genes: a Bayesian approach to evaluate
causality. Am J Hum Genet, 62(6): 1516-24.
Ramus, S.J. et al., 1997. A breast/ovarian cancer patient with germline mutations in both BRCA1 and BRCA2. Nat Genet,
15(1): 14-5.
Shayeghi, M. et al., 1998. Heterozygosity for mutations in the ataxia telangiectasia gene is not a major cause of radiotherapy
complications in breast cancer patients. Br J Cancer, 78(7): 922-7.
Sommer, S.S. et al., 2002. Elevated frequency of ATM gene missense mutations in breast cancer relative to ethnically matched
controls. Cancer Genet Cytogenet, 134(1): 25-32.
Sommer, S.S. et al., 2003. ATM missense mutations are frequent in patients with breast cancer. Cancer Genet Cytogenet,
145(2): 115-20.
Spurdle, A.B., Hopper, J.L., Chen, X., McCredie, M.R.E., Giles, G.G., Newman, B., Chenevix-Trench, G. Khanna, K.K., 2002.
No evidence for association of ataxia-telangeictasia mutated gene T2119C and C3161C amino acid sustitution variants with
risk of breast cancer. Breat Cancer Research, 4: R15.
Stankovic, T. et al., 1998. ATM mutations and phenotypes in ataxia-telangiectasia families in the British Isles: expression of
mutant ATM and the risk of leukemia, lymphoma, and breast cancer. Am J Hum Genet, 62(2): 334-45.
Swift, M., Chase, C.L. and Morrell, D., 1990. Cancer predisposition of ataxia-telangiectasia heterozygotes. Cancer Genet
Cytogenet, 46(1): 21-7.
Szabo C.I., Schutte M., Broeks A., Houwing-Duistermaat J.J., Thorstenson Y.R., Durocher F., Oldenburg R.A., Wasielewski
M., Odefrey F., Thompson D., Floore A.N., Kraan J., Klijn J.G., van den Ouweland A.M., Wagner T.M., Devilee P., Simard
J., van 't Veer L.J., Goldgar D.E., Meijers-Heijboer H. Are ATM mutations 7271T-->G and IVS10-6T-->G really high-risk
breast cancer-susceptibility alleles? Cancer Res. 2004 Feb 1;64(3):840-3.
Teraoka S.N.. Malone K.E., Doody D.R., Suter N.M., Ostrander E.A., Daling J.R., Concannon P.. Increased frequency of ATM
mutations in breast carcinoma patients with early onset disease and positive family history. Cancer. 2001 Aug 1;92(3):479-
87.
Thompson, D., Easton, D.F. and Goldgar, D.E., 2003. A full-likelihood method for the evaluation of causality of sequence
variants from family data. Am J Hum Genet, 73(3): 652-5.
Thorstenson, Y.R. et al., 2003. Contributions of ATM mutations to familial breast and ovarian cancer. Cancer Res, 63(12):
3325-33.
Vorechovsky, I. et al., 1996. The ATM gene and susceptibility to breast cancer: analysis of 38 breast tumors reveals no
evidence for mutation. Cancer Res, 56(12): 2726-32.
Wang, V.W., Bell, D.A., Berkowitz, R.S. and Mok, S.C., 2001. Whole genome amplification and high-throughput allelotyping
identified five distinct deletion regions on chromosomes 5 and 6 in microdissected early-stage ovarian tumors. Cancer Res,
61(10): 4169-74.
Wooster, R. et al., 1993. Absence of linkage to the ataxia telangiectasia locus in familial breast cancer. Hum Genet, 92(1): 91-4.
Document Outline
Wyszukiwarka
Podobne podstrony:
The Relationship between Twenty Missense ATM Variants and Breast Cancer Risk The Multiethnic Cohort
Variants in the ATM gene and breast cancer susceptibility
Predictors of perceived breast cancer risk and the relation between preceived risk and breast cancer
Established breast cancer risk factors by clinically important
Population Based Estimates of Breast Cancer Risks Associated With ATM Gene Variants c 7271T4G and c
Variants in the ATM gene associated with a reduced risk of contralateral breast cancer
Perceived risk and adherence to breast cancer screening guidelines
RAD51C Germline Mutations in Breast and Ovarian Cancer Cases from High Risk Families
Missense Variants in ATM in 26,101 Breast Cancer Cases an 29,842 Controls
Rare, Evolutionarily Unlikely Missense Substitutions in ATM Confer Increased Risk of Breast Cancer
Single nucleotide polymorphism D1853N of the ATM gene may alter the risk for breast cancer
Cancer Risk According to Type and Location of ATM Mutation in Ataxia Telangiectasia Families
A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer
Breast and other cancers in 1445 blood relatives of 75 Nordic patients with ataxia telangiectasia
INTERNET USE AND SOCIAL SUPPORT IN WOMEN WITH BREAST CANCER
Quality of life of 5–10 year breast cancer survivors diagnosed between age 40 and 49
03 Antibody conjugated magnetic PLGA nanoparticles for diagnosis and treatment of breast cancer
Spectrum of ATM Gene Mutations in a Hospital based Series of Unselected Breast Cancer Patients
więcej podobnych podstron