
2001;61:7608-7615.
Cancer Res
Thilo Dörk, Regina Bendix, Michael Bremer, et al.
Unselected Breast Cancer Patients
Gene Mutations in a Hospital-based Series of
ATM
Spectrum of
Updated Version
http://cancerres.aacrjournals.org/content/61/20/7608
Access the most recent version of this article at:
Cited Articles
http://cancerres.aacrjournals.org/content/61/20/7608.full.html#ref-list-1
This article cites 66 articles, 18 of which you can access for free at:
Citing Articles
http://cancerres.aacrjournals.org/content/61/20/7608.full.html#related-urls
This article has been cited by 27 HighWire-hosted articles. Access the articles at:
E-mail alerts
related to this article or journal.
Sign up to receive free email-alerts
Subscriptions
Reprints and
Department at
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Department at
To request permission to re-use all or part of this article, contact the AACR Publications

[CANCER RESEARCH 61, 7608 –7615, October 15, 2001]
Spectrum of ATM Gene Mutations in a Hospital-based Series of Unselected Breast
Cancer Patients
1
Thilo Do¨rk,
2
Regina Bendix, Michael Bremer, Dirk Rades, Karin Klo¨pper, Marion Nicke, Britta Skawran,
Ariadne Hector, Paria Yamini, Diana Steinmann, Sabine Weise, Manfred Stuhrmann, and Johann H. Karstens
Department of Biochemistry and Tumour Biology, Clinic of Obstetrics and Gynecology [T. D., R. B., K. K., A. H., P. Y., D. S.], Department of Radiation Oncology [R. B., M. B.,
D. R., M. N., J. H. K.], Institute of Human Genetics [M. N., B. S., M. S.], and Department of Haematology and Oncology [D. S.], Medical School Hannover, D-30659 Hannover;
and Center for Child Neurology, Hospital Gerresheim, D-40225 Du¨sseldorf [S. W.], Germany
ABSTRACT
Blood relatives of patients with the inherited disease ataxia telangiec-
tasia (A-T) have an increased susceptibility for breast cancer. We there-
fore looked for sequence alterations of the ATM gene in a large hospital-
based series of unselected breast cancer patients. The whole ATM coding
sequence was analyzed in genomic DNA samples from a core group of 192
consecutive breast cancer cases to define the spectrum of ATM gene
mutations. Common sequence alterations were then screened in the whole
series of 1000 breast cancer patients and in 500 random individuals. In the
core group, 21 distinct sequence alterations were identified throughout the
ATM coding region, and 1 common splicing mutation was uncovered in
intron 10. Almost half of the breast cancer patients (46%) were heterozy-
gotes for 1 of 16 different amino acid substitutions, and three patients
(1.6%) carried a truncating mutation. These data indicate that
⬃1 in 50
German breast cancer patients is heterozygous for an A-T-causing muta-
tion. In our extended series, the most common A-T mutation 1066-6T
3G
was disclosed in 7 of 1000 (0.7%) breast cancer patients. Transcript
analyses indicated that the loss of exon 11 in the ATM mRNA was the
pathogenic consequence of this splicing mutation, which produced a
<10% of full-length ATM mRNA and ATM protein in a homozygous A-T
patient. We also found an excess of rare missense substitutions in the
breast cancer cohort compared with random individuals (7.9% versus
5.3% of alleles; odds ratio
ⴝ 1.6; P < 0.01). One missense substitution,
S707P in exon 15, was two times more frequent in breast cancer patients
(odds ratio
ⴝ 2.4; 95% confidence interval, 1.0–5.8) and five times more
frequent in patients with bilateral disease than in random individuals
(P
< 0.001). We conclude that a large variety of distinct ATM mutations
and variants exist among breast cancer patients, some of which can
contribute to the etiology and progression of the malignancy. Screening
for frequent A-T mutations such as the 1066-6
3G splice site substitution
can be effective to prospectively identify A-T heterozygotes in an uns-
elected cancer patient population.
INTRODUCTION
Several risk factors for breast cancer have been defined, including
age, family history, hormonal factors, and radiation exposure (1–3).
Apart from the two familiar breast cancer genes BRCA1 and BRCA2,
it is thought that mutations in genes with lower penetrance may
explain much of the hereditary predisposition to breast cancer (3). One
candidate is ATM, the gene mutated in A-T
3
(4, 5). The ATM gene
consists of 66 exons encoding a large protein kinase that orchestrates
the recognition and repair of radiation-induced DNA double strand
breaks (5–9). Several oncoproteins are regulated by ATM, including
the tumor suppressors p53 and BRCA1 (6, 10). A-T patients have a
high incidence of cancer (11–13), and some adult patients with A-T
have been reported to develop breast cancer (13, 14).
Life expectancy is reduced in A-T heterozygotes, who may account
for
⬃1% of the general population, because of an increased appear-
ance of age-related disorders (15). Several epidemiological studies
have provided strong evidence for an increased frequency of malig-
nancies, particularly breast cancer, among blood relatives of patients
with A-T (11, 16 –23). A-T heterozygotes appear to have a risk of
breast carcinoma that is
⬃3.8 times greater than that of noncarriers,
leading to the estimation that carriers of an ATM gene mutation may
account for 6.6% of all breast cancer cases (20). Direct molecular
examination of selected breast cancer cohorts outside of A-T families
has led to conflicting, albeit not mutually exclusive, results (3, 24 –
33). In a recent report, Broeks et al. (32) detected seven germline ATM
mutations among 82 breast cancer cases who had either early-onset
disease or bilateral breast cancer; the authors concluded that truncat-
ing germline mutations of ATM contribute to breast cancer suscepti-
bility. By contrast, FitzGerald et al. (26) detected ATM mutations in
only 2 of 401 (0.5%) breast cancer patients with onset
⬍40 years and
concluded that truncating mutations of ATM do not predispose to
early-onset breast cancer. Within A-T families, the relative risk of
breast cancer appears to be highest, i.e., 6 –7-fold increased, in obli-
gate A-T heterozygotes 50 – 69 years of age (20), a range similar to the
median age at onset of breast cancer in the general population. We
initiated a population-based study to define the spectrum and elucidate
the clinical relevance of ATM gene mutations in a large series of
unselected breast cancer patients treated at the same hospital.
PATIENTS AND METHODS
Patients. Peripheral EDTA blood samples were collected, after written
informed consent had been obtained, from 1000 consecutive breast cancer
patients who received postoperative radiotherapy at the Medical School Han-
nover from September 1995 to April 1999. All patients were residents of
Lower Saxony, a region in the north of Germany. Median age at onset of breast
cancer was 57 years in this patient cohort (range, 27– 85 years). Of these
patients, 26.9% had developed breast cancer below the age of 50 years and 7%
below the age of 40 years; 6.7% had bilateral breast cancer. The involved
breast was irradiated postoperatively with a 6 MV photon beam of a linear
accelerator with a mean total dose of 52 Gy (range, 45–54 Gy; single dose, 1.8
Gy), followed by a boost with electrons (10 –14 Gy) to the tumor bed in 6.8%
of the patients. The ipsilateral periclavicular lymph nodes were irradiated in
14% of patients (total dose 45 Gy, single dose 1.8 Gy). Only 1 of the 1000
patients showed severe acute toxicity, scored as grade III according to common
toxicity criteria (34), and 2 developed severe late reactions related to local
radiotherapy, which were classified as grade III and IV, respectively, according
to the LENT-SOMA score criteria (36), a relatively low proportion which may
be related to the sequential application of chemotherapy and radiotherapy.
Several of the patients (28.1%) reported at least one blood relative with breast
cancer. Fourteen patients had been identified as carriers of a frequent BRCA1
or BRCA2 gene mutation in a parallel study (data not shown); these patients
were left within the cohort to keep the study group unbiased and to take into
account the possibility of double heterozygosity. Genomic DNA was extracted
from leukocytes of all patients and served as the primary source for mutational
screening. For comparison, a series of 500 genomic DNA samples were
Received 4/30/01; accepted 8/14/01.
The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with
18 U.S.C. Section 1734 solely to indicate this fact.
1
Part of this study was funded by a research grant from the Medical School Hannover
(Grant 19570006).
2
To whom requests for reprints should be addressed, at Department of Biochemistry
and Tumour Biology, Clinic of Obstetrics and Gynecology, Medical School Hannover,
OE 6410, Podbielskistrasse 380, D-30659 Hannover, Germany. Phone: 49 511 906 3649;
Fax: 49 511 906 3433; e-mail: thilo.doerk.oststadt@klinikum-hannover.de.
3
The abbreviations used are: A-T, ataxia telangiectasia; OR, odds ratio; CI, confidence
interval; SSCP, single-strand conformation polymorphism.
7608

collected from random blood donors from the same geographic region who had
been anonymized to keep confidentiality. These samples served merely to
determine the frequency of selected gene alterations in the general population
from Lower Saxony; they were not strict controls because the individuals were
not matched by age and sex, i.e., they were generally younger than the cases,
and their health status remained unknown.
Total RNA was obtained by acid guanidinium-phenol extraction (38) from
isolated WBCs of selected patients and controls for further characterization of
particular mutations. Lymphoblastoid cell lines were established from a few
selected breast cancer patients and from an A-T child homozygous for the
1066-6T
3G mutation according to a previously described protocol (39).
Methods. Mutation analysis of all coding exons and flanking intron se-
quences of the ATM gene was performed in the genomic DNA samples
obtained from the first 192 consecutive breast cancer patients. In brief, PCR
products were obtained using exon-flanking primer pairs (40), digested with
appropriate restriction enzymes to generate fragments 150 –300 bp in length,
and subjected to SSCP analysis on 40-cm-long 5– 6% nondenaturing poly-
acrylamide gels supplemented with 5% glycerol (41). After electrophoresis in
0.8
⫻ Tris-borate-EDTA at 40 W for 5 h in a cold room, gels were fixed in 10%
acidic acid, and bands were visualized by conventional silver staining. Addi-
tional samples from 35 unrelated German A-T patients were run in parallel to
confirm a detection rate of 75% of A-T mutations with this exon-scanning
approach (data not shown). PCR products with an aberrant migration on the
SSCP gel were sequenced on both strands using the BigDye Terminator Cycle
Sequencing Kit and an ABI 310 sequencer (Perkin-Elmer) to identify the
underlying sequence alteration. Using mutation-specific restriction enzyme-
based screening assays, we subsequently screened for frequent mutations and
variants in the total cohort of 1000 breast cancer patients and in 500 anony-
mous blood donors from the general population of Lower Saxony. We used
AlwI to test for the presence of the P1054R substitution, MboII to test for the
presence of the L1420F substitution, and RsaI to test for the presence of the
1066-6T
3G mutation. The S707P substitution was screened using the primers
5
⬘-TAAGGCAAAGCATTAGGTACTTG-3⬘ and 5⬘-TTTCTCCTTCCTAA-
CAGTTTACC-3
⬘ followed by Bsu36I digestion. A specific mismatch primer
(5
⬘-GGAGGATCAGTCATCCATGAATGTA-3⬘) was used in combination
with the reverse primer (5
⬘-GGAACAATCCTAAAAGGCTATAC-3⬘) to
screen for the F858L substitution by RsaI digestion. Similarly, splicing muta-
tion 3576G
3A was screened using mismatch primer 5
⬘-GGATTAGAACCT-
CACCTCCTGAAAAA-3
⬘ together with the reverse primer 5⬘-CCTAGTCT-
TAAATAAGTGCCACTC-3
⬘, followed by digestion with EcoNI. The
substitutions D1853N and D1853V were screened for and distinguished by
SSCP analyses of exon 39 PCR products.
We investigated the effect of mutation 1066-6T
3G on splicing, using RNA
samples obtained from the lymphoblastoid cell lines of a homozygous A-T
patient (HA141) and of unrelated individuals who did not carry this mutation,
as well as from peripheral blood lymphocytes of three breast cancer patients
heterozygous for the mutation. Total RNA was reverse-transcribed using
random hexamer primers and a First-strand-cDNA synthesis Kit (Amersham/
Pharmacia). One-fifth of the cDNA served as the template for a subsequent
PCR using primers 5
⬘-GATCTGCTAGTGAATGAGATAAGTC-3⬘ and 5⬘-
CATGAAGGTCTGCAGGCTGACCCA-3
⬘, with the forward primer being
fluorescein labeled (annealing at 57°C; 30 cycles). The labeled PCR products,
659 bp for product containing exon 11 and 489 bp for product lacking exon 11,
were separated in a denaturing 5% polyacrylamide gel on an A.L.F. sequencer
(Amersham/Pharmacia), and their relative quantities were determined by Frag-
ment Manager 1.2 software (Amersham/Pharmacia). Values given for the
homozygous patient are the mean of three measurements from either of two
different RNA preparations.
Expression of ATM protein was determined by Western blot analyses of
lymphoblastoid cell extracts from the 1066-6T
3G homozygote as well as
from unrelated controls, including one obligate noncarrier (HA169) from an
A-T family. Cell pellets were lysed essentially as described previously (10),
and protein concentrations were determined by the Bradford method (Bio-
Rad). Forty
g of total protein per lane were separated on 4.2% SDS-
polyacrylamide gels, followed by blotting onto Hybond C-extra nitrocellulose
membrane (Amersham) and overnight incubation with a 1:1000 dilution of
monoclonal antibody 2C1 raised against the COOH-terminal portion of ATM
(amino acids 2577–3056; GeneTex; Ref. 42). After incubation with secondary
antibody, signals were detected by enhanced chemiluminescence (Amersham)
followed by autoradiography. The same blot was subsequently probed with a
1:500 dilution of the monoclonal antibody Ab-2 raised against the catalytic
subunit of DNA-dependent protein kinase (Oncogene Research), which served
as the internal control for loading and integrity of total protein.
Clinical data were evaluated retrospectively from all available patient
records. Patients were grouped by their ATM genotype, and the characteristics
of patients and tumors were compared by
2
tests. Results were considered to
be significant for P
⬍ 0.05, or where multiple testing with six independent
mutation genotypes was performed, for P
⬍ 0.008 following Bonferroni’s
correction. CIs (95% or 99%) of ORs were calculated by
2
-based approxi-
mation using the Win Episcope 1.0 software package.
RESULTS
Mutation Scanning of the ATM Gene. We performed a mutation
analysis of genomic PCR products amplified from DNA samples of
192 consecutive breast cancer patients by SSCP analysis of all coding
exons and flanking intron sequences of the ATM gene. Subsequent
direct sequencing of PCR products with an aberrant migration iden-
tified a total of 21 different sequence alterations within the ATM
coding region (Table 1). One of the two truncating mutations was a
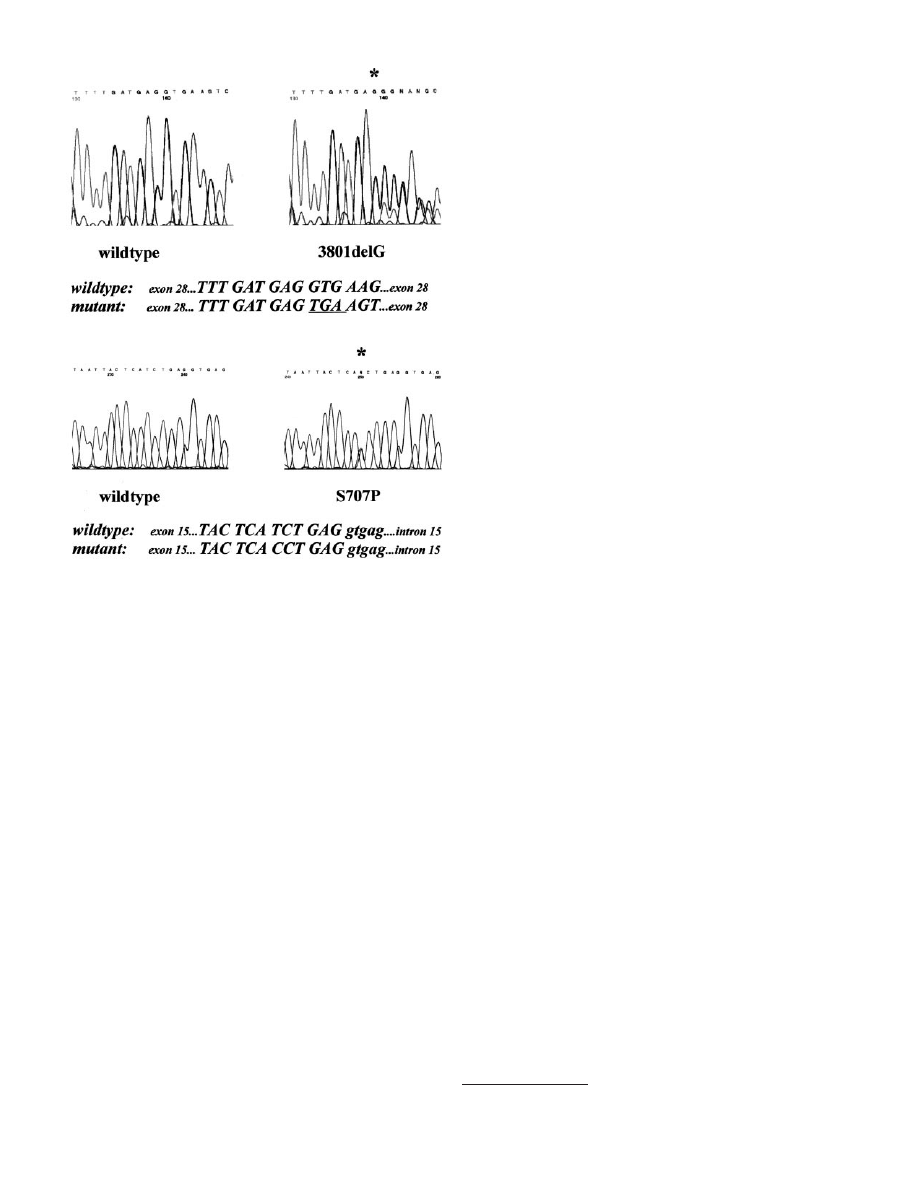
frameshift deletion, 3801delG in exon 28 of the ATM gene (Fig. 1),
which was detected in a single patient with bilateral breast cancer.
This woman had been diagnosed by age 50 for her first breast cancer
and had received postoperative local regional radiation therapy after
mastectomy. By age 69, she had developed a second primary of the
contralateral breast, which was irradiated after breast-conservative
surgery. She died from cancer by the age of 74 years.
An additional 16 exonic alterations were amino acid substitutions
dispersed throughout the whole coding region (Table 1), classifying
amino acid substitutions as the major type of sequence alteration
within the ATM open reading frame. Homozygotes were identified for
the amino acid substitution D1853N, and compound heterozygosity
was found in five patients that also involved the frequent D1853N
polymorphism. One patient was a compound heterozygote for the
D1853N and D1853V substitutions, and two patients carried the
Table 1 Mutations of the ATM gene in 192 breast cancer patients
Mutations are designated according to the recommended nomenclature (67). Nucleo-
tides are numbered according to the published ATM cDNA sequence (8), beginning with
the first nucleotide of the start codon.
Mutation
Nucleotide
Location
No. of patients
Frameshift mutation
3801delG
Deletion of G at
3801–3802
Exon 28
1 Het
a
Splicing mutation
1066-6T
3G
T
3G at 1066-6
Intron 10
2 Het
Amino acid substitutions
S49C
C
3G at 146
Exon 5
3 Het
I550V
A
3G at 1648
Exon 13
1 Het
P604S
C
3T at 1810
Exon 14
1 Het
S707P
T
3C at 2119
Exon 15
5 Het
F858L
b
T
3C at 2572
Exon 19
4 Het
P1054R
C
3G at 3161
Exon 24
9 Het
L1420F
C
3T at 4258
Exon 31
8 Het
V1570A
T
3C at 4709
Exon 33
1 Het
S1691R
A
3C at 5071
Exon 36
2 Het
D1853N
G
3A at 5557
Exon 39
51 Het, 4 Hom
D1853V
A
3T at 5558
Exon 39
3 Het
A2274T
G
3A at 6820
Exon 49
1 Het
G2287A
G
3C at 6860
Exon 49
1 Het
C2464R
T
3C at 7390
Exon 52
2 Het
S2592C
C
3G at 7775
Exon 54
1 Het
G2772R
G
3A at 8314
Exon 59
1 Het
Synonymous substitutions
735C/T
C
3T at 735
Exon 9
2 Het
1020C/A
C
3A at 1020
Exon 10
1 Het
2193C/T
C
3T at 2193
Exon 16
1 Het
4578C/T
C
3T at 4578
Exon 32
16 Het, 4 Hom
a
Het, heterozygote; Hom, homozygote.
b
F858L is linked with P1054R on the same allele.
7609
GERMLINE ATM MUTATIONS IN BREAST CANCER
S707P substitution apparently in trans with D1853N. The A2274T
and the G2772R substitutions were similarly found in patients who
were heterozygous for the D1853N polymorphism, but the phase
could not be deduced in these single cases.
An additional four exonic alterations were synonymous sequence
variants (Table 1). Because these do not change the coding sequence,
they were considered neutral variants, although the 735C
3T substi-
tution may be able to enhance the alternative splicing of ATM exon 9
(43). In addition, several intronic alterations were identified, including
the common polypyrimidine tract mutation 1066-6T
3G, which could
be shown to cause aberrant splicing that results in a premature stop
codon (see below). Altogether, 89 (46%) of the breast cancer patients
in our initial study cohort were carriers of at least one sequence
change that affects the coding potential of the ATM gene, but the
physiological impact of these substitutions for ATM function could
not easily be assessed for many of these alterations.
Characterization of the Splicing Mutation 1066-6T
3G. The
most frequent truncating A-T mutation in our sample of breast cancer
patients was a splicing mutation, polypyrimidine tract substitution
1066-6T
3G within the acceptor splice site of intron 10 of the ATM
gene. Two patients in our core group of 192 breast cancer cases were
heterozygous for the intronic transversion, but when we extended our
screening to the whole cohort, using an RsaI restriction enzyme based
assay, 5 additional breast cancer cases were identified, raising the total
number to 7 in 1000 (0.7%). In addition, this mutation was observed
in 3 of 500 (0.6%) random individuals with unknown phenotype from
the general population of Lower Saxony.
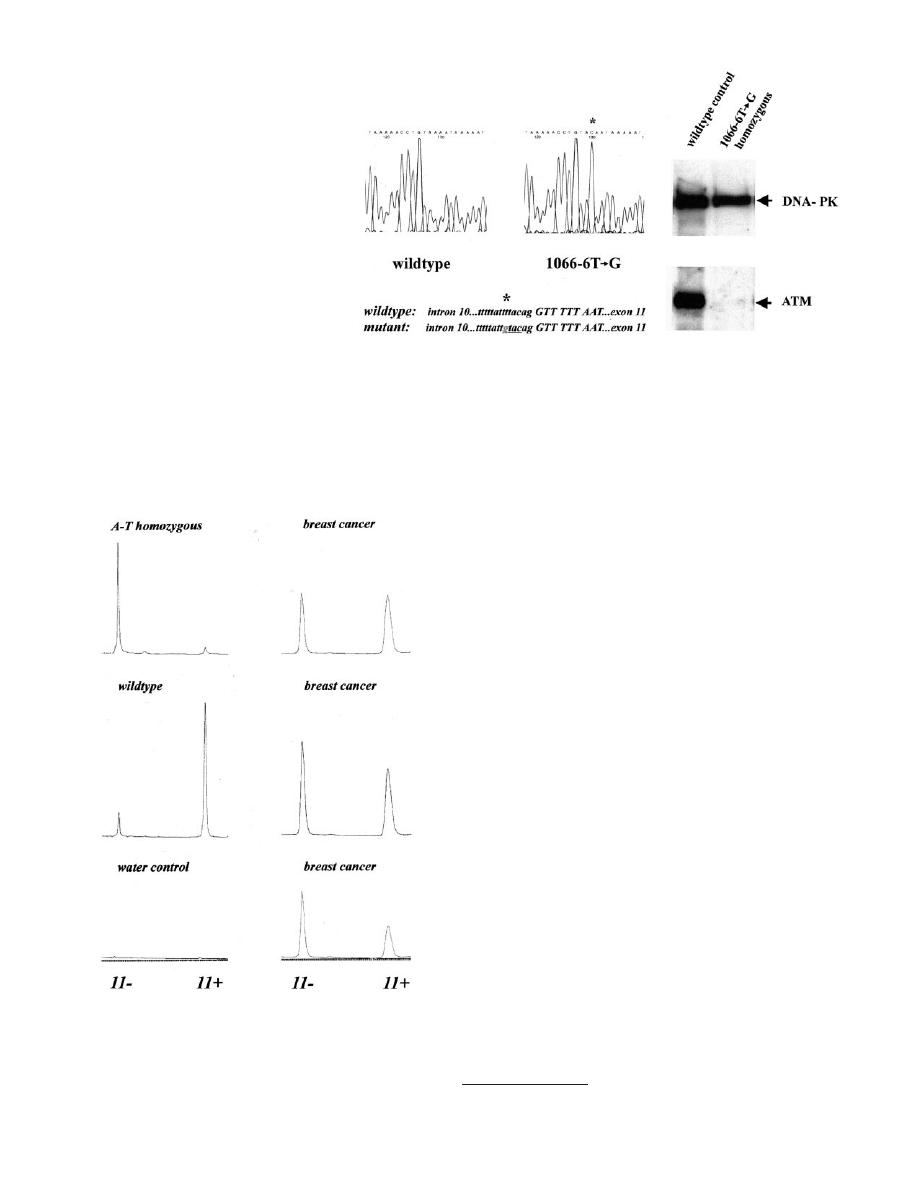
We had initially identified the 1066-6T
3G mutation in the ho-
mozygous state as the disease-causing mutation in a German A-T
patient of Turkish descent (Fig. 2). The 10-year-old boy developed
ataxia at 2 years of age and now presents all typical symptoms of
classical A-T, including telangiectasia, IgA deficiency, chromosomal
instability, elevated AFP concentration, and the recent occurrence of
non-Hodgkin lymphoma. To confirm the pathogenic effect of the
subtle splice site alteration, we established a lymphoblastoid cell line
from this patient and assessed the residual protein and RNA levels of
ATM expression. Only traces of full-length ATM protein could be
detected in the patient’s cell line by Western blot analyses using an
antibody against the COOH-terminal portion of ATM (Fig. 2). To
determine the underlying mechanism more closely, we performed a
quantitative analysis of ATM mRNA splicing. Repeated reverse tran-
scription-PCR analyses confirmed the loss of exon 11 in 93
⫾ 4% of
ATM mRNA transcripts in this cell line (Fig. 3). A parallel analysis of
several cell lines and tissues obtained from individuals without this
mutation showed that the alternative splicing of exon 11 usually
occurs in 5–15% in normal cells (Fig. 3).
4
When we examined the ATM transcripts in primary lymphocytes
isolated from three unrelated breast cancer patients of our study who
were heterozygous for the mutation, the proportions of exon 11
skipping were 40 – 60% (Fig. 3). Thus, the 1066-6T
3G mutation is a
common A-T mutation in German breast cancer patients, and ATM
inactivation in these patients occurs in the range expected for A-T
heterozygotes. The clinical characteristics of breast cancer in all seven
identified heterozygotes are compiled in Table 2. Six patients received
postoperative radiotherapy without severe acute complications. After
a follow-up of 3 years, two of the patients had died from cancer,
whereas the other five were alive, and late radiation-related toxicities
did not exceed grade II according to LENT-SOMA score criteria.
Frequency and Clinical Relevance of Other Common ATM
Gene Alterations. Apart from the 1066-6T
3G mutation, the more
common ATM gene mutations in our breast cancer cohort were of the
missense type. To gain more insight into whether some of these
changes predispose to breast cancer, we screened our total group of
1000 unselected breast cancer patients and a cohort of 500 random
blood donors from the general population of Lower Saxony for the
presence of the more frequent ATM substitutions (Table 3). No
difference was observed in the frequency distribution of the common
polymorphism D1853N between cases and random individuals (allele
frequency, 0.13). However, the rarer missense substitutions with a
carrier frequency
⬎1% were, as a group, more prevalent in the breast
cancer cohort than in the comparison group (7.9% versus 5.3% of
alleles; P
⬍ 0.01). The OR of the cumulative frequencies of these
amino acid substitutions between cases and random individuals was
1.6 (99% CI, 1.1–2.6; Table 3). This difference remained significant
after we excluded the hypothesis-generating cohort of the first 192
patients. The most prominent trend was observed for the S707P
substitution in exon 15 of ATM, which had a
⬎2-fold increased
prevalence in the breast cancer cohort compared with random blood
donors (OR
⫽ 2.4; 95% CI, 1.0–5.8; Table 3). One of the patients had
a sister who also was affected by breast cancer, and she also had
inherited the S707P allele. Tendencies were less pronounced for the
missense substitutions L1420F, P1054R, and F858L, and similar to
the D1853N polymorphism, no trend was observed in case of the
D1853V substitution (Table 3).
Although their frequencies as well as the identification of homozy-
4
Our unpublished data.
Fig. 1. Mutations of the ATM gene in patients with bilateral breast cancer. Top, direct
sequencing of frameshift mutation 3801delG in exon 28 of the ATM gene. Top left,
wild-type control sequence; top right, heterozygosity for 3801delG. The deletion of one
of two guanines at nucleotides 3801–3802 (
ⴱ) creates a frameshift and a premature stop
codon (underlined). Bottom, direct sequencing of missense substitution S707P in exon 15
of the ATM gene. Bottom left, wild-type control sequence; bottom right, heterozygosity for
S707P. The T
3C substitution at nucleotide 2119 (
ⴱ) creates the codon CCT for proline.
7610
GERMLINE ATM MUTATIONS IN BREAST CANCER
gotes for the F858L, P1054R, and L1420F substitutions in our study
indicate that these missense mutations do not represent classical A-T
mutations, this does not preclude their possible cancer-predisposing
role (the patient homozygous for the F858L-P1054R double mutant
allele suffered from local recurrences, and the patient homozygous for
L1420F developed metachronous bilateral breast cancer). We thus
retrospectively analyzed clinical data recorded for heterozygous
breast cancer patients carrying the D1853N, D1853V, L1420F,
P1054R, and S707P alleles, respectively, and compared them with
those of a random sample of patients who do not carry these substi-
tutions. The patient characteristics as stratified by genotype are sum-
marized in Table 4. No differences were observed regarding family
history of breast cancer or estrogen receptor status of the tumor (data
not shown). Age at diagnosis and occurrence of bilateral breast cancer
were similar in D1853N homozygotes and heterozygotes, D1853V
heterozygotes, L1420F heterozygotes, P1054R heterozygotes, and
noncarriers. A higher proportion of node-positive patients was ob-
served in D1853N homozygotes (P
⬍ 0.01), P1054R heterozygotes
(P
⬍ 0.01), and D1853V heterozygotes (P ⬍ 0.01). These findings
leave open the possibility that these ATM variants, although not
classical A-T mutations, could modulate the course and prognosis of
breast carcinoma. In a comparison of clinical characteristics of pa-
tients carrying the S707P substitution with those of noncarriers, we
failed to observe differences regarding the age at onset, but we found
a markedly higher proportion of axillary node-positive patients (17 of
26; P
⬍ 0.001) and a higher proportion of bilateral breast cancer (5 of
26; P
⫽ 0.02) among the S707P heterozygotes. Altogether, the S707P
substitution was five times more frequent in patients with bilateral
breast cancer (P
⬍ 0.001) than in our random sample from the general
population.
DISCUSSION
We investigated the whole ATM coding sequence and flanking
untranslated regions in a hospital-based sample of 192 consecutive
breast cancer patients; we also screened the most common ATM gene
alterations in a total of 1000 breast cancer patients and 500 controls.
In contrast to previous studies, our sample was, to our best knowledge,
not selected by age, family history, or radiation-related adverse effects
and therefore did not preclude the characterization of low-penetrance
mutations. We used genomic DNA for mutational screening to avoid
possible pitfalls related to nonsense-mediated mRNA decay, and we
used SSCP analysis as an unbiased scanning method to detect both
truncating and nontruncating alterations. Three of the 192 unselected
patients (1.6%) carried truncating ATM gene mutations, the most
frequent mutation type in classical A-T (40, 44, 45).
5
The single-base
deletion 3801delG is a known disease-causing mutation in patients
with A-T, which has previously been observed in British, German,
and Spanish A-T families (Refs. 14, 40, and data not shown), indi-
5
ATM gene mutation database (http://www.vmresearch.org/atm.htm).
Fig. 2. Identification of splicing mutation 1066-6T
3G in a
homozygous A-T patient. Left panel, direct genomic sequencing
of exon 11 and the flanking sequence of intron 10. Sequencing of
the antisense strand is shown. The sequence on the left is the
wild-type sequence; the sequence on the right is from a A-T
patient homozygous for the 1066-6T
3G mutation. The location
of the substitution within the polypyrimidine tract of intron 10 is
written below (
ⴱ). The mutation creates a RsaI recognition se-
quence (underlined). Right panel, Western blot analysis of lym-
phoblastoid cell extracts from an obligate non-A-T carrier (left
lane) and the homozygous A-T patient (right lane). DNA-PKcs
(470 kDa; top band) served as internal loading and quality
control. Only traces of ATM protein (370 kDa; bottom band)
were detectable in the patient compared with the control.
Fig. 3. Exon skipping in carriers of the ATM splicing mutation 1066-6T
3G. Relative
peak areas correspond to the relative amounts of product with and without ATM exon 11.
One representative experiment is shown for each investigated sample. Top left, homozy-
gous A-T patient [exon 11(
⫹), 7%; exon 11(⫺) 93%]; middle left, noncarrier control
[exon 11(
⫹), 88%; exon 11(⫺), 12%]; bottom left, control reaction without RNA; top
right, breast cancer patient 1, heterozygous carrier [exon 11(
⫹), 60%; exon 11(⫺), 40%];
middle right, breast cancer patient 2, heterozygous carrier [exon 11(
⫹), 53%; exon 11(⫺),
47%]; bottom right, breast cancer patient 3, heterozygous carrier [exon 11(
⫹), 44%; exon
11(
⫺), 56%].
7611
GERMLINE ATM MUTATIONS IN BREAST CANCER

cating that it is a common A-T mutation. The splicing mutation
1066-6T
3G was found twice in our initial cohort and represents
another frequent A-T mutation as discussed below. With the appro-
priate adjustment for the 75% detection rate of our screening method,
it follows that
⬃1 in 50 breast cancer patients appears to be heterozy-
gous for a truncating ATM mutation. This value is intermediate
between previous studies that had focused on early-onset breast can-
cer (26, 32) and is consistent with the view that the number of A-T
heterozygotes among unselected breast cancer patients exceeds the
estimated carrier frequency in the general population.
In our extended screening, the polypyrimidine tract substitution
1066-6T
3G was a frequent A-T splicing mutation. This mutation has
independently been reported by Broeks et al. (32) to be associated
with breast cancer in the Netherlands. Our results indicate that
⬃1 in
140 German breast cancer patients is heterozygous for this mutation.
These results were unexpected because previous studies had not
uncovered 1066-6T
3G as a major mutation in German or Dutch A-T
families (35, 40). However, studies of A-T patients are often ham-
pered by confounding effects, such as small sample sizes, ethnic
heterogeneity, and consanguinity, and therefore the mutational spec-
trum in A-T may not accurately reflect the frequency distribution of
ATM gene mutations in the general population. Although the 1066-
6T
3G mutation occurs at a poorly conserved position within the
branch/acceptor splice site of intron 10, it is clearly a pathogenic
mutation for the following reasons. (a) It was initially identified in the
homozygous state as the only detected ATM mutation in a patient with
a classical course of A-T and with deficiency of ATM protein. (b) It
led to extensive skipping of exon 11 in ATM mRNA transcripts from
the homozygous A-T patient’s cell line (93%) as well as in lympho-
cyte samples from three heterozygous breast cancer patients (40 –
60%). The loss of exon 11 results in a frameshift and premature
termination codon, as do the vast majority of A-T mutations, and this
is not a prominent alternative splicing event in normal cells (Refs. 37,
46, and this study). (c) Lymphoblastoid cell lines established from our
breast cancer patients heterozygous for the 1066-6T
3G mutation
have been shown to exhibit increased cellular radiosensitivity as
assessed by whole chromosome painting and by micronucleus forma-
tion tests (47, 48). (d) Cosegregation of the 1066-6T
3G mutation
with breast cancer has been demonstrated in some breast cancer
families. Thus, the 1066-6T
3G substitution now appears to be the
most common truncating mutation in Northern European A-T het-
erozygotes. In our small sample, the frequency of 1066-6T
3G was
not different between patients (0.7%) and random individuals (0.6%),
indicating a need for further validation studies with larger sample
sizes and with defined age-matched cancer-free individuals as con-
trols. Additional insight can also be gained from screening this mu-
tation in large breast cancer families to determine the age-dependent
penetrance and the size of the relative risks for breast cancer and other
malignancies that are associated with heterozygosity for this frequent
A-T mutation.
Table 2 Characteristics of breast cancer patients carrying A-T splicing mutation 1066-6T
3G
Patient
Age at
diagnosis,
(years)
Follow-up
(months)
Family
history
a
Histology
TNM
b
Radiotherapy
c
Treatment volume/
Total dose
Acute toxicity
(CTC)
Late toxicity
(LENT-SOMA)
EL
69
18
0
Invasive ductal
pT
1c
,pN
0
,M
0
Breast, 50.4 Gy
Grade I
Grade I
HR
35
19
2
Invasive ductal
pT
2(m)
,pN
1biii
,M
0
Breast, 50.4 Gy;
periclavicular, 45 Gy
Grade I
Grade 0
KS
63
13
0
Invasive ductal;
invasive tubular
pT
1c
,pN
0
,M
0
Breast, 50.4 Gy
Grade I
Grade 0
ME
36
24
d
1
Invasive ductal
pT
2
,pN
1biii
,M
0
Refused radiotherapy
EL
54
15
e
0
Invasive ductal
pT
x
,pN
1biii
,M
0
Breast, 52.2 Gy,
periclavicular, 45 Gy
Grade II
Grade I
MW
50
34
3
Invasive ductal
pT
1c
,pN
0
,M
0
Breast, 54 Gy
Grade I
Grade I
ES
62
Invasive tubular
pT
1c
,pN
0
,M
0
None
74
28
0
Invasive ductal
rcT
0
,pN
1biii
,M
0
Axillary/periclavicular, 50.4 Gy
Grade I
Grade 0
a
Family history is given as the number of first- and second-degree relatives with breast cancer.
b
Tumors are characterized according to the TNM classification (68) with one patient (HR) harboring a multifocal carcinoma, and one (ES) suffering from a regional recurrence
at 74 years.
c
None of the A-T heterozygotes showed radiotherapy-related toxicities higher than grade II.
d,e
Two patients died from cancer at age
d
38 years and
e
56 years.
Table 3 Prevalence of ATM missense substitutions in breast cancer patients and in the general population
Mutation
No. of carriers
a
(%)
P
b
OR
c
(95% CI)
Breast cancer (n
⫽ 1000)
Controls (n
⫽ 500)
S707P
28 heterozygotes (0.03)
6 heterozygotes (0.01)
0.05
2.4 (1.0–5.6)
F858L
d
35 heterozygotes (0.04)
13 heterozygotes (0.03)
0.35
1.4 (0.7–2.7)
1 homozygote (
⬍⬍0.01)
P1054R
63 heterozygotes (0.06)
24 heterozygotes (0.05)
0.24
1.4 (0.8–2.2)
1 homozygote (
⬍⬍0.01)
L1420F
50 heterozygotes (0.05)
17 heterozygotes (0.03)
0.16
1.5 (0.9–2.7)
1 homozygote (
⬍⬍0.01)
D1853V
12 heterozygotes (0.01)
4 heterozygotes
e
(0.01)
0.97
1.0 (0.3–3.0)
⬍0.01
f
1.6
f
(1.2–2.2)
D1853N
235 heterozygotes (0.24)
74 heterozygotes
e
(0.23)
0.79
1.0 (0.8–1.4)
12 homozygotes (0.01)
4 homozygotes
e
(0.01)
0.97
1.0 (0.3–3.0)
a
Carrier frequencies are given as numbers of heterozygotes or homozygotes, respectively, with the corresponding percentages given in parentheses.
b
P was calculated from the comparison of allele frequencies to account for both heterozygotes and homozygotes.
c
ORs and 95% CIs are shown for each substitution separately as well as for the whole group of rare missense mutations, excluding the common D1853N polymorphism. Note that
the comparison group is not composed of age-matched controls and that the given ratios do not represent the relative risks conferred by each of the substitutions.
d
F858L is linked with P1054R on the same allele and therefore was excluded from the calculation. None of the carriers tested positive for splicing mutation 3576G
3A, an A-T
mutation known to reside on a P1054R-F858L haplotype (40).
e
Total number of controls tested for codon 1853 variants was 325.
f
P and OR for cumulative analysis of non-D1853N missense substitutions.
7612
GERMLINE ATM MUTATIONS IN BREAST CANCER

The pathogenicities of the 16 identified nontruncating mutations
cannot accurately be predicted at the present stage. Any formal test of
the association between A-T heterozygosity and breast cancer in a
population-based sample is hampered not only by the very large
sample sizes required, but also by the large number and variety of
missense mutations whose allelic effects on protein function and DNA
repair capacity are unknown at present. Although none of the mis-
sense substitutions in our study targeted a residue known to be crucial
for ATM function, 12 of the 16 substitutions affected residues that are
identical in murine atm, the exceptions being I550V, F858L, V1570A,
and S1691R (49). Only five of the amino acid substitutions were
located in the COOH-terminal third of the protein, where a cluster of
missense substitutions had previously been implicated in malignancy
(50) and which harbors the conserved phosphatidylinositol 3
⬘-kinase
signature motifs and a putative domain shared by members of the
FRAP, ATM, and TRRAP subfamilies (8, 51).
Eight of the 16 missense substitutions were found to be polymor-
phisms, including the D1853N and the P1054R substitutions, which
have been proposed as genetic modifiers of cancer penetrance (52,
53). Although the most frequent polymorphism, D1853N, showed the
same prevalence in cases and random individuals, we found a signif-
icant excess of the less common missense substitutions in our breast
cancer patients. A similar observation has been reported by others in
a recent study of patients in the United States with early-onset breast
cancer (54). The S707P substitution in our study appeared to be
associated with high-risk breast carcinoma, characterized by a positive
axillary nodal status and an increased risk of contralateral breast
cancer; however, it is too frequent to be a classical A-T mutation.
Indeed, S707P has not been found as an A-T mutation on its own (40,
45), but a three-amino acid mutation involving the S707P substitution
has been detected as the disease-causing lesion in a single A-T patient
(55). Possible explanations for our findings thus include a reduced
penetrance similar to missense substitutions reported in other tumor
suppressor genes (56 – 60), a specific dominant-negative effect (61),
linkage disequilibrium with an unidentified locus, or an association by
chance. The substitution P1054R, which has been discussed as a
candidate mutation in cancer patients (24, 52, 62, 63), was only
slightly more frequent in our breast cancer group than in the compar-
ison cohort. On the other hand, homozygotes for the F858L-P1054R
and L1420F substitutions have been identified only among our breast
cancer patients thus far, and moderate risks cannot be excluded.
Several other missense substitutions seem to be rare or even private
changes, which will make it difficult to obtain rapid answers from
case-control association studies or from the identification of homozy-
gotes. Quantitative in vitro and in vivo expression analyses are re-
quired and have been initiated to further characterize these amino acid
substitutions and their potential effects on ATM protein stability
and/or function.
None of the patients in our study who were heterozygous for ATM
gene alterations had a higher-degree acute or late normal tissue
reaction related to radiotherapy, indicating that there is no clinically
recognized contraindication for postoperative radiotherapy in A-T
heterozygous patients. This finding is consistent with previous reports
on A-T heterozygotes with breast cancer (25, 26, 64) and with the
absence of truncating ATM mutations in cancer patients selected by
severe acute radiation reactions (27–30, 65). It does not exclude,
however, that A-T heterozygotes may be more susceptible to the
carcinogenic effect of ionizing radiation because of an increased
intrinsic cellular radiosensitivity (19, 66). In the present study, the
frameshift mutation 3801delG was uncovered in one patient who had
developed a contralateral breast cancer almost 20 years after receiving
radiotherapy for the first tumor, and the S707P missense substitution
also was more frequent in patients with bilateral disease. The occur-
rence of bilateral breast cancer in A-T heterozygotes has been docu-
mented by other authors (25, 32), but even if one mutant ATM allele
predisposes affected individuals to the development of a radiation-
induced second malignancy, alternative treatment or omission of
radiotherapy for cancer in A-T heterozygotes is not recommended and
can be counterproductive (64). Further investigations are needed to
address the effectiveness of strategies to reduce radiation doses or
modify therapy for A-T heterozygotes.
In summary, we have identified and characterized a heterogeneous
spectrum of ATM gene alterations in a large hospital-based cohort of
unselected breast cancer patients. The identification of frequent mu-
tations in our population will simplify screening and enable prospec-
tive studies in a clinical research setting. The results presented here
provide a basis for future investigations of the functional and clinical
impact of ATM gene variations and the magnitude of the relative
cancer risk conferred by each of the several identified substitutions.
ACKNOWLEDGMENTS
We thank Christine Volkmann, Hildegard Frye, Andrea Korte, Wolfgang
Ku¨hnau, Elisabeth Katja Ortmann, Philip Wobst, Andrea Wiedenroth, Kerstin
Potthast, Bertha Guiterrez, Uta Go¨lnitz, Cornelia Siebrands, and Claudia
Sto¨ckmann for contributions to the DNA extractions and/or mutation analysis.
We also thank Marianne Twardowski, Dieter Schnalke, and Anja Hermann for
early contributions to the ascertainment of patient samples and clinical data.
Table 4 Clinical characteristics of breast cancer patients carrying common ATM missense substitutions
A total of 571 breast cancer patients were grouped according to their ATM genotypes as D1853N heterozygotes, D1853N homozygotes, D1853V heterozygotes, L1420F
heterozygotes, P1054R heterozygotes, S707P heterozygotes, or others (i.e., patients without any of these substitutions).
D1853N het
a
(n
⫽ 171)
D1853N hom
(n
⫽ 14)
D1853V het
(n
⫽ 10)
L1420F het
(n
⫽ 50)
P1054R het
(n
⫽ 52)
S707P het
(n
⫽ 26)
Others
(n
⫽ 248)
Age at diagnosis, median (range), years
58 (27–85)
58 (33–84)
62 (35–91)
58 (31–76)
55 (34–80)
56 (31–76)
56 (27–85)
Tumor stage
T
1
99 (0.58)
4 (0.29)
5 (0.5)
22 (0.44)
34 (0.67)
12 (0.46)
147 (0.60)
T
2
62 (0.36)
8 (0.57)
2 (0.2)
22 (0.44)
15 (0.29)
11 (0.42)
80 (0.32)
T
3/4
10 (0.06)
2 (0.14)
3 (0.3)
4 (0.08)
2 (0.04)
3 (0.12)
19 (0.08)
Tumor grade
G1
10 (0.06)
1 (0.07)
1 (0.1)
2 (0.05)
2 (0.04)
0 (0)
17 (0.08)
G2
78 (0.45)
6 (0.43)
4 (0.4)
24 (0.55)
31 (0.63)
15 (0.58)
112 (0.54)
G3
84 (0.49)
6 (0.43)
5 (0.5)
18 (0.41)
16 (0.33)
11 (0.42)
80 (0.38)
Axillary nodal status
Negative (N
0
)
123 (0.72)
5 (0.36)
2 (0.2)
29 (0.60)
26 (0.50)
9 (0.35)
165 (0.69)
Positive (N
1–3
)
48 (0.28)
9 (0.64)
8 (0.8)
19 (0.40)
26 (0.50)
17 (0.65)
b
73 (0.31)
Contralateral breast cancer
7 (0.04)
1 (0.07)
1 (0.10)
1 (0.02)
4 (0.08)
5 (0.19)
c
16 (0.06)
a
het, heterozygote; hom, homozygote.
b,c
Comparison between groups revealed
b
that the proportion of patients with positive axillary nodal status (N
1–3
) was higher in S707P heterozygotes than in others (
2
⫽ 13.8;
P
⬍ 0.001; df, 1), and
c
that the proportion of bilateral breast cancer was also higher in S707P heterozygotes (
2
⫽ 5.3; P ⫽ 0.02; df, 1). No differences were observed regarding family
history of breast cancer or estrogen receptor status of the tumor (data not shown).
7613
GERMLINE ATM MUTATIONS IN BREAST CANCER

We gratefully acknowledge Professor Ralf Hass and Professor Christof Sohn
for their support.
REFERENCES
1. Martin, A-M., and Weber, B. L. Genetic and hormonal risk factors in breast cancer.
J. Natl. Cancer Inst. (Bethesda), 92: 1126 –1135, 2000.
2. Goss, P. E., and Sierra, S. Current perspectives on radiation-induced breast cancer.
J. Clin. Oncol., 16: 338 –347, 1998.
3. Nathanson, K. N., Wooster, R., and Weber, B. L. Breast cancer genetics: what we
know and what we need. Nat. Med., 7: 552–556, 2001.
4. Boder, E., and Sedgwick, R. P. Ataxia telangiectasia: a familial syndrome of pro-
gressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary
infection. Pediatrics, 21: 526 –554, 1958.
5. Lavin, M., and Shiloh, Y. The genetic defect in ataxia telangiectasia. Annu. Rev.
Immunol., 15: 177–222, 1997.
6. Kastan, M. B., and Lim, D. S. The many substrates and functions of ATM. Nat. Rev.
Mol. Cell Biol., 1: 179 –186, 2000.
7. Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle,
D. A., Smith, S., Uziel, T., Sfez, S., Ashkenazi, M., Pecker, I., Frydman, M., Harnik,
R., Patanjali, S. R., Simmons, A., Clines, G. A., Sartiel, A., Gatti, R. A., Chessa, L.,
Sanal, O., Lavin, M. F., Jaspers, N. G. J., Taylor, A. M. R., Arlett, C. F., Miki, T.,
Weissman, S. M., Lovett, M., Collins, F. S., and Shiloh, Y. A single ataxia telangi-
ectasia gene with a product similar to PI-3 kinase. Science (Wash. DC), 268:
1749 –1753, 1995.
8. Savitsky, K., Sfez, S., Tagle, D. A., Ziv, Y., Sartiel, A., Collins, F. S., Shiloh, Y., and
Rotman, G. The complete sequence of the coding region of the ATM gene reveals
similarity to cell cycle regulators in different species. Hum. Mol. Genet., 4: 2025–
2032, 1995.
9. Platzer, M., Rotman, G., Bauer, D., Uziel, T., Savitsky, K., Bar-Shira, A., Gilad, S.,
Shiloh, Y., and Rosenthal, A. Ataxia telangiectasia locus: sequence analysis of 184 kb
of human genomic DNA containing the entire ATM gene. Genome Res., 7: 592– 605,
1997.
10. Gatei, M., Scott, S. P., Filippovitch, I., Soronika, N., Lavin, M. F., Weber, B., and
Khanna, K. K. Role for ATM in DNA damage-induced phosphorylation of BRCA1.
Cancer Res., 60: 3299 –3304, 2000.
11. Boder, E., and Sedgwick, R. P. Ataxia-telangiectasia. A review of 101 cases. In:
Walsh, G (ed.), Little Club Clinics in Developmental Medicine, No. 8, Cerebellum,
Posture and Cerebral Palsy, pp. 110 –118. London: The National Spastics Society and
Heinemann Medical Books, Ltd., 1963.
12. Hecht, F., and Hecht, B. K. Cancer in ataxia-telangiectasia patients. Cancer Genet.
Cytogenet., 46: 9 –19, 1990.
13. Taylor, A. M. Ataxia telangiectasia genes and predisposition to leukaemia, lymphoma
and breast cancer. Br. J. Cancer, 66: 5–9, 1992.
14. Stankovic, T., Kidd, A. M., Sutcliffe, A., McGuire, G. M., Robinson, P., Weber, P.,
Bedenham, T., Bradwell, A. R., Easton, D. F., Lennox, G. G., Haites, N., Byrd, P. J.,
and Taylor, A. M. ATM mutations and phenotypes in ataxia-telangiectasia families in
the British Isles: expression of mutant ATM and the risk of leukemia and lymphoma,
and breast cancer. Am. J. Hum. Genet., 62: 334 –345, 1998.
15. Su, Y., and Swift, M. Mortality of carriers of ataxia-telangiectasia mutations in
ataxia-telangiectasia families. Ann. Intern. Med., 133: 770 –778, 2000.
16. Swift, M., Sholman, L., Perry, M., and Chase, C. Malignant neoplasms in the families
of patients with ataxia-telangiectasia. Cancer Res., 36: 209 –215, 1976.
17. Swift, M., Reitnauer, P. J., Morrell, D., and Chase, C. L. Breast and other cancers in
families with ataxia-telangiectasia. N. Engl. J. Med., 316: 1289 –1294, 1987.
18. Pippard, E. C., Hall, A. J., Barker, D. J. P., and Bridges, P. Cancer in homozygotes
and heterozygotes of ataxia-telangiectasia and xeroderma pigmentosum in Britain.
Cancer Res., 48: 2929 –2932, 1988.
19. Swift, M., Morrell, D., Massey, R. B., and Chase, C. L. Incidence of cancer in 161
families affected by ataxia-telangiectasia. N. Engl. J. Med., 325: 1831–1836, 1991.
20. Athma, P., Rappaport, R., and Swift, M. Molecular genotyping shows that ataxia-
telangiectasia heterozygotes are predisposed to breast cancer. Cancer Genet. Cyto-
genet., 92: 130 –134, 1996.
21. Inskip, H. M., Kinlen, L. J., Taylor, A. M. R., and Arlett, C. F. Risk of breast cancer
and other cancers in heterozygotes for ataxia-telangiectasia. Br. J. Cancer, 79:
1304 –1307, 1999.
22. Janin, M., Andrieu, N., Ossian, K., Lauge, A., Croquette, M-F., Griscelli, C., Debre,
M., Bressac-de-Pailleret, B., Aurias, A., and Stoppa-Lyonnet, D. Breast cancer risk in
ataxia telangiectasia (AT) heterozygotes: haplotype study in French AT families.
Br. J. Cancer, 80: 1042–1045, 1999.
23. Olsen, J. H., Hahnemann, M. J., Borresen-Dale, A-L., Brondum-Nielsen, K.,
Hammarstro¨m, L., Kleinerman, R., Ka¨a¨ria¨inen, H., Lo¨nnqvist, T., Sankila, R.,
Seersholm, N., Tretli, S., Yuen, J., Boice, J. D., Jr., and Tucker, M. Cancer in
patients with ataxia-telangiectasia and in their relatives in the Nordic countries.
J. Natl. Cancer Inst. (Bethesda), 93: 121–127, 2001.
24. Vorechovsky, I., Luo, L., Lindblom, A., Negrini, M., Webster, A. D. B., Croce, C. M.,
and Hammarstro¨m, L. ATM mutations in cancer families. Cancer Res., 56: 4130 –
4133, 1996.
25. Ramsay, J., Birrell, G., and Lavin, M. Breast cancer and radiotherapy in ataxia-
telangiectasia heterozygote. Lancet, 347: 1627, 1996.
26. FitzGerald, M. G., Bean, J. M., Hedge, S. R., Unsal, H., MacDonald, D. J., Harkin,
D. P., Finkelstein, D. M., Isselbacher, K. J., and Haber, D. A. Heterozygous ATM
mutations do not contribute to early onset of breast cancer. Nat. Genet., 15: 307–310,
1997.
27. Appleby, J. M., Barber, J. B. P., Levine, E., Varley, J. M., Taylor, A. M. R.,
Stankovic, T., Heighway, J., Warren, C., and Scott, D. Absence of mutations in the
ATM gene in breast cancer patients with severe responses to radiotherapy. Br. J.
Cancer, 76: 1546 –1549, 1997.
28. Shayeghi, M., Seal, S., Regan, J., Collins, N., Barfoot, R., Rahman, N., Ashton, A.,
Moohan, M., Wooster, R., Owen, R., Bliss, J. M., Stratton, M. R., and Yarnold,
J. Heterozygosity for mutations of the ataxia telangiectasia gene is not a major cause
of radiotherapy complications in breast cancer patients. Br. J. Cancer, 78: 922–927,
1998.
29. Clarke, R. A., Goozee, G. R., Birrell, G., Fang, Z. M., Hasnain, H., Lavin, M., and
Kearsley, J. H. Absence of ATM truncations in patients with severe acute radiation
reactions. Int. J. Radiat. Oncol. Biol. Phys., 41: 1021–1027, 1998.
30. Ramsay, J., Birrell, G., and Lavin, M. Testing for mutations of the ataxia telangiec-
tasia gene in radiosensitive breast cancer patients. Radiother. Oncol., 47: 125–128,
1998.
31. Izatt, L., Greenman, J., Hodgson, S., Ellis, D., Watts, S., Scott, G., Jacobs, C.,
Liebmann, R., Zvelebi, M. J., Mathew, C., and Solomon, E. Identification of germline
missense mutations and rare allelic variants in the ATM gene in early-onset breast
cancer. Genes Chromosomes Cancer, 26: 286 –294, 1999.
32. Broeks, A., Urbanus, J. H. M., Floore, A. N., Dahler, E. C., Klijn, J. G. M., Rutgers,
E. J. T., Devilee, P., Russell, N. S., van Leeuwen, F. E., and van’t Veer, L.
ATM-heterozygous germline mutations contribute to breast cancer-susceptibility.
Am. J. Hum. Genet., 66: 494 –500, 2000.
33. Shafman, T. D., Levitz, S., Nixon, A. J., Gibans, L-A., Nichols, K. E., Bell, D. W.,
Ishioka, C., Isselbacher, K. J., Gelman, R., Garber, J., Harris, J. R., and Haber, D. A.
Prevalence of germline truncating mutations in ATM in women with a second breast
cancer after radiation therapy for a contralateral tumor. Genes Chromosomes Cancer,
27: 124 –129, 2000.
34. Trotti, A., Byhardt, R., Stetz, J., Gwede, C., Corn, B., Fu, K., Gunderson, L.,
McCormick, B., Morris, M., Rich, T., Shipley, W., and Curran, W., for the Radiation
Therapy Oncology Group. Common toxicity criteria: version 2.0, an improved
reference for grading the acute effects of cancer treatment: impact on radiotherapy.
Int. J. Radiat. Oncol. Biol. Phys., 47: 13– 47, 2000.
35. Broeks, A., de Klein, A., Floore, A. N., Muijtjens, M., Kleijer, W. J., Jaspers, N. G.,
and van’t Veer, L. ATM germline mutations in classical ataxia-telangiectasia patients
in the Dutch population. Hum. Mutat., 12: 330 –337, 1998.
36. RTOG/EORTC Late Effects Working Group. Late effects of normal tissues consen-
sus conference; LENT SOMA scale for all anatomic sites. Int. J. Radiat. Oncol. Biol.
Phys., 31: 1049 –1091, 1995.
37. Savitsky, K., Platzer, M., Uziel, T., Gilad, S., Sartiel, A., Rosenthal, A., Elroy-Stein,
O., Shiloh, Y., and Rotman, G. Ataxia-telangiectasia: structural diversity of untrans-
lated sequences suggests complex post-transcriptional regulation of ATM gene ex-
pression. Nucleic Acids Res., 25: 1678 –1684, 1997.
38. Chomczynski, P., and Sacchi, N. Single-step method of RNA isolation by acid
guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162: 156 –
159, 1987.
39. Neitzel, H. A routine method for the establishment of permanent growing lympho-
blastoid cell lines. Hum. Genet., 73: 320 –326, 1986.
40. Sandoval, N., Platzer, M., Rosenthal, A., Do¨rk, T., Bendix, R., Skawran, B.,
Stuhrmann, M., Wegner, R. D., Sperling, K., Banin, S., Shiloh, Y., Baumer, A.,
Bernthaler, U., Sennefelder, H., Brohm, M., Weber, B. H. F., and Schindler, D.
Characterization of ATM gene mutations in 66 ataxia telangiectasia families.
Hum. Mol. Genet., 8: 69 –79, 1999.
41. Orita, M., Suzuki, Y., Sekiya, T., and Hayashi, K. Rapid and sensitive detection of
point mutations and DNA polymorphisms using polymerase chain reaction. Genom-
ics, 5: 874 – 879, 1989.
42. Chen, G., and Lee, E. Y-H. P. The product of the ATM gene is a 370 kDa nuclear
phosphoprotein. J. Biol. Chem., 271: 33693–33697, 1996.
43. Laake, K., Jansen, L., Hahnemann, J. M., Brøndum-Nielsen, K., Lo¨nnqvist, T.,
Ka¨a¨ria¨inen, H., Sankila, R., La¨ndesma¨ki, A., Hammarstro¨m, L., Yuen, J., Tretli, S.,
Heiberg, A., Olsen, J. H., Tucker, M., Kleinerman, R., and Børresen-Dale, A.
Characterization of ATM mutations in 41 Nordic families with ataxia telangiectasia.
Hum. Mutat., 16: 232–246, 2000.
44. Gilad, S., Khosravi, R., Shkedy, D., Uziel, T., Ziv, Y., Savitsky, K., Rotman, G.,
Smith, S., Chessa, L., Jorgensen, T. J., Harnik, R., Frydman, M., Sanal, O., Portnoi,
S., Goldwicz, Z., Jaspers, N. G. J., Gatti, R. A., Lenoir, G., Lavin, M. F., Tatsumi, K.,
Wegner, R. D., Shiloh, Y., and Bar-Shira, A. Predominance of null mutations in
ataxia-telangiectasia. Hum. Mol. Genet., 5: 433– 439, 1996.
45. Concannon, P., and Gatti, R. A. Diversity of ATM gene mutations detected in patients
with ataxia-telangiectasia. Hum. Mutat., 10: 100 –107, 1997.
46. Teraoka, S., Telatar, M., Becker-Catania, S., Liang, T., Onegut, S., Tolun, A., Chessa,
L., Sanal, O., Bernatowska, E., Gatti, R. A., and Concannon, P. Splicing defects in the
ataxia-telangiectasia gene, ATM: underlying mutations and phenotypic conse-
quences. Am. J. Hum. Genet., 64: 1617–1631, 1999.
47. Neubauer, S., Do¨rk, T., Bendix, R., Bremer, M., Rades, D., Karstens, J. H., Stumm,
M., and Sauer, R. Chromosomal and clinical radiosensitivity in breast cancer patients
with different ATM gene mutations. Med. Genet., 12: 94, 2000.
48. Speit, G., Trenz, K., Schu¨tz, P., Bendix, R., and Do¨rk, T. Mutagen sensitivity of
human lymphoblastoid cells with a BRCA1 mutation in comparison to ataxia telan-
giectasia heterozygote cells. Cytogenet. Cell Genet., 91: 261–266, 2000.
49. Pecker, I., Avraham, K. B., Gilbert, D. J., Savitsky, K., Rotman, G., Harnik, R.,
Fukao, T., Schro¨ck, E., Hirotsune, S., Tagle, D. A., Collins, F. S., Wynshaw-Boris,
A., Ried, T., Copeland, N. G., Jenkins, N. A., Shiloh, Y., and Ziv, Y. Identification
and chromosomal localisation of Atm, the mouse homolog of the ataxia-telangiectasia
gene. Genomics, 35: 39 – 45, 1996.
7614
GERMLINE ATM MUTATIONS IN BREAST CANCER

50. Vorechovsky, I., Luo, L., Dyer, M. J. Catovsky, D., Amlot, P. L., Yaxley, J. C.,
Foroni, L., Hammarstrom, L., Webster, A. D., and Yuille, M. A. Clustering of
missense mutations in the ataxia telangiectasia gene in a sporadic T-cell leukaemia.
Nat. Genet., 17: 96 –99, 1997.
51. Bosotti, R., Isacchi, A., and Sonnhammer, E. L. FAT: a novel domain in PIK related
kinases. Trends Biochem. Sci., 25: 225–227, 2000.
52. Larson, G. P., Zhang, G., Ding, S., Foldenauer, K. F., Udar, N., Gatti, R. A., Neuberg,
D., Lunetta, K. L., Ruckdeschel, J. C., Longmate, J., Flanagan, S., and Krontiris, T. G.
An allelic variant at the ATM locus is implicated in breast cancer susceptibility.
Genet. Testing, 1: 165–170, 1998.
53. Maillet, P., Chappuis, P. O., Vaudan, G., Dobbie, Z., Muller, H., Hutter, P., and
Sappino, A. P. A polymorphism in the ATM gene modulates the penetrance of
hereditary non-polyposis colorectal cancer. Int. J. Cancer, 88: 928 –931, 2000.
54. Teraoka, S. N., Malone, K. E., Doody, D. R., Suter, N. M., Ostrander, E. A., Daling, J.
R., and Concannon, P. Increased frequency of ATM mutations in breast carcinoma
patients with early onset disease and positive family history. Cancer, 92: 479 – 487, 2001.
55. Vorechovsky, I., Luo, L., Prudente, S., Chessa, L., Russo, G., Kanariou, M., James,
M., Negrini, M., Webster, A. D. B., and Hammarstro¨m, L. Exon-scanning mutation
analysis of the ATM gene in patients with ataxia-telangiectasia. Eur. J. Hum. Genet.,
4: 352–355, 1996.
56. Otterson, G. A., Modi, S., Nguyen, K., Coxon, A. B., and Kaye, F. J. Temperature-
sensitive, RB mutations linked to incomplete penetrance of familial retinoblastoma in
12 families. Am. J. Hum. Genet., 65: 1040 –1046, 1999.
57. Varley, J. M., McGown, G., Thorncroft, M., James, L. A., Margison, G. P., Forster,
G., Evans, D. G. R., Harris, M., Kelsey, A. M., and Birch, J. M. Are there low-
penetrance p53 alleles? Evidence from childhood adrenocortical tumors. Am. J. Hum.
Genet., 65: 995–1006, 1999.
58. Healey, C. S., Dunning, A. M., Dawn Teare, M., Chase, D., Parker, L., Burn, J.,
Chang-Claude, J., Mannermaa, A., Kataja, V., Huntsman, D. G., Pharoah, P. D. P.,
Luben, R. N., Easton, D. F., and Ponder, B. A. J. A common variant in BRCA2 is
associated with both breast cancer risk and prenatal viability. Nat. Genet., 26:
362–364, 2000.
59. Lamlum, H., Tassan, N. A., Jaeger, E., Frayling, I., Sieber, O., Bin Reza, F., Eckert,
M., Rowan, A., Barclay, E., Atkin, W., Williams, C., Gilbert, J., Cheadle, J., Bell, J.,
Houlston, R., Bodmer, W., Sampson, J., and Tomlinson, I. Germline APC variants in
patients with multiple colorectal adenomas, with evidence for the particular impor-
tance of E1317Q. Hum. Mol. Genet., 9: 2215–2221, 2000.
60. Rebbeck, T. R., Walker, A. H., Zeigler-Johnson, C., Weisburg, S., Martin, A-M.,
Nathanson, K. L., Wein, A. J., and Malkowicz, S. B. Association of HPC2/ELAC2
genotypes and prostate cancer. Am. J. Hum. Genet., 67: 1014 –1019, 2000.
61. Gatti, R. A., Tward, A., and Concannon, P. Cancer risk in ATM heterozygotes: a
model of phenotypic and mechanistic differences between missense and truncating
mutations. Mol. Genet. Metab., 68: 419 – 423, 1999.
62. Stankovic, T., Weber, P., Stewart, G., Bedenham, T., Murray, J., Byrd, P. J., Moss,
P. A., and Taylor, A. M. Inactivation of ataxia-telangiectasia mutated gene in B-cell
chronic lymphocytic leukaemia. Lancet, 353: 26 –29, 1999.
63. Vorechovsky, I., Ortmann, E. K., Steinmann, D., and Do¨rk, T. Missense mutations at
ATM gene and cancer risk. Lancet, 353: 1276, 1999.
64. Weissberg, J. B., Huang, D-D., and Swift, M. Radiosensitivity of normal tissues in
ataxia-telangiectasia heterozygotes. Int. J. Radiat. Oncol. Biol. Phys., 42: 1132–1136,
1998.
65. Oppitz, U., Bernthaler, U., Schindler, D., Sobeck, A., Hoehn, H., Platzer, M.,
Rosenthal, A., and Flentje, M. Sequence analysis of the ATM gene in 20 patients with
RTOG grade 3 or 4 acute and/or late tissue radiation side effects. Int. J. Radiat. Oncol.
Biol. Phys., 44: 981–988, 1999.
66. West, C. M., Elyan, S. A., Berry, P., Cowan, R., and Scott, D. A comparison of the
radiosensitivity of lymphocytes from normal donors, cancer patients, individuals with
ataxia telangiectasia (AT) and AT heterozygotes. Int. J. Radiat. Biol., 68: 197–203,
1995.
67. Beaudet, A. L., and Tsui, L-C. A suggested nomenclature for designating mutations.
Hum. Mutat., 2: 245–248, 1993.
68. Sobin, L. H., and Wittekind, C. (eds.). International Union against Cancer (UICC).
TNM Classification of Malignant Tumours, Ed. 5. New York: Wiley-Liss, 1997.
7615
GERMLINE ATM MUTATIONS IN BREAST CANCER
Wyszukiwarka
Podobne podstrony:
Functional and Computational Assessment of Missense Variants in the Ataxia Telangiectasia Mutated (A
Variants in the ATM gene associated with a reduced risk of contralateral breast cancer
Risk of Cancer by ATM Missense Mutations in the General Population
Predictors of perceived breast cancer risk and the relation between preceived risk and breast cancer
A nonsense mutation (E1978X) in the ATM gene is associated with breast cancer
Population Based Estimates of Breast Cancer Risks Associated With ATM Gene Variants c 7271T4G and c
ATM Gene Founder Haplotypes and Associated Mutations in Polish Families with Ataxia Telangiectasia
Cancer Risk According to Type and Location of ATM Mutation in Ataxia Telangiectasia Families
Impact of resuscitation system errors on survival from in hospital cardiac arrest
In hospital cardiac arrest Is it time for an in hospital chain of prevention
A recurrent mutation in type II collagen gene causes Legg Calvé Perthes disease in a Japanese family
Impact of resuscitation system errors on survival from in-hospital cardiac arrest, MEDYCYNA, RATOWNI
Genome wide mapping of gene–microbiota interactions in susceptibility to autoimmune skin blistering
Evaluation of the role of Finnish ataxia telangiectasia mutations in hereditary predisposition to br
Continuous mechanical chest compression during in hospital cardiopulmonary resuscitation of patients
Proton Magnetic Resonance Spectroscopy of the Medial Prefrontal Cortex in Patients With Deficit Schi
Rare, Evolutionarily Unlikely Missense Substitutions in ATM Confer Increased Risk of Breast Cancer
Single nucleotide polymorphism D1853N of the ATM gene may alter the risk for breast cancer
więcej podobnych podstron