Marissa C. Rosen
Thursday, March 13, 2003
Asymmetric Synthesis of α-Amino Acids: New Twists on Old Ideas
Chiral α–amino acids are important biological molecules. They are the building blocks
of proteins, and the twenty proteogenic
L
-amino acids are ubiquitous to all living
organisms on earth. Because of their biological importance, they were some of the first
chiral synthetic targets.
α–Amino acids can exist in either the
D
or
L
form. The majority of
naturally occurring amino acids are
L
-amino acids; however, some
D
-
amino acids occur naturally. For example, the peptidoglycan that
makes up the cell wall of the bacterium Staphylococcus aureus
contains
D
-isoglutamate and
D
-alanine. The twenty proteogenic
amino acids are
L
-amino acids and all except cysteine have the S
absolute configuration at the α–carbon.
Natural and non-natural amino acids have found many uses in
organic chemistry. They can be used as chiral starting materials for natural product total
synthesis, or as chiral auxiliaries, catalysts or catalyst ligands. In the field of protein
engineering, non-natural amino acids can be incorporated into proteins in order to study
protein structure and function They have also been used in order to create drugs or other
bioactive molecules that will not be degraded as quickly as natural amino acids by
protease enzymes.
Although enzymatic syntheses and resolutions are viable methods for the production of
naturally occurring chiral α–amino acids, they are often not useful for synthesizing non-
natural
D
-amino acids or amino acids with non-natural side chains. An alternate method
is the formation of diastereomeric salts of optically impure amino acid mixtures, followed
by chiral resolution. However, this method is time consuming and therefore expensive,
and not all mixtures are amenable to this approach. In general,
L
-amino acids can often
be obtained by biological methods; however non-natural
D
-amino acids and
D
- or
L
-
amino acids with unusual side chains cannot. For that reason, synthetic routes to these
molecules are necessary.
There are 4 general methods of chiral α–amino acid synthesis.
1
The first is selective
addition of a carboxylic acid equivalent to the pro-chiral α–carbon of an imine, as in the
asymmetric Strecker reaction. Also, hydrogen can be added asymmetrically to a
prochiral α–carbon of a di-dehydro amino acid, as in the Knowles Monsanto synthesis.
The Corey-Link reaction involves asymmetric addition of hydride to a ketone amino acid
precursor. Various asymmetric derivitizations of glycine involve creating a nucleophile
or electrophile at the α–carbon of a glycine equivalent and subsequent asymmetric
addition of an R group to that center. There are, of course, many other routes to chiral α-
amino acids, such as use of Evans chiral auxiliary
2
, electrophilic amination of enolates,
H
2
N
CO
2
H
R
H
H
2
N
CO
2
H
R
H
(S)-
L
-amino acid
(R)-
D
-amino acid
Figure 1: Designations
of amino acids
nucleophilic amination of α–substituted acids, and enzymatic syntheses, including
enzymatic resolution
3
or enzyme-catalyzed bond-forming reactions
The Strecker Synthesis
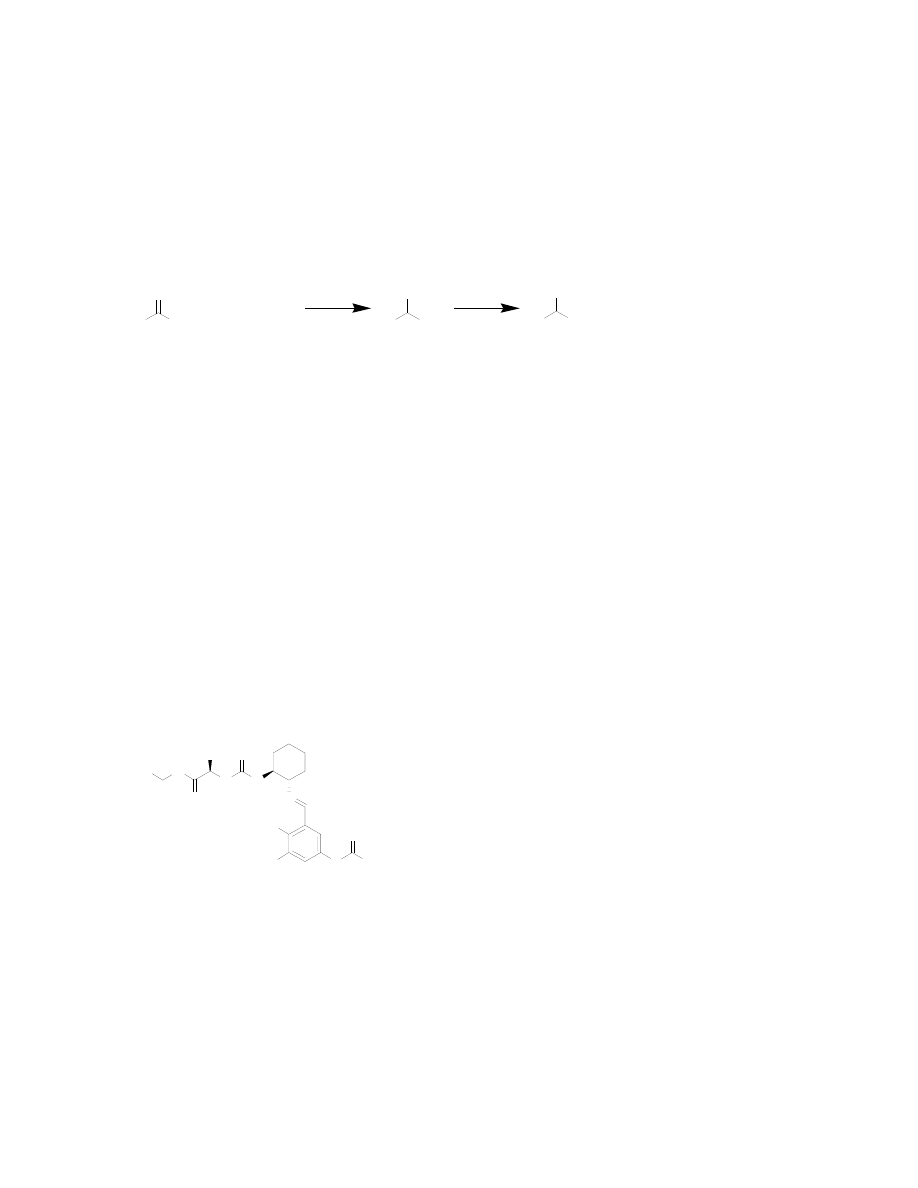
The Strecker reaction was developed by Adolph Strecker in 1850.
4
It is a three-
component reaction between an aldehyde, an amine and hydrogen cyanide to form an α–
amino nitrile. The nitrile can then be hydrolyzed to the free acid, resulting in a racemic
α–amino acid (Figure
2). The Strecker
synthesis is one of the
most convenient
methods for
synthesizing amino acids and much research has focused on the development of
asymmetric versions of this reaction.
The first asymmetric Strecker reaction was reported over one hundred years after the
introduction of the racemic version.
5
In this reaction, a chiral α-methylbenzylamine was
condensed with an aldehyde to form a chiral imine. The chiral center was able to direct
the addition to the imine with enantiomeric excesses from 9-58%, depending on the side
chain. After hydrolysis of the nitrile group, the chiral auxiliary is then destructively
removed to yield the free amino acid. While this method is not useful today, it represents
the initial attempts in an important field of asymmetric Strecker reactions.
The first non-metal catalyzed asymmetric Strecker synthesis, reported by Lipton and co-
workers, used a cyclic dipeptide catalyst to achieve high yields and enantiomeric excess.
6
In particular, phenylglycine and p-methoxyphenylglycine were synthesized in >99% and
96% ee, respectively. However, electron-poor aromatic side chains and aliphatic side
chains give essentially racemic products with this catalyst, making it useful only in the
synthesis of certain types of amino acids. This work was soon followed by Jacobsen and
co-workers report of on another chiral, non-metal
catalyst for the asymmetric Strecker synthesis.
7,8
Jacobsen’s catalyst was based on a tridentate Schiff
base scaffold that would be amenable to solid phase
synthesis. The catalyst was synthesized on solid
support and optimized through a series of parallel
libraries. The final catalyst was superior to that
previously described by Lipton et. al. because it was
capable of catalyzing the formation of aliphatic α–
amino nitriles with significant enantiomeric excesses (80-90%). The conversion of
ketimines to α,α-disubstituted amino acids was also achieved with this catalyst.
9
Because
it can be synthesized in 5 steps with 80% overall yield and no chromatographic
separation and the resin-bound catalyst has been shown to be recyclable at least 10 times
with no loss of yield or enantiomeric excess, Jacobsen’s Schiff base catalyst should find
use in other labs, and possibly in industry.
10
R
H
O
NH
3
HCN
R
CN
NH
2
R
CO
2
H
NH
2
+
+
(+/-)
Figure 2: The Strecker synthesis of amino acids
Ph
H
N
N
H
N
H
N
HO
O
O
O
t
Bu
O
O
t
Bu
t-Bu
Figure 3: Jacobsen's Schiff base catalyst
Recently, the laboratory of Kobayashi reported a novel zirconium catalyst that could
effect the enantioselective synthesis of α-aminonitriles.
11
While Kobayashi’s catalyst
gave high yields and enantiomeric excesses for aromatic imines and improved yields and
enantiomeric excesses for aliphatic imines over the previously discussed catalysts, its
large molecular weight (1671.95 amu) endows this catalyst with very poor atom
economy. In a related work the authors report the first efficient, asymmetric three
component Strecker reaction that is amenable to industrial use, using the same zirconium
catalyst.
12
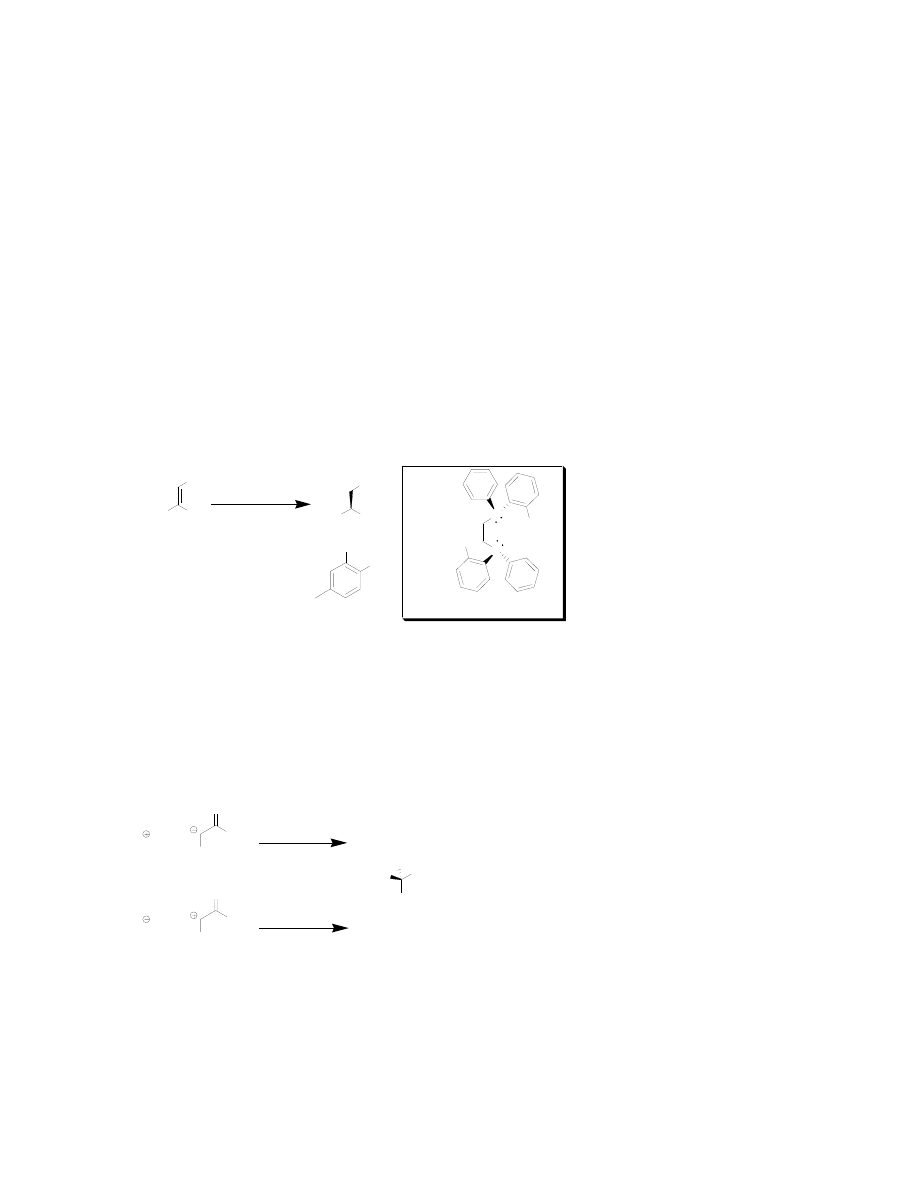
Knowles-Monsanto Synthesis
William S. Knowles won the Nobel prize in 2001, along with Noyori and Sharpless, for
his pioneering work in the development of asymmetric hydrogenation of di-
dehydroamino acids. Knowles and co-workers began this project while trying to develop
an efficient industrial process to synthesize
L
-DOPA that did not involve time-consuming
resolutions and recycling. This led to the development of DiPAMP, a chiral phosphine
ligand for asymmetric hydrogenations with rhodium metal (Figure 4).
13
This new chiral
phosphine rhodium catalyst
was used in Monsanto’s
synthesis of L-DOPA (a
phenylalanine derivative
that is used in the treatment
of Parkinson’s disease) and
has since had widespread
use outside of the
laboratory in which it was developed. This catalyst gives high yields and enantiomeric
excesses, and the availability of both enantiomers of DiPAMP (R,R and S,S) affords
either enantiomer of amino acid.
Asymmetric Derivitizations of Glycine and Glycine Equivalents
The two general approaches for derivatizing glycine are nucleophilic and electrophilic.
Both involve the use of a chiral auxiliary to direct the formation of the new stereogenic
center (Figure 5).
14
Nucleophilic
derivatizations are much more common in
the literature and will therefore be the focus
of this abstract. Some of the first efficient
examples of a nucleophilic derivatization
came from the labs of Schöllkopf.
15
In these
examples, a chiral amino acid is cyclized
with glycine to form a bis-lactam.
Treatment of the bis-lactam with
Meerwein’s salt forms a bis-lactim ether,
which can be deprotonated at the α-carbon to form an enolate. The chiral center on the
essentially planar bis-lactim anion directs the enolate to attack from the opposite side.
Chiral auxiliaries derived from
L
-t-leucine
16
,
L
- alanine
15
,
L
-valine
17
and
L
-O,O-dimethyl-
NR
2
χ
O
NR
2
χ
O
(enolate)
(cation equivalent)
Nucleophilic
Electrophilic
R
R
+
+
NH
2
CO
2
H
R
H
Figure 5: Types of Derivatizations of Glycine
(R,R)-DiPAMP
HO
2
C
NH
2
HO
2
C
NH
2
R
R
H
2
, Rh(L)(cod)
P
P
OCH
3
H
3
CO
ligand =
Figure 4: Knowles-Monsanto Synthesis
L
-DOPA: R=
OH
OH
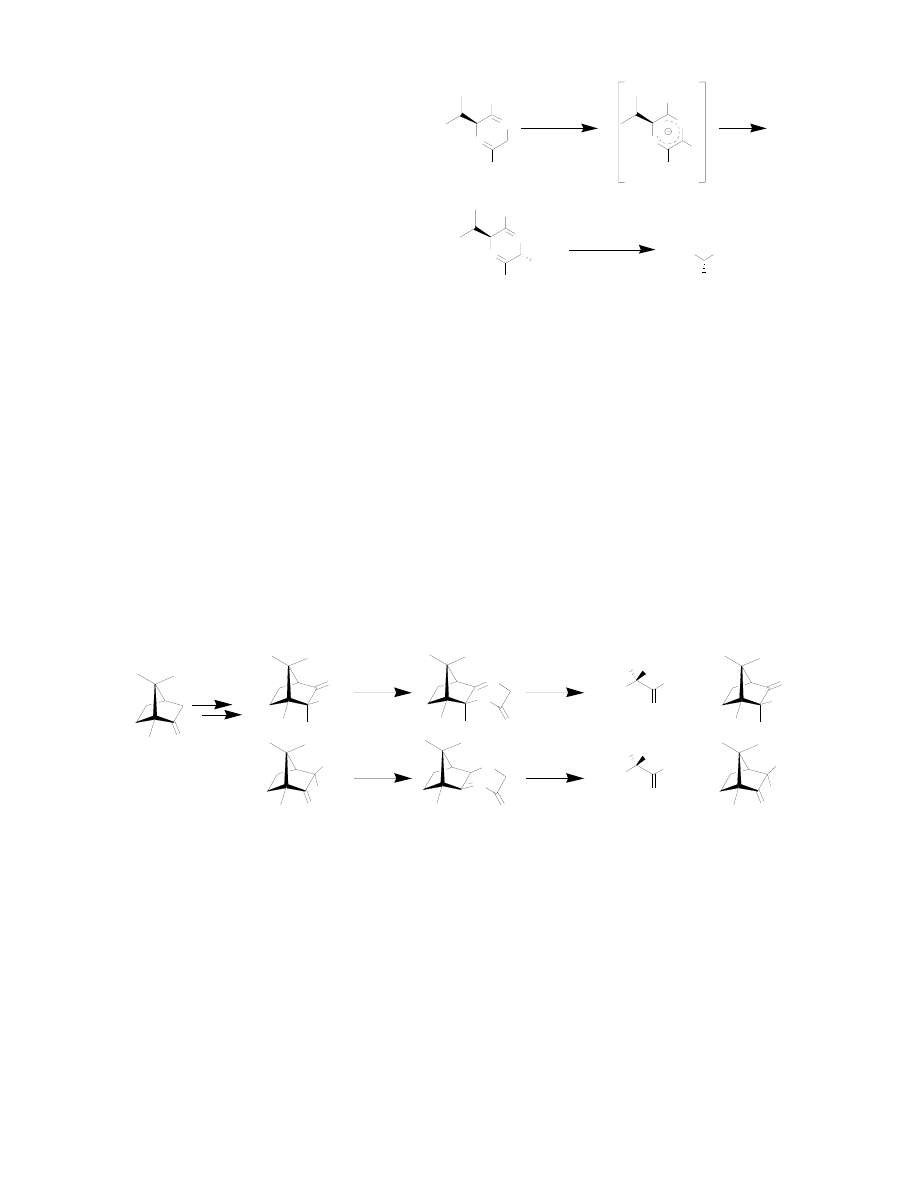
α-methyldopa
18
have all shown good
yields and good to excellent
enantiomeric excesses, depending on the
nucleophilic substrate. Bis-lactim ethers
can also be used to synthesize α, α–
disubstituted amino acids, by cyclizing
the chiral auxiliary amino acid with
racemic alanine instead of gylcine.
Again, the chiral auxiliary directs the
enolate to attack from the opposite face
to yield the disubstituted amino acid in good yields and enantiomeric excesses in most
cases.
19
Williams and co-workers developed a similar system in which a cyclic lactone consisting
of a glycine equivalent and a chiral auxiliary was deprotonated to form the enolate. The
chiral auxiliary acts to direct the enolate to attack from the opposite side to form the
amino acid precursor. The real strength of this methodology is that different cleavage
methods can be used to obtain either the N-protected amino acid or the zwiterionic free
amino acid.
20
α, α–Disubstituted amino acids could also be synthesized by this method.
21
Lu et. al. recently published another example of a chiral glycine enolate in which a chiral
auxiliary derived from (+)-R-camphor was coupled with glycine to give the chiral
template (Figure 7).
22
This could then be deptrotonated at the α-carbon to form the
enolate. The chiral auxiliary directs the formation of the endo product in high
diastereomeric excesses. Both enantiomers of amino acids are accessible through two
different chiral auxiliaries, both derived from the inexpensive (+)-R-camphor.
23
Corey-Link Reaction
The Corey-Link reaction involves the application of the CBS reduction, originally
applied to the reduction of carbonyls, to chiral α-amino acid synthesis.
24,25
This sequence
involves first the stereoselective reduction of a substituted trichloromethyl ketone to the
corresponding (R)-secondary alcohol with the (S)-oxazaborolidine CBS reduction
catalyst, with > 97:3 enantioselectivity in most cases. The alpha-trichloromethyl alcohols
can be converted to (S)-amino acids in four steps high overall yields. This methodology
is useful for preparing α-amino acids with a wide range of side chains, from aryl and
napthyl to cyclic and acyclic aliphatic groups.
O
H
OH
N
H
O
O
O
H
OH
H
2
N
OH
O
H
R
+
O
N
O
H
2
N
OH
O
R
H
+
O
OH
H
O
OH
H
D
-amino acid
L
-amino acid
O
(+)-(R)-Camphor
Figure 7: Chiral Tricyclic Iminolactone Derived from (1R)-(+)-Camphor
N
N
OMe
OMe
1. n-BuLi
2. RCH
2
X
N
N
OMe
OMe
CH
2
R
0.25 N HCl
H
2
N
CO
2
Me
CH
2
R
+
L
-Val-OMe
N
N
OMe
OMe
H
Figure 6: Bis-Lactim Ether Glycine Enolates
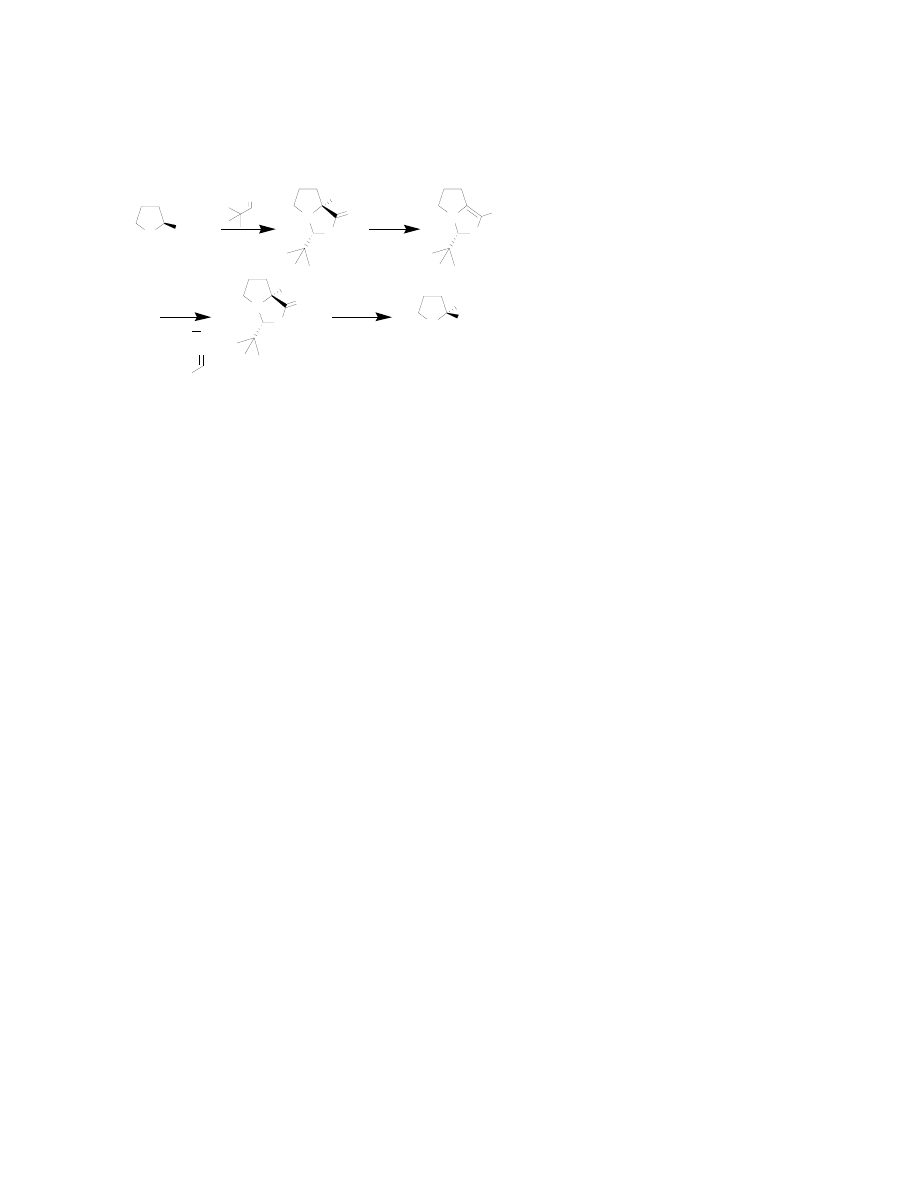
Seebach’s Self-Reproduction of Chirality
A very interesting example of
amino acid synthesis comes from
the laboratory of Seebach, in which
a chiral amino acid starting
material such as
L
-proline is used.
Although the chirality of the
original stereogenic center of the
amino acid is lost in enolate
formation, that stereocenter is
directed back to its original
conformation (Figure 8).
27
In an
example of this methodology,
L
-
proline is condensed with pivalaldehyde to yield a single diastereomer of the bicyclic
aminal. The stereogenic α-carbon of proline directs the formation of the (S)
conformation at the newly formed stereogenic center. The aminal is then deprotonated to
form the enolate, thereby erasing the stereochemistry at proline’s α-carbon. However,
upon nucleophilic attack by the enolate to an electrophlie (such as an alkyl halide or
aldehyde), the (S) stereocenter directs the reproduction of the original (S) conformation at
the proline’s α-carbon. This methodology can be used to synthesize other, non-cyclic
amino acid derivatives besides proline.
28
Conclusions
There are many methods available for the synthesis of chiral α-amino acids, and the large
amounts of time and effort that has gone into the development of these methods attests to
the importance of chiral α-amino acids as synthetic targets today. The different
approaches presented herein have various strengths and weaknesses, but together they
present an arsenal that is capable of synthesizing a wide range of natural and non-natural
chiral α-amino acids.
References
(1) Williams, R.M. Synthesis of optically Acitve α–Amino Acids, Vol 7 of Organic
Cemistry Series; Baldwin, J.E.; Magnus, P.D. (Eds.); Pergamon Press, Oxford 1989.
(2) Evans, D.A.; Weber, A.E. J. Am. Chem. Soc. 1986, 108, 6757. “Asymmetric Glycine
Enolate Aldol Reactions: Synthesis of Cyclosporine’s Unusual Amino Acid MeBmt”
(3) Crich, J.Z.; Brieva, R.; Marquart, P.; Bu, R.L.; Flemming, S.; Sih, C.J. J. Org. Chem.
1993, 58, 3252. “Enzymic Asymmetric Synthesis of α–Amino Acids. Enantioselective
Cleavage of 4-Substituted Oxazolin-5-ones and Thazolin-5-ones”
(4) Strecker, A. Liebigs Ann. Chem. 1850, 75, 27-45.
N
H
CO
2
H
O
N
O
H
O
LDA
THF
-78 C
°
N
O
OLi
N
O
E
O
R X
or
R
O
E
+
E
+
=
92%
15-48%
HBr
N
H
CO
2
H
E
Figure 9: Self-Reproduction of Chirality

(5) Harada, K. Nature, 1963, 200, 1201. “Asymmetric Synthesis of α–Amino Acids by
the Strecker Synthesis
(6) Iyer, M.S.; Gigstad, K.M.; Namdev, N.D.; Lipton, M. J. Am. Chem. Soc. 1996, 118,
4910. “Asymmetric Catalysis of the Strecker Amino Acid Synthesis by a Cyclic
Dipeptide”
(7) Sigman, M.S.; Jacobsen, E.N. J. Am. Chem. Soc. 1998, 120, 4901. “Schiff Base
Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel
Synthetic Libraries”
(8) Sigman, M.S.; Vachal, P.; Jacobsen, E.N. Angew. Chem. Int. Ed. 2000, 39, 1279. “A
General Catalyst for the Asymmetric Strecker Reaction”
(9) Vachal, P.; Jacobsen, E.N. Org. Lett. 2000, 2, 867. “Enantioselective Catalytic
Addition of HCN to Ketoimines. Catalytic Synthesis of Quaternary Amino Acids”
(10) Su, J.T.; Vachal, P.; Jacobsen, E.N. Adv. Synth. Catal. 2001, 343, 197. “Practical
Synthesis of a Soluble Schiff Base Catalyst for the Asymmetric Strecker Reaction”
(11) Ishitani, H., Komiyama, S., Kobayashi, S. Angew. Chem. Int. Ed. 1998, 37, 3186.
“Catalytic, Enantioselective Synthesis of α-Aminonitriles with a Novel Zirconium
Catalyst”
(12) Kobayashi, S; Ishitani, H. Chirality 2000, 12, 540. “Novel Binuclear Chiral
Zirconium Catalysts Used in Enatnioselective Strecker Reactions”
(13) Vineyard, B.D.; Knowles, W.S.; Sabacky, M.J.; Bachman, G.L.; Weinkauff, D.J.
J. Am.Chem. Soc. 1977, 99, 5946. “Asymmetric Hydrogenation. Rhodium Chiral
Bisphosphine Catalyst”
(14) Williams, R.M., Im, M. Tetrahedron Lett. 1988, 29, 6075. “Asymmetric Synthesis
of α-Amino Acids: Comparison of Enolate vs. Cation Functionalization of N-Boc-5,6-
diphenyl-2,3,5,6-tetrahydro-4H-1,4-oxazin-2-ones”
(15) Schöllkopf, U.; Hartwig, W.; Groth, U. Angew. Chem. Int. Ed. Engl. 1979, 18, 863.
“Enantioselective Synthesis of α-Methyl- α-aminocarboxylic Acids by Alkylation of the
Lactim Ether of cyclo-(
L
-Ala-
L
-Ala)”
(16) Schöllkopf, U.; Neubauer, H-J. Synthesis. 1982, 861. “Asymmetric Synthesis via
Hetercyclic Intermediates. 12. enantioselective Synthesis of (R)-α-Amino Acids using
tert-Leucine as chiral Auxilliary Reagent”

(17) Schöllkopf, U., Groth, U., Deng, D. Angew. Chem. Int. Ed. Engl. 1981, 20, 798.
“Enantioselective Synthesis of (R)-Amino acids Using
L
-Valine as a Chiral Agent”
(18) Schöllkopf, U., Nozulak, J.; Groth, U. Synthesis 1982, 868. “Asymmetric Syntheses
via Heterocyclic Intermediates. 15.Enantioselective Synthesis of (R)-(-)-Beta-
Hydroxyvaline Using
L
-Valine or (S)-O,O-Dimethyl-Alpha-Methyldopa as Chiral
Auxiliary Reagents”
(19) Schöllkopf, U., Groth, U., Westphalen, K-O., Deng, D. Synthesis, 1981, 969.
“Asymmetric Syntieses via Heterocyclic Intermediates; VIII. Enantioselective synthesis
of (R)-α-Methyl-α-amino Acids Using
L
-Valine as Ciral Auxiliary Reagent”
(20) Williams, R.M., Sinclair, P.J., Zhai, D., Chen, D. J. Am. Chem. Soc. 1988, 110,
1547. “Practical Asymmetric Syntheses of α-Amino Acids through Carbon-Carbon Bond
Constructions son Electrophilic Glycine Templates.”
(21) Williams, R.M., Im, M-N. J. Am. Chem. Soc. 1991, 113, 9276. “Asymmetric
Synthesis of Monosubstituted and α,α-Disubstituted α-Amino Acids via
Diastereoselective Glycine Enolate Alkylations”
(22) Xu, P-F., Chen, Y-S., Lin, S-I., Lu, T-J. J. Org. Chem. 2002, 67, 2309. “Chiral
Tricyclic Iminolactone Derived from (1R)-(+)-Camphor as a Glycine Equivalent for the
Asymmetric Synthesis of α-Amino Acids”
(23) Xu, P-F., Lu, T-J. J. Org. Chem. 2003, 68, 658. “Selective Synthesis of Either
Enantiomer of α-Amino Acids by Switching the Regiochemistry of the
TricyclicIminolactones Prepared from a Single Chiral Source”
(24) Corey, E.J.; Bakshi, R.K.; Shibata, S. J. Am. Chem. Soc. 1987, 109, 5551. “Highly
Enantioselective Borane Reduction of Ketones Catalyzec by Chiral Oxazaborolidines.
Mechanism and Synthetic Implications”
(25) Corey E.J., Link, J.O. J. Am. Chem. Soc. 1992, 114, 1906. “A General, Catalytic,
and Enantioselective Synthesis of α-Amino Acids”
(26) Seebach, D., Boes, M., Naef, R., Schweizer, B.W., J. Am. Chem. Soc. 1983, 105,
5390. “Alkylation of Amino Acids without Loss of the Optical Activity: Preparation of α-
Substituted Proline Derivatives. A Case of Self-Reproduction of Chirality”
(27) Seebach, D., Weber, T., Helv. Chim. Acta 1984, 67, 1650. “193.
Hydroxyalkylierungen von Cystein über das Enolat von (2R,5R)-2(tert-Butyl)-1-aza-3-
oxa-7-thiabicyclo[3.3.0]octan-4-on und unter Selbstreproduktion des
Chiralitätszentrums”
Wyszukiwarka
Podobne podstrony:
efficient synthesis of tryptamine heterocycles 6 (8) 1167 1171 (1977) [R 1977 08 1167]
efficient synthesis of tryptamine heterocycles 6 (8) 1167 1171 (1977) [R 1977 08 1167]
Total synthesis of batrachotoxinin A
Highly selective synthesis of menthols from citral in a one step process
Total syntheses of ( ) terpestacin
Concise Large Scale Synthesis of Psilocin and Psilocybin (Shirota, Hakamata & Goda)
brainwashing a synthesis of the russian textbook on psychopolitics
Syntheses, structural and antimicrobial studies of a new N allylamide
Synthetic Drugs of?use
A Review of Energetic Materials Synthesis
Polypeptide Synthesis, Ring Opening Polymerization of alfa Amino Acid N Carboxyanhydrides
Genetic Methods of Polymer Synthesis
Edgar Rice Burroughs Martian Tales 09 Synthetic Men Of Mars
Survey of Tetrodotoxin synthesis
The Organic Chemistry of Drug Synthesis VOLUME 1 DANIEL LEDNICER
Polypeptide Synthesis, Ring Opening Polymerization of alfa Amino Acid N Carboxyanhydrides
Burroughs, Edgar Rice Mars 09 Synthetic Men of Mars
więcej podobnych podstron