Wskazania do skierowania pacjenta (rodziny)
do Poradni Genetycznej
-‐ Każda choroba genetycznie uwarunkowana lub o podejrzewanej e9ologi genetycznej.
-‐ Choroba o niewyjaśnionej e9ologi powtarzająca się w rodzinie u dwóch lub więcej
osób.
-‐ Wrodzona wada rozwojowa lub zespół wad (także wówczas, gdy jest to pierwszy
przypadek wady rozwojowej w rodzinie).
-‐ Upośledzenie umysłowe lub opóźnienie rozwoju psycho-‐motorycznego (nawet jeśli
jest to pierwszy przypadek w rodzinie).
-‐ Zaburzenia determinacji i różnicowania płci oraz rozwoju płciowego.
-‐ Osoby w wieku rozrodczym, narażone na działanie szkodliwych czynników
mutagennych. Ciężarne eksponowane na czynniki teratogenne (np. infekcje
wirusowe,
niektóre leki, alkohol i inne).
-‐ Pary małżeńskie z niepowodzeniami rozrodu (dwa lub więcej poronienia samoistne,
martwe porody lub niepłodność małżeńska).
-‐ Kobiety powyżej 35 roku życia, planujące potomstwo.

Model dziedziczenia autosomalnego
dominującego

Model dziedziczenia autosomalnego
recesywnego

Model dziedziczenia sprzężonego z chromosomem X
(cechy sprzężone recesywnie)

Model dziedziczenia sprzężonego z chromosomem X
(cechy sprzężone dominująco)
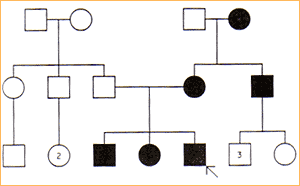
Model dziedziczenia mitochondrialnego
Metody diagnostyki prenatalnej
Metody nieinwazyjne:
-‐ ultrasonografia
-‐ oznaczanie specyficznych substancji pochodzenia płodowego obecnych
w surowicy krwi matki
-‐ badanie komórek i DNA pochodzenia płodowego obecnych w krążeniu
matczynym
Metody inwazyjne:
-‐ amniocenteza
-‐ biopsja kosmówki
-‐ kordocenteza
q ocena stanu płodu
q w ciążach podwyższonego ryzyka wykluczenie wady rozwojowej i/lub
choroby uwarunkowanej genetycznie
q wykrycie wady rozwojowej i/lub choroby uwarunkowanej genetycznie
w przypadku których interwencja lekarska w okresie życia wewnątrzmacicznego
stwarza szanse uratowania dziecka lub zmniejsza ryzyko powikłań okresu
okołoporodowego
q wykrycie u płodu wad wrodzonych, w przypadku których istnieje szansa uratowania
dziecka pod warunkiem interwencji lekarskiej bezpośrednio po urodzeniu
q wykrycie wad letalnych
Ultrasonografia
Zaleca się przynajmniej 3-‐krotne wykonanie w czasie trwania ciąży:
• 11 – 14 tydzień
• ok. 20 tygodnia
• ok. 30 tygodnia
Cele:
• potwierdzenie wieku ciążowego
• ocena żywotności płodu
• ocena ilości płodów
• diagnostyka wad płodu
• ocena przezierności fałdu karkowego (11-‐14 tydzień ciąży)
Przezierność karkowa (NT -‐ nuchal translucency)
rośnie wraz z wiekiem ciążowym
a tym samym długością ciemieniowo-‐siedzeniową
(CRL-‐ crown-‐rump lenght)
Normy:
CRL = 45 mm (11 Hbd); mediana wynosi 1.2 mm
CRL = 84 mm (13+6 Hbd); mediana wynosi 1.9 mm
Ryzyko indywidualne obliczamy mnożąc wartość ryzyka wstępnego dla
danej pacjentki (wynikającego z jej wieku oraz wieku ciążowego) przez
różnicę między wartością NT zmierzoną a medianą dla danego CRL
Badanie NT pozwala zidentyfikować około 72% płodów z zespołem Downa
(odsetek wyników fałszywie dodatnich 5%)
NT = 3 mm -‐ ryzyko trisomi podwyższone ponad ryzyko wynikające z wieku matki 3 razy
NT = 4 mm -‐ ryzyko trisomi podwyższone ponad ryzyko wynikające z wieku matki 18 razy
NT = 5 mm -‐ ryzyko trisomi podwyższone ponad ryzyko wynikające z wieku matki 28 razy
NT > 5 mm -‐ ryzyko trisomi podwyższone ponad ryzyko wynikające z wieku matki 36 razy
Inne przyczyny zwiększenia grubości fałdu karkowego:
q niewydolność płodowego układu krążenia związana z wadą serca i/lub dużych
naczyń
q zastój krwi żylnej spowodowany uciskiem
q nieprawidłowy lub opóźniony rozwój układu limfatycznego
q niedokrwistość płodowa
q hipoproteinemia
q infekcje płodu powodujące niedokrwistość lub niewydolność krążenia
Brak lub niedorozwój kości nosowej u płodu
jako marker aberracji chromosomowych
Brak kości nosowej stwierdza się:
q u 67% płodów z trisomią 21
q u 55% płodów z trisomią 18
q u 34% płodów z trisomią 13
q u 11% płodów z monosomią X (zespół Turnera)
q u 7% płodów z triploidią
Wady wrodzone a aberracje chromosomowe
Wskazania do wykonania inwazyjnej diagnostyki prenatalnej z
oceną kariotypu płodu:
q zesp. Dandy-‐Walkera (1/1000) – występuje w ok.50 zespołach
genetycznych (40% aberracje chromosomowe)
q cys9c hygroma (torbielowate struktury w okolicy potyliczno-‐
szyjnej)
– 75% aberracje chromosomowe (gł.zespół Turnera)
q brak ciała modzelowatego (1/1000) -‐ wystepuje w ok.100
zespołach
genetycznych , w tym trisomi 13 i 18
q małogłowie (1/1000) – 15% aberracje chromosomowe (trisomia 13,
delecja 4p i 5p)
q przepuklina przeponowa (1/3000) – 20% aberracje chromosomowe
q przepuklina sznura pępowinowego (1/3000) -‐ 60% aberracje
chromosomowe
Wady wrodzone a aberracje chromosomowe
Wskazania do wykonania inwazyjnej diagnostyki prenatalnej z
oceną kariotypu płodu:
q wady serca (5-‐10/1000) -‐ 5% aberracje chromosomowe
q zarośnięcie przełyku (1/3000) -‐ 4% aberracje chromosomowe
q hipotrofia płodu -‐ 1% aberracje chromosomowe (trisomia 21,
triploidia)
q wodonercze -‐ 3% aberracje chromosomowe
Ryzyko aberracji chromosomowych wzrasta wraz z ilością wad
wrodzonych
Badania przesiewowe surowicy krwi matki
I trymestr ciąży:
Badania przesiewowe w 1. trymestrze (test PAPP-‐A):
1. Wolna podjednostka beta-‐hCG
2. PAPP-‐A
Test PAPP-‐A wraz z pomiarem NT (wykonywane między 11 a 14 tygodniem ciąży):
-‐ dla trisomi 21 (tzn. zespołu Downa) – wykrywalność wynosi 86,3%, przy
odsetku wyników fałszywie dodatnich równym 5%
-‐ dla wszystkich aberracji chromosomowych wykrywalność sięga 90%, przy 6%
wyników fałszywie dodatnich
Polegają na oznaczeniu markerów biochemicznych w surowicy krwi kobiet
ciężarnych:
-‐ Alfa-‐fetoproteina (AFP)
-‐ Podjednostka beta ludzkiej gonadotropiny kosmówkowej (beta-‐hCG)
-‐ Nieskoniugowany estriol (uE3)
-‐ PAPP-‐A
-‐ Inhibina A
Badania przesiewowe surowicy krwi matki
II trymestr ciąży:
Test potrójny (wykonywany między 15 a 19 tygodniem ciąży):
Test potrójny -‐ polega na określeniu stężeń podjednostki beta-‐hCG, alfa-‐fetoproteiny
(AFP) i nieskoniugowanego estriolu (uE3).
Wykrywa ono 50-‐75% ciąż z trisomią 21 (zespołem Downa), a fałszywie dodatnie wyniki
otrzymuje się w 5% przypadków. Przy uwzględnieniu NT, wykrywalność sięga 85-‐90%
przy odsetku wyników fałszywie dodatnich wynoszącym 5%.
Patologie ciąży, w jakich obserwuje się wzrost lub spadek stężeń markerów
biochemicznych, badanych testem potrójnym.
q podjednostka beta-‐hCG – wyższe stężenie w ciąży z trisomią 21, zaśniadzie
groniastym, ciąży mnogiej;
-‐ niższe stężenie w ciąży z trisomią 18, ciąży obumarłej
q nieskoniugowany estriol (uE3) – niższe stężenie w ciąży z trisomią 21,
bezmózgowiem, aplazją lub hipoplazją nadnerczy
Badania przesiewowe surowicy krwi matki
q AFP
Patologia płodu, w której podwyższone jest stężenie AFP w surowicy:
1) otwarte wady cewy nerwowej
2) ubytki ściany brzucha
3) cys$c hygroma
4) atrezja przewodu pokarmowego
5) niektóre wady układu pokarmowego
6) choroby skóry ( epidermolysis bul osa simplex, aplasia cu$s congenita)
Inne przyczyny matczyno-‐płodowe wzrostu AFP w surowicy:
-‐ ciąża mnoga, ciąża obumarła, wady łożyska lub pępowiny, immunizacja Rh
Choroby matki, w których podwyższone jest stężenie AFP:
-‐ guzy wątroby, ostre zapalenie wątroby
Badania przesiewowe surowicy krwi matki
Komórki i DNA pochodzenia płodowego we krwi kobiety ciężarnej
q We krwi ciężarnych krąży pewna ilość komórek płodowych (komórki trofoblastu,
pierwotne erytrocyty jądrzaste, granulocyty) dostępnych badaniu. Ograniczeniami metody
są jednak mała ilość komórek pochodzenia płodowego we krwi ciężarnej oraz trudności
w odróżnieniu komórek matczynych od komórek płodowych.
q Odsetek komórek pochodzenia płodowego w pobranej próbce krwi kobiety ciężarnej
może zostać zwiększony poprzez zastosowanie metod automatycznego sortowania
komórek: MACS ( magne$c cel sor$ng) lub FACS ( fluorescence ac$vated cel sor$ng). Na
uzyskanych komórkach można następnie przeprowadzać badania cytogenetyczne techniką
FISH. Czułość metody podobna jest do czułości biochemicznych testów przesiewowych.
q We krwi kobiet ciężarnych występują także niewielkie ilości wolnego DNA pochodzenia
płodowego, który może być wykorzystany do badań genetycznych techniką PCR.
Amniopunkcja (amniocenteza)
-‐ wykonywana jest między 15-‐18 tygodniem ciąży
-‐ pobranie 15-‐20 ml płynu owodniowego
-‐ ryzyko utraty ciąży (poronienia) w związku z badaniem wynosi 0,5-‐1%
Amniocenteza jest możliwa do wykonania także między 10 a 14 tygodniem ciąży
(amniocenteza wczesna), ale wówczas ryzyko poronienia wzrasta do 2%, a
ryzyko wystąpienia wad kończyn (m. in. stóp końsko-‐szpotawych u płodu) jest
wysokie.
Amniopunkcja (amniocenteza)
Wskazania:
1) Podwyższone ryzyko urodzenia dziecka z aberracją chromosomową:
-‐ wiek ciężarnej powyżej 35 lat
-‐ urodzenie dziecka z aberracją chromosomową z poprzedniej ciąży
-‐ nosicielstwo translokacji lub innej aberracji chromosomowej u jednego z rodziców
-‐ stwierdzenie w USG patologi płodu sugerującej występowanie aberracji
chromosomowej
-‐ nieprawidłowy wynik testów biochemicznych (test PAPP-‐A, test potrójny) – ryzyko
aberracji u płodu >1:200
2) Urodzenie dziecka z wadą cewy nerwowej z poprzedniej ciąży
Biopsja kosmówki
-‐ wykonywana jest między 10-‐14 tygodniem ciąży
-‐ pobranie 5-‐10 g tkanki
-‐ oczekiwanie na wynik 1-‐3 tygodnie
-‐ ryzyko poronienia około 2 %
Stwierdzono związek między biopsją kosmówki wykonywaną przed 10 tygodniem
ciąży, a występowaniem wad ubytkowych kończyn płodu oraz niedorozwojem
żuchwy i języka.
Biopsja kosmówki
Wskazania:
-‐ podobne jak w przypadku amniocentezy z wyjątkiem podwyższonego
ryzyka urodzenia dziecka z wadą cewy nerwowej
-‐ CVS jest metodą z wyboru w przypadku molekularnej diagnostyki
prenatalnej chorób monogenowych
Kordocenteza
-‐ wykonywana od 17 tygodnia ciąży do momentu porodu
-‐ pobranie 0,5-‐1,0 ml krwi płodu z żyły pępowinowej
-‐ ryzyko poronienia ok. 1,0-‐1,5%
-‐ istnieje ryzyko zanieczyszczenia krwią matki i otrzymania błędnego wyniku
kariotypu płodu
Kordocenteza
Wskazania:
-‐ wykrycie w USG po 18. tygodniu ciąży wad sugerujących możliwość aberracji
chromosomowych u płodu
-‐ uzyskanie materiału do badań cytogenetycznych (np. weryfikacja
mozaikowatości) lub badań molekularnych
-‐ diagnostyka i leczenie konfliktu serologicznego, ocena morfologi krwi płodu
-‐ diagnostyka prenatalna genetycznie uwarunkowanych chorób krwi
Nowoczesne metody badań cytogenetycznych
w diagnostyce prenatalnej
FISH (fluorescencyjne hybrydyzacja in situ):
·∙ stosowana od lat 80-‐tych XX wieku
·∙ obok analizy chromosomów umożliwia także badanie jąder w stadium interfazy
·∙ zastosowanie sond specyficznych dla chromosomów pary 13, 18, 21, X i Y
umożliwia szybką diagnostykę najczęściej występujących aneuploidi
chromosomowych
·∙ wymaga pozyskania niewielkiej ilości amniocytów bądź komórek kosmówki
·∙ wyniki badania uzyskuje się w przeciągu 24 godzin
Nowoczesne metody badań cytogenetycznych
w diagnostyce prenatalnej
Array-‐CGH (porównawcza hybrydyzacja genomowa
z wykorzystaniem mikromacierzy):
·∙ DNA pacjenta (wyizolowany z amniocytów płynu owodniowego) i DNA
referencyjny (kontrolny) wyznakowane na różne kolory są hybrydyzowane
do płytek mikromacierzy zawierających fragmenty genomowego DNA
·∙ umożliwia detekcję niewielkich zmian ilościowych (duplikacje, delecje) w obrębie
chromosomów pacjenta
·∙ wysoka rozdzielczość (1 mln -‐ 500 tys. par zasad)
Nowoczesne metody badań cytogenetycznych
w diagnostyce prenatalnej
QF-‐PCR (ilościowy, fluorescencyjny PCR):
·∙ fragmenty DNA (powtórzenia mikrosatelitarne) podlegają amplifikacji, znakowaniu
fluorescencyjnemu a ilość kopi mierzona jest podczas rozdziału elektroforetycznego
·∙ umożliwia szybką detekcję aneuploidi chromosomowych z wykorzystaniem DNA
wyizolowanego z amniocytów lub komórek kosmówki
·∙ technika umożliwia również identyfikację przypadków disomi jednorodzicielskiej
(UPD)
Nowoczesne metody badań cytogenetycznych
w diagnostyce prenatalnej
MLPA (ang. Mul$plex Liga$on-‐dependent Probe Amplifica$on )
-‐ metoda oparta o reakcję ligacji odpowiednich sond połączoną z reakcją amplifikacji.
-‐ polega na amplifikacji nie łańcucha DNA, lecz sondy dodawanej do badanej próbki.
Pojedyncza sonda zawiera dwa różne oligonukleotydy, z których każdy wiąże się ze
starterem PCR. Każda z sond wiąże się specyficznie z wybranym miejscem sekwencji
Liczba otrzymanych w wyniku reakcji PCR sond zależy od liczby odpowiadających
sekwencji w badanym DNA.
-‐ równoczesna ocena ilościowa obecności ilości kopi wieloeksonowych genów.
Na podstawie zmian stosunków ilościowych poszczególnych fragmentów można
wnioskować o delecjach bądź duplikacjach odpowiednich odcinków genów.
Wyszukiwarka
Podobne podstrony:
DO TEL! Wskazania do skierowania pacjenta genetyka
DO TEL! Wskazania do skierowania pacjenta genetyka
DO TEL! Wskazania do skierowania pacjenta genetyka
genetyka, ćw 6 geny, 6 Techniki oparte na PCR do diagnozowania chorów genetycznych i uchwycenia zmie
rola edukacyjna pielegniarki w cukrzycy-konspekt, Konspekt do edukacji pacjenta
Adaptacja człowieka do roli pacjenta
1.7Wskazania do wykonania PGD, Genetyka
wstep do cytogenetyki, 4 ROK, GENETYKA KLINICZNA
KONSPEKT DO?UKACJI PACJENTÓW NA TEMAT ŻYWIENIA W CHOROBACH SERCA
Czynności do kąpieli pacjenta w łóżku, Studium medyczne
Analiza czynników wpływających na powrót do pracy pacjentów po aloplastyce całkowitej stawu biodrowe
Genetyka - opracowanie do egz, Materiały =), Genetyka
KONSPEKT DO NAUCZANIA PACJENTA Z CUKRZYCĄ TYPU I, Konspekty, Cukrzyca typ 1 (indywidualna edukacja)
Czynności do kąpieli pacjenta pod prysznicem, Studium medyczne
Terminy do egzaminu 2009, genetyka
więcej podobnych podstron