Chemia analityczna (analityka) - dyscyplina rozwijająca metody i narzędzia pozw. uzyskać informacje o składzie i strukturze materii.
Odp. na podstawowe pytania:
- co? ( analiza jakościowa)
- ile? ( analiza ilościowa)
- w jakiej postaci? (specjacja)
ANALITYKA - interdyscyplinarna nauka zajmująca się tworzeniem i praktycznym wykorzystaniem metod pozwalających na określenie ze znaną precyzją i dokładnością składu chemicznego układów materialnych.
Analityka dzieli się na:
1.Analitykę teoretyczną opracowanie nowych metod i technik oznaczania wraz z aparaturą oraz metodyką
2.Analitykę stosowaną:
•naukowo-badawczą
•medyczno-biologiczną
•kontrolno-pomiarową
•środowiskową łącznie z monitoringiem
Przedmiotem analityki jest:
•opr. metodyki niezbędnej do uzyskania informacji o składzie badanej próbki;
•pozyskiwanie inf. o rodzaju i ilości składników włącznie z ich przestrzennym uporząd. i rozmieszczeniem, a także zmianami w czasie;
•wynikiem badań analitycznych jest informacja uzyskiwana poprzez materialne lub energetyczne oddziaływanie na badany obiekt.
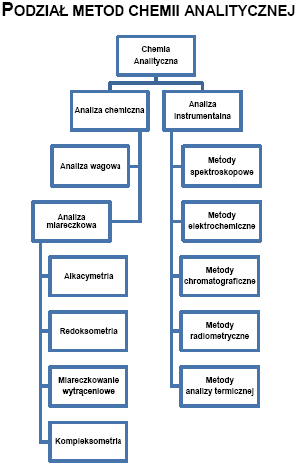
Metody analizy chemicznej można podzielić w zależności od typu reakcji na metody:
•Analizy wagowej (grawimetrycznej), w której wykorzystuje się reakcje wytrącania trudno rozpuszczalnych osadów. Osad zawierający oznaczany analit wydziela się z roztworu, suszy się lub praży, a następnie waży się na wadze analitycznej.
Zachodzące reakcje wytrącania i masy osadu określają ilość oznaczanego osadu.
•Analizy miareczkowej polegają na pomiarze objętości roztworu - titrantu dodaw. powoli z biurety w procesie miareczkowania. Reaguje on z oznaczanym analitem, a koniec reakcji ustala się przy pomocy odpowiedniego wskaźnika.
W oparciu o objętość zużytego roztworu titrantu i jego stężenie wylicza się ilość analitu.
Wskaźniki pH - sub. org., słabe kwasy lub zasady organiczne, których barwa zależy od stężenia jonów H3O+ (jony mają inne zabarwienie niż cząsteczki niezdys.)
Barwa zależy od stosunku stężeń formy zdys. i niezdys.
W r-rze zawsze istnieją obie formy odmiennie zabarwione
• ZAKRES WSKAŹNIKOWY - przedział pH w którym zachodzą widoczne zmiany barwy roztworu wskaźnika
----------------------------------------
Analizę Miareczkowa dzielimy
(W zależności od typu reakcji):
1.Alkacymetrię - wykorzystuje reakcje zobojętniania kwas-zasada i dzieli się :
•Alakimetrię -oznaczanie substancji przez miar. mianowanym r-rem zasady
•Acydemetrię - oznaczanie substancji przez miar. mianowanym r-rem kwasu
2.Redoksometrię - wykorzystuje reakcje utlenienia-redukcji i ogólnie stosuje się do oznaczania reduktorów i utleniaczy.
Dzieli się na:
Oksydymetria - oznaczanie substancji przez miareczkow. roztworami utleniaczy.
Reduktometria oznaczanie substancji przez miar. reduktorami: ferometria (FeSO4) tytanometria (TiCl3) askorbinometria
3.Miareczkowanie strąceniowe - wykorzystuje reakcje wytrącania trudno rozpuszczalnych osadów w wyniku łączenia jonów titrantu i substancji oznaczanej. Najbardziej znane:
•argentometria AgNO3, NH4SCN
•merkurometria Hg2(NO3)2
4.Kompleksometrię -
polega na reakcji tworzenia rozpuszczalnych, słabo zdysocjowanych (trwałych) związków kompleksowych.
_________________________
ETAPY PROCESU ANALITYCZNEGO :
Źródła błędów w procesie analitycznym:
- Pobieranie i przyg.próbki 67%
- pomiar i kalibracja 30%
- obróbka danych 10%
Czas wykonania procesu:
- Pobieranie i przygotowanie próbki 67%
- obróbka danych 27%
- pomiar 10%
PRÓBKA REPREZENTATYWNA-
porcja materiału pobrana, obrabiana i przechowywana w ten sposób, by jej skład chemiczny był najbardziej zbliżony do przeciętnego, średniego składu całkowitej ilości analizowanego materiału.
Zasadnicze czynniki warunkujące reprezentatywność próbki;
• pobrana próbka musi być dostatecznie duża
• pobrana losowo
• należy zapewnić niezmienność składu na wszystkich etapach postępowania.
• próbka powinna być doskonale jednorodna.
Sposoby poboru próbek reprezentatywnych
1.Metody sedymentacyjne- próbkę analitów zbiera się w wyniku procesu swobodnej migracji analitu do powierzchni zbierającej
2.Metody izolacyjne- próbkę zbiera się do pojemnika o określonej objętości
3. Metody aspiracyjne- próbkę analitów pobiera się przepuszczając strumień będący medium przez pułapkę (np. rurka sorpcyjna)
Procedura pobierania próbek jest wielostopniowa:
1.Badany obiekt
2.Próbka pierwotna poddana homogenizacji
3. Próbka laboratoryjna- przygotowana z próbki ogólnej, reprezentująca właściwości partii produktu, przeznaczona do prowadzenia analiz
4. Próbki analityczne (najczęściej trzy i każdą oddzielnie poddajemy kolejnym etapom postępowania analitycznego) -część produktu wydzielona z próbki laboratoryjnej przeznaczona w całości do 1 oznaczenia Efekt końcowy:
3 niezależne wyniki oznaczeń
Przyg. próbek do analizy:
1.przeprowadzenie pr. do r-ru
- Rozpuszczenie
- Roztwarzanie - reakcja chemiczna między rozpuszczalnikiem a substancją rozpuszczoną w wyniku czego powstaje inne indywiduum
•może być przeprowadzone:
-w rozcieńczonych kwasach
-w stężonych kwasach
-poprzez stapianie z topnikami
2.wydzielanie, rozdzielanie i zatężanie analitu
Metody pomiarowe w analizie chemicznej są mniej lub bardziej selektywne.
W wielu analizach przeszkadzają substancje towarzyszące. (np. Strącanie, wymiana jonowa)
3. maskowanie czynników zakłócających pomiar
4.derywatyzacja analitu - polega na otrzymywaniu związków pochodnych badanego, o korzystniejszej charakterystyce fizykochemicznej, np. o większej lotności, bardziej intensywnym zabarwieniu.
METODY POMIARU ANALITYCZNEGO:
Bezwzględne: nie wymagają wzorcowania i są oparte na reakcjach chemicznych, przebiegających całkowicie i ze znaną stechiometrią (miareczkowanie - V titranta, gazometria - Vgazu, Kulometria - Ładunek)
Względne: Wymaga kalibracji względem znanych wzorców, większość metod instr. opiera się na pomiarach względnych gdy mierzony parametr jest funkcją stężenia analitu Y=f(c) zwykle Y=ac
Y-wielkośc mierzona;
c-stężenie analitu;
a-wsp. Proporcjonalności
POMIARY WZGLĘDNE:
1.Metoda krzywej kalibracyjnej (wzorcowej)
Y = ac +b
y-wart. mierzona
c - st. analitu
a - wsp. proporcjonalności
•Krzywa kalibracyjna ma ograniczony zakres prostoliniowości
•po dopasowaniu funkcji wyliczamy jej wartość pomiędzy znanymi punktami
•duży wpływ matrycy na mierzone wartości
• wpływ matrycy można ograniczyć poprzez dodanie:
-do wzorców roztworów buforowych i regulację siły jonowej roztworów
- lub do próbki substancji maskujących
2.Metoda dodawania wzorca
Muszą być spełnione następujące warunki: - nachylenie krzywej kalibracyjnej musi być stałe
(a=const) -wielkość b=0 w równaniu Y=ac+b
-do analizowanej próbki zawierającej analit dodajemy kilkakrotnie wzorzec i mierzymy wielkość sygnału Y dla próbki pierwotnej i po każdym dodaniu wzorca.
-stężenie badanej próbki odczytuje się z przecięcia prostej z osią odciętych na lewo od punktu zerowego
•metoda ekstrapolacyjana; prognozowanie wartości pewnej zmiennej poza zakresem, dla którego mamy dane, Metoda ta eliminuje wpływ matrycy na przebieg wzorcowania, bo wszystkie pomiary są dokonywane w tym samym środowisku
•Analit- oznaczany składnik.
•Interferenty- składniki utrudniające w procesie oznaczania analitów.
•Matryca - pozostałe składniki, stanowią zazwyczaj największą część próbki do analizy.
•Metoda analityczna - sposób wykrywania lub oznaczania składnika próbki.
•Granica wykrywalności- najmniejsza ilość lub najmniejsze stężenie substancji możliwe do wykrycia za pomocą danej metodyki z określonym prawdop.
•Granica oznaczalności- najmniejsza ilość lub najmniejsze stężenie substancji możliwe do ilościowego oznaczenia …
Efekty interferencyjne - wpływ substancji towarzyszących analitowi (matrycy) na sygnał analityczny. Efekty interferencyjne mogą być: dodatnie (zwiększają sygnał analitu o danym stężeniu) lub ujemne (zmniejszają sygnał analitu o danym stężeniu).
- efekt addytywny - w przypadku oddziaływania stałej ilości interferenta na zmienne ilości analitu zmiany sygnału mogą mieć stałą wartość.
- efekt multiplikatywny wielkość proporcjonalną do stężenia analitu
Granica wykrywalności (LOD)
-Jest najmniejszym stężeniem analitu przy którym istnieje pewność jego obecności w próbce.
-Ściśle powiązana z poziomem szumów stosowanego urządzenia pomiarowego ( przyjmuje się, że jej wartość to trzykrotność tego poziomu szumu).
Granica oznaczalności (LOQ)
- zwykle wielokrotność granicy wykrywalności
Najczęściej : LOQ = 3 x LOD
•Czułość- pojęcie określające, jaka najmniejsza różnica zawartości analitu może być stwierdzona za pomocą konkretnej metodyki (jest to nachylenie wykresu kalibracyjnego : sygnał w funkcji stężenia).
•Próba ślepa (zerowa)- próba wykonywana w warunkach identycznych jak analiza badanej próbki, ale bez dodawania substancji oznaczanej
Selektywność metody- możliwość jej zastosowania do wykrywania lub oznaczania tylko niewielkiej liczby składników
•Zakres pomiarowy - zakres wartości (stężeń analitu), w którym błąd urządzenia pomiarowego jest poniżej założonego.
•Liniowość- przedział zakresu pomiarowego metodyki analitycznej, w którym sygnał wyjściowy jest proporcjonalny do oznaczanego stężenia analitu.
Błąd przypadkowy oznaczenia
-tym mniejszy im mniejszy rozrzut sygnałów wokół prostej regresji
-tym mniejszy im większa jest czułość oznaczenia
- tym mniejszy im więcej wzorców zastosowano do sporządzenia wykresu i maleje ze wzrostem liczby pomiarów n dla każdego z punktów na wykresie
-błędy te są różne w różnych fragmentach wykresu kalibracyjnego
Kryteria wyboru metody analitycznej:
1.Cel
•Rodzaj analizowanego materiału, w tym rodzaj matrycy
•Dostępna wielkość próbki
•Poziom zawartości oznaczanego składnika
•Dop. czas trwania analizy
•Wymagana dokładność i precyzja wyniku (czułość)
2. Koszty wykonania analizy
•Aparatura, robocizna, koszty materiałowe
3. Stan wyposażenia laboratorium
•posiadana aparatura
•doświadczenie personelu
•wzorce anal. odczynniki
A także:
•Jakie techniki analityczne mamy do dyspozycji?
•Która z dostępnych metod gwarantuje najbardziej precyzyjny wynik?
•Ile oznaczeń zaplanowano?
•Jakie będą koszty, w tym koszty wyposażenia, odczynników?
•Jaki jest czas przewidywany na rozwiązanie problemu?
•Jakie dodat. info uzyskamy od zleceniodawcy?
Podział metod analitycznych:
- klasyczne m. chemiczne (wagowe i miareczkowe)
- met. Instumentalne:
1.Metody elektrochemiczne - związane z efektami towarzyszącymi przepływowi prądu przez badany r-r lub spowodowane reakcjami na elektrodach zan. w r-rze
(potencjometria)
2.Metody spektroskopowe (optyczne) - związane z oddz. prom. elektromagn. z materią.
(spektroskpia)
3.Metody chromatograficzne- korzystające z rozdzielania badanych mieszanin w układzie faza stacjonarna -faza ruchoma i oznaczenia rozdzielonych składników dowolna metodą
POTENCJOMETRIA
Zasada potencjometrii polega tym, że potencjał elektryczny odpowiednio dobranej elektrody zależy od składu roztworu, w którym ta elektroda jest zanurzona.
prawo Nersta
•Każdy pomiar potenc. polega na pomiarze SEM ogniwa zbudowanego z 2 elektrod zanurzonych w analizowanym roztworze.
Ogniwo pomiarowe skł. się z:
- elektrody pomiarowej Ep (wskaźnikowej), której potencjał zależy od stężenia (aktywności) oznaczanego jonu
- elektrody odniesienia Eo (porównawczej, która posiada stały potencjał niezależny od stężenia oznaczanego jonu
z - l. elektronów wymienianych w reakcji połówkowej
Zasady prawidłowej pracy w potencjometrii:
1.Elektrodę jonoselektywna zanurzyć w roztworze jonów oznaczanych na godzinę przed pomiarem-kondycjonowanie.
2. Dodać do r-rów wzorcowych i badanych odpowiedni bufor w celu: zachowania stałej siły jonowej, zamaskowania jonów przeszkadzających, ustabilizowania pH.
3.Wszystkie pomiary należy przeprowadzić w stałej temperaturze.
4. Całą serię pomiarów należy przeprowadzić dla próbek o tej samej objętości, elektrody zanurzać w roztworze na taką samą głębokość, r-ry mieszać z taka sama szybkością.
5. Przed każdą serią pomiarową należy skalibrować elektrodę tzn. wyznaczyć dla co najmniej 3 r-rów wartość SEM ogniwa.
Miarecz. potencjometryczne
Polega na rejestrowaniu SEM ogniwa pomiarowego po dodaniu każdej porcji titranta.
Notujemy SEM, a następnie przedstawiamy je w układzie SEM=f(Vtitr.)
ZASTOSOWANIE POTENCJOM.
•Bezpośrednie oznaczanie niektórych jonów za pomocą elektrody jonoselektywnej
•Pomiary pH- pehametria
•Potencjometryczne wykrywanie punku końcowego w różnych miareczkowaniach
•Można stosować wszędzie tam gdzie jest wymagana:
- duża prędkość pomiaru,
-duża selektywność pomiaru
-możliwość automatyzacji
-konieczność pracy w przepływie
-prostota wykonania.
ELEKTRODY ODNIESIENIA
•standardowa elektroda wodorowa SEW
•Nasycona elektroda kalomelowa NEK Hg/ HgCl2(s)/KCl
• elektroda chlorosrebrowa : Ag/AgCl(s)/KCl
- z pojedynczym płaszczem
- z podwójnym płaszczem
ELEKTRODY POMIAROWE
•Funkcję elektrod pomiarowych pełnią obecnie elektrody jonoselektywne (membranowe) ich potencjał zależy wyłącznie od aktywności (stężenia) tylko jednego jonu.
•Na powierzchni zewnętrznej membrany stykającej się z badanym roztworem ustala się potencjał zależny od przesunięcia procesu wymiany jonowej między membraną a roztworem.
Ze względu na rodzaj membrany wyróżniamy następujące rodzaje EJ:
1.Z membranami stałymi: szklane
homogeniczne
heterogeniczne
2.Z membranami ciekłymi: z wymieniaczami jonowymi
z nośnikami obojętnymi
3. Elektrody specjalne: enzymatyczne gazowe ( do oznaczania SO2, CO2, NH3)
Potencjał elektrod jonoselektywnych opisuje zależność Nikolskiego;
E=Eo+(RT/nF) ln( aj + Σ Kij ajn/z)
Eo - standardowy potencjał elektrody
R - stała gazowa
T - temperatura
F - stała Faradaya
n - wartościowość jonu i, na który czuła jest elektroda
z- wartościowość jonu przeszkadzającego j
Kij-wsp. selektywności elektrody czułej na jon i względem jonu j, jest miarą nieselektywności elektrody im większa jego wartość tym elektroda jest mniej selektywna.
Elektrody pomiarowe specjalne: elektrody czułe na gazy np. NH3, CO2, SO2 H2S, NO, NO2 (czułe na produkty analizowanego gazu z wodą) np.
NH3+H2O OH-+ NH4+
SO2+H2O H++ HSO3-
CO 2+H2O H++ HCO3-
W wyniku reakcji CO2 z wodą powstają jony H+ wywołujące zmianę potencjału elektrody szklanej.
Elektrody te zb. są z elektrody szklanej umieszczonej w zbiorniczku z roztworem buforowym. R-r buforowy jest oddzielony od badanego r-u membraną półprzepuszczalną przepuszczajcą selektywnie oznaczany gaz. Gaz przedostaje się przez membranę do buforu i zmienia się pH buforu.
W elektrodzie są dwie membrany:
-membrana półprzepuszczalna dla gazów
-membrana czuła na jony wodorowe
Elektrody pomiarowe specjalne: enzymatyczne
Podstawa działania to reakcja elektrody jonoselektywnej z prostymi jonami, które powstały w wyniku działania enzymu na analizowane związki.
Elektrodę enzymatyczną uzyskuje się przez pokrycie klasycznej elektrody jonoselektywnej warstwą membranowa zawierającą enzym
------------------------------------
SPEKTROSKOPIA zajmuje się oddziaływaniem pomiędzy promieniowaniem elektromagn. a materią.
Promieniowanie elektromagn. ma dwojaką naturę:
falową i korpuskularną.
Można je opisać jako falę i jako strumień fotonów.
Fali można przyporządkować następujące wielkości:
- długość fali prom. λ[nm]
- częstość prom. (liczba drgań na sekundę) [Hz=s-1] = c/λ
- liczba falowa ( liczba fal na cm) [cm-1] = 1/λ
c-pr. rozchodzenia się prom. w próżni c=2,998·108[m/s]
Podział spektroskopii
1. Wg długości fali prom.:
IR 750-106 nm
VIS 400-750 nm
UV 10-400 nm
2. Wg efektów oddziaływania promieniowania z materią:
•Rozproszenie promieniowania -nefelometria i turbidymetria
energia promieniowania jest niedopasowana do różnicy energii pomiędzy dwoma stanami energetycznymi atomu lub cząsteczki.
•Absorpcja promieniowania-energia promieniowania jest równa różnicy energii pomiędzy dwoma stanami energetycznymi atomu lub cząsteczki.
• Emisja promieniowania- wzbudzony atom lub cząsteczka dążąc do stanu równowagi oddaje nadmiar energii w postaci promieniowania
3. układ materialny
•Spektroskopia atomowa
•Spektroskopia cząsteczkowa
Formy energii występujące w cząsteczce:
I. Energia translacji ulega zmianom w sposób ciągły
Jednakże energia dostarczana cząsteczkom może być magazynowana na szereg innych sposobów (Energia poniższych ruchów ulega zmianom w sposób nieciągły, poprzez pochłonięcie i oddanie kwantu hν):
II. Energia rotacji - wprowadza cząsteczki w ruch obrotowy
III. Energia oscylacji - wprowadza cząsteczki w ruchy drgające
IV. Energia wzbudzania elektronów - powoduje przeniesienie elektronów z niższych poziomów energetycznych na wyższe
poziomy energetyczne
Prawo Lamberta-Beera:
Absorbancja promieniowania monochromatycznego przechodzącego przez jednorodny ośrodek jest proporcjonalna do grubości warstwy i stężenia roztworu.
A= log ( Io / Ix) = ε·l ·c A=f(c)
A - absorbancja - zdolność pochłaniania promieniowania
c - stężenie roztworu przez, który przechodzi promieniowanie [mol/dm3]
k - współczynnik absorpcji gdy c [g/cm3]
ε -molowy współczynnik absorpcji gdy c [mol/dm3]
Odstępstwa od prawa Lamberta-Beera:
1.Czynniki chemiczne:
•Reakcje w roztworze przy wzroście stężenia (kondensacja, polimeryzacja, hydroliza)
•Tworzenie się związków kompleksowych
•Częściowa dysocjacja substancji rozpuszczonej
•Mętność roztworu
2.Czynniki aparaturowe
•Brak monochromatyczności promieniowania
•Obecność promieniowania rozproszonego
Spektroskopia cząsteczkowa UV - VIS
•Absorpcja promieniowania w zakresie widzialnym i nadfiolecie zależy głównie od liczby i rozmieszczenia elektronów w cząsteczce lub jonie.
•W zakresie 180-800 nm absorbują promieniowanie elektromagnetyczne tylko substancje zawierające chromofory:
•substancje nieorganiczne -większość związków metali przejściowych
•substancje organiczne -związki nasycone nie wykazują absorpcji
Chromofory są to takie ugrupowanie atomów w cząsteczce, które zawierają wiązania podwójne lub potrójne sprzężone tzn. rozdzielone tylko jednym wiązaniem pojedynczym
Zalety metody:
•Czułość
•Precyzja i dokładność
•Selektywność oznaczeń
•Prosta
•Niedroga
Zastosowanie:
Absorpcja UV- oznaczanie węglowodorów aromatycznych i innych związków organicznych, a także metali ziem rzadkich
Absorpcja VIS- stosuje się do oznaczeń barwnych soli nieorganicznych oraz związków organicznych
Źródło promieniowania:
VIS-lampy wolframowe
UV- lampy deuterowe lub wodorowe IR-włókno Nernsta lub Globar- żarzące się ciała stale
---------------------------------------
Precyzja- stopień zgodności między wynikami uzyskanymi w trakcie analizy danej próbki z zastosowaniem określonej procedury analitycznej;
Charakteryzuje rozrzut uzyskanych wyników oznaczeń wokół wartości średniej.
Określana na podstawie wartości obliczonego odchylenia standardowego dla danej serii pomiarowej
Dokładność- jest to stopień zgodności między uzyskanym wynikiem pomiaru
(pojedynczego!) a wartością rzeczywistą (oczekiwaną).
Poprawność (Prawdziwość) - stopień zgodności wyniku oznaczenia (jako średniej arytmetycznej obliczonej na podstawie serii pomiarów) z wartością oczekiwaną.
Precyzja pośrednia - określa długoterminowe odchylenie procesu pomiarowego, do którego wykorzystuje się odchylenie standardowe serii pomiarów uzyskanych w danym laboratorium w kilkutygodniowym okresie. (pojęcie szersze od powtarzalności).
Powtarzalność- wyznacza się na podstawie wartości obliczonego odchylenia standardowego serii pomiarów (analizie poddaje się próbki o różnej zawartości analitu i różnym składzie matrycy) przeprowadzonych :
- w danym laboratorium,
- przez danego analityka
- z wykorzystaniem danego urządzenia pomiarowego
- w krótkim czasie.
Wartość tego odchylenia standardowego można obliczyć:
-przeprowadzenie co najmniej 9 niezależnych oznaczeń w całym zakresie pomiarowym (np.3 niezależne oznaczenia na 3 poziomach stężeń)
-przeprowadzenie 6 niezależnych oznaczeń analitu w próbkach wzorcowych na poziomie stężenia odpowiadającego stężeniu w próbce
-przeprowadzenie 6 niezależnych oznaczeń dla analitów występujących w 3 różnych matrycach i na 2 lub 3 poziomach stężeń.
Odtwarzalność- wyznacza się na podstawie wartości obliczonego odchylenia standardowego wyników uzyskanych przez różne laboratoria (badania międzylaboratoryjne)
Rozstęp R- różnica miedzy wartością maksymalną a minimalną R=xmax-xmin
Odchylenie standardowe (s) SD czyli pierwiastek kwadratowy z wariancji, definiowane jako miara rozproszenia uzyskanych poszczególnych wartości oznaczeń wokół wartości średniej.
Wariancja s2 - średnia arytmetyczna sumy kwadratów odchyleń poszczególnych wartości od średniej arytmetycznej zbiorowości
Względne odchylenie standardowe RSD- otrzymuje się przez podzielenie wartości odchylenia standardowego przez wartość średnią; niezależne od jednostek pomiaru, bez miana.
Współczynnik zmienności CV
Stosuje się go w przypadku porównania zróżnicowania: -kilku zbiorowości pod względem tej samej cechy -tej samej zbiorowości pod względem kilku różnych cech
Przedział ufności - przedział, w którym wartość rzeczywista znajduje się z góry założonym prawdopodobieństwem określanym także mianem poziomu ufności. Poziom ufności przyjmuje wartości p=0,95 i p=0,99 co oznacza, że na 100 wyników 95 lub 99 znajduje się w przedziale ufności.
•Przy dużej liczbie pomiarów n>20 przedział ufności wyznacza się z rozkładu normalnego xśr 1,96√n dla
p= 0,95 xśr 2,58√n dla p=0,99
•Przy małej liczbie oznaczeń n<20 przedział ufności wyznacza się z rozkładu Studenta xśr t·s
Test t-Studenta umożliwia porównanie: -wyników analiz ze znaną wartością rzeczywistą (porównanie istotności różnić między wartością średnią xśr a wartością zadaną (wartość odniesienia np. wartość certyfikowana) - dwóch średnich uzyskanych przez analizę jednej próbki dwiema różnymi metodami, w dwóch laboratoriach, przez dwie różne osoby
karty Shewharta - (wykres)
-do sprawdzenie stabilności wyników uzyskanych w danym laboratorium
-pozwalają monitorować przebieg procesu
-umożliwiają szybkie i proste dostrzeżenie nieprawidłowości i szybkie podjęcie odpowiednich działań
Procedura prowadzenia karty: 1.Wykonać 10-20 pomiarów dla próbki wzorcowej 2.Obliczyć wartość średnią xśr i wartość odchylenia standardowego s przy czym obie te wartości należy wyznaczyć dla serii nieobciążonych tzn. po wstępnym odrzuceniu wyników odbiegających 3.Zweryfikować hipotezę o statystycznie nieistotnej różnicy między uzyskaną wartością średnią a wartością oczekiwaną (test t-Studenta) czyli - obliczyć t
-odczytać z tablic rozkładu
t-Studenta wartość tkr testu dla przyjętego poziomu istotności p oraz liczby stopni swobody f=n-1
-porównać wartości t i t kr :
-jeżeli t< t kr to uzyskane wyniki nie różnią się w sposób statystycznie istotny
W przypadku gdy t< t kr przystąpić do sporządzania karty -nanieść na kartę obliczone wartości (na osi OX nanieść kolejne numery pomiarów natomiast na osi OY wartość średnią -zaznaczyć na wykresie linię centralną LC)
-wykreślić określone statystycznie granice kontrolne ( jedną po każdej stronie linii centralnej)
-dolną i górną granicę kontrolną (ostrzegania) UCL (GGL) i LCL (DGL) =xśr 2s
-dolną i górną granicę działania (granicę dopuszczalności) (GGS) i (DGS) =xśr 3s
BŁĘDY PRZYPADKOWE -niewielkie -przyczyna ich występowania nie jest znana
( np. przypadkowe straty, zmęczenie analityka, niestabilność pracy aparatu)
-wielkość i kierunek błędów nie wykazują określonej prawidłowości
-są to błędy różne co do znaku (dodatnie i ujemne)
-są skutkiem sumowania błędów elementarnych
-w celu zmniejszenia ich wpływu na średnią arytmetyczną powtarza się dane oznaczenie kilkakrotnie i oblicza odrzucając wyniki odbiegające od pozostałych
-są ściśle związane z precyzją metody
BŁĘDY SYSTEMATYCZNE -mają charakter stały
-powodują zmianę sygnału zawsze w jednym kierunku
-mają ściśle określoną przyczynę (błąd przyrządu, zanieczyszczenie odczynników), którą można ustalić i usunąć, ewentualnie wprowadzić poprawkę
-przyczyną jest jednokierunkowe, stałe działanie powodujące stałą zmianę wyników oznaczeń
-związany z dokładnością i poprawnością metody pomiarowej
rodzaje Błędów systematyczne:
1.Błąd metodyczny- związany z daną metodą analityczną 2.Błędy operacyjne -spowodowane niewłaściwym wykonaniem czynności analitycznych przez np. nieświadomość analityka
3.Błędy aparaturowe-niewłaściwe działanie przyrządu pomiarowego
Błąd gruby - znacznie odbiega od pozostałych błędów
-spowodowany przyczyną działającą przejściowo np. niewłaściwe pobranie i przechowywanie próbki, użycie wadliwie działającego przyrządu, niewymieszanie roztworu przed pobraniem pipetą do analizy, pomyłki w obliczeniach, pomyłki w odczytach na wadze, biurecie
-w rezultacie błędu grubego otrzymuje się wynik kilkakrotnie inny od wartości rzeczywistej
-błąd gruby powoduje powstanie wyniku wątpliwego, który nie powinien być uwzględniany w obliczeniu średniej
- aby uniknąć błędu grubego należy wykonać co najmniej dwa oznaczenia równoległe
Niepewność pomiaru- jest parametrem określającym przedział wokół wartości przyjętej jako wynik pomiaru, w którym na założonym poziomie prawdopodobieństwa można spodziewać się wystąpienia wartości oczekiwanej.
Standardowa niepewność pomiaru u(xi)- niepewność pomiaru przedstawiona i obliczona jako odchylenie standardowe
Niepewność względna u(xi)/xi -stosunek standardowej niepewności do wartości wielkości mierzonej ( niepewność wyniku zmiennej xi podzielona przez wartość xi).
Złożona standardowa niepewność pomiaru uc (wyniku oznaczenia) standardowa niepewność oznaczenia, której wartość uwzględnia niepewności standardowe parametrów wpływających na wynik analizy ( np. niepewność wzorca, niepewność stałych użytych do obliczeń itp.). Obliczana na podstawie prawa propagacji :
Względna złożona niepewność standardowa uc(y)/y - niepewność wyniku pomiaru pośredniego podzielona przez wartość y
Niepewność rozszerzona U- wielkość określająca przedział w którym można z założonym prawdopodobieństwem ( poziom istotności) oczekiwać wystąpienia wartości oczekiwanej. Niepewność rozszerzona powstaje przez pomnożenie niepewności całkowitej (złożonej standardowej niepewności pomiaru) przez współczynnik rozszerzenia k.
Współczynnik rozszerzenia k - wartość liczbowa użyta do wymnożenia złożonej standardowej niepewności pomiaru w celu uzyskania rozszerzonej niepewności, zależy od przyjętego poziomu istotności, (np. dla 95% wynosi 2), mnożnik wybieramy zwykle z przedziału 2-3.
U=k ·uc
Wyszukiwarka
Podobne podstrony:
referat pierwsza pomoc kop, pierwsza pomoc
kop ład
T4 PŁACE I WYNAGRODZENIA W KOP
Catapillar kop 336D 385C długi zasięg
Kop czyli budowa społeczeństwa obywatelskiego
13 bezpieczeństwa i higieny pracy, prowadzenia ruchu kop posp
kop-lad maly sprzet
KOP SROP
KONSTYTUCJA RZECZYPOSPOLITEJ POLSKIEJ kop
kop lad maly sprzet
kop 9 50
KOP ZGARNIAKOWA
~$kop w gruntach słabonośnych Mrozu
Zal 2 Reg KOP karta oceny merytorycznej, Fundusze Unijne
Catapillar kop 301 6C 301 8C mini
Zal 3 Reg KOP KARTA OCENY MERYT. WNIOSKU, Fundusze Unijne
dane kop lad komatsu
Zal 1 Reg KOP Deklaracja bezstronnosci i poufnosci, Fundusze Unijne
KOP biul 3 2013
więcej podobnych podstron