KOROZJA
Korozją nazywamy proces niszczenia materiałów, najczęściej metali wskutek chemicznego lub elektrochemicznego oddziaływania środowiska.
Podstawowym etapem korozji metali jest reakcja oksydacyjno-redukcyjna, w wyniku której metal jest utleniany i ze stanu metalicznego przechodzi w odpowiedni związek chemiczny, w danych warunkach termodynamicznie trwały, lub przechodzi do roztworu w postaci jonu.
Korozja chemiczna metalu występuje wtedy, gdy reakcji nie towarzyszy przepływ ładunku elektrycznego przez metal lub elektrolit. Przykładem tej korozji są reakcje metali z aktywnymi gazami, np tlenem, związkami siarki lub z fluorowcami, zachodzące w wysokiej temperaturze.
W procesie korozji elektrochemicznej następuje przepływ elektronów przez granicę metal-elektrolit w wyniku powstania i działania ogniw galwanicznych. Korozja elektrochemiczna ma miejsce w środowiskach zawierających wodę (wilgoć) i elektrolity.
Miarą podatności metali na korozję są ich potencjały elektrodowe. Tendencję metali do samorzutnych procesów korozji określa szereg napięciowy metali.
Szereg standardowy
|
Szereg praktyczny pH=6,0
|
||
Metal |
mV |
Metal |
mV |
miedź |
+345 |
miedź |
+140 |
nikiel |
-230 |
nikiel |
+118 |
żelazo |
-440 |
aluminium |
-169 |
cynk |
-760 |
żelazo |
-350 |
aluminium |
-1600 |
cynk |
-823 |
Najbardziej rozpowszechniona jest korozja żelaza, czyli jego rdzewienie. Podczas rdzewienia zachodzą reakcje utleniania żelaza, wymagające obecności tlenu i wilgoci. Miejsca na powierzchni metalu w których zachodzi proces utleniania są określane jako obszary anodowe.
W pośrednich stadiach reakcji bierze też udział dwutlenek węgla. Sumaryczny zapis reakcji ma postać:
4Fe + 3O2 + 6H2O → 4Fe(OH)3
Wodorotlenek żelaza nie jest jedynym produktem rdzewienia. Rdza składa się z tlenku żelaza(III) o różnym stopniu uwodnienia, któremu przypisujemy wzór Fe203.xH2O. Powstawanie tych produktów przebiega przez liczne etapy pośrednie, wśród których wyróżniamy m. i n.
Fe → Fe2+ + 2e
Fe2+ → Fe3+ + 2e
Reakcjom utlenienia towarzyszą reakcje redukcji - najczęściej reakcja redukcji tlenu zawartego w roztworze elektrolitu:
O2 + 2H2O + 4e → 4OH-
lub w warunkach braku kontaktu z gazowym tlenem redukcja wody:
2H2O + 2e → H2 + 2OH-
W roztworach kwaśnych zachodzi redukcja jonów wodorowych:
2H+ + 2e → H2
W wyniku reakcji redukcji w pobliżu obszarów katodowych następuje alkalizacja środowiska korozji.
Korozja elektrochemiczna
Podstawową przyczyn korozji w niezbyt wysokich temperaturach jest powstawanie i istnienie ogniw galwanicznych, czyli obszarów różniących się miedzy sobą potencjałem oksydacyjno-redukcyjnym. W takim układzie, zgodnie z poznanymi wcześniej zasadami działania ogniw galwanicznych, zachodzi reakcja oksydacyjno-redukcyjna, podczas której anoda ulega roztwarzaniu, czyli korozji, a na katodzie wydziela się odpowiedni produkt redukcji. Szybkość reakcji zależy m.in. od różnicy potencjałów stykających się metali, od temperatury oraz od rodzaju i stężenia elektrolitu.
Ogniwo korozyjne różni się od typowego ogniwa galwanicznego tym, że elektrody metalowe są zwarte, a elektrony nie przepływają przez obwód zewnętrzny.
W obserwowanych w praktyce procesach korozji elektrochemicznej wyróżniamy różne rodzaje ogniw: stykowe, stężeniowe, naprężeniowe i temperaturowe.
Ogniwa stykowe
Przyczyną powstania makroogniwa stykowego jest zetknięcie dwóch różnych metali z sobą i z wspólnym elektrolitem. W układzie przedstawionym na rysunku (rys.1) atomy żelaza, pierwiastka o niższym potencjale normalnym niż miedź (mniej szlachetnego), przechodzą do elektrolitu zgodnie z równaniem reakcji;
Fe → Fe2+ + 2e
Uwalniane elektrony są zużywane w biegnącym równolegle procesie katodowym, którym jest albo wydzielanie wodoru z elektrolitu albo redukcja tlenu.
Sumaryczna reakcja korozji żelaza z wydzielaniem wodoru ma postać;
Fe + 2H+→Fe2+ + H2
Reakcja utleniania jest tu oddzielona od reakcji redukcji - roztwarzaniu żelaza towarzyszy wydzielanie wodoru na powierzchni miedzi.
Na powierzchni stopów o strukturze wielofazowej tworzą się mikroogniwa stykowe. Ziarna kryształów różnych faz zachowują się jak kolejne elektrody ogniwa galwanicznego (rys.2).
Wszystkie stopy wielofazowe są zatem mniej odporne na korozję niż stopy jednofazowe.
Korozji ulegają także miejsca styku różnych metali (śruby, nity, miejsca lutowania itp.).
Stężeniowe ogniwa tlenowe
Korozja jest przyspieszona na tych obszarach metalu, który styka się z elektrolitem o mniejszym stężeniu tlenu, ponieważ powstaje wtedy stężeniowe ogniwo tlenowe. Przykładem takiego mechanizmu może być korozja płytki żelaza pod kroplą wody lub konstrukcji żelaznej pod warstwą wilgotnego piasku (rys.3).
Tlen ma swobodny dostęp do krawędzi kropli, natomiast pod środkową (centralną) częścią kropli ilość tlenu jest ograniczona jego rozpuszczalnością w wodzie. Na krawędziach kropli następuje reakcja według równania:
2H2O + O2 + 4e → 4OH-
podczas której są pobierane elektrony z metalu. Jest to katodowa redukcja tlenu. Pobieranie elektronów z żelaza jest równoznaczne z jego utlenianiem, swobodna wędrówka elektronów w masie metalu powoduje, ze utlenianie żelaza następuje w obszarach o mniejszym dostępie tlenu, czyli pod kroplą. Tam będzie zlokalizowana anodowa reakcja utleniania:
Fe -2e → Fe2+
Małe jony żelaza są ruchliwe w roztworze elektrolitu, dyfundują ku krawędziom kropli, gdzie łączą się z jonami OH - w praktycznie nierozpuszczalny Fe(OH)2, który pod wpływem tlenu powietrza ulega powolnemu utlenieniu do Fe(OH)3,. Rdzą pokrywa się przede wszystkim katodowa część płytki, która nie uległa korozji.
Zgodnie z opisaną zasadą, miejscami przyspieszonej korozji będą miejsca zgrzewania czy nitowania części metalowych, szczeliny i pęknięcia metalu, gdzie gromadzi się wilgoć, a dopływ tlenu jest utrudniony. Główka gwoździa wbitego w wilgotny mur wystawiona na działanie powietrza pokrywa się rdzą, lecz korozji ulega ostrze, które ma utrudniony kontakt z tlenem.
Opisane stężeniowa ogniwo tlenowe jest jednym z możliwych rodzajów korozyjnych ogniw stężeniowych. Korozyjne ogniwa stężeniowe występują wszędzie tam, gdzie jednorodny metal styka, się z elektrolitem o różnym stężeniu. Korozja lokalizuje się zawsze w tych miejscach, gdzie metal styka się z elektrolitem o mniejszym stężeniu, czyli na anodzie ogniwa stężeniowego.
Inne ogniwa korozyjne
Występowanie ogniw powodujących korozję obserwuje się również w materiałach chemicznie jednorodnych, nawet przy braku różnicy stężeń w roztworze. Przyczyną różnicy potencjałów nie jest wtedy styk różnych jakościowo materiałów ani stan elektrolitu, ale istnienie obszarów, w których występują naprężenia mechaniczne, lub innych obszarów o zwiększonej energii wewnętrznej, jak np. defekty struktury krystalicznej i granice ziaren. W takim obszarze, mającym nadmiar energii, metal wykazuje obniżony potencjał elektrodowy i zachowuje się względem pozostałej części metalu jak anoda. Korozji ulegają miejsca działania dużych sił i miejsca, gdzie naprężenia wprowadzono przez obróbkę mechaniczną - korozja naprężeniowa (rys.4)
Przyspieszona korozja przebiega też na granicy ziaren polikryształu; nazywamy ją korozją międzykrystaliczną (rys. 5).
Ogniwo galwaniczne stanowi też zanurzony do roztworu elektrolitu metal, którego części mają różne temperatury (np. części wymienników ciepła). Zjawisko to także przyspiesza korozję.
Pasywność
Korozja jest typową reakcją na granicy faz i jej szybkość zależy od natury powstających produktów. Jeżeli warstwa produktów reakcji korozji jest przewodząca, porowata, nie przylega ściśle do metalicznego podłoża i kontakt reagentów z powierzchnią metalu nie jest utrudniony, to korozja może doprowadzić do całkowitego zniszczenia metalu. Natomiast jeśli wytworzona warstwa produktów korozji jest szczelna, wówczas szybkość reakcji jest ograniczona szybkością dyfuzji reagentów przez tę warstwę. Szybkość dyfuzji zmniejsza się ze wzrostem grubości warstwy i po pewnym czasie korozja zostaje zahamowana a rozpatrywany metal ulega pasywacji.
Pasywność jest to stan wyższej odporności metalu na korozję niż wynika to z jego pozycji w szeregu napięciowym metali
Rozróżniamy
pasywność chemiczną ( często samorzutną) i
pasywność mechaniczną
Pasywność chemiczna związana jest z obecnością na powierzchni metalu ( np. Al., Cr, Ni, Mo, Ti) cienkiej niewidocznej gołym okiem (10-100Å) warstewki tlenkowej lub chemisorbowanej warstewki tlenu ew. pasywujących jonów, co powoduje silne przesunięcie potencjału elektrodowego metalu w kierunku dodatnim
Pasywność mechaniczna związana jest z obecnością na powierzchni metalu grubej, mniej lub bardziej porowatej warstwy soli nieprzewodzącej- np. warstwy chromianowe lub fosforanowe na cynku i stali, powłoki oksydacyjne na stali.
W większości przypadków warstewki te otrzymuje się sztucznie. Jedną z metod pasywacji jest zanurzanie metalu podatnego na pasywację w odpowiedniej kąpieli utleniającej. Wówczas reakcja utleniania (korozji) jest hamowana na etapie tworzenia warstewki, która może być odporna także w innych ośrodkach. Jako przykład można podać pasywację żelaza czy aluminium w kwasie azotowym(V), stężonym kwasie siarkowym(VI) lub roztworach chromianów(VI). Innym ze sposobów uzyskania stanu pasywnego jest wytworzenie warstewki poprzez anodowe utlenianie metalu pasywującego się w odpowiednim roztworze.
Podobnie jak żelazo, pasywacji ulega chrom. Pasywacja tego pierwiastka jest łatwiejsza, tzn. zachodzi przy niższej wartości potencjału i przy niższej gęstości prądu, a szybkość roztwarzania w zakresie pasywnym jest także mniejsza. Możliwa jest, zatem pasywacja tego pierwiastka w ośrodkach o mniejszej zdolności utleniającej. Z dodatkiem chromu i niklu (ok. 18%Cr i 8%Ni) wytwarza się stale nierdzewne i kwasoodporne, Są to stale łatwo pasywujące się i dlatego dobrze odporne na działanie dość agresywnych kwaśnych środowisk korozyjnych.
Wpływ czynników zewnętrznych na szybkość korozji
Korozja elektrochemiczna przebiega szybciej, jeżeli środowisko otaczające metal zwiększa agresywność roztworu elektrolitu. Tak działają np. obecne w atmosferze gazy, jak SO2, NO2, CO2, które z parą wodną i tlenem tworzą na powierzchni metali kwasy. Cząsteczki sadzy i pyłów osadzają się na powierzchni metalu i ułatwiają powstawanie lokalnych ogniw. W ośrodkach miejskich i przemysłowych korozja metali zakopanych w ziemi przyspieszona jest prądami błądzącymi, powstającymi w pobliżu kabli elektrycznych i uziemionych urządzeń energetycznych.
Występujące w glebie bakterie siarkowe utleniają siarkę do kwasu siarkowego, który przyspiesza korozję. W środowisku beztlenowym, występują bakterie, które redukują siarczany do siarczków oddziałujących korozyjnie na metale.
Często korozja jest przyspieszona przez zespół czynników wspólnie oddziałujących. Ochrona metali przed korozją musi więc także działać wielokierunkowo.
Badanie zjawiska korozji metali metodami elektrochemicznymi
Proces korozji elektrochemicznej polega na sprzężeniu reakcji anodowego roztwarzania metalu z katodową reakcją redukcji depolaryzatora. Każdej reakcji elektrodowej odpowiada przepływ, przez granicę faz metal - elektrolit, odpowiednich prądów katodowych jk i anodowych ja. W warunkach gdy metal nie jest polaryzowany przez zewnętrzne źródło prądu, prądu suma wszystkich prądów anodowych i katodowych jest równa zero. Korodujący w warunkach obwodu otwartego metal przyjmuje wówczas potencjał stacjonarny zwany potencjałem korozji Ekor a prąd wymiany płynący przez granicę faz jest prądem korozji jkor = ja + ja , ja= - jk..
Przebieg procesu korozji metalu można więc zbadać przy pomocy krzywych prąd - napięcie zwanych krzywymi polaryzacji i = f(E), zarejestrowanych przy pomocy techniki woltamperometrycznej. Technika ta bardzo często stosowana w badaniach korozyjnych polega na kontrolowanych (najczęściej narastających liniowo) zmianach potencjału elektrody z materiału korodującego i pomiarze prądu płynącego w badanym układzie przy zadanym potencjale Na postawie kształtu i przebiegu charakterystyk w obszarze polaryzacji anodowej można określić zakres potencjałów przy którym występuje aktywne roztwarzanie metalu, wytwarzanie pasywnych warstw ochronnych, czy rozwój korozji wżerowej. Charakterystyki te są pomocne także w badaniach kinetyki i mechanizmu reakcji korozji oraz w doborze metod ochrony przeciwkorozyjnej.
Do pomiaru niezbędny jest specjalny układ elektrochemiczny, tzw. potencjostat, w którym obok możliwości zadawania potencjału pomiędzy metalem - WE i przeciwelektrodą - CE np. z platyny istnieje możliwość kontroli potencjału elektrody metalowej za pomocą trzeciej elektrody - elektrody odniesienia - RE o stałym potencjale (Rys 6).
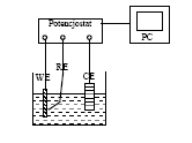
Rys. 6 Schemat układu pomiarowego
Metoda ta jest użyteczna przy badaniu zjawiska pasywności metali. Rys. 7 przedstawia krzywą polaryzacji anodowej żelaza w kwasie siarkowym.
W układzie badanym, przy braku potencjału zewnętrznego ustala się potencjał korozyjny, przy którym zachodzi samorzutny proces korozji metalu. Zwiększając potencjał metalu wymuszamy procesy utleniania (polaryzacja w kierunku anodowym), których intensywność określa natężenie lub gęstość prądu (natężenie prądu na jednostkę powierzchni elektrody) płynącego w obwodzie - obszar aktywny. Gęstość prądu wzrasta stopniowo ze wzrostem potencjału, a przy powierzchni elektrody wzrasta stężenie produktu korozji - siarczanu żelaza(II) - FeSO4. Powstająca w kontakcie z elektrodą dyfuzyjna warstwa graniczna wkrótce zawiera nasycony roztwór FeSO4, z którego wytrąca się nie przewodząca, lecz porowata warstwa stałego siarczanu żelaza(II). Wartość prądu korozji aż do potencjału ok. 0,6 V stabilizuje się, ponieważ korozja przebiega w warunkach polaryzacji dyfuzyjnej. Przy potencjale około +0,6 V wzgl. NEW gęstość prądu raptownie spada, ponieważ porowata warstwa siarczanu żelaza zanika a wydziela się tlenek żelaza (III) Fe2O3* nH2O, tworzący cienką, nieporowatą warstewkę pasywną. Potencjał przy którym metal korodujący przechodzi ze stanu aktywnego rozpuszczania w stan pasywny nosi nazwę potencjału pasywacji Epas. Warstwa pasywna istnieje na powierzchni żelaza do potencjału o wartości około 1.4 V- obszar pasywny, po czym ulega rozpuszczeniu, a szybkość roztwarzania żelaza ponownie wzrasta proporcjonalnie do gęstości prądu - obszar ten nosi nazwę obszaru transpasywacji.
Żelazo jest podatne na anodową pasywację w ośrodku o własnościach utleniających. Jeśli obniżymy stężenie kwasu żelazo roztwarza się w stanie aktywnym, ponieważ własności utleniające kwasu są zbyt słabe, aby pasywacja była możliwa. Szeroki zakres plateau dla krzywej woltamperometrycznej otrzymanej dla Fe w 0,1 M H2SO4 związany jest z występowaniem polaryzacji dyfuzyjnej związanej z równowagowym transportem jonów żelaza do roztworu.
.
Rys. 7. Krzywa polaryzacji anodowej żelaza.
Fe w 1M H2SO4 , pasywuje się
----- Fe w 0,1M H2SO4 , roztwarza się aktywnie
Równanie krzywej polaryzacji
Jeśli obydwie reakcje korozji -anodowa i katodowa biegną w warunkach polaryzacji aktywacyjnej, to zależność gęstości prądu katodowego lub anodowego od potencjału (równanie krzywej polaryzacji) podaje równanie Tafela:
ia = iao exp , ik =- ikoexp,
gdzie iao, iko - gęstości prądów wymiany, Ea0 , Ek0 - potencjały normalne. Przy potencjale korozji (E = Ekor) ikor = ia = - ik , tak więc:
ikor = iao exp= ikoexp
Jeżeli potencjał nieco różni się od potencjału korozji ; E = Ekor +ΔE to i = ia+ ik , wówczas po połączeniu równań (4) i (5) otrzymujemy :
i = ikor
Po zróżniczkowaniu uzyskujemy tzw. równanie Sterna- Geary'ego:
Wyznaczanie szybkości korozji w pomiarach woltamperometrycznych
Szybkość korozji z prądem korozji łączy I prawo Faraday`a.
v = kor
Najstarszą historycznie metodą wyznaczania szybkości korozji w oparciu o dane polaryzacyjne jest metoda ekstrapolacji tzw. tafelowskich odcinków krzywej polaryzacji do potencjału korozyjnego. Estrapolację można przeprowadzić graficznie w układzie log = f(E). Dla uzyskania tej zależności elektrodę polaryzuje się w dość szerokim zakresie potencjałów, poza obszar wzajemnego nakładania się reakcji anodowej i katodowej procesu korozyjnego. Charakterystyki prądowo-napięciowe rejestruje się, stosując bardzo niskie prędkości liniowego narastania potencjału 0,1-10m/s.
W warunkach niewielkiej polaryzacji, w pobliżu potencjału korozji (tzw. obszar nietafelowski), gdy reakcja anodowa i katodowa są kontrolowane aktywacyjnie, gęstość prądu korozji ikor z rezystancją polaryzacyjną wiąże równanie Sterna- Gaery`ego
Metoda wyznaczania prądu korozji poprzez pomiar rezystancji polaryzacyjnej elektrody korodującej, w obszarze nietafelowskim, nosi nazwę metody polaryzacji liniowej. Interpretacja danych pomiarowych, polega na dopasowaniu danych doświadczalnych do teoretycznych krzywych polaryzacji otrzymanych dla różnych parametrów ba i bk. Uzyskane w ten sposób przybliżone wartości współczynników równania krzywej polaryzacji służą do wyznaczania wartości ikor z równania Sterna-Geary'ego.
Obok metod graficznych przydatne są tu metody algebraiczne polegające na rozwiązywaniu równań kwadratowych, wywodzących się z równania krzywej polaryzacji przy uwzględnieniu różnej ilości punktów pomiarowych.
Szybki rozwój technik komputerowych otworzył nową drogę do rozwiązywania problemu wyznaczania parametrów korozyjnych na podstawie analizy danych doświadczalnych. Celem rozwijanych procedur obliczeniowych jest wyznaczenie takich parametrów równania krzywej polaryzacji, aby było ono spełnione dla dowolnej ilości punktów doświadczalnych. Stosowane są głównie metody iteracyjne, prowadzące najczęściej do minimalizacji funkcji
F( ikor , ba , bk ) =
n - liczba punktów pomiarowych, iieksp , ii teor - gęstość prądu dla i-tego punktu pomiarowego lub wyznaczona z równania krzywej polaryzacji.
Czas wyznaczania parametrów równania korozyjnego tą metoda jest bardzo krótki, programy obliczeniowe są coraz doskonalsze i obejmują coraz większą ilość punktów doświadczalnych ..
Wielkość prądu korozji zależy od wielu czynników, w tym od składu badanego metalu, składu i parametrów środowiska korozyjnego, stanu powierzchni elektrody korodującej, geometrii celki pomiarowej, obecności śladów substancji inhibitujących. Wyniki pomiarów prądów korozji można więc praktycznie porównywać między sobą tylko jeśli pomiary były przeprowadzone w tych samych warunkach.
Metody elektrochemiczne cechuje duża szybkość i dokładność. Umożliwiają one przede wszystkim określenie zmian szybkości korozji w czasie, stanowią istotną pomoc w badaniach kinetyki i mechanizmu różnego rodzaju reakcji korozyjnych. Zastosowanie tych metod przyczyniło się do wyraźnego postępu w zakresie badań podstawowych procesów korozji oraz do rozwoju technik zabezpieczeń antykorozyjnych.
Ochrona przed korozją
Ograniczenie szkodliwych skutków korozji odbywa się przez :
wytwarzanie powłok ochronnych izolujących powierzchnie metalu od czynników atmosferycznych,
unikanie takich kompozycji materiałowych, które umożliwiają powstawanie ogniw lub mikroogniw stykowych,
zastosowanie, ochrony galwanicznej,
zastosowanie inhibitorów zwalniających szybkość reakcji towarzyszących korozji w roztworach.
Zapobieganie powstawaniu ogniw korozyjnych
Ponieważ ogniwa stykowe są poważną przyczyną korozji, należy unikać bezpośredniego kontaktu różnych metali oraz wystawiania na działanie czynników korodujących części metalowych, w których występują podwyższone naprężenia i inne niezrównoważone siły. Podstawowym sposobem zapobiegania korozji jest stosowanie zamiast zwykłej stali odpowiednich gatunków stali nierdzewnej, mającej strukturę jednofazową. Taką strukturę ma stal nierdzewna zawierająca znaczne ilości chromu i niklu, np. 18% Cr i 8% Ni, która nie powoduje występowania na powierzchni metalu mikroogniw międzyfazowych i nie jest podatna na korozję międzykrystaliczną.
Powłoki ochronne
Powłoki ochronne są najstarszym i najprostszym sposobem zapobiegania korozji. Ze względu na rodzaj stosowanego materiału rozróżnia się
powłoki organiczne (farby, lakiery i powłoki z tworzyw sztucznych),
powłoki ceramiczne czyli emalie,
powłoki tlenkowe i
powłoki metaliczne.
Każda z nich ma pewne wady i zalety i musi być indywidualnie dostosowana do potrzeb.
Farby, lakiery i warstwy tworzyw sztucznych są mało odporne mechanicznie i nie nadają się do eksploatacji w wysokiej temperaturze.
Emalie są kruche i utrudniają wymianę ciepła, ale mogą być używane w szerokim zakresie temperatur.
Wykorzystanie powłok tlenkowych w ochronie przeciwkorozyjnej ogranicza się do metali ulegających pasywacji, jak np. glin, chrom i tytan. Rola wymienionych powłok ochronnych polega wyłącznie na izolacji powierzchni metalu od kontaktu z czynnikami korodującymi.
Funkcje powłok metalicznych nakładanych elektrolitycznie lub innymi technikami są bardziej złożone i zależne od rodzaju użytych materiałów.
Metaliczna warstwa ochronna może być wytwarzana z metalu bardziej lub mniej szlachetnego od metalu chronionego, tzn. metal chroniony może leżeć w szeregu napięciowym poniżej lub powyżej metalu chronionego.
Rozpatrzmy sytuację, w której element żelazny jest pokryty warstwą cynku. Szczelna powłoka cynku chroni żelazo od dostępu tlenu i wilgoci. W wyniku pojawienia, się rysy lub szczeliny w powłoce cynkowej powstaje typowe ogniwo stykowe, w którym rolę katody spełnia żelazo, a rolę anody cynk (rys. 8a).
Cynk zatem ulega reakcji utleniania, która jest zahamowana wskutek pasywacji. W rozpatrywanym ogniwie żelazo jest katodą, czyli jest chronione przed korozją przez samą obecność cynku, a nie na skutek szczelności warstwy cynkowej .
Powłoka ochronna z metalu szlachetniejszego od żelaza, np. miedzi lub cyny, jest skuteczna tylko wtedy, kiedy idealnie cała powierzchnia żelaza jest nią okryta- Rys. 8b. W chwili wytworzenia szczeliny zaczyna działać ogniwo stykowe, w którym żelazo jest anodą podlegającą przyspieszonej korozji. Na miedzianej katodzie, która dysponuje nadmiarem elektronów, następuje redukcja tlenu według równania reakcji
O2 + 2H2O +4e → 4OH-
Ochrona elektrochemiczna
Zasadę działania, ogniw galwanicznych wykorzystuje się do odwrócenia mechanizmów korozji. Tego rodzaju zjawisko jest obserwowane w działaniu pokryć galwanicznych z metalu mniej szlachetnego od podłoża (np. żelazo pokryte cynkiem omówione wyżej) Tę samą zasadę można wykorzystać w sposób jeszcze prostszy. Mianowicie płytki z metalu mało szlachetnego, np. magnezu lub cynku, umieszcza się na podziemnych przewodach instalacji wodnej, na kadłubach statków lub we wnętrzach zbiorników wodnych Rys. 9a.
Płytki spełniają rolę anod w utworzonych ogniwach, ulegają szybkiej korozji chroniąc jednocześnie podstawowe urządzenie. Zastępowanie skorodowanych płytek nowymi jest zabiegiem prostym i tanim. Tego rodzaju zapobieganie korozji nazywany ochroną anodową. Ażeby taka anodowa ochrona działała skutecznie, płytki z mniej szlachetnego metalu nie mogą być umieszczone w zbyt dużych odstępach i musi być zapewniony bardzo dobry kontakt między płytką a metalem.
Drugi rodzaj ochrony elektrochemicznej, nazywany katodowym, polega na połączeniu chronionego obiektu z ujemnym biegunem słabego źródła napięcia stałego Rys. 9b. Elektrony doprowadzone do części chronionej przed korozją powodują, że staje się ona katodą wytworzonego układu elektrochemicznego.
Ochrona za pomocą inhibitorów
Inhibitory zmniejszające szybkość korozji są czasem nazywane ujemnymi katalizatorami. Dodawany np. do chłodnic samochodowych Na2CrO4, powoduje wytworzenie powierzchniowej warstwy tlenku chromu (III) i podwyższa potencjał elektrochemiczny metalu. Inhibitory organiczne - substancje dodawane zwykle do smarów i olejów, adsorbując się na powierzchni zmniejszają szybkość korozji elektrochemicznej.
Ćwiczenie 1
a) Badanie wpływu różnych czynników na korozję stali .
Wykonanie ćwiczenia
12 probówek umieszczonych w statywie napełnić kolejno:
wodą destylowaną
wodą z kranu
wodą z kranu zakwaszoną paroma kroplami 0.1M H2SO4
wodą z kranu zakwaszoną paroma kroplami 0.1M H2SO4 z dodatkiem paru kropel 3% wody utlenionej
3% NaCl
3% NaCl z dodatkiem paru kropel 3% wody utlenionej
0.1M NaOH
0.1M NaOH z dodatkiem paru kropel 3% wody utlenionej
0.1M H2SO4
0.1M H2SO4 z dodatkiem paru kropek 3% wody utlenionej
3% NaCl + 0.1% K2Cr2O7
0.1M H2SO4 +0.1% K2Cr2O7
12 gwoździ stalowych kolejno:
oczyścić papierem ściernym
odtłuścić przez zanurzenie w trójchloroetylenie (30 s),
opłukać przez zanurzenie w alkoholu metylowym,.
opłukać bieżącą zimną wodą z kranu,
trawić przez zanurzenie w 15% roztworze kwasu siarkowego (15 s)
Tak przygotowane gwoździe niezwłocznie wrzucić po jednym do każdej z probówek.
Obserwować zmiany zachodzące w roztworach przez okres 30min.
Następnie do każdej probówki dodać parę kropel roztworu rodanku potasowego KSCN.
Uwaga - w obecności jonów żelaza (III) w roztworze tworzy się czerwono zabarwiony związek Fe(SCN)3- tzw. smocza krew. Jony żelaza (II) nie dają tej reakcji! Można proces utleniania Fe2+ do Fe3+ tlenem powietrza przyspieszyć wytrząsając probówkę zatkaną palcem lub dodając do probówek w których nie uzyskano różowego zabarwienia po kropli H2O2 - probówki nr 1,2,3,5,7,9,11,12. Jeśli różowe zabarwienie nadal nie jest wyraźne mamy pewność że proces korozji przebiega w danych warunkach powoli.
Wyniki obserwacji zanotować w tabelce nr 1, ocenić intensywność korozji żelaza w danym środowisku i wyjaśnić ich przyczynę
b) Badanie działania ogniw stykowych
Rozłożyć kilka spinaczy biurowych w literę S -Następnie, przygotować po jednym w następujący sposób:
1 - kilkakrotnie zgiąć w środku
2 - owinąć w środkowej części drutem miedzianym
3 - owinąć w środkowej części paseczkiem folii aluminiowej
4 - owinąć w środkowej części paseczkiem cynku
5 - cześć spinacza sklepać silnie młotkiem na podkładce ze stali
Przygotowane spinacze umieścić w szalce Petriego ustawionej na zimnym podłożu i ostrożnie wlać lekko ciepły roztwór odczynnika ferroksylowego.. Nie poruszać szalką dopóki odczynnik nie zastygnie w żel. Po upływie 15-20 min opisać zaobserwowane zmiany w tab. nr 2
Uwaga:
Odczynnik ferroksylowy przygotowuje się następująco:
Namoczyć w 100cm3 wody destylowanej 1g agaru, dodać 0,1g fenoloftaleiny w 5cm3 alkoholu oraz 0,05g K3Fe(CN)6 i 3g NaCl.. Roztwór ogrzać prawie do wrzenia i przesączyć na gorąco przez sączek z bibuły. Po lekkim ostudzeniu dodawać bardzo ostrożnie kroplami 0,01M NaOH aż do uzyskania słabego bladoróżowego zabarwienia.
Powstawanie jonów żelaza (II) w miejscach anodowych można śledzić za pomocą K3Fe(CN)6 - tworzy się zielononiebieska plama błękitu pruskiego. Fenoloftaleina jest wskaźnikiem który w obecności jonów OH- barwi się na różowo - sygnalizacja miejsc katodowych.
Tabela nr 1
Nr |
Skład roztworu |
Reakcja z KSCN- ocena intensywności zabarwienia |
Ocena intensywności korozji |
Wyjaśnienie zjawiska |
1 |
Woda destylowana
|
|
|
|
2 |
Woda z kranu
|
|
|
|
3 |
Zakwaszona woda z kranu
|
|
|
|
4 |
Zakwaszona woda z kranu + H2O2 |
|
|
|
5 |
3 % NaCl
|
|
|
|
6 |
3 % NaCl + H2O2
|
|
|
|
7 |
0,1 M NaOH
|
|
|
|
8 |
0,1 M NaOH + H2O2
|
|
|
|
9 |
0,1 M H2SO4
|
|
|
|
10 |
0,1 M H2SO4 + H2O2
|
|
|
|
11 |
3% NaCl + 0,1 % K2Cr2O7
|
|
|
|
12 |
0.1M H2SO4 +0,1 % K2Cr2O7 |
|
|
|
Tabela nr 2
Sposób przygotowania spinacza
|
Rozmieszczenie miejsc katodowych i anodowych ( pokaż na rysunku) |
Wyjaśnienie zjawiska korozji |
kilkakrotnie zgięty w środku
|
|
|
owinięty w środkowej części drutem miedzianym
|
|
|
owinięty w środkowej części paseczkiem folii aluminiowej
|
|
|
owinięty w środkowej części paseczkiem blachy cynkowej
|
|
|
Ćwiczenie 2
Badanie szybkości korozji metodami elektrochemicznymi
a) rejestracja krzywej polaryzacji stali w roztworach kwasu siarkowego
Do celki pomiarowej nalać roztworu 0,1 M H2SO4, zanurzyć elektrodę badaną wykonaną ze stali -WE, chlorko-srebrową elektrodę odniesienia - RE, i przeciwelektrodę wykonaną z platyny- CE. Elektrody podłączyć do potencjostatu ATLAS 81 zgodnie z oznaczeniem na obudowie przyrządu.
Podłączyć przyrząd i połaczony z nim generator do sieci. Włączyć komputer i uruchomić program sterujący pomiarem pol_91.exe. Otworzyć na pasku zadań u góry ekranu okienko options i w powstałej liście zadań otworzyć okienko measurement history. Uruchomić polecenie Get Erest i poczekać aż przyrząd zmierzy i wyświetli w okienku initial potential wartość Ekor w mV. Zaznaczyć jako wartość początkową potencjału dla pomiarów wartość Ekor poprzez zaznaczenie opcji start with stacionary potential. Wybrać liniowe narastanie potencjału - ramp sweet. Uruchomić polecenie Edit steps i wprowadzić wartość potencjału końcowego pomiarów Destination potential - 1500 mV, czas utrzymywania potencjału startowego - hold time - O sek.i wprowadzić wartość kroku pomiarów reading density -10 mV. Uruchomić polecenie sweep rate i ustalić szybkość liniową narastania potencjału na 100mV/min. Każdorazowo zakończyć proces określania warunków pomiaru poprzez kliknięcie OK. Uruchomić w pasku zadań okienko curve, wybrać opcję nowy pomiar - new i wypełnić etykietę pomiarów. Kliknięcie OK. uruchamia pomiar. Po zakończeniu pomiaru kliknąć ok. i po otwarciu okienka curve poleceniem save zapisać wyniki na dysku w katalogu podanym przez prowadzącego ćwiczenia. Uruchomić w pasku zadań okienko graph, polecenie view i wydrukowac uzyskaną krzywą polaryzacji poleceniem hardcopy. Po zakończeniu opróżnić i opłukać naczynko, opłukać wodą elektrody. Następnie napełnić celkę 1M roztworem H2SO4 i przeprowadzić pomiar w analogiczny sposób. Otrzymane krzywe polaryzacji opisać zaznaczając wartość Ekor, Epas, zakres potencjałów obszaru aktywnego i pasywnego.
b) wyznaczanie szybkości korozji stali w roztworach kwasu siarkowego metodą polaryzacji liniowej
Do celki pomiarowej nalać roztworu 0,1 M H2SO4, zanurzyć elektrodę badaną wykonaną ze stali -WE, chlorko-srebrową elektrodę odniesienia - RE, i przeciwelektrodę wykonaną z platyny- CE. Elektrody podłączyć do potencjostatu ATLAS 81 zgodnie z oznaczeniem na obudowie przyrządu.
Podłączyć przyrząd i połaczony z nim generator do sieci. Włączyć komputer i uruchomić program sterujący pomiarem pol_91.exe. Otworzyć na pasku zadań u góry ekranu okienko options i w powstałej liście zadań measurement history. Uruchomić polecenie Get Erest i poczekać aż przyrząd zmierzy i wyświetli w okienku initial potential czyli wartość Ekor w mV. Przyjąć jako wartość początkową potencjału dla pomiarów wartość E = Ekor -20mV wpisując odpowiednią wartość w okienku. Wybrać liniowe narastanie potencjału - ramp sweet. Uruchomić polecenie Edit steps i wprowadzić wartość potencjału końcowego pomiarów Destination potential - E=Ekor+20mV, czas utrzymywania potencjału startowego - hold time - O sek.i wprowadzić wartość kroku pomiarów reading density -1 mV. Uruchomić polecenie sweep rate i ustalić szybkość liniową narastania potencjału na 10mV/min. Każdorazowo zakończyć proces określania warunków pomiaru poprzez kliknięcie OK. Uruchomić w pasku zadań okienko curve, wybrać opcję nowy pomiar - new i wypełnić etykietę pomiarów. Kliknięcie OK. uruchamia pomiar. Po zakończeniu pomiaru kliknąć ok. i po otwarciu okienka curve poleceniem save zapisać wyniki na dysku w katalogu podanym przez prowadzącego ćwiczenia. Uruchomić w pasku zadań okienko mathematics, polecenie fitting, wybrać równanie polaryzacji aktywacyjnej- activation polarisation i komendą fit uruchomić procedurę dopasowania krzywej polaryzacji. Po zakończeniu działania programu kliknąć ok., zatwierdzając wynik, przejść do okienka Report i po przez kliknięcie polecenia hardcopy wydrukować otrzymane wyniki w postaci równania krzywej polaryzacji. Po zakończeniu opróżnić i opłukać naczynko, opłukać wodą elektrody. Następnie napełnić celkę 1M roztworem H2SO4 i przeprowadzić pomiar w analogiczny sposób.
Po przeprowadzeniu pomiarów porównać szybkości korozji stali w obydwu roztworach w warunkach stacjonarnych i przy E = 1000mV.