CHEMIA ORGANICZNA I - laboratorium
Daniel Prasał
rok I, nr 13
SPIS TREŚCI:
1. Krystalizacja 1
2. Destylacja 6
3. Dibenzylidenoaceton 9
4. Cykloheksanon 10
5. Rozdział mieszanin przez ekstrakcję. Destylacja pod zmniejszonym ciśnieniem 11
6. Chromatografia kolumnowa i cienkowarstwowa 13
7. Chlorek tert - butylu (2 - chloro - 2 metylopropan) 16
8. Mrówczan etylu 18
9. p - Acetylotoluidyna (N - acetylo - p - toluidyna) 19
10. p - nitroacetanilid 20
11. 1,4 - di - tert - butylobenzen 21
Krystalizacja
Krystalizacja jest jedną z podstawowych i najczęściej stosowanych technik laboratoryjnych. Wykorzystuje się ją do oczyszczania (krystalizacja prosta) i rozdzielania (krystalizacja frakcjonowana) substancji stałych, wydzielających się z roztworów w odpowiednich rozpuszczalnikach w postaci krystalicznej. Oczyszczanie przez krystalizację opiera się na dwóch podstawach. Pierwsza to różnica rozpuszczalności substancji oczyszczanej i obecnych zanieczyszczeń w rozpuszczalniku stosowanym do krystalizacji. Druga to zależność rozpuszczalności oczyszczanej substancji od temperatury. Najczęściej wykorzystywany wariant krystalizacji polega na rozpuszczeniu oczyszczanej substancji w podwyższonej temperaturze, temperaturze następnie ochłodzeniu roztworu.
Ochłodzenie powoduje przesycenia roztworu. i w sprzyjających warunkach, wykrystalizowanie substancji oczyszczanej. Zastosowana ilość rozpuszczalnika zazwyczaj jest zbyt duża, aby roztwór stał się przesycony w stosunku do składników zanieczyszczających oczyszczaną substancję. Pozostają one zatem w roztworze (ługu macierzystym). W przeciwnym przypadku, gdy zanieczyszczenia są znacznie mniej rozpuszczalne w stosowanym rozpuszczalniku niż oczyszczana substancja, zostaję usunięte przez sączenie gorącego roztworu.
Ze względu na różnice w stosowanej metodyce pracy w laboratorium, rozróżnia się krystalizację z wody i krystalizację z rozpuszczalników organicznych. Odpowiedni dobór rozpuszczalnika do krystalizacji zapewnia uzyskanie dobrych wyników. Oczyszczany związek winien być dobrze rozpuszczalny w stosowanym rozpuszczalniku na gorąco, a słabo rozpuszczalny na zimno. Natomiast obecne zanieczyszczenia powinny być bądź doskonale rozpuszczalne w tym rozpuszczalniku na zimno, bądź bardzo słabo rozpuszczalne na gorąco. Temperatura wrzenia rozpuszczalnika powinna być niższa od temperatury topnienia substancji oczyszczanej.
Usuwanie obecnych często zanieczyszczeń barwnych odbywa się przez adsorpcję na węglu aktywnym (w przypadku krystalizacji z wody lub alkoholi; w przypadku używania rozpuszczalników niepolarnych stosuje się tlenek glinu). Dodany adsorbent odsącza się na gorąco.
Zdarza się, że krystalizacja nie następuje natychmiast po ochłodzeniu roztworu. Czasami wystarcza wówczas potrząsnąć kolbę z roztworem przesyconym lub zainicjować krystalizację przez dodanie kilku kryształów oczyszczanej substancji. Często stosowanym sposobem inicjowania krystalizacji jest pocieranie bagietkę wewnętrznych ścianek naczynia z roztworem do krystalizacji.
Stopień czystości ciał stałych można ocenić badając ich temperaturę topnienia. Substancje czyste topnieją w bardzo małym przedziale temperatury, zaś zanieczyszczone przeciwnie. Wraz ze wzrostem stopnia zanieczyszczenia substancji jej temperatura topnienia zazwyczaj obniża się. Zjawisko to, znane jako depresja temperatury topnienia, wykorzystywane jest do potwierdzenia identyczności próbek krystalicznych przez oznaczenie temperatury topnienia mieszaniny próbki badanej z próbką wzorcową. Brak depresji temperatury topnienia uważany jest za dowód identyczności porównywanych próbek.
Przeprowadzanie krystalizacji stwarza pewne problemy związane z bezpiecznym wykonaniem tej operacji. Należy zwrócić szczególną uwagę na palność rozpuszczalników stosowanych do krystalizacji (z wyjątkiem wody i chlorowcopochodnych takich jak CCl4 i CHCl3) oraz na ich toksyczność. Z tych względów w operacji sączenia na gorąco, dopuszczalne jest wyłącznie pod sprawnie działającym wyciągiem z dala od źródeł otwartego płomienia. Dodawanie węgla aktywnego, który zawsze zawiera dużą ilość zaadsorbowanego powietrza, powoduje zazwyczaj silne pienienie się gorącego roztworu. Dlatego, węgiel należy dodawać ostrożnie, do nieco przestudzonego roztworu i z dala od źródeł ognia. W każdym momencie należy unikać wdychania par rozpuszczalnika.
Ćwiczenie składa się z dwóch części. Celem obu jest oczyszczenie otrzymanej próbki związki. W części pierwszej przez krystalizację z wody, w części drugiej przez krystalizację z etanolu. Należy zwrócić szczególną uwagę na kolejność wykonywanych czynności, prawidłowe zmontowanie aparatury (zwłaszcza w części drugiej) oraz na prawidłową pracę z pompką wodną. Istotne jest także opanowanie umiejętności oznaczania temperatury topnienia.
Część pierwsza: Krystalizacja z wody
Substraty: acetanilid - 3g
Opis wykonania ćwiczenia:
Zestawiamy sprzęt wg rys.1. Do kolby wsypujemy substancję przeznaczoną do krystalizacji oraz nalewamy niewielką ilość wody. Wrzucamy kamyczek wrzenny i rozpoczynamy ogrzewanie. Gdy zawartość kolby zawrze, sprawdzamy, czy pozostała jeszcze nierozpuszczona substancja. Jeśli tak, dodajemy porcjami wodę, każdorazowo odsuwając płaszcz grzejny oraz upewniając się czy substancja rozpuściła się w tej ilości wrzącego rozpuszczalnika. Po osiągnięciu rozpuszczenia przerywamy ogrzewanie. Następnie dodajemy około 0,5g węgla aktywnego i ponownie ogrzewamy zawartość kolby do wrzenia przez około 3 minuty.
Gorącą mieszaninę sączymy przez sączek karbowany do kolby stożkowej (rys.2. lejek przed sączeniem powinien być ogrzany w suszarce). Wylot kolby zakrywamy odstawiamy do ochłodzenia (krystalizacji) najpierw w temperaturze pokojowej, a następnie w lodzie.
Uzyskane kryształy odsączamy na lejku Buchnera lub Schotta. Do ilościowego przeniesienia substancji z kolby na lejek używamy ługu macierzystego z kolby ssawkowej. Po odsączeniu, osad dokładnie przemywamy na lejku zimną wodą (podczas przemywania pompka wodna powinna być odłączona od kolby ssawkowej). Dokładnie odciskamy kryształy i pozostawiamy na lejku do wyschnięcia bez odłączania pompki wodnej. Suche kryształy przenosimy na wytarowaną szalkę Petriego i ważymy. Oznaczamy temperaturę topnienia.
Rys.2. Zestaw do sączenia
Część pierwsza: Krystalizacja z rozpuszczalnika organicznego
Substraty: p - nitroanilina - 3g
Opis wykonania ćwiczenia:
Montujemy aparaturę wg rys.3. Do kolby wlewamy niewielką ilość etanolu i wsypujemy rozdrobnioną substancję przeznaczoną do krystalizacji.
Wrzucamy kamyczek wrzenny, montujemy chłodnicę zwrotną i rozpoczynamy ogrzewanie. Gdy zawartość kolby zawrze, sprawdzamy, czy pozostała jeszcze nierozpuszczona substancja. Jeśli nie dodajemy porcjami etanolu (przez chłodnicę), każdorazowo upewniając się czy substancja rozpuściła się w użytej ilości wrzącego rozpuszczalnika. Po osiągnięciu rozpuszczenia, dodajemy jeszcze 10% całkowitej objętości użytego etanolu i przerywamy ogrzewanie. Gdy wrzenie ustanie, dodajemy 0,5 g węgla aktywnego (po zdjęciu chłodnicy) i ponownie ogrzewamy zawartość kolby do wrzenia przez około 3 minuty. Gorącą mieszaninę sączymy przez sączek karbowany do kolby stożkowej (rys.2. lejek powinien być uprzednio ogrzany w suszarce). Wylot kolby zakrywamy papierem i odstawiamy do
Rys.3. Zestaw do krystalizacji z rozpuszczalnika organicznego
ochłodzenia (krystalizacji), najpierw w temperaturze pokojowej, a następnie w lodówce.
Wykrystalizowaną substancję odsączamy na lejku Buchnera lub Schotta. Do ilościowego przeniesienia substancji z kolby na lejek używamy ługu macierzystego z kolby ssawkowej. Po odsączeniu, osad dokładnie przemywamy na lejku zimną wodą (podczas przemywania pompka wodna powinna być odłączona od kolby ssawkowej). Dokładnie odciskamy kryształy i pozostawiamy na lejku do wyschnięcia bez odłączania pompki wodnej. Suche kryształy przenosimy na wytarowaną szalkę Petriego i ważymy. Oznaczamy temperaturę topnienia.
Opracowanie ćwiczenia.
Po przeprowadzeniu powyżej opisanych czynności w pierwszej części ćwiczenia otrzymałem substancję krystaliczną - acetanilid. Po wykrystalizowaniu i wysuszeniu masa oczyszczanego związku wynosiła 1,07g. Natomiast wydajność procesu krystalizacyjnego:
![]()
Czystość otrzymanej substancji sprawdziłem na podstawie jej temperatury topnienia, która obejmowała zakres 110 - 1130C. Natomiast wartość tablicowa wynosi 1140C. Depresja temperaturowa - choć niewielka - świadczy o istniejących zanieczyszczeniach w otrzymanym acetanilidzie.
W drugiej części doświadczenia oczyszczanym związkiem jest p - nitroanilina. Masa, jaką otrzymałem po krystalizacji i osuszeniu wynosiła 1,72g. Wydajność tego procesu:
![]()
Temperatura topnienia p - nitroanilina obejmowała zakres 139 - 1130C. Podobnie jak w powyższym przypadku, obniżenie i rozszerzenie zakresu temperaturowego spowodowane jest zanieczyszczeniami, które znajdowały się w przekrystalizowanej substancji.
Wnioski.
Zastosowana technika oczyszczania substancji jest jedną z najczęściej wykorzystywanych (o ile jest to możliwe) ze względu na prostotę, niski koszt a przede wszystkim ze względu na dużą wydajność i skuteczność. Mimo braku wprawy udało mi się otrzymać substancje dostatecznie czyste, jednak z niezadowalającą wydajnością, szczególnie w przypadku acetanilidu.
DESTYLACJA
Destylacja jest jedną z najstarszych i najczęściej stosowanych technik laboratoryjnych służących do oczyszczania (destylacja prosta) lub rozdzielania (destylacja frakcyjna, rektyfikacja) cieczy.
Oczyszczanie przez destylację polega na odparowaniu lotnych cieczy, a następnie skropieniu par. Warunkiem oczyszczenia i rozdzielenia substancji lotnych jest dostateczna różnica temperatur wrzenia substancji oczyszczanej i obecnych zanieczyszczeń. Zanieczyszczenia nielotne usuwane są niejako automatycznie.
Efektywność rozdziału przez destylację zależy jednak również od innych czynników, np. konstrukcji aparatury, szybkości prowadzenia procesu, a także specyficznych własności cieczy rozdzielanych (tworzenie azeotropów).
Temperatura wrzenia cieczy pod określonym ciśnieniem zewnętrznym (mierzona w ściśle określony sposób, np. w ebuliometrach) jest jej fizyczną cechą charakterystyczną. Wartość jej różni się zazwyczaj od wartości odczytywanej na termometrze umieszczonym w aparaturze i potocznie określanej jako temperatura wrzenia. Wartość odczytywania z termometru (a także szybkość i kierunek jej zmian) stanowi raczej orientacyjny wskaźnik jednorodności aktualnie destylującej frakcji, wpływający na decyzje prowadzącego destylację co do zmiany odbieralnika lub zakończenia procesu. Jedynie podczas destylacji niemal czystych cieczy wartość ta jest stała i może być uważana za temperaturę wrzenia. Dlatego podajemy zawsze zakres temperatur destylacji danej frakcji. Im węższy jest ten zakres w stosunku do objętości frakcji, tym bardziej jednorodna jest zebrana ciecz.
Dobrym sposobem oceny czystości frakcji jest pomiar jej współczynnika załamania światła i porównanie otrzymanej wartości z podaną w literaturze. Niestety nie jest to jednak metoda doskonała.
Przeprowadzenie destylacji stwarza pewne problemy związane Z bezpiecznym wykonaniem tej operacji. Należy zwrócić szczególną uwagę na palność większości cieczy organicznych oraz na ich toksyczność. Z tego względu kolbę destylacyjną nie należy ogrzewać bezpośrednio nad palnikiem. Takie ogrzewanie powoduje nierównomierne ogrzanie kolby i może doprowadzić do jej pęknięcia, a w konsekwencji — do pożaru. Chłodzenie par powinno być dostatecznie efektywne (chłodnica dobrana do temperatury wrzenia destylowanej cieczy). Użycie kamyczka wrzennego (kawałek porowatej porcelany lub fajansu) skutecznie zabezpiecza przed przegrzewaniem cieczy.
Wiele substancji organicznych nie może być poddanych destylacji w normalnych warunkach ze względu na rozkład (nawet wybuchowy) w temperaturze wrzenia lub niższej. Takie ciecze destyluje się wykorzystując technikę destylacji pod zmniejszonym ciśnieniem.
Ćwiczenie składa się z dwóch części. W pierwszej celem ćwiczenia jest oczyszczenie otrzymanej próbki cieczy przez destylację.
Wykonując ćwiczenie należy zwrócić szczególną uwagę na opanowanie umiejętności prawidłowego montażu aparatury, podziału destylatu na frakcje oraz pomiaru współczynnika załamania światła.
Część pierwsza: Destylacja prosta
Substraty: zanieczyszczony etanol 25 cm3
Opis wykonania ćwiczenia:
Rys.4. Zestaw do destylacji prostej
Aparaturę do destylacji zwykłej przedstawia rys.4. Przed montażem wlewamy do kolby etanol przeznaczony do destylacji. Wrzucamy kamyczek wrzenny, montujemy aparaturę, uruchamiamy przepływ wody przez chłodnicę i rozpoczynamy ogrzewanie, obserwując wskazania termometru. Pierwsza porcja destylatu, przechodząca przez chłodnicę przy ciągle rosnących wskazaniach termometru stanowi przedgon i zbieramy ją w całości do pierwszego odbieralnika. Po ustaleniu się temperatury (destylacja zazwyczaj wtedy ulega znacznemu przyspieszeniu) zmieniamy odbieralnik i zbieramy frakcję główną. Destylację kontynuujemy do momentu, gdy temperatura wskazywana przez termometr zacznie opadać (czasami oznaką końca frakcji głównej jest dalszy wzrost temperatury). Zmieniamy wówczas odbieralnik i zbieramy pogon.
Mierzymy objętość frakcji głównej. Notujemy początkową i końcową temperaturę wrzenia tej frakcji oraz mierzymy jej współczynnik załamania światła, porównując go z wartością podaną w literaturze.
Część druga: Destylacja frakcyjna
Substraty: mieszanina heksan - toluen 20 cm3
Opis wykonania ćwiczenia:
Rys.5. Zestaw do destylacji frakcyjnej (rektyfikacji)
Aparaturę przedstawia rys.5. Przed montażem wlewamy do kolby mieszaninę przeznaczoną do rozdestylowania. Wrzucamy kamyczek wrzenny, montujemy aparaturę, uruchamiamy przepływ wody przez chłodnicę i rozpoczynamy ogrzewanie. Destylację prowadzimy powoli, podnosząc temperaturę płaszcza grzejnego i obserwując wskazania termometru.
Zbieramy pięć frakcji, zmieniając kalibrowane odbieralniki w zależności od zmian temperatury wskazywanej przez termometr. Dla każdej frakcji notujemy początkową i końcową temperaturę wrzenia, oznaczamy współczynnik załamania światła, oraz mierzymy jej objętość. Następnie określamy skład frakcji w procentach objętościowych, korzystając z prostej wzorcowej, wyznaczonej przez pomiary współczynnika załamania światła czystego toluenu i heksanu.
Opracowanie ćwiczenia.
W części pierwszej powyższego ćwiczenia dokonałem oczyszczenia skażonego etanolu. Otrzymany destylat odbierany był w przedziale temperatur 77 - 800C. Temperatura wrzenia czystego etanolu wynosi 78,30C, zatem zawiera się w wyżej wymienionym zakresie. Objętość czystego etanolu po destylacji wynosiła 22,4 cm3. Jego czystość sprawdziłem na podstawie współczynnika załamania światła, który wynosił 1,3587. Wartość tablicowa podaje 1,3594. Wydajność przeprowadzonej destylacji wynosi:
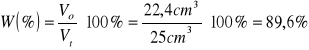
W drugiej części przeprowadziłem destylację frakcyjną w celu rozdzielenia mieszaniny toluenu z heksanem. Zebrane przeze mnie frakcje charakteryzowały się następującymi właściwościami:
tw = 69 - 740C; nD = 1,3785; V = 4,8 cm3
tw = 74 - 800C; nD = 1,4073; V = 5,1 cm3
tw = 80 - 870C; nD = 1,4528; V = 4,7 cm3
tw = 87 - 960C; nD = 1,4731; V = 4,9 cm3
tw = 96 - 1070C; nD = 1,4913; V = 5,0 cm3
Znając współczynniki załamania światła poszczególnych frakcji można na podstawie prostej wzorcowej obliczyć zawartość procentową składników we wszystkich frakcjach:
Powyższa prosta wzorcowa opisana jest następującym równaniem:
y = 0,0012x + 1,3726
Znając powyższe równanie oraz współczynniki załamania wszystkich frakcji można policzyć zawartość procentową każdej z nich:
toluen: 4,92%; heksan: 95,08%
toluen: 28,92%; heksan: 71,08%
toluen: 66,83%; heksan: 33,17%
toluen: 83,75%; heksan: 16,25%
toluen: 98,92%; heksan: 1,08%
W ten sposób na drodze destylacji frakcyjnej otrzymałem pięć próbek o różnej zawartości rozdzielanych składników. Pierwszą z nich jest niemalże czysty heksan, kolejne są mieszaninami o odpowiednim składzie, natomiast ostatnia to prawie czysty toluen.
Wnioski.
Zastosowane techniki laboratoryjne oczyszczania i rozdzielania cieczy są jednymi z najbardziej wydajnych i skutecznych spośród dostępnych metod. Jest ona prosta szybka ale niestety na większą skalę dosyć kosztowna.
DIBENZYLIDENOACETON
Synteza dibenzylidenoacetonu jest przykładem reakcji Claisena-Schmidta, która polega na kondensacji aldolowej aldehydu aromatycznego z alifatycznym aldehydem lub ketonem i następnym samorzutnym odwodnieniu pośredniego aldolu. Produktem jest , - nienasycony związek karbonylowy. W przypadku reakcji benzaldehydu z acetonem powstaje najpierw benzylidenoaceton, który może reagować z drugą cząsteczką benzaldehydu dając dibenzylidenoaceton.
Zachodzące reakcje:
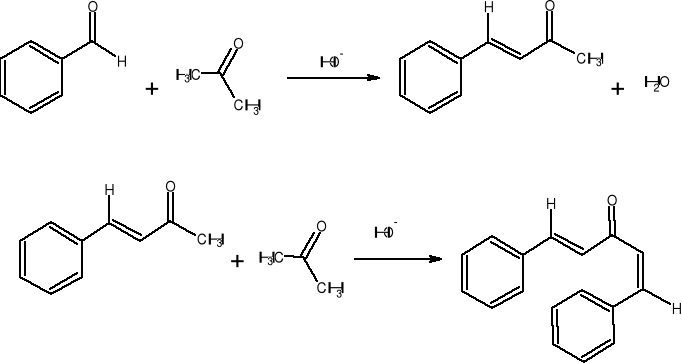
Opis wykonania ćwiczenia.
W kolbie okrągłodennej o pojemności 250 cm3 umieszcza się zimny roztwór 7 g NaOH w 70 cm3 wody destylowanej i 55 cm3 etanolu i kolbę umieszcza w łaźni z zimną wodą. Osobno w kolbce Erlenmeyera przygotowuje się mieszaninę 2,6 cm3 acetonu i 7,1 cm3 aldehydu benzoesowego destylowanego. Do roztworu NaOH w kolbie dodaje się około połowę mieszaniny acetonu i benzaldehydu mieszając zawartość kolby ruchem kolistym. Po 15 minutach dodaje się pozostałą część mieszaniny i po wymieszaniu pozostawia na pół godziny w lodówce. Wydzielony osad odsącza się pod zmniejszonym ciśnieniem, przemywa wodą do niemal obojętnego odczynu i dokładnie odciska. Surowy produkt poddajemy krystalizacji z etanolu.
Opracowanie ćwiczenia.
Otrzymany produkt stanowią żółte kryształy. Masa otrzymanej substancji wynosiła 4,86g. Aby obliczyć wydajność reakcji należy policzyć liczbę moli każdego z substratów porównać i do obliczeń uwzględnić ten, którego było w niedomiarze, ponieważ cała jego ilość ulegnie reakcji. Do doświadczenia użyto następujących ilości substratów:
aldehyd benzoesowy: V = 7,1 cm3 (7,4053g, 0,0699 mola)
aceton: V = 2,6 cm3 (2,0394g, 0,0352 mola)
Ponieważ w reakcji uczestniczą jeden mol acetonu i dwa mole aldehydu, zatem w niedomiarze był aldehyd. Na tej podstawie można policzyć, jaką ilość dibenzylidenoacetonu otrzymalibyśmy z 7,4053 grama aldehydu benzoesowego. Zatem teoretyczna masa produktu wynosiłaby 8,17 grama. Natomiast masa otrzymanego produktu wynosiła 4,86 grama (M = 234 g/mol). Zatem wydajność reakcji:
![]()
Czystość otrzymanego produktu sprawdziłem na podstawie temperatury topnienia, która obejmowała zakres 104 - 1090C. Wartość tablicowa podaje 1120C.
CYKLOHEKSANON
W praktyce laboratoryjnej do utleniania alkoholi stosuje się najczęściej: związki chromu (VI), dwutlenek manganu lub nadmanganian potasowy. Utlenianie alkoholi pierwszorzędowych prowadzi do aldehydów, drugorzędowych — do ketonów, natomiast alkohole trzeciorzędowe nie ulegają utlenieniu w tych warunkach.
Utlenianie cykloheksanolu do cykloheksanonu za pomocą dwuchromianu sodu w środowisku kwaśnym zachodzi szybko i z dobrą wydajnością.
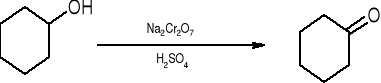
Istotnym warunkiem wydajnego przeprowadzenia tej reakcji jest przestrzeganie wskazanego zakresu temperatury.
Opis wykonania ćwiczenia.
W kolbie stożkowej o pojemności 250 cm3 umieszcza się 60g drobno potłuczonego lodu. Mieszając dodaje się ostrożnie 20 cm3 stężonego kwasu siarkowego następnie 20,6 cm3 cykloheksanolu i chłodzi się w łaźni lodowej. Oddzielnie, w kolbie stożkowej o pojemności 100 cm3 przygotowuje się roztwór 25 g dwuchromianu sodu w 15 cm3 wody i przenosi do biurety. Mierzy się temperaturę roztworu cykloheksanolu i kiedy jest niższa od 15°C dodaje się z biurety 1 cm3 roztworu dwuchromianu sodu. Obserwuje się temperaturę i barwę mieszaniny. Dalsze porcje roztworu utleniacza dodaje się z taką szybkością, aby temperatura nie przekroczyła 450C.
W trakcie wkraplania należy delikatnie wstrząsnąć kolbą, aby zapobiec miejscowemu przegrzaniu roztworu. Dodawanie odczynnika trwa zwykle około 20 minut. Mieszaninę poreakcyjną przenosi się do kolby destylacyjnej o pojemności 250 cm3 i po zmontowaniu zestawu do destylacji destyluje możliwie szybko, ogrzewając kolbę płaszczem grzejnym. Gdy temperatura osiągnie 100°C zbiera się jeszcze 10 — 15 cm3 destylatu i proces przerywa się. Destylat przenosi się do rozdzielacza, dodaje 15g chlorku sodu i wytrząsa do całkowitego rozpuszczenia. Warstwę cykloheksanonu oddziela się i suszy bezwodnym siarczanem magnezu w ciągu 15 — 20 minut. Osuszony cykloheksanon sączy się przez karbowany sączek do kolby destylacyjnej o pojemności 50 cm3 i destyluje, zbierając frakcję wrzącą w zakresie 146 — 152°C. Otrzymuje się około 12g produktu.
Opracowanie ćwiczenia.
Otrzymana ciecz - cykloheksanon - poddana została destylacji z parą wodną ze względu na wysoką temperaturę wrzenia. Objętość uzyskanego produktu wynosiła 5,1 cm3. Do reakcji użyłem 20,3 cm3, cykloheksanolu, czyli 19,65 grama. Otrzymana masa cykloheksanonu wynosiła 4,52 grama. Zatem wydajność reakcji:
![]()
Czystość uzyskanego cykloheksanonu sprawdziłem na podstawie współczynnika załamania światła. Dla badanej substancji wynosił on 1,4489, natomiast literatura podaje wartość 1,4513.
Wnioski.
Niska wydajność reakcji spowodowana jest prawdopodobnie dużymi stratami produktu w czasie destylacji, natomiast zaniżony współczynnik załamania światła świadczy o niedostatecznym osuszeniu cykloheksanonu po destylacji.
ROZDZIAŁ MIESZANIN PRZEZ EKSTRAKCJĘ. DESTYLACJA POD ZMNIEJSZONYM CIŚNIENIEM.
Ekstrakcja jest techniką laboratoryjną, której celem jest przeniesienie (wyciągnięcie) substancji z jej roztworu w jednym rozpuszczalniku do innego nie mieszającego się z pierwszym. Ideą jest bądź zmiana rozpuszczalnika na dogodniejszy (np. łatwiejszy do odparowania — często stosowana ekstrakcja roztworów wodnych rozpuszczalnikami organicznymi), bądź selektywne usunięcie jednego ze składników mieszaniny z jej roztworu, a zatem rozdział tej mieszaniny Istnieje wiele sposobów przeprowadzenia ekstrakcji, różniących się stosowaną aparaturą. Wariant wykorzystywany w ćwiczeniu (najczęściej stosowany w laboratorium) polega na wytrząsaniu w rozdzielaczu roztworu z kolejnymi porcjami rozpuszczalnika ekstrahującego i następnym rozdzieleniu obu raz. Jak wynika z teorii korzystne jest kilkakrotne powtórzenie tej operacji z kolejnymi porcjami czystego rozpuszczalnika ekstrahującego. Wytrząsanie ma na celu zapewnienie możliwie dużej powierzchni granicy raz, co przyspiesza osiąganie równowagi termodynamicznej określonej przez współczynnik podziału substancji pomiędzy dwa rozpuszczalniki.
Przeprowadzenie ekstrakcji wiąże się z pewnymi zagadnieniami dotyczącymi bezpieczeństwa pracy. Podczas wytrząsania mieszaniny w szczelnie zamkniętym rozdzielaczu, na skutek wielu zjawisk, ciśnienie w jego wnętrzu zmienia się (najczęściej wzrasta). Nieostrożne otwarcie rozdzielacza może spowodować wyrzucenie jego zawartości, co grozi poważnymi konsekwencjami od pożaru w przypadku ekstrakcji rozpuszczalnikami palnymi do poparzeń stosowanym w ćwiczeniu roztworem wodorotlenku potasowego, a zawsze powoduje utratę części mieszaniny rozdzielanej. Dlatego ciśnienie w rozdzielaczu należy możliwie często wyrównywać przez otwarcie kranu (rozdzielacz trzymany ukośnie, wylot skierowany w pustą część pomieszczenia). Podczas rozdzielania faz, zawsze pod wylotem rozdzielacza powinno znajdować się naczynie o odpowiedniej pojemności.
Przeprowadzane w ćwiczeniu zobojętnianie wykonujemy z zachowaniem koniecznych środków ostrożności.
Ćwiczenie składa się z dwóch części. Celem części pierwszej jest rozdzielenie mieszaniny kwasu benzoesowego i benzoesanu etylu. W tej części ćwiczenia wykorzystuje się różnicę współczynników podziału rozdzielanych związków pomiędzy chlorek metylenu i wodny roztwór wodorotlenku potasowego. Należy zwrócić szczególną uwagę na rozróżnienie fazy organicznej i wodnej. W drugiej części ćwiczenia, polegającej na oczyszczeniu obu składników mieszaniny wyjściowej, stosuje się krystalizację z wody do oczyszczenia kwasu benzoesowego oraz destylację pod zmniejszonym ciśnieniem do oczyszczenia benzoesanu etylu.
Stosowana w tej części ćwiczenia technika destylacji pod obniżonym ciśnieniem nie odbiega zbytnio od destylacji pod ciśnieniem atmosferycznym. Jedyna istotna różnica polega na stosowaniu dokładnie uszczelnionej aparatury. W czasie destylacji w aparaturze jest utrzymywane obniżone ciśnienie, którego wartość zależy od rodzaju stosowanej pompy próżniowej (w ćwiczeniu stosuje się pompkę wodną pozwalającą obniżyć ciśnienie do około 15 mmHg). Kamyczek wrzenny jest przy tej technice destylacji zastąpiony kapilarą sięgającą do dna kolby. Zastosowanie destylacji pod zmniejszonym ciśnieniem pozwala na obniżenie temperatury wrzenia oczyszczanego benzoesanu etylu.
Znacznie ostrzejsze niż podczas destylacji pod ciśnieniem atmosferycznym, są wymagania bezpieczeństwa pracy. Groźba implozji powoduje, że wszystkie elementy aparatury muszą być uprzednio sprawdzone pod względem wytrzymałości na ciśnienie. Niedopuszczalne jest stosowanie kolb destylacyjnych i odbieralników innych niż okrągłodenne. Podczas pracy obowiązuje noszenie okularów ochronnych. Wszelkie zauważone usterki szkła elementów aparatury należy natychmiast zgłosić prowadzącemu. Należy przestrzegać zasad pracy z pompką wodną.
Opis wykonania ćwiczenia.
Rozdzielaną mieszaninę rozcieńczamy za pomocą 50 cm3 chlorku metylenu i umieszczamy w rozdzielaczu gruszkowym o pojemności około 250 cm3. 250 cm Sporządzamy 5% - owy roztwór wodorotlenku potasowego. Dodajemy 15 cm3 tego roztworu do rozdzielacza i dokładnie wytrząsamy. Po rozdzieleniu warstw, dolną warstwę spuszczamy do kolby A, górną do kolby B. Zawartość kolby A ekstrahujemy jeszcze dwukrotnie roztworem KOH, łącząc warstwy górne w kolbie B. Połączone frakcje B zakwaszamy mieszając rozcieńczonym kwasem solnym (1:1) do pH = 1. Odsączamy oddzielony osad na lejku Buchnera lub Schotta, przemywamy wodą, odciskamy i przekrystalizowujemy z wody. Po odsączeniu, przemyciu i wysuszeniu kryształów na lejku Buchnera lub Schotta ważymy substancję i oznaczamy temperaturę topnienia.
Pozostałość po ekstrakcji (kolba A) przemywamy w rozdzielaczu wodą, dokładnie oddzielamy i pozostawiamy do suszenia nad bezwodnym siarczanem sodowym.
Destylacja pod zmniejszonym ciśnieniem.
Montujemy aparaturę według rys.6. (wszystkie połączenia szlifowi muszą być nasmarowane smarem próżniowym lub wazeliną).
Rys.6. Zestaw do destylacji pod zmniejszonym ciśnieniem
Zawartość kolby A po odsączeniu środka suszącego odparowujemy na wyparce w celu usunięcia chlorku metylenu. Pozostałość wlewamy do kolby destylacyjnej. Zestawiamy aparaturę, uruchamiamy pompkę wodną i oczekujemy na ustalenie się ciśnienia w aparaturze, kontrolując pracę kapilary. Gdy ciśnienie ustali się, rozpoczynamy ogrzewanie. Temperaturę podnosimy powoli, obserwując wskazania termometru. Pierwsza porcja destylatu, przechodząca przez chłodnicę przy ciągle rosnących wskazaniach termometru stanowi przedgon. Po ustaleniu się temperatury i ciśnienia (destylacja ulega przy tym przyśpi - zmieniamy odbieralnik i zbieramy frakcję główną. Destylację kontynuujemy do momentu, gdy temperatura wskazywana przez termometr zacznie spadać (czasami oznaką końca frakcji głównej jest dalszy wzrost temperatury).
Po zakończeniu destylacji odcinamy aparaturę od źródła próżni, zamykając zawór łączący aparaturę z pompką wodną. Czekamy na lekkie ostygnięcie kolby destylacyjnej i wyrównujemy ciśnienie przez wyjęcie kapilary lub termometru (uwaga: należy to robić powoli i ostrożnie, podtrzymując jednocześnie odbieralnik).
Mierzymy objętość frakcji głównej oraz oznaczamy jej współczynnik załamania światła.
Opracowanie ćwiczenia.
Otrzymany przez rozdzielenie kwas benzoesowy ma postać białych kryształów, których temperatura topnienia obejmuje zakres 117 - 1210C. Wartość tablicowa wynosi 1220C. Masa uzyskanego kwasu benzoesowego wynosi 1,35g. Masę, jaką otrzymałem do ćwiczenia wynosiła 3,2g. Zatem wydajność ekstrakcji kwasu benzoesowego wynosi:
![]()
Po destylacji pod zmniejszonym ciśnieniem uzyskałem benzoesan etylu, którego objętość wynosiła 12,7 cm3.
Objętość uzyskanego przez destylację pod zmniejszonym ciśnieniem benzoesanu etylu wynosiła 16,8 cm3. Zatem jego masa wynosiła 17,51 grama. Do wykonania ćwiczenia otrzymałem 20 cm3 benzoesanu etylu czyli 20,84 grama. Zatem wydajność przeprowadzonej destylacji wynosi:
![]()
CHROMATOGRAFIA KOLUMNOWA I CIENKOWARSTWOWA
Metoda rozdzielania składników mieszaniny, w której wykorzystuje się różnice we współczynnikach podziału tych składników między dwie fazy, nosi nazwę chromatografii. Wspólną cechą metod chromatograficznych jest obecność fazy ruchomej (roztwór ciekły lub mieszanina gazów) zawierającej rozdzielaną mieszaninę, która przepływa wzdłuż fazy nieruchomej (ciało stałe lub ciecz osadzona na powierzchni i w porach ciała stałego). Na granicy faz zachodzą procesy fizykochemiczne (adsorpcja chemiczna lub fizyczna, podział lub wymiana jonowa), które w różnym stopniu opóźniają szybkość przesuwania się poszczególnych składników mieszaniny, co powoduje ich rozdział.
W zależności od warunków i metodyki rozdziału wyróżnia się: chromatografię kolumnową, cienkowarstwową, bibułową, wstępującą, zstępującą, itd.
Chromatografia kolumnowa.
W chromatografii kolumnowej podział składników mieszaniny pomiędzy stały adsorbent i rozpuszczalnik pozwala na jej rozdzielenie i wyodrębnienie poszczególnych składników. Adsorbentami najczęściej używanymi, są tlenek glinowy (kwaśny, zasadowy lub obojętny) i żel krzemionkowy o odpowiedniej aktywności, które umieszczone są w pionowej, szklanej rurze. Kolumnę (rurę) dobiera się tak, aby była ona napełniona mniej więcej do połowy wysokości. Im większy jest stosunek wysokości kolumny do jej średnicy, tym lepszy osiąga się rozdział. Zwykle jednak stosunek ten wynosi od 20:1 do 10:1, ponieważ zbyt wysoka warstwa adsorbentu stawiałaby przepływającemu roztworowi zbyt duży opór hydrauliczny. Na ogół do chromatografii kolumnowej używa się od 20 do 50 gramów adsorbenta na każdy gram rozdzielanej mieszaniny. Zazwyczaj używa się kolumn zaopatrzonych w kran, który musi być szczelny (kranu nie należy smarować, aby uniknąć zanieczyszczaniu rozdzielanych substancji smarem).
Adsorbent w kolumnie umieszcza się na tamponie z waty, zwykle pokrytym warstwę czystego piasku. Kolumna musi być tak napełniona, aby nie pozostały w niej pęcherzyki powietrza.
Nie zaleca się napełniania kolumny suchym adsorbentem, gdyż nie uzyskuje się wtedy równego ułożenia napełnienia i rozdział jest mało precyzyjny.
Prawidłowe napełnienie kolumny można przeprowadzić w następujący sposób: do kolumny nalewa się rozpuszczalnik (około 2/3 jej wysokości) i umieszcza się w jej dolnym końcu watę. Po usunięciu pęcherzyków powietrza wsypuje się cienkim strumieniem adsorbent bezpośrednio do rozpuszczalnika. Inna metoda napełnienia kolumny polega na wlaniu papki adsorbenta z rozpuszczalnikiem. Podczas dodawania adsorbenta reguluje się wypływ rozpuszczalnika u dołu kolumny, tak jednak, aby adsorbent przez cały czas był przykryty warstwą rozpuszczalnika. Zebrany rozpuszczalnik zawraca się na szczyt kolumny. Po osadzeniu się adsorbenta, ostukuje się kawałkiem węża gumowego na końcu bagietki aż adsorbent przestanie osiadać. Przez cały czas należy zwracać uwagę na to, aby warstwa adsorbenta była przykryta warstwą rozpuszczalnika. Górną powierzchnię adsorbenta należy przykryć piaskiem i małym krążkiem bibuły, co zapobiega unoszeniu adsorbenta podczas wprowadzania roztworu na kolumnę.
Możliwość rozdziału na kolumnie sprawdza się uprzednio metodą chromatografij cienkowarstwowej. Podobnie można dobrać odpowiedni adsorbent i rozpuszczalnik lub układ rozpuszczalników.
Ustalono kilka ogólnych reguł, którymi można się kierować przy doborze rozpuszczalnika (eluenta):
1/ Wzrost polarności rozpuszczalnika zwiększa jego zdolność do wymywania (eluowania) substancji z polarnego adsorbenta.
2/ Im bardziej polarne substancje poddaje się rozdziałowi, tym bardziej polarny rozpuszczalni i tym mniej aktywny adsorbent należy stosować
3/ Jeżeli do rozdzielania poszczególnych składników mieszaniny trzeba użyć rozpuszczalników o różnej polarności to dodaje się drugi rozpuszczalnik (bardziej polarny od pierwszego, ponieważ zbyt nagłe podwyższenie polarności mieszaniny może spowodować zlewanie się rozwiniętych uprzednio pasm.
Do przygotowanej kolumny wprowadza się rozwór rozdzielanych substancji w możliwie małej ilości rozpuszczalnika. Wskazane jest użycie tego samego rozpuszczalnika, którym napełniono kolumnę. Jeżeli substancje rozpuszczają się słabo, można wprowadzić je na kolumnę w postaci zawiesiny. Po wlaniu roztworu do kolumny otwiera się lekko kran i czeka aż poziom roztworu obniży się do poziomu adsorbenta. Czynność tę należy wykonać możliwie powoli i dokładnie. Następnie wlewa się do kolumny rozpuszczalnik i rozpoczyna rozwijanie chromatogramu. Szybkość wypływu cieczy z kolumny powinna by początkowo mała (kropla na sekundę), a następnie można ją zwiększyć do kilku kropel na sekundę. W tym czasie uwidaczniają się oddzielne pasma składników. Przemywanie kolumny rozpuszczalnikiem prowadzi się dalej. W ten sposób roztwory poszczególnych substancji spływają do odbieralników.
Jeśli substancje nie są zabarwione, to ich rozdział można śledzić obserwując kolumnę w świetle nadfioletowym, lub (częściej) kontrolując skład frakcji za pomocą chromatografii cienkowarstwowej. Frakcje o identycznym składzie łączy się ze sobą. Frakcje zawierające kilka składników (tzw. międzyfrakcje) poddaje się ponownemu rozdzieleniu (na nowej kolumnie) lub, jeśli jest ich mało i składniki nie są zbyt cenne, odrzuca. Z roztworu substancje wydziela się przez odparowanie rozpuszczalnika na wyparce obrotowej. Pełne oczyszczenie wydzielonych substancji wymaga jeszcze zwykle krystalizacji lub destylacji.
Chromatografia cienkowarstwowa.
Chromatografia cienkowarstwowa (TLC—ang. thin layer chromatography) jest specjalną odmianą chromatografii, w której fazą stałą jest cienka (0,25 - 2,0 mm) warstwa adsorbenta, np. tlenku glinowego lub żelu krzemionkowego, naniesiona na płytkę szklaną lub inny materiał (folia aluminiowa, folia z tworzywa sztucznego). Pozwala ona na rozdział bardzo małych ilości substancji (5-100 g) w celach analitycznych, lub nieco większych (do 2 g) w celach preparatywnych.
Stosuje się ją do celów diagnostycznych (badanie składu mieszanin), do rozdzielania preparatywnego oraz do oznaczeń ilościowych.
W celu identyfikacji substancji przy rozdziale chromatogra!icznym wprowadzono współczynnik RF:
![]()
gdzie ls - odległość przebyta przez substancję; lr - odległość przebyta przez czoło rozpuszczalnika
Jeżeli doświadczenie przeprowadza się w stałych warunkach) to wartość ta w danym układzie chromatograficznym jest dla danej substancji stała i charakterystyczna.
Jako komorę do rozwijania płytek można stosować słoik szklany lub zlewkę przykrytą szkiełkiem zegarkowym.
Roztwory badanych substancji nanosi się na płytkę kapilarami, w odległości 1—2 cm od brzegu lub od siebie, starając się, aby naniesione plamki miały jak najmniejsze średnice. Tak przygotowaną płytkę roztworów naniesionymi plamkami substancji wzorcowych i roztworów badanych wstawia ostrożnie się do komory, na której dnie znajduje się warstewka około 0,5 cm rozpuszczalnika i przykrywa komorę. Gdy rozpuszczalnik osiągnie odpowiednią wysokość (6—10 cm od punktu startu) wyjmuje się płytkę z komory, oznacza czoło rozpuszczalnika i suszy.
W celu wykrycia wszystkich związków zawartych w mieszaninie, rozwiniętą i wysuszoną płytkę poddaje się działaniu odczynnika wywołującego (tj. związku, który reaguje ze wszystkimi możliwymi składnikami mieszaniny, np. pary jodu, stężony kwas siarkowy) lub ogląda w świetle UV. Prawie wszystkie związki organiczne, poza węglowodorami nasyconymi i chlorowcopochodnymi tworzą z parami jodu barwne kompleksy o charakterystycznych barwach. Wywoływanie parami jodu przeprowadza się w szczelnych komorach z kilkoma kryształami jodu wewnątrz. Po wywołaniu płytkę należy opisać I obliczyć wartości RF dla poszczególnych plam.
Celem niniejszego ćwiczenia jest rozdzielenie mieszaniny azobenzenu i o—nitroaniliny przy użyciu chromatografii kolumnowej, kontrolując ten proces za pomocą chromatografii cienkowarstwowej.
Opis wykonania ćwiczenia.
Pod wyciągiem montuje się aparaturę wg rys.7. stosując kolumnę o długości około 80 cm i średnicy 1,5 do 2 cm, zaopatrzoną w kran.
Kolumnę napełnia się mieszaniną heksan(lub eter naftowy)—
—octan etylu 95:5 oraz adsorbentem, którym jest 30 g tlenku glinu, stosując wskazania podane we wstępie. Rozdzielaną mieszaninę rozpuszcza się w probówce w minimalnej ilości benzenu i wprowadza na kolumnę. Rozwija się mieszaniną heksan (eter naftowy)—octan etylu 95:5, zawracając początkowo eluent na szczyt kolumny. Gdy barwny pierścień zbliży się do dołu kolumny należy rozpocząć zbieranie frakcji (kilku) do numerowanych kolb Erlenmeyera. Gdy wyciek z pomarańczowego stanie się bezbarwny zmienia się eluent na mieszaninę heksan(eter naftowy)—octan etylu 70:30 i zbiera się najpierw frakcję pośrednią., a następnie frakcję (kilka) drugiego składnika mieszaniny.
Otrzymane frakcje bada się przy pomocy chromatografii cienkowarstwowej korzystając ze wskazówek podanych we wstępie.
Rys.7. Kolumna chromatograficzna
Opracowanie ćwiczenia.
Po rozdzieleniu i odparowaniu rozpuszczalnika uzyskałem dwie substancje - azobenzen i o-nitroanilinę, których czystość sprawdziłem na podstawie chromatografii cienkowarstwowej i temperatur topnienia. Obie substancje nie wykazały śladów zanieczyszczeń na płytce. Temperatury topnienia mieściły się odpowiednio w zakresach:
- azobenzen: 63 - 680C (wartość tablicowa: 680C),
- o-nitroanilina: 67 - 720C (wartość tablicowa: 71,50C).
Do doświadczenia użyte było po 0,3 grama każdego ze związków. Po rozdzieleniu i zważeniu otrzymałem 0,28g azobenzenu i 0,29 o-nitroaniliny. Zatem odpowiednie wydajności wynoszą:
![]()
![]()
Wnioski.
Jak widać wydajność tego procesu jest bardzo wysoka co czyni tę metodę idealną do rozdziału mieszanin, jeżeli tylko pozwalają na to właściwości składników mieszaniny (różnica polarności). Czystość obu substancji również jest zadowalająca.
CHLOREK TERT - BUTYLU
Chlorek tert—butylu można otrzymać przez podstawienie grupy hydroksylowej chlorem w alkoholu tert—butylowym za pomocą chlorowodoru, chlorku tionylu lub chlorowych związków fosforu (np. PCl5). Do najprostszych z metod należy reakcja podstawienia nukleofilowego z użyciem kwasu solnego. Alkohol tert-butylowy reaguje łatwo z kwasem solnym, podobnie jak i inne alkohole trzeciorzędowe.
Alkohole drugorzędowe reagują z kwasem solnym o wiele trudniej. a najtrudniej alkohole pierwszorzędowe.
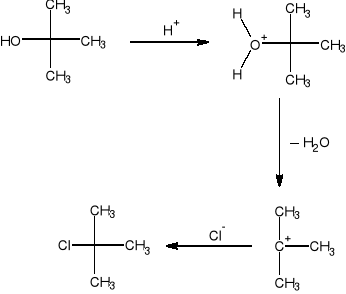
Opis wykonania ćwiczenia.
W rozdzielaczu o pojemności 250 cm3 umieszcza się 32 cm3 alkoholu tert—butylowego i 85 cm3 stężonego kwasu solnego oraz 10 g bezwodnego chlorku wapnia (dodanie bezwodnego CaCl2 zwiększa gęstość warstwy kwasowej i ułatwia rozdzielenie warstw, co polepsza nieco wydajność reakcji) a następnie mieszaninę wytrząsa się co pewien czas w ciągu 20 minut. Po każdym wytrząśnięciu wyjmuje się korek z rozdzielacza dla wyrównania ciśnienia. Mieszaninę pozostawia się na kilka minut do wyraźnego rozdzielenia warstw, po czym dolną kwaśną warstwę spuszcza
Rys.8. Rozdzielacz
się i odrzuca. Warstwę górną chlorku tert—butylu przemywa się 20 cm3 5%-owego roztworu wodorowęglanu sodu, następnie 20 cm3 wody, przenosi do kolby stożkowej o pojemności 100 cm3 i suszy, dodając 5 g bezwodnego siarczanu magnezu (lub 5g bezwodnego chlorku wapnia). Osuszoną ciecz sączy się przez fałdowany sączek umieszczony w lejku stożkowym do kolby okrągłodennej O pojemności 100 cm3 dodaje kamyczki wrzenne i destyluje z użyciem kolumny Vigreaux, zbierając frakcję wrzącą w temperaturze 49 - 510C.
Opracowanie ćwiczenia.
Otrzymany produkt jest bezbarwną cieczą o współczynniku załamania światła równym 1,3826. Wartość tablicowa wynosi 1,3856. Pomiar współczynnika prowadzony był w temperaturze 220C. Objętość uzyskanego chlorku tert - butylu wyniosła 9,7 cm3. Do ćwiczenia zużyte zostało 32 cm3 alkoholu tert - butylowego, czyli 0,3378 mola. Uwzględniając stechiometrię reakcji można stwierdzić, że liczba moli produktu jest taka sama jak substratu. Otrzymana w wyniku doświadczenia masa chlorku tert - butylu wynosi 7,67 grama. Teoretyczna masa jaką powinniśmy otrzymać wynosi 29,23 grama. Zatem wydajność reakcji:
![]()
Wnioski.
Bardzo niska wydajność przeprowadzonej reakcji spowodowana jest najprawdopodobniej dużymi stratami w czasie wytrząsania mieszaniny reakcyjnej (nieszczelny rozdzielacz) oraz zbyt krótkim czasem przeprowadzenia tego procesu. Rozbieżności we współczynnikach załamania światła spowodowane są pozostałą w produkcie wodą oraz różnica temperatury (wartość tablicowa odnosi się do temperatury 200C).
MRÓWCZAN ETYLU
Mrówczan etylu, podobnie jak i inne estry kwasów karboksylowych, najprościej jest syntezować w reakcji estryfikacji, tj. przez ogrzewanie kwasu mrówkowego z alkoholem etylowym:
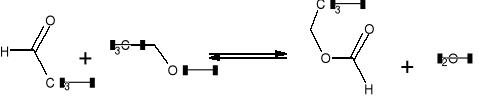
Ponieważ kwas mrówkowy jest kwasem mocnym, reakcja przebiega łatwo bez dodawania kwasu mineralnego. W celu zwiększenia wydajności reakcji powstający ester oddestylowuje się w sposób ciągły z mieszaniny reakcyjnej. Prócz tego, dla ułatwienia przebiegu reakcji, używa się krystalicznego chlorku wapniowego.
Opis wykonania ćwiczenia.
Mieszaninę 15 cm3 85% - owego kwasu mrówkowego, 19 cm3 bezwodnego alkoholu etylowego i 3,7g krystalicznego chlorku wapniowego (CaCl2 H2O) wprowadza się do kolby okrągłodennej o pojemności 100 cm3 zaopatrzonej w kolumnę Vigreux. Na górnej części kolumny destylacyjnej umieszcza się nasadkę destylacyjną z termometrem oraz chłodnicę Liebiga. Jako odbieralnik stosuje się kolbę okrągłodenną lub stożkową o pojemności 100 cm3 osadzoną za pomocą przedłużacza destylacyjnego na końcu chłodnicy. Odbieralnik chłodzi się w łaźni lodowej.
Mieszaninę w kolbie okrągłodennej ogrzewa się powoli na łaźni wodnej, tak, aby ciecz w ciągu 0,5 godz. doprowadzić do łagodnego wrzenia. Podczas destylacji temperatura nie powinna przekroczyć 53—55°C. Destylacja estru trwa około 1 godziny.
W odbieralniku zbiera się około 23 g (25 cm3 surowego produktu, który oczyszcza się przez powtórną destylację. W tym celu należy przygotować zestaw do destylacji zwykłej. Surowy ester umieszcza się w kolbie okrągłodennej o pojemności 100 cm3, dodaje 3,3g bezwodnego węglanu potasowego, wrzuca kilka drobnych kawałków porowatej porcelany i destyluje z łaźni wodnej zbierając frakcję w granicach 53—54°C. Uzyskuje się około 22 g czystego mrówczanu etylu.
Rys.9. Zestaw do destylacji prostej
Opracowanie ćwiczenia.
Po destylacji zanieczyszczonego estru otrzymałem 20,5 cm3 czystego produktu, którego wartość współczynnika załamania światła idealnie zgadzała się z wartością tablicową i wynosiła 1,3597. Do reakcji użyłem 11,8 cm3 kwasu mrówkowego oraz 19,8 cm3 alkoholu etylowego. Porównując liczbę moli obu reagentów i uwzględniając stechiometrię reakcji można stwierdzić, iż kwas mrówkowy znajdował się w niedomiarze. Liczba moli kwasu wynosiła 0,3111 mola, zatem liczba moli otrzymanego estru powinna wynosić tyle samo. Masa otrzymanego estru powinna wynosić 23,05 grama. Natomiast otrzymana masa wynosiła 18,99 grama. Zatem wydajność reakcji:
![]()
Wnioski.
Otrzymany ester charakteryzuje się wysoką czystością. Wydajność reakcji również jest duża, co świadczy o poprawnie przeprowadzonym ćwiczeniu.
N - acetylo - p - toluidyna
Reakcje acylowania amin należą do reakcji substytucji nukleofilowej. Spośród wielu możliwych reakcji acylowania w syntezie organicznej wykorzystuje się najczęściej reakcje acylowania i benzoilowania.
Związkami acylującymi mogą być chlorki kwasowe, bezwodniki i kwasy karboksylowe, a niekiedy i inne pochodne kwasów karboksylowych (np. keteny).
p - Acetylotoluidynę można - otrzymać p - toluidynę bezwodnikiem octowym:
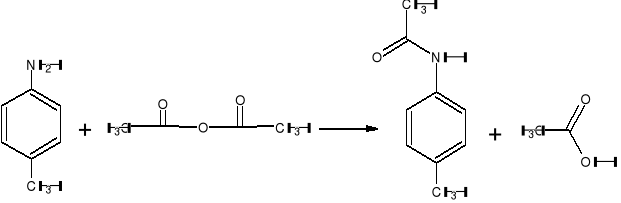
Opis wykonania ćwiczenia.
W kolbie okrągłodennej o pojemności 100 cm3 zaopatrzonej w chłodnicę zwrotną i wkraplacz, rozpuszcza się 5,4g p - toluidyny w 12,5 cm3 toluenu, ogrzewając kolbę. Po rozpuszczeniu aminy odstawia się płaszcz grzejny i do ogrzanego roztworu wkrapla się bezwodnik octowy z taką szybkością, aby temperatura mieszaniny reakcyjnej nie przekraczała 85° — 90° C. Po wkropleniu całej ilości bezwodnika utrzymuje się jeszcze mieszaninę w tej temperaturze przez 15 — 20 minut. Po oziębieniu krystalizuje p-acetylotoluidyna. Krystaliczny produkt odsącza się, przemywa kilkoma cm3 toluenu i po osuszeniu, jeżeli jest zanieczyszczony, przekrystalizowuje się go z etanolu.
Opracowanie ćwiczenia.
Otrzymany produkt posiada postać żółtych kryształów. Temperatura topnienia tego związku obejmuje zakres 146 - 1530C. Wartość tablicowa podaje 1530C. Masa zużytej do reakcji p-toluidyny wynosiła 5,23 g czyli 0,0489 mola. Natomiast masa otrzymanej p-acetylotoluidyny 4,66 grama. Teoretyczna masa jaką powinniśmy otrzymać wynosi 7,28 grama. Zatem wydajność reakcji:
![]()
Próba z roztworem wodorotlenku miała wynik negatywny, ponieważ p-acetylotoluidyna należy do grupy amidów i posiada charakter zasadowy.
Próba z roztworem kwasu ukazała, iż p-acetylotoluidyna ma bardzo słabe właściwości zasadowe, gdyż tylko w niewielkim stopniu uległa rozpuszczeniu.
Wnioski.
Otrzymany produkt wykazuje się dostateczną czystością, o czym świadczy niewielka rozbieżność temperatury topnienia. Obniżona wydajność reakcji spowodowana jest prawdopodobnie stratami podczas krystalizacji oraz całego procesu doświadczalnego.
p - nitroacetanilid
Acetanilid pod wpływem mieszaniny stężonego kwasu azotowego i stężonego kwasu siarkowego ulega reakcji nitrowania, polegającej na podstawieniu atomu wodoru w pierścieniu grupą nitrową - NO2-. Reakcja ta jest reakcją podstawienia elektrofilowego, a czynnikiem atakującym jest kation nitroniowy powstający w reakcji:
![]()
![]()
![]()
Głównym produktem reakcji jest p-nitroacetanilid:
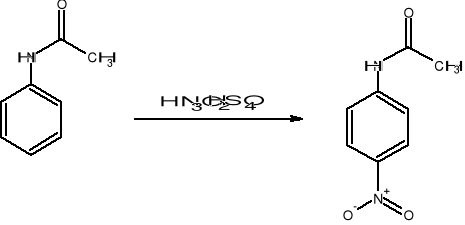
Opis wykonania ćwiczenia.
W zlewce o pojemności 100 cm3 umieszcza się 3,35 g dobrze sproszkowanego w moździerzu, suchego acetanilidu oraz 3,4 cm3 lodowatego kwasu octowego i energicznie mieszając, wprowadza 6,8 cm3 stęż. kwasu siarkowego. Mieszanina rozgrzewa się samorzutnie i powstaje przeźroczysty roztwór. Zlewkę umieszcza się w mieszaninie chłodzącej lodu z solą, a zawartość miesza mechanicznie. Nad zlewką umocowuje się wkraplacz zawierający oziębioną mieszaninę 1,5 cm3 stęż. kwasu azotowego i 2 cm3 stęż. kwasu siarkowego. Gdy temperatura roztworu spadnie do 0 — 2°C, zaczyna się stopniowo wprowadzać mieszaninę kwasów, przy czym temperatura nie powinna przekroczyć 10°C. Po dodaniu całej ilości mieszaniny nitrującej zlewkę wyjmuje się z mieszaniny oziębiającej i pozostawia na 0,5 godz. w temp. pokojowej. Następnie zawartość zlewki wylewa się do 30 g pokruszonego lodu (lub 60 cm3 zimnej wody), podczas czego wydziela się surowy p—nitroacetanilid. Po 15 min. odsącza się go na lejku Buchnera pod zmniejszonym ciśnieniem, przemywa starannie zimną wodą aż do całkowitego usunięcia kwasów (sprawdzić odczyn wody z przemycia) i dobrze odciska. Otrzymany jasnożółty produkt krystalizuje się z około 100cm etanolu (niewielką próbkę surowego produktu oraz przesączu po krystalizacji zachować do badań chromatograficznych), po ochłodżeniu sączy pod zmniejszonym ciśnieniem, przemywa niewielką ilością zimnego alkoholu i suszy w temp. 60 — 80°C. Wydajność około 2,5 g.
Opracowanie ćwiczenia.
Otrzymany produkt posiada postać bezbarwnych kryształów. Zmierzona temperatura topnienia obejmuje zakres 211 - 2160C. Wartość tablicowa: 215 - 2160C. Masa uzyskanego produktu wynosi 3,64 grama czyli 0,020204 mola. Do reakcji zużyłem 3,32 grama czyli 0,024635 mola acetanilidu. Stechiometria reakcji wskazuje na to, że z 0,024635 mola acetanilidu powinienem otrzymać tyle samo moli p - nitroacetanilidu. Zatem teoretyczna masa p - nitroacetanilidu wynosi 4,43 grama. Wydajność przeprowadzonej reakcji wynosi:
![]()
Czystość otrzymanego p - nitroacetanilidu sprawdziłem za pomocą chromatografii cienkowarstwowej (TLC). Współczynnik RF wynosi:
RF = ![]()
Wnioski.
Jako, że na płytce nie zaobserwowałem innych niż pochodzącej od produktu plamek mogę stwierdzić, iż otrzymany p - nitroacetanilid jest dostatecznie czysty, o czym świadczy również porównywalna z wartością tablicową temperatura topnienia.
1,4 - di - tert - butylobenzen
Synteza 1,4 - di - tert - butylobenzenu, jest przykładem reakcji Friedela - Craftsa, polegającej na alkilowaniu lub acylowaniu związku aromatycznego halogenkiem alkilu lub acylu w obecności AlCl3. Mechanizm reakcji polega na elektrofilowym ataku karbokationu (w tym przypadku tert—butylowego), wytworzonego przez reakcję RX z AlCl3. Pierwotnie tworzy się produkt mono - alkilowy, który przy nadmiarze halogenku (chlorku tert - butylu) ulega dalszemu alkilowaniu, dając produkt dwupodstawiony (zwykle mieszaninę izomerów para i orto). Zachodzące reakcje:
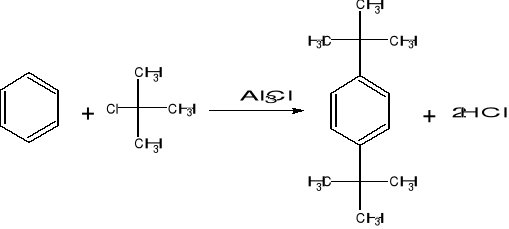
Do syntezy należy stosować wyłącznie bezwodny chlorek glinu w postaci jasno - kremowego lub żółtego proszku, który był przechowywany w szczelnym naczyniu (jest silnie higroskopijny). Jeśli chlorek glinu jest w formie grudek, należy rozgnieść je w suchym moździerzu. Aparatura i substraty użyte do syntezy muszą być bezwzględnie suche. Ze względu na wydzielający się chlorowodór oraz wysoką toksyczność benzenu syntezę należy prowadzić pod sprawnie działającym wyciągiem.
Opis wykonania ćwiczenia.
W suchej kolbie okrągłodennej o pojemności 100 cm3 umieszcza się 11,5 cm3 chlorku tert — butylu oraz 4,4 cm3 benzenu. Kolbę zabezpiecza się rurką z bezwodnym CaCl2. Osobno w małej kolbce Erlemayera odważa się 0,5 g bezwodnego AlCl3 (chronić przed wilgocią!). Do mieszaniny w kolbie wsypuje się łopatką metalową około 1/8 ilości odważonego AlCl3 (szczyptę). Po krótkim czasie rozpoczyna się reakcja, której towarzyszy wydzielanie się gazowego HCl. Od czasu do czasu miesza się zawartość kolby ruchem kolistym. W miarę jak reakcja ulega uspokojeniu dodaje się łopatką dalsze porcje AlCl3. Dodawanie chlorku glinu trwa około 20 minut. Gdy mieszanina zakrzepnie od wydzielonego produktu, przerywa się dodawanie chlorku glinu (jeśli mieszanina nie zakrzepnie po dodaniu całego AlCl3 należy nieco zwiększyć jego ilość). Do zakrzepniętej masy dodaje się 25 cm3 eteru etylowego, a następnie ostrożnie wkrapla 10 cm3 wody mieszając lekko zawartość kolby ruchem kolistym. Roztwór początkowo mętnieje, a następnie staje się klarowny. Mieszaninę przenosi się do rozdzielacza, warstwę wodną odrzuca a eterową przemywa wodą (10 cm3). Po dokładnym oddzieleniu wody roztwór eterowy suszy się przez godzinę nad bezwodnym Na2SO4 lub MgSO4 (około 1 g),mieszając od czasu do czasu .Następnie sączy się do małej kolby okrągłodennej przemywając środek suszący eterem (5 — 10 cm3 Otrzymany roztwór produktu zatęża się z łaźni wodnej zbierając destylat w cylindrze miarowym. Po oddestylowaniu ok. 15 cm3 eteru przerywa się destylację i zawartość kolby ochładza w lodówce (zamrażalniku). W razie potrzeby krystalizację produktu zainicjować można pocierając bagietką o ścianki naczynia. Produkt odsącza się na lejku Schotta dokładnie odciska i pozostawia na lejku, przepuszczając powietrze, aż do całkowitego wyschnięcia. Otrzymuje się około 7 g produktu.
Opracowanie ćwiczenia.
Syntetyzowany produkt otrzymałem w postaci drobnych bezbarwnych kryształów. Temperatura topnienia obejmowała zakres 76 - 790C. Wartość tablicowa: 780C. Masa uzyskanego produktu wynosi 6,85 grama, czyli 0,0359920 mola. Do reakcji zużyłem 4,4 cm3 benzenu (3,84 grama - 0,0492162 mola), oraz 11,4 cm3 chlorku tert - butylu (9,60 grama - 0,1036923 mola). Zatem w niedomiarze znajdował się benzen i na tej podstawie policzona zostanie wydajność. Teoretyczna masa jaką powinienem otrzymać wynosi 9,37 grama. Wydajność przeprowadzonej reakcji wynosi:
![]()
Wnioski.
Otrzymany produkt wykazuje się dostateczną czystością, o czym świadczy niewielka rozbieżność temperatury topnienia.
DANIEL PRASAŁ
5
toluen
heksan
0 %
0 %
50 %
50 %
100 %
100 %
![]()
1
2
3
4
5
M = 106,12 g/mol
M = 234,29 g/mol
M = 94,11 g/mol
M = 93,10 g/mol
M = 74,08 g/mol
M = 62,07 g/mol
M = 46,07 g/mol
M = 107,15 g/mol
M = 102,09 g/mol
M = 149,19 g/mol
M = 135,16 g/mol
M = 180,16 g/mol
M = 92,57 g/mol
M = 190,32 g/mol
M = 78,11 g/mol
Wyszukiwarka
Podobne podstrony:
Lista 11, Polibuda, Podstawy Chemii Organicznej, Chemia Organiczna Laborki, Listy zadań
Synteza octanu n-butylu, Biotechnologia PWR, Semestr 3, Podstawy chemii organicznej - Laboratorium (
Zajecia 4, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Zajecia 3, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Instrukcja NMR, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organic
Lista 5, Polibuda, Podstawy Chemii Organicznej, Chemia Organiczna Laborki, Listy zadań
więcej podobnych podstron