1.5.2.2. Opium
Opium jest wysuszonym na powietrzu mlecznym sokiem niedojrzałych torebek nasiennych (makówek) maku lekarskiego (Papaver somniferum). Zawiera ponad 20 alkaloidów, których proporcje mogą być bardzo zmienne. Głównym alkaloidem jest morfina; z pozostałych ważniejszych alkaloidów towarzyszących należy wymienić: noskapinę (dawniej nazywaną narkotyną), kodeinę, papawerynę, tebainę i narceinę.
Opium bywa jeszcze bardzo rzadko stosowane pod postacią nalewki opiumowej w celu uspokojenia motoryki jelitowej w biegunkach. Ze względu na zawartość innych alkaloidów, zwłaszcza papaweryny, podawanie opium prowadzi do zaparcia atonicznego (w przeciwieństwie do zaparcia spastycznego - wywoływanego przez morfinę).
1.5.2.3. Silnie działające leki opioidowe stopnia 3. wg WHO
Morfina jest od samego początku standardową substancją opioidową. Może być podana na drodze pozajelitowej (parenteralnie, np. dożylnie, podskórnie, nadtwardówkowo), doustnie pod postacią preparatów retard oraz doodbytniczo.
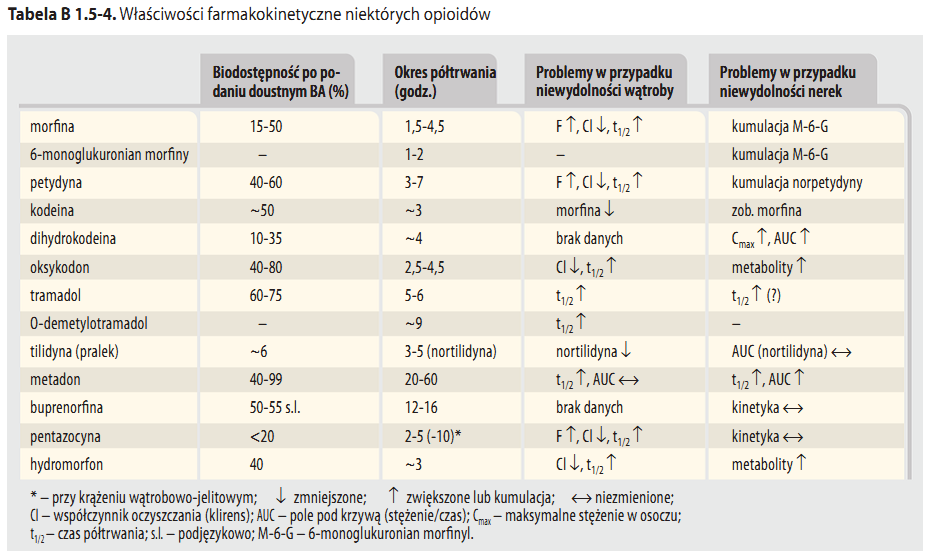
Wchłanianie z przewodu pokarmowego następuje względnie powoli. Ponadto morfina charakteryzuje się silnym efektem pierwszego przejścia. Biodostępność po podaniu doustnym preparatów niemających przedłużonego działania wynosi ok. 25%, tymczasem biodostępność związków typu retard wzrasta do 40%). Jednakże morfina już w trakcie pierwszego pasażu przez wątrobę ulega w znaczącym stopniu przekształceniu w 6-glukuronian morfiny, który wykazuje podobne działanie agonistyczne wobec receptorów μ (zob. ryc. B 1.5-11). Głównym metabolitem jest nieczynny 3-glukuronian morfiny. Wydalanie następuje głównie przez nerki pod postacią glukuronianów. Okres półtrwania wynosi 2-3 godziny. Pojedyncza dawka przy podaniu dożylnym (powoli) wynosi 5-10 mg, przy podaniu podskórnym 10-30 mg, a przy podaniu doustnym (postać retard) 30-60 mg. U chorych z niewydolnością nerek kumulacji ulega czynny 6-glukuronian morfiny. Dlatego też należy zmniejszyć dawkę morfiny lub lepiej zastosować inny lek opioidowy (zob. tab. 1.5-4).
Diamorfina (heroina, diacetylomorfina) ulega szybkiej hydrolizie przez 6-monoacetylomorfinę do morfiny. Heroina i 6-monoacetylomorfina pokonują barierę krew-mózg szybciej niż morfina, gdyż są względnie lipofilne. W następstwie szybkiej penetracji heroiny do ośrodkowego układu nerwowego bardzo łatwo powoduje ona uzależnienie. Ponieważ może być w prosty sposób i z dużą wydajnością wytworzona z morfiny, a ponadto jest od niej wyraźnie silniejsza - stosowana jest nielegalnie jako środek odurzający. W większości krajów wytwarzanie i rozprowadzanie heroiny jest prawnie zakazane (z wyjątkiem wykorzystania w specjalnych programach odwykowych, zob. powyżej). Natomiast w Anglii diamorfina jest nadal stosowana jako lek przeciwbólowy. W ostatnim okresie podejmowane są badania modelowe nad wykorzystaniem heroiny do substytucji u pacjentów z ciężkimi postaciami uzależnienia od opioidów.
Hydromorfon jest 7,5 razy silniejszy niż morfina. Ze względu na silny efekt pierwszego przejścia - biodostępność po podaniu doustnym preparatu typu retard wynosi w przybliżeniu 40%. Główny metabolit 3-glukuronian hydromorfonu nie ma działania przeciwbólowego. Okres półtrwania wynosi w przybliżeniu 3 godziny.
Lewometadon jest prawie 4-krotnie silniejszy od morfiny i dłuższy jest także jego okres działania. W równoważnych dawkach przeciwbólowych (ekwianalgetycznych) jego objawy niepożądane wydają się nieco słabsze. Również objawy abstynencyjne rozwijają się wolniej i są mniej nasilone. Po zażyciu doustnym lek jest dobrze wchłaniany, a jego biodostępność jest niemal całkowita. Okres półtrwania jest indywidualnie zmienny i wynosi między 20 a 60 godzin (w pojedynczych przypadkach nawet do 100 godzin).
Oksykodon działa nieco silniej od morfiny. Biodostępność po podaniu doustnym uzależniona jest od postaci galenowej i jest większa o 40-80% od morfiny. Czas połowiczej eliminacji wynosi 3-56 godzin.
Ostatnio do stosowania dopuszczony został preparat złożony typu retard zawierający oksykodon wraz z naloksonem (TARGIN; 10 mg oksykodonu z 5 mg naloksonu lub 20 mg oksykodonu z 10 mg naloksonu). Nalokson może połączyć się w sposób antagonistyczny z receptorami opioidowymi znajdującymi się po stronie wewnętrznej ściany jelit i w ten sposób częściowo zapobiega zaparciom indukowanym opioidami. Po wchłonięciu nalokson w wyniku pierwszego przejścia przez wątrobę ulega niemal całkowitemu rozkładowi i dlatego nie znosi przeciwbólowego działania oksykodonu. Ponieważ jednak oksykodon osiąga receptory opioidowe od strony zewnętrznej jelit - koniecze jest przeprowadzenie dalszych obserwacji, aby stwierdzić, jak dalece klinicznie korzystna jest tego rodzaju kombinacja obu leków.
Petydyna działa około 5 razy słabiej od morfiny. Jej biodostępność oceniana jest na poziomie 50%, a okres półtrwania - na 2-6 godzin. Ważnym metabolitem jest norpetydyna, mająca właściwości drgawkorodne. Ponieważ norpetydyna - po wielokrotnym podaniu petydyny - ulega kumulowaniu w organizmie, petydyna nie nadaje się do stosowania w długotrwałej terapii. Zasada ta jest ważna szczególnie w przypadku pacjentów z niewydolnością nerek.
Piritramid wykazuje podobnie silne działanie jak morfina i stosowany jest często w Niemczech - z powodu względnie dobrej sterowalności - w dożylnej terapii bólów pooperacyjnych („miareczkowanie bólu”). Jego okres półtrwania wynosi 4-10 godzin. W handlu niedostępna jest doustna postać tego leku.
Buprenorfina, endoetylenowa pochodna morfinianu, jest częściowym agonistą receptorów opioidowych μ i ma do nich wysokie powinowactwo. Stąd też wykazuje prawie 40 razy silniejsze działanie przeciwbólowe niż morfina (duża moc). Jednakże charakteryzuje się niższą aktywnością wewnętrzną niż pełny agonista jakim jest morfina, co powoduje, że nigdy nie osiąga maksymalnego efektu analgetycznego (skuteczności) jaki uzyskuje się w przypadku morfiny (efekt pułapowy - ceiling). Buprenorfina działa również agonistycznie z małym powinowactwem względem receptorów N/OFQ (zob. powyżej) oraz antagonistycznie na receptory opioidowe κ. Zasadniczo buprenorfina ma podobny potencjał uzależniający jak inne opioidy i dlatego do niej również odnoszą się odpowiednie przepisy dotyczące środków odurzających.
Poza terapią bólu buprenorfina- podobnie jak metadon - stosowana jest w leczeniu substytucyjnym osób uzależnionych od narkotyków. Ze względu na duże powinowactwo do receptorów μ i silne wiązanie między agonistą a receptorem - heroina jest wypierana z receptorów lub też nie może związać się z receptorami μ, czego skutkiem jest niewywołanie (lub też w niewielkim zakresie) objawów euforii.
Ze względu na właściwości lipofilne buprenorfina jest dobrze wchłaniana, lecz w związku ze względnie wysokim efektem pierwszego przejścia jej biodostępność po podaniu doustnym jest niewielka (ok. 15%). Tymczasem biodostępność po podaniu podjęzykowym (lingwetki) wynosi ok. 55%, a okres połowiczej eliminacji 12-16 godzin.
Ponieważ buprenorfina dość powoli oddziela się (dysocjuje) od receptora, w przypadku przedawkowania lub też zatrucia, aby znieść depresję ośrodka oddechowego, konieczne są względnie duże dawki czystych antagonistów opioidowych w rodzaju naloksonu.
Pentazocyna jest czystym (pełnym) agonistą receptorów opioidowych κ, mającym dodatkowe działanie antagonistyczne względem receptorów opioidowych μ, oraz wykazuje efekt pułapowy. Należy do leków przeciwbólowych z grupy benzomorfanów, w których pierścień C morfinanu jest jeszcze częściowo zachowany. Pentazocyna posiada jedną trzecią siły przeciwbólowego działania morfiny. W przeciwieństwie do tej ostatniej podwyższa ciśnienie tętnicze i przyspiesza czynność serca. W związku z antagonistycznym działaniem wobec receptorów μ - zmianie terapii z pentazocyny na czystego agonistę (np. morfinę) towarzyszy początkowo zmniejszenie skuteczności czystego agonisty. Z kolei w przypadku zmiany z pełnego agonisty na pentazocynę mogą ujawnić się objawy odstawienia (abstynencyjne). Stąd też pentazocyna nie jest żadnym zamiennym środkiem dla osób uzależnionych. Ze względu na nasilony efekt pierwszego przejścia jej biodostępność sięga jedynie ok. 20%. Jej okres półtrwania szacowany jest na2-4 godz.
Dożylne stosowanie fentanylu, alfentanylu, sufentanylu i remifentanylu zostało omówione w rozdz. B 1.7.2.6. Tabletki do ssania (lizaki) z fentanylem stosowane są w terapii bólów przebijających.
Przezskórne systemy terapeutyczne. W przewlekłej terapii bólu (np. w chorobie nowotworowej) dostępne są pochodzące od różnych producentów plastry zawierające fentanyl lub buprenorfinę - pod postacią tzw. przezskórnych systemów terapeutycznych (TTS). Po założeniu pierwszego plastra upływa około 12-24 godzin, zanim osiągnięte zostanie odpowiednio wysokie stężenie leku w osoczu, a tym samym - zanim wystąpi działanie przeciwbólowe. Później stężenia fentanylu w osoczu utrzymują się na względnie stałym poziomie przy zmienianiu plastra co trzy dni. Zaparcia i sedacja występują w przypadku przezskórnego podawania fentanylu rzadziej niż przy doustnym stosowaniu morfiny. U chorych z podwyższoną temperaturą ciała (np. w stanach gorączkowych, przy użyciu elektrycznych poduszek, po korzystaniu z sauny, solarium itp.) zwiększa się resorpcja substancji aktywnej przez skórę, w wyniku czego częściej mogą być obserwowane typowe dla opioidów objawy niepożądane. Zawierające fentanyl plastry są dostępne w 5 różnych zakresach dawkowania (szybkość uwalniania odpowiednio: 12, 25, 50, 75 i 100 μg/godz.), natomiast plastry z buprenorfiną w 3 zakresach dawkowania (szybkość uwalniania: 35, 52,5 i 70 μg/godz.).
1.5.2.4. Słabo działające leki opioidowe stopnia 2. wg WHO
Kodeina stosowana jest jako lek przeciwbólowy niemal wyłącznie w terapii skojarzonej z nieopioidowymi lekami przeciwbólowymi. Efekt przeciwbólowy wiąże się z demetylacją cząsteczki kodeiny (za pomocą CYP2D6) do morfiny (w 10%). W przypadku osób z wolnym metabolizmem (slow metabolizers) CYP2D6 (polimorfizm genetyczny) działanie przeciwbólowe leku może być niewystarczające; z kolei u osób z bardzo szybkim metabolizmem (ultra rapid metabolizers) może dojść do zatrucia morfiną. Biodostępność po zażyciu doustnym wynosi 40-60%, a jej okres półtrwania 2-3 godziny.
Dihydrokodeina jest prawie trzykrotnie silniejsza w zakresie działania przeciwbólowego niż kodeina. W wątrobie ulega przemianie m.in. do dihydromorfiny, która cechuje się znacznym potencjałem uzależniającym. DHC charakteryzuje się nasilonym efektem pierwszego przejścia (biodostępność rzędu 20%), a jej okres półtrwania wynosi 3-5 godzin.
Tilidyna jest prolekiem. Wykazuje bardzo słabe działanie agonistyczne. Właściwą substancją działającą jest nortilidyna - powstająca w wyniku oksydatywnej demetylacji w obrębie atomu azotu. Tilidyna stosowana jest w stałych proporcjach w preparatach łączonych z naloksonem (50 mg tilidyny + 4 mg naloksonu). U podstaw tego rodzaju terapii skojarzonej leży następująca przesłanka: tilidyna ulega aktywacji w wyniku pierwszego przejścia przez wątrobę, z kolei nalokson jest w wyniku silnego efektu pierwszego przejścia inaktywowany. W przypadku pozajelitowego podania preparatu (w przebiegu uzależnienia) lub też spożycia nadmiernych dawek leku drogą doustną - nalokson prowadzi do zantagonizowania działania (nor)tilidyny i tym samym zapobiega nadużywaniu. Stosowanie takich skojarzonych preparatów u pacjentów z niewydolnością wątroby nie jest wskazane, gdyż powstaje wówczas zbyt mało skutecznej nortilidyny, a równocześnie nalokson przy pierwszym przejściu przez wątrobę nie ulegnie pełnej dezaktywacji. Stosowanie preparatu jest natomiast możliwe u osób z niewydolnością nerek (zob. tab. B 1.5-4).
Praktycznie nie są znane przypadki uzależnienia od tego typu terapii skojarzonej opartej na połączeniu tilidyny z naloksonem w postaci retard.
Tramadol dostępny jest na rynku pod postacią racemiczną i należy do najczęściej stosowanych na świecie opioidów. Jest on słabym agonistą receptorów opioidowych μ [ok. 1/6000 w porównaniu z morfiną; (+)-tramadol >> (-)-tramadol]. O-demetolotramadol (metabolit M1) posiada wprawdzie jeszcze większe powinowactwo do receptorów opioidowych μ niż substancja macierzysta, lecz działanie przeciwbólowe tramadolu wiąże się również z hamowaniem wychwytu zwrotnego monoamin (noradrenaliny, serotoniny). (+)-Tramadol hamuje głównie wychwyt zwrotny serotoniny, podczas gdy (-)-tramadol wpływa na wychwyt noradrenaliny. Możliwa do osiągnięcia (maksymalna) siła działania jest wyraźnie słabsza niż w przypadku morfiny, lecz z drugiej strony działanie depresyjne na ośrodek oddechowy a także wpływ uzależniający są również wyraźnie słabsze. Tramadol - szczególnie w dużych dawkach - dość często wywołuje nudności i/lub wymioty - prawdopodobnie w wyniku zahamowania wychwytu zwrotnego serotoniny.
Tramadol w przypadku podania doustnego jest wchłaniany w 95%, jego doustna biodostępność szacowana jest na poziomie 70%, a okres półtrwania wynosi ok. 5 godzin. Ulega procesowi N- lub O-demetylacji, po czym jest sprzęgany z kwasem glukuronowym lub siarkowym.
Wyszukiwarka
Podobne podstrony:
NLPZ (2), Stomatologia UMED, Farmakologia, 07 - przeciwbólowe [opioidy i NLPZ]
Gobol - Leki przeciwbolowe, Stomatologia UMED, Farmakologia, 07 - przeciwbólowe [opioidy i NLPZ]
Gobol - NLPZ, Stomatologia UMED, Farmakologia, 07 - przeciwbólowe [opioidy i NLPZ]
pytania - test zaliczeniowy u Kubickiej-Musiał, Stomatologia UMED, Periodontologia, egzaminy, testy
pytania stomatopatie całość, Stomatologia UMED, Protetyka, pytania, egzaminy
materiały stos w stom zach, Stomatologia UMED, Zachowawcza
wykład3 - Eutanazja, Stomatologia UMED, prawo medyczne, prawo i etyka w stomatologii 2010-2011
Cwiczenia III rok , Stomatologia UMED, Protetyka, Inne, Protetyka
wstęp stomatologia, Stomatologia UMED, rehabilitacja, materiały
EGZAMIN FARMAKOLOGIArozwiazana do wydrukuKOLOR, Stomatologia, Inne, Farmakologia, Farmakologia
Konkretne opracowanie, stomatologia umed, biologia
Farmakognozja, 07 Śluzy
Gieldy interna, Stomatologia UMED, Interna, prelekcje interne I, Interna- Giełdy z egzaminu
chirurgia szczękowa - notatki ze wszystkiego, Stomatologia UMED, Chirurgia, chirurgia stomatologiczn
Blad lekarski, Stomatologia UMED, prawo medyczne, prawo i etyka w stomatologii 2010-2011
więcej podobnych podstron