
Podstawy genetyki klasycznej
Co to jest ALLEL?
•
Poszczególne geny mogą występować w dwu lub więcej „ odmianach”. Takie „odmiany”
nazywamy ALLELAMI.
•
Człowiek posiada jednocześnie najwyżej dwa różna allele tego samego genu. (2n)
•
Wpływ alleli na siebie: a) dominacja - jeden z dwu alleli może „maskować”
drugi allel, tak że nie ujawnia się on fenotypowo.
•
Allel, który ujawnia się, nazywamy allelem dominującym (oznaczamy dużą literą np. A), a
ten maskowany allelem recesywnym (oznaczamy mała literą np. a)
Homozygoty – osobniki, które posiadają dwa allele tego samego rodzaju (aa lub AA)
Heterozygoty – osobniki, które posiadają różne allele (aA)
I Prawo Mendla
Prawo czystości gamet
Każdy organizm (diploidalny 2n) posiada w genomie dwa allele warunkującą daną cechę. W
wyniku mejozy do gamet przechodzi po jednym allelu (1n) z pary.
Mendel opracował to prawo analizując dziedziczenie barwy kwiatu grochu.
W pokoleniu F1 – wszystkie kwiaty czerwone, w pokoleniu F2 – stosunek wynosi 3:1 (3 -
czerwone, 1- biały)
I prawo Mendla
Krzyżówka dwóch odmian grochu o kwiatach białych i czerwonych
Pokolenie F1 wszystkie kwiaty są czerwone
Pokolenie F2 stosunek fenotypów 3:1
II Prawo Menda
Zasada niezależnej segregacji różnych cech
Różne cechy dziedziczą się niezależnie od siebie. Na przykład barwa nasion grochu dziedziczy się
niezależnie od rodzaju ich powierzchni.
Zasada ta ma zastosowanie do cech leżących na różnych chromosomach.
II prawo Mendla
W pokoleniu F1 wszystkie nasiona żółte i gładkie.
W pokoleniu F2 stosunek wynosi (9:3:3:1)
Kodominacja – nie ma stosunku dominacja - recesywność, produkty dwu różnych alleli mają
odbicie w fenotypie (np. grupy krwi allel IA i IB)
Allel i jest recesywny w stosunku do IA i IB
Grupy krwi posiadają trzy rodzaje alleli:
IA, IB, i
Dominacja niezupełna –
allel dominujący nie maskuje całkowicie recesywnego,
- w pokoleniu F1 wszystkie kwiaty dziwaczka są różowe,
- w pokoleniu F2 1:2:1
1-czerwony, 2-różowy, 1-biały
Chromosomowa teoria dziedziczności
Geny znajdują się na chromosomach i ułożone są liniowo ( jeden za drugim ) w określonej
kolejności.
Każdy gen zajmuje na chromosomie określone miejsce zwane locus.
Różne chromosomy zawierają różne liczby genów.
Geny umieszczone na jednym chromosomie są ze sobą sprzężone, razem się dziedziczą (wchodzą
do gamety)
Sprzężenie genów nie jest całkowite ponieważ zachodzi crossing-over.
.

Crossing-over - allele tego samego genu mogą „zamieniać się miejscami” na chromosomach
homologicznych. Prawdopodobieństwo wystąpienia „zamiany” jest tym większe, im geny na
chromosomie są od siebie bardziej oddalone.
Badając częstość procesu crossing-over zachodzącego między poszczególnymi genami można
określić kolejność ich ułożenia na chromosomie. Tak można stworzyć tzw. mapy chromosomów.
Crossing-over ; zachodzi podczas mejozy, powstawanie komórek rozrodczych
Epistaza
Zjawisko tłumiącego działania genu (z jednej pary alleli) na jakąś cechę uwarunkowaną inną parą
alleli. Gen hamujący nazywamy epistatycznym, a maskowany hipostatycznym.
Fenotyp bombajski jako przykład epistazy. Przy braku genu H nie dochodzi do wytworzenia
cząsteczki prekursorowej antygenów grupowych krwi A lub B. Homozygoty hh mają grupę krwi 0,
mimo, że posiadają gen dla antygenów grupowych krwi A lub B.
Poligeny
To geny z różnych par alleli, zajmujące różne loci w chromosomach, wpływające na wytworzenie
tej samej cechy. Efekty ich działania się sumują.
U człowieka poligenami uwarunkowane są takie cechy jak wzrost, IQ, barwa skóry
Plejotropia
Uwarunkowanie przez jeden zmutowany gen kilku pozornie nie związanych ze sobą cech
fenotypowych.
Zespół Marfana –zaburzenia w tworzeniu się kolagenu. Wtórnie dochodzi do zmian w układzie
kostnostawowym, w gałce ocznej i układzie krążenia.
Efekty plejotropowe obserwujemy również w albiniżmie, fenyloketonurii, galaktozemii.
Zespół Marfana
Komplementacja - geny dopełniające się
Dopełniające się działanie produktów dwu różnych genów.
U groszku pachnącego, mamy dwa geny warunkujące barwę czerwoną (A i B). U roślin tych musi
wystąpić allel dominujący A i allel dominujący B równocześnie aby powstała czerwona barwa
kwiatu.
U człowieka komplementacja dotyczy genów warunkujących kolor włosów i skóry
Diagnostyka prenatalna
Badania prenatalne;
Nieinwazyjne:
a) USG
b) Test potrójny
c) oznaczenie a-fetoproteiny
d) badanie komórek płodowych izolowanych z krwi matki
Inwazyjne:
a) Amniocenteza
b) Biopsja kosmówki
c) Kordocenteza
d) Fetoskopia
USG genetyczne
Wskazania do diagnostyki prenatalnej.
Wiek matki 35 lat
Poprzednie dziecko z wadą wrodzoną, późne poronienie, śmierć noworodka
Obecność aberracji chromosomowej u rodzica lub poprzedniego dziecka
Wady cewy nerwowej w poprzednich ciążach i nieprawidłowe stężenie α-fetoproteiny
Obecność w rodzinie wad sprzężonych z płcią i chorób metabolicznych
Nieprawidłowy obraz zarodka w badaniu USG
Narażenie na działanie czynników teratogennych

Kariotyp człowieka
DNA jest upakowane w określonej liczbie chromosomów
Każdy chromosom zbudowany jest z dwu chromatyd połączonych centromerem
Centromer – miejsce przyczepienia wrzeciona podziałowego, zawiera charakterystyczne sekwencji
DNA i kompleks białek zwanych kinetochorem
Wyróżniamy chromosomy metacentryczne, submetacentryczne, akrocentryczne i telocentryczne
Rodzaje chromosomów
Telomery
Telomery – końcowe fragmenty chromosomów niezbędne do utrzymania ich stabilności
Są zbudowane z powtórzonych sekwencij typu –TTAGGG-
U noworodka długość telomerów wynosi 6-10 tys. nukleotydów i skraca się przy każdym podziale
komórki
Po nadmiernym skróceniu telomerów komórki przestają się dzielić
Nie skracają się w komórkach płciowych, macierzystych czy komórkach szpiku
U człowieka wyodrębniono 22 pary chromosomów oraz chromosomy płci
Prawidłowy kariotyp to 46XX lub 46XY
Podział chromosomów na 7 grup (od A do G)
Gr A; 1,2,3 - metacentryczne
Gr B; 4,5 - duże, submetacentryczne
Gr C; 6 -12 i X – średnie, submetacentryczne
Gr D; 13-15- duże, akrocentryczne
Gr E; 16-18- małe, submetacentryczne
Gr F: 19-20 - najmniejsze, metacentryczne
Gr G; 21,22,Y – małe, akrocentryczne
Kariotyp ; zestaw chromosomów występujących w komórce somatycznej o charakterystycznej
liczbie i morfologii.
Prążkowy wzór chromosomów uzyskujemy dzięki odpowiedniemu barwieniu. Odzwierciedla on
nierównomierną kondensację chromatyny.
Najczęściej są to prążki G, po barwieniu barwnikiem Giemzy.
- Ciemne prążki zawierają zasady AT, silnie skondensowaną heterochromatynę i specyficzne
białka niehistonowe bogate w siarkę.
- Prążki jasne zawierają głównie GC, rozluźniona, aktywna euchromatyna
Prążki Q – barwienie fluorescencyjne, silne wybarwienie rejonów bogatych w pary AT,
chromosomu Y, satelity i centromery chromosomów akrocentrycznych
Prążki C – barwienie Ba(OH)2 i odczynnikiem Giemzy wybarwienie rejonów cenrtomerów oraz
rejony przycentromerowe (przewężenia wtórne)
Prążki R –różne techniki otrzymywania
Odwrotność prążków G; pary AT-ciemne, pary GC jasne. Możliwe ujawnienie drobnych aberacji.
Barwienie Ag-NOR –barwienie organizatorów jąderka w rejonach satelit
Barwienie DA/DAPI
Wskazania do oznaczenia kariotypu
Występowanie cech fenotypowych charakterystycznych dla określonego zespołu chromosomowego
Występowanie zespołu wad rozwojowych i/lub cech dysmorfii ze współistnieniem opóźnienia
rozwoju psychoruchowego.
Niepowodzenia rozrodu; poronienia samoistne lub urodzenia dzieci z wadami rozwojowymi,
niepłodność o nieznanym pochodzeniu
Brak cech dojrzewania płciowego
Pierwotny lub wtórny brak miesiączki
Znaczny niedobór wzrostu o nieznanej etiologii u kobiet
Nieprawidłowa budowa zewnętrznych narządów płciowych, obojnactwo
Występowanie strukturalnej aberracji chromosomowej w rodzinie
u rodziców dzieci obarczonych aberracją

u potomstwa nosicieli zrównoważonej aberracji chromosomowej
u rodziców i rodzeństwa nosicieli zrównoważonej aberracji chromosomowej
Hodowla tkankowa
Przygotowanie hodowli; pobranie krwi obwodowej, dodanie soli sodowej heparyny
Do płynnej pożywki dodajemy;
Glukozę, L-glutaminę (stymulacja wzrostu komórek)
Surowicę FCS (Fetal Bovine Serum) w ilości 10 -30 % w pożywce
Antybiotyki i fungicydy
Mitogeny; Fitohemaglutynina (PHA) stymuluje wzrost limfocytów T , a wirus Epsteina-Barr i LPS-
lipopolisacharyd z E.coli –limfocyty B
Czynniki wzrostu
Warunki hodowli –temperatura 37 C, pH 7,2-7,4
Długość trwania hodowli ok. 72h
Dodanie inhibitora mitoz – Colcemid (analog kolchicyny)
Dodanie do pożywki roztworu hipotonicznego (0,075 M KCL)
Utrwalenie napęczniałych komórek 100% metanolem i lodowatym kwasem octowym w
proporcjach 3:1
Umieszczenie zawiesiny na szkiełku podstawowym
Barwienie
MITOZA I MEJOZA CYKL KOMÓRKOWY
Faza G1
Komórki dwukrotnie mniejsze
Zwiększona ruchliwość i wrażliwość na bodźce
Wzrasta ilość białek, RNA itp. Zwiększa się masa i objętość komórek
Długość fazy G1 decyduje o długości całego cyklu
W późnej fazie G1 znajduje się punkt restrykcyjny R , komórka która przekroczy ten punkt musi
przejść do następnej fazy cyklu.
Faza syntezy DNA (S)
Trwa przeważnie tyle samo ok. 8 godzin ( wyjątek np. bruzdkowanie)
Euchromatyna jest syntetyzowana na początku fazy
Heterochromatyna jest syntyetyzowana pod koniec fazy S
Masa i objętość komórki zwiększa się w porównaniu do fazy G1.
Faza G2
Bezpośrednie przygotowanie do mitozy, synteza białek wrzeciona podziałowego (tubuliny),
cykliny B
Nadprodukcja składników błony komórkowej
Aktywacja kinazy białkowej CDK, która później odpowiada za zanik błony jądrowej i
kondensację chromosomów
Faza G0
Komórki funkcjonują , ale tracą zdolność dzielenia się.
Komórki zawierają bardziej skondensowaną chromatynę, inne rodzaje RNA i białka
Czas trwania jest różny od kilku dni do kilku miesięcy lub dłużej
Komórki mogą przechodzić z fazy G0 do G1, np.
Limfocyty T i B pod wpływem mutagenów
komórki wątroby,
komórki nowotworowe (cytostatyki eliminują komórki nowotworowe w różnych stadiach, poza
tymi w fazie G0, po pewnym czasie komórki w fazie G0 wchodzą do cyklu dając wznowę
nowotworowi)
MITOZA
Proces podziału komórki, w którym z jednej komórki powstają dwie komórki o takiej samej

liczbie chromosomów , jak komórka macierzysta.
Składa się z kariokinezy i cytokinezy
Mitotycznie dzielą się komórki somatyczne – budujące ciało organizmu.
Mitoza składa się z faz:
PROFAZA
następuje zanik błony jądrowej,
chromatyna kondensuje i przekształca się w chromosomy,
każdy chromosom składa się z dwóch chromatyd (pojedynczych cząsteczek DNA), połączonych
centromerem
centriole rozchodzą się parami do biegunów komórki
powstają włókna wrzeciona podziałowego – kariokinetycznego.
rozpad jąderka i otoczki jądrowej
METAFAZA
chromosomy ustawiają się w płaszczyźnie równikowej komórki, powstaje płytka metafazalna
Silna kondensacja chromatyny, rozdział każdego chromosomu na dwie chromatydy
Mikrotubule wrzeciona podziałowego łączą się z chromosomami w miejscu kinetochoru
ANAFAZA
rozdzielenie chromatyd każdego z chromosomów i przemieszczanie ku biegunom komórki, poprzez
skurcz włókien wrzeciona,
każde włókno przyciąga jedną chromatydę z chromosomu.
TELOFAZA
nici DNA rozplątują się,
zanikają wrzeciona kariokinetyczne,
odtwarzana jest otoczka jądrowa
w jednej komórce widoczne są dwa jądra komórkowe.
intensywna synteza rRNA
defosforylacja nukleoliny – odtwarzanie jąderka
CYTOKINEZA
powstaje bruzda podziałowa, która pogłębia się aż do momentu zetknięcia się i rozdzielenia
dwóch komórek.
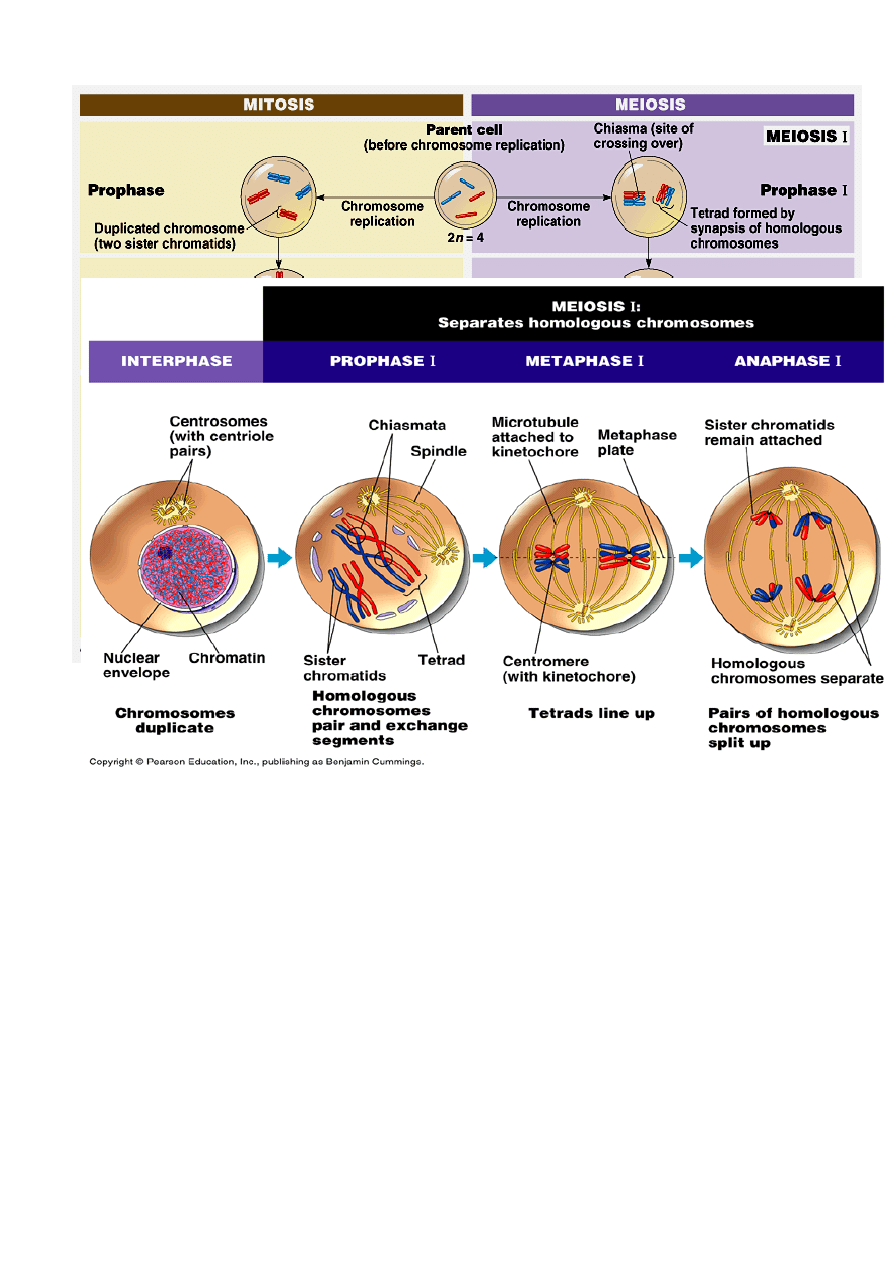
MEJOZA
Proces podziału komórki w którym z jednej komórki diploidalnej (2n) powstają komórki
haploidalne (n).
Podczas mejozy zachodzi wymieszanie materiału genetycznego
CROSSING – OVER
Mejoza dotyczy tylko powstawania komórek rozrodczych
Mejoza składa się z faz:
PROFAZA I
LEPTOTEN – powstaje wrzeciono podziałowe, chromosomy zaczynają się różnicować,
Chromosomy przybierają postać długich cienkich nitek. Chromosomy mają rodzaj białkowego
rdzenia, który stanowi rdzeń , powstaje kompleks synaptonemalny
Każdy chromosom mejotyczny jest przyczepiony na końcu do otoczki jądra za pośrednictwem
płytki przyczepowej (istnieje do stadium diakinezy)
PROFAZA I
ZYGOTEN – połączenie chromosomów homologicznych w biwalenty,
PACHYTEN – proces CROSSING – OVER wymiana między dwiema niesiostrzanymi
chromatydami chromosomów homologicznych, na przebiegu tworzącego się kompleksu
synaptonemalnego powstają węzły rekombinacyjne

* CROSSING - OVER
zachodzi wówczas, gdy chromatydy chromosomów homologicznych splatają się ze sobą, i
wymieniają się fragmętami
Chiazma to miejsce w którym zachodzi crossing-over
.
DIPLOTEN – zakończenie procesu CROSSING – OVER rozdzielenie chromosomów
homologicznych, Kompleks białkowy rozpuszcza się. Chromosomy ulegają dekondensacji i
następuje synteza RNA.
DIAKINEZA- zmniejszenie syntezy RNA, Kondensacja chromosomów i oddzielenie ich od
otoczki jądrowej
METAFAZA I
ustawienie biwalentów w płaszczyźnie równikowej.
Wytwarzanie wrzeciona podziałowego
Każdy chromosom biwalentny składa się z 4 chromatyd i ma tylko dwa centromery
ANAFAZA I
Z każdego chromosomu biwalentnego do biegunów komórki przesuwa się jeden chromosom
pochodzący od ojca lub od matki, wybór chromosomów jest przypadkowy
TELOFAZA I
powstają dwa jądra potomne o haploidalnej liczbie chromosomów, ale z diploidalną ilością DNA
Część chromosomów pochodzi od ojca a część od matki
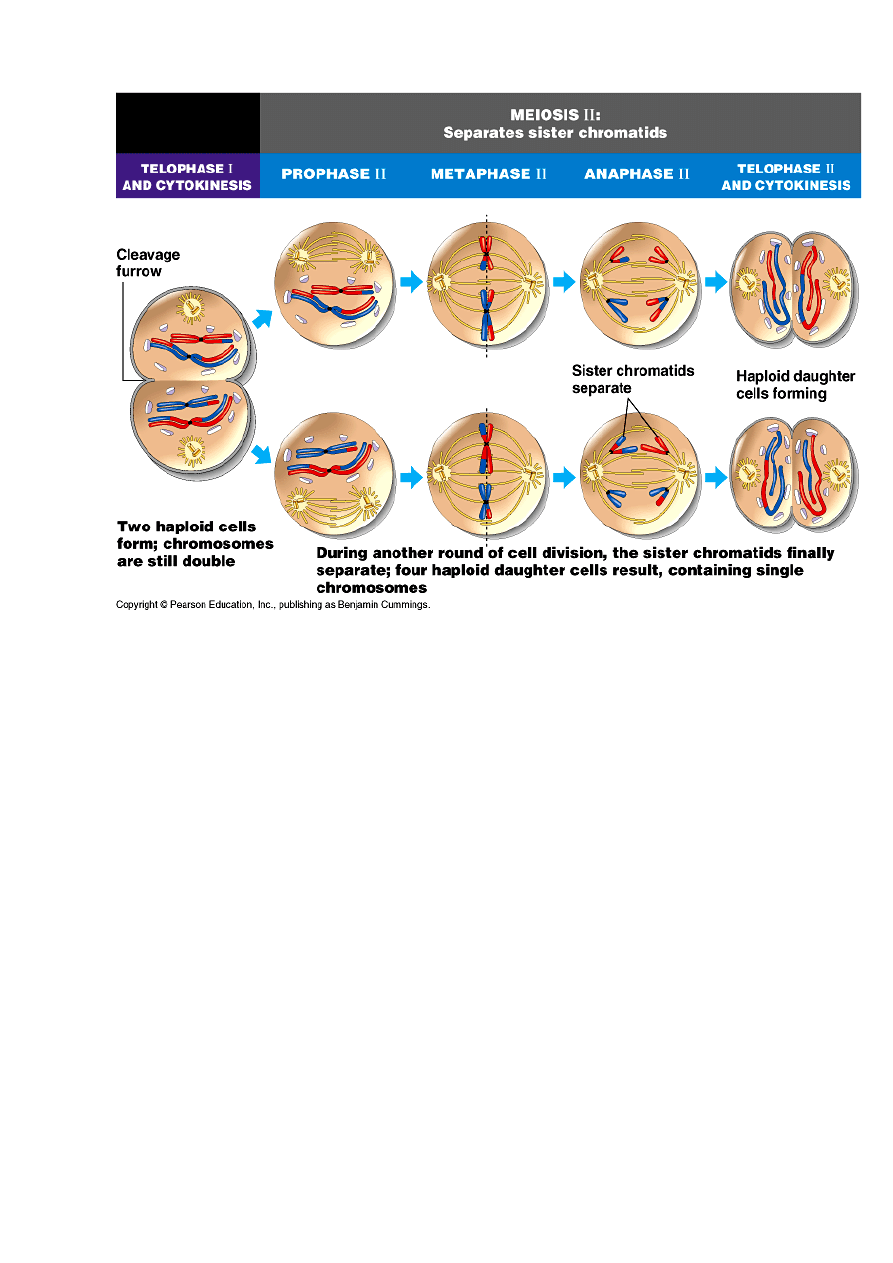
Cytokineza i profaza drugiego podziału mejotycznego bez syntezy DNA
II PODZIAŁ MEJOTYCZNY
przebiega podobnie do podziału mitotycznego;
Profaza Metafaza Anafaza Telofaza
AMITOZA
to przypadkowy rozpad komórki macierzystej na komórki potomne, często niezdolne do
samodzielnego funkcjonowania.
MITOZA
prowadzi do powstania dwóch komórek potomnych o takiej samej liczbie chromosomów jak
komórka macierzysta,
składa się z czterech faz (profazy, metafazy, anafazy i telofazy),
dotyczy komórek somatycznych.
Budowa DNA
DNA jest liniowym nierozgałęzionym polimerem zbudowanym z nukleotydów
Każdy nukleotyd zbudowany jest z
reszty fosforanowej,
cukru pentozy,
zasady azotowej
Wyróżniamy 4 rodzaje nukleotydów różniące się zasadami azotowymi
A - adenina
T - tymina
C – cytozyna
G – guanina
Budowa DNA
DNA jest liniowym nierozgałęzionym polimerem zbudowanym z nukleotydów
Zasady azotowe
Pirymidyny zbudowane z pojedynczego pierścienia aromatycznego; należą tu cytozyna C i tymina
Puryny zawierają dwa pierścienie aromatyczne; należą tu adenina A i guanina G
Zasady azotowe są syntetyzowane w komórkach de novo
Nukleotydy mogą być syntetyzowane de novo lub uwalniane w wyniku hydrolizy kwasów
nukleinowych
Zasady azotowe różnią się między sobą obecnością określonych grup funkcyjnych.
W DNA dwie nici łączą się ze sobą za pomocą wiązań wodorowych
Zasada komplementarności
adenina z tyminą wiąże się za pomocą dwóch wiązań wodorowych,
cytozyna z guaniną za pomocą trzech wiązań wodorowych.
Deoksyryboza
Cukier 2’- deoksyryboza jest zbudowana z pięciu atomów węgla, ma budowę cykliczną , jest to
struktura przestrzenna przypominająca kształtem kopertę.
Przy węglu 2’ grupa – OH zastąpiona jest -H
Grupa fosforanowa
Reszta fosforanowa łączy ze sobą dwa nukleotydy w łańcuchu DNA za pomocą wiązań 3’→ 5′
-fosfodwuestrowych
Łańcuch DNA jest zwyczajowo zapisywany w orientacji 5′ → 3’
Wiązanie 3’→ 5′ -fosfodwuestrowe powstaje podczas biochemicznej syntezy DNA z udziałem
polimerazy DNA
Nukleotydy a nukleozydy
Nukleotyd= zasada azotawa + cukier+ grupa fosforanowa
Nukleozyd = zasada azotowa + cukrem
Kwasy nukleinowe powstają przez łączenie się ze sobą kolejnych nukleotydów poprzez wiązania
fosfodiestrowe między atomem 3’ i 5′ drugiego nukleotydu
Przestrzenne ułożenie DNA
B-DNA najczęściej występująca forma - skręt prawostronny, zgodny z ruchem wskazówek zegara.

Występuje duży rowek i mały rowek. Wewnątrz tych rowków są charakterystyczne układy atomów
które umożliwiają przyłączenie się do DNA określonych białek regulatorowych.
Z-DNA lewoskrętna, cząsteczka jest dłuższa i węższa niż w przypadku B- DNA. Dodatnio
superhelikalny DNA
A- DNA helisa jest szersza i krótsza niż w B-DNA Rozluźniony DNA
Nukleosom i co dalej ?
DNA nawinięty jest na nukleosomy zbudowane z białek histonowych. Nukleosom ma kształt dysku
i jest zbudowany z ośmiu białek
Aminokwasy mają ładunek dodatni, dzięki czemu przyciągają DNA
Wtórne zwinięcie DNA - ciasno upakowane nukleosomy.
Tworzenie struktur wyższego rzędu, biorą tu udział białka niehistonowe.
Największe upakowanie w metafazie - współczynnik upakowania 8000 do 10000 razy
Chromosom
Rodzaje chromatyny: heterochromatyna, euchromatyna
Centromer
telomer fragment chromosomu zlokalizowany na jego końcu, który zabezpiecza go przed
uszkodzeniem podczas kopiowania. Telomer skraca się podczas każdego podziału komórki .
Budowa RNA – kwas rybonukleinowy
Cukier występujący w RNA to ryboza
Twarzą się wiązania między
G a C (trzy wiązania wodorowe)
między A i U (dwa wiązania wodorowe)
Rodzaje RNA
m RNA – kodujacy RNA
r RNA - rybosomalny RNA
t RNA - transportujący RNA
m RNA
Powstaje na bazie DNA w wyniku transkrypcji.
Po zakończeniu syntezy białka szybko ulega destrukcji
Na końcu 5 ’ znajduje się czapeczka, jest ona ważna w czasie transportu mRNA i sn RNA z jądra
do cytoplazmy
Czapeczka bierze udział w translacji na etapie tworzenia kompleksu inicjacji
Czapeczka spełnia funkcje ochronne względem mRNA poprzez ograniczenie działania 5’-
Spliceosom - kompleks białek i RNA, który bierze udział w wycinaniu intronów z pre-mRNA w
procesie splicingu. W skład klasycznego spliceosomu wchodzi pięć małych jądrowych
nukleoprotein (snRNP, czyli białko + snRNA), zwanych U1, U2, U4, U5 i U6. Wycina on introny
mające sekwencję GU na 5'-końcu i AG na 3'-końcu.
Spliceosom alternatywny (typu U12) wycina introny zaczynające się od AU i kończące się AC.
Również zawiera pięć rodzajów snRNP. Są to U11, U12, U4atac, U6atac, oraz -U5
m RNA
Sekwencja kodująca – decydująca o składzie wytwarzanych podczas translacji białek
Pre-m RNA zawiera introny i eksony, w czasie procesu dojrzewania dochodzi do wycięcia intronów
(splicing)
Dojrzałe mRNA łączy się z określonymi białkami jeszcze w jądrze komórkowym tworząc
informosom i w tej postaci przechodzą do cytoplazmy. Białka te pełnią funkcję ochronną
Na końcu 3 ′ znajduje się ogon poliA 200-250 powtórzeń nukleotydu adeniny. Są one dodane
niezależnie od matrycy przez polimerazę poli(A) chroni przed degradacją przez 3’- egzonukleazy,
bierze udział w procesie translacji.
tRNA
Transportujący RNA to małe cząsteczki zbudowane z 74-95 nukleotydów,
struktura przypomina liść kończyny. Zawiera 3 pętle i 4 dwuniciowe ramiona. Trójwymiarowa

struktura tRNA przypomina literę L.
Ważne miejsca;
miejsce akceptorowe tu przyłącza się aminokwas zakończone ACC
antykodon (trzy nukleotydy w drugiej pętli tRNA) wiąże się z kodonem na nici mRNA
tRNA
t RNA przenosi zaktywowane aminokwasy z cytoplazmy na kompleks rybosom-mRNA
W tRNA występują zmodyfikowane nukleotydy np. dihydrourydyna, pseudourydyna, inozyna,
rybotymidyna
Geny dla t RNA występują w wielu kopiach
rRNA
Jest częścią składową rybosomów (50-70%) na których dochodzi do syntezy białek na bazie mRNA
rRNA nie tylko decyduje o kształcie i wielkości rybosomów ale również o rozmieszczeniu białek
rybosomalnych.
Rybosom jest zbudowany z dużej i małej podjednostki
Geny rRNA kodują pre rRNA, które powstaje w jąderku dzięki działaniu polimerazy RNA1
U człowieka geny rRNA są w rejonach jąderkotwórczych (nitki satelitonośne) chromosomów
akrocentrycznych
Występują w tandemowo powtórzonych grupach - 100 lub więcej kopii
r RNA
Cząsteczki pre rRNA mają stałą sedymentacji 47S a potem 45S
Cięcie przez rybonukleazy
Budowa rybosomu
Budowa białek
Białka to cząsteczki zbudowane z aminokwasów.
Na bazie jednego genu (fragment DNA) powstaje jedno konkretne białko.
Aminokwas zbudowany jest z atomu węgla, gr. aminowej (-NH 2) , gr. karboksylowej (- COOH),
atomu wodoru i ze zmiennym łańcuchem bocznym R.
Wyróżniamy 20 aminokwasów .
Dzielimy je na grupy np.
A. Aminokwasy obojętne, niepolarne łańcuchy boczne
B. Aminokwasy obojętne, polarne łańcuchy boczne
C. Aminokwasy obdarzone ładunkiem elektrycznym ; zasadowe - ładunek dodatni, kwasowe -
ładunek ujemny
Aminokwasy alifatyczne (kl. I)- z łańcuchem węglowym bez grup dodatkowych, nie mają w
łańcuchach bocznych atomów N, O, S ani pierścieni
Aminokwasy zawierające siarkę (kl.II)cysteina odgrywa ważną role w stabilizacji struktury białek
ze względu na zdolność tworzenia mostków dwusiarczkowych
Aminokwasy aromatyczne - zawierające pierścień (kl.III), prolina – kl.VII inna budiwa
pierścienia, przyczyna zagięć w łańcuchu białkowym
Aminokwasy z grupą hydroksylową –OH obojętne (kl.IV) :
Aminokwasy kwaśne (kl. V)
Aminokwasy zawierające grupę karboksylową –COOH:
Aminokwasy zawierające azot w tym lizyna i arginina to aminokwasy zasadowe (kl. VI)
Aminokwasy zawierające azot:
Grupa karboksylowa jednego aminokwasu łączy się z grupą następnego aminokwasu tworząc
wiązanie peptydowe.
Grupa aminowa uważana jest jako początek wiązania peptydowego. Wiązanie peptydowe
Zaangażowane są we wszystkie procesy chemiczne zachodzące w organizmie
Enzymy katalizują reakcje chemiczne w komórkach cięcie wiązań, łączenie cząsteczek itp.
Umożliwiają transport małych czasteczek, jonów

Towarzyszą wzrostowi komórek, podziałom (mitoza, mejoza), różnicowaniu się komórek (różnice
w ekspresji genów).
Pierwszorzędowa struktura białka liniowa cząsteczka zbudowana z aminokwasów o ściśle
określonej sekwencji. Tworzenie się wiązań dwusiarczkowych między cysteinami
Drugorzędowa struktura białka, przestrzenne ułożenie aminokwasów względem siebie. Główne
struktury to α-helisa i β-harmonijka
Trzeciorzędowa struktura białka trójwymiarowa zdeterminowana składem aminokwasów.
Reszty aminokwasowe. bardzo odległe od siebie w strukturze pierwszorzędowej mogą po
„zwinięciu się białka” znajdować się obok siebie.
Czwartorzędowa struktura przestrzenne ułożenie podjednostek białka.
Replikacja DNA
Replikacja DNA – synteza DNA. Podczas replikacji każda nić DNA służy jako matryca do syntezy
nowej nici (replikacja semikonserwatywna).
Udział wielu enzymów współdziałających ze sobą.
Topoizomeraza rozplata skręt helisy DNA
Helikazy rozdzielają dwie nici tworząc widełki replikacyjne.
Polimerazy dokonują syntezy na podstawie dostępnej matrycy i mają możliwość naprawy drobnych
błędów replikacji
Prymazy inicjują syntezę starterów RNA dla fragmentów Okazaki.
Ligaza łączy w czasie replikacji fragmenty Okazaki w jedną nić.
Białka inicjacyjne- rozpoznają miejsce startu
Cały kompleks tych białek nazywamy replisomem
Synteza DNA odbywa się w czasie trwania fazy S cyklu komórkowego
Rozpoczyna się ona w wielu miejscach na raz tzw. replikonach.
Synteza przebiega w obu kierunkach równocześnie, aż sąsiednie replikony połączą się
Tworzą się widełki replikacyjne, obie nici służą jako matryca
Nić DNA jest rozplatana za pomocą topoizomeraz
Replikacja zachodzi w sposób ciągły tylko na jednej z nici od 5’do 3 ’ nić wiodąca
Na drugiej nici od 3′do 5′ nić opóźniona składa się z fragmentów Okazaki. Do inicjacji wymagane
są krótkie fragmenty RNA (startery) syntetyzowane przez prymazę. Po zainicjowaniu syntezy
fragmentu Okazaki RNA jest usuwane, dobudowywane jest DNA. Sąsiadujące fragmenty DNA są
łączone przez ligazę.
Enzym polimeraza DNA jest złożony z kilku podjednostek. Wyróżniamy różne typy polimeraz.
Polimeraza-α, β,γ
Podczas replikacji polimeraza może usuwać powstałe błędy
Replikacja przebiega z zachowaniem zasady komplementarności A –T, C-G (Rys. A)
Kod genetyczny określa zasady według których na podstawie kolejności nukleotydów w DNA
powstaje białko
Sekwencja DNA przepisana jest na mRNA na zasadzie komplementarności A-U, G-C
Jeden aminokwas jest kodowany przez trójkę kolejnych nukleotydów tzw. kodon
Każdy kodon a jest ich 64, odpowiada jednemu aminokwasowi, ale jeden aminokwas może mieć
kilka kodonów
Wyróżniamy kodony AUG (metionina) kodon startowy i kodony stop czyli UAA, UAG, UGA
Cechy kodu genetycznego
Trójkowy; 1 aminokwas trzy nukleotydy
Uniwersalny; dany kodon jest właściwy dla określonego aminokwasu u wszystkich organizmów
żywych
Niezachodzący; dany nukleotyd może być tylko w jednym kodonie

Bezprzecinkowy; nie ma przerw między trójkami i dlatego kod odczytywany jest w sposób ciągły
Wieloznaczny (zdegenerowany); jeden aminokwas może być zapisany przez więcej niż jeden
kodon.
Ogólna budowa genów
W większości genów są fragmenty kodujące eksony i fragmenty niekodujące introny. Introny są
wycinane z pre-mRNA a sąsiednie eksony są ze sobą łączone.
Pierwszy i ostatni ekson zazwyczaj posiadają sekwencje które nie ulegają translacji
Sekwencja intronu rozpoczyna się od motywu GT miejsce donorowe a kończy motywem AG
miejsce akceptorowe.
Dojrzały mRNA zawiera czapeczkę (zmieniony nukleotyd 7 –metyloguanina) i fragment poliA
(poliadenylowy)
Przed miejscem gdzie rozpoczyna się transkrypcja znajduje się rejon promotorowy, mogą się tam
przyłączać białka regulatorowe i to decyduje o tym czy dany gen ulegnie ekspresji czy nie.
Transkrypcja - przepisanie pojedyńczej nici DNA (o orientacji od końca 3’do 5’) do pre-mRNA
to pierwszy etap ekspresji genów
Tworzy się kompleks transkrypcyjny
Polimeraza RNA rozpoznaje specyficzne miejsce w którym podwójna helisa otwiera się i zaczyna
się rozwijać rozpoczyna się synteza RNA inicjacja
Polimeraza RNA klasy II wiąże się z amanityną pochodząca z muchomora sromotnikowego
elongacja.
Po przesunięciu się polimerazy RNA DNA zwija się w podwójną helisę
Terminacja – polimeraza RNA odłącza się od DNA
Transkrypcja rozpoczyna się tuż przed sekwencją genu w miejscu zwanym promotorem (przyłącza
się tam polimeraza RNA)
INICJACJA TRANSKRYPCJI
Rejon promotorowy leży przed miejscem startu transkrypcji
25-30 nukleotydów przed jest TATA box
Tworzenie kompleksu transkrypcyjnego; Przyłączenie TBP (TATA binding protein), a następnie
TFIID(czynnika asocjującego z TBP), zmiana konformacji DNA
Przyłączenie czynnika TFIIA i TFIIB
przyciąganie i łączenie kompleksu pol II i TFIIF
Przyłączenie TFIIE a następnie TFIIH
Kompleks zajmuje obszar na DNA od
-30 do + 30 nukleotydu (razem 60)
Przebieg transkrypcji
Translacja
Podczas translacji sekwencja kodonów w mRNA jest przekładana na odpowiednią sekwencję
aminokwasów.
Istnieje faza odczytu trójek nukleotydów (ramka odczytu) która zaczyna się od kodonu startowego
Translacja odbywa się w rybosomach umieszczonych w cytoplaźmie do których przyłącza się
mRNA
Do mRNA przyłączają się kolejne tRNA z odpowiednim antykodonem komplementarnym do
kodonu. Na drugim końcu tRNA znajduje się specyficzny dla tego kodonu aminokwas jest on
następnie przyłączany do nici białka (wiązanie peptydowe).
.
Translacja składa się z czterech faz:
aktywacji inicjacji elongacji terminacji
W aktywacji właściwy aminokwas jest dołączany do właściwego tRNA za pomocą wiązania

estrowego , powstałego przez reakcję grup karboksylowej aminokwasu i grupy OH przy końcu 3’
tRNA. Taki zespół określa się mianem aminoacylo-tRNA.
.
Inicjacja translacji ma miejsce, kiedy mała podjednostka rybosomu przyłącza się do końca
5' mRNA. Do małej podjednostki przyłącza się duża podjednostka rybosomu. Na podjednostce 50s
uaktywniają się dwa miejsca:
P - miejsce peptydowe i A - miejsce akceptorowe.
Pierwszy aminoacylo-tRNA ustawia się w miejscu P.
.
Elongacja ma miejsce, kiedy następny aminoacylo-tRNA przyłącza się do rybosomu w miejscu A.
Następnie proces translacji zachodzi na zasadzie komplementarności kodonu mRNA
z antykodonem na tRNA.
Rybosom i tRNA są tak ukształtowane, aby dwa aminokwasy, przyłączone do tRNA zajmujące w
rybosomie miejsca A i P znajdowały się blisko siebie. Dzięki temu zachodzi reakcja między grupą
aminową i karboksylową - dwa aminokwasy łączą się.
.
Ten proces - tworzenie wiązań peptydowych jest katalizowany przez peptydylotransferazę
- rybozym (rRNA) wchodzący w skład rybosomu.
Po syntezie, tRNA szybko zwalnia miejsce P i wraca do cytoplazmy, z kolei aminoacylo-tRNA
ulega przesunięciu z miejsca A na miejsce P. Proces ten nazywamy translokacją. Jednocześnie
przesuwa się także mRNA. Wielkość tego przesunięcia wynosi zawsze trzy nukleotydy
Na miejsce A nasuwa się nowy tRNA zawierający antykodon odpowiadający kolejnemu kodonowi
na mRNA. Proces elongacji powtarza się aż do napotkania przez podjednostkę mniejszą rybosomu
w miejscu A kodonu stop (UAA, UAG lub UGA). Tych trójek kodonowych, w normalnych
warunkach, nie koduje żaden tRNA.
.
terminacja translacji. Łańcuch polipeptydowy zostaje uwolniony do cytoplazmy, t RNA zostaje
oddzielone od mRNA, a rybosom rozpada się na podjednostki, które mogą zostać ponownie
wykorzystane do inicjacji translacji kolejnego mRNA.
W fazie wydłużania mamy powtarzające się po sobie reakcje;
1. rozpoznanie kodonu przez tRNA
2. przyłączenie peptydu do kolejnej reszty aminokwasowej wiązanie peptydowe
3. przesunięciem się rybosomu o trzy nukleotydy wzdłuż mRNA
Terminacja następuje po napotkaniu na nici mRNA kodonu stop
Powstały polipeptyd opuszcza rybosom, który ulega dysocjacji na dwie podjednostki
Fałdowanie polipeptydu
Powstanie struktury trzeciorzędowej
Do modyfikacji translacyjnych zaliczają się:
Obróbka proteolityczna - proteazy usuwają zbędne fragmenty
N-acetylacja, N-metylacja, N-formylacja - dołączenie grupy acetylowej, metylowej i metioniny
Hydroksylacja - dołączenie grupy hydroksylowej -OH\
Fosforylacja - aktywacja białka przez dołączenie grupy fosforylowej
Defosforylacja- dezaktywacja przez odłączenie grupy fosforylowej
dołączenie lipidów i metali
Glikozylacja - enzymatyczne przyłączenie reszt cukrowych do białka
Ubikwitynacja - przyłączenie ubikwityny i późniejsza degradacja białka

2. Mutacje strukturalne :
1. Duplikacja powielenie genów
2. delecja utrata genów
3. inwersja przestawienie kolejności genów
4. translokacja przeniesienie genów na inny chromosom
5. chromosom kolisty
6. izochromosom niewłaściwy podział centromeru
Delecje
Delecje powstają na skutek pęknięcia chromosomu i następującej utraty materiału genetycznego;
dotyczą zwykle dość dużej liczby genów i wywołują charakterystyczne zespoły chorobowe; są
możliwe do zobaczenia pod mikroskopem
Delecja terminalna powstaje, kiedy dochodzi do pojedynczego pęknięcia chromosomu i utraty
materiału z końca chromosomu
Delecja interstycjalna jest efektem dwóch pęknięć chromosomu i utraty materiału pomiędzy nimi
Zespół cri-du-chat
Delecja krótkiego ramienia 5p chromosomu
cechy kliniczne są związane z regionami chromosomu
piskliwy płacz dziecka w okresie niemowlęcym występuje w przypadku utraty proksymalnego
5p.15.3
pozostałe charakterystyczne cechy zespołu występują przy utracie małego regionu w obrębie
centralnego 15p,12.2
zespół cechuje się upośledzeniem umysłowym (średnie IQ około 35), małą głową i dość
charakterystycznym wyrazem twarzy
częstość występowania dużych wad rozwojowych jest zmienna
chorzy rzadko dożywają pełnoletności
Mikrodelecje to podtypy delecji chromosomów, które można zaobserwować tylko w barwionych
chromosomach lub przy zastosowaniu metod genetyki molekularnej
generalnie obejmują delecje całej serii sąsiadujących genów
Inwersje
są one rzadko przyczyną choroby u nosiciela inwersji, ale inwersja która przerywa gen czynnika
krzepliwości VIII, jest przyczyną poważnej hemofilii A
Rodzice z inwersjami i ich dzieci
Translokacja – wymiana materiału genetycznego pomiędzy niehomologicznymi chromosomami
przynajmniej 1/500 osób jest nosicielem translokacji zrównoważonej
Translokacje wzajemne są powodowane przez dwa pęknięcia na różnych chromosomach, z
następującą wymianą materiału
Translokacje robertsonowskie to takie, gdzie dwa krótkie ramiona dwóch niehomologicznych
chromosomów zostają utracone, a ramiona długie łączą się w centromerze, tworząc pojedynczy
chromosom
Translokacja robertsonowska chromosomów 14 i 21
w zależności od segregacji w gametach matki,
potomstwo może mieć:
-trisomię 21 (zespół Downa)
-kariotyp prawidłowy
-translokację zrównoważoną z prawidłowym fenotypem
-monosomię 21
-płody z trisomią 14 i monosomią 14 nie przeżywają do porodu
(należy zwrócić uwagę, że te trisomie i monosomie są genetycznie identyczne z tymi, które
powstały w wyniku nondusjunkcji, ponieważ tylko długie ramiona tych chromosomów zawierają
istotny materiał genetyczny)

Chromosom kolisty
często ulegają utracie, co powoduje monosomię chromosomową w niektórych komórkach
zostały opisane przynajmniej w jednym przypadku dla każdego autosomu człowieka
chromosomy pierścieniowe 13 i 14 wywołują zespół związany z upośledzeniem umysłowym
zespół pierścieniowego chromosomu 20 powodujący padaczkę i upośledzenie umysłowe
Izochromosomy
powstają w rezultacie podziału wzdłuż osi prostopadłej do zwykłej osi podziału chromosomu
posiadają dwie kopie jednego ramienia i żadnej kopii drugiego ramienia
.
3. Mutacje genomowe (chromosomowe) – zaburzenia segregacji
euploidia zwiększony cały zestaw chromosomów np. z 2n na 4n
Aneuploidia zmiana liczby jednego z chromosomów
Monoasomia (1 kopia zamiast 2)
Nullisomia (0 kopi zamiast 2)
Trisomia (3 kopie zamiast 2)
MOZAIKOWATOŚĆ
•
Obecność u danej osoby dwóch lub więcej linii komórkowych, które powstały z pojedynczej
zygoty
•
Zazwyczaj zygota ma trisomię, a prawidłowa linia powstaje w następnym podziale
mitotycznym
•
Rzadziej zygota jest prawidłowa (2n) a trisomiczna (2n+1) linia komórkowa jest wynikiem
nondysjunkcji chromosomów w następnym podziale mitotycznym, powstaje wtedy również
linia z monosomią (2n-1), która jednak zazwyczaj ginie
•
Obecność prawidłowej linii komórkowej łagodzi zazwyczaj objawy kliniczne np. ok. 1%
pacjentów z trisomią 21 ma mozaikę prawidłowej i trisomicznej linii komórkowej (47,XX,
+21/46, XX)
MOZAIKOWATOŚĆ GONADALNA
Nieprawidłowa linia występuje tylko w gonadach
Osoba o pozornie prawidłowym kariotypie
Może mieć wysokie ryzyko posiadania dzieci z abberacją chromosomową
ZESPOŁY ABBERACJI LICZBOWYCH AUTOSOMÓW
ZESPÓŁ DOWNA
Obecność dodatkowego chromosomu 21
Występuje 1:700 urodzeń
Ok. 60% zarodków i płodów ulega samoistnemu poronieniu
20% płodów rodzi się martwych
Prawdopodobieństwo wystąpienia z. Downa znacznie wzrasta wraz z wiekiem matki
ZESPÓŁ DOWNA objawy
Niedorozwój umysłowy
Niski wzrost
Płaska potylica
Szeroka, płaska twarz
Skośne oczy
Krótki nos
Wady serca
Krótkie szerokie dłonie i stopy
Hipotonia mięśniowa
Małpia bruzda

ZESPÓŁ PATAU
dodatkowy chromosom 13
występuje u 1:8000-1:10000 urodzeń
90% żywo urodzonych dzieci nie przeżywa 1 roku
Trisomia 13 (75%)
Translokacja niezrównoważona (20%)
Kariotyp mozaikowy (5%)
ZESPÓŁ EDWARDSA
Trisomia chromosomu 18
Występuje u 1:5000 urodzeń
90% żywo urodzonych dzieci nie przeżywa 1 roku
Kariotyp mozaikowy występuje w niewielkim odsetku przypadków
ZESPÓŁ CRI-DU-CHAT
występuje u 1:50000 – 1:100000
delecja terminalna krótkiego ramienia chromosomu 5
niekiedy powstaje chromosom pierścieniowy
zrównoważona translokacja fragmentu chromosomu 5 u rodziców (20%)
Objawy: upośledzenie umysłowe, mała głowa, niedorozwój żuchwy, małe uszy, zniekształcenia
twarzoczaszki, hipotonia mięśniowa, charakterystyczny płacz dziecka, zaburzenia mowy
ZABURZENIA HETEROCHROMOSOMÓW
Zespół Turnera 45,X
Zespół kobiety 47,XXX
Zespół Klinefeltera 47,XXY
Zespół mężczyzny 46,XX
ZESPÓŁ TURNERA
Objawy:
•
zaburzenia wzrastania (mniejsze rozmiary ciała, niski wzrost)
•
specyficzny fenotyp morfologiczny (obrzęk dłoni i stóp; trójkątna twarz; krótka, szeroka
szyja z podłużnym fałdem skórnym, klatka piersiowa szeroka, narządy płciowe zewnetrzne
niedorozwinięte)
•
wady narządów wewnętrznych (ukł.krążenia u 15%, nerek, kośćca)
•
pierwotna niewydolność jajników (hipoplazja jajników, spłaszczona macica, pierwony brak
miesiączki i pierwotna bezpłodność)
•
specyficzny fenotyp rozwoju i zachowania (trudności w nauce, dyslekcja, IQ w normie)
ZESPÓŁ KLINEFELTERA
Objawy:
brak charakterystycznych objawów przed okresem dojrzewania,
wysoki wzrost, wygląd ciała nieco kobiecy,
ginekomastia
zmiany degeneracyjne w kanalikach nasiennych,
brak spermatogenezy, pierwotna bezpłodność
czasami wtórny zanik jąder
nieznaczne upośledzenie umysłowe i obniżenie IQ
ZESPÓŁ MĘŻCZYZNY 46,XX
Występowanie 1:20000
Hipotezy rozwoju:
Translokacja części lub całego chromosomu Y na ramię krótkie chromosomu X
Utrata chromosomu Y w komórkach zarodka (47,XXY) we wczesnym rozwoju
Mutacja genu związanego z różnicowaniem płciowym
OBOJNACTWO PRAWDZIWE
u jednego osobnika występują tkanki jądra oraz jajnika
defekt molekularny nie jest w pełni poznany (mutacje genów odwrócenia płci)

kariotypy: 46,XX (56%), 46,XY (23%) oraz mozaikowatość 46,XX/46,XY (21%)
OBOJNACTWO RZEKOME
Jest tylko jeden typ gonad a cechy płciowe zewnętrzne i wewnętrzne są charakterystyczne
dla płci przeciwnej
OBOJNACTWO RZEKOME ŻEŃSKIE
występują jajniki
maskulinizacja narządów płciowych zewnętrznych
Przyczyny nadmiaru androgenów (źródło):
płód (wrodzony przerost nadnerczy, blok steroidogenezy nadnerczowej)
matka (guzy hormonalnie czynne nadnerczy i jajników lub leki hormonalne przyjmowane w ciąży)
OBOJNACTWO RZEKOME MĘSKIE
występują jądra
niepełny rozwój narządów płciowych w kierunku męskim (obojnacze lub żeńskie)
kariotyp 46,XY – płeć często trudna do określenia
Przyczyny
hipoplazja lub brak komórek Leydiga
wrodzony defekt syntezy testosteronu
nieprawidłowe działanie androgenów (np. zespół niewrażliwości na androgeny)
Aberracje strukturalne
Niezrównoważone – rearanżacja powoduje dodanie lub utratę materiału chromosomowego
Zrównoważone – rearanżacja nie powoduje dodania lub utraty materiału chromosomowego
Zmiany struktur mogą być spowodowane:
ustawieniem się chromosomów homologicznych nieprawidłowo w linii podczas mejozy
nie naprawionymi lub źle naprawionymi pęknięciami chromosomów podczas mejozy lub mitozy
prawdopodobieństwo pęknięcia może się zwiększyć w przypadku obecności pewnych szkodliwych
czynników zwanych klastogenami (promieniowanie jonizujące, pewne zakażenia wirusowe,
niektóre środki chemiczne)
RODZAJE NIEPRAWIDŁOWOŚCI CHROMOSOMALNYCH U
ZARODKÓW
ZESPÓŁ DOWNA mechanizm powstawania
Hipoteza „chiazm-hormon”
Związek między hormonalnie kontrolowanym tempem przebiegu mejozy a czasem zaniknięcia
chiazm, z wiekiem u kobiet zmienia się poziom hormonów i długość cyklu miesięcznego, co
prowadzi do zwolnienia tempa przebiegu mejozy, zaburzenia hormonalne mają wpływ na
zaburzenia czynności mitochondriów, co wiąże się z dysfunkcją wrzeciona podziałowego
RYZYKO POWTÓRNEGO URODZENIA DZIECKA Z Z.DOWNA
ZESPÓŁ WOLFA-HIRSCHHORNA
Występowanie 1:50000
Delecja terminalna (46,XX,del(4)(p16.3) lub interstycjalna krótkich ramion chromosomu 4
translokacje
czasami powstaje chromosom pierścieniowy (46,XX,r4)
Objawy: niska masa urodzeniowa, niedorozwój psychiczny i fizyczny, małogłowie, szeroka nasada
nosa, niedorozwój żuchwy, układ ust w tzw. rybie usta, duże małżowiny uszne, zaburzenia budowy
narządów płciowych
ZESPÓŁ ANGELMANA
Objawy:
trudności w ssaniu, mały przyrost masy ciała,
zaburzenia mowy
opóźnienie i zaburzenie chodu, charakterystyczny chód „zespół szczęśliwej kukiełki”
Wyszukiwarka
Podobne podstrony:
Genetyczny id 187377 Nieznany
Genetyka 5 id 187498 Nieznany
Algorytmy genetyczne 2 id 57672 Nieznany (2)
genetyka) id 187384 Nieznany
genetyka0006 id 187629 Nieznany
cw 2 genetyka id 100363 Nieznany
genetyka0001 id 187624 Nieznany
CHOROBY GENETYCZNEp id 115004 Nieznany
genetyka0005 id 187628 Nieznany
genetyka0002 id 187625 Nieznany
Medicus Genetyka 1 id 292099 Nieznany
genetyka0004 id 187627 Nieznany
Poradnictwo genetyczne id 18765 Nieznany
genetyka0003 id 187626 Nieznany
choroby genetyczne id 114957 Nieznany
Genetyczny id 187377 Nieznany
Notatki na egzamin genetyka id Nieznany
więcej podobnych podstron