
1
CHEMIA ANALITYCZNA
PROGRAM ĆWICZEŃ LABORATORYJNYCH
(ANALIZA KLASYCZNA Z ELEMENTAMI ANALIZY INSTRUMENTALNEJ)
Analiza wagowa
1.
Jedno z trzech oznaczeń tj.
Ba – BaSO
4
,
Fe – Fe
2
O
3
,
Ni – Ni(Hdmg)
2
.
Analiza miareczkowa
Wyznaczanie pojemności naczyń miarowych
1. Wyznaczanie pojemności kolby miarowej (100 mL) i pipety (25 mL); (ćwiczenie to należy
zacząć w trakcie analizy wagowej).
Alkacymetria
1.
Sporządzanie bezwęglanowego roztworu 0,1 M NaOH i mianowanie tego roztworu
za pomocą wodoroftalanu potasu.
2.
Oznaczanie kwasu solnego.
Redoksometria
1.
Sporządzanie mianowanego roztworu 0,0166 M KBrO
3
(z odważki).
2.
Sporządzanie i mianowanie roztworu 0,1 M Na
2
S
2
O
3
(roztwór Na
2
S
2
O
3
należy sporządzić
wcześniej, aby ustaliło się jego stężenie). Mianowanie za pomocą roztworu KBrO
3
.
3.
Oznaczanie tlenu rozpuszczonego w wodzie wodociągowej metodą Winklera.
Kompleksometria
1.
Sporządzanie mianowanego 0,01 M roztworu EDTA (z odważki).
2.
Oznaczanie twardości wody.
Wprowadzenie do analizy instrumentalnej
Potencjometria pośrednia
1.
Oznaczenie procentowej zawartości kwasu octowego w handlowym occie metodą
miareczkowania potencjometrycznego (potencjometria pośrednia) oraz miareczkowania
klasycznego (roztworem NaOH wobec fenoloftaleiny).
Spektrofotometria
1. Oznaczenie żelaza metodą rodankową.`

2
ZASADY WYDAWANIA I WYKONYWANIA ĆWICZEŃ
Zadania z analizy wagowej wydawane są do zlewek o pojemności 250 mL
w przypadku oznaczania baru i żelaza, lub 400 mL (wąska, wysoka zlewka) w przypadku
oznaczenia wagowego niklu. Wykonuje się jedno oznaczenie z całości otrzymanego
roztworu.
Zadania z metod miareczkowych są wydawane do małych zlewek (25 lub 50 mL).
Roztwór ze zlewki przenosi się ilościowo do kolby miarowej o pojemności 100 mL
(o wyznaczonej pojemności), uzupełnia wodą destylowaną do kreski i dokładnie miesza.
Z tego roztworu pobiera się trzy porcje pipetą (o wyznaczonej pojemności) do kolbek
stożkowych (erlenmayera) i wykonuje trzy równoległe oznaczenia. Wynik końcowy
jest średnią z trzech oznaczeń.
Wyjątki: w kilku przypadkach zadania są wydawane do innych naczyń. Dotyczy
to oznaczenia twardości wody oraz procentowej zawartości kwasu octowego w occie.
Szczegóły są podane w instrukcji wykonania danego oznaczenia.
ZLEWKI MUSZĄ BYĆ BEZWZGLĘDNIE CZYSTE, SUCHE Z ZEWNĄTRZ
I PODPISANE (PISAKIEM) IMIENIEM, NAZWISKIEM ORAZ NUMEREM SALI.
Aby otrzymać zadanie należy wraz ze zlewką przedłożyć preparatkę z prośbą o wydanie
zadania podpisaną przez asystenta. W przypadku braku podpisu asystenta zadania nie będą
wydawane.
LITERATURA
1. Skoog D.A., West D. M. , Holler F. J., Crouch S. R., Podstawy chemii analitycznej,
PWN 2006
2.
Cygański A., Chemiczne metody analizy ilościowej, Warszawa, WNT, 1999, wyd. 5.
3.
Rokosz A., Wprowadzenie do chemii analitycznej, Kraków, UJ, 1980.
4.
Minczewski J., Marczenko Z., Chemia analityczna, t.1 i 2, Warszawa, PWN, 1985; 1997
2001 wyd. zmienione.
5.
Minczewski J., Marczenko Z., Chemia analityczna (analiza instrumentalna), t.3,
Warszawa, PWN, 1985.
6.
Szczepaniak W., Metody instrumentalne w analizie chemicznej, Warszawa, PWN, 1996;
2002 wyd.4.
7.
Cygański A., Ptaszyński B., Krystek J., Obliczenia w chemii analitycznej, Warszawa,
WNT, 2000.
3
ANALIZA WAGOWA
Kilka uwag ogólnych dotyczących pokoju wagowego
1.
W pokoju wagowym powinny być tylko te osoby, które zajmują się ważeniem.
Nie należy robić kolejek do wag!
2.
Należy zachować ciszę i nie otwierać okien ani nie zostawiać otwartych drzwi,
aby zachować stałą temperaturę w pokoju wagowym.
3.
Nie wolno stawiać na konsoli (obok wag analitycznych): eksykatorów, zlewek,
słoików z odczynnikami oraz żadnych innych naczyń z wyjątkiem naczyniek
wagowych, które powinny być postawione na szalce Petriego.
4.
Wszystkie wymienione naczynia oraz odczynniki zostawiamy na stole. Odczynniki
wsypujemy do naczyniek również na stole. Nie wolno wsypywać odczynników
do naczyniek obok wagi ani tym bardziej na wadze! Odczynniki wsypujemy
odpowiednimi łyżeczkami (każdy odczynnik indywidualną łyżeczką), nie zamieniamy
łyżeczek i chronimy je przed zanieczyszczeniem oraz przed zanieczyszczeniem
odczynników.
Ważenie na wadze analitycznej WA-31
Waga analityczna WA-31: granica obciążalności – 200 g;
dokładność ważenia – 0,0001 g
Rzadko jednak odważamy na wagach analitycznych masy powyżej 100 g, gdyż
ważenie zbyt dużych mas może spowodować uszkodzenie wagi i zmniejszenie jej czułości.
Dlatego naczyńka wagowe, w których odważamy substancję powinny być jak najlżejsze.
Wagi analityczne, którymi posługujemy się na ćwiczeniach są to wagi półautomatyczne;
odważniki są zawieszone na odpowiednich haczykach, nakładamy je za pomocą pokręteł
(umieszczonych w podstawie wagi), na odpowiednie dźwignie aby zrównoważyć masę ciała
znajdującego się na szalce.
1.
Waga powinna być prawie zawsze zaaretowana! Odaretowujemy ją zawsze powoli
i bardzo ostrożnie za pomocą odpowiedniej dźwigni znajdującej się w podstawie wagi,
tylko wtedy gdy waga jest w równowadze, tj. przed ważeniem gdy na szalce nie ma
nic, jak również nie nałożono odważników, oraz gdy ciało ważone jest dokładnie
zrównoważone odważnikami.
2.
Nakładanie i zdejmowanie odważników jak również ciała ważonego należy
wykonywać zawsze na wadze zaaretowanej!!!
3.
Drzwiczki wagi otwieramy tylko w celu włożenia na szalkę przedmiotu ważonego
oraz jego zdjęcia z szalki. W czasie ważenia powinny być zamknięte.
4.
Odważniki nakładamy za pomocą pokręteł. Pokrętłami z lewej strony podstawy wagi
nakładamy odważniki gramowe (dziesiątki i jednostki g), z prawej – odważniki
miligramowe (setki i dziesiątki mg). Każdy zakres zarówno z lewej jak i z prawej
strony ma odrębne pokrętło. Nie można używać dwóch pokręteł jednocześnie.
Najmniejszy odważnik jaki możemy nałożyć jest równy 0,01 g tj. 10 mg (drugie
miejsce po przecinku). Trzecie i czwarte miejsce (tj. tysięczne i dziesięciotysięczne

4
części grama) odczytujemy na skali, która jest podświetlona i po odaretowaniu wagi
dokładnie widoczna, w przedniej jej części.
5.
Nie wolno odważać żadnych substancji bezpośrednio na szalce lub na papierku.
Wszystko odważamy w zamkniętych naczyńkach wagowych lub w tyglach. Naczynia
stawiane na szalce muszą być bezwarunkowo czyste i suche o temperaturze pokoju
wagowego. Naczyńka, w których odważamy substancje powinny być możliwie lekkie
(o czym była mowa wyżej). Naczyńka wagowe, kładziemy na szalce chwytając
je szczypcami, lub przez pasek z papieru nigdy palcami. Tygle zawsze chwytamy
szczypcami.
Uwaga: Naczyńka wagowe, które mamy do dyspozycji, należy zabezpieczyć i tak
przechowywać aby nie zamieniać pokrywek, gdyż każde naczyńko ma fabrycznie
dopasowaną pokrywkę, która je szczelnie zamyka. Najlepiej już przed użyciem (także
przed myciem) oznaczyć tym samym numerem naczyńko i pokrywkę. Po umyciu
naczyniek i wysuszeniu ich w suszarce, umieszczamy je w eksykatorze. Naczyńka
wagowe stawiamy zawsze na szalce Petriego (zarówno w suszarce – w czasie suszenia,
jak i w eksykatorze – studzenie i przechowywanie oraz obok wagi – przed ważeniem).
6.
Przed przystąpieniem do ważenia sprawdzamy „położenie zerowe wagi”, delikatnie
ją odaretowując. Zanim odaretujemy sprawdzamy czy szalka jest czysta oraz czy nie
pozostawiono po poprzednim ważeniu nałożonych odważników. Po odaretowaniu
odczekujemy kilkanaście sekund aż ustali się równowaga. Jeśli zero skali nie pokrywa
się z pionową kreską na „szybce” możemy to skorygować za pomocą śrubki
znajdującej się poniżej „szybki”. Jeśli przesunięcie zera jest zbyt duże (trzy lub więcej
kresek) nie korygujemy ponieważ śrubka ma ograniczony zakres działania,
lecz zapisujemy faktyczne przesunięcie zera i po zważeniu ciała wprowadzamy
poprawkę do uzyskanego wyniku ważenia. Jeżeli np. przesunięcie zera przed
ważeniem wynosi (-5) tj. –0,0005 g do wyniku końcowego dodajemy 0,0005 g; jeżeli
przesunięcie zera wynosi (+5) tj. +0,0005 g, wartość tę odejmujemy od wyniku
końcowego.
7.
Przed ważeniem na wadze analitycznej najlepiej ustalić przybliżoną masę ciała
ważonego, ważąc je na wadze technicznej. Dotyczy to szczególnie ważenia
określonych próbek, których wielkość ustalamy dosypując lub odsypując substancji.
Dosypywania i odsypywania substancji do naczyniek wagowych nigdy nie wolno
robić na wadze analitycznej, ani też obok wagi. Próbkę do ważenia
przygotowujemy zawsze na stole. Nawet jeśli znamy masę ciała, które mamy zważyć
na
wadze analitycznej,
po
nałożeniu
odpowiednich
odważników,
nigdy
nie odaretowujemy wagi zdecydowanie i szybko, lecz ostrożnie i powoli obserwując
kierunek przesuwania się skali.
Ogólna zasada: Jeśli skala przesuwa się szybko („ucieka”) w kierunku wartości
ujemnych oznacza to że należy odjąć odważników. Jeżeli skala przesuwa się szybko
w kierunku dodatnich wartości należy dodać odważników. W obu przypadkach
nie odaretowujemy wagi całkowicie (do końca), lecz tylko częściowo i na krótko aby
sprawdzić czy odważniki równoważą ciało. Jeśli skala przesuwa się powoli można
odaretować wagę do końca i po ustaleniu się równowagi odczytać masę ciała
ważonego.
8.
Jeżeli ważymy ciało od razu na wadze analitycznej, bez wcześniejszego zważenia
na wadze technicznej to postępujemy w następujący sposób: Nakładając odważniki
5
zaczynamy od najmniejszego odważnika górnego zakresu tj. od 10 g. Nakładamy więc
10 g i delikatnie uchylamy aretaż wagi na tyle aby zaobserwować kierunek
przesuwania się skali. Jeżeli 10 g jest za mało, dokładamy następne 10 g, i znowu
uchylamy aretaż, jeśli 20 g jest także za mało dodajemy następny odważnik 10 g
i sprawdzamy jak poprzednio. Jeżeli nałożonych 30 g jest za dużo zdejmujemy 10 g
i „przechodzimy” na zakres jednostek gramów. Ponieważ masa ciała zawiera się
pomiędzy 20 a 30 g nakładamy 5 g aby sprawdzić czy masa jest pomiędzy 20 i 25 g,
czy 25 i 30 g. I tak dodając lub odejmując po jednym gramie musimy znaleźć dwa
kolejne odważniki jednogramowe, które stanowią niedomiar i nadmiar w stosunku
do masy ciała ważonego. Zostawiamy więc ten odważnik, który stanowi niedomiar
(przy uchylonym aretażu skala przesuwa się w kierunku +) i „przechodzimy”
na kolejny zakres tj. 0,1 g (nakładamy odważniki miligramowe zaczynając od 500
mg). Np. jeżeli stwierdzimy, że nałożone odważniki 24 g stanowią nadmiar, a 23 g
niedomiar, aby zrównoważyć masę ważonego ciała, nakładamy 23,500 g. I znowu
dodajemy lub odejmujemy po 0,1 g (tj. po 100 mg) tak aby znaleźć dwa kolejne
odważniki z tego zakresu, których jest nadmiar lub niedomiar w stosunku
do ważonego ciała i jak poprzednio „zafiksować” ten zakres. Z kolei „przechodzimy”
na najniższy zakres tj. rząd 0,01 g (setne części) nakładając 0,05 g = 50 mg.
I podobnie dodajemy lub odejmujemy po 0,01 g (tj. po 10 mg) tak aby znaleźć dwa
kolejne odważniki 10 miligramowe po położeniu których jest ich za dużo lub za mało
w stosunku do ważonego ciała. Zostawiamy odważnik stanowiący pewien niedomiar
i odaretowujemy wagę do końca a po ustaleniu się równowagi odczytujemy na skali
„trzecie „ i „czwarte” miejsce tj. dokładną masę ważonego ciała. W czasie
sprawdzania czy nałożony odważnik jest odpowiedni, aretaż wagi jedynie uchylamy,
tzn. odaretowujemy wagę częściowo a nie do końca, lecz na tyle aby zaobserwować
kierunek przesuwania się skali. Zawsze zaczynamy od zakresu najwyższego
i przechodzimy kolejno do najniższego „zafiksowując” każdy zakres.
9.
Po skończonym ważeniu i zapisaniu masy ważonego ciała zdejmujemy ciało ważone
i odważniki (oczywiście przy zaaretowanej wadze), po czym delikatnie wagę
odaretowujemy i sprawdzamy „zero”. Jeśli „zero wagi” uległo przesunięciu należy
dokonać korekty (tj. wprowadzić poprawkę opisaną w p. 6) i powtórzyć ważenie,
nakładając znaną już wartość odważników oraz sprawdzić i upewnić się czy wartość
ostatniego miejsca jest prawidłowo odczytana i zapisana.
„Zero wagi” przed ważeniem i po ważeniu powinno być takie samo.
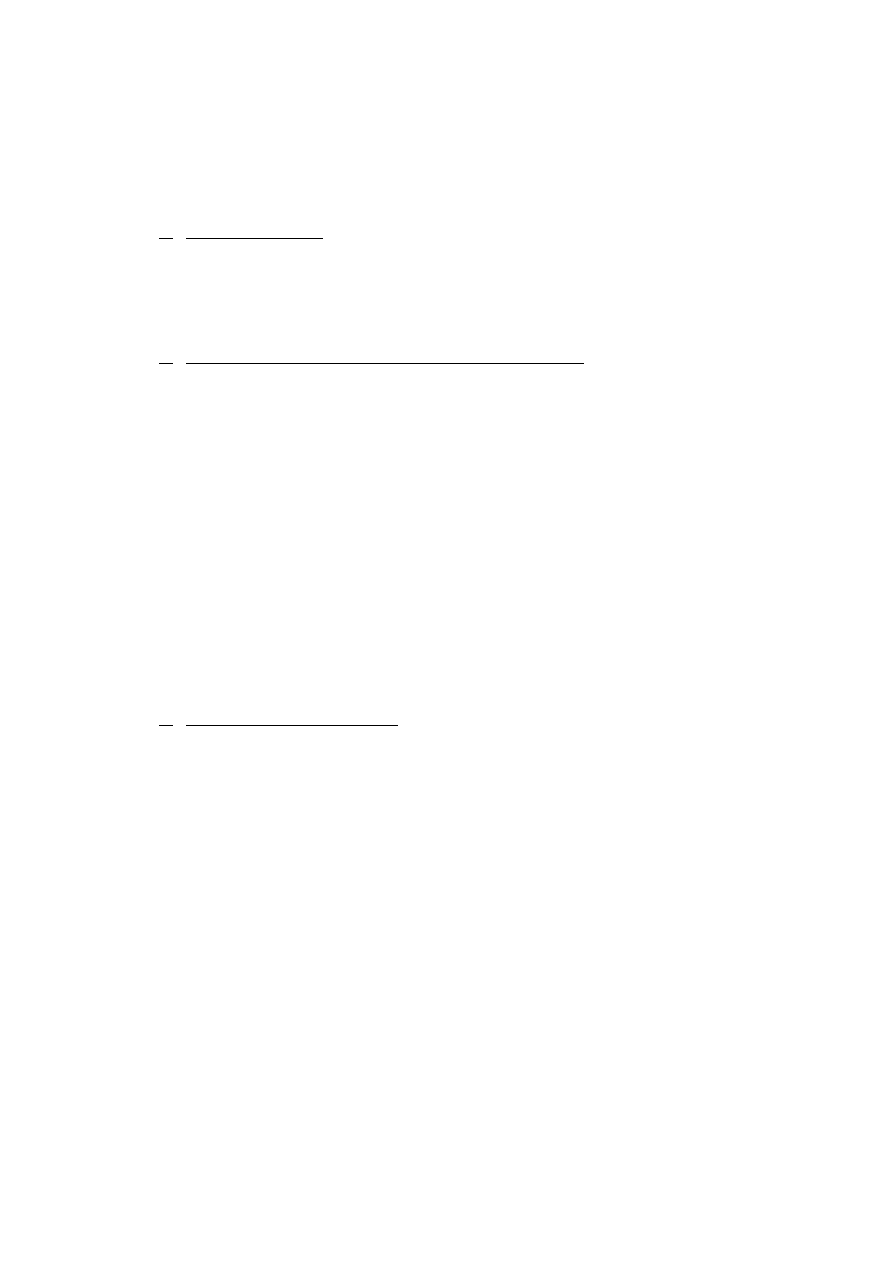
6
Podstawowe czynności w analizie wagowej
1.
Ogrzewanie cieczy
Roztwory ogrzewane zabezpiecza się przed przegrzaniem i wyrzuceniem ze zlewki
w razie nagłego wrzenia, przez umieszczenie w zlewce z ogrzewanym roztworem pręcika
szklanego i nakrycie zlewki szkiełkiem zegarkowym. Pozostawiając zlewkę z wytrąconym
osadem i roztworem na łaźni wodnej, również należy przykryć ją szkiełkiem zegarkowym.
2.
Wytrącanie osadu i sprawdzanie całkowitości strącenia
Roztwór w zlewce przygotowany do wytrącenia osadu (tzn. według przepisu:
rozcieńczony, ogrzany, zakwaszony, itp.) zadaje się odczynnikiem strącającym, wlewając go
ostrożnie, tak aby spływał po pręciku szklanym z szybkością wskazaną w przepisie
i dokładnie miesza się pręcikiem. Jeżeli nie jest podana inna wskazówka w przepisie,
odczynnika strącającego dodaje się dotąd aż osad przestanie się wytrącać. Czasem jest to
trudne do zaobserwowania w trakcie dodawania odczynnika; w takim przypadku należy
odczekać aż osad opadnie na dno i wtedy dodać ostrożnie nową porcję odczynnika
(po ściance zlewki) tak aby nie zmącić osadu i obserwować czy jeszcze zachodzi wytrącanie.
Niezależnie od tego zawsze sprawdza się całkowitość strącenia po rozpoczęciu sączenia.
W tym celu po spłynięciu z sączka pierwszej części przesączu dodaje się do niego
odczynników strącających (wszystkich, jeżeli było ich więcej, np. dimetyloglioksym
i amoniak przy oznaczaniu niklu) i jeżeli osad się jeszcze wytrąca w przesączu, wówczas –
po spłynięciu całej cieczy z sączka – krótko przemywa się sączek, przenosi ilościowo
przesącz do zlewki z osadem, przemywa zlewkę wodą dołączając ją do całości i kontynuuje
się wytrącanie. Jeżeli w pierwszej porcji przesączu nie pojawia się osad, można kontynuować.
3.
Umieszczanie sączka w lejku
Do sączenia osadów w analizie wagowej stosuje się lejki szybkosączące, z długą
wąską nóżką. Sączek ilościowy, z bibuły pozostawiającej nieznaczną ilość popiołu, dobiera
się do postaci osadu, co zawsze jest podane w przepisie. Rozróżniamy sączki twarde
– stosowane do sączenia osadów krystalicznych; oraz średnie i miękkie – do sączenia osadów
koloidalnych. Wielkość użytego sączka zależna jest od ilości osadu a nie od wielkości lejka.
Należy jednak zwrócić uwagę aby sączek nie wystawał powyżej brzegu lejka. Jeżeli
nie dysponuje się sączkiem o odpowiednich rozmiarach, należy wziąć sączek większy, złożyć
dwukrotnie pod kątem prostym (wzdłuż średnicy) i odpowiednio przyciąć jego brzeg,
następnie po włożeniu do lejka, sprawdzić czy powstały kąt odpowiada kątowi ścian lejka;
jeżeli nie – dopasowuje się go przez odpowiednie przesunięcie zagięcia sączka. Następnie
odrywa się kawałeczek sączka (rys. 1) w celu jego lepszego przylgnięcia do ścian lejka,
wkłada się sączek do lejka, zwilża wodą destylowaną i przyciska do ścian lejka tak, aby jego
brzegi przylegały dokładnie do szkła. W końcu napełnia się sączek wodą destylowaną
i sprawdza czy nóżka lejka wypełnia się całkowicie wodą, co przyspiesza proces sączenia.
Oderwany kawałeczek sączka zachowuje się, i po przeniesieniu całego osadu na sączek,
zbiera się nim resztki osadu ze ścian zlewki i dołącza do całości na sączku.

7
Rys. 1. Sposób przygotowania sączka
4.
Przemywanie osadów i przenoszenie ich na sączek
W większości oznaczeń, na początku osad przemywa się przez dekantację. Polega
to na tym, że zlewa się roztwór znad osadu starając się zostawić cały osad na dnie zlewki.
Następnie przemywa się osad kilka razy w zlewce małymi porcjami (10 – 20 ml) cieczy
przemywającej. Zarówno dodawanie cieczy do osadu jak i zlewanie roztworu na sączek
prowadzi się po pręciku szklanym. Po każdorazowym dodaniu roztworu przemywającego
miesza się go z osadem, a następnie odczekuje aż osad opadnie na dno i zlewa się na sączek
ciecz przemywającą. Przemywanie takie wykonuje się około 3 razy. Za czwartym razem
po dodaniu wody lub odpowiedniego roztworu przemywającego, osad miesza się razem
z roztworem i przenosi po pręciku na sączek, uważając aby roztwór z osadem nie był
rozpryskiwany poza sączek. Zarówno sam roztwór jak i roztwór z osadem należy wlewać
na sączek około 1 cm poniżej jego brzegów. Po spłynięciu przesączu do podstawionej
pod lejek zlewki dolewa się nowa porcję cieczy. Po zakończeniu sączenia zbiera się resztki
osadu z pręcika i ze ścian zlewki kawałeczkiem bibuły oderwanej z sączka (rys. 1) i dołącza
do osadu na sączku.
Po przemyciu osadu według przepisu danego oznaczenia, zawsze należy sprawdzić
czy przemycie jest dobre. W tym celu zbiera się wprost z lejka do małej zleweczki, probówki
lub na szkiełko zegarkowe, kilka kropel przesączu i zadaje odczynnikiem pozwalającym
stwierdzić brak lub obecność jonów przeszkadzających w oznaczeniu. Jeżeli jonów tych
w przesączu brak (brak reakcji charakterystycznych), oznacza to, że osad jest dobrze
przemyty.
5.
Przenoszenie sączka do tygla
Po przemyciu osadu i całkowitym spłynięciu cieczy przemywającej podważa się
sączek szpatułką w miejscu gdzie jest potrójna warstwa bibuły (uważając aby nie dotknąć
osadu) i odchyla się go tak, aby móc uchwycić go za jego zewnętrzną stronę bez osadu.
Następnie składa się sączek tak jak pokazano na rys. 2 i umieszcza w tyglu porcelanowym
(wyprażonym uprzednio do stałej masy w takiej samej temperaturze jak osad) w ten sposób,
aby część sączka z osadem znajdowała się na górze.
8
Rys. 2. Sposób składania sączka z osadem
6.
Suszenie, spalanie sączka i prażenie osadu
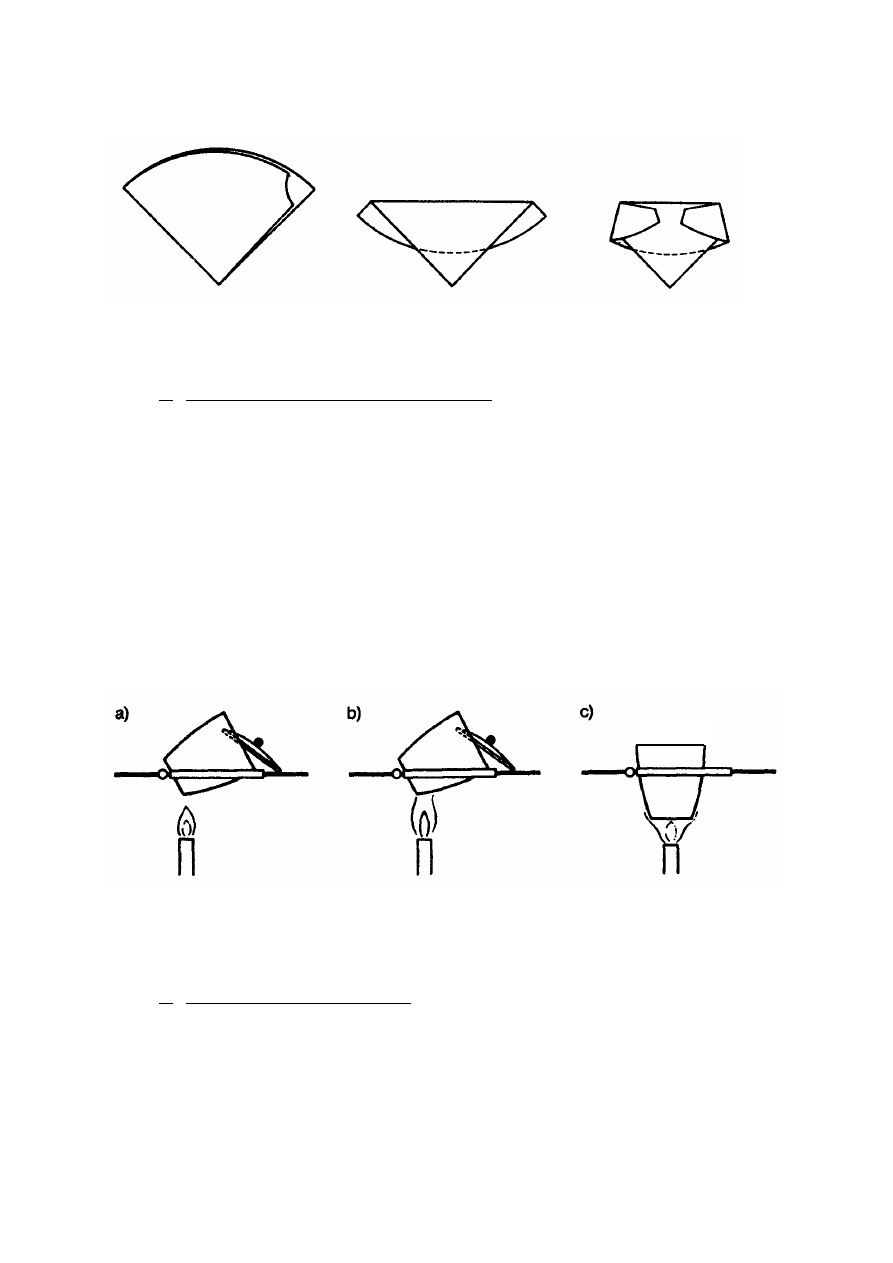
Tygiel z sączkiem wstawia się na szalce Petriego do suszarki i suszy, po czym ustawia
się pochyło na trójkącie porcelanowym i częściowo nakrywa pokrywką (rys. 3). Tygiel
ogrzewa się bardzo małym płomieniem palnika (wachlując palnikiem), tak aby sączek
zwęglał się powoli i nie zapalił się płomieniem. Należy obserwować zwęglanie się sączka
i w razie zapalenia się go płomieniem, natychmiast zamknąć dopływ powietrza do tygla przez
całkowite przykrycie go pokrywką.
Po zwęgleniu sączka należy zwiększyć płomień i powoli spalać aż w tyglu pozostanie
tylko osad. Wówczas tygiel umieszcza się na trójkącie pionowo i praży przez pół godziny,
otwarty lub przykryty pokrywką w zależności od danego oznaczenia, które opisuje dokładnie
przepis. Następnie tygiel zdejmuje się z trójkąta i umieszcza w eksykatorze. Uwaga! Tygiel
zawsze chwyta się szczypcami, których końcówki muszą być idealnie czyste.
Rys. 3. Etapy spalania sączka; a) suszenie i zwęglanie, b) spalanie, c) prażenie osadu
7.
Ważenie i dokładność obliczeń
Eksykator z gorącym tyglem pozostawia się w pokoju wagowym na dwie godziny
w celu osiągnięcia temperatury pokoju wagowego i wagi. Wagi pozostające do dyspozycji
studentów w naszym laboratorium (pokoju wagowym) są bardzo czułe na różnice temperatur
wagi i przedmiotu ważonego, dlatego zaleca się pozostawienie eksykatora z gorącym tyglem
na dwie godziny w pokoju wagowym, aczkolwiek różne podręczniki polecają 0,5 lub
1 godzinę. Czas taki jest zbyt krótki dla uzyskania na naszych wagach powtarzalnych
i rzetelnych wyników ważenia. Eksykatory z gorącymi tyglami należy postawić
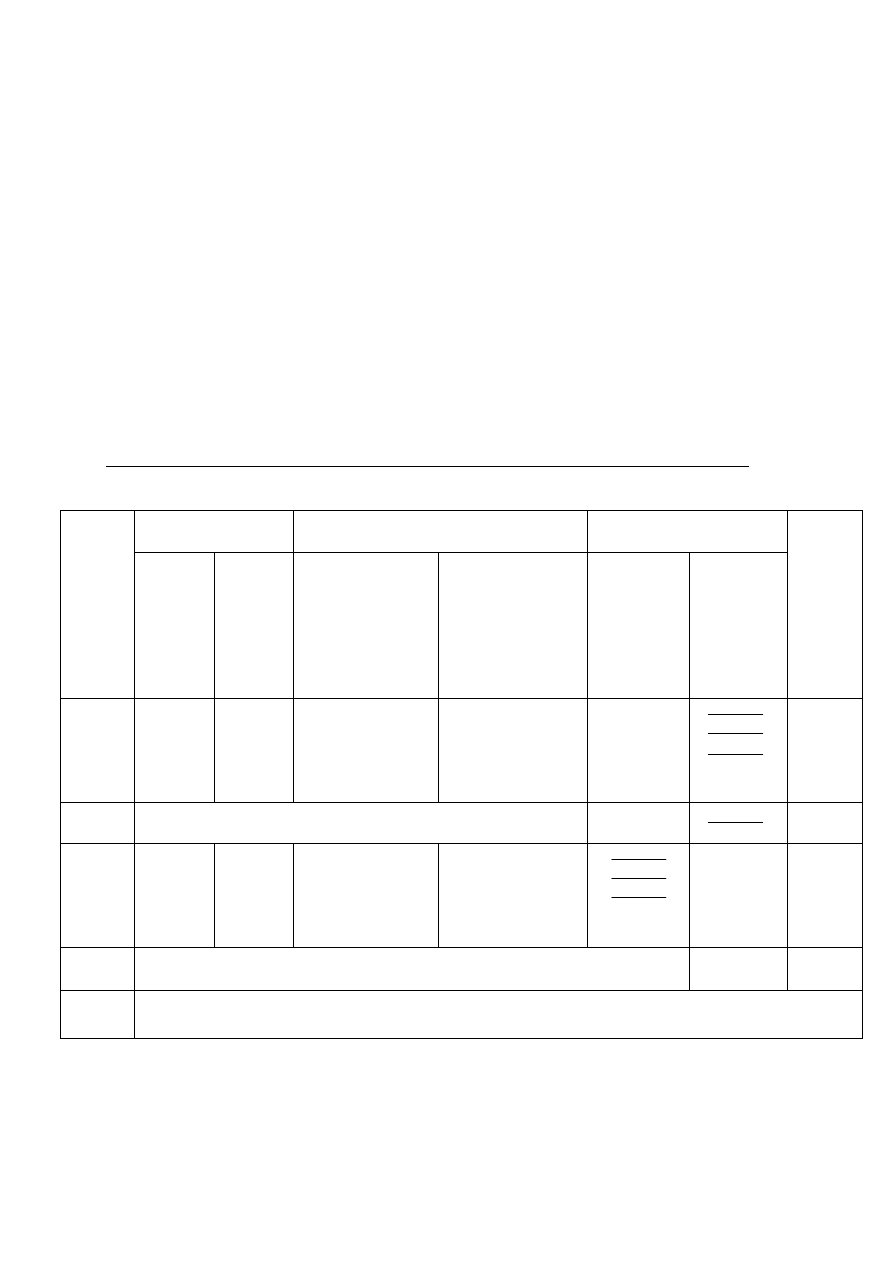
9
na oddzielnym, odpowiednio oznaczonym stole i po 1 godzinie przenieść je na drugi stół
(w środku pokoju wagowego). Umożliwi to wszystkim studentom uzyskanie poprawnych
wyników ważenia i oszczędzi zbędnej pracy wielokrotnego prażenia tygla. Po dwóch
godzinach tygiel przenosi się szczypcami na wagę i waży. Następnie tygiel z osadem praży
się ponownie pół godziny i powtarza powyższe czynności aż uzyska się stałą masę. Jeżeli
w dwóch kolejnych ważeniach masy nie różnią się więcej niż 0,0003 g, można uznać,
ż
e uzyskało się stałą masę.
(Uwaga! przy bardzo dokładnych analizach wymagana jest idealna zgodność, jednakże
w naszych warunkach wykonywania oznaczeń taka „zgodność” jest wystarczająca).
Wyniki analizy należy podawać w gramach z dokładnością do czwartego miejsca
po przecinku. Wykonując poszczególne mnożenia nie należy zaokrąglać otrzymanych liczb
do mniejszej ilości miejsc.
Wyniki ważenia należy zebrać w następującej tabeli:
Tabela 1. Warunki przygotowania i wyniki ważenia tygla podczas oznaczenia wagowego
PRAŻENIE
STUDZENIE
WAŻENIE
DATA
początek
(godzina)
koniec
(godzina)
początek i koniec
studzenia
pod oknem
(godzina)
początek i koniec
studzenia na stole
laboratoryjnym
(godzina)
masa
pustego
tygla [g]
masa tygla
z osadem
[g]
UWAGI
...
...
...
stała masa pustego tygla [g]
...
...
...
stała masa tygla z osadem [g]
masa osadu [g]:

10
BaSO
4
BaSO
4
BaSO
4
BaSO
4
Oznaczanie baru w postaci BaSO
4
Oznaczenie polega na wytrąceniu BaSO
4
kwasem siarkowym (VI) z roztworów soli
baru, zakwaszonych kwasem solnym. Podczas wytrącania zachodzi reakcja:
Ba
2+
+ SO
4
2-
= BaSO
4
(↓)
Odsączony osad BaSO
4
praży się w tyglu porcelanowym w temperaturze 600 – 900˚C
do stałej masy i waży na wadze analitycznej. W czasie prażenia osad BaSO
4
nie zmienia
swego składu.
Odczynniki:
stęż. (lub 2 M) roztwór HCl;
0,1 M roztwór H
2
SO
4
;
ok. 0,01 M roztwór H
2
SO
4
(do przemywania osadu).
Wykonanie:
Otrzymany roztwór soli baru rozcieńczyć w zlewce do objętości 150 – 200 mL,
zakwasić kwasem solnym tak, aby stężenie HCl wynosiło 0,05 – 0,1 M (dodać 6 mL 2M HCl)
i ogrzać do wrzenia. Do gorącego roztworu dodawać po kropli gorący 0,1 M H
2
SO
4
,
energicznie mieszając roztwór, aż do całkowitego wytrącenia osadu siarczanu baru. Następnie
zlewkę z wytrąconym osadem przykrytą szkiełkiem zegarkowym ogrzewać na łaźni wodnej
około 1 godziny w temperaturze 80 – 90
0
C. Po opadnięciu osadu na dno zlewki dodać
ostrożnie (po ściance zlewki) jeszcze kilka kropli 0,1 M H
2
SO
4
w celu sprawdzenia
całkowitości strącenia. Brak zmętnienia świadczy o całkowitym wytrąceniu jonów Ba
2+
.
Wytrącony osad w roztworze macierzystym pozostawić do następnego dnia. Osad sączyć
przez ilościowy sączek (twardy) z bibuły filtracyjnej, przemywając go trzy razy przez
dekantację ok. 0,01 M H
2
SO
4
a następnie na sączku wodą zakwaszoną kilkoma kroplami
rozcieńczonego H
2
SO
4
, a na końcu czystą wodą. Przemywanie prowadzić aż do całkowitego
usunięcia jonów Cl
-
(reakcja z AgNO
3
w obecności HNO
3
). Po przesączeniu i przemyciu
osadu, sączek z osadem wyjąć z lejka, złożyć jak podano na rys. 2, umieścić w wyprażonym
do stałej masy tyglu porcelanowym i wysuszyć w suszarce. Następnie tygiel z sączkiem
ustawić na trójkącie pochyło, częściowo przykryć pokrywką i powoli spalać sączek,
ogrzewając tygiel bardzo małym płomieniem palnika, nie dopuszczając do powstania
płomienia w tyglu. Po spaleniu sączka ustawić tygiel pionowo i prażyć pełnym płomieniem
palnika (temp. 600 – 900
°
C) przez około 45 minut. Następnie gorący tygiel z osadem
przenieść do eksykatora, pozostawić w pokoju wagowym na około 2 godziny i zważyć.
Tygiel z osadem ponownie prażyć około 0,5 godziny, wystudzić, jak poprzednio, i zważyć.
Czynność tę powtarzać aż do uzyskania stałej masy osadu. Masę baru (m
Ba
) obliczyć z masy
BaSO
4
(m ):
m
Ba
= [M
Ba
/ M ] · m = 0,5885 · m [g]
Uwaga!
Jeżeli wyprażony osad BaSO
4
ma barwę szaro-zieloną, świadczy to o częściowej redukcji
BaSO
4
do BaS. W takim przypadku należy zwrócić się do asystenta prowadzącego zajęcia
i pod jego kontrolą przeprowadzić BaS w BaSO
4
przy pomocy stężonego H
2
SO
4
.

11
Fe
2
O
3
Fe
2
O
3
Fe
2
O
3
Fe
2
O
3
Oznaczanie żelaza w postaci Fe
2
O
3
Z
roztworów
soli
ż
elaza(III)
pod
działaniem
amoniaku
wytrąca
się
trudnorozpuszczalny wodorotlenek żelaza (III) (a właściwie uwodniony tlenek Fe
2
O
3
·xH
2
O).
Reakcję wytrącania można zapisać następująco:
Fe
3+
+ 3NH
3 aq
→
Fe(OH)
3
(↓) + 3NH
4
+
(2Fe
3+
+ 6NH
3 aq
→
Fe
2
O
3
·xH
2
O(↓) + 6NH
4
+
)
W czasie prażenia osad traci wodę i przechodzi w Fe
2
O
3
. Z masy Fe
2
O
3
oblicza się masę
ż
elaza.
Odczynniki:
stęż. roztwór HCl;
roztwór NH
3 aq
(1:1) tj. 1 obj. NH
3 aq
stęż. + 1 obj. wody.
Wykonanie:
Otrzymany roztwór rozcieńczyć w zlewce wodą destylowaną do objętości
150 – 200 mL, zakwasić 12 mL 2 M HCl i ogrzewać do wrzenia. Do gorącego roztworu
dodawać po kropli, mieszając, amoniaku o stężeniu 1:1, aż do wystąpienia wyraźnego jego
zapachu w roztworze. Roztwór z osadem przykryć szkiełkiem zegarkowym pozostawić
na łaźni wodnej aż do opadnięcia osadu, po czym sprawdzić całkowitość strącenia dodając
ostrożnie do klarownego roztworu kilka kropli amoniaku. Następnie przesączyć gorący
roztwór przez miękki sączek, starając się jak największą ilość osadu pozostawić w zlewce.
Osad przemyć 3-4 krotnie gorącą wodą przez dekantację, następnie przenieść cały osad
na sączek i przemywać gorącą wodą do całkowitego usunięcia jonów Cl
-
(reakcja z AgNO
3
w obecności HNO
3
). Nieprzemytego osadu nie należy pozostawiać na sączku, gdyż pęka
on wysychając, a woda ścieka pęknięciami nie przemywając osadu. Po przemyciu osadu
i dobrym odcieknięciu, wyjąć sączek z lejka, złożyć, umieścić w tyglu porcelanowym
wyprażonym do stałej masy i suszyć w suszarce. Po wysuszeniu tygiel z sączkiem umieścić
pochyło na trójkącie, częściowo przykryć pokrywką i powoli spalać sączek bardzo małym
płomieniem palnika, nie dopuszczając do zapalenia się sączka. Po spaleniu sączka tygiel
ustawiać pionowo i prażyć pełnym płomieniem palnika Meckera (temp. 800 – 1000
°
C) przez
około 45 minut w otwartym tyglu. Następnie pozostawić tygiel w eksykatorze (w pokoju
wagowym) na dwie godziny i zważyć. Osad w tyglu prażyć ponownie około 0,5 godziny
i po ostudzeniu, jak poprzednio, zważyć. Czynności te powtarzać aż do uzyskania stałej masy
osadu.
Masę żelaza (m
Fe
) obliczyć z masy Fe
2
O
3
(m ):
m
Fe
= [2M
Fe
/ M ] · m = 0,6994 · m [g]
Uwagi!
1.
W temperaturze wyższej niż 1000
°
C Fe
2
O
3
może ulec redukcji do Fe
3
O
4
. Wówczas
osad z brunatno-czerwonego przechodzi w czarny, a wyniki oznaczenia są za niskie.
2.
Jeżeli nie jest wiadome, że całe żelazo w próbce występuje w postaci soli żelaza(III),
należy sprawdzić obecność jonów Fe(II) i jeśli są obecne utlenić je za pomocą H
2
O
2
do Fe(III)

12
Ni(Hdmg)
2
Ni(Hdmg)
2
Ni(Hdmg)
2
Ni(Hdmg)
2
Oznaczanie niklu w postaci dimetyloglioksymianu niklu
W roztworze amoniakalnym dimetyloglioksym, H
2
dmg, wytrąca jony Ni
2+
w postaci
czerwonego osadu wewnątrzkompleksowej soli dimetyloglioksymianu niklu Ni(Hdmg)
2
,
według reakcji:
2C
4
H
8
O
2
N
2
+ Ni
2+
+ 2NH
3 aq
→
Ni(C
4
H
7
O
2
N
2
)
2
(↓) + 2NH
4
+
Osad odsącza się, przemywa gorącą wodą i suszy w temperaturze 110 – 120
°
C (w tej
temperaturze osad nie zmienia składu podczas suszenia).
Odczynniki:
1%-owy alkoholowy roztwór dimetylogliksymu;
2 M roztwór NH
3 aq
;
stęż. roztwór HCl.
Wykonanie:
Analizowany roztwór nie powinien zawierać więcej niż 0,05 – 0,1 g Ni, ponieważ
powstający osad jest bardzo lekki i zajmuje dużą objętość.
Otrzymany roztwór rozcieńczyć w zlewce do objętości około 200 mL, zakwasić 6 mL
2 M HCl i ogrzewać do około 80
°
C. (Uwaga! Po ogrzaniu roztworu należy zgasić palnik lub
odsunąć go daleko od zlewki z roztworem). Do gorącego roztworu dodawać 1%-owy
alkoholowy roztwór dimetyloglioksymu, unikając dużego nadmiaru (na 0,05 g Ni dodać 30
ml roztworu dimetyloglioksymu). Po dodaniu odpowiedniej ilości odczynnika strącającego
nie obserwujemy osadu z powodu kwaśnego środowiska. Następnie, mieszając roztwór,
dodawać do niego kroplami 2M amoniak, (wtedy pojawia się osad) aż do wystąpienia
wyraźnego jego zapachu w badanym roztworze i całkowitego wytrącenia osadu. Zlewkę
z wytraconym osadem przykryć szkiełkiem zegarkowym i pozostawić na łaźni wodnej
o temperaturze 60 – 70
°
C przez około 0,5 – 1 godziny. Następnie odsączyć osad przez tygiel
szklany G4 lub S3 wysuszony do stałej masy w temperaturze 110 – 120
°
C. W przesączu
sprawdzić całkowitość strącenia przez dodanie niewielkiej ilości 1% roztworu
dimetyloglioksymu i dodatkowe zalkalizowanie amoniakiem. Osad przemyć ciepłą wodą
(o temperaturze 60
°
C) do całkowitego wymycia jonów Cl
-
(reakcja z AgNO
3
w obecności
HNO
3
). Sączek z osadem suszyć przez 1 godzinę w temperaturze 110 – 120
°
C, następnie
pozostawić w eksykatorze w pokoju wagowym na 1 godzinę aż do uzyskania temperatury
pokoju wagowego, i zważyć. W przypadku sączków szklanych 1 godzina studzenia jest
wystarczająca. Jeżeli jednak w czasie ważenia zaobserwujemy niestabilność wskazań, czas
studzenia należy przedłużyć do 1,5 godz.. Sączek z osadem suszyć ponownie przez około 0,5
godziny, pozostawić do ostygnięcia, jak poprzednio, i zważyć. Suszenie sączka z osadem
prowadzić do uzyskania jego stałej masy. Masę niklu (m
Ni
) obliczyć z masy osadu
dimetyloglioksymianu niklu m :
m
Ni
= [M
Ni
/ M ] · m = 0,2032 · m [g]

13
ANALIZA MIARECZKOWA
WYZNACZANIE POJEMNOŚCI I KALIBROWANIE NACZYŃ MIAROWYCH
W analizie miareczkowej bardzo istotne znaczenie ma dokładne odmierzanie objętości
cieczy, które przeprowadza się za pomocą naczyń miarowych.
Objętość roztworu może być wyrażana w dm
3
lub litrach. XII Konferencja Miar w 1964 r.
określiła litr jako jednostkę objętości równą 1 dm
3
. Litr znalazł się w wykazie legalnych
jednostek miar nie należących do układu SI, które mogą być stosowane bez ograniczeń
terminowych. A zatem, litr może być używany pełnoprawnie z dm
3
. Obecnie obowiązuje
zależność:
1L = 1 dm
3
= 10
-3
m
3
.
W chemii analitycznej dużo wygodniejsze jest wyrażanie objętości w litrach i tak jest
ona wyrażana w Polskich Normach. 1L = 1000 mL.
Poprzednio występowała zależność: 1L = 1,000028 dm
3
. Różnica, jaka wynika z tych
dwóch zależności wynosi 28 mg/kg, jest więc bardzo mała i poprzednio miała znaczenie tylko
przy bardzo dokładnych pomiarach. Podczas pomiarów, w których ta różnica odgrywałaby
rolę, należy stosować dm
3
.
Przed użyciem naczyń miarowych należy sprawdzić ich pojemność, gdyż często nie są
one dokładnie cechowane i deklarowana pojemność różni się od rzeczywistej. A zatem,
sprawdzenie pojemności naczyń miarowych polega na wyznaczeniu rzeczywistej
ich pojemności, natomiast kalibrowanie na wyznaczeniu i cechowaniu objętości
odpowiadającej deklarowanej pojemności naczynia. Z kalibracją związane jest wyznaczenie
poprawek kalibracyjnych, tj. różnicy między deklarowaną a rzeczywistą pojemnością
naczynia miarowego. Różnicę tę wyraża się najczęściej w procentach i nazywa się błędem
kalibracji.
Sprawdzenie pojemności naczyń miarowych dokonuje się przez zważenie wody
destylowanej, wypełniającej naczynie do kreski (dolny menisk wody powinien stykać się
z kreską), przy czym woda powinna mieć temperaturę pokoju, w którym wykonujemy
ważenie w celu sprawdzenia pojemności naczynia. Należy odróżnić sprawdzanie pojemności
na wlew i na wylew. Pojemność kolb miarowych sprawdza się na wlew, a pipet i biuret na
wylew. Różnica między pojemnością ustaloną na wlew i na wylew wynika z objętości
cieczy, która pozostaje na ściankach naczynia po wylaniu z niej roztworu.
Przy sprawdzaniu pojemności i kalibrowaniu naczyń miarowych należy uwzględnić
następujące poprawki:
1.
Poprawka na zmianę gęstości wody ze zmianą temperatury.
2.
Poprawka na stratę masy ciała ważonego w powietrzu. Poprawka ta wynika z faktu,
ż
e ważenie wykonujemy w powietrzu a nie w próżni.
3.
Poprawka na zmianę objętości naczynia ze zmianą temperatury powietrza.
Z wymienionych poprawek największe znaczenie ma poprawka związana ze zmianą
gęstości wody wraz z temperaturą.
Sumaryczne wartości poprawek dla różnych temperatur i obliczona masa wody, którą
zajmuje w kolbie miarowej pojemność dokładnie 1 litra w temp. 20
o
C, podaje tabela 1.
W tabeli tej
ΣΣΣΣ∆∆∆∆
m (kolumna 3) oznacza sumaryczną wartość (w gramach) wszystkich
trzech w/w poprawek w zależności od temperatury. Kolumna 4 podaje masę wody zajmującej
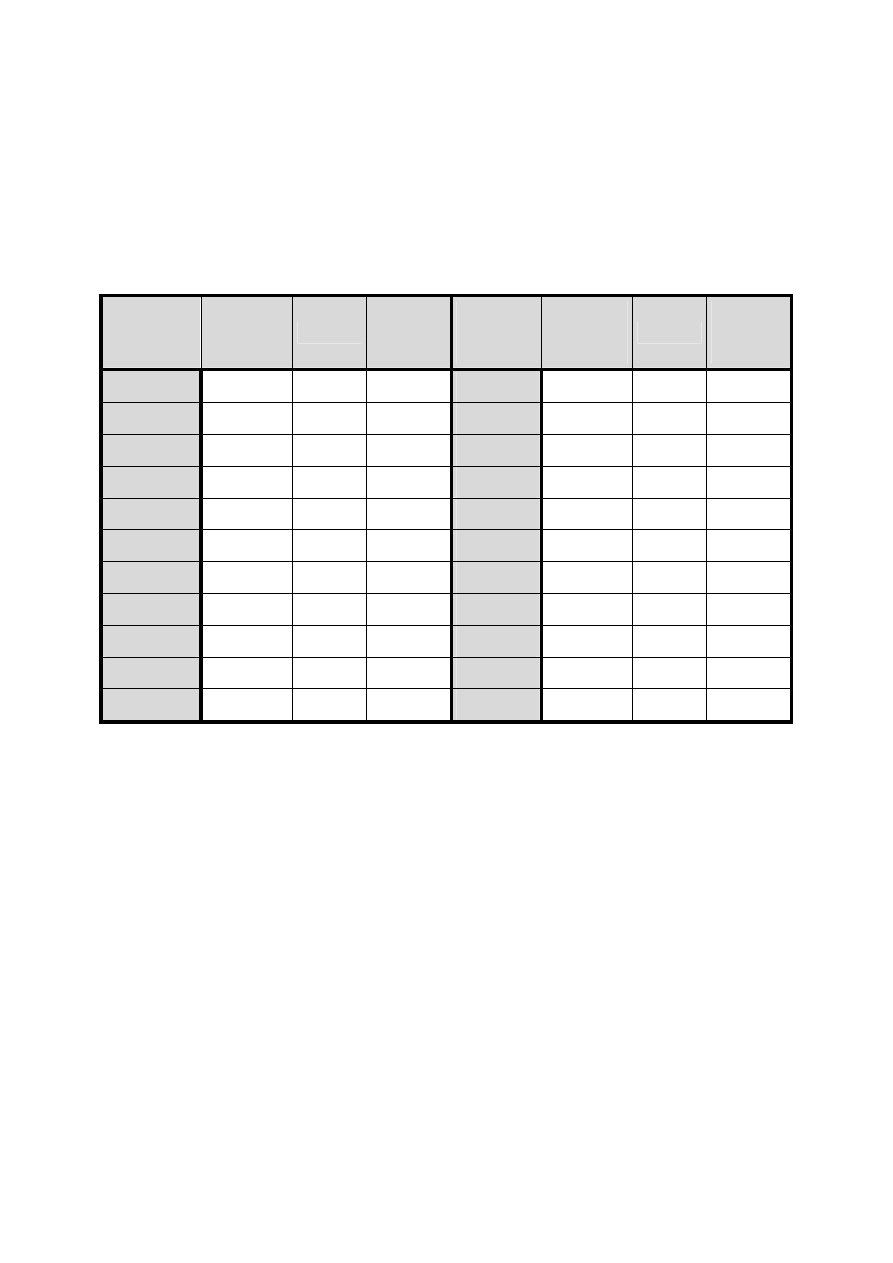
14
w temperaturze 20˚C objętość 1 L w zależności od temperatury pomiaru. Aby obliczyć
pojemność kolby na podstawie masy wody w niej zawartej należy wyznaczoną masę wody
pomnożyć przez 1000/(1000 -
ΣΣΣΣ∆∆∆∆
m).
Tabela 2. Masa wody zajmującej w temperaturze 20°C objętość 1L w zależności od
temperatury pomiaru
Tempera-
tura
°C
Gęstość
wody
g/mL
ΣΣΣΣ
∆∆∆∆
m
Masa
wody
g
Tempera-
tura
°C
Gęstość
wody
g/mL
ΣΣΣΣ
∆∆∆∆
m
Masa
wody
g
15
0,99913
2,07
997,93
26
0,99682
4,07
995,93
16
0,99897
2,20
997,80
27
0,99655
4,31
995,69
17
0,99880
2,34
997,66
28
0,99627
4,55
995,45
18
0,99862
2,49
997,51
29
0,99598
4,82
995,18
19
0,99843
2,65
997,35
30
0,99568
5,08
994,92
20
0,99823
2,83
997,17
31
0,99537
5,38
994,62
21
0,99802
3,00
997,00
32
0,99506
5,65
994,35
22
0,99780
3,20
996,80
33
0,99473
5,94
994,06
23
0,99757
3,41
996,59
34
0,99400
6,25
993,75
24
0,99733
3,62
996,38
35
0,99406
6,55
993,45
25
0,99708
3,84
996,16
Wyznaczanie pojemności pipet i kolb miarowych
Studenci w pracowni analizy ilościowej wyznaczają pojemność kolb (100 mL) i pipet
(25 mL) miarowych.
Pojemność kolby miarowej na wlew sprawdza się przez wyznaczenie masy wody
destylowanej zawartej w kolbie. Waży się najpierw kolbę pustą, idealnie suchą, a następnie
napełnioną wodą do kreski (szyjka nad kreską powinna być sucha), na wadze technicznej
o dokładności odczytu masy 0,01 g. Czynności te powtarza się kilka razy (co najmniej pięć
razy). Po każdym zważeniu kolby z wodą należy odlać trochę wody, dopełnić ponownie
i zważyć. Należy za każdym razem zmierzyć temperaturę wody. Wyniki należy zebrać w
tabeli 3.
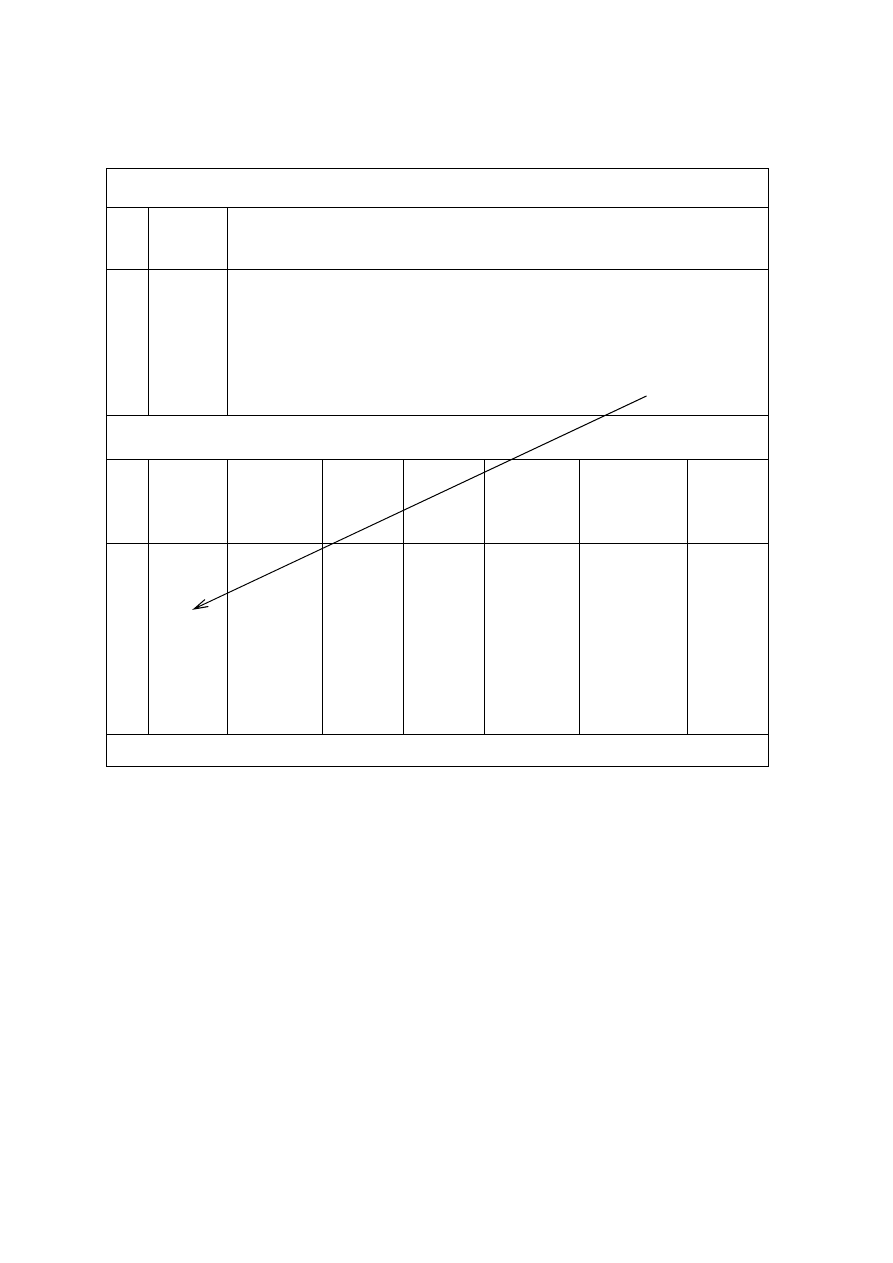
15
Tabela 3. Wyznaczenie pojemności kolby miarowej na 100 mL
Wyznaczenie masy pustej kolby
lp.
masa
pustej
kolby [g]
1
2
3
4
5
średnia masa pustej kolby [g]: ..................
Wyznaczenie pojemności kolby
lp.
ś
rednia
masa
pustej
kolby [g]
masa
kolby
z wodą
[g]
masa
wody
[g]
tempe-
ratura
wody
[°C]
poprawka
Σ
∆
m
[g]
pojemność
kolby* [mL]
Uwagi
1
2
3
4
5
6
7
8
średnia pojemność kolby [mL]:
* V
kolby
= m
wody
⋅
1000/(1000 -
Σ
∆
m) [mL]
W celu wyznaczenia pojemności pipety na wylew wyznacza się masę wylanej z niej
wody. Zgodnie z zasadami posługiwania się pipetą, do uprzednio zważonego naczyńka
wagowego wlewa się zawartość wody z pipety i zamknięte naczyńko waży się na wadze
analitycznej.
Z uwagi na to, że nie dysponujemy odpowiednio dużymi naczyńkami wagowymi,
przy sprawdzaniu pojemności pipety, porcje wody wylane z pipety będziemy ważyć w
kolbie miarowej o pojemności 100 mL. W tym celu ważymy pustą kolbę (w tym przypadku
nie musi być wewnątrz sucha) na wadze technicznej o dokładności odczytu 0,01 g,
a następnie z porcją wody wlanej prawidłowo z pipety. Do tej ilości dodajemy kolejne porcje
wody, ważąc po każdej prawidłowo wlanej porcji. W ten sposób bardzo szybko możemy
wykonać 4 pomiary (pierwsza seria pomiarów). Sprawdzenie pojemności pipety powtarzamy
w opisany sposób (druga seria) uzyskując 8 pomiarów. Różnica między zgodnymi wynikami
powinna być mniejsza niż 0,05%. W czasie wykonywania powyższych czynności
kontrolujemy temperaturę używanej wody. Najlepiej jest wlać pewną ilość wody do dużej
16
zlewki, zanurzyć w niej termometr i zapisywać temperaturę wody, którą ważymy. Wyniki
należy zebrać w tabeli 4.
Posługując się pipetą należy pamiętać, że odmierzane roztwory powinny spływać z pipety
zawsze w taki sam sposób, w jaki woda spływała przy sprawdzaniu lub kalibrowaniu pipety.
Szczególnie ściśle musi być przestrzegany ustalony czas czekania (20 s) po spłynięciu
odpowiedniej ilości wody, lub roztworu z pipety. Znając masę wody spuszczonej z pipety
oraz jej temperaturę oblicza się pojemność pipety, korzystając z danych zawartych w tabeli 2.
Tabela 4. Wyznaczenie pojemności pipety miarowej na 25 mL
Seria I
lp.
masa kolby
przed
dodaniem
porcji wody
z pipety [g]
masa
kolby po
dodaniu
porcji
wody
z pipety
[g]
masa
wody
[g]
tempe-
ratura
wody
[°C]
poprawka
Σ
∆
m
[g]
pojemność
pipety*
[mL]
Uwagi
1
2
3
4
m
m + V
p
m + 2V
p
m + 3V
p
m + V
p
m + 2V
p
m + 3V
p
m + 4V
p
Seria II
lp.
masa kolby
przed
dodaniem
porcji wody
z pipety [g]
masa
kolby po
dodaniu
porcji
wody
z pipety
[g]
masa
wody
[g]
tempe-
ratura
wody
[°C]
poprawka
Σ
∆
m
[g]
pojemność
pipety* [mL]
Uwagi
1
2
3
4
m
m + V
p
m + 2V
p
m + 3V
p
m + V
p
m + 2V
p
m + 3V
p
m + 4V
p
średnia pojemność pipety [mL]:
* V
pipety
= m
wody
⋅
1000/(1000 -
Σ
∆
m) [mL]
Często w analizie stosuje się określoną kolbę wraz z określoną pipetą. Na przykład
otrzymany roztwór do oznaczenia przenosi się ilościowo do kolby miarowej na 100 mL
i po rozcieńczeniu wodą destylowaną (lub innym rozpuszczalnikiem) do kreski oraz
dokładnym wymieszaniu roztworu, pobiera się pipetą do poszczególnych oznaczeń po 25 mL
roztworu. W takiej sytuacji należy określić współmierność kolby z pipetą. W tym celu
wyznaczoną objętość (lub masę) wody w kolbie (na wlew) dzieli się przez wyznaczoną

17
objętość (lub masę) wody zawartej w pipecie (na wylew). Należy przy tym zwracać
uwagę na temperaturę wody stosowanej podczas pomiarów. Jeżeli temperatura wody
w czasie pomiarów jest stała aby wyznaczyć współmierność kolby z pipetą wystarczy
obliczyć stosunek mas wody w kolbie i pipecie, bez uwzględniania poprawek.
Wyznaczenie współmierności jest bardzo ważne. Dokładny wynik oznaczenia danego
składnika pomnożony przez źle wyznaczoną współmierność prowadzi do złego wyniku
analizy.
Użytkowanie kolb miarowych i pipet
Istnieją pewne ustalone zasady posługiwania się naczyniami miarowymi. Ścisłe ich
przestrzeganie zmniejsza możliwości błędów, jakie są związane z użytkowaniem tych naczyń.
Kolba miarowa służy do sporządzania roztworów o określonym stężeniu oraz
do rozcieńczania roztworów. Sporządzając roztwór przenosi się ilościowo odważoną
substancję, z naczynia wagowego do kolby przez lejek z długą nóżką, spłukując dokładnie
naczyńko i lejek. Po przeniesieniu ilościowym substancji do kolby, miesza się zawartość
ruchem okrężnym do całkowitego rozpuszczenia substancji. Następnie dolewa się wody
mieszając cały czas ruchem okrężnym. Po dopełnieniu zawartości do kreski tak aby najniższy
punkt menisku zetknął się z kreską na szyjce kolby, zamyka się kolbę szczelnym, suchym
korkiem i miesza się roztwór odwracając kilkanaście razy kolbę z zawartością dnem do góry.
Należy przy tym zwracać uwagę, aby za każdym odwróceniem powietrze przechodziło
od korka do dna kolby i odwrotnie. Ciecz, którą napełnia się kolbę, powinna mieć temperaturę
bliską 20˚C. W czasie napełniania kolby i pobierania z niej roztworu pipetą temperatura
roztworu nie powinna się zmieniać. Dopełniając kolbę miarową do kreski nie należy zwilżać
szyjki kolby powyżej kreski. Dopełnianie kolby kończy się dodając ciecz wkraplaczem
lub pipetą.
Uwaga: Każda kolba powinna być zamknięta dokładnie dopasowanym korkiem niezależnie
od tego czy w niej jest jakiś roztwór, czy jest pusta.
Prawidłowy sposób pipetowania. Dokładnie oczyszczoną, przemytą wodą
destylowaną i suchą z zewnątrz pipetę zanurza się dolnym końcem w cieczy pipetowanej,
zasysa ciecz do ok. 1/5 pojemności pipety, zatyka pipetę palcem wskazującym, zmienia się
położenie pipety na poziome i dokładnie przemywa wewnątrz pipetę cieczą pipetowaną.
Następnie ciecz tę się wylewa (spuszcza). Czynność tę powtarza się jeszcze jeden lub dwa
razy, po czym przystępuje się do właściwego pipetowania.
Suchą z zewnątrz pipetę zanurza się w roztworze na taką głębokość, aby podczas
wciągania roztworu nie zassać powietrza w wyniku obniżenia się poziomu cieczy w naczyniu,
z którego pipetuje się. Następnie zasysa się roztwór do pipety, napełniając ją nieco powyżej
kreski i zatyka się pipetę palcem wskazującym. Wyjmuje się pipetę z kolby (lub innego
naczynia zawierającego pipetowany roztwór), osusza z zewnątrz kawałkiem bibuły i ustawia
się pipetę w pozycji pionowej dotykając jej dolnym końcem suchego pomocniczego naczynia
szklanego. Zwalniając nieco palec zamykający pipetę spuszcza się powoli nadmiar roztworu
z pipety aż do ustalenia dolnego menisku cieczy na wysokości kreski na szyjce pipety
(oko musi być na poziomie obserwowanej kreski) po czym zamyka się szczelnie dociskając
palec wskazujący. Tak napełnioną pipetę roztworem przenosi się nad przygotowane naczynie
(np. kolbę stożkową w przypadku miareczkowania), dotyka się jej końcem ścianki tego
naczynia (przechylonego tak aby pipeta była w pozycji pionowej) i usuwając palec
wskazujący spuszcza się zawartość. Po swobodnym spłynięciu roztworu z pipety trzyma się
jeszcze pipetę w tej samej pozycji pionowej przez 20 s aż spłynie cały roztwór zawarty
w pipecie. Odrywa się pipetę od ścianki naczynia z pozostałą niewielką ilością roztworu

18
w końcówce pipety. Nie wolno wydmuchiwać ani strząsać resztek cieczy z pipety, nie należy
również dotykać końcem pipety powierzchni cieczy.
METODY MIARECZKOWE
Analiza miareczkowa jest działem analizy ilościowej, której podstawą jest
miareczkowanie. Miareczkowanie to czynność polegająca na dodawaniu titranta tj. roztworu
zawierającego reagent o znanym stężeniu, do roztworu zawierającego jeden lub więcej
oznaczanych składników. Roztwór titranta dodaje się z biurety stopniowo, małymi porcjami
(miarami), stąd nazwa – analiza miareczkowa.
Aby móc oznaczyć daną substancję w roztworze trzeba znaleźć sposób, który pozwoli łatwo
wyznaczyć punkt, w którym cały oznaczany składnik przereagował z titrantem. W tym
punkcie należy zakończyć miareczkowanie i zmierzyć (odczytać na biurecie) objętość titranta.
Punkt
miareczkowania
(objętość
titranta),
który
odpowiada
(teoretycznie)
stechiometrycznemu przereagowaniu oznaczanego składnika z dodawanym titrantem nazywa
się punktem równoważności miareczkowania (PR). Istnieją różne sposoby pozwalające na
ustalenie tego punktu. Punkt miareczkowania (objętość titranta), w którym wystąpi
odpowiednia zmiana, świadcząca o osiągnięciu lub nieznacznym przekroczeniu punktu
równoważności nazywa się punktem końcowym miareczkowania (PK). W idealnym
przypadku, do którego dążymy, punkt równoważności pokrywa się z punktem końcowym.
Znając
objętość
roztworu
titranta
odpowiadającą
punktowi
końcowemu
miareczkowania (PK) oraz jego dokładne stężenie, na podstawie stechiometrii reakcji będącej
podstawą miareczkowania, wyznacza się zawartość (stężenie) oznaczanego składnika.
Czasami do wyznaczenia PK wykorzystuje się krzywą miareczkowania. Krzywa
miareczkowania jest obrazem graficznym zależności pomiędzy pewnym parametrem
charakteryzującym przebieg miareczkowania (np. pH) a objętością dodanego titranta
wyrażoną w ml.
Ze względu na typ reakcji zachodzącej podczas miareczkowania, pomiędzy oznaczaną
substancją a roztworem titranta, metody miareczkowe dzielimy na:
-
alkacymetrię, która opiera się na reakcjach zobojętniania (kwas-zasada)
i obejmuje dwa działy: alkalimetrię (oznaczanie substancji przez
miareczkowanie
mianowanym
roztworem
zasady)
oraz
acydymetrię
(oznaczanie substancji przez miareczkowanie mianowanym roztworem
kwasu);.
-
redoksometrię, która opiera się na reakcjach utlenienia i redukcji, i obejmuje
dwa działy: oksydymetrię (oznaczanie substancji przez miareczkowanie
mianowanymi roztworami utleniaczy), reduktometrię (oznaczanie substancji
przez miareczkowanie mianowanymi roztworami reduktorów). W obu działach
wyróżnia się dodatkowo kilka grup metod miareczkowych, których nazwy
tworzy się od nazwy stosowanego titranta, np. manganometria, jodometria;
-
miareczkowanie strąceniowe, które opiera się na reakcji wytrącania trudno-
rozpuszczalnych osadów w wyniku łączenia jonów titranta i oznaczanej
substancji;
-
kompleksometrię, która opiera się na tworzeniu rozpuszczalnych, słabo-
zdysocjowanych (trwałych) związków kompleksowych; najważniejszym jej
działem jest kompleksonometria, w której titrantami są roztwory
kompleksonów tworzących z metalami kompleksy chelatowe.

19
Ze względu na sposób prowadzenia miareczkowania można wyróżnić dwa sposoby:
bezpośredni i pośredni.
Miareczkowanie bezpośrednie polega na tym, że oznaczana substancja reaguje
bezpośrednio – stechiometrycznie i szybko z dodawanym titrantem. W miareczkowaniu tym
używa się jednego roztworu mianowanego – titranta.
Miareczkowanie pośrednie polega na dobraniu takiej substancji trzeciej, która
reagując stechiometrycznie i ilościowo z oznaczanym składnikiem tworzy nowy związek,
reagujący następnie stechiometrycznie z titrantem.
Szczególnym rodzajem miareczkowania pośredniego jest miareczkowanie odwrotne.
Polega ono na tym, że do badanego roztworu dodaje się odmierzoną ilość roztworu
mianowanego (titrant I) w nadmiarze, a następnie nadmiar tego odczynnika odmiareczkowuje
się innym odpowiednio dobranym roztworem mianowanym (titrant II). Potrzebne są więc
dwa roztwory mianowane. Miareczkowanie odwrotne stosuje się w przypadku wolno
przebiegających reakcji lub gdy trudno jest dobrać odpowiedni wskaźnik do miareczkowania
bezpośredniego.
SPORZĄDZANIE I MIANOWANIE ROZTWORÓW WZORCOWYCH
Roztwory odczynników o dokładnie znanym stężeniu używane do miareczkowania
jako titranty, nazywamy roztworami wzorcowymi, mianowanymi, podstawowymi lub
standardowymi. Roztwory wzorcowe otrzymujemy w dwojaki sposób:
1.
Przez dokładne odważenie substancji, której roztwór sporządzamy i rozpuszczenie jej
w wodzie (lub innym rozpuszczalniku) tak, aby otrzymać ściśle określoną objętość
roztworu.
2.
Przez sporządzenie roztworu danej substancji o przybliżonym stężeniu i zmianowanie
go za pomocą odpowiedniej substancji wzorcowej.
Ad. 1. Jeżeli substancja, której roztwór wzorcowy (mianowany) chcemy sporządzić, jest
wystarczająco czysta i trwała (tzn. spełnia wymagania stawiane substancjom wzorcowym),
wówczas miano roztworu „nastawiamy” przez odważenie odpowiedniej porcji tej substancji
na wadze analitycznej, ilościowe przeniesienie jej do kolby miarowej, rozpuszczenie
w wodzie i dopełnienie do żądanej objętości. Wyznaczone w ten sposób miano nazywa się
bezwzględnym
Uwaga! Rozpuszczaniu substancji towarzyszą często efekty cieplne egzo- lub endotermiczne,
dlatego przed ostatecznym dopełnieniem roztworu w kolbie miarowej do kreski należy
doprowadzić do wyrównania temperatur roztworu i otoczenia. Należy również dokładnie
wymieszać zawartość kolby. (Przed dopełnieniem kolby do kreski mieszamy jej zawartość
ruchem okrężnym, zaś po dopełnieniu, odwracamy wielokrotnie zamkniętą szczelnie korkiem
kolbę dnem do góry i na dół).
Ad. 2. Jeżeli substancja, której roztwór wzorcowy (mianowany) chcemy sporządzić, nie ma
odpowiedniego stopnia czystości, lub jest higroskopijna, czy też po rozpuszczeniu roztwór
zmienia stężenie, wówczas odważamy tę substancję na wadze technicznej i sporządzamy jej
roztwór o stężeniu przybliżonym. Roztwór ten mianujemy zaraz po sporządzeniu lub po
odstaniu przez odpowiedni okres czasu potrzebny do ustalenia się stężenia. Mianowanie
roztworu polega na kilkakrotnym zmiareczkowaniu tym roztworem porcji odpowiedniej
substancji wzorcowej. Miano roztworu wzorcowego wyznaczone przez zmiareczkowanie
substancji wzorcowej tylko wtedy jest bezwzględne, gdy punkt końcowy miareczkowania
pokrywa się w granicach błędu doświadczalnego z punktem równoważności. Nie zawsze tak
bywa. Miano wyznaczone wtedy nazywamy roboczym i jest ono obarczone błędem
systematycznym, związanym z daną metodą oznaczenia miareczkowego.

20
Uwaga! Roztwory mianowane przechowuje się w butelkach szczelnie zamkniętych, często
z ciemnego szkła, aby zabezpieczyć przed działaniem światła, lub z tworzywa sztucznego.
Roztworów mianowanych odlanych z butelki do biurety lub zlewki a nie zużytych nie wlewa
się z powrotem do butelki, w której są przechowywane.
Ogólne zasady mianowania
Należy podkreślić z naciskiem, że od dokładności zmianowania roztworów titrantów
zależy dokładność oznaczeń miareczkowych przy użyciu tych roztworów. Dokładność
i precyzja nastawiania miana powinny być większe niż zwykłych oznaczeń miareczkowych.
Aby to osiągnąć stosuje się następujące zasady:
1.
Należy stosować odpowiednio dobraną, o sprawdzonej czystości, substancję
wzorcową.
2.
Odważki substancji wzorcowej stosowanej do mianowania roztworów powinny być
odpowiednio duże, tak aby błąd względny ważenia był jak najmniejszy.
3.
Objętość mianowanego roztworu, zużyta do zmiareczkowania porcji (odważki)
substancji wzorcowej, nie powinna być zbyt mała (najlepiej 40 – 50 mL tak aby błąd
względny wyznaczenia tej wielkości był niewielki.
4.
Miareczkowanie należy powtórzyć kilkakrotnie (3 – 5 razy), przez co zmniejsza się
błąd przypadkowy mianowania. W miarę możności należy unikać nastawiania miana
roztworu przez miareczkowanie próbek (odmierzonych porcji) innego roztworu
wzorcowego, np. nastawianie miana roztworu NaOH na roztwór HCl. Przy takim
sposobie mianowania błędy przypadkowe są większe.
Substancje wzorcowe
Substancje wzorcowe są to substancje o odpowiednich właściwościach służące bądź
to do sporządzania roztworów titrantów, których miano jest dokładnie znane bezpośrednio
z odważonej ilości tej substancji, bądź też do mianowania roztworów wzorcowych
służących jako titranty. Przykładem substancji wzorcowej służącej do sporządzenia
roztworu mianowanego przez odważenie odpowiedniej porcji tej substancji
i rozpuszczenie jej w odpowiedniej ilości wody w kolbie miarowej, może być bromian(V)
potasu, KBrO
3
. Natomiast szczawian sodu, Na
2
C
2
O
4
, jest przykładem substancji
wzorcowej stosowanej do mianowania roztworu manganianu(VII) potasu, przez
zmiareczkowanie tym roztworem odważek Na
2
C
2
O
4
.
Najważniejsze wymagania dotyczące właściwości substancji wzorcowych są
następujące:
1.
Ilościowy przebieg właściwej dla danej substancji wzorcowej reakcji chemicznej;
2.
Łatwość otrzymania substancji wzorcowej w stanie wysokiej czystości;
3.
Trwałość w warunkach laboratoryjnych – substancja wzorcowa nie powinna być
higroskopijna oraz nie powinna wietrzeć;
4.
Duża masa molowa;
5.
Dobra rozpuszczalność w wodzie;
6.
Uniwersalność tj. możliwość wykorzystania tej substancji jako wzorca w różnych
działach analizy miareczkowej. Substancją, która w dużej mierze spełnia ten warunek
jest wodorojodan(V) potasu, KH(IO
3
)
2
. Substancja ta może służyć do mianowania
roztworów zasad:
KH(IO
3
)
2
+ NaOH → KIO
3
+ NaIO
3
+ H
2
O

21
tiosiarczanu(VI) sodu:
KH(IO
3
)
2
+ 10KI +11HCl → 6I
2
+ 6H
2
O + 11KCl
oraz azotanu(V) srebra: po zredukowaniu do jodku i usunięciu nadmiaru czynnika
redukującego, otrzymany roztwór jodku o dokładnie znanym stężeniu może być
zastosowany do mianowania roztworów azotanu(V) srebra, a także do mianowania
roztworów KMnO
4
.
ALKACYMETRIA
Alkacymetria jest działem analizy miareczkowej, opartym na reakcji kwas-zasada.
Obejmuje dwie grupy metod: alkalimetrię (oznaczanie substancji przez miareczkowanie
mianowanym roztworem zasady) i acydymetrię (oznaczanie substancji przez miareczkowanie
mianowanym roztworem kwasu).
W zależności od własności miareczkowanego związku i titranta rozróżnia się
następujące przypadki miareczkowania alkacymetrycznego:
-
miareczkowanie mocnego kwasu mocną zasadą,
-
miareczkowanie mocnej zasady mocnym kwasem,
-
miareczkowanie słabego kwasu mocną zasadą,
-
miareczkowanie słabej zasady mocnym kwasem,
-
miareczkowanie słabego kwasu słabą zasadą i odwrotnie; ten przypadek nie
ma praktycznego zastosowania, gdyż miareczkowanie za pomocą mocnego
kwasu lub mocnej zasady daje zawsze lepsze wyniki,
-
miareczkowanie wieloprotonowych kwasów (zasad) oraz mieszanin kwasów
(zasad).
Do wyznaczania PK miareczkowania alkacymetrycznego stosuje się wskaźniki
kwasowo - zasadowe (wskaźniki pH). Są to przeważnie związki organiczne – słabe kwasy lub
słabe zasady organiczne, które zmieniają swoją barwę w określonym zakresie pH roztworu.
Całkowita zmiana barwy wskaźnika (tzw. zakres zmiany barwy wskaźnika) występuje
w zakresie dwóch jednostek pH. Niektóre, bardziej czułe wskaźniki odznaczają się mniejszym
zakresem zmiany barwy, a mniej czułe – większym. Charakterystykę najczęściej stosowanych
wskaźników alkacymetrycznych można znaleźć w poradnikach chemicznych lub
w podręcznikach chemii analitycznej.
W idealnym przypadku wskaźnik powinien zmieniać zabarwienie dokładnie
w punkcie równoważności. Takie dobranie wskaźnika jest najczęściej niemożliwe.
W praktyce stosuje się zasadę, według której zakres zmiany barwy wskaźnika powinien
znajdować się wewnątrz skoku miareczkowania lub co najmniej częściowo pokrywać się
ze skokiem miareczkowania. Skokiem miareczkowania nazywa się gwałtowną zmianę
wartości pH w pobliżu punktu równoważności. Skok miareczkowania zależy od stężeń
roztworu miareczkowanego i titranta. Im bardziej stężone są roztwory, tym większy jest skok
miareczkowania. Skok miareczkowania zależy również od mocy miareczkowanego kwasu
(zasady). Im mocniejszy miareczkowany kwas (zasada) tym skok miareczkowania jest
większy. W przypadku miareczkowania słabego kwasu (zasady) skok miareczkowania nie
występuje przy pH=7, lecz jest przesunięty w obszar alkaliczny lub kwaśny. Przesunięcie
to jest tym większe im słabszy jest miareczkowany analit. W przypadku miareczkowania
mocnego kwasu mocną zasadą skok miareczkowania przypada na zakres pH 4,3 – 9,7,
pozwala to na zastosowanie jako wskaźnika zarówno oranżu metylowego (zakres zmiany
barwy 3,1 – 4,4), czerwieni metylowej (zakres zmiany barwy 4,4 – 6,3) jak i fenoloftaleiny
(zakres zmiany barwy: 8,0 – 9,8). W przypadku miareczkowania np. słabego kwasu mocną

22
zasadą skok miareczkowania przypada na zakres pH 7,7 – 9,7, więc z wyżej wymienionych
wskaźników do wyznaczenia PK można zastosować tylko fenoloftaleinę.
ALKALIMETRIA
Sporządzanie i mianowanie roztworu NaOH o stężeniu 0,1 mol/L
Mianowany roztwór wodorotlenku sodu nie powinien zawierać węglanów. Roztwór
taki sporządza się przez rozcieńczenie odpowiedniej porcji stężonego (50%) roztworu NaOH
wodą destylowaną, świeżo wygotowaną nie zawierającą CO
2
.
1.
Pobrać ostrożnie pipetą około 6 mL stężonego, klarownego roztworu NaOH
i rozcieńczyć wodą destylowaną świeżo wygotowaną do objętości 1000 mL w kolbie
miarowej.
UWAGA: Stężony roztwór NaOH zawiera osadzony na dnie Na
2
CO
3
, dlatego należy
uważać, aby przy pobieraniu nie mącić tego roztworu. Przygotowany roztwór chronić
przed dostępem powietrza.
2.
Sporządzony roztwór po wymieszaniu i wyrównaniu jego temperatury z temperaturą
otoczenia, zmianować za pomocą odpowiednio czystego wodoroftalanu potasu.
Podstawą mianowania jest reakcja:
a)
Odważyć na wadze analitycznej trzy odważki wodoroftalanu potasu o masach
odpowiadających masie odważki optymalnej (odważka optymalna jest to masa
substancji wzorcowej jaką należy odważyć, aby podczas mianowania zużyć
taką objętość titranta, która odpowiada 80% objętości nominalnej biurety).
Optymalna odważka KHC
8
H
4
O
4
przy mianowaniu roztworu NaOH z użyciem
biurety o pojemności 50 mL wynosi około 0,8 g. Ponieważ wodoroftalan
potasu posiada dużą masę molową, odważki tej substancji o masach 0,4 – 0,5 g
zapewniają wystarczającą dokładność mianowania roztworu NaOH.
b)
Rozpuścić każdą porcję (odważkę) w około 40 - 50 mL wody destylowanej
przenosząc ilościowo do kolby stożkowej (zw. Kolbą Erlenmayera), dodać
po 3 - 4 krople fenoloftaleiny (wskaźnik) i miareczkować roztworem NaOH,
którego miano się ustala, do pojawienia się różowego zabarwienia. Wyniki
zestawić w tabeli 5.
c)
Obliczyć stężenie molowe zasady według wzoru:
c
NaOH
=
22
,
204
v
1000
m
⋅
⋅
[mol/L]
gdzie: m – masa odważki wodoroftalanu potasu [g],
v – objętość roztworu NaOH zużyta do miareczkowania [mL],
204,22 – masa molowa wodoroftalanu potasu [g/mol].
+ H
2
O
COOK
COONa
COOH
COOK
+ NaOH

23
v · c · 36,461· W
1000
Tabela 5. Mianowanie roztworu NaOH za pomocą wodoroftalanu.
Masa [g]
Numer
naczynka
pustego
naczynka
naczyńka z
wodoroftalanem
wodoroftalanu
V
NaOH
[ml]
C
NaOH
[mol/L]
średnie stężenie NaOH [mol/L]
Roztwór wodorotlenku sodu można także zmianować za pomocą zmianowanego
uprzednio kwasu solnego. W tym celu należy odmierzyć kalibrowaną pipetą co najmniej trzy
porcje kwasu solnego o znanym stężeniu i zmiareczkować wodorotlenkiem sodu w obecności
wskaźnika „5,1” do zmiany barwy z czerwonej na zieloną. Jest to jednak sposób rzadziej
stosowany, ponieważ jest mniej dokładny.
Wskaźnik „5,1” jest to mieszanina zieleni bromokrezolowej i czerwieni metylowej. Roztwory
te są 0,1% i zmieszane w stosunku 3 : 2. Wskaźnik ten zmienia barwę przy pH równym 5,1.
Oznaczanie kwasu solnego HCl
Kwas solny jest kwasem mocnym i miareczkując go roztworem mocnej zasady
do wyznaczenia punktu końcowego miareczkowania można stosować szereg wskaźników,
m.in. wskaźnik „5,1”. Podczas miareczkowania zachodzi następująca reakcja:
HCl + NaOH
→
NaCl + H
2
O
Odczynniki:
mianowany 0,1 M roztwór NaOH;
wskaźnik „5,1”.
Wykonanie:
Otrzymany roztwór przenieść ilościowo do skalibrowanej kolby miarowej na 100
mL, dopełnić wodą destylowaną do kreski, wymieszać, a następnie pobrać kalibrowaną pipetą
trzy porcje po 25 mL do kolb stożkowych i miareczkować mianowanym roztworem NaOH
w obecności wskaźnika „5.1” do zmiany barwy z czerwonej na zieloną.
Zawartość kwasu solnego w próbce obliczyć ze wzoru:
m
HCl
= [g]

24
gdzie: v – objętość NaOH zużyta do miareczkowania [mL],
c – stężenie NaOH [mol/L],
36,461 – masa molowa HCl [g/mol],
W – współczynnik współmierności kolby i pipety.
REDOKSOMETRIA
Redoksometria jest działem analizy miareczkowej opartym na reakcjach utleniania
i redukcji. Metody oksydymetryczne służą do bezpośredniego oznaczania substancji
o charakterze redukującym, stosowany w tych metodach titrant jest odczynnikiem
o właściwościach utleniających. Do tej grupy metod należą manganometria, bromianometria,
chromianometria. Metody reduktometryczne służą do oznaczania substancji o właściwościach
utleniających, miareczkowania prowadzi się odczynnikiem redukującym. Do tej grupy metod
zalicza się jodometrię.
Zwykle przed miareczkowaniem redoks trzeba przeprowadzić oznaczaną substancję
w postać zredukowaną (jeśli miareczkuje się roztworem utleniacza) lub w postać utlenioną
(jeśli miareczkuje się roztworem reduktora). Najczęściej stosuje się takie reduktory lub
utleniacze, które następnie można łatwo można usunąć z roztworu. Jako reduktory stosuje się:
dwutlenek siarki, chlorek cyny(II) i amalgamaty metali (w kolumnie Jonesa). Jako substancje
utleniające stosuje się: nadtlenek wodoru, brom, bizmutan sodu.
BROMIANOMETRIA
Bromianometria należy do oksydymetrycznych metod miareczkowania, w której jako
titrant wykorzystywany jest mianowany roztwór bromianu(V) potasu, KBrO
3
.
W kwaśnym środowisku bromian jest silnym utleniaczem i reaguje z substancjami
redukującymi np. As(III), Sb(III), Sn(II), Fe(II) tworząc w pierwszym etapie bromki,
a w następnym reaguje z powstałymi bromkami, utleniając je do bromu.
I etap BrO
3
−
+ 6H
+
+ 6e → Br
−
+ 3H
2
O (1)
II etap BrO
3
−
+ 5Br
−
+ 6H
+
→ 3Br
2
+ 3H
2
O (2)
Z powyższych równań wynika, że współczynnik równoważności bromianu w reakcji
utleniania wynosi 1/6. Gdy reakcja utleniania substancji miareczkowanej bromianem
zachodzi szybko, wówczas miareczkowanie wykonuje się bezpośrednio KBrO
3
W ten sposób
można oznaczyć wspomniane wyżej metale. Punkt końcowy miareczkowania rozpoznaje się
po odbarwieniu odpowiedniego wskaźnika barwnego, np. czerwieni metylowej, oranżu
metylowego. Pierwsza kropla nadmiaru bromianu reaguje z obecnymi w roztworze jonami
bromkowymi tworząc brom (równanie 2), który reaguje z barwnikiem w sposób
nieodwracalny odbarwiając go.
Zastosowanie bromianu jako titranta stosuje się również w miareczkowaniach
odwrotnych. Metodą tą można oznaczyć wiele związków organicznych aromatycznych,
np. fenol, anilinę oraz związki organiczne z podwójnym wiązaniem. Substancje te zadaje się
bromkiem w środowisku kwaśnym oraz znaną ilością mianowanego roztworu bromianu.
Wydziela się w tej reakcji brom, który podstawia atomy wodoru w pierścieniu benzenowym,
lub przyłącza się do wiązania podwójnego. Nadmiar bromu oznacza się pośrednio, przez
odmiareczkowanie wydzielonego jodu, po uprzednim zadaniu roztworu jodkiem potasu.

25
KBrO
3
Br
2
+ 2 I
−
→ I
2
+ 2Br
−
(3)
Wydzielony jod miareczkuje się mianowanym roztworem tiosiarczanu(VI) sodu, Na
2
S
2
O
3
,
zgodnie reakcją:
I
2
+ 2 S
2
O
3
2−
→ S
4
O
6
2−
+ 2 I
−
(4)
Punkt końcowy reakcji tiosiarczanu z jodem określa się używając kleiku skrobiowego jako
wskaźnika, który dodaje się do roztworu pod koniec miareczkowania. Jod tworzy ze skrobią
związek addycyjny o barwie granatowej, który w punkcie końcowym rozkłada się (roztwór
miareczkowany odbarwia się).
Sporządzanie mianowanego roztworu KBrO
3
o stężeniu 0,01666 mol/L
Bromian(V) potasu należy do substancji wzorcowych (podstawowych) stosowanych
w analizie chemicznej. Związek ten może być otrzymany w bardzo czystej postaci o składzie
zgodnym ze wzorem chemicznym. Mianowany roztwór bromianu(V) potasu sporządza się
przez odważenie na wadze analitycznej odpowiedniej ilości KBrO
3
, ilościowe przeniesienie
odważonej porcji do kolby miarowej, rozpuszczenie tej soli w wodzie i rozcieńczenie
do odpowiedniej objętości. Roztwory bromianu(V) potasu są trwałe i mogą być
przechowywane przez okres około 10 miesięcy nie zmieniając miana.
Mianowany roztwór bromianu (V) potasu może służyć do zmianowania roztworu
tiosiarczanu(VI) sodu, stosowanego jako titrant w jodometrii.
Jak wynika z reakcji (2) 1 molowi bromianu odpowiada 6 moli atomowych bromu
lub jodu (współczynnik równoważności bromianu w reakcji utleniania wynosi 1/6).
Przygotowujemy więc roztwór o stężeniu: 1/60(KBrO
3
) = 0,01666 mol/L.
Masa molowa bromianu(V) potasu wynosi 167,000 g/mol W celu przygotowania 250 mL
mianowanego roztworu bromianu(V) potasu o stężeniu ok. 0,01666 mol/L należy odważyć
na wadze analitycznej ok. 0,695 g odpowiedniej czystości KBrO
3
(wysuszonego
w temperaturze 150ºC), przenieść ilościowo do kolby miarowej, rozpuścić sól w wodzie
i dopełnić wodą do 250 mL.
Stężenie otrzymanego roztworu należy obliczyć ze wzoru:
c =
250
,
0
M
m
⋅
[mol/L]
gdzie: m – masa odważki bromianu(V) potasu [g],
167,001– masa molowa bromianu(V) potasu [g/mol]
0,250 – objętośc roztworu [L].
JODOMETRIA
Metody jodometryczne należą do najważniejszych metod analizy miareczkowej
ze względu na szerokie zastosowania praktyczne oraz możliwość dokładnego ustalenia PK
miareczkowania.
Oznaczenia
jodometryczne
można
przeprowadzać
za
pomocą
miareczkowania bezpośredniego i pośredniego. W miareczkowaniu bezpośrednim titrantem
jest mianowany roztwór jodu, w miareczkowaniu pośrednim – mianowany roztwór

26
tiosiarczanu(VI) sodu. Jodometrycznie można oznaczać zarówno substancje utleniające jak
i redukujące, gdyż kierunek reakcji:
I
2
+ 2 e ↔ 2 I
−
(E
0
= 0,535 V)
zależy od wartości potencjału utleniającego drugiego układu obecnego w roztworze oraz
często od stężenia jonów wodorowych.
Substancje o potencjale utleniającym niższym od potencjału układu I
2
/I
−
miareczkuje
się bezpośrednio mianowanym roztworem jodu. W ten sposób można oznaczać wiele
reduktorów, np. siarczki, siarczany(IV), tiosiarczany, As(III), Sn(II). Substancje o potencjale
utleniającym wyższym od potencjału układu I
2
/I
−
utleniają jony I
−
do wolnego jodu, I
2
, który
odmiareczkowuje się mianowanym roztworem Na
2
S
2
O
3
:
I
2
+2S
2
O
3
2−
→
2 I
−
+ S
4
O
6
2−
Tą metodą oznacza się wiele utleniaczy, np. bromiany(V), jodany(V), dichromiany(VI),
manganiany(VII), chlor, nadtlenek (di)wodoru, Fe(III), Ce(IV).
Wskaźnikiem stosowanym w jodometrii jest skrobia (w postaci kleiku skrobiowego),
która, tworząc z jodem związek addycyjny, barwi się na kolor granatowo fioletowy.
Duża lotność jodu może być przyczyną dużych błędów. Z tego powodu
miareczkowania jodometryczne przeprowadza się w obecności dużego nadmiaru jodku
potasu, gdyż nadmiar jonów jodkowych przesuwa równowagę reakcji w kierunku tworzenia
się nielotnego jonu trójjodkowego: I
2
+ I
−
→
I
3
−
.
Sporządzanie roztworu Na
2
S
2
O
3
o stężeniu 0,1 mol/L
Tiosiarczanu sodu, Na
2
S
2
O
3
·5H
2
O, nie można traktować jako substancji wzorcowej,
gdyż hydrat ten nie zachowuje stałej odpowiadającej wzorowi ilości wody krystalizacyjnej.
Ponadto, po sporządzeniu roztworu jego stężenie zmienia się przez kilkanaście dni, wskutek
reakcji tiosiarczanu z kwasem węglowym, zawartym w wodzie destylowanej:
S
2
O
3
2−
+ H
+
→
HSO
3
−
+ S(↓)
Wpływ na stężenie roztworu tiosiarczanu mają również bakterie zawarte w tym roztworze.
Aby sporządzić 500 mL 0,1 mol/L roztworu tiosiarczanu sodu odważa się na wadze
technicznej około 12,5 g Na
2
S
2
O
3
·5H
2
O, rozpuszcza w wodzie destylowanej w kolbie
o pojemności 0,5 L i po rozpuszczeniu uzupełnia wodą do kreski. Wskazane jest używanie
wody wygotowanej, nie zawierającej CO
2
. (Masa molowa Na
2
S
2
O
3
·5H
2
O wynosi
248,174g/mol).
Mianowanie roztworu należy wykonać po upływie około dwóch tygodni od jego
sporządzenia. Jeżeli w roztworze pojawi się osad przed mianowaniem należy roztwór
przesączyć. Stężenie roztworu Na
2
S
2
O
3
określa się dokładnie na podstawie reakcji z takimi
substancjami wzorcowymi jak jod, I
2
, jodan(V) potasu, KIO
3
, bromian(V) potasu, KBrO
3
,
dichromian(VI) potasu, K
2
Cr
2
O
7
, heksacyjanożelazian(III) potasu, K
3
[Fe(CN)
6
], mianowany
roztwór manganianu(VII) potasu, KMnO
4
.
Najczęściej miano roztworu tiosiarczanu nastawia się na jod sublimowany. W tym
celu przygotowuje się odważkę odpowiednio suchego, czystego jodu, którą odważa się
w naczyńku wagowym zawierającym stężony roztwór jodku potasu. W stężonym roztworze
jodku potasu jod rozpuszcza się natychmiast i unika się w ten sposób ewentualnych strat jodu

27
Na
2
S
2
O
3
KBrO
3
Na
2
S
2
O
3
Na
2
S
2
O
3
KBrO
3
KBrO
3
KBrO
3
Na
2
S
2
O
3
spowodowanych jego lotnością (I
2
+ I
−
→
I
3
−
). Następnie naczyńko z zawartością zsuwa się
ostrożnie po ściance do kolby stożkowej zawierającej roztwór 1 g jodku potasu w 100 mL
wody i po wymieszaniu natychmiast miareczkuje się roztworem tiosiarczanu do odbarwienia
roztworu, dodając pod koniec kleiku skrobiowego.
Nastawianie miana roztworu Na
2
S
2
O
3
na mianowany roztwór KBrO
3
Mianowanie
roztworu
Na
2
S
2
O
3
za
pomocą
bromianu(V)
potasu
polega
na odmiareczkowaniu roztworem tiosiarczanu sodu, jodu wydzielonego w reakcji jonów
bromianowych z jonami jodkowymi w środowisku silnie kwaśnym, analogicznie jak z jonami
Br
-
(reakcja 2). Nastawianie miana można przeprowadzić miareczkując bądź odważki KBrO
3
,
bądź dokładnie odmierzone porcje roztworu bromianu. Reakcja między jonami
bromianowymi a jodkowymi zachodzi zgodnie z równaniem:
BrO
3
−
+ 6I
−
+ 6H
+
→ 3I
2
+ Br
−
+3H
2
O (5)
Wydzielony jod odmiareczkowuje się roztworem Na
2
S
2
O
3
(reakcja 4).
Odczynniki:
mianowany 0,01666 M roztwór KBrO
3
;
stały KBr;
stały KJ;
2M roztwór HCl;
kleik skrobiowy.
Wykonanie:
Pipetą o sprawdzonej pojemności (25 mL) pobrać porcję mianowanego roztworu
KBrO
3
przenieść do kolby stożkowej (zamykanej doszlifowanym korkiem) o pojemności
250 mL, rozcieńczyć wodą destylowaną do ok. 50 mL, dodać 0,5 g bromku potasu, 2 g jodku
potasu uraz 15 mL 2 M HCl. Wymienione odczynniki dodawać kolejno przez lejek, spłukując
go dokładnie wodą destylowaną po każdym dodanym odczynniku. Kolbę zamknąć korkiem,
(zawartość zamieszać ruchem poziomym) odczekać 5 min., po czym zmiareczkować
wydzielony jod roztworem tiosiarczanu sodu w obecności kleiku skrobiowego jako
wskaźnika. Kleik skrobiowy dodać pod koniec miareczkowania – gdy barwa roztworu
miareczkowanego stanie się jasnożółta. Mianowanie powtórzyć co najmniej trzy razy.
Stężenie roztworu Na
2
S
2
O
3
obliczyć ze wzoru:
v · 6 · c
c = [mol/L]
v
gdzie:
c
- stężenie roztworu tiosiarczanu sodu [mol/L],
c
- stężenie roztworu bromianu [mol/L],
v
- objętość roztworu tiosiarczanu sodu [mL],
v
- objętość roztworu bromianu [mL] odmierzona pipetą o sprawdzonej pojemności.
Uwaga: jeżeli w przygotowanym roztworze Na
2
S
2
O
3
wytrąci się siarka, przed przystąpieniem
do mianowania należy roztwór przesączyć przez sączek Bűchnera.

28
Oznaczanie tlenu rozpuszczonego w wodzie metodą Winklera
Tlen rozpuszczony w wodzie pochodzi głównie z powietrza. W pewnych przypadkach
ź
ródłem tlenu może być także proces fotosyntezy roślin wodnych. W zależności
od zawartości tlenu zmieniają się właściwości korozyjne wody.
Do oznaczenia zawartości rozpuszczonego tlenu w wodzie stosuje się najczęściej
miareczkową metodę Winklera i jej modyfikacje.
Zasada oznaczenia:
Tlen rozpuszczony w wodzie utlenia w środowisku alkalicznym wodorotlenek
manganu(II) do związków manganu czterowartościowego:
Mn
2+
+ 2OH
−
= Mn(OH)
2
(↓)
2Mn(OH)
2
+O
2
→
2MnO(OH)
2
(↓)
W środowisku kwaśnym jony manganu(IV) utleniają jony jodkowe z jodku potasu
do wolnego jodu w ilości równoważnej zawartości tlenu w wodzie:
2MnO(OH)
2
+ 8H
+
→
2Mn
4+
+ 6H
2
O
2Mn
4+
+ 4I
−
→
2Mn
2+
+2I
2
Wydzielony jod oznacza się miareczkowo tiosiarczanem(VI) sodu wobec skrobi, zgodnie
z reakcją:
2I
2
+ 4S
2
O
3
2−
→
2S
4
O
6
2−
+ 4I
−
a z ilości zużytego tiosiarczanu oblicza się zawartość tlenu.
Oznaczaniu tlenu przeszkadzają zawarte w wodzie substancje utleniające
i redukujące. Substancje utleniające mogą dodatkowo utleniać jony jodkowe, natomiast
substancje redukujące mogą reagować z wydzielonym w reakcji jodem. Wpływ substancji
przeszkadzających usuwa się przez dodanie odpowiednich odczynników, np. azydku sodu,
manganianu(VII) potasu, chloranu(I) sodu.
Istotną sprawą jest właściwe pobranie próbki wody do oznaczania tlenu. Pobrana
próbka nie może stykać się z powietrzem i powinna być jak najszybciej zanalizowana.
Próbkę wody należy pobrać do skalibrowanej (o znanej pojemności) butelki lub kolby
stożkowej zamykanej szczelnie doszlifowanym korkiem.
Odczynniki:
40% roztwór MnSO
4
;
alkaliczny roztwór KI sporządzony przez rozpuszczenie w wodzie podwójnie
destylowanej 50g NaOH i 15g KI, a następnie rozcieńczenie roztworu wodą
destylowaną do objętości 100 mL;
H
2
SO
4
stęż.;
mianowany roztwór Na
2
S
2
O
3
(o stężeniu około 0,025 mol/L); roztwór ten
otrzymuje się przez czterokrotne rozcieńczenie mianowanego roztworu 0,1M
Na
2
S
2
O
3
;
kleik skrobiowy.

29
Wykonanie:
W celu pobrania próbki wody należy podłączyć wąż z obojętnego tworzywa do kurka
i wprowadzić wylot węża aż do dna kolby stożkowej (butelki). Napełnić kolbę stożkową
przepuszczając przez nią strumień wody o objętości równej kilku jej pojemnościom.
Po usunięciu wszystkich pęcherzyków, które mogą przylegać do ścianek, zamknąć dokładnie
kolbę stożkową korkiem tak aby wewnątrz nie było pęcherzyków powietrza, a następnie
możliwie szybko związać rozpuszczony tlen. W tym celu dodać pipetą włożoną do dna
kolby, kolejno, 2 mL 40% roztworu MnSO
4
i 2 mL alkalicznego roztworu KI (za każdym
razem pipetę powoli wyciągać z kolby). Kolbę szczelnie zamknąć tak, aby wewnątrz nie było
pęcherzyków powietrza. Ponieważ podczas zamykania kolby część wody wypłynie
z naczynia, kolbę należy wczesniej umieścić w krystalizatorze lub większej zlewce.
Po zamknięciu, kolbę odwrócić kilkanaście razy dnem do góry w celu dokładnego
wymieszania zawartości i odstawić na około 25-30 minut w celu opadnięcia osadu. Jeżeli
osad nie opada należy zawartość kolby ponownie wymieszać i odstawić do opadnięcia osadu.
Gdy utworzony osad MnO(OH)
2
osiądzie na dnie naczynia (osad ten powinien zajmować
najwyżej jedną trzecią część kolby), zdekantować znad osadu ostrożnie (bez zmącenia) około
100 mL klarownej cieczy. Zwrócić uwagę czy część osadu nie przylega do szlifu korka lub
szyjki. W przypadku przylegania niewielkiej ilości osadu do korka lub szyjki naczynia należy
ostrożnie powoli zdekantować ciecz tak, aby cząstki osadu zatrzymały się na szkle,
a następnie dokładnie spłukać małą ilością wody destylowanej szyjkę i korek. Do
pozostałości w kolbie dodać szybko 2 mL (odmierzone cylindrem) stężonego H
2
SO
4
, kolbę
zamknąć, a następnie mieszać tak długo, aż cały osad rozpuści się, a jod zostanie
równomiernie rozprowadzony. Zmiareczkować wydzielony jod roztworem tiosiarczanu(VI)
sodu o stężeniu około 0,025 mol/L, dodając pod koniec miareczkowania 1 – 2 mL kleiku
skrobiowego.
Zawartość tlenu wyrażoną w mg/l obliczyć ze wzoru:
32 · v· c · 1000
C
O2
= [mg/L]
4 (V
0
- 4)
gdzie:
c
– stężenie roztworu Na
2
S
2
O
3
[mol/L]
,
v – objętość Na
2
S
2
O
3
zużyta do miareczkowania [mL],
32 – masa molowa tlenu [g/mol];
(V
0
- 4) – objętość analizowanej próbki [mL] pomniejszona o sumę objętości dodanego
roztworu siarczanu manganu(II) (2mL) i alkalicznego roztworu KI (2mL);
liczba 4 w mianowniku związana jest ze stechiometrią reakcji zachodzących podczas
oznaczania: w reakcji tlenu z jonami Mn
2+
i dalszej jonów Mn
4+
z jonami jodkowymi jednej
cząsteczce (dwuatomowej) tlenu odpowiada 4 atomy jodu.

30
KOMPLEKSOMETRIA
Podstawą metod kompleksometrycznych są reakcje kompleksowania różnego typu,
w wyniku których powstają rozpuszczalne i słabo zdysocjowane kompleksy. W zależności od
rodzaju tworzącego się kompleksu, miareczkowania kompleksometryczne można podzielić na
takie, w których tworzą się kompleksy niechelatowe, utworzone przez ligandy
jednofunkcyjne i kompleksy chelatowe, utworzone przez ligandy wielofunkcyjne
(wielokleszczowe). W tym drugim przypadku stosuje się często określenie : miareczkowanie
chalatometryczne.
Przykładem miareczkowania kompleksometrycznego niechelatometrycznego jest
argentometryczne oznaczanie cyjanków oraz oznaczenia merkurymetryczne, np.w reakcji
rtęci z jonami chlorkowymi powstaje słabo zdysocjowany chlorek rtęci(II):
Hg
2+
+ 2Cl
−−−−
→ HgCl
2
,
a z jonami jodkowymi powstaje najpierw rozpuszczalny kompleks:
Hg
2+
+ 2I
−−−−
→ HgI
4
2-
,
który z nadmiarem jonów rtęci(II) tworzy trudno rozpuszczalny związek, HgI
2
.
Hg
2+
+ HgI
4
2
−−−−
→ 2HgI
2
(↓)
Pojawienie się tego związku (zmętnienie roztworu) wskazuje na przekroczenie punktu
równoważności reakcji (miareczkowania).
Istotny rozwój kompleksometrii spowodowało odkrycie przez Schwarzenbacha,
w latach 1945-1952, kompleksotwórczych właściwości kwasów aminopolikarboksylowych,
których najczęściej stosowanym przedstawicielem jest kwas etylenodiaminotetraoctowy.
Kwas ten i jego sól dwusodowa są oznaczane skrótem EDTA, pochodzącym od angielskiej
nazwy ethylenediaminetetraacetic acid.
EDTA reaguje z jonami metali w stosunku molowym 1:1, niezależnie
od wartościowości jonu metalu. Sześć atomów ligandowych jednej cząsteczki kwasu
etylenodiaminotetraoctowego (2 atomy azotu i 4 atomy tlenu) wysyca koordynacyjnie atom
metalu. Kompleksy EDTA z metalami są bezbarwne lub barwne, jeżeli metal wchodzący
w skład kompleksu ma właściwości chromoforowe (np. żelazo, chrom, miedź, nikiel).
Zastępując wzór kwasu etylenodiaminotetraoctowego
HOOC H
2
C CH
2
COOH
N – CH
2
– CH
2
– N
HOOC H
2
C CH
2
COOH
skrótem H
4
Y, jego reakcję z jonami metali w środowisku obojętnym można napisać w postaci
ogólnego równania:
M
n+
+ H
2
Y
2
−−−−
→ MY
(n-4)+
+ 2H
+
Podstawowym
odczynnikiem
(titrantem)
w
kompleksometrycznej
analizie
miareczkowej
jest
sól
dwusodowa
kwasu
etylenodiaminotetraoctowego,

31
etylenodiaminotetraoctan disodu, oznaczana podobnie jak sam kwas skrótem EDTA. Sól ta
jest znana pod nazwą kompleksonu III (Na
2
H
2
Y),
NaOOC H
2
C CH
2
COOH
N – CH
2
– CH
2
– N
HOOC H
2
C CH
2
COONa
Kwas etylenodiaminotetraoctowy zwany kompleksonem II, w odróżnieniu od swej
soli sodowej praktycznie nie rozpuszcza się w wodzie, dlatego jego zastosowanie jako titranta
jest ograniczone.
W zależności od wartości pH, EDTA tworzy prawie ze wszystkimi jonami metali
wielowartościowych trwałe, rozpuszczalne kompleksy chelatowe, w których stosunek
M : EDTA wynosi 1:1. Dzięki wynalezieniu wielu barwnych wskaźników, umożliwiających
wyznaczenie końca reakcji kompleksowania jonów podczas miareczkowania mianowanymi
roztworami EDTA, kompleksometria znalazła zastosowanie do oznaczania wielu metali –
w miareczkowaniu bezpośrednim i odwrotnym, oraz niemetali – w miareczkowaniu
pośrednim. Mianowane roztwory EDTA można stosować w stężeniach od 0,1 M do 0,001 M,
co umożliwia oznaczanie pierwiastków w szerokich granicach stężeń
Miareczkowanie bezpośrednie polega na dodawaniu mianowanego roztworu EDTA
do roztworu zawierającego badany kation oraz odpowiednie odczynniki maskujące
i utrzymujące wartość pH na ustalonym poziomie przy czym do wyznaczenia punktu
końcowego miareczkowania stosuje się metalowskaźniki. W ten sposób oznacza się m.in.
glin, żelazo(III), wapń, cer, magnez, cynk, kadm, miedź(II), nikiel, kobalt.
Miareczkowanie odwrotne stosuje się w przypadku, gdy jony oznaczanego metalu
zbyt wolno ulegają kompleksowaniu lub gdy w roztworach o pH koniecznym
do miareczkowania wytracają się one w postaci osadu. W takich przypadkach
do miareczkowanego roztworu wprowadza się nadmiar mianowanego roztworu EDTA
oraz odpowiedni roztwór buforowy i nadmiar odczynnika odmiareczkowuje się mianowanym
roztworem jonu metalu. Metodą tą oznacza się również metale, które tworzą trwałe
kompleksy z EDTA, lecz nie reagują ze wskaźnikami (np. tal) lub takie kationy, które tworzą
zbyt trwałe kompleksy ze wskaźnikami (takie jak kompleksy kobaltu, niklu i glinu z czernią
eriochromową T).
Miareczkowanie podstawieniowe stosuje się w przypadku oznaczania jonów metali,
które tworzą z EDTA kompleksy bardziej trwałe od np. kompleksu magnezu z EDTA.
Przykładem tego typu miareczkowania jest oznaczanie wapnia wobec czerni eriochromowej.
Jeżeli do analizowanego roztworu doda się kompleksu Mg-EDTA, w wyniku reakcji
wymiany uwalnia się ilość jonów magnezu równoważna ilości oznaczanego kationu.
Uwolnione jony magnezu miareczkuje się bezpośrednio mianowanym roztworem EDTA,
przy czym zachodzi następująca reakcja wymiany:
Ca
2+
+ MgY
2
−−−−
→ Mg
2+
+ CaY
2
−−−−
Pośrednie oznaczanie anionów polega na tym, że oznaczany anion strąca się
roztworem odpowiedniego kationu, o znanym stężeniu, dodanego w ściśle określonej
objętości. Nadmiar użytego roztworu kationu w przesączu po oddzieleniu osadu
odmiareczkowuje się mianowanym roztworem EDTA. W ten sposób oznacza się na przykład
siarczany, stosując do ich strącania roztwór chlorku baru.

32
m
M
EDTA
⋅
0,500
Przygotowanie 0,01 M roztworu EDTA
W miareczkowaniach kompleksometrycznych najczęściej stosuje się roztwór EDTA
o stężeniu 0,01 mol/l. Wybór stężenia odczynnika zależy od zawartości oznaczanego jonu
w badanym roztworze. Spośród bardziej stężonych roztworów EDTA stosuje się roztwory:
0,1; 0,05; 0,02 i 0,01 M; spośród roztworów rozcieńczonych – 0,005; 0,002 i 0,001 M.
Roztwór EDTA przygotowuje się przez rozpuszczenie odpowiedniej odważki
dwuwodnej dwusodowej soli, Na
2
H
2
Y·2H
2
O, (nazwa handlowa tej soli to wersenian
dwusodowy), w wodzie zdejonizowanej. Sól ta wysuszona do stałej masy w temperaturze
80
o
C jest trwała w dużym zakresie wilgotności powietrza; natomiast w temperaturze
120 – 140
o
C traci obie cząsteczki wody, lecz jako bezwodna jest higroskopijna. Jeżeli miano
roztworu EDTA otrzymanego z odważki nie jest pewne (np. nie jest znana czystość użytego
preparatu), nastawia się je na roztwór wzorcowy metalu, np. magnezu, cynku, bizmutu,
miedzi.
Masa molowa Na
2
H
2
Y· 2H
2
O wynosi 372,24 g/mol. W celu przygotowania 500 mL
0,01 M roztworu należy odważyć na wadze analitycznej ok. 1,86 g EDTA (o odpowiedniej
czystości), przenieść ilościowo do kolby miarowej na 500 mL i po rozpuszczeniu dopełnić
wodą destylowaną do kreski. Do sporządzania roztworu należy stosować wodę
zdejonizowaną (dwukrotnie destylowaną). Z masy odważki obliczyć stężenie otrzymanego
roztworu ze wzoru:
c
EDTA
= [mol/L]
gdzie:
m – masa odważki EDTA [g];
M
EDTA
– masa molowa EDTA [g/mol].
0,500 – objętość roztworu [L].
Mianowane roztwory EDTA przechowuje się najczęściej w butelkach polietylenowych.
Uwaga: przed sporządzeniem roztworu EDTA nale.ży upewnić się, że woda nie zawiera
kationów wapnia lub magnezu. W tym celu należy wykonać slepą próbę; do ok. 25 mL wody
dodać 2 mL buforu amonowego o pH=10 oraz szczyptę czerni eriochromowej T.
Oznaczanie twardości wody
Twardością wody nazywamy właściwości wody wynikające z obecności w wodzie
przede wszystkim jonów wapnia i magnezu, oraz innych jonów wielowartościowych, takich
jak żelaza, glinu i cynku, występujących w wodzie w znacznie mniejszej ilości.
Na twardość całkowitą wody składa się sumaryczna zawartość Ca i Mg przeliczona
na tlenek wapnia. Część twardości całkowitej stanowi twardość węglanowa odpowiadająca tej
części wapnia i magnezu, która występuje w wodzie w postaci wodorowęglanów. Jest to tzw.
twardość przemijająca, gdyż po zagotowaniu wody następuje rozkład wodorowęglanów,
strąca się wtedy CaCO
3
i część MgCO
3
(jest lepiej rozpuszczalny).
Twardość trwała, niewęglanowa, jest związana z obecnością w wodzie chlorków,
siarczanów(VI) i azotanów.

33
winnoczerwony bezbarwny niebieski
Twardość wody wyraża się w stopniach niemieckich lub francuskich.
1 stopień niemiecki to 10 mg CaO w 1 litrze wody lub 1 część wagowa CaO w 100 000
częściach wagowych wody. 1 stopień francuski to 10 mg CaCO
3
w 1 litrze wody lub 1 część
wagowa CaCO
3
w 100 000 częściach wagowych wody.
Twardość węglanową wody oznacza się acydymetrycznie, miareczkując próbkę wody
kwasem solnym wobec oranżu metylowego.
Twardość całkowitą oznacza się kompleksometrycznie za pomocą EDTA. Podczas
oznaczania wobec czerni eriochromowej T w środowisku buforu amonowego o pH=10
zachodzą następujące reakcje:
Mg
2+
+ HF
2
−
→
MgF
−
+ H
+
oraz MgF
−
+ H
2
Y
2
−
→
MgY
2
−
+ HF
2
−
+ H
+
gdzie:
HF
2-
– zdysocjowana forma czerni eriochromowej T w jakiej występuje wskaźnik w
roztworze o pH=10;
H
2
Y
2-
– anion EDTA .
Analogiczne reakcje zachodzą z udziałem wapnia.
Odczynniki:
bufor amonowy o pH = 10;
czerń eriochromowa T (w postaci stałej; czysty wskaźnik zmieszany z NaCl
w stosunku 1 : 200).
Na zadanie przygotować zlewkę o pojemności 400 mL, suchą również wewnątrz.
Wykonanie:
Z otrzymanej próbki pobrać bez rozcieńczania trzy porcje po 50 mL, kalibrowaną
pipetą, do kolb stożkowych. Do każdej porcji dodać po ok. 2 mL roztworu buforowego
o pH=10, szczyptę czerni eriochromowej T i miareczkować mianowanym 0,01 M roztworem
EDTA do uzyskania czysto niebieskiego zabarwienia.
Twardość wody w stopniach niemieckich obliczyć ze wzoru:
tw. wody =
V
100
56,08
c
v
⋅
⋅
⋅
[˚ N]
gdzie: v – objętość roztworu EDTA zużytego do miareczkowania [mL],
c – stężenie roztworu EDTA [mol/L],
V – objętość próbki miareczkowanej wody [mL] (dwie objętości skalibrowanej
pipety),
56,08 – masa molowa CaO [g/mol].
Przyjmujemy, że 1 mL wody = 1 g.
34
WPROWADZENIE DO ANALIZY INSTRUMENTALNEJ
POTENCJOMETRIA
Metody potencjometryczne wykorzystują zależność między stężeniem (a ściślej
aktywnością) oznaczanego jonu w roztworze i potencjałem elektrycznym odpowiedniej
elektrody. Ponieważ potencjał elektrody względem roztworu nie jest dostępny bezpośrednim
pomiarom, wykonuje się pomiar siły elektromotorycznej (SEM) ogniwa, którego jednym
półogniwem jest elektroda wskaźnikowa, której potencjał jej zależy od stężenia
oznaczanego jonu), a drugim – elektroda porównawcza, której potencjał ma wartość stałą.
Obie elektrody są w kontakcie z badanym roztworem. Metody potencjometryczne polegają
więc na pomiarze siły elektromotorycznej (SEM) ogniwa złożonego z dwu elektrod
zanurzonych do badanego roztworu. Mierzona SEM zależy w określony sposób od stężenia
w roztworze oznaczanego składnika. Za zmianę SEM odpowiedzialna jest jedna z elektrod,
elektroda wskaźnikowa.
Metody potencjometryczne dzielą się na dwie grupy:
1.
Metody bezpośrednie polegające na wyznaczeniu stężenia oznaczanego składnika
na podstawie wartości SEM ogniwa, którego kalibracji dokonano za pomocą próbek
wzorcowych. Należą tu pomiary pH roztworów oraz oznaczenia za pomocą elektrod
jonoselektywnych.
2.
Metody pośrednie stosowane są do wyznaczania punktu końcowego miareczkowania –
jest to tzw. miareczkowanie potencjometryczne. W miareczkowaniu tym wyznacza się
zmiany SEM odpowiedniego ogniwa spowodowane dodawaniem mianowanego roztworu
odczynnika miareczkującego, związane ze zmianą stężenia analitu.
Rodzaje elektrod
W pomiarach potencjometrycznych bardzo ważną rolę odgrywają elektrody,
które ze względu na mechanizm działania można podzielić na cztery grupy:
1.
Elektrody pierwszego rodzaju, czyli elektrody odwracalne w stosunku do kationu;
są to elektrody składające się z metalu lub gazu w równowadze z roztworem
zawierającym jony tego metalu.
2.
Elektrody drugiego rodzaju, są to elektrody odwracalne w stosunku do anionu
tworzącego z metalem elektrody trudno rozpuszczalny związek.
3.
Elektrody trzeciego rodzaju, elektrody te tworzą metale w równowadze z roztworem
nasyconym dwoma trudno rozpuszczalnymi elektrolitami o tym samym anionie.
Kation jednego z elektrolitów jest kationem metalu elektrody, drugi kation znajduje
się w roztworze w nadmiarze.
4.
Elektrody utleniająco-redukujące, w elektrodach tych obojętny chemicznie metal (Pt,
Au) jest zanurzony w roztworze zawierającym substancje zarówno w formie
utlenionej, jak i zredukowanej.
5.
Elektrody membranowe zwane jonoselektywnymi (ISE)i. Wspólną ich cechą jest to, że:
-
elektrodowo czynną częścią elektrody jest membrana,
-
różnica potencjałów na granicy faz membrana/roztwór spowodowana jest reakcją
wymiany jonowej między jonami z roztworu a jonami z membrany.
Potencjał elektrod pierwszego rodzaju ustala się zgodnie z następującym
rozumowaniem. Metal M zanurzony do roztworu jego soli wykazuje tendencję przechodzenia
do roztworu w postaci jonów M
n+
. Tendencji tej przeciwstawia się dążenie jonów M
n+
do wydzielania się z roztworu w postaci zredukowanej M. W rezultacie ustala się równowaga

35
między metalem zanurzonym w roztworze a jego jonami obecnymi w roztworze. Równowagę
tę można zapisać równaniem:
M ↔ M
n+
+ ne
lub w postaci ogólnej:
red ↔ utl + ne
przy czym „red” oznacza postać zredukowaną, „utl” – postać utlenioną, n – liczbę elektronów
biorących udział w reakcji.
Zjawiska zachodzące na granicy faz metal – roztwór są źródłem potencjału, którego wielkość
określa równanie Nernsta:
E = E
0
nF
RT
+
[ ]
[ ]
red
utl
ln
(1)
w którym E
0
oznacza potencjał normalny elektrody, R – stałą gazową, T – temperaturę
bezwzględną roztworu, n – liczbę elektronów biorących udział w reakcji, F – stałą Faradaya.
(nawiasy kwadratowe oznaczają stężenia molowe).
Równanie (1) wyraża potencjał dowolnej elektrody. Jest to wielkość, której absolutnej
wartości nie można ani zmierzyć, ani obliczyć teoretycznie. Można natomiast wyrazić
liczbowo wielkość potencjału danej elektrody w stosunku do potencjału innej elektrody,
mierząc siłę elektromotoryczną (SEM) ogniwa utworzonego z badanej elektrody i elektrody
porównawczej. Za równy zeru został przyjęty umownie potencjał normalnej elektrody
wodorowej i w stosunku do niego wyznacza się potencjały innych elektrod.
Równanie (1) można przekształcić, wprowadzając liczbowe wartości stałych R, T i F
(dla temperatury 25
°
C) oraz zastępując logarytm naturalny logarytmem dziesiętnym:
[ ]
[ ]
red
utl
n
E
E
log
059
.
0
0
+
=
(2)
Bardziej ścisłe równanie, wyrażające potencjał elektrody, powinno zawierać aktywności
zamiast stężeń. Gdy postacią zredukowaną jest metal elektrody, zgodnie z zasadą, że faza
stała ma aktywność równą 1, uproszczony wzór Nernsta (2) przyjmie postać:
[ ]
+
+
=
n
M
n
E
E
log
059
.
0
0
(3)
Jest to przypadek elektrody pierwszego rodzaju.
Powstawanie potencjału na granicy elektroda – roztwór dla elektrod pierwszego,
drugiego, trzeciego i czwartego rodzaju jest wynikiem reakcji redoksowych. Powstawanie
potencjału na granicy elektroda – roztwór w elektrodach jonoselektywnych, jak już
wspomniano, jest wynikiem wymiany jonowej pomiędzy membraną a roztworem.

36
Aparatura potencjometryczna
Aparatura do pomiarów potencjometrycznych składa się z dwóch zasadniczych części:
-
dwóch elektrod (wskaźnikowej i porównawczej) w kontakcie z badanym roztworem,
co stanowi ogniwo o określonym SEM,
-
przyrządu pomiarowego siły elektromotorycznej ogniwa (SEM) (woltomierza o dużym
oporze wejściowym lub zestawu do pomiaru SEM metodą kompensacyjną).
Pomiary potencjometryczne polegają na określeniu potencjału lub zmian potencjału elektrody
wskaźnikowej, której potencjał zależy od stężenia oznaczanego jonu. Potencjał elektrody
porównawczej jest stały w warunkach prowadzenia pomiaru.
Elektrody wskaźnikowe
Pod pojęciem elektrody wskaźnikowej rozumiemy takie półogniwo, którego potencjał
zależy zgodnie z równaniem Nernsta od stężenia (ściślej aktywności) oznaczanego jonu. Jako
elektrody wskaźnikowe mogą być użyte wszystkie wymienione już elektrody, a więc
elektrody I, II, III rodzaju, elektrody utleniająco-redukujące oraz elektrody jonoselektywne
(ISE).
Elektrody porównawcze
Dobra elektroda porównawcza powinna posiadać następujące właściwości:
-
stałość potencjału na przestrzeni długiego czasu w warunkach pomiaru;
-
odtwarzalność potencjału i brak histerezy temperaturowej;
-
łatwość sporządzenia z materiałów i odczynników dostępnych w każdym laboratorium;
-
mały opór elektryczny;
-
odporność mechaniczną niezbędną przy częstym użyciu.
Jako elektroda porównawcza największe znaczenie teoretyczne ma normalna elektroda
wodorowa (NEW). Jest to blaszka platynowa pokryta czernią platynową, omywana wodorem
pod ciśnieniem 760 mm Hg i zanurzona w roztworze kwasu solnego o aktywności równej 1:
Pt, H
2
(760 mm Hg) H
+
(a
H+
). Reakcje zachodzące na tej elektrodzie można przedstawić
równaniem analogicznym do równania opisującego procesy zachodzące na elektrodach
metalowych.
H
2
↔ 2H
+
+ 2e
Nie jest ona jednak wygodna w użyciu i w praktyce stosuje się najczęściej nasyconą
elektrodę kalomelową (NEK) lub chlorosrebrową. Obie są elektrodami drugiego rodzaju.
Elektroda kalomelowa
Elektrodę kalomelową stanowi drut platynowy będący w kontakcie z rtęcią metaliczną
pokrytą warstwą chlorku rtęci(I) Hg
2
Cl
2
(kalomelu), zanurzoną w nasyconym roztworze
chlorku potasu. Półogniwo takie można zapisać: Hg, Hg
2
Cl
2(s)
nas. KCl.
Przemiany zachodzące na elektrodzie można przedstawić równaniem:
2Hg + 2Cl
−
↔ Hg
2
Cl
2
+ 2e
Elektroda chlorosrebrowa
Elektrodę chlorosrebrową stanowi drut srebrny pokryty warstewką chlorku srebra zanurzony
w roztworze zawierającym jony Cl
-
, pochodzące z chlorku potasu lub kwasu solnego.
Schemat takiego półogniwa można zapisać: Ag, AgCl
(s)
KCl.
Przemiany zachodzące na elektrodzie można przedstawić równaniem: Ag + Cl
−
↔ AgCl + e.

37
Elektrody jonoselektywne
Elektrodami jonoselektywnymi (ISE – Ion Selective Electrode) nazywa się elektrody, których
potencjał zależy liniowo od logarytmu aktywności danego jonu w roztworze (w określonym
przedziale stężeń). Zależność prostoliniowa utrzymuje się na ogół w zakresie kilku rzędów
stężenia (3 – 4). Wspólną ich cechą jest to, że na ich potencjał ma wpływ nie tylko stężenie
jonu oznaczanego, lecz także stężenia innych jonów. Elektrody te są zaopatrzone
w membranę jonowymienną, która oddziela odpowiednie półogniwo od roztworu badanego.
Na potencjał elektrody membranowej składa się potencjał międzyfazowy na granicy faz
membrana-roztwór, uwarunkowany wymianą jonową między roztworem i membraną, oraz
potencjał dyfuzyjny, wynikający z procesów zachodzących wewnątrz membrany, szczególnie
w jej warstwie przylegającej do roztworu.
Elektrody jonoselektywne różnią się szczegółami konstrukcyjnymi, m.in. stanem skupienia
fazy tworzącej membranę. Elektrody te dzielimy na:
-
elektrody ze szklanymi membranami – elektrody szklane,
-
elektrody ze stałymi membranami, (membrany homogeniczne – monokrystaliczne
i heterogeniczne – polikrystaliczne),
-
elektrody z membranami ciekłymi,
-
elektrody z podwójnymi membranami – elektrody czułe na gazy i elektrody
enzymatyczne.
Elektroda szklana
Elektroda szklana – jonoselektywna elektroda czuła na jony wodorowe, jest to
półogniwo w którym membrana jest wykonana ze specjalnego gatunku szkła sodowego.
Zwykle jest to wąska rurka szklana zakończona cienkościenną membraną w kształcie bańki.
Wewnątrz znajduje się roztwór buforowy o dokładnie znanym pH, zawierający chlorki.
W roztworze tym jest zanurzona porównawcza (wyprowadzająca) elektroda wewnętrzna
o stałym potencjale, zwykle chlorosrebrowa, która posiada wyprowadzenie na zewnątrz.
W przypadku elektrody szklanej różnica potencjałów między szkłem i roztworem stykającym
się z nim, zależy od pH tego roztworu. Zależność ta daje się wyrazić wzorem słusznym
dla odwracalnej elektrody wodorowej (równanie 4).
H
sz
a
nF
RT
E
E
ln
0
+
=
(4)
gdzie: E
0
sz
– potencjał normalny charakterystyczny dla danego rodzaje szkła.
Elektroda szklana zawiera dwie powierzchnie graniczne szkło – roztwór i gdyby E
0
sz
było
jednakowe dla obu tych powierzchni, to różnica potencjałów między roztworami po obu
stronach szklanej membrany zależna byłaby tylko od stosunku aktywności jonów
wodorowych w tych roztworach.
nF
RT
E
=
ln
HII
HI
a
a
(5)
Potencjał elektrody szklanej zależy jedynie od aktywności jonów wodorowych w roztworze,
do którego elektroda została zanurzona. Zależność tę wyraża wzór analogiczny do wzoru (4),
w którym
E
0
sz
jest stałą zależną od rodzaju elektrody wewnętrznej i pH elektrolitu
wewnętrznego. Jeżeli elektrodę szklaną zanurzyć do roztworu takiego jaki znajduje się
wewnątrz elektrody i jako elektrodę porównawczą zastosować elektrodę taką samą jak
we wnętrzu bańki, to SEM utworzonego ogniwa zgodnie ze wzorem (5) powinna równać się

38
zeru. Praktycznie okazuje się jednak, że występuje pewna niewielka SEM równa ± 2 mV.
Wielkość ta jest nazwana potencjałem asymetrii, który spowodowany jest nieidentycznością
powierzchni zewnętrznej i wewnętrznej szklanej membrany (
E
0
sz
różne dla obu powierzchni).
Potencjał asymetrii ulega zmianom w okresie eksploatacji elektrody i dlatego elektrodę
szklaną należy kalibrować we wzorcowych roztworach buforowych o znanych wartościach
pH, sporządzając krzywą kalibracyjną, która jest charakterystyką elektrody.
Wyznaczenie charakterystyki elektrody szklanej polega na ustaleniu dla niej zależności SEM
od pH szeregu wzorcowych roztworów buforowych tj. określeniu zakresu jej stosowalności
i nachylenia. Jeżeli zależność ta przebiegałaby w myśl równania (4), to charakterystyka
elektrody byłaby linią prostą w całym zakresie pH. Faktycznie dla najczęściej stosowanych
elektrod prostoliniowość występuje w zakresie pH 1 – 9 a często do 12 pod warunkiem,
ż
e roztwór nie zawiera zbyt dużo soli metali alkalicznych. Odchylenie od prostoliniowości
w roztworach silnie kwaśnych i silnie alkalicznych wynika z niesłuszności wzoru (4) w tych
warunkach. Odstępstwa od tego wzoru związane są ze zmianą aktywności wody jako
rozpuszczalnika w stężonych roztworach oraz z dochodzenia do głosu innych mechanizmów
ustalania się różnicy potencjałów między szkłem i roztworem niż za pośrednictwem samych
tylko jonów wodorowych.
Nachylenie charakterystyki elektrody określa zmianę potencjału danej elektrody
spowodowaną dziesięciokrotną zmianą aktywności (stężenia) jonu na który czuła jest dana
elektroda. Dla elektrody szklanej jest to zmiana jej potencjału na jednostkę pH [mV/pH].
Dla elektrod czułych na jony jednowartościowe teoretyczne nachylenie charakterystyki, tzw.
Nernstowskie nachylenie wynosi 59,15 mV na dekadę zmiany aktywności (stężenia) tych
jonów w temperaturze 25
0
C (298 K).
MIARECZKOWANIE POTENCJOMETRYCZNE
Miareczkowanie potencjometryczne polega na mierzeniu różnicy potencjałów między
elektrodą wskaźnikową i elektrodą porównawczą po dodaniu każdej porcji odczynnika
miareczkującego. Jest ono możliwe do wykonania wówczas, gdy dobierze się elektrodę
wskaźnikową, która będzie reagowała na zmiany stężenia składnika oznaczanego
lub odczynnika miareczkującego, zachodzące podczas miareczkowania. Dodawanie
odczynnika miareczkującego powoduje zmiany stężenia składnika oznaczanego. Początkowo
względne zmiany stężenia oznaczanych jonów są niewielkie i zmiany potencjału również są
niewielkie. Natomiast w pobliżu punktu równoważności następuje skok potencjału. Krzywa
miareczkowania, przedstawiająca zależność potencjału od objętości titranta, jest analogiczna
do krzywej miareczkowania w alkacymetrii, redoksometrii i kompleksometrii. Różnica
między
miareczkowaniem
prowadzonym
metodą
klasyczną
i
miareczkowaniem
potencjometrycznym polega na wyznaczeniu punktu końcowego miareczkowania (PK).
Miareczkowanie potencjometryczne jest sposobem detekcji punktu końcowego. W metodach
wizualnych, każdy typ reakcji, a nawet każda analiza, wymagała zastosowania
odpowiedniego wskaźnika. Miareczkowanie potencjometryczne ma bardziej ogólne
zastosowanie, można je zastosować do oznaczania roztworów zabarwionych, mętnych oraz
do oznaczania kilku składników obok siebie w jednym miareczkowaniu.

39
CH
3
COOH
v · c · 60,053
1000
Oznaczanie procentowej zawartości CH
3
COOH w handlowym occie metodą
miareczkową, z wizualną i potencjometryczną detekcją PK
Oznaczenie procentowej zawartości kwasu octowego w occie handlowym wykonuje
się alkalimetrycznie (titrantem jest NaOH) wyznaczając punkt końcowy wizualnie wobec
fenoloftaleiny (miareczkowanie słabego kwasu mocna zasadą) oraz potencjometrycznie.
Oznaczenie opiera się na reakcji:
CH
3
COOH + NaOH
→
CH
3
COONa + H
2
O
Na zadanie należy przygotować kolbę o pojemności 250 mL (ćwiczenie wykonuje się
parami).
Miareczkowanie wizualne
Odczynniki:
mianowany 0,1M NaOH;
fenoloftaleina.
Wykonanie:
Z otrzymanej próbki (rozcieńczonej uprzednio do 250 mL) roztworu pobrać
skalibrowaną pipetą 3 porcje po 25 mL do kolb stożkowych, rozcieńczyć wodą destylowaną
do ok. 50 mL, dodać kilka kropli wskaźnika – fenoloftaleiny i miareczkować mianowanym
roztworem ok. 0,1mol/L NaOH do jasno różowej barwy.
Zawartość kwasu obliczyć ze wzoru:
m = [g]
gdzie: v – objętość roztworu NaOH zużytego do miareczkowania [mL],
c – stężenie roztworu NaOH [mol/L],
60,053 – masa molowa CH
3
COOH [g].
Z podanego wzoru oblicza się zawartość kwasu octowego [g] w objętości „v
p
” mL próbki
(objętość pipety o wyznaczonej objętości) pobranej do miareczkowania. Zawartość tę należy
przeliczyć na 100 mL próbki i wynik podać w procentach wagowo-objętościowych (masa
substancji na 100 mL roztworu).
Miareczkowanie potencjometryczne
Odczynniki:
mianowany 0,1M NaOH;
Aparatura
:
pH-metr, na przykład N-517 z odczytem cyfrowym lub N-512 – wychyleniowy;
elektroda szklana (elektroda wskaźnikowa) i nasycona elektroda kalomelowa
(elektroda porównawcza), lub elektroda kombinowana (wskaźnikowa i porównawcza
jako ogniwo zespolone);
mieszadło magnetyczne, mieszadełko.
Wykonanie:
Miareczkowanie z potencjometryczną detekcją PK wykonać 3 razy w następujący
sposób. Z otrzymanej próbki roztworu (tej samej, w której oznaczano kwas octowy metodą
wizualną) pobrać pipetą (o sprawdzonej objętości) 25 mL roztworu do zlewki o pojemności
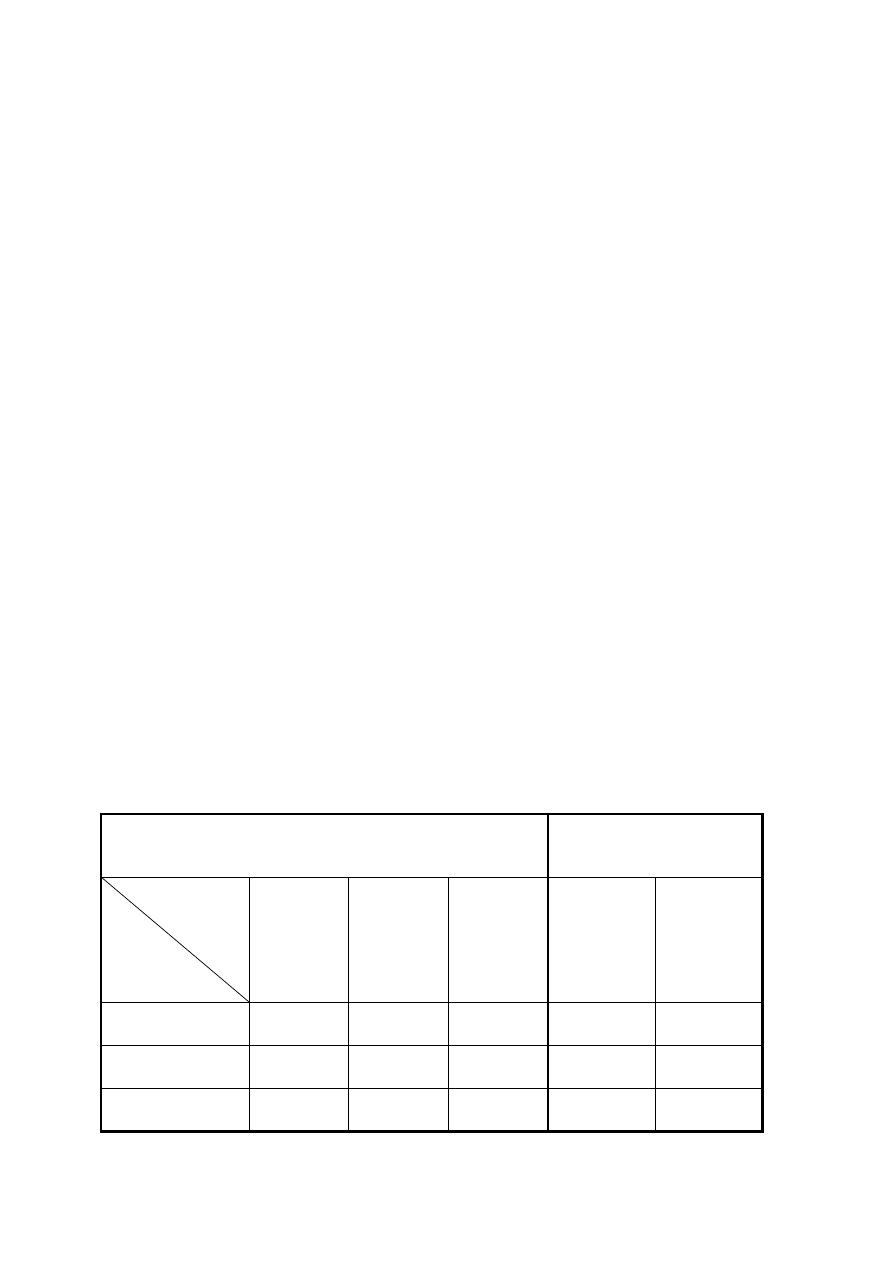
40
100 mL. Zanurzyć w roztworze elektrodę kombinowaną. Jest to ogniwo zespolone złożone
z elektrody wskaźnikowej – szklanej (czułej na jony wodorowe) i elektrody porównawczej,
elektrody chlorosrebrowej. Podłączyć elektrodę do odpowiedniego gniazda pH-metru. W tym
przypadku pH-metr będzie stosowany jako miliwoltomierz. Elektrodę należy umocować
w odpowiednim uchwycie i zanurzyć do roztworu tak aby klucz elektrody porównawczej
był zanurzony. Zainstalować mieszadło magnetyczne, sprawdzić czy mieszający magnes
nie uderza o elektrodę lub ściany zlewki. Nad zlewką ustawić biuretę z roztworem NaOH
(mianowanym ok. 0,1 mol/L). Po włączeniu mieszadła magnetycznego dodawać po 0,5 mL
roztworu NaOH, notując każdorazowo stan biurety i odpowiadające mu wskazania pH-metru
w mV. W miareczkowaniu tym dodawać cały czas tj. od początku do zakończenia
miareczkowania po 0,5 mL titranta. Miareczkowanie przerwać po znacznym przekroczeniu
PK, tak aby uzyskać „pełny” przebieg krzywej miareczkowania. Drugie i trzecie
miareczkowanie wykonać tak aby w zakresie skoku potencjału uzyskać na krzywej
miareczkowania dokładniejsze „wypunktowanie”. Należy więc dodawać, tak jak
w pierwszym miareczkowaniu po 0,5 ml NaOH, z chwilą kiedy 0,5 mL porcja roztworu
NaOH wywoła większą różnicę potencjału, następne porcje zmniejszyć do 0,1 mL, po czym
przy małych różnicach potencjału zwiększyć dodawane porcje titranta z powrotem do 0,5 mL.
Miareczkowanie przerwać gdy kolejne porcje NaOH wywołują niewielkie różnice potencjału.
Zakres objętości, w której należy zagęścić punkty na krzywej ustala się na podstawie
pierwszego miareczkowania oraz konsultacji z asystentem Po zakończeniu miareczkowania
elektrodę opłukać starannie wodą i osuszyć bibułą.
Wyniki pomiarów zestawić w tabelach zawierających kolumny z wartościami: v
NaOH
[mL],
SEM [mV],
∆
SEM [mV],
∆
SEM/
∆
v [mV/mL].
Z otrzymanych danych narysować wykresy zależności SEM[mV] – v[mL] oraz
∆
SEM/
∆
v [mV/mL] – v [mL] (wykres pierwszej pochodnej) i z wykresów odczytać objętości
NaOH odpowiadające PK miareczkowania. Wyznaczyć także objętości NaOH odpowiadające
PK miareczkowania metodą rachunkową Hahna. Objętości wyznaczone trzema metodami
z miareczkowania potencjometrycznego oraz z miareczkowania wobec fenoloftaleiny
zestawić w tabeli (wzór podany poniżej) i dokonać interpretacji otrzymanych wyników.
Obliczyć %-ową zawartość kwasu octowego w otrzymanej próbce i porównać
z wynikiem uzyskanym w miareczkowaniu wizualnym.
Miareczkowanie potencjometryczne
Miareczkowanie wobec
fenoloftaleiny
Metoda
wyznaczania
v PK
Nr
próbki
Metoda
graficzna
stycznych
v PK [mL]
Metoda
graficzna
I pochodnej
v PK [mL]
Metoda
Hahna
v PK [mL]
Nr
Próbki
v PK
[mL]
1
1
2
2
3
3

41
Zasada metody Hahna
Metoda Hahna jest jedną z najlepszych i najprostszych metod wyznaczania PK.
Dodając w czasie miareczkowania stałe porcje titranta i notując odpowiadające im wartości
potencjału oraz ich różnice ustala się największy przyrost potencjału, ∆E
max
oraz dwa kolejne
co do wielkości przyrosty potencjału ∆E
1
i ∆E
2
, leżące po obu stronach ∆E
max
. Na podstawie
tych wartości oblicza się poprawkę a [mL] określoną wzorem:
a = ∆vּ q
a
w którym ∆v oznacza objętość odczynnika dodawanego w jednej porcji (∆v = const),
q
a
– mnożnik obliczany ze wzoru:
1
2
2 E
E
q
∆
∆
=
Poprawkę a należy dodać do liczby mililitrów v odpowiadającej ∆E
1
, jeżeli ∆E
1
znajduje się
przed ∆E
max
, lub odjąć od v mililitrów odpowiadającej ∆E
max
, jeżeli ∆E
1
występuje
po ∆E
max
.
Metodą Hahna można wyznaczyć PK miareczkowania z dobrą dokładnością, jeżeli wartości
przyrostów, ∆E
max
, ∆E
1
, i ∆E
2
, spełniają następujące warunki:
5
.
2
1
max
≥
∆
∆
=
E
E
Q
i
1
2
2 E
E
q
∆
∆
=
≥
0,25 (najlepiej, jeżeli wynosi ok. 0,5)
Jeżeli warunki te nie są spełnione dla wartości odczytanych, można łączyć ze sobą wyniki
odpowiadające kolejnym ∆v, np. po dwa, lub trzy (a nawet więcej), tak aby otrzymać wartości
∆E
max
, ∆E
1
i ∆E
2
spełniające powyższe warunki. Wartość współczynnika q
a
ma większe
znaczenie niż wartość Q.
Zaletą metody Hahna jest to, że pozwala wyznaczyć dokładnie PK miareczkowania
nawet przy zastosowaniu stosunkowo dużych porcji odczynnika, np. 0,5 mL.

42
SPEKTROFOTOMETRIA
Spektrofotometria jest techniką instrumentalną, w której do celów analitycznych
wykorzystuje się przejścia energetyczne zachodzące w cząsteczkach, spowodowane absorpcją
promieniowania elektromagnetycznego w zakresie nadfioletu (UV, 200-380 nm), widzialnym
(VIS, 380-780 nm) lub bliskiej podczerwieni (0,78-30000
µ
m). W analizie nieorganicznej
zastosowanie znajduje głównie spektrofotometria UV i VIS.
Metodą spektrofotometrii UV-VIS można oznaczać substancje organiczne (np. wiele
związków posiadających wiązanie
π
lub elektrony n, w tym węglowodory aromatyczne,
aldehydy, ketony, kwasy i aminy) i nieorganiczne (np. pierwiastki ziem rzadkich, ozon, SO
2
)
wykazujące absorpcję w nadfiolecie, związki absorbujące promieniowanie w zakresie
widzialnym, w tym barwne związki organiczne (barwniki) i barwne sole metali (np. KMnO
4
,
CuSO
4
) oraz substancje, których formy absorbujące promieniowanie uzyskuje się na drodze
reakcji chemicznych. Do celów tych najczęściej wykorzystuje się reakcje kompleksowania.
Opracowano wiele procedur oznaczeń kationów metali w formie barwnych związków
kompleksowych z ligandami organicznymi.
Analiza ilościowa metodą spektrofotometrii UV-VIS oparta jest na pomiarze
absorbancji A
λ
badanego roztworu przy określonej długości fali
λ
i wykorzystaniu prawa
Lamberta-Beera, zgodnie z którym:
A
λ
=
ε
λ
⋅
l
⋅
c
gdzie
ε
λ
jest współczynnikiem absorpcji przy długości fali
λ
, l grubością warstwy
absorbującej, a c stężeniem analitu w badanym roztworze.
Absorbancję definiuje się jako:
I
I
log
=
A
0
gdzie I
0
to natężenie promieniowania padającego na ośrodek absorbujący a I to natężenie
promieniowania po przejściu przez ośrodek absorbujący.
Prawo Lamberta-Beera
dotyczy
absorpcji promieniowania przez roztwory i można je sformułować następująco:
je
ż
eli
współczynnik absorpcji rozpuszczalnika jest równy zeru, to absorbancja wi
ą
zki
promieniowania monochromatycznego przechodz
ą
cej przez jednorodny roztwór jest wprost
proporcjonalna do st
ęż
enia c roztworu i do grubo
ś
ci warstwy absorbuj
ą
cej l.
Zależność pomiędzy absorbancją a stężeniem analitu, w warunkach gdy badany układ
spełniania prawo Lamberta-Beera ma charakter prostoliniowy i można ją wykorzystać
do wyznaczenia stężenia analitu w próbce. Prawo Lamberta-Beera stosuje się do roztworów
rozcieńczonych, bowiem przy większych stężeniach wartość współczynnika absorpcji zależy
zwykle od stężenia oznaczanej substancji. Na wykresie obrazującym zależność absorbancji
roztworu od jego stężenia uwidacznia się to w zakrzywieniu linii kalibracyjnej w zakresie
wyższych stężeń w górę lub w dół, które określa się odpowiednio jako dodatnie lub ujemne
odstępstwa od prawa Lamberta-Beera. Odstępstwa te mogą być natury chemicznej
lub instrumentalnej. Odstępstwa natury chemicznej występują, gdy zachodzą oddziaływania
cząsteczek substancji rozpuszczonej pomiędzy sobą (dysocjacja asocjacja) lub z cząsteczkami
rozpuszczalnika natomiast zasadniczym czynnikiem aparaturowym powodującym odstępstwa
od prawa jest niedostateczna monochromatyzacja promieniowania.

43
Prawo Lamberta-Beera odnosi się do przypadku, gdy w roztworze znajduje się jedna
substancja absorbująca.
Je
ż
eli w roztworze jest wi
ę
cej substancji, które absorbuj
ą
promieniowanie przy wybranej długo
ś
ci fali, to absorbancja tego roztworu jest równa sumie
absorbancji jego poszczególnych składników:
A = A
1
+ A
2
+ A
3
+ … + A
n
= (
ε
1
c
1
+
ε
2
c
2
+
ε
3
c
3
+ … +
ε
n
c
n
)
⋅
l
gdzie
ε
1
,
ε
2
,
ε
3
i
ε
n
to współczynniki absorpcji odpowiednich substancji obecnych
w roztworze, c
1
, c
2
, c
3
i c
n
oznaczają stężenia tych substancji, a l jest grubością warstwy
absorbującej. Powyższa zależność to
prawo addytywno
ś
ci absorbancji
, z którego korzysta się
w spektrofotometrycznej analizie układów wieloskładnikowych.
Kalibracja
W spektrofotometrii do wyznaczenia stężenia oznaczanej substancji wykorzystuje się
zwykle pomiar absorbancji roztworu zawierającego dany analit. W zależności od środowiska
chemicznego, w którym znajduje się analit, jak również ustalonych warunków pomiarowych
wartość sygnału mierzonego dla danego stężenia analitu może ulegać zmianie i stąd nie może
jednoznacznie świadczyć o ilości analitu w próbce. Dlatego konieczne jest przeprowadzanie
kalibracji danego oznaczenia.
Istnieją różne metody kalibracji, do najczęściej stosowanych należy metoda serii
wzorców. W metodzie serii wzorców przygotowuje się roztwory wzorcowe, czyli syntetyczne
roztwory o znanych, ściśle ustalonych stężeniach analitu. Po poddaniu tych roztworów
pomiarom, przedstawia się otrzymane wyniki w układzie współrzędnych sygnał (w przypadku
spektrofotometrii jest to zazwyczaj absorbancja) – stężenie analitu, a następnie dopasowuje
się do nich określoną funkcję (zazwyczaj linię prostą). Pomiary wykonuje się zwykle przy
określonej długości fali, najczęściej odpowiadającej maksimum absorpcji oznaczanej
substancji oraz względem roztworu odniesienia (tzw. ślepej próby), czyli roztworu
przygotowanego
analogicznie
jak
roztwory
wzorcowe
i
roztwór
próbki,
lecz
nie zawierającego analitu. Prostoliniowy przebieg zależności A=f(c) świadczy o spełnieniu
przez badany układ prawa Lamberta-Beera. Wyznaczenie współczynnika kierunkowego
tej prostej umożliwia obliczenie współczynnika absorpcji oznaczanej substancji. W celu
wyznaczenia stężenia analitu w próbce rejestruje się odpowiadający jej sygnał i odnosi się go
do linii kalibracyjnej. Roztwór próbki powinien być przygotowany w taki sposób,
by maksymalny zakres stężeń analitu w roztworach wzorcowych obejmował przewidywane
jego stężenie w próbce.
Aparatura
Do badania absorpcji promieniowania elektromagnetycznego w nadfiolecie i zakresie
widzialnym widma służą spektrofotometry UV-VIS. Schemat blokowy spektrofotometru
przedstawiono na rysunku 1.
Rys. 1. Schemat blokowy spektrofotometru
Ź
ródło
promieniowania
ciągłego
Monochromator
Roztwór
badany
Detektor
Miernik
Lampa
wolframowa,
halogenowa
Pryzmat,
siatka
dyfrakcyjna
Uchwyt
i kuweta
Fotokomórka,
fotopowielacz,
fotodioda
Galwanometr,
mikroprocesor

44
Do pomiarów wykorzystywany będzie spektrofotometr Spekol 11 firmy Carl Zeiss
Jena. Spekol 11 wyposażony jest w halogenową żarówkę projektorową stanowiącą źródło
promieniowania, monochromatorem jest precyzyjna siatka dyfrakcyjna (651 szczelin/mm),
zaś do detekcji promieniowania służą dwie fotokomórki: niebieskoczuła (340-620 nm) oraz
czerwonoczuła (620-850 nm). Aparat wyposażony jest we wskaźnik cyfrowy.
Spektrofotometryczne oznaczenie Fe(III) metodą rodankową
Jony rodankowe (tiocyjanianowe) w niezbyt kwaśnym środowisku reagują z Fe(III)
tworząc czerwono zabarwione kompleksy żelazowo-rodankowe (
λ
max
=480 nm). W wyniku
stopniowej reakcji kompleksowania w roztworze mogą powstać kompleksy Fe(SCN)
2+
,
Fe(SCN)
2
+
itd., aż do Fe(SCN)
6
3-
. Stężenia reagentów i pH środowiska decydują, które
z kompleksów przeważają w roztworze. W roztworach o mikrogramowych stężeniach Fe(III)
przeważa pierwszy kompleks w szeregu. Kwasowość roztworu powinna być co najmniej taka,
aby nie dopuścić do hydrolizy jonów Fe(III), która zaczyna się już przy pH ok. 3.
Metodą
rodankową
można
oznaczać
Fe(III)
lub
całkowitą
zawartość
ż
elaza,
po wcześniejszym utlenieniu Fe(II). Oznaczaniu przeszkadzają aniony tworzące z Fe(III)
trwałe kompleksy: fluorki, fosforany, cytryniany i szczawiany. Przeszkadzają również jony
metali tworzących w warunkach reakcji barwne kopmpleksy (Co, Mo, Bi, Ti).
Odczynniki:
roztwór podstawowy soli Fe
3+
o stężeniu 1 mg Fe
3+
/mL;
0,1% roztwór HCl;
0,1 M roztwór HCl;
20 % roztwór KSCN.
Przygotowanie roztworów wzorcowych i sporz
ą
dzenie wykresu kalibracyjnego
Roztworem wykorzystywanym do przygotowania serii roztworów wzorcowych
jest roboczy roztwór wzorcowy soli Fe
3+
o stężeniu 10
µ
g Fe
3+
/mL sporządzony przez 100-
krotne rozcieńczenie roztworu podstawowego żelaza(III) o stężeniu 1mg Fe
3+
/mL 0,1%-wym
roztworem HCl. Roztwór roboczy należy przygotować w kolbie o pojemności 100 mL
po wcześniejszym jej przepłukaniu 0,1% roztworem HCl.
W celu sporządzenia serii roztworów wzorcowych do wyznaczenia zależności
kalibracyjnej 6 kolbek miarowych o pojemności 50,00 mL należy przepłukać 0,1M
roztworem HCl. Do każdej z nich odpipetować następujące objętości roboczego wzorcowego,
tj. o stężeniu 10
µ
g/mL: 0 (ślepa próba), 2,00; 4,00; 6,00; 8,00 i 10,00 mL, następnie dodać
5,0 mL 20% roztworu tiocyjanianu potasu* i uzupełnić do kreski 0,1 M roztworem HCl.
Roztwory dokładnie wymieszać, następnie kolejno przelać do kuwety i zmierzyć ich
absorbancję przy długości fali
λ
=480 nm stosując jako odnośnik roztwór ślepej próby.
Na podstawie uzyskanych wyników sporządzić wykres kalibracyjny.

45
c · v
1000
Oznaczanie
ż
elaza w badanej próbce
Otrzymany roztwór przenieść ilościowo do skalibrowanej kolby miarowej na 100 mL,
dopełnić wodą destylowaną do kreski, wymieszać, a następnie pobrać kalibrowaną pipetą trzy
porcje po 25 mL do kolbek miarowych o pojemności 50,00 mL. Do każdej kolbki dodać 5,0
mL 20% roztworu tiocyjanianu potasu* i uzupełnić do kreski 0,1 M roztworem HCl.
Roztwory dokładnie wymieszać, następnie kolejno przelać do kuwety i zmierzyć ich
absorbancję przy długości fali
λ
=480 nm stosując jako odnośnik roztwór ślepej próby.
Odczytać stężenie żelaza(III) w barwnym roztworze z krzywej kalibracyjnej i obliczyć
oznaczaną ilość żelaza (w mg) ze wzoru:
x = [mg]
gdzie:
c – stężenie żelaza odczytane z krzywej kalibracyjnej (
µ
g/mL),
v – objętość badanego roztworu żelaza (tu 50,00 mL).
Zawartość żelaza w otrzymanej próbce wynosi: m
Fe
= x·W [mg] (W - współmierność kolby
z pipetą).
*Uwaga:
Kompleks żelazo-tiocyjanian jest nietrwały, dlatego należy dodawać tiocyjanian do
wszystkich próbek (zarówno wzorcowych jak i analizowanych) bezpośrednio przed
pomiarem absorbancji. Pomiary spektrofotometryczne wzorców i analizowanych
próbek należy wykonać bezpośrednio po sobie.

46
WYKAZ OBOWIĄZUJĄCYCH SPRAWOZDAŃ
1.
Analiza wagowa, oznaczanie Ba / Fe / Ni.
2.
Wyznaczanie pojemności naczyń miarowych: kolby miarowej na 100 mL i pipety na
25 mL.
3.
Alkacymetria. Sporządzanie bezwęglanowego 0,1 M roztworu NaOH i jego
mianowanie za pomocą wodoroftalanu potasu.
4.
Alkalimetryczne oznaczanie kwasu solnego.
5.
Redoksometria. Jodometria. Bromianometria. Sporządzanie mianowanego roztworu
0,1 M KBrO
3
.
6.
Sporządzanie i mianowanie 0,1 M roztworu Na
2
S
2
O
3
.
7.
Oznaczanie tlenu w wodzie metodą Winklera.
8.
Kompleksometria. Sporządzanie mianowanego 0,01M roztworu EDTA.
9.
Oznaczanie twardości wody.
10.
Potencjometria i miareczkowanie potencjometryczne. Oznaczanie procentowej
zawartości CH
3
COOH w handlowym occie z wizualną i potencjometryczną detekcją
PK.
11.
Spektrofotometria. Oznaczanie żelaza metodą rodankową.
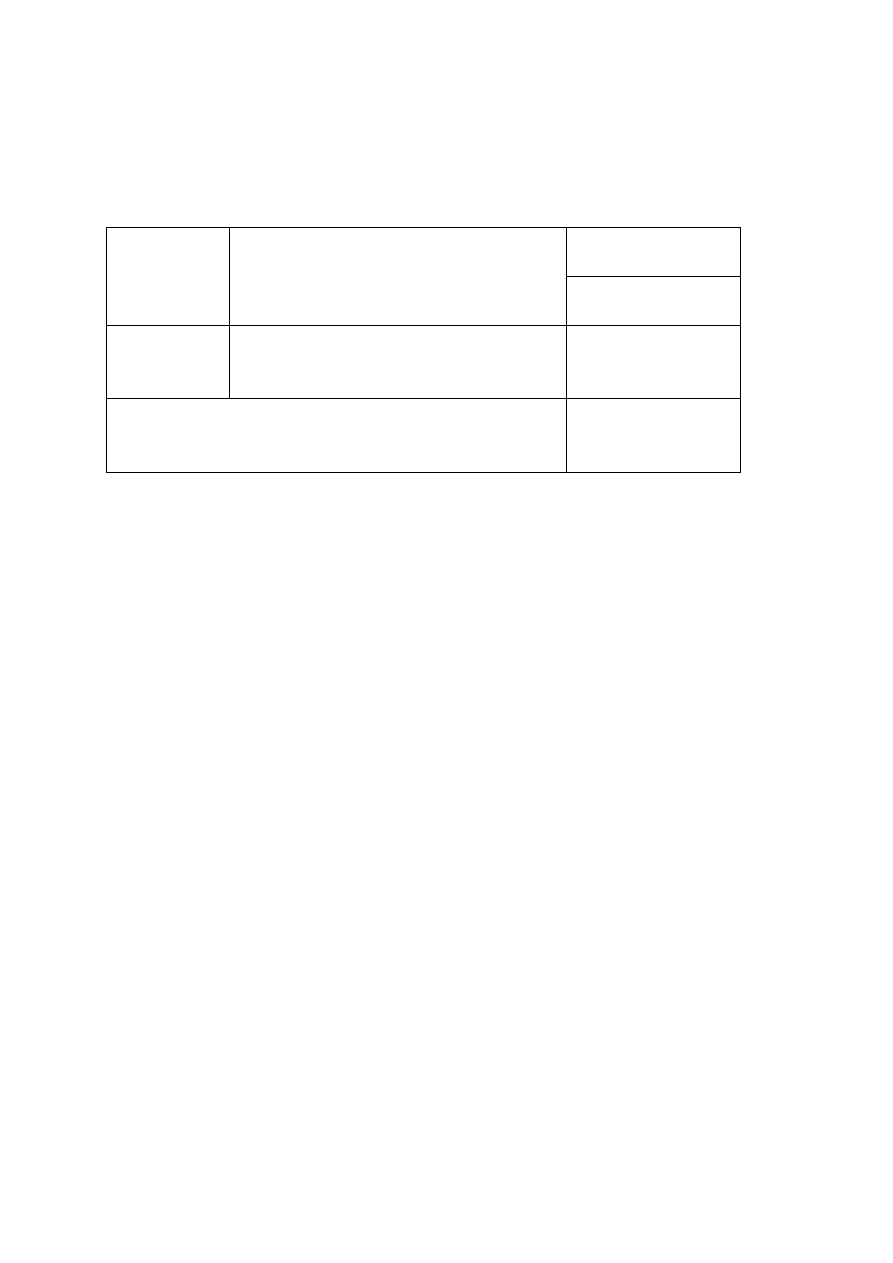
47
S C H E M A T S P R A W O Z D A N I A
data wykonania
ćwiczenia
numer
ćwiczenia
dział analizy i temat ćwiczenia
data oddania
sprawozdania
grupa
Imię i nazwisko
nazwisko
sprawdzającego
Uwagi:
Ocena
I.
Wstęp teoretyczny (m.in. zasada oznaczenia, równanie reakcji)
II.
Część doświadczalna
1.
Stosowane odczynniki
2.
Wykonanie oznaczenia
3.
Wyniki oznaczenia
III.
Opracowanie wyników
1.
Obliczenie wyniku analizy
2.
Oszacowanie niepewności pomiaru
3.
Podanie wyniku końcowego wraz z niepewnością
IV. Podsumowanie (cel ćwiczenia i jego realizacja, indywidualne
spostrzeżenia i obserwacje)
Wyszukiwarka
Podobne podstrony:
Chemia kliniczna kontrola id 11 Nieznany
Chemia kliniczna kontrola id 11 Nieznany
CHEMIA SA,,DOWA WYKLAD 7 id 11 Nieznany
chemia pp pr odp klucz(1) id 11 Nieznany
CHEMIA SA,,DOWA WYKLAD 4 id 11 Nieznany
CHEMIA SA,,DOWA WYKLAD 7 id 11 Nieznany
charakterystyka plazincow id 11 Nieznany
chemia fizyczna lab id 112228 Nieznany
blok 2 skrypt id 90327 Nieznany (2)
chemia okiem niechemika id 1126 Nieznany
blok 3 skrypt id 90351 Nieznany (2)
analityczna test id 59602 Nieznany (2)
chemia fizyczna egzamin id 1122 Nieznany
chemia maj 2005 id 112453 Nieznany
md skrypt id 290151 Nieznany
chemia przykladowe zad id 11281 Nieznany
AiSD skrypt id 53503 Nieznany (2)
Chemia analityczna skrypt
więcej podobnych podstron