
1. Wstęp
1.1. Historyczny rozwój materiałów
Człowiek od zarania dziejów wykorzystywał, a z czasem przetwarzał, materiały dla zdobycia
pożywienia, zwiększenia swego bezpieczeństwa i zapewnienia sobie odpowiedniego poziomu
życia. Śledząc dzieje cywilizacji ludzkiej można dojść do przekonania, że o jej rozwoju decyduje
w dużej mierze rozwój materiałów i towarzyszący temu rozwój sił wytwórczych. Świadczy o
tym niewątpliwie między innymi nazwanie różnych okresów w dziejach ludzkości od
materiałów decydujących wówczas o warunkach życia, np. epoki: kamienia, brązu, żelaza.
Również wdrożenie różnych wynalazków stało się możliwe dopiero po udostępnieniu
odpowiednich materiałów. Przykładowo już w notatkach Leonardo da Vinci z piętnastego wieku
znaleziono szkic helikoptera, lecz śmigłowiec wyprodukowano dopiero w latach czterdziestych
dwudziestego wieku. Statki kosmiczne dawno opisano w literaturze, a niezbędnych obliczeń
dokonano już w pierwszym dziesięcioleciu dwudziestego wieku, gdy pierwszy sztuczny satelita
Ziemi wystartował z sukcesem dopiero pod koniec lat pięćdziesiątych, a pierwszy prom
kosmiczny w latach siedemdziesiątych tego wieku.
1.2. Materiały konstrukcyjne
Metaloznawstwo jest nauką o budowie, właściwościach i metodach badań metalicznych
materiałów konstrukcyjnych, tzn. używanych do produkcji maszyn, urządzeń i konstrukcji.
Zadaniem tej dziedziny wiedzy technicznej jest określanie wpływu zmiany warunków
zewnętrznych, w tym również wywołanej procesami technologicznymi, na budowę tworzywa
oraz ustalanie zależności pomiędzy składem i budową tworzywa a jego właściwościami.
Zrozumienie tych zależności wymaga znajomości elementarnych mikroprocesów zachodzących
w materiale pod wpływem zmian temperatury, obciążenia i innych czynników zewnętrznych.
Zdefiniowanie pojęcia struktury (budowy) materiału zależy od przyjętej skali obserwacji.
W skali podmikroskopowej (atomowej) rozpatruje się strukturę krystaliczną, tj. przestrzenny
rozkład cząstek materii (atomów, jonów, cząsteczek), typ i symetrię sieci przestrzennej, rozkład
cząstek materii w komórce zasadniczej, jej wymiary wreszcie. Współczesne metody
eksperymentalne w zasadzie nie umożliwiają bezpośredniej obserwacji poszczególnych atomów,
a tylko pewnych ich zgrupowań (np. strefy G P). Jednak pośrednio metodami dyfrakcji
rentgenowskiej lub elektronowej wymienione cechy struktury krystalicznej można określić i
zmierzyć ze znaczną dokładnością.
W skali mikroskopowej rozpatruje się podstrukturę, tj. strukturę rzeczywistą kryształu albo
ziarna. Struktura rzeczywista obejmuje granice, orientację i rozmiary bloków oraz defekty
struktury krystalicznej. Bezpośrednią obserwację podstruktury, zwłaszcza granic
wąskokątowych lub dyslokacji, umożliwia mikroskopia elektronowa z wykorzystaniem techniki
folii.
Wreszcie w skali mikroskopowej albo makroskopowej mówi się odpowiednio o mikrostrukturze
albo makrostrukturze. Jej opis obejmuje w materiałach jednofazowych kształt, wielkość i
orientację poszczególnych ziarn, a w materiałach wielofazowych ponadto rodzaj, udział i
wzajemne usytuowanie faz składowych. W obu przypadkach opis obejmuje również ewentualne
wady materiałowe: wtrącenia niemetaliczne (kształt i rozkład wydzieleń), pęknięcia, pory itp.
Metody makroskopii oraz mikroskopii świetlnej i elektronowej umożliwiają bezpośrednią
obserwację makrostruktury i mikrostruktury materiału oraz przy wykorzystaniu odpowiednich
wskaźników jej ilościowy opis.
Pełny opis struktury wymaga więc znajomości składu chemicznego materiału (faz
składowych), struktury krystalicznej, podstruktury i mikrostruktury. Warto pamiętać, że
struktura materiału jest stabilna w określonych warunkach zewnętrznych (temperatura,
ciśnienie). Zmiana tych warunków może wywołać w materiale przemianę fazową i w
konsekwencji zmianę struktury, a więc i właściwości. Analogiczny skutek można uzyskać
poddając materiał odpowiednim procesom technologicznym; w tym zakresie szczególnie
efektywne są obróbka plastyczna i obróbka cieplna.
Do cech materiału o szczególnym znaczeniu użytkowym należą właściwości mechaniczne.

JW
2
Rozumie się przez nie zespół cech (granica sprężystości, wytrzymałość, twardość)
określających wytrzymałość oraz (granica plastyczności, wydłużenie, przewężenie, udarność)
charakteryzujących plastyczność materiału. Spośród nich najczęściej wytrzymałość albo granica
plastyczności są podstawą obliczeń podczas projektowania. Decydując o wymiarach przekroju
elementów, niezbędnych do przenoszenia przewidywanych obciążeń, wespół z ciężarem
właściwym przesądza o gabarycie i ciężarze konstrukcji.
Zespół cech umożliwiających zachowanie, niezmiennych w czasie, właściwości materiału,
jak odporność na korodujące lub mechaniczne (erozja, kawitacja) działanie środowiska oraz
mechaniczne działanie (ścieranie) współpracujących elementów, wreszcie odporność na
działanie podwyższonej temperatury decydują o niezawodności i trwałości konstrukcji.
Przez właściwości technologiczne rozumie się podatność materiału do określonych technik
wytwarzania, jak odlewanie (lejność), spawanie (spawalność), obróbka plastyczna (ciągliwość,
tłoczność), obróbka skrawaniem (skrawalność), obróbka cieplna (hartowność) itp. Właściwości
te, przy uwzględnieniu wielkości produkcji, decydują o wyborze optymalnej technologii, a w
połączeniu z ceną materiału o koszcie konstrukcji.
Specjalne właściwości fizyczne, np. temperatura topnienia, rozszerzalność cieplna,
przenikalność magnetyczna itp., czy chemiczne, np. odporność na utlenianie w wysokiej
temperaturze, odporność na działanie określonej substancji chemicznej itp., w konkretnych
przypadkach przesądzają wybór materiału, usuwając na dalszy plan właściwości mechaniczne,
technologiczne oraz cenę.
Ogromna liczba współcześnie stosowanych materiałów konstrukcyjnych stwarza wielorakość
kryteriów klasyfikacyjnych. Jedną z najogólniejszych jest klasyfikacja oparta na charakterze
dominującego wiązania działającego między cząstkami materii. Z tego punktu widzenia
wyróżnia się materiały:
metaliczne o wiązaniu metalicznym,
ceramiczne o wiązaniu kowalencyjnym albo jonowym,
polimeryczne, w których działa wiązanie kowalencyjne (w obrębie makrocząsteczek) i
siły Van der Waalsa (między makrocząsteczkami).
Materiały metaliczne, tj. metale techniczne i ich stopy, należą do grupy tworzyw
krystalicznych. Charakteryzują się bardzo dobrymi właściwościami wytrzymałościowymi i
plastycznymi, dobrą przewodnością elektryczną i cieplną oraz zróżnicowaną odpornością na
korozję. Odznaczają się na ogół dobrymi właściwościami technologicznymi oraz łatwością
nadawania im (stopy metali) bardzo różnorodnych właściwości fizycznych i chemicznych. Wadą
materiałów metalicznych jest na ogół duży ciężar właściwy. Stanowią one podstawowe
tworzywo na wyroby przemysłu maszynowego oraz na konstrukcje metalowe.
Materiały ceramiczne należą w zasadzie do tworzyw krystalicznych, jakkolwiek mogą mieć
pewien udział fazy amorficznej. Cechuje je duża twardość i kruchość. Przeważnie są izolatorami
elektrycznymi i cieplnymi, o znacznej odporności na korozję. Wadą ich są złe właściwości
technologiczne, przez co wymagają specjalnych technik przetwarzania. Właściwości
predystynują materiały ceramiczne do specjalnych zastosowań, np. do wyrobu elementów
żaroodpornych, elektroizolacyjnych, termoizolacyjnych oraz jako specjalne materiały
narzędziowe (ostrza narzędzi skrawających, środki ścierne i polerskie).
Materiały polimeryczne, tj. tworzywa sztuczne, należą do grupy tworzyw amorficznych.
Odznaczają się stosunkowo dobrymi właściwościami mechanicznymi, są elektroizolatorami oraz
są bardzo odporne na działanie czynników chemicznych. Zaletą ich jest mały ciężar właściwy, a
wadą - mała odporność na działanie temperatur przekraczających 200-300° C (organiczne
związki węgla z wodorem i tlenem). Aktualnie obserwuje się ogromny wzrost zastosowań
tworzyw sztucznych, coraz skuteczniej konkurujących z materiałami metalicznymi w zakresie
elementów maszyn i zdecydowanie wypierających metale i szkło w zakresie opakowań, albo
metale i drewno w zakresie elementów wystroju wnętrz i taboru komunikacyjnego. Jednym z
powodów wzrostu produkcji tworzyw sztucznych jest możliwość wydatnego powiększenia ich
cech mechanicznych przez tzw. zbrojenie kompozyty), np. włóknami metalicznymi lub
ceramicznymi (szkło, węgiel).

3
JW
2. Struktura materiałów
2.1. Budowa atomu
Każdy przedmiot, zarówno wytworzony przez przyrodę, jak i będący dziełem pracy ludzkiej,
utworzony jest z materii, która za pomocą procesów chemicznych lub fizycznych można
rozłożyć na proste składniki, zwane pierwiastkami. Składniki te nie ulegają zmianie w żadnych
reakcjach chemicznych.
Najmniejsza cząstka pierwiastka jest atom, będący skupieniem jeszcze drobniejszych cząstek
materii, zwanych elementarnymi. Cząstkami tymi są elektrony, protony i neutrony. Teorię
budowy pojedynczego izolowanego atomu opracowano z wykorzystaniem mechaniki falowej.
Mechanika falowa umożliwia opisanie zachowania się elektronów w atomach i kryształach.
Każdy atom składa się z części wewnętrznej, tj. tzw. jądra i części zewnętrznej - powłok
elektronowych. Średnice wszystkich atomów są bardzo małe i zawierają się w granicach od
0,106 nm dla azotu do 0,58 nm dla fransu. Znacznie mniejsze rozmiary maja jądra atomów
zbudowane z protonów i neutronów (średnice rzędu 0,0001 nm), a najmniejsze są średnice
elektronów, które zawsze wynoszą 0,000 002 8 nm, czyli 2,8 • 10
-12
mm.
Podstawowa cecha atomu jest jego masa atomowa (ciężar atomowy), wyrażona jednostką
względna w stosunku do 1/16 masy atomu tlenu (umownie przyjęto masę jednego atomu tlenu
na 16 jednostek). Praktycznie za masę atomu przyjmuje się masę jego jądra (tj. protonów i
neutronów) gdyż masa elektronów jest bardzo mała i wynosi 9,009 • 10
-28
g, tzn. 0,000 55 część
masy atomu wodoru. Masa protonów wynosi 1,6 • 10
-24
g, tzn. jest równa masie atomu wodoru
(w jednostkach względnych ma masę równa 1), masa neutronów jest zbliżona do masy
protonów.
Elektrony krążą dookoła jądra z bardzo duża prędkością po ściśle określonych eliptycznych
orbitach (torach) i zawierają w sobie jeden elementarny ładunek elektryczności ujemnej,
równoważąc w ten sposób dodatnio naładowane jądro o ładunku równym sumie ładunków
wszystkich elektronów, tak że atom jako całość jest elektrycznie obojętny. Trzeba jednak
podkreślić, że elementarne ładunki dodatnie, równe co do wielkości elementarnym ładunkom
ujemnym zawartym w elektronach, mają tylko protony, toteż ich liczba w atomie zawsze
odpowiada liczbie elektronów. Natomiast liczba neutronów nie zawierających ładunku
elektrycznego jest różna.
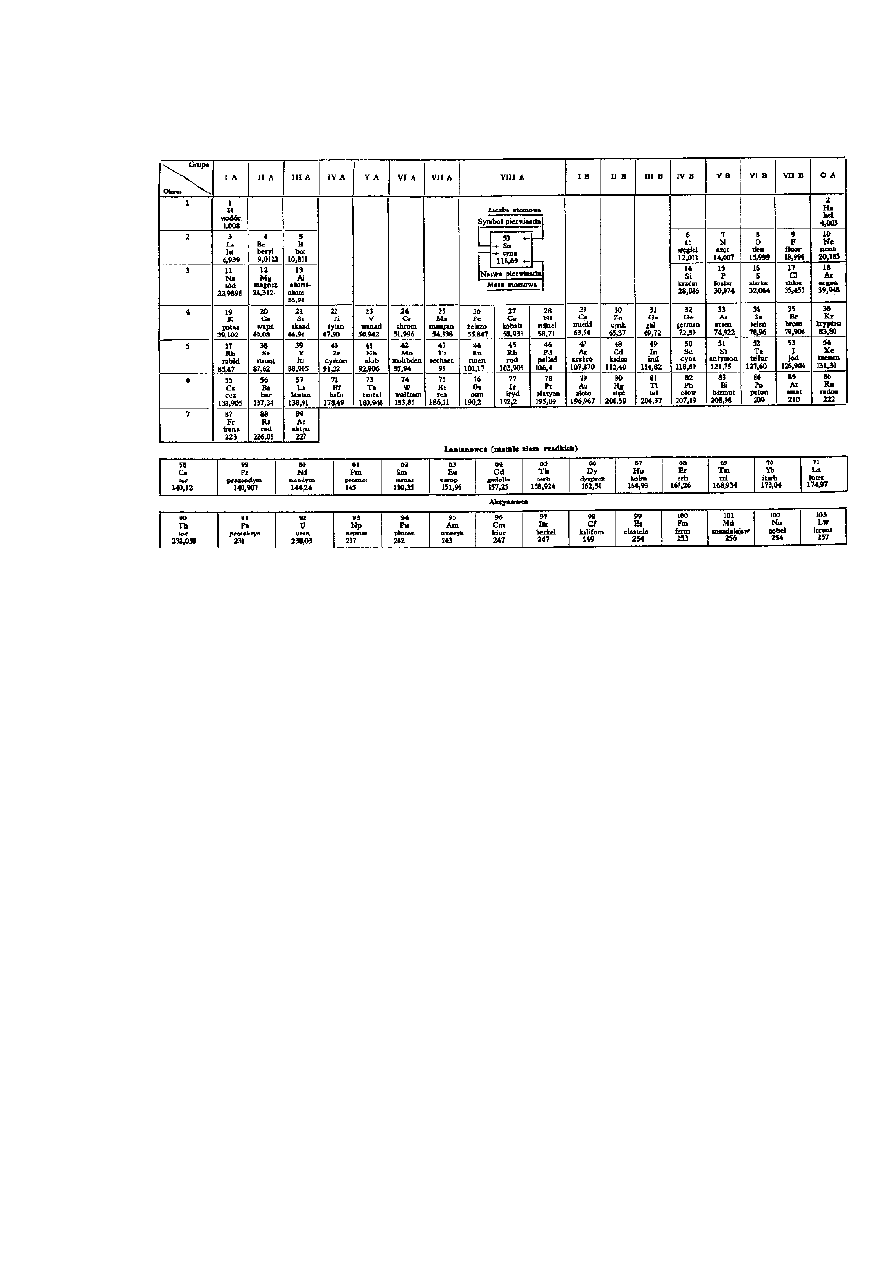
Obecnie znanych jest 105 różnych pierwiastków chemicznych, przy czym ostatnich
trzynaście zostało wytworzonych sztucznie. Wszystkie te pierwiastki można uszeregować w tzw.
układ okresowy
1)
wg wzrastających (z małymi wyjątkami) ciężarów atomowych, od
najlżejszego wodoru do pierwiastków najcięższych. Każdy z nich ma swoją kolejną liczbę
porządkową określającą zarówno wielkość ładunku elektrycznego jądra, a więc liczbę protonów,
jak i miejsce w układzie okresowym, co jednocześnie ustala własności chemiczne pierwiastka i
większość własności fizycznych (temperaturę topnienia, przewodnictwo elektryczne, budowę
wewnętrzną, własności magnetyczne itd.).
Okresowy układ pierwiastków w postaci obecnie najczęściej stosowanej przedstawiono na
rys. 2.1. Jak widać, w niektórych przypadkach pierwiastek następny ma niższą masę atomową od
poprzedniego (np. potas i argon, nikiel i kobalt itd.). Dzieje się tak dlatego, że większość
pierwiastków jest mieszaniną swych izotopów
2)
o różnych masach atomowych równych liczbom
całkowitym. Występujący np. w potasie w największej ilości izotop ma masę atomową 39,
odpowiedni izotop argonu - 40, w wyniku czego mieszanina izotopów argonu ma większą masę
atomową, niż mieszanina izotopów potasu.
Z tego samego powodu masa atomowa pierwiastków mających izotopy wyraża się
przeważnie liczbą ułamkową (izotopy występują w różnych stosunkach).
Układ okresowy w pionowych kolumnach grupuje pierwiastki o podobnych własnościach. W
wierszach poziomych, zwanych okresami, umieszczone są z lewej metale, z prawej niemetale;
1) Pierwszy zauważył to chemik rosyjski Dymitr Mendelejew (1834 — 1907).
2) Izotopy — odmiany tego samego pierwiastka, rożnącę się tylko ilością neutronów w jądrze, np. nikiel ma 5
izotopów o masach atomowych równych 58, 60, 61, 62 i 64, tzn. zawierających po 28 protonów i odpowiednio 30,
32, 33, 34 i 36 neutronów.
4
JW
do metali zalicza się wszystkie pierwiastki znajdujące się w układzie okresowym na lewo od
galu, indu i talu, do niemetali — pierwiastki znajdujące się na prawo od arsenu, antymonu i
bizmutu.
Rys. 2.1 Układ okresowy pierwiastków
. Natomiast pierwiastki znajdujące się w kolumnach IIIb, IVb i Vb zajmują miejsce pośrednie,
gdyż bez zastrzeżeń nie można ich zaliczyć ani do jednej, ani do drugiej grupy.
Jak wspomniano, elektrony poruszają się wokół jądra atomu po określonych torach, tworząc tzw.
powfoki i podpowfoki elektronowe. Pierwiastki pierwszego okresu (H, Hę) mają tylko jedną
powłokę elektronową, która nie może zawierać więcej niż 2 elektrony (podpowłoka s).
Pierwiastki drugiego okresu (Li, Be, B itd.) mają dwie powłoki elektronowe; pierwsza zawiera 2
elektrony, druga — 1-8 (1-2 na podpowłoce s i 0-6 na podpowłoce p). Pierwiastki trzeciego
okresu (Na, Mg, Al itd.) pierwszą i drugą powłokę mają taką samą, jak ostatni pierwiastek
drugiego okresu — neon, ale poza tym mają jeszcze trzecią powłokę elektronową, złożoną z
dwóch podpowłok: s, na której znajduje się 1-2 elektronów, i p, na której znajduje się 0-6
elektronów, przy czym maksymalną ilość elektronów zawiera atom ostatniego pierwiastka tego
okresu - argonu, którego liczba atomowa wynosi 18.
Strukturę elektronową atomów omówionych pierwiastków zapisuje się następująco:
1. H- 1s
1
2. He – 1s
2
3. Li – 1s
2
2s
1
………………
7. N – 1s
2
2s
2
2p
3
……………….
10. Ne - 1s
2
2s
2
2p
6
11. Na- 1s
2
2s
2
2p
6
3s
1
…………………….
18. Ar - 1s
2
2s
2
2p
6
3s
2
3p
6
,
gdzie: l, 2, 3 ... są głównymi liczbami kwantowymi, określającymi poszczególne powłoki
elektronowe K, L, M, ...; s, p, ... poboczne liczby kwantowe określające podpowłoki
elektronowe; indeksy górne oznaczają liczbę elektronów w danej pod-powłoce elektronowej.
Rozbudowa zewnętrznych powłok elektronowych atomów pierwiastków czwartego okresu
przebiega nieco inaczej. Rozpoczynający ten okres potas ma zewnętrzny elektron umieszczony

5
JW
w podpowłoce 4s zamiast 3d:
19. K - 1s
2
2s
2
2p
6
3s
2
3p
6
4s
1
podobnie
20. Ca- 1s
2
2s
2
2p
6
3s
2
3p
6
4s
2
Dopiero poczynając od skandu następuje rozbudowa podpowłoki 3d (mieszczącej maksymalnie
10 elektronów), poprzedzona całkowitym lub częściowym (chrom) zapełnieniem podpowłoki 4s:
21. Sc - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
1
4s
2
22. Ti - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
2
4s
2
………………………………
26. Fe - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
6
4s
2
27. Co - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
7
4s
2
28. Ni - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
8
4s
2
29. Cu - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
10
4s
1
Pierwiastki o rozbudowującej się podpowłoce d nazywają się pierwiastkami przejściowymi
lub metalami przejściowymi.
Pozostałe pierwiastki tego okresu po wypełnieniu podpowłok 3d i 4s rozbudowują
podpowłokę 4p, osiągając dla kryptonu konfigurację 4s
2
4p
6
, charakterystyczną dla gazu
szlachetnego. Struktury elektronowe atomów pierwiastków piątego okresu rozbudowują się tak
jak czwartego, tzn. w kolejności 5s, 4d i 5p.
Okres szósty zawiera 32 pierwiastki. Cez, bar i lantan mają elektrony zewnętrzne
rozmieszczone kolejno w podpowłokach 6s i 5d:
57. La - 1s
2
2s
2
2p
6
3s
2
3p
6
3d
10
4s
2
4p
6
4d
10
5s
2
5p
6
5d
1
6s
2
Kolejne jednak pierwiastki rozbudowują podpowłokę 4f, mieszczącą 14 elektronów. Te
pierwiastki (od ceru do lutetu) nazywa się lantanowcami lub pierwiastkami ziem rzadkich.
Dopiero po wypełnieniu podpowłoki 4f następuje uzupełnienie podpowłoki 5d, a później 6p.
Ostatni pierwiastek tego okresu radon jest gazem szlachetnym, gdyż ma konfigurację elektronów
zewnętrznych 6s
2
6p
6
.
W okresie siódmym trzy pierwsze pierwiastki frans, rad i aktyn mają zewnętrzne elektrony
rozmieszczone kolejno w podpowłokach 7s i 6d. Począwszy od toru następuje rozbudowa
podpowłoki 5f. Pierwiastki zawierające tę podpowłokę nazywają się aktynowcami,
Od struktury elektronowej, a zwłaszcza od konfiguracji elektronów powłoki zewnętrznej
zależą własności chemiczne pierwiastków. Atomy z powłoką zewnętrzną całkowicie wypełnioną
elektronami mają kulistą chmurę elektronów, są chemicznie obojętne i w przyrodzie występują
jako jednoatomowe. Atomy z powłoką zewnętrzną niecałkowicie wypełnioną elektronami mają
mniej symetryczną powłokę elektronową i są skłonne do łączenia się w cząsteczki (np. wodór).
Elektrony krążące w zewnętrznej powłoce nazywa się elektronami walencyjnymi lub
wartościowości. Od nich zależą wiązania atomowe. Atom z niecałkowicie wypełnioną
zewnętrzną powłoką, dążąc do uzyskania struktury elektronowej zbliżonej do struktury gazu
szlachetnego, oddaje swoje lub przyłącza elektrony walencyjne innego atomu. Pierwiastki
oddające swe elektrony walencyjne nazywa się elektrododatnimi, przyłączające elektrony -
elektroujemnymi.
Metale mają l lub 2 elektrony w zewnętrznej powłoce elektronowej i są zawsze pierwiastkami
elektrododatnimi. Najbardziej typowe metale, jak sód, potas, miedź, srebro i złoto, mają tylko
jeden elektron walencyjny. Do metali zalicza się także pierwiastki przejściowe i pierwiastki ziem
rzadkich. Szczególna struktura elektronowa pierwiastków przejściowych wyjaśnia m.in. ich
wysoką temperaturę topnienia oraz zdolność tworzenia trwałych związków z węglem i azotem.
Pierwiastki mające trzy, cztery lub pięć elektronów walencyjnych (np. aluminium, cyna,
bizmut) mają własności amfoteryczne i mogą zarówno oddawać, jak i przyłączać elektrony
walencyjne. Pierwiastki o sześciu lub siedmiu elektronach walencyjnych są typowymi
niemetalami.
6
JW
2.2. Siły spójności
W zbiorach atomów tworzących większe skupienia materii występują określone siły
przyciągania i odpychania. Bardzo trafną hipotezę co do natury tych połączeń postawił jeszcze
Newton (1704 r.), który napisał: „ja raczej wnoszę ze spójności tych ciał, że cząstki przyciągają,
się wzajemnie pewną silą, która jest niezwykle duża, gdy cząstki się stykają przy małych
odległościach... Są więc w przyrodzie czynniki, które powodują, ze cząstki zlepiają się wskutek
bardzo silnego przyciągania". Twierdzenie to w ciągu 300 lat nie straciło swojej aktualności.
Cząsteczki podlegające wiązaniu mogą być atomami określonego pierwiastka np. (H, N, 0) lub
związku (np. CO). Każdy pierwiastek zależnie od temperatury i ciśnienia może istnieć w trzech
stanach skupienia, tj. gazowym, ciekłym i stałym. W stanie gazowym odległości między
atomami są duże rzędu dziesiątków średnic atomowych, w stanach ciekłych i stałym atomy
znajdują się blisko siebie. Dlatego te oba stany nazywają się stanami skondensowanymi. Zawsze
w przypadku połączenia się atomów ze sobą muszą między nimi działać określone siły, które
zależą przede wszystkim od typu wiązania. Siły te mają zarówno charakter odpychający, jak i
przyciągający, ale przy braku działania sił zewnętrznych równoważą się ze sobą, w wyniku
czego ustala się między atomami równowagowa odległość, przy której siła wzajemnego
oddziaływania jest równa zeru. Jeśli jednak na taki układ działa siła zewnętrzna, usiłująca
zbliżyć lub oddalić atomy, powstają siły międzyatomowe przeciwdziałające temu działaniu
nazywane siłami spójności. Wielkość tych sił decyduje o wytrzymałości mechanicznej ciał
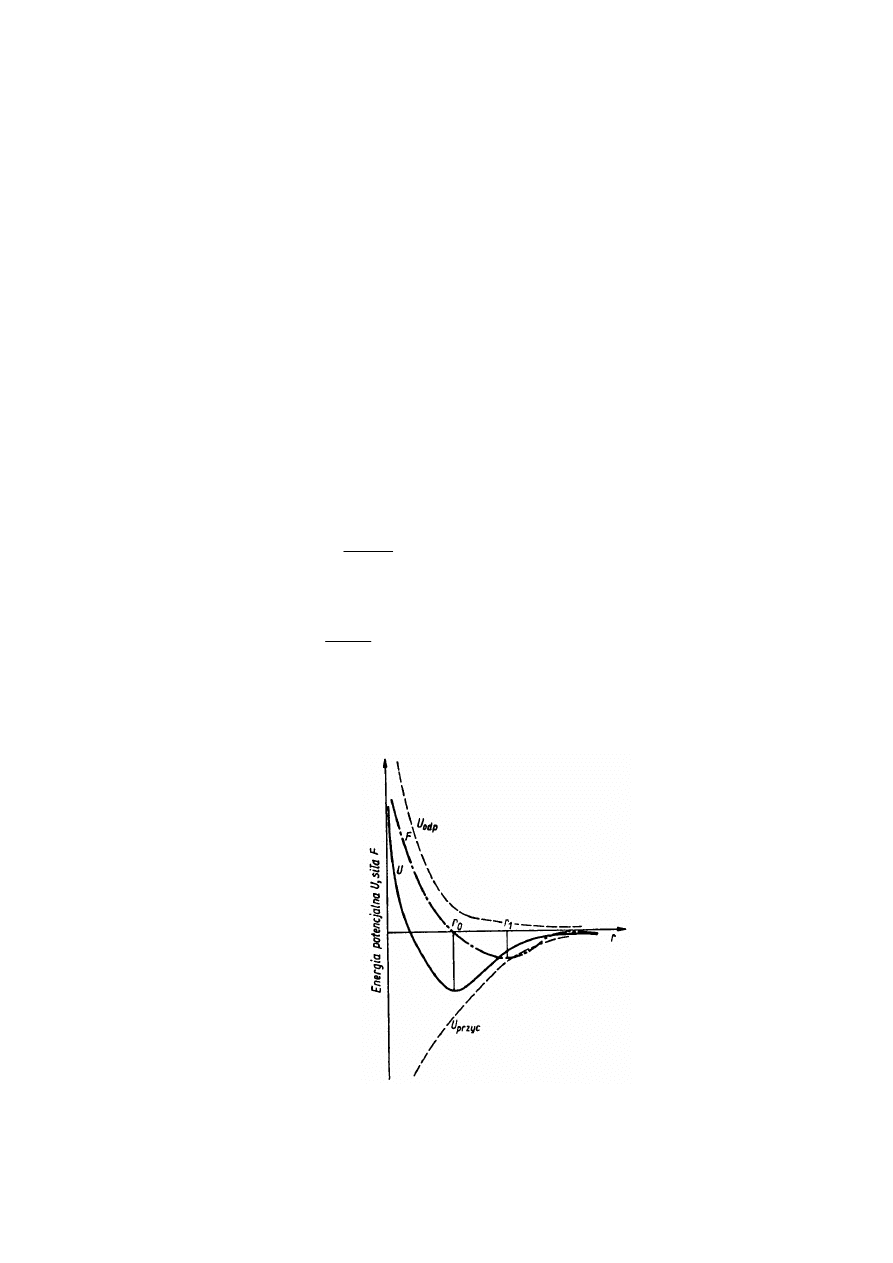
stałych. Rozważania teoretyczne wykazały, że siła przyciągania F
p
występująca między
atomami przy ich łączeniu, jest określona następującym wzorem ogólnym:
natomiast siła odpychająca
gdzie: a, b, m, n – stałe zależne od rodzaju kryształu, r – odległość między atomami (znak „–„
oznacza przyciąganie, a „+” odpychanie)
Rys. 2.2. Siły F(r) i energia potencjalna U(r) wzajemnego oddziaływania między atomami
w funkcji odległości międzyatomowej r
1
+
−
=
m
r
b
p
F
1
+
=
n
r
a
o
F

7
JW
Ponieważ przy zmniejszaniu odległości r siły odpychania zwiększają się szybciej niż siły
przyciągania przy oddalaniu, wykładnik potęgowy m < n
Siła wypadkowa
Jeżeli wzrasta r o dr, to siła F wykonuje pracę Fdr kosztem zmniejszenia energii potencjalnej
wzajemnego oddziaływania U. Możemy napisać zależność Fdr = —dU, czyli F = —(dU/dr).
Stąd energia potencjalna w funkcji odległości międzyatomowej dwuatomowego układu będzie
równa
gdzie: A = a/n, B = b/n
Na rysunku przedstawiono zależność zmiany siły wypadkowej F i energii potencjalnej
wzajemnego oddziaływania U w funkcji r. Z wykresu tego wynikają następujące wnioski:
l. Gdy r jest bardzo duże, wówczas zarówno siła wzajemnego oddziaływania, jak i energia U są
równe zeru
2. Zmniejszanie r powoduje powstanie siły przyciągającej, która początkowo zwiększa się, a po
przekroczeniu wartości r
1
maleje, osiągając wartość zero przy odległości równowagowej r
o
Jednocześnie energia U maleje, osiągając minimum przy r = r
o
3. Dalsze zmniejszanie r (poniżej r
o
) powoduje powstanie siły odpychającej i zwiększenie
energii U
2.3. Rodzaje wiązań
2.3.1.Wiązanie jonowe
Wiązanie jonowe jest typowe dla kryształów jonowych, które z reguły są przeźroczyste, a
ich przewodnictwo elektryczne jest bardzo małe. Cechuje je dość duża wytrzymałość
mechaniczna i twardość oraz wysoka temperatura topnienia, a także mają one tendencję do
łupliwości wzdłuż określonych płaszczyzn krystalograficznych, co świadczy o kierunkowym
charakterze wiązania.
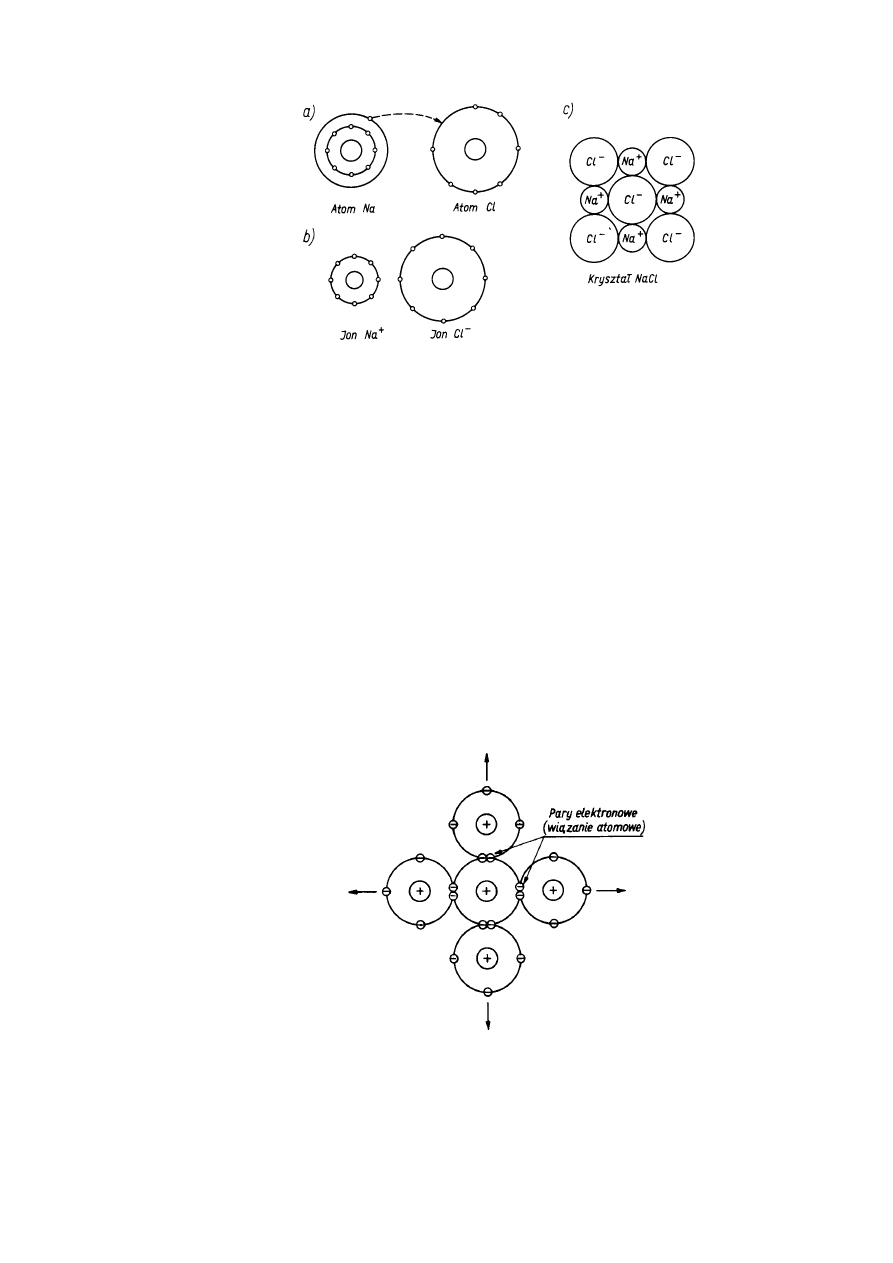
Wiązanie jonowe jest spowodowane dążeniem różnych atomów do tworzenia trwałych 8-
elektronowych konfiguracji gazów szlachetnych przez uwspólnienie elektronów. Tak na
przykład w przypadku tworzenia cząsteczki NaCl atom sodu, mający na zewnętrznej orbicie l
elektron, oddaje go atomowi chloru, stając się jonem dodatnim, a atom chloru, mający na
ostatniej orbicie 7 elektronów, po dołączeniu dodatkowego elektronu staje się jonem ujemnym.
Możemy to zapisać symbolicznie
Na + Cl
⇒ Na
+
+ C1
-
Wiązanie to oparte jest na przyciąganiu kulombowskim, jakie powstaje między dwoma
przeciwnymi ładunkami. Utworzona cząsteczka związku NaCl jest elektrycznie obojętna, ale ma
zaznaczone bieguny elektryczne (czyli jest dipolem), co umożliwia jej łączenie się z innymi
cząsteczkami i tworzenie kryształu. Na rysunku 2.3 pokazano schemat utworzenia jonów Na
+
i
Cl
-
oraz kryształu NaCl. Do kryształów jonowych zaliczamy halogenki metali alkalicznych
(sole) oraz wiele materiałów ceramicznych stosowanych w technice.
1
1
+
−
+
=
+
=
m
r
b
n
r
a
p
F
o
F
F
m
r
B
n
r
A
U
−
=
8
JW
Rys. 2.3. Schemat powstawania wiązania jonowego w NaCI: a) przejście elektronu z
atomu Na do Cl, b) utworzone jony, c) kryształ NaCI
2.3.2. Wiązania atomowe
Wiązanie atomowe (zwane także kowalentnym lub homopolarnym) występuje w cząsteczkach
gazów dwuatomowych (H
2
, Cl
2
, O
2
, N
2
), niektórych pierwiastkach stałych (C - diament, Ge, Si,
Sn-
α) i związkach (SiC), a także w polimerach. Jest to wiązanie silne i kierunkowe. Energia
wiązania w diamencie wynosi 710 kJ/mol, a w SiC 1,18 MJ/mol. Wiązanie tworzy się zgodnie z
teorią Levisa-Kossela, na skutek dążenia atomów do tworzenia trwałych 2- lub 8-elektronowych
konfiguracji gazów szlachetnych, dzięki powstawaniu par wiążących, których liczba zależy od
grupy układu okresowego N (reguła 8-N). Elektrony przechodzą od jednego do drugiego atomu,
zamieniając je w jony dodatnie, które są przyciągane przez elektrony znajdujące się między
nimi. Wiązanie to można przedstawić schematycznie następująco
H
.
+
.
H = H : H
: Cl
.
+
.
C1: = Cl : Cl
Rys.2.4. Wiązanie atomowe w krzemie (brak elektronu w parze stanowi dziurę, dodatkowy
elektron jest elektronem przewodnictwa)
W tlenie występują dwie pary wiążące, w azocie - trzy, w węglu, krzemie i germanie po
cztery (rys. 2.4). Wiązanie atomowe może być zinterpretowane przy użyciu zasad mechaniki
falowej (obliczenia wykonali Heitler i London 1927 r.). Zgodnie z zakazem Pauliego elektrony
w parach mają takie same trzy liczby kwantowe i różnią się spinem. Przy antyrównoległych

9
JW
spinach energia układu zmniejsza się, osiągając minimum przy równowagowej odległości
atomów w cząsteczce. W niektórych przypadkach (np. w diamencie) wiązanie atomowe
powstaje w wyniku hybrydyzacji (zmieszania) stanów elektronowych 2s i 2p. Dzięki temu
powstaje konfiguracja 1s
2
2s
1
2p
3
zamiast konfiguracji 1s
2
2s
2
2p
2
co umożliwi utworzenie czterech
równorzędnych wiązań z sąsiednimi atomami o antyrównoległych spinach elektronów 2s i 2p.
W przypadku gdy łączą się różne atomy za pomocą par elektronowych mamy do czynienia
z wiązaniem atomowym spolaryzowanym. Na skutek różnego oddziaływania elektronów z
rdzeniami atomów powstają dipole i wiązanie ma wtedy charakter pośredni między atomowym i
jonowym. Nie jest to jednak regułą. W niektórych przypadkach (np. w metanie CH
4
)
symetryczny rozkład wiązań powoduje, że wypadkowy moment dipolowy jest równy zeru.
Istnieją związki (np. NH
4
), w których obydwa elektrony tworzące parę pochodzą od tego samego
atomu. W tym przypadku atom azotu może za pomocą wolnej pary elektronowej przyłączyć
dodatkowy jon H
+
. Takie wiązanie nazywa się koordynacyjnym.
2.3.3. Wiązanie van der Waalsa
Wiązanie van der Vaalsa jest bardzo słabe (energia wiązań wynosi 100 - 1500 J/mol) i
bezkierunkowe. Siły van der Waalsa działają w skroplonych gazach szlachetnych i między
łańcuchami polimerów. Przyczyną powstawania tych sił jest nierównomierny rozkład ładunków
w chmurach elektronowych. Pewna polaryzacja jest w tym przypadku wynikiem wzajemnego
oddziaływania atomów. Chwilowe dipole indukują dipole w sąsiednich atomach. Wiązania van
der Waalsa występują wraz z innymi w kryształach molekularnych, które składają się z
cząsteczek o wiązaniach kowalentnych zespolonych ze sobą siłami van der Waalsa. Przykładem
mogą być zestalone gazy (H, F, Cl, N) oraz kryształy jodu, siarki, selenu i telluru.
2.3.4. Wiązanie metaliczne
Wiązanie metaliczne występuje między atomami metali w skondensowanych stanach skupienia.
Istota tego wiązania wynika z teorii swobodnego elektronu (Drudego-Lorentza). Dzięki
niskiemu potencjałowi jonizacyjnemu elektronów, po zbliżeniu się atomów do siebie, następuje
oderwanie się elektronów wartościowości od rdzeni atomowych i utworzenie gazu
elektronowego, w którym zachowują się jako cząstki swobodne. Poruszają się one między
jonami i wiążą je na zasadzie elektrostatycznego przyciągania. Wiązanie metaliczne należy do
wiązań silnych (energia wiązania jest pośrednia miedzy jonowym a atomowym) i jest
bezkierunkowe. Poza tym typowymi własnościami metali są: dobre przewodnictwo elektryczne i
cieplne, ciągliwość i metaliczny połysk. Z własnościami gazu elektronowego jest także związane
charakterystyczne dla metali zwiększenie oporności ze wzrostem temperatury. Dotychczas brak
jest uniwersalnej teorii wyjaśniającej związek między budową elektronową a strukturą oraz
własnościami określonych metali.
Wielu badaczy przyjmuje, że wiązanie metaliczne jest podobne do wiązania kowalentnego
(Ormont, Pauling). Na przykład w sodzie atomy po zbliżeniu mogą utworzyć wiązanie za
pomocą pary elektronów 3s o różnych spinach. Następne elektrony mogą przejść na poziom 3p,
gdyż na skutek hybrydyzacji ich energie są zbliżone. Muszą one jednak ulegać ciągłej wymianie
z sąsiednimi atomami. Można więc powiedzieć, że istota wiązania jest atomowa, chociaż jest
utrzymana, zakładana w teorii swobodnych elektronów, możliwość ich ruchu od atomu do
atomu. Stąd wiązanie metaliczne bywa traktowane jako nienasycone wiązanie atomowe (z
niedoborem elektronów), w którym duża liczba atomów jest połączona przez uwspólnienie
elektronów wartościowości. Są także znane inne teorie; np. Wignera i Seitza, umożliwiająca
wyliczenie wartości energii wiązań w metalach alkalicznych, nie sprawdza się jednak ona w
przypadku innych metali).
10
JW
2. 4. Struktury krystaliczne metali
2.4.1. Elementy krystalgrafii
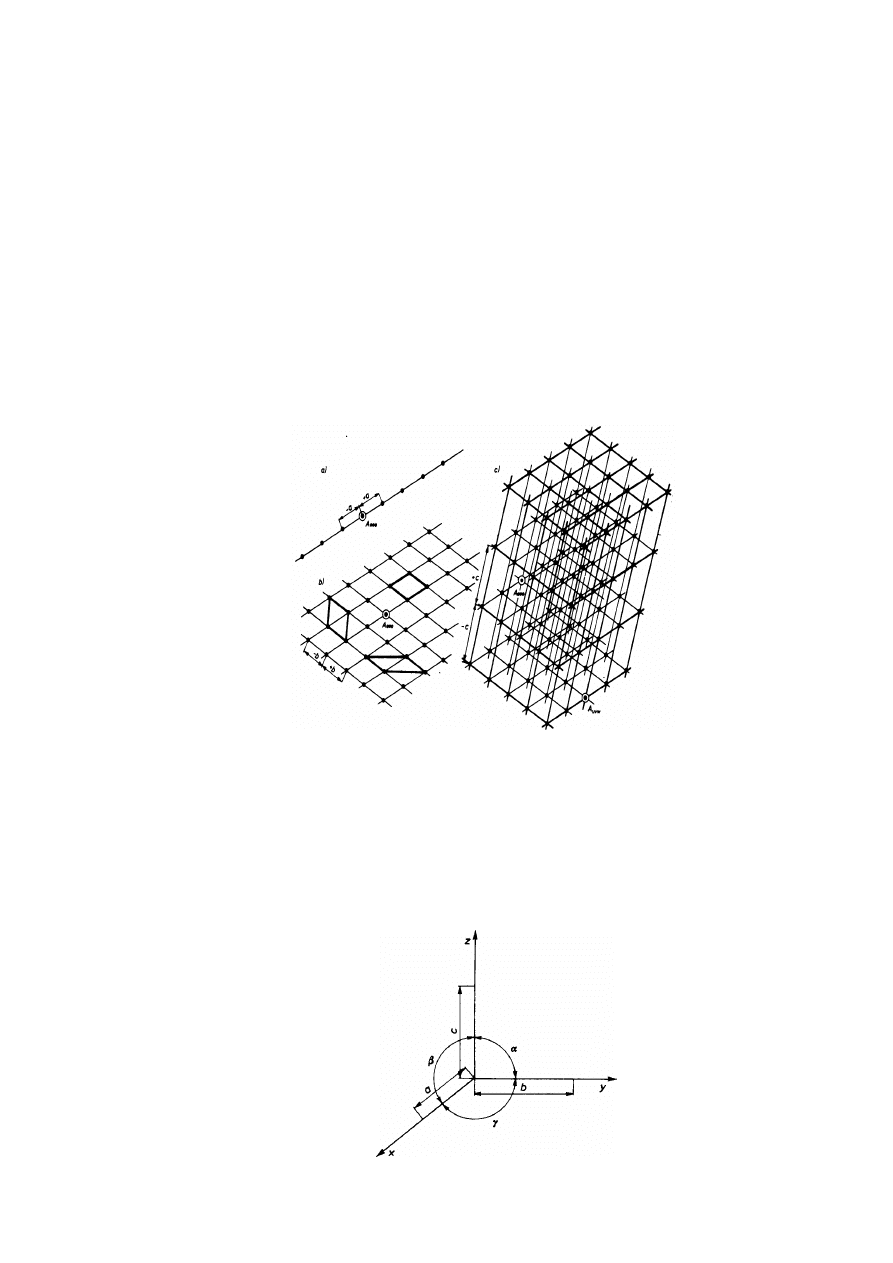
Materiały krystaliczne w pewnych warunkach przyjmują kształt charakterystycznych,
regularnych brył geometrycznych — kryształów. Budową kryształów zajmuje się krystalografia,
traktująca materię jako zbiorowisko atomów o kształcie sztywnych kul, rozmieszczonych
statycznie (nieruchomo) w przestrzeni. Odwzorowaniem przestrzennego rozmieszczenia atomów
jest sieć przestrzenna, utworzona przez powtórzenie w trzech kierunkach podstawowego
elementu, zwanego komórką zasadniczą albo sieciową. Jednoznaczne określenie komórki
zasadniczej umożliwia więc odwzorowanie sieci przestrzennej.
Przekształcenie geometryczne polegające na przesunięciu w określonym kierunku dowolnego
tworu geometrycznego o stały wektor, nazywa się translacją. Translacje - a i + a punktu -A
000
prowadzą do utworzenia prostej sieciowej (rys. 2. 5a). Translacje - b i + b prostej w kierunku
nierównoległym do niej tworzą płaszczyznę sieciową (rys. 2. 5b). Wreszcie translacje - c i + c
płaszczyzny w kierunku nierównoległym do niej tworzą sieć przestrzenną (rys. 2. 5c), której
punkty A
uvw
translacyjnie identyczne z punktem wyjściowym /A
000
stanowią węzły sieci.
Rys.2. 5. Konstrukcja sieci przestrzennej. Translacje: a) punktu, b) prostej, c) płaszczyzny
Do jednoznacznego zdefiniowania komórki zasadniczej (sieci przestrzennej) w ogólnym
przypadku konieczna jest znajomość trzech wektorów translacji: a, b, c, zwanych stałymi
(parametrami) sieciowymi oraz trzech kątów sieciowych:
α
,
β
,
γ
. Przyjęty w krystalografii do
opisu komórki zasadniczej układ współrzędnych przedstawiono na rys. 2.6. Istotną cechą sieci
przestrzennej (układu węzłów, prostych i płaszczyzn sieciowych) jest symetria.
Rys. 2.6. Krystalograficzny układ współrzędnych

11
JW
Wyróżnia się trzy podstawowe elementy symetrii: płaszczyznę, oś i środek. Płaszczyzna
symetrii dzieli kryształ na dwie części, stanowiące wzajemne lustrzane odbicie. Oś symetrii jest
osią obrotu, dookoła której obracając kryształ o pewien kąt otrzymuje się identyczne położenie
wszystkich elementów kryształu jak przed obrotem. Zależnie od wartości kąta obrotu mogą być
osie symetrii dwukrotne (180°), czterokrotne (90°) itp. Oś o największej krotności jest główną
osią symetrii — jej kierunek wyznacza orientację kryształu w przestrzeni. Wreszcie środek
symetrii jest punktem, względem którego wszystkie elementy kryształu po obrocie o 180°
zajmują położenia identyczne jak przed obrotem. Wszystkie płaszczyzny i osie symetrii
kryształu przecinają się w środku jego symetrii. Z tego powodu symetrię wynikającą z opisanych
elementów nazwano punktową. Możliwe są różnorodne kombinacje podstawowych elementów
tworzące złożone elementy symetrii.
Symetria sieci jest cechą znacznie ważniejszą od geometrycznego kształtu komórki
zasadniczej, ponieważ bryły o różnym kształcie, np. sześcian i ośmiościan, mają identyczne
elementy symetrii. Na podstawie symetrii punktowej możliwe sieci przestrzenne zalicza się do
jednego z siedmiu układów krystalograficznych, których charakterystykę podano w tabl. 1.
W określonym układzie krystalograficznym komórka zasadnicza może mieć różny kształt (przy
stałej objętości), jak to przedstawiono poglądowo na rys. 2
.
5 b
TABLICA 1. Charakterystyka układów krystalograficznych
Parametry komórki zasadnicze
Krotność elementów
Środek
osie
płaszczyzny –
prostopadłe do osi
Układ
krystalograficzny
stale
sieciowe
kąty sieciowe
parametry
charakte-
rystyczne
2-kr. 3-kr. 4-kr. 6-kr. 2-kr. 4-kr. 6-kr.
trojskośny
a
≠
b
≠
c
α ≠ β ≠ γ
a, b, c
α, β, γ
- - - - - - - 1
jednoskośny
a
≠
b
≠
c α = γ = 90°, β
a, b, c,
β
l - - - l - - 1
rombowy
a
≠
b
≠
c α ≠ β ≠ γ = 90° a, b, c
3 - - - 3 - - 1
romboedryczny
a= b= c
α = β = γ
a,
α
3 l —
— 3 - - 1
tetragonalny
a= b, c
α = β = γ = 90° a, c/a
4 - 1 - 4 1 - 1
heksagonalny
a = b, c α = β = 90°, γ =
120°
a, c/a
6 - - l 6 - 1 1
regularny
a = b = c
α = β = γ = 90° a
6 4 3 - 6 3 —
1
W określonym układzie krystalograficznym, zależnie od pozycji obsadzonych atomami,
wyróżnia się typy komórki zasadniczej, oznaczane symbolami:
• prostą (P) o obsadzonych atomami tylko narożach komórki,
• centrowanej podstawie (C) o dodatkowo obsadzonych atomami środkach podstaw,
• płaskocentryczną (F) o dodatkowo obsadzonych atomami środkach wszystkich ścian,
• przestrzenniecentryczną (I) o dodatkowo obsadzonym atomem środku komórki,
• złożoną o dodatkowo obsadzonym atomami wnętrzu komórki.
Bravais (1848 r.) drogą translacji prostych komórek zasadniczych poszczególnych układów
krystalograficznych wyprowadził 14 typów sieci, tzw. translacyjnych (rys. 2.7). Systematyka
Bravais'ego wskazuje, który z wymienionych typów komórek zasadniczych możliwy jest w
określonym układzie krystalograficznym.
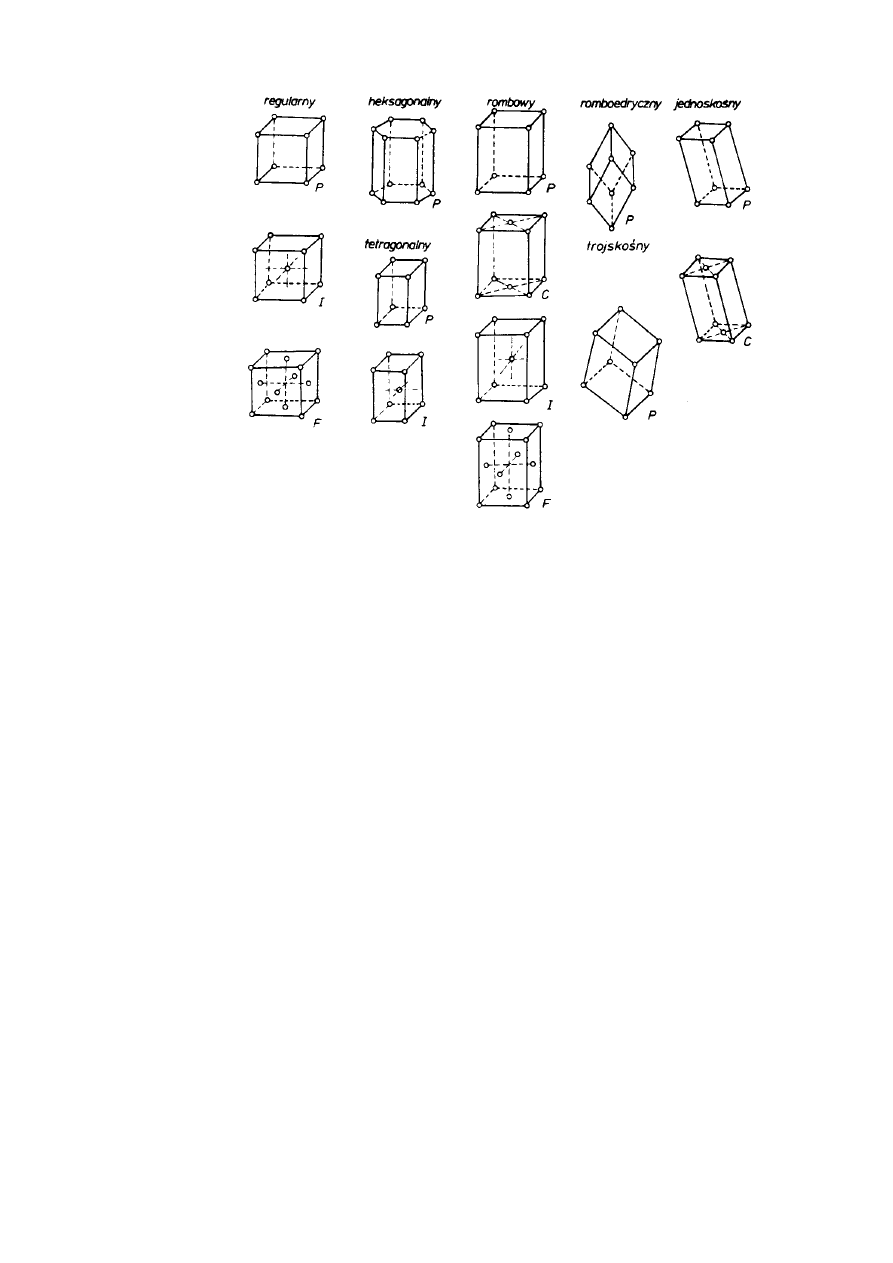
12
JW
Rys.2.7. Sieci translacyjne Bravais'ego
Niezależnie od układu krystalograficznego i typu komórki zasadniczej sieć przestrzenną
charakteryzują następujące wielkości:
Liczba koordynacyjna ( l.k..). Jest to liczba najbliższych i równoodległych atomów od
dowolnego atomu sieci. Odpowiada ona liczbie wiązań atomu, czyli stanowi
energetyczną miarę trwałości struktury krystalicznej.
Liczba atomów (l.a.) przypadająca na komórkę zasadniczą. Jest to wielkość
charakteryzująca w pewien sposób wielkość komórki zasadniczej.
Wypełnienie komórki zasadniczej (w.). Jest to stosunek objętości atomów (kul) do
objętości komórki zasadniczej (bryły), charakteryzujący gęstość atomową struktury
krystalicznej.
Opis położenia płaszczyzn i kierunków krystalograficznych umożliwiają wskaźniki Millera.
Płaszczyzna sieciowa odcina na osiach współrzędnych odcinki stanowiące ułamki lub
wielokrotności stałych sieciowych (rys.2.8). Wskaźniki Millera płaszczyzny (hkl) są
odwrotnościami tych odcinków, z uwzględnieniem znaku odpowiadającego zwrotowi osi,
wyrażonymi w liczbach całkowitych. Jeżeli płaszczyzna jest równoległa do osi, to punkt jej
przebicia przez tę oś leży w nieskończoności, której odwrotność jest zerem. Jeżeli odwrotności
odcinków są liczbami ułamkowymi, to sprowadza się je do liczb całkowitych, mnożąc przez
wspólną najmniejszą wielokrotną.
Sposób wyznaczania wskaźników Millera płaszczyzny wyjaśnia przykład (rys.2.7).
Płaszczyzna P odcina na osiach współrzędnych x, y, z odcinki odpowiednio 3, - 2, 4.
Odwrotności odcinków 1/3, -1/2, 1/4 po pomnożeniu przez wspólną najmniejszą wielokrotną 12
prowadzą do wskaźników Millera płaszczyzny P (4-63).
W szczególnym przypadku układu heksagonalnego często stosuje się układ współrzędnych
x
1
, x
2
,x
3
, z. Wskaźniki Millera-Bravais'ego płaszczyzny (hkil) wyznacza się analogicznie,
pamiętając, że w układzie heksagonalnym równoważne osie x spełniają zależność wektorową x
1
+ x
2
= -x
3
, z której wynika równoważność wskaźników h + k = - i. Należy pamiętać, że
wskaźniki, np. (111), opisują wszystkie płaszczyzny sieciowe równoległe. Grupę płaszczyzn
równoważnych krystalograficznie, np. płaszczyzny sześcianu w sieci regularnej (100), (010),
(001), (-100), (0-l0), (00-1) opisuje się wskaźnikami jednej z nich w nawiasach klamrowych, np.
{100}.
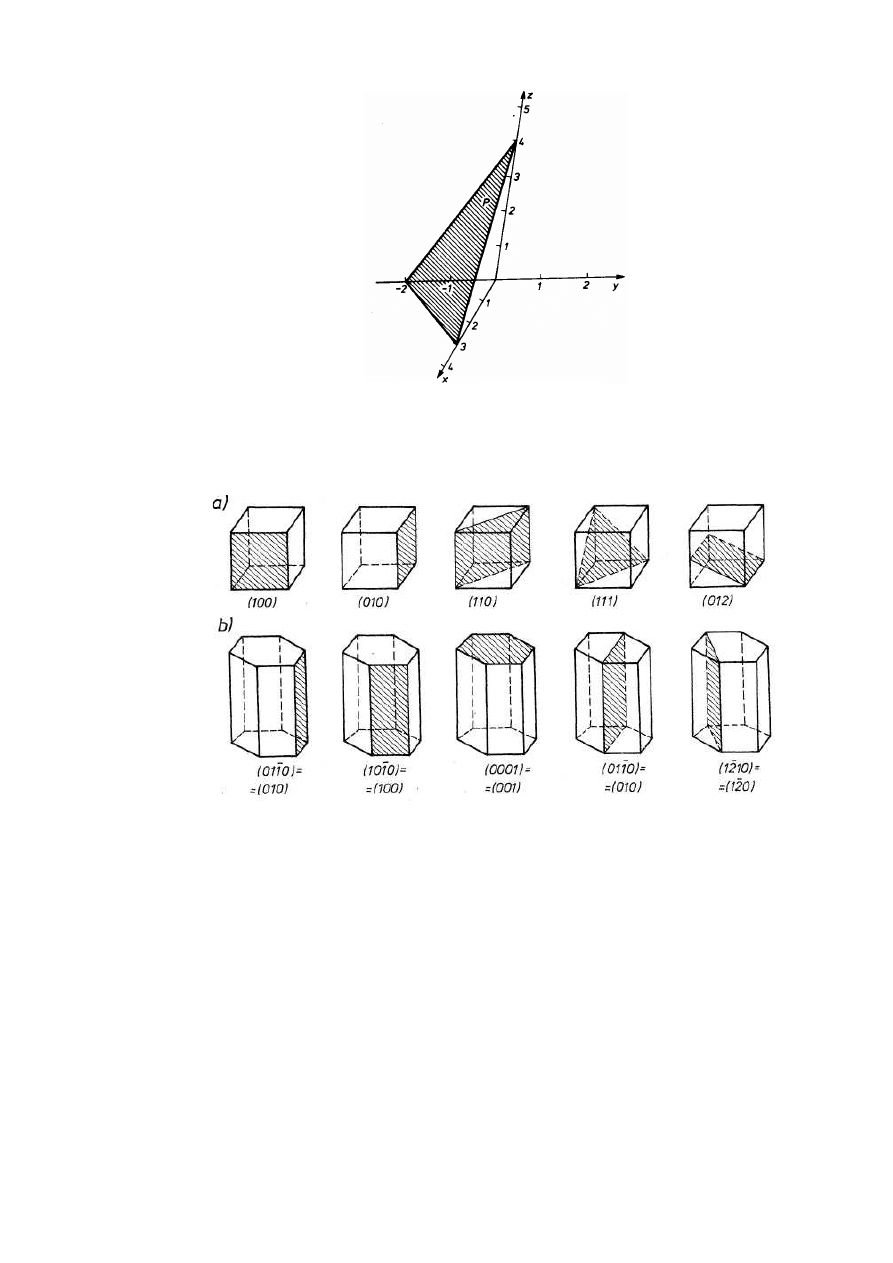
13
JW
Rys. 2.8. Wyznaczanie wskaźników Millera płaszczyzny sieciowej.
Na rysunku 2.9 pokazano oznaczenia typowych płaszczyzn sieciowych układów
regularnego i heksagonalnego.
Rys.2.9. Wskaźniki ważniejszych płaszczyzn: a) Millera układu regularnego, b) Millera-
Bravais'ego układu heksagonalnego
Prosta sieciowa przechodzi przez dwa węzły sieci. Wskaźniki Millera prostej [uvw] są
współrzędnymi węzła sieci położonego najbliżej początku układu współrzędnych, przez który
przechodzi prosta po takim przesunięciu równoległym, że przechodzi również przez początek
układu. Współrzędne węzła — wskaźniki Millera prostej — przedstawia się z uwzględnieniem
znaku odpowiadającego zwrotowi osi jako całkowite wielokrotności stałych sieciowych.
Równoważny krystalograficznie zespół prostych sieciowych opisuje się wskaźnikami jednej z
nich w nawiasach ostrych, np. <111>. Na rysunku 2.10 pokazano oznaczenie kilku prostych
sieciowych w układach regularnym i heksagonalnym. W szczególnym przypadku sieci
regularnej proste sieciowe prostopadłe do płaszczyzn sieciowych i odwrotnie mają jednakowe
wskaźniki Millera. Tak na przykład kierunek [100] jest prostopadły do płaszczyzny (100), a np.
płaszczyzna (111) jest prostopadła do kierunku [111].
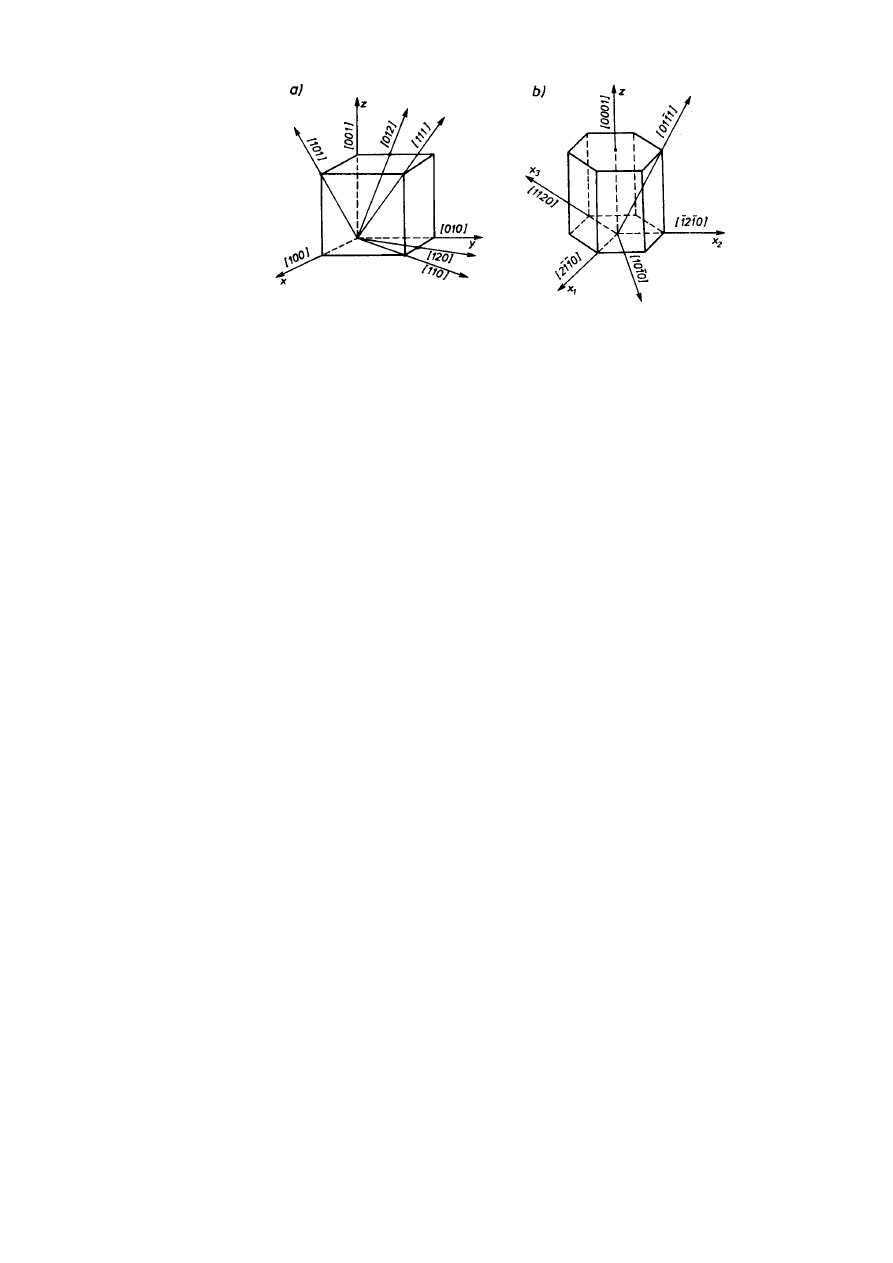
14
JW
Rys.2.10. Wskaźniki ważniejszych kierunków: a) Millera układu regularnego,
b) Millera-Bravais'ego układu heksagonalnego
1.3.1. Struktury metaliczne
W strukturach metalicznych węzły sieci obsadzone są rdzeniami atomowymi. Rdzeniem
atomowym jest atom pozbawiony pewnej liczby elektronów wartościowości, czyli kation. W
przypadku metali jednowartościowych rdzeń atomowy jest jednowartościowym kationem,
natomiast w przypadku metali o większej wartościowości rdzeń atomowy zazwyczaj różni się
swoim ładunkiem od wartościowości kationu w roztworze wodnym. Uwolnione elektrony, tzw.
gaz elektronowy, poruszają się w określonych obszarach ruchem bezładnym (analogicznie jak
cząsteczki gazu doskonałego), przenosząc się z powłoki jednego na powłokę drugiego rdzenia
atomowego.
Między dodatnimi ładunkami rdzeni atomowych i ujemnymi ładunkami elektronów
swobodnych działają silne przyciągające siły elektrostatyczne — bezkierunkowe wiązanie
metaliczne, zapewniające spójność materiału.
Znaczna energia wiązania metalicznego (200 -800 kJ/mol) zapewnia dużą trwałość materiału,
tj. przeważnie wysoką temperaturę topnienia i wrzenia, dużą wytrzymałość i twardość, a m. in.
dzięki bezkierunkowości dobrą plastyczność. Obecność elektronów swobodnych zapewnia
elektronowy charakter przewodnictwa, a więc dobre przewodnictwo elektryczne, o ujemnym
współczynniku temperaturowym (przewodność elektryczna zmniejsza się ze wzrostem
temperatury) oraz dobre przewodnictwo cieplne i stosunkowo dużą rozszerzalność cieplną.
Wymienione cechy materiałów metalicznych są wyraźne w stanie stałym, znacznie słabsze w
stanie ciekłym, a zanikają całkowicie w stanie gazowym. Ponadto pewne pierwiastki w
odpowiednich warunkach przyjmują modyfikacje odznaczające się lub pozbawione wiązania
metalicznego. Z tego powodu wiązanie metaliczne i wywołane nim właściwości traktuje się jako
stan metaliczny, w którym materiał może się znajdować w odpowiednich warunkach
zewnętrznych (temperatura, ciśnienie).
1.3.2. Metale
Przeważająca większość metali odznacza się jedną z trzech struktur: Al. - płaskocentryczną
układu regularnego, A2 - przestrzenniecentryczna układu regularnego albo A3 - zwarta
(złożoną) układu heksagonalnego.
Struktura Al, oznaczana również symbolem RCS (rys. 2.11a), jest najgęściej wypełniona
(74,04°/o) o liczbie atomów La. = 8 • 1/8 + 6 • 1/2 = 4 i 1.k. = 12. Dowolny atom " w sieci
otoczony jest dwunastoma sąsiadami w najmniejszej, jednakowej odległości a
√2/2 = 0,707a
(rys. 2.11b). W strukturze najgęściej wypełnione atomami są cztery płaszczyzny ośmiościanu
{111}, a w każdej z nich trzy kierunki <110>, w których atomy są do siebie styczne (rys. 2.11c).
Puste przestrzenie między atomami tworzą tzw. luki. Luki oktaedryczne (większe)
zlokalizowane w środku komórki i na środkach jej krawędzi są otoczone sześcioma atomami,
tworzącymi ośmiościan foremny (rys. 2.11d). Luki tetraedryczne (mniejsze) zlokalizowane na
przekątnych komórki w odległościach a
√3/4 = 0,433a od naroży, otoczone są czterema atomami
tworzącymi czworościan foremny (rys. 2.11e).
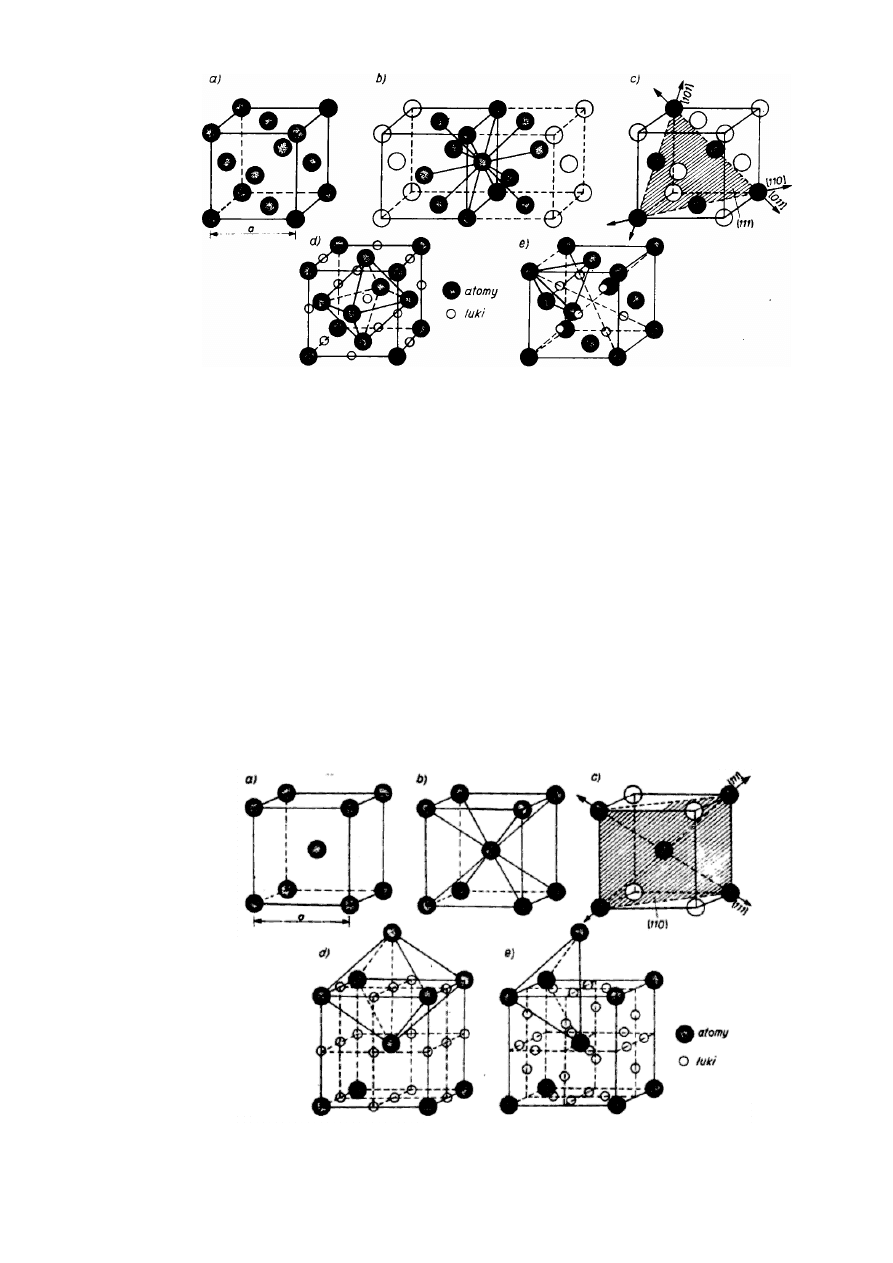
15
JW
Rys.2.11. Struktura Al: a) komórka zasadnicza, b) koordynacja sieci, c) płaszczyzny i
kierunki gęsto wypełnione, d) luki oktaedryczne, e) luki tetraedryczne
Ponieważ z warunku styczności wynika promień atomu R = a/2
√2 = 0,353a, w lukach
oktaedrycznych może się pomieścić kula (atom) o promieniu r =0,414R, a w tetraedrycznych —
o promieniu r = 0,225R.
Struktura A2, oznaczana również symbolem RCP (rys.2.12a), ma mniejsze wypełnienie
(68,02°/o) przy liczbie atomów l.a. = 8 • 1/8 +1 = 2 i Lk. = 8. Dowolny atom sieci otoczony jest
ośmioma sąsiadami, w najmniejszej, jednakowej odległości a
√3/2 = 0,866a (rys. 2.12b).
Najgęściej obsadzonych atomami, ale o odmiennym rozkładzie w porównaniu ze strukturą Al,
jest sześć płaszczyzn przekątnych komórki zasadniczej {ll0}, a w każdej są dwa kierunki <111>
gęstego ułożenia stycznych do siebie atomów (rys. 2.12c). Struktura A2 ma analogiczne jak
struktura Al rodzaje luk, lecz mniejszych, pomimo mniejszego wypełnienia sieci. Z warunku
styczności wynika promień atomu R = a
√/3/4 = 0,433a. Luki oktaedryczne zlokalizowane na
środkach ścian i na środkach krawędzi komórki zasadniczej (rys. 2.12d) mieszczą atomy o
promieniu zaledwie r = 0,l54R. Luki tetraedryczne zlokalizowane na każdej ścianie komórki w
połowie odległości między środkiem krawędzi i środkiem ściany (rys. 2.12e) mieszczą atomy o
promieniu r = 0,291R.
Rys. 2.12. Struktura A2: a) komórka zasadnicza, b) koordynacja sieci, c) płaszczyzny i
kierunki gęsto wypełnione, d) luki oktaedryczne, e) luki tetraedryczne
16
JW
Rys.2.13. Struktura A3: a) komórka zasadnicza, b) koordynacja sieci, c) płaszczyzny i
kierunki gęsto wypełnione, d) luki oktaedryczne, e) luki tetraedryczne
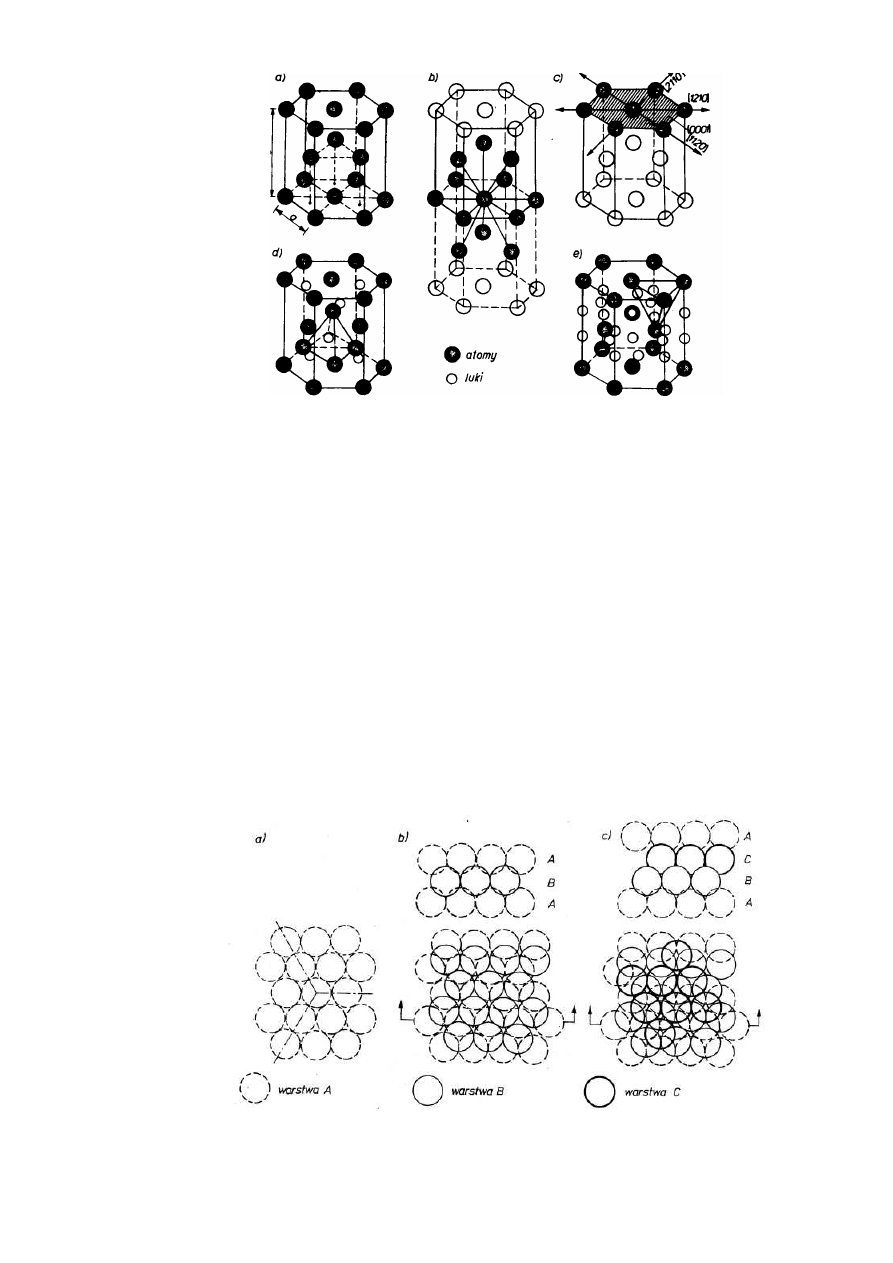
Struktura A3 albo HZ (rys. 2.13a) o identycznym wypełnieniu jak Al (74,04 °/o) ma l.a. = 12
..
1/6 +2
.
1/2 +3 = 6 i l.k. = 12. Dowolny atom sieci otoczony jest dwunastoma sąsiadami w
jednakowej, najmniejszej odległości a (rys. 2.13b). Najgęściej obsadzona atomami jest
płaszczyzna podstawy (0001), w której są trzy kierunki gęstego ułożenia stycznych atomów
<1120> (rys. 2.13c). Promień atomowy dla tej struktury wynosi R = 0,5a. Luki oktaedryczne
(rys. 2.13d) i tetraedryczne (rys. 2.13e) mieszczą atomy o identycznych średnicach jak w
strukturze Al, odpowiednio r = 0,414R i r = 0,225R.
Opisana idealna struktura A3, charakteryzująca się stycznością atomów w trzech kolejnych
płaszczyznach (0001) jest możliwa tylko przy stosunku stałych sieciowych c/a =
√8/3 = 1,633.
Spośród metali o tej strukturze większość w przybliżeniu spełnia ten warunek (c/a = 1,56
÷
1,63). Wyjątki stanowią cynk i kadm o wartościach stosunku c/a odpowiednio 1,86 i 1,89.
Anomalie te są spowodowane elipsoidalnym zniekształceniem w kierunku osi c powłok
elektronowych atomów oraz działaniem między płaszczyznami (0001) dodatkowego wiązania
kowalencyjnego. Dla obu tych struktur liczbę koordynacyjną poprawnie) jest zapisywać w
postaci 6 + 6, bowiem, od dowolnego atomu sieci w jednakowej najmniejszej odległości a jest
sześć atomów, natomiast sześć pozostałych jest w jednakowej, nieco większej odległości
√a
2
/3+c
2
/4.
Rys. 2.14. Układ płaszczyzn gęstego ułożenia: a) płaszczyzna heksagonalna, b) struktura
A3, c) struktura Al

17
JW
Struktury A l i A3 są do siebie podobne. Mianowicie, w obu występują płaszczyzny (o różnej
orientacji krystalograficznej) z jednakowym gęstym ułożeniem atomów, o heksagonalnej
symetrii (rys. 2.14a). Aby warunek najgęstszego ułożenia atomów był spełniony, druga
płaszczyzna heksagonalna musi mieć środki atomów przesunięte w stosunku do pierwszej,
natomiast trzecia może mieć środki atomów pokrywające się z pierwszą (rys. 2.14b) albo jeszcze
bardziej przesunięte (rys. 2.14c).Kolejność płaszczyzn heksagonalnych w strukturze A3 (0001)
odpowiada pierwszemu wariantowi, co można przedstawić symbolicznie zapisem AB AB AB ...,
a w strukturze A1 (111) – drugiemu wariantowi co przedstawia zapis ABC ABC ABC
...Stosunkowo nieliczne metale mają odmienne struktury. Złożone struktury układu regularnego
mają odmiany alotropowe (por. punkt 3.4) manganu: Mn
α o l.a. = 58 atomów (typ A12) i Mnβ o
l.a. = 20 atomów (typ A13). Cyna
β (biała) i ind mają złożone struktury układu tetragonalnego
(typ A5). Rtęć w stanie stałym (poniżej -38,8°C) ma strukturę układu romboedrycznego, o
prostej komórce zasadniczej (typ A 10). Pierwiastki o słabiej zaznaczonych cechach
metalicznych, jak As, Sb, Bi, Se, Te, mają różne struktury układu heksagonalnego, w których
obok wiązania metalicznego współistnieje również wiązanie kowalencyjne.
Wśród struktur metalicznych obserwuje się wyraźną prawidłowość: im większe są symetria,
liczba koordynacyjna i wypełnienie sieci, tym wyraźniejsze są cechy metaliczne materiału.
Sieć regularna płasko-centryczna występuje w większości metali (m.in. w żelazie
γ
1)
, aluminium,
niklu, miedzi, srebrze, złocie, platynie, palladzie, ołowiu i berylu). Metale te wykazują szczególnie
dobrą plastyczność na gorąco, a niektóre także na zimno, i są bardzo dobrymi przewodnikami ciepła
i prądu elektrycznego.
Sieć regularna przestrzennie centryczna występuje w takich metalach, jak żelazo
α, chrom,
wolfram, molibden, wanad, tantal, niob, sód, potas itd. Metale tej grupy są mniej ciągliwe niż
metale grupy pierwszej i nadają się przeważnie tylko do obróbki plastycznej na gorąco.
Sieć heksagonalna przestrzennie centryczna występuje m.in. w magnezie, tytanie
α, cyrkonie α,
kobalcie
α, cynku, kadmie i rtęci. Metale o sieci heksagonalnej mają znacznie gorsze własności
plastyczne niż metale o sieci regularnej i tylko niektóre z nich mogą być obrabiane plastycznie na
gorąco i na zimno.
Sieć tetragonalna występuje w białej cynie, galu i indzie. Metale o sieci tetragonalnej cechuje
niska twardość i niska temperatura topnienia.
Trzeba podkreślić, że w omówionych typach sieci krystalizuje większość metali.
Budowa wewnętrzna ma bardzo duży wpływ na własności metalu, zarówno chemiczne, jak fizyczne
(tabl. 2.2) i mechaniczne. Z chemicznych własności uzależniona jest od niej przede wszystkim
odporność metalu na korozję, z fizycznych — przewodność cieplna i elektryczna, z mechanicznych
— wytrzymałość, plastyczność twardość.
T a b l i c a 2.2
Niektóre własności fizyczne ważniejszych metali
Parametr sieci, nm
Pierwiastek
Typ sieci
w temp.
°C
a
c
Tempera
tura
topnieni
a °C
Gęstość
w 20°C
g/cm
3
Uwagi
Żelazo
α
A2
20
2,8605
-
1534
7,87 do 910 i pow. 1390°C
Żelazo
γ
A1
950
3,649
-
-
-
910 ÷ 1390°C
Aluminium
A1
25
4,0414
-
660
2,70
Magnez
A3
25
3,2030 5,2002
650
1,74
Miedź
A1
18
3,6074
-
1083
8,96
Nikiel
A1
20
3,5169
-
1453
8,90
Kobalt
α
A3
20
2,507
4,081
-
8,9
do 400°C
Kobalt
β
A1
3,537
-
1495
-
pow. 400°C
Tytan
α
A3
20
2,9503 4,6831
-
4,54
do 882°C
Tytan
β
A2
900
3,283
-
1668
-
pow. 882°C
18
JW
Pojedyncze kryształy metali mają zazwyczaj bardzo małe wymiary, toteż każdy przedmiot
składa się z dużej liczby kryształów. Budowa taka nosi nazwę wielokrystalicznej lub
polikrystdlicznej. Z różnych przyczyn, o których będzie mowa dalej, poszczególne kryształy nie
mogą w zgrupowaniu wielokrystalicznym przybierać prawidłowego kształtu, lecz są mniej lub
więcej zdeformowane. Takie kryształy o nieprawidłowym kształcie nazywa się ziarnami lub
krystalitami.
Różnice między poszczególnymi ziarnami metali polegają na różnej orientacji przestrzennej
ich sieci krystalicznych. Orientacja ta jest na ogół przypadkowa i z jednakowym
prawdopodobieństwem może mieć w przestrzeni dowolny kierunek. Wskutek obróbki plastycznej
na zimno (np. walcowania) można jednak uzyskać jednokierunkową orientację ziarn (tzw. teksturę
walcowania). Oczywiście własności mechaniczne metalu w obu przypadkach będą zupełnie różne,
przy czym przy jednokierunkowej orientacji ziarn będą się zmieniać zależnie od kierunku badania.
Jest to spowodowane faktem, że różne płaszczyzny i kierunki sieci krystalicznej są obsadzone
atomami niejednakowo. Na przykład w elementarnej komórce sieci regularnej przestrzennie
centrycznej o parametrze a, każda ścianka o powierzchni a
2
obsadzona jest przez l atom (atomy
leżące w wierzchołkach należą jednoczesne do 4 komórek elementarnych, więc (4·1/4 = 1),
natomiast w płaszczyźnie przekątnej komórki o powierzchni a
2
• √2 znajdują się 2 atomy (4·1/4 +
1 atom środkowy). Mimo różnicy w wielkościach powierzchni, wyraźnie widać, że drugi płaszczyzna
jest gęściej obsadzona atomami. Ilustruje to rys. 2.15.
Rys. 2.15. Płaszczyzny krystalograficzne w elementarnej komórce sieci regularnej przestrzennie
centrycznej: a) obsadzona jednym atomem, b) obsadzona dwoma atomami
Podobnie dzieje się w przypadku innych sieci krystalicznych, tak że własność wszystkich
pojedynczych kryształów metali zmieniają się zależnie od kierunki badania. Zjawisko to nazywa się
anizotropią. W przypadku budowy wielokrystalicznej anizotropia występuje szczególnie wyraźnie, gdy
ziarna mają jednakową orientację. Nie występuje natomiast w przypadku różnej orientacji
poszczególnych ziarn i wtedy mówi się, że metal jest izotropowy. Na przykład monokryształ żelaza
α o
sieci regularnej przestrzennie centrycznej charakteryzuje wartość współczynnika sprężystości wzdłużnej E
= 135 000
÷ 290 000 MPa, zależnie od kierunku badania. To samo żelazo o budowie wielokrystalicznej
wykazuje wartość E = 214 000 MPa.

19
JW
2. 4. Defekty struktur krystalicznych
Jak już powiedziano wyżej, siły międzyatomowe warunkujące spójność metalu są siłami
przyciągania i odpychania. Trwałe rozłączenie atomów, czyli wywołanie złomu metalu jest
uwarunkowane działaniem siły rozciągającej większej od maksymalnej wypadkowej sił
między atomowych.
Wartość tej siły i krytyczną odległość odpowiadającą granicznemu odkształceniu
sprężystemu można wyliczyć zarówno dla dwóch wyodrębnionych atomów, jak i całego
kryształu, przy założeniu doskonałej jego budowy sieciowej. W tym drugim przypadku
teoretyczne naprężenie rozciągające potrzebne do pokonania sił spójności wynosi ok. 100 000
MPa, a graniczne odkształcenie sprężyste — ok. 10%.
Jak jednak stwierdzono doświadczalnie, rzeczywiste wartości zarówno naprężeń
rozrywających, jak i odkształceń sprężystych, są 100-1000 razy mniejsze od teoretycznych.
Ta rozbieżność między obliczeniami teoretycznymi a wynikami pomiarów wielu własności
metali nasunęła wniosek, potwierdzony następnie doświadczalnie, że struktura rzeczywistych
kryształów nie jest doskonała i zawiera pewne wady, wywołujące określone
nieprawidłowości budowy i wpływające na ich własności. Stwierdzono również, że niektóre
własności metali (np. gęstość, ciepło właściwe, współczynnik rozszerzalności cieplnej) nie są
wrażliwe na strukturę i nie zmieniają się ani na skutek nieprawidłowej struktury sieciowej
pojedynczego kryształu, ani na skutek obecności w nim domieszek obcych atomów, a w
przypadku budowy wielokrystalicznej nie zależą od wielkości ziarn.
Większość jednak własności metali, a przede wszystkim wytrzymałość i plastyczność,
odporność na korozję, przewodność elektryczna i przenikalność magnetyczna, wyraźnie
zależy od struktury. Wpływają na nie zarówno wszelkie nieprawidłowości struktury
sieciowej, jak i wielkość ziarn rozłożenie ich granic.
Nieprawidłowości struktury sieciowej spotykane w rzeczywistych strukturach
krystalicznych można podzielić na trzy grupy:
• defekty punktowe,
• defekty liniowe,
• defekty złożone.
Defektami punktowymi nazywa się zakłócenia budowy krystalicznej umiejscowione
wokół punktu. Najprostszym defektem tego typu jest brak atomu w węźle sieci przestrzennej,
zwany wakansem albo luką.
Wakanse powstają przede wszystkim wskutek drgań cieplnych sieci, które są tym większe,
im wyższa jest temperatura. Przy określonej amplitudzie drgań atom może wypaść ze swego
średniego położenia w węźle sieci i zająć pozycję międzywęzłową. Powstaną wówczas
jednocześnie dwa defekty punktowe: wakans i atom wtrącony między węzłowo. Oba
wywołują lokalne zakłócenie budowy sieciowej, gdyż obecność wakansu powoduje większe
od normalnego zbliżenie sąsiednich atomów (rys. 2.15b), natomiast atom wtrącony powoduje
rozsunięcie sąsiednich atomów na odległość większą od normalnej. Opisany defekt nosi
nazwę defektu Frenkla i może powstawać tylko w strukturach metali alkalicznych, w których
odległości między atomami są wystarczająco duże, by atom mógł zająć pozycję
międzywęzłową (rys. 2.15b). Natomiast w zwarcie wypełnionych sieciach krystalicznych
tworzą się, defekty punktowe, polegające na powstawaniu wakansu i wywędrowaniu atomu,
który ten wakans utworzył, na powierzchnię kryształu. Ten typ defektu nazywa się defektem
Schottky'ego i jest powszechny w kryształach metali – rys. 2.15a. Wakanse powstające w
sieci mogą wędrować wewnątrz kryształu przez zamianę miejsc z węzłami obsadzonymi
atomami. Mogą wywędrować na powierzchni kryształu, co prowadzi do zmniejszenia się
ogólnej liczby wakansów. Mogą wreszcie się łączyć, tworząc tzw. zgrupowania wakansów.
Liczba wakansów w metalu w stanie równowagi termodynamicznej, w temperaturze
otoczenia jest stosunkowo niewielka, wzrasta jednak bardzo szybko przy podwyższeniu
temperatury. Ponieważ defekty tego typu odgrywają istotną rolę w procesach dyfuzyjnych, w
wielu przypadkach dąży się do uzyskania zwiększone liczby wakansów również w

20
JW
temperaturze otoczenia, poprzez szybkie przechłodzenie metalu z wysokich temperatur,
obróbkę plastyczną na zimno (tj. w temperaturach niższych od temperatury rekrystalizacji
danego metalu) lub bombardowaniu ciężkimi cząsteczkami alfa.
Rys.2.15. Punktowe defekty sieci krystalicznej wywołane drganiami cieplnymi: a) defekt
Schottky’ego, b) defekt Frenkla
Punktowe defekty sieci tworzą również znajdujące się w niej obce atomy. Możliwe są tu
następujące przypadki. Jeśli obcy atom ma średnicę atomową dużo mniejszą od średnicy
atomowej atomów metalu, to zajmuje on położenie między węzłowe, wywołując lokalne
rozsunięcie sąsiednich atomów i powiększenie parametrów sieci (rys.2.16b). W typowych
sieciach krystalicznych metali przestrzenie międzywęzłowe są niewielkie, toteż położenie
międzywęzłowe mogą zajmować w nich tylko atomy azotu, wodoru, węgla i boru, mające
najmniejsze średnice atomowe. Wtrącone atomy innych pierwiastków mogą zajmować
wyłącznie pozycje węzłowe zastępując atomy metalu podstawowego. W tym przypadku
rodzaj zniekształcenia sieci krystalicznej zależy od tego czy obcy atom ma mniejszą, czy
większą średnicę od atomu metalu podstawowego (rys. 2.16b i c). Jeśli większą — występuje
lokalne rozsunięcie sąsiednich atomów (powiększenie parametrów sieci), jeśli mniejszą —
lokalne zbliżenie atomów (zmniejszenie parametrów sieci)
Rys.2.16. Defekty punktowe: a) wakans, b) atom międzywęzłowy, c) atom obcy
węzłowy, d) atom obcy międzywęzłowy
Odkształcenie sieci wywołane wakansem polega na kontrakcji, a atomem
międzywęzłowym — na ekspansji. Atom obcy węzłowy powoduje kontrakcję, jeżeli jego
promień jest mniejszy, albo ekspansję, jeżeli jego promień jest większy od promienia atomu
bazowego, natomiast atom obcy międzywęzłowy zawsze powoduje ekspansję sieci.
Wzajemne oddziaływanie defektów punktowych, przy większym ich stężeniu
Defektami liniowymi nazywa się zakłócenia budowy krystalicznej, które w jednym
kierunku mają wymiar kilku odległości atomowych, a w drugim — całego ziarna lub znacznej
jego części. Rozróżnia się dwa zasadnicze rodzaje defektów liniowych: dyslokację
krawędziową i dyslokację śrubową. Pierwszą odkryli w 1934 i Taylor, Orowan i Polanyi,
drugą w l939 r.— Burgers.
Dyslokację krawędziową wywołuje obecność w przestrzennej sieci krystaliczne
dodatkowej półpłaszczyzny obsadzonej atomami (zw. ekstrapłaszczyzną), które krawędź
stanowi dowolna linia brzegowa, nazywana linią dyslokacji. W zależność od usytuowania

21
JW
dodatkowej półpłaszczyzny rozróżnia się dyslokację dodatnią, oznaczoną symbolem -
⊥
i
ujemną, oznaczoną symbolem
T
(pionowa kreska w symbolu dyslokacji oznacza dodatkową
półpłaszczyznę, pozioma — płaszczyznę poślizgu). Na rysunku 2.17 pokazano dyslokację
krawędziową dodatnią
Rys. 2.17. Schemat dyslokacji krawędziowej w krysztale o sieci regularnej: a) przekrój
poprzeczny kryształu zawierającego dyslokację dodatnią, b) perspektywiczny obraz
rozmieszczenia atomów wokół dyslokacji dodatniej; AB — płaszczyzna poślizgu
Wokół linii dyslokacji istnieje pole naprężeń sprężystych; ściskających w części kryształu
zawierającej dodatkową półpłaszczyznę (odległości między sąsiednimi atomami są mniejsze
od stałych sieciowych), rozciągających — w pozostałej części kryształu (odległości między
sąsiednimi atomami są większe od stałych sieciowych). Wynika z tego, że wokół dyslokacji
krawędziowej występuje jednocześnie postaciowe i objętościowe odkształcenie kryształu.
Dyslokacje krawędziowe charakteryzują się określonymi własnościami dynamicznymi, m.in.
mają możliwość poruszania się w płaszczyźnie poślizgu pod wpływem naprężeń
wewnętrznych lub zewnętrznych, w wyniku czego następuje poślizg części kryształu wzdłuż
określonej płaszczyzny sieciowej. Obliczono, że naprężenie potrzebne do wywołania
przesuwania się dyslokacji jest bardzo małe, rzędu l MPa pod warunkiem, że siły wiązań w
krysztale nie zależą od kierunków.
Dyslokacje krawędziowe mogą się przemieszczać w krysztale również przez wspinanie,
polegające na odłączeniu się atomów od krawędzi dodatkowej półpłaszczyzny i ich migracji
do wakansów (rys. 2.18). Oczywiście możliwe jest także zjawisko odwrotne, polegające na
opuszczaniu pozycji węzłowych przez atomy i ich dołączaniu do krawędzi półpłaszczyzny.
Przemieszczanie się dyslokacji krawędziowych przez wspinanie zależy od ilości wakansów w
krysztale i zachodzi bardziej intensywnie w temperaturach podwyższonych, np. podczas
pełzania metali. Innym przejawem własności dynamicznych jest przyciąganie się dyslokacji
różnoimiennych i odpychanie się dyslokacji jednoimiennych. W pierwszym przypadku
możliwa jest anihilacja (zanik) dyslokacji, jeśli leżą one w tej samej płaszczyźnie poślizgu lub
w płaszczyznach równoległych.
Określone oddziaływanie występuje także między dyslokacjami krawędziowymi atomami
obcych pierwiastków znajdujących się w metalu. Atomy o większych średnicach zajmujące
położenia węzłowe oraz atomy o małych średnicach zajmujące położenia międzywęzłowe
(węgiel, azot, wodór) migrują do rozciągniętej strefy kryształu, leżącej bezpośrednio pod
krawędzią dodatkowej półpłaszczyzny. Natomiast atomy o małych średnicach, zajmujące
położenia węzłowe migrują do ściskanej części kryształu, gdzie zastępując większe atomy
metalu osnowy, obniżają energię odkształcenia kryształu
Drugim prostym rodzajem dyslokacji jest dyslokacja śrubowa, wyznaczająca granicę
między przesuniętą i nieprzesuniętą częścią kryształu. Granica ta przebiega równolegle do
kierunku poślizgu a nie prostopadle, jak to ma miejsce w przypadku dyslokacji krawędziowej.
Dyslokację śrubową najlepiej wyjaśnić na perspektywicznym modelu fragmentu kryształu,
którego jedna część jest przesunięta względem drugiej o jedną odległość atomową (rys. 2.19).
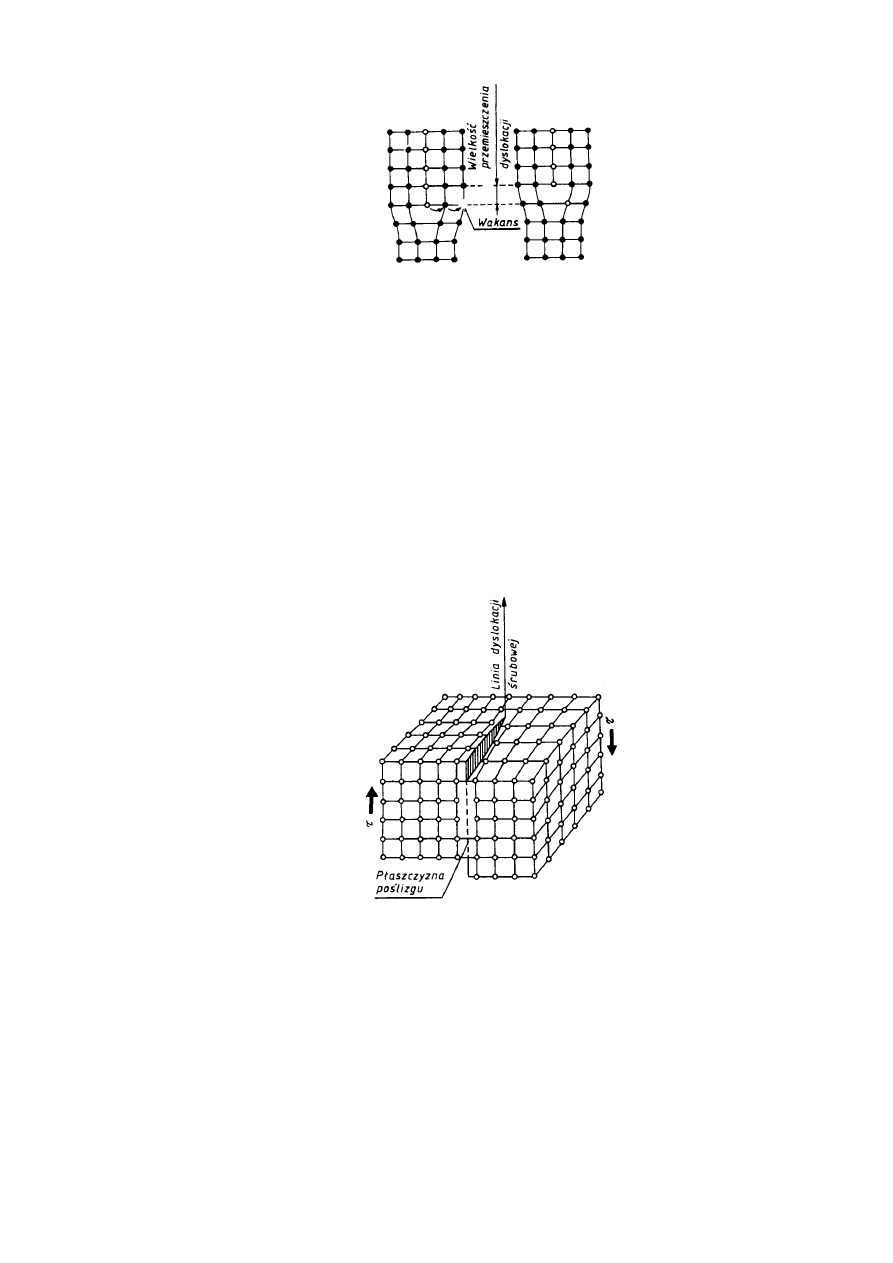
22
JW
Rys. 2.18.
Schemat przemieszczania się dyslokacji krawędziowej przez wspinanie
W wyniku tego przesunięcia poszczególne płaszczyzny atomowe przekształcają się w
powierzchnie śrubowe. Rozróżnia się dyslokacje prawo-skrętne wywołujące poślizg w
kierunku pokazanym na rys. 2.19, i dyslokacje lewo-skrętne wywołujące poślizg w kierunku
przeciwnym.
Podobnie jak dyslokacje krawędziowe, dyslokacje śrubowe mogą przemieszczać się przy
małych naprężeniach stycznych, jeśli w płaszczyźnie poślizgu nie ma przeszkód hamujących
ich ruch. W przypadku obecności takich przeszkód (np. obcych atomów), naprężenie
potrzebne do uruchomienia dyslokacji jest tym większe, im mniejsza jest odległość między
sąsiednimi przeszkodami. Zjawisko to ma oczywisty wpływ na własności wytrzymałościowe
stopów.
Równoległe dyslokacje śrubowe jednoimienne odpychają się, różnoimienne —
przyciągają. Te ostatnie mogą się także w określonych przypadkach anihilować.
Rys. 2.19. Schemat dyslokacji śrubowej
Dyslokacjom śrubowym nie towarzyszy objętościowe odkształcenie kryształu. Dlatego
wokół nich nie występuje wybiorcze rozmieszczenie atomów obcych pierwiastków.
Proste typy dyslokacji występują w sieci krystalicznej rzadko. Większość dyslokacji
stanowi kombinację dyslokacji krawędziowych i śrubowych.
Omówione defekty dotyczyły zakłóceń budowy sieci krystalicznej występujących w
pojedynczym krysztale. Metale i stopy techniczne, jak już wiadomo, są jednak materiałami
wielokrystalicznymi, złożonymi z wielkiej liczby ziarn. Orientacja krystalograficzna tych
ziarn jest w zasadzie chaotyczna (rys. 2.20), toteż na granicy ziarn spotykają się różnie
zorientowane sieci przestrzenne, ukierunkowane względem siebie pod dużymi kątami,
wynoszącymi najczęściej kilkanaście do kilkudziesięciu stopni (dlatego granice ziarn nazywa
się także granicami dużego kąta). Jest rzeczą oczywistą, że ułożenie atomów na granicy ziarn

23
JW
jest uzależnione od działania obu stykających się sieci krystalograficznych, w wyniku czego
stanowi pewną mikrostrukturę przejściową, nie odpowiadającą orientacji ani jednego, ani
drugiego ziarna - rys. 2.21. Ta przejściowa struktura o grubości kilku odległości
międzyatomowych jest strukturą zakłóconą, tym bardziej, że na granicach ziarn grupują się
również wszelkie zanieczyszczenia, które nawet w najczystszych metalach występują w
pewnych ilościach. W rezultacie granice ziarn mają wyższą wytrzymałość niż inne ziarna,
natomiast niższy potencjał elektrochemiczny, a więc mniejszą odporność chemiczną,
objawiająca się m.in. łatwiejszym trawieniem na zgładach metalograficznych.. Łączna energia
granic osiąga minimum w przypadku ziarn o kształcie (w przekroju) foremnych
sześcioboków i prostoliniowych granicach. Ziarna o liczbie boków (w przekroju) mniejszej
od sześciu mają granice wypukłe, a o liczbie boków większej od sześciu — granice wklęsłe
(rys. 2.22).
Rys. 2.20. Schemat orientacji krystalicznej Rys. 2.21. Schemat zakłócenia budowy
poszczególnych ziarnach metalu krystalicznej na granicy ziarn metalu
Tendencja do uzyskania stanu stabilnego o minimum energii granic objawia się ich
migracją, tzw. rozrostem ziarn. Siły napędowej migracji dostarcza energia cieplna, toteż
rozrost ziarn następuje w podwyższonej temperaturze. Najbardziej ruchliwe są niesprzężone
granice szerokokątowe.
Rys. 2.22. Kształt granic przekroju ziarna: a) równowagowy, b) i c) nierównowagowy,
d) zanik ziarna (przy rozroście): 1
÷ 4 — kolejne stadia
Ruch granicy odbywa się zawsze w kierunku środka jej krzywizny, jako rezultat
elementarnych przeskoków atomów przez granicę w kierunku przeciwnym, ma więc
charakter dyfuzyjny. Określony kierunek ruchu granicy sprawia, że rozrost ziarn jest
selektywny. Mianowicie, rozrastają się ziarna o liczbie boków większej od sześciu, natomiast
zanikają ziarna o liczbie boków mniejsze) od sześciu, jak to przedstawiono poglądowo na rys.
2.22. Rozrostowi ziarn towarzyszy zmniejszenie ich liczby. Migracja stosunkowo stabilnych
granic wąskokątowych ma charakter ruchu bezdyfuzyjnego, którego siłą napędową jest
obciążenie zewnętrzne.
Czynnikiem hamującym ruch granic szerokokątowych (rozrost ziarn) są dyspersyjne
wydzielenia obcej fazy, ale ich skuteczność jest ograniczona do temperatury, w której ulegają
rozpuszczeniu w osnowie. Tak na przykład wydzielenia azotka aluminium (AlN) w stali

24
JW
hamują rozrost ziarna do temperatury 950 -1050°C.
Granice wąskokątowe powstają podczas krystalizacji jako rezultat zrastania się gałęzi
dendrytów oraz w stanie stałym podczas wygrzewania metalu uprzednio odkształconego
plastycznie. Granice szerokokątowe niesprzężone są rezultatem dużej liczby zarodków
krystalizacji podczas krzepnięcia. Tworzą się również podczas wygrzewania metalu
uprzednio odkształconego plastycznie (zdrowienie). Wreszcie granice szerokokątowe
sprzężone powstają głównie podczas przemian fazowych w stanie stałym (np. wydzielanie z
przesyconego roztworu stałego), a granice bliźniacze — podczas odkształcenia plastycznego.
Granice ziam nie są jedynymi defektami złożonymi, występującymi w materiałach
polikrystalicznych. Okazało się, że pojedyncze ziarno składa się z dużej liczby drobnych
bloków (o wymiarach liniowych ok. 0,000 01 cm) usytuowanych względem siebie pod
niewielkimi kątami, wynoszącymi najczęściej kilka minut. Bloki te nazywa się blokami
mozaiki, a strukturę ziarna z nich złożoną - strukturą mozaikową. Granice bloków mozaiki
utworzone są przez ugrupowania jednoimiennych dyslokacji krawędziowych (rys. 2.23)
towarzyszą im więc naprężenia sprężyste o analogicznym zasięgu, jak w tych dyslokacjach
Odkrycie dyslokacji umożliwiło wyjaśnienie dwojakiego wpływu defektów sieci
krystalicznej na wytrzymałość kryształu .
Z jednej strony defekty sieci krystalicznej osłabiają kryształ, a odkształcenie plastyczne
jest wynikiem przemieszczania się w nim dyslokacji bądź już istniejących, bądź powstających
podczas odkształcania (czemu sprzyjają niektóre inne defekty sieciowe).
Z drugiej jednak strony wiadomo, że wytrzymałość pojedynczych kryształów jest
mniejsza niż materiałów polikrystalicznych, ponieważ zaburzenia budowy sieciowej na
granicach ziarn umacniają metal. Wiadomo też, że kryształy zawierające dużą liczbę
defektów są bardziej wytrzymałe od kryształów z małą liczbą defektów. Dzieje się tak
dlatego, że w przypadku dużej liczby defektów sieciowych ruch dyslokacji jest hamowany na
skutek wzajemnego przecinania się dyslokacji (powstają dyslokacje nie tylko równolegle do
siebie, ale również umiejscowione w różnych płaszczyznach i o różnych kierunkach), ich
grupowania się, a także obecności przeszkód w postaci innych defektów sieciowych, np.
obcych atomów.
Rys. 2.23. Schemat zakłócenia budowy krystalicznej na granicy bloków mozaiki: a)
przed połączeniem się bloków, b) po połączeniu (widoczne pionowe ugrupowanie
jednoimiennych dyslokacji krawędziowych
Wynika z tego, że wytrzymałość rzeczywista metali zmniejsza się wraz ze zwiększaniem
liczby (gęstości) dyslokacji i innych defektów sieciowych, tylko do pewnej granicy i po
osiągnięciu minimalnej wartości, przy tzw. krytycznej gęstości dyslokacji zaczyna ponownie
wzrastać. Zależność między rzeczywistą wytrzymałością metalu a liczbą defektów jego sieci
krystalicznej pokazano na rys. 2.24.
Wynika z tego również, że warunkiem podwyższenia wytrzymałości metalu jest albo
całkowite usunięcie z niego wszelkich nieprawidłowości budowy krystalicznej, albo
zwiększenie oporu ruchu dyslokacji poprzez wytworzenie w nim odpowiedniej liczby
dyslokacji i innych defektów.
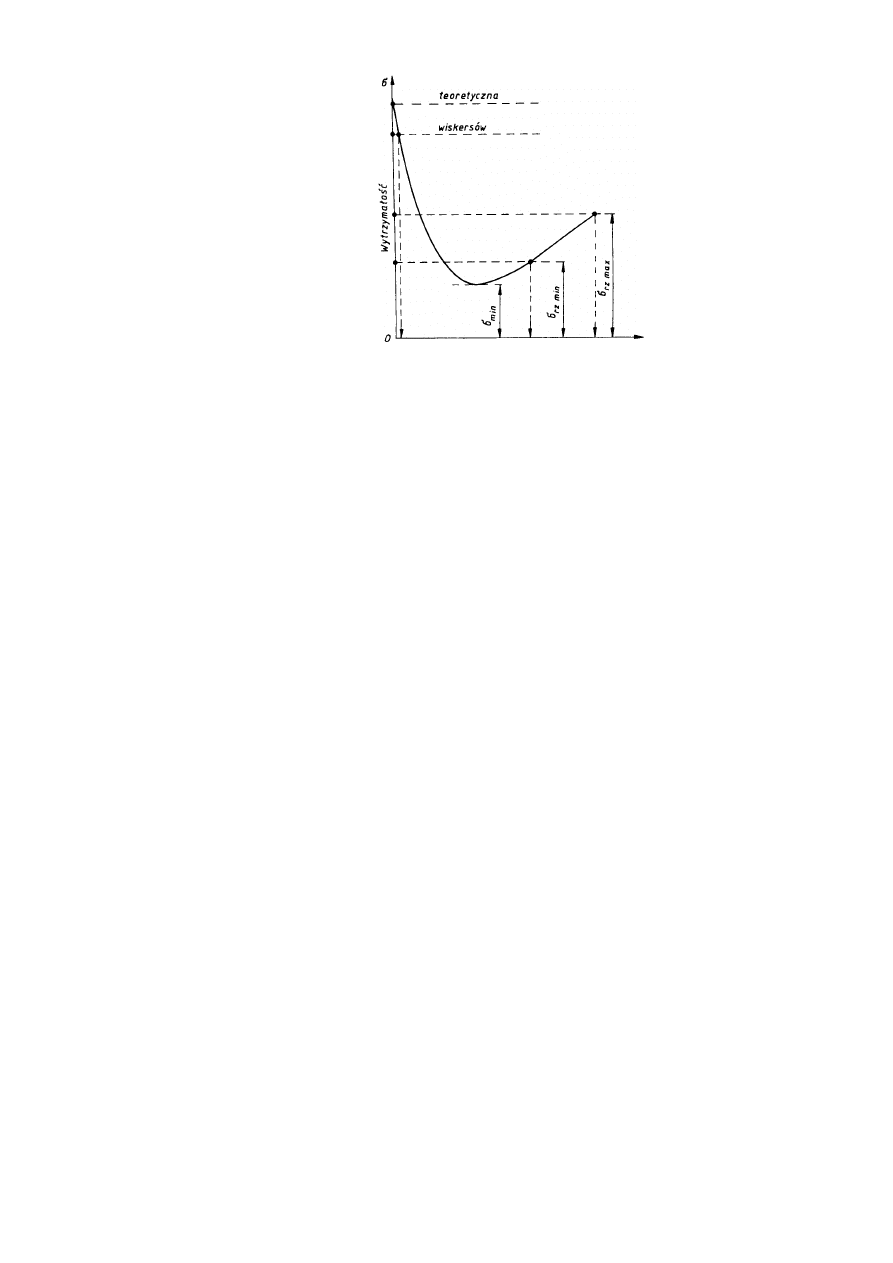
25
JW
Rys.2.24. Zależność między wytrzymałością metalu a liczbą defektów sieci
krystalicznej;
σ
rzmin
i
σ
rzmax
wytrzymałości rzeczywistych metali i stopów technicznych
Pierwsza możliwość oznacza zbliżanie się do wytrzymałości teoretycznej i została
potwierdzona doświadczalnie na otrzymywanych specjalnymi metodami kryształach
włoskowych (z angielskiego zwanych także wiskersami), o strukturze krystalicznej zbliżonej
do doskonalej. Włoskowe kryształy żelaza wykazują wytrzymałość na rozciąganie około
1330 MPa, miedzi - około 3000 MPa, cynku -około 2250 MPa, podczas gdy wytrzymałość na
rozciąganie tych samych metali uzyskanych zwykłymi metodami (a więc zawierających
defekty sieciowe) wynosi odpowiednio około 300, 220 i 180 MPa. Kryształy włoskowe nie
wykazują poślizgów, zrywają się bez widocznego odkształcenia plastycznego. Jednak ich
małe rozmiary (średnica kilku mikrometrów, długość do kilkunastu milimetrów)
uniemożliwiają w obecnej chwili praktyczne wykorzystanie w technice.
Natomiast druga możliwość podwyższenia wytrzymałości metalu, polegająca na
wytworzeniu w nim optymalnej gęstości dyslokacji i innych defektów, jest powszechnie w
praktyce wykorzystywana.
Metod zwiększania liczby defektów sieciowych w metalach i stopach jest wiele. Jedną z
najczęściej stosowanych jest odkształcanie metalu na zimno, czyli jego zgniot, drugą —
tworzenie stopów, czyli tworzyw metalicznych uzyskiwanych najczęściej przez stopienie dwu
lub więcej metali lub metalu z niemetalami. Powstawanie dyslokacji podczas odkształcania na
zimno odkryli niezależnie od siebie uczeni Frank i Read, stąd źródła powstawania tych
dyslokacji nazwane zostały źródłami Franka-Reada. Według ich teorii potwierdzonej
doświadczeniem przyjmuje się, że w metalu nieodkształconym istnieje przestrzenny układ
dyslokacji, czym niektóre z nich są w pewnych miejscach unieruchomione. Istnienie takich -
unieruchomionych w dwóch punktach dyslokacji jest oczywiście możliwe również w
płaszczyźnie poślizgu, tzn. w płaszczyźnie, w której następuje przesunięcie-się jednej części
kryształu względem drugiej (będącej zwykle płaszczyzną najgęściej obsadzoną atomami).
Na rys. 2.25 przedstawiono linię dyslokacji utwierdzonej w węzłach A i B, prz czym
płaszczyzna rysunku odpowiada płaszczyźnie poślizgu. Jeśli w płaszczyznie poślizgu działa
naprężenie styczne
τ,
linia dyslokacji zaczyna się wyginać tworząc w pierwszej fazie
półokrąg, a następnie dwie przeciwnie zorientowane spirale. Powiększanie się tych spirali
doprowadza do ich zetknięcia w punktach C i C’ podziału na dwie dyslokacje: zewnętrzną —
tworzącą zamkniętą pętlę i wewnętrzną — łączącą węzły kotwiczące A i B. Dyslokacja
zewnętrzna rozrasta się aż do osiągnięcia granic kryształu lub bloku, a dyslokacja
wewnętrzna, utwierdzona między węzłami A i B, wyginając się pod wpływem naprężeń
stycznych daje początek kolejnej pętli linii dyslokacji.
Teoretycznie źródło Franka-Reada może wytworzyć nieskończenie wiele pętli linii
dyslokacji, praktycznie jednak rozrastające się linie dyslokacji napotykając różne przeszkody,
będące defektami strukturalnymi i rozwój ich jest hamowany.
Liczba defektów struktury
26
JW
Rys. 2.25. Kolejne etapy tworzenia się pętli dyslokacji ze źródła Franka-Reada; strzałki
pokazują kierunek ruchu linii.
W zależności od rodzaju przeszkody źródło może zaniknąć całkowicie albo odnowić się
pod wpływem większych naprężeń ścinających. W miarę zwiększania ilości dyslokacji
wzrasta wytrzymałość metalu i jego twardość, maleją zaś jego własności plastyczne.
Zmieniają się również niektóre własności fizyczne: np. maleje przewodność elektryczna i
przenikalność magnetyczna wzrasta siła koercji i histereza magnetyczna, a także wzrasta
potencjał elektrochemiczny (metal zgnieciony jest mniej odporny na korozję niż metal bez
zgniotu).
27
JW
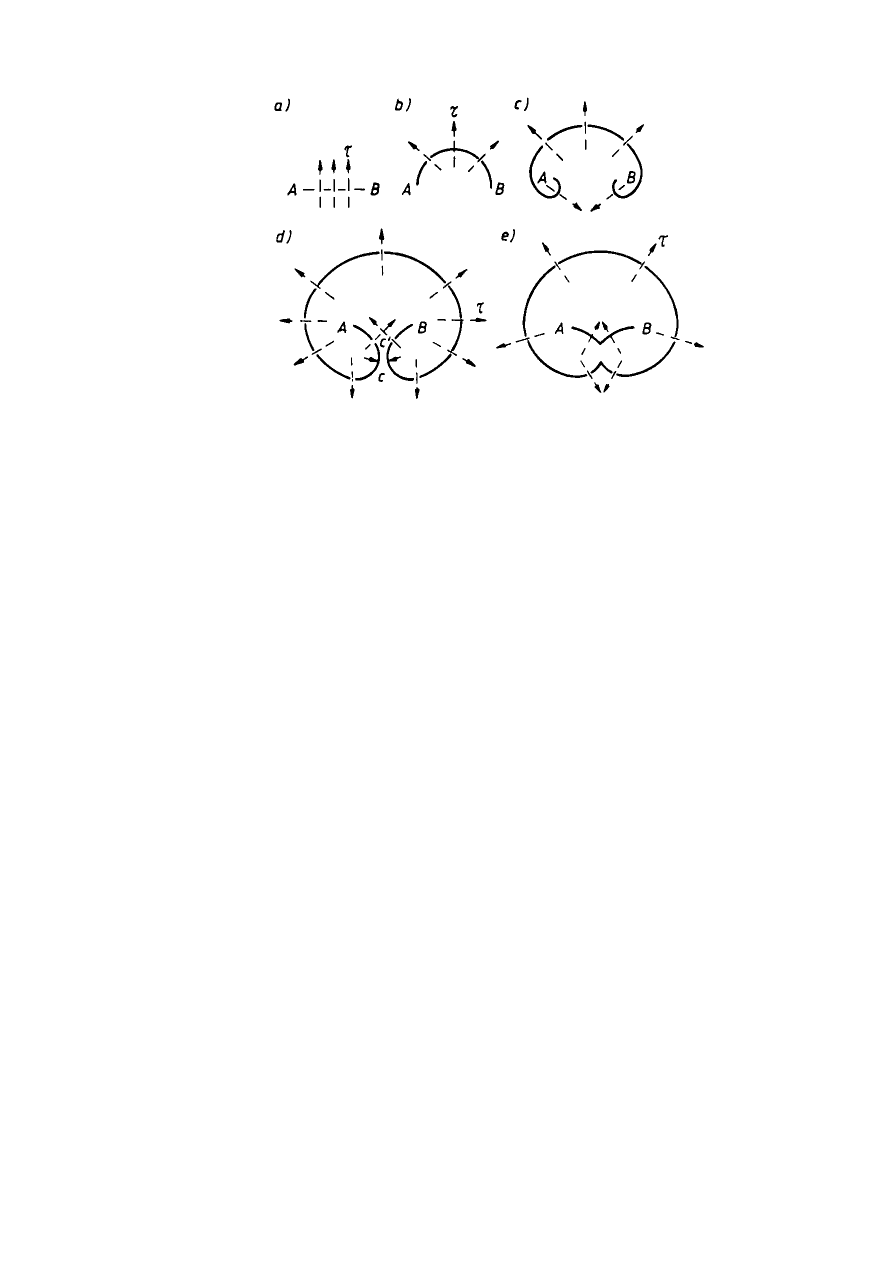
2.5. Krystalizacja metali
Proces przejścia ze stanu ciekłego w stan stały, w czasie którego następuje krzepnięcie
ciekłego metalu w postaci kryształów, nosi nazwę krystalizacji. Aby krystalizacja mogła się
rozpocząć, procesowi temu musi towarzyszyć zmniejszenie się energii swobodnej układu. Jest to
możliwe wówczas, gdy temperatura ciekłego metali spadnie nieco poniżej temperatury
krystalizacji (T
s
) tj. temperatury równowagi faz; ciekłej i stałej. Temperaturę, w której
praktycznie zaczyna się krystalizacja, nazywamy rzeczywistą temperaturą krystalizacji (T
p
).
Natomiast różnicę między teoretyczną a rzeczywistą temperaturą krystalizacji nazywamy
stopniem przechłodzenia (p).
Krzywe chłodzenia. Rozpatrując krzywe przedstawiające zmianę temperatury w funkcji
czasu podczas chłodzenia ciekłego metalu (rys. 2.26) obserwujemy początkowo ciągły spadek
temperatury, natomiast po osiągnięciu temperatury krystalizacji na krzywej temperatura-czas
zjawia się poziomy odcinek, gdyż odpływ ciepła zaczyna być kompensowany przez
wydzielające się ciepło krystalizacji (pochłonięte w czasie procesu topnienia). Po zakończeniu
krystalizacji zakrzepły metal stygnie i temperatura ponownie zaczyna się obniżać w sposób
ciągły. Krzywa l na rys. 2.26 przedstawia teoretyczne zmiany temperatury w czasie krystalizacji,
natomiast krzywa 2 — rzeczywisty przebieg tego procesu wskazujący na występowanie
przechłodzenia p.
Rys.2.26. Krzywe chłodzenia (temperatura w funkcji czasu) podczas krystalizacji metalu
W przypadku niektórych metali może wystąpić silne przechłodzenie w stanie ciekłym i w
pierwszym momencie krystalizacji ciepło krystalizacji zaczyna gwałtownie się wydzielać, co
powoduje raptowne podwyższenie temperatury przechłodzonego metalu, która zbliża się do
temperatury teoretycznej (krzywa 3).
Zarodkowanie. W procesie krystalizacji wyodrębnia się dwa elementarne procesy: tworzenie
się zarodków krystalizacji oraz wzrost tych zarodków. Obydwa te procesy przebiegają
jednocześnie, a ich wynikiem jest utworzenie się kryształów. Ze względu na warunki pojawiania
się zarodków krystalizacji rozróżnia się zarodkowanie homogeniczne i heterogeniczne.
W przypadku zarodkowania homogenicznego, zarodkami krystalizacji są grupy atomów fazy
ciekłej, stanowiące zespoły bliskiego uporządkowania. Muszą one osiągnąć wielkość krytyczną,
co na ogół wymaga dużych przechłodzeń. W ciekłych metalach na ogół występują zbyt małe
przechłodzenia (ok. 1°C), aby możliwe było zarodkowanie homogeniczne. Jedynie metal
rozdrobniony na bardzo małe krople można silnie przechłodzić nawet o 300°C, dzięki czemu w
pojedynczych kroplach występują warunki umożliwiające zarodkowanie homogeniczne. W
czystych metalach zarodki i ciecz mają jednakowy skład chemiczny, natomiast w stopach
zagadnienie staje się bardziej złożone, ponieważ z warunków równowagi w danej temperaturze
wynika, że zarodki i roztwór ciekły różnią się znacznie składem.
W przypadku zarodkowania heterogenicznego, powstawanie zarodków następuje na
powierzchniach fazy stałej stykającej się z cieczą. Zarodkowanie następuje na powierzchniach
ścian naczynia, na drobnych cząstkach stałych zawieszonych w cieczy, jak wtrącenia
niemetaliczne, nierozpuszczone zanieczyszczenia itp. Zarodkowanie może następować również
na warstewce stałych tlenków znajdującej się na powierzchni ciekłego metalu. W takich
warunkach krystalizacja przebiega przy znacznie mniejszym przechłodzeniu niż w przypadku
zarodkowania homogenicznego.
28
JW
Wzrost fazy stałej. Podczas wzrostu zarodka krystalicznego szybkość nawarstwiania się
atomów na poszczególnych ściankach kryształu jest różna i zależy od jego struktury
krystalicznej. Badania w tym zakresie prowadził Bravais, który sformułował następującą regułę:
szybkość wzrostu ściany kryształu jest odwrotnie proporcjonalna do jej gęstości atomowej. Z
reguły tej wynika, że szybko rosnące ściany będą wykazywały tendencję do zaniku, natomiast
wolno rosnące (najgęściej upakowane) - tendencję do wzrostu. Reguła Bravais'go jest zgodna z
doświadczeniem dla olbrzymiej większości kryształów.
Powierzchnia międzyfazowa między cieczą a już utworzoną fazą stałą może się nieco inaczej
kształtować, jeśli np. występuje spadek temperatury równocześnie w kierunku cieczy i fazy
stałej. Może to zaistnieć, jeśli ciecz zostanie znacznie przechłodzona, a na granicy
międzyfazowej wydziela się ciepło krystalizacji podwyższające temperaturę w tym obszarze.
Przykładowo szybki wzrost kryształu np. od punktu A do B (rys. 2.27) zostaje w pewnym
momencie zahamowany wydzielającym się ciepłem krzepnięcia i zanikiem przechłodzenia.
Kryształ wzrasta w innym miejscu dostatecznego przechłodzenia, np. od punktu C do D, aż do
zaniku przechłodzenia wydzielającym się ciepłem krzepnięcia. Warunki takie sprzyjają tzw.
wzrostowi dendrytycznemu, czyli tworzeniu się rozgałęzionych kryształów (dendron po grecku
oznacza drzewo). Rozrastający się i w ten sposób kryształ nazywa się dendrytem.
W przypadku metali o sieci sześciennej kierunki wzrostu kryształów są takie, że gałęzie
dendrytów są do siebie prostopadłe.
Kryształy powstające podczas krystalizacji mają zazwyczaj regularny kształt dopóki otoczone
są cieczą, później jednak na skutek stykania się z sobą i zrastania ulegają zniekształceniu. Z tego
względu zewnętrzny kształt kryształów metalu, nie jest regularny. Anizotropia krystalizacji i
przechłodzenie uwarunkowane warunkami krzepnięcia doprowadza do tworzenia się
zróżnicowanej struktury pierwotnej – rys. 2.28. Odlewy z form piaskowych posiadają ziarna
poliedryczne, podczas gdy w odlewach z form metalowych dominują ziarna słupkowe. Należy
podkreślić, że ziarna te posiadają identyczną strukturę krystaliczną i różnią się tylko kształtem
zewnętrznym.
Rys.2.27. Schemat wzrostu dendrytu
Rys.2.28. Tworzenie się struktury pierwotnej podczas krystalizacji
29
JW
Krystalizacja wlewka. Proces krystalizacji przebiegający w warunkach rzeczywistych staje
się bardziej złożony wskutek wpływu różnych czynników ubocznych. Na przykład przy
odlewaniu dużych wlewków stalowych do wlewnicy kryształy rosną najszybciej w kierunku
prostopadłym do jej ścianek, tj. w kierunku najintensywniejszego doprowadzenia ciepła.
Schemat struktury takiego wlewka jest przedstawiony na rys. 2.29. Rozróżnić w nim można trzy
główne strefy: strefę kryształów zamrożonych, strefę kryształów słupkowych i strefę kryształów
równoosiowych.
Kryształy zamrożone powstają na skutek nagłego zetknięcia się ciekłego metalu ze
ściankami wlewnicy, co powoduje raptowny spadek temperatury, znaczne przechłodzenie i
powstanie dużej liczby zarodków. W rezultacie strefa ta ma strukturę drobnoziarnistą.
Rys. 2.29. Schemat struktury wlewka stalowego; a) l — strefa kryształów zamrożonych; 2
— strefa kryształów słupkowych; 3 — strefa kryształów równoosiowych, b) – rozkład
siarki, c) przekrój prostopadły do osi wlewka
W trzeciej strefie tworzą się kryształy równoosiowe, gdyż w środkowej części wlewka nie
zaznacza się już określony kierunek odpływu ciepła, a temperatura krzepnącego metalu niemal
całkowicie się wyrównuje.
Wzajemne rozmieszczenie wymienionych trzech stref w objętości wlewka ma duże znaczenie
praktyczne, gdyż wzdłuż miejsc styku np. stref kryształów słupkowych mogą często powstawać
pęknięcia podczas walcowania wlewka.
c)
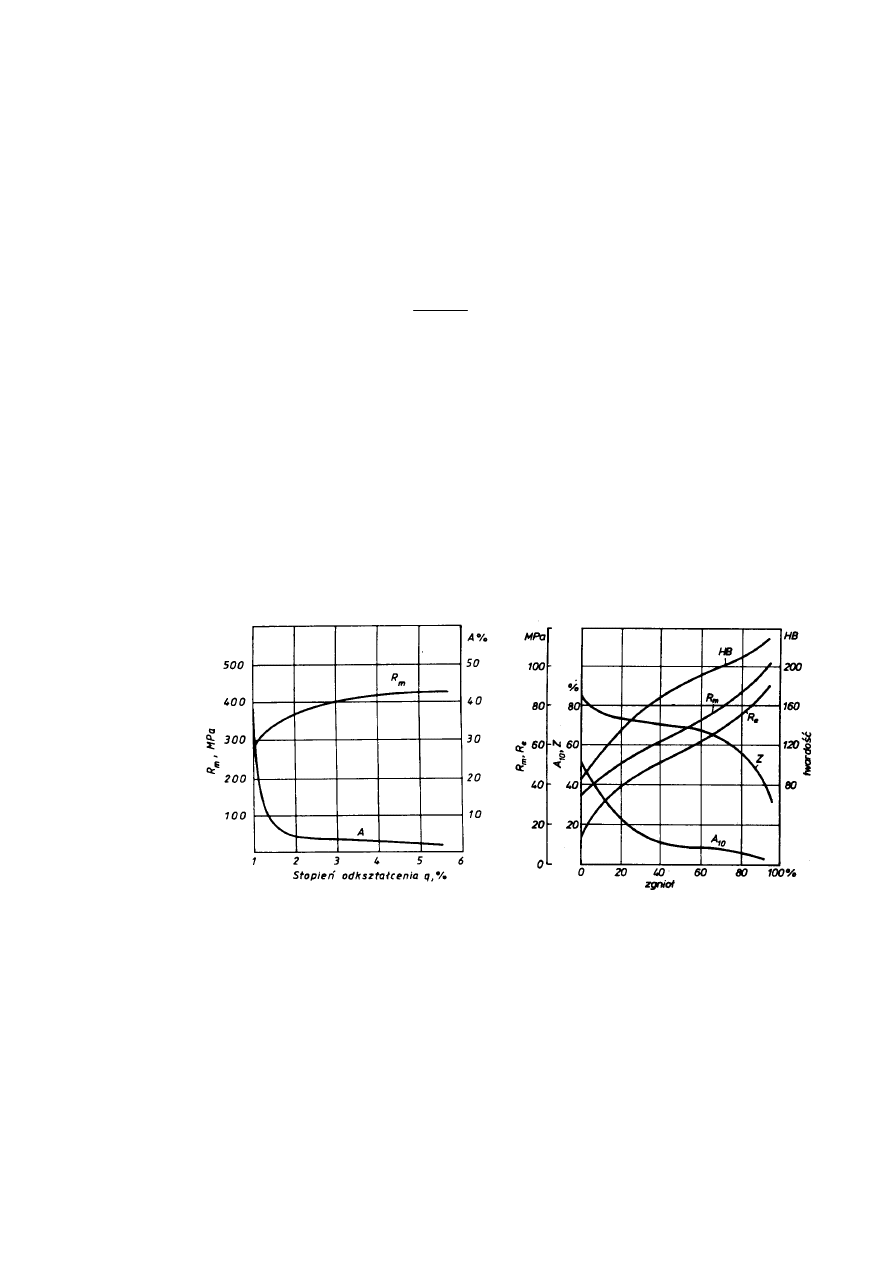
30
JW
2.6. Odkształcenie plastyczne i rekrystalizacja metali
2.6.1. Skutki odkształcenia plastycznego
Odkształcenie plastyczne metalu, które powstaje podczas deformacji na zimno, powoduje
znaczną zmianę jego własności fizycznych i mechanicznych. Zmiany te objawiają się przede
wszystkim wzrostem twardości i wytrzymałości przy jednoczesnym spadku własności
plastycznych (rys. 2.30), oraz obniżeniem przewodności elektrycznej i gęstości.
Wielkość odkształcenia plastycznego określa ilościowo tzw. stopień odkształcenia
plastycznego q, który np. dla walcowania można wyrazić jako procentową zmianę przekroju
materiału.
gdzie : S
o
– przekrój początkowy
S
1
– przekrój końcowy
Wzrost twardości i wytrzymałości związany z odkształceniem plastycznym ma duże znaczenie i
w pewnych przypadkach jest wykorzystywany w celu umocnienia materiału.
Często jednak zachodzi konieczność przywrócenia materiałom ich własności, jakie miały przed
odkształceniem plastycznym np. w celu obniżenia twardości lub uzyskania odpowiednich
własności fizycznych, takich jak np. dobra przewodność elektryczna (jest to bardzo istotne np. w
procesie ciągnienia drutów miedzianych przeznaczonych na przewody elektryczne). Obniżenie
twardości i zwiększenie plastyczności odkształconego metalu oraz przywrócenie innych
własności "fizycznych można uzyskać przez wyżarzanie, które polega na wytrzymaniu
odkształconego materiału przez pewien okres czasu w podwyższonej temperaturze, zwykle
powyżej jednej trzeciej bezwzględnej temperatury topnienia
a)
b)
Rys. 2.30. Zmiana własności mechanicznych: a) – miedzi, b) – mosiądzu (35% Zn) w
zależności od stopnia odkształceni, plastycznego
Odkształcenie plastyczne na zimno powoduje wzrost gęstości dyslokacji. Dla większości
metali gęstość ta wzrasta od wartości ok. l0
6
-10
8
dyslokacji na cm
2
typowej dla stanu
wyżarzonego, do 10
11
÷ l0
12
dyslokacji na cm
2
, w przypadki dużego odkształcenia plastycznego.
Ponieważ odkształcenie plastyczne jest związane z ruchem dyslokacji, występowanie
zjawiska utwardzenia oznacza, że w odkształconym metalu następuje wzrost oporu dla ruchu
dyslokacji. Opór ten rośnie wraz ze wzrostem gęstości dyslokacji, które blokują się nawzajem.
Część dyslokacji zostaje utwierdzona w kryształach i wywołuje wewnętrzne naprężenia, które
przeciwdziałają przemieszczaniu się innych dyslokacji. W konsekwencji powoduje to obniżenie
plastyczności i umocnienie materiału.
Wskutek odkształcenia plastycznego i związanych z nim poślizgów, zachodzących w
poszczególnych ziarnach, w metalu pojawia się tzw. tekstura, czyli określona orientacja
%
100
0
1
0
⋅
−
=
S
S
S
q

31
JW
krystalograficzna ziaren związana z kierunkiem odkształcenia. Stopień steksturowania metalu
wzrasta ze stopniem odkształcenia plastycznego.
Zmiany w strukturze metalu, jakie powstają w wyniku odkształcenia plastycznego można
stwierdzić najwyraźniej za pomocą takich metod, jak np. mikroskopia optyczna, mikroskopia
elektronowa i dyfrakcja promieni X.
Za pomocą badań metalograficznych można stwierdzić odkształcenie ziarn i pojawienie się
pasm poślizgu (rys. 2.31). Natomiast transmisyjna mikroskopia elektronowa umożliwia
obserwację zmian rozkładu i gęstości dyslokacji.
Wzrost gęstości dyslokacji zwiększa energię wewnętrzną sieci krystalicznej, gdyż wzrasta
stopień zaburzenia regularnego rozmieszczenia atomów. Stan odkształcenia plastycznego jest w
związku z tym termodynamicznie nietrwały w stosunku do stanu wyżarzonego. W konsekwencji
odkształcony plastycznie metal będzie wykazywał tendencję do powrotu do stanu o mniejszej
energii swobodnej, tj, do stanu bardziej uporządkowanego. Powrót ten jednak na ogół nie może
zachodzić w sposób samorzutny, lecz jedynie w temperaturach podwyższonych, w których mogą
mieć miejsce procesy aktywowane cieplnie, takie jak dyfuzja, poślizg poprzeczny i wspinanie się
dyslokacji. Dlatego, aby utwardzony przez odkształcenie plastyczne metal zmiękczyć i
przywrócić mu inne własności, jakie przedtem wykazywał, konieczne jest jego podgrzanie do
odpowiedniej temperatury
Rys. 2.31. Pasma poślizgu w austenitycznej stali chromowo-niklowej (18% Cr, 8% Ni)
odkształconej plastycznie przez rozciąganie. Próbka nietrawiona. Powiększenie 800x
W czasie usuwania skutków odkształcenia plastycznego przez wyżarzanie można wyróżnić
trzy procesy, które kolejno zachodzą w odkształconym plastycznie metalu:
• zdrowienie,
• rekrystalizacja
• rozrost ziarna
3.6.2. Zdrowienie
W czasie wygrzewania odkształconego plastycznie metalu można zaobserwować, że
W
pewnej temperaturze następuje usunięcie zniekształceń sieci krystalicznej. Objawia się to tym, że
linie dyfrakcyjne na rentgenogramach (otrzymanych metodą proszkową) rozmyte wskutek
deformacji sieci, stają się znowu wyraźne i ostre. Zjawisko to nosi nazwę zdrowienia.
Zanikowi zniekształceń sieci krystalicznej towarzyszy częściowe usunięcie skutków
odkształcenia plastycznego. Następuje pewne podwyższenie przewodności elektrycznej oraz
częściowy spadek umocnienia.
Minimalna temperatura, w której można stwierdzić te zjawiska, określana jest jako
temperatura zdrowienia.
Proces zdrowienia związany jest ze zmianą rozmieszczenia i gęstości defektów sieci
krystalicznej, głównie wakansów i dyslokacji. W odkształconym na zimno metalu istnieje gęsta
sieć dyslokacji, która powstała w wyniku poślizgów i wzajemnego oddziaływania dyslokacji. W
czasie zdrowienia następuje przemieszczanie i zmiana uporządkowania dyslokacji, co powoduje
zmniejszenie energii zmagazynowanej w odkształcanej sieci. Proces ten jest aktywowany
cieplnie.
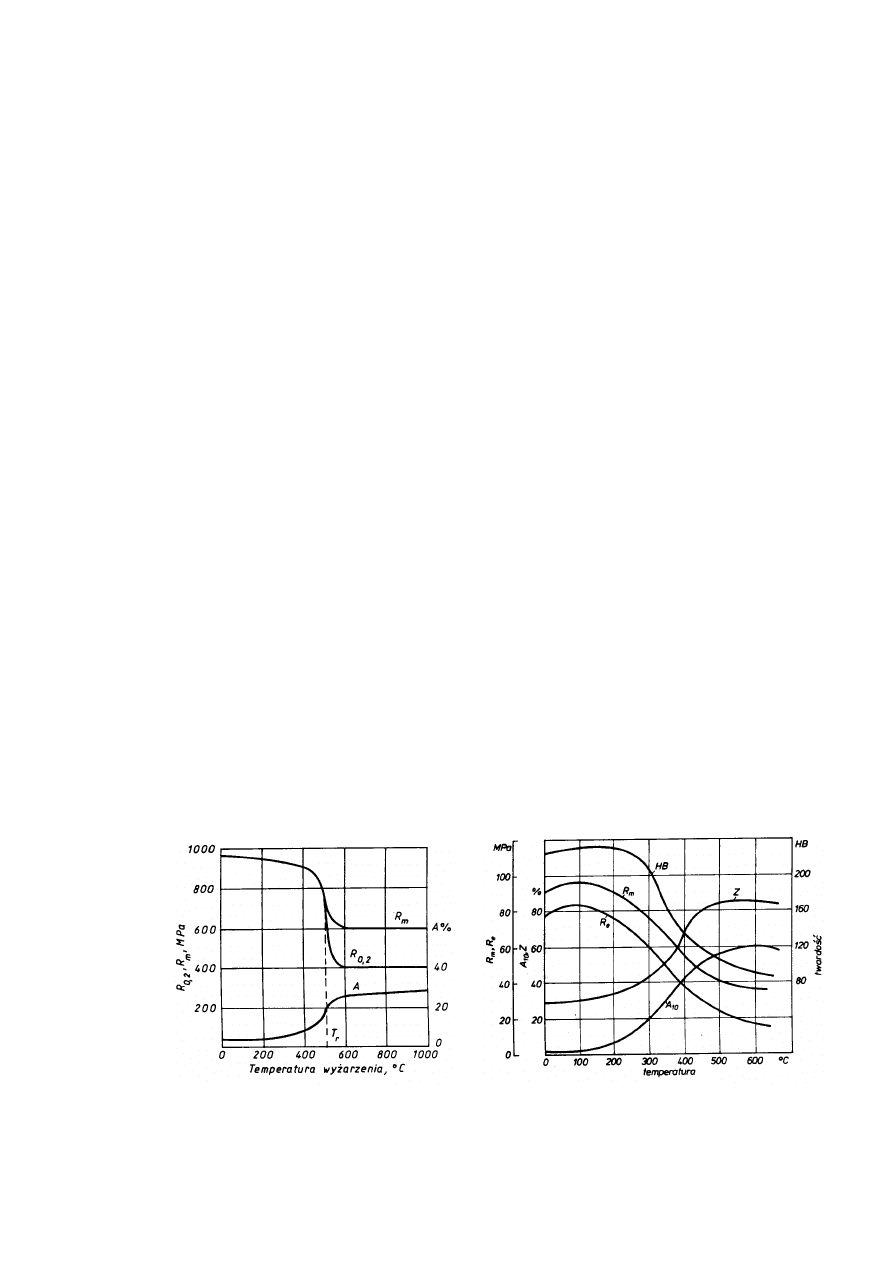
32
JW
3.6.3. Rekrystalizacja
Jeśli odkształcony na zimno metal będzie poddawany dalszemu wygrzewaniu, to w pewnej
określonej temperaturze, wyższej od temperatury zdrowienia, zaczną powstawać zarodki nowych
nieodkształconych ziarn metalu. Nowe ziarna rozrastają się kosztem ziarn odkształconych i po
pewnym czasie wszystkie stare ziarna zostają zastąpione przez nowe.
Zjawisko to nosi nazwę rekrystalizacji, zwane jest również rekrystalizacją pierwotną.
Orientacja krystalograficzna nowych ziarn różni się znacznie od orientacji ziarn starych, kosztem
których powstają ziarna nieodkształcone. Wynika stąd, że sieć krystaliczna nowych ziarn nie jest
koherentna z siecią ziarn odkształconych (tzn. nie jest z nią związana i nie jest do niej
dopasowana), a proces rekrystalizacji polega na przemieszczaniu się (migracji) wysokokątowych
granic ziarn oddzielających nowe kryształy od odkształconych ziarn osnowy.
Temperatura rekrystalizacji. Najniższa temperatura, w jakiej zachodzi proces
rekrystalizacji, nazywana jest temperaturą rekrystalizacji. Temperatura ta jest charakterystyczna
dla danego metalu lub stopu i zależy głównie od dwóch czynników:
a) od uprzedniego stopnia odkształcenia plastycznego, tj. im wyższy był jego stopień, tym
niższa będzie temperatura rekrystalizacji; b) od czystości metalu.
Porównując temperaturę rekrystalizacji z temperaturą topnienia dla różnych metali można
stwierdzić, że zachodzi pomiędzy nimi prosta proporcjonalność. Dla metali technicznie czystych
w przypadku dużych odkształceń plastycznych występuje zależność
T
r
= 0,3
÷ 0,4 T
top
gdzie: T
r
— temperatura rekrystalizacji,
T
top
— bezwzględna temperatura topnienia.
Temperatura rekrystalizacji dla stopów jest wyższa niż dla metali technicznie czystych i w
niektórych przypadkach dochodzi do 0,8 T
top
.. Natomiast dla metali o wysokiej czystości
temperatura rekrystalizacji jest bardzo niska i wynosi 0,1
÷ 0,2 T
top
. Wartości te są słuszne w
przypadku dużych stopni odkształcenia plastycznego, natomiast dla małych odkształceń
plastycznych mogą być znacznie wyższe. Procesowi rekrystalizacji towarzyszą znaczne zmiany
własności mechanicznych odkształconego metalu. W wyniku wyżarzania rekrystalizującego
twardość i wytrzymałość maleją, osiągając wartości właściwe dla materiału przed
odkształceniem plastycznym. Jednocześnie rekrystalizacja przywraca w pełni własności
plastyczne metalu. Na rysunku 2.32 przedstawiona jest zmiana wytrzymałości na rozciąganie
(R
m
, granicy plastyczności (R
02
) i wydłużenia A odkształconego plastycznie żelaza, w zależności
od temperatury wyżarzania. W pewnym wąskim zakresie temperatur widoczny jest
charakterystyczny spadek wytrzymałości i wzrost plastyczności. Temperaturę T
r
odpowiadającą
punktom przegięcia krzywych, przyjmuje się umownie jako temperaturę rekrystalizacji
a)
b)
Rys. 2.32. Zmiana własności mechanicznych odkształconego plastycznie: a) żelaza;T
r
- umowna
temperatura rekrystalizacji, b) mosiądzu (35% Zn) w zależności od temperatury wyżarzania
Podobnie jak R
m
, zmienia się również twardość odkształconego plastycznie metalu
wyżarzonego w różnych temperaturach.
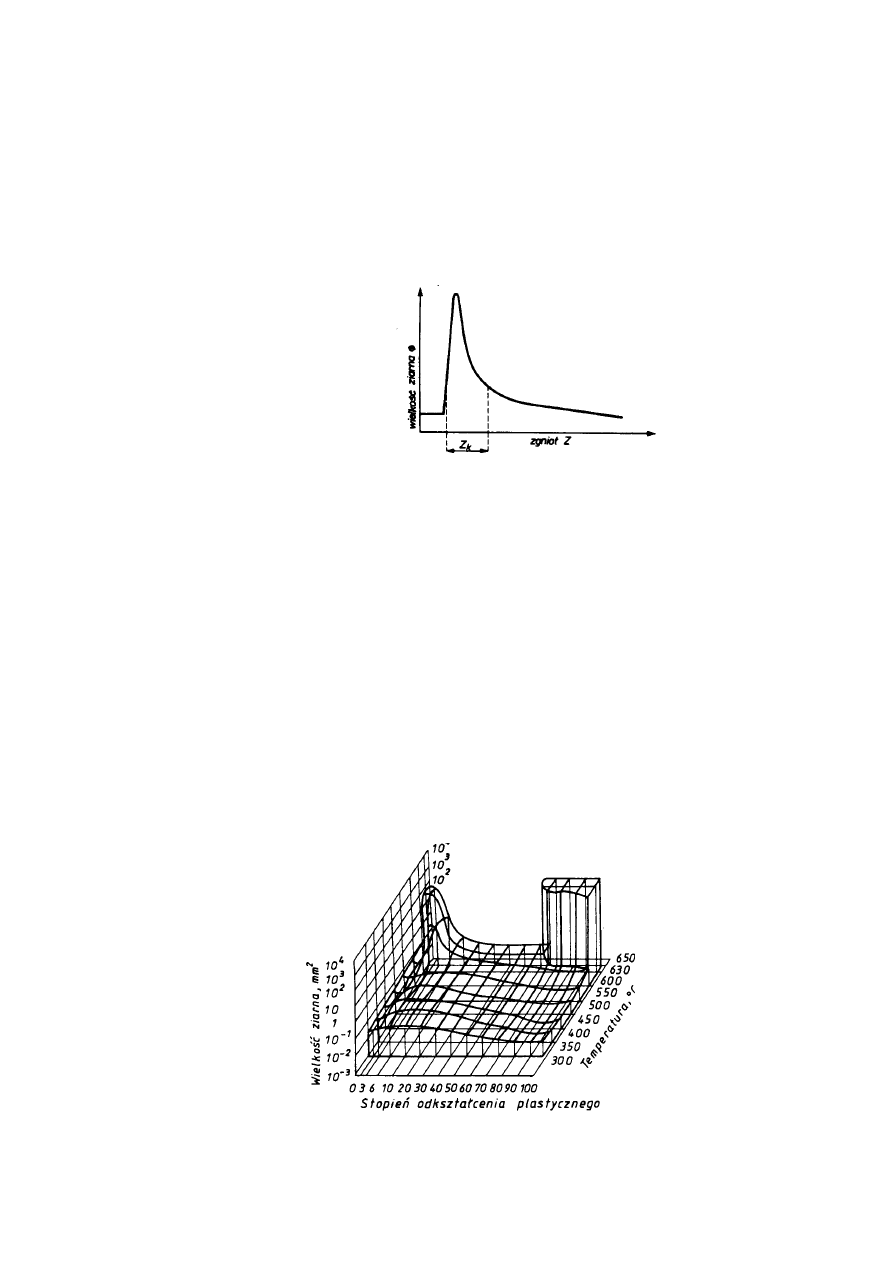
33
JW
Wielkość ziarna po rekrystalizacji. Wielkość ziarna powstałego po rekrystalizacji zależy
przede wszystkim od następujących czynników:
• uprzedniego stopnia odkształcenia plastycznego na zimno,
• temperatury wyżarzania,
• czasu wyżarzania.
Wraz ze wzrostem czasu wyżarzania w danej stałej temperaturze wzrasta wielkość ziarna.
Dlatego aby określić wpływ odkształcenia plastycznego i temperatury wyżarzania na wielkość
ziarna, przyjmuje się pewien stały czas wygrzewania. Stopień odkształcenia plastycznego,
któremu metal został poddany przed wyżarzaniem-wpływa bardzo silnie na wielkość ziarna po
rekrystalizacji (rys. 2.33).
Rys. 2.33. Wpływ stopnia odkształcenia plastycznego na wielkość ziarna po rekrystalizacji
Dla każdego metalu istnieje w zakresie stosunkowo małych odkształceń plastycznych pewien
charakterystyczny stopień tego odkształcenia, zwany krytycznym odkształceniem plastycznym -
q
kr
, który powoduje w czasie rekrystalizacji w wysokiej temperaturze wyjątkowo silny rozrost
ziarna.
Krytyczne odkształcenie plastyczne dla większości metali waha się w granicach od ok. l do 10%.
W wielu przemysłowych procesach technologicznych polegających np. na walcowaniu na
zimno i wyżarzaniu międzyoperacyjnym, występowanie krytycznego odkształcenia plastycznego
jest zjawiskiem niepożądanym, gdyż daje materiał o strukturze gruboziarnistej o odpowiednich
własnościach mechanicznych oraz skłonny do pęknięć.
Istotnym czynnikiem wpływającym na wielkość ziarna po rekrystalizacji jest również
temperatura wyżarzania. Im wyższa jest ta temperatura, tym większe ziarno otrzymuje się w
wyniku rekrystalizacji, przy stałym czasie wyżarzania. Zależność wielkości ziarna metalu po
rekrystalizacji jednocześnie od temperatury wyżarzania i od stopnia odkształcenia plastycznego
Dla aluminium wykres taki pokazano na rys. 2.33. Na wykresie tym w zakresie wysokich
Rys. 2.33. Przestrzenny wykres rekrystalizacji dla aluminium przedstawiający wielkość ziarna w
funkcji odkształcenia plastycznego i temperatury wyżarzania

34
JW
temperatur wyżarzania oprócz omówionego wyżej krytycznego odkształcenia plastycznego,
występuje również drugi bardzo wyraźny obszar rozrostu ziaren w zakresie dużych odkształceń
plastycznych.
Obszar ten pojawia się w przypadku niektórych metali i jest związany z rozrostem ziarna (na
skutek tzw. rekrystalizacji wtórnej) oraz z teksturą deformacji metalu tj. ukierunkowaniem
struktury, które silnie się zaznacza przy dużych odkształceniach plastycznych.
3.6.4. Rozrost ziarna
Wyżarzanie metalu w wysokich temperaturach (już po zakończeniu procesu rekrystalizacji)
powoduje, jak już wspomniano, rozrost ziarna Głównym czynnikiem rządzącym tym procesem
jest napięcie powierzchniowe występujące na granicach ziarn, związane z wyższą energią
swobodną atomów znajdujących się na powierzchni ziarn w porównaniu z energią atomów
znajdujących się wewnątrz nich. W konsekwencji w materiale polikrystalicznym będzie
występowała tendencja do zmniejszenia powierzchni ziarn, a więc do ich rozrostu, gdyż
związane to jest z obniżeniem energii swobodnej materiału. Proces rozrostu ziarna odbywa się
przez pochłanianie małych ziarn przez większe.
Czynnikiem hamującym rozrost ziarna są m.in. zanieczyszczenia metalu, wydzielenia innych
faz oraz obecność obcych cząstek o dużej dyspersji celowo wprowadzonych do metalu w celu
umocnienia i nadania mu określonych własności mechanicznych.
3.6.5. Techniczne znaczenie rekrystalizacji
Wyżarzanie rekrystalizujące jest szeroko stosowane przy wytwarzaniu takich półwyrobów,
jak: blachy, rury, pręty, druty, kształtowniki itp., które są poddawane obróbce plastycznej na
zimno. Ponieważ odkształcenie plastyczne umacnia metal, nie można w jednej operacji nadać
wyrobom ostatecznego kształtu lub wymiarów. Metal umocniony na skutek odkształcenia
plastycznego tak dalece traci własności plastyczne, że nie odkształca się dalej, lecz pęka.
Dlatego konieczne jest międzyoperacyjne wyżarzanie rekrystalizujące, które zmiękcza i
uplastycznia metal. Jeżeli odkształcenie plastyczne metalu przeprowadza się w temperaturze
wyższej od temperatury rekrystalizacji, to proces taki nosi nazwę obróbki plastycznej na gorąco.
W czasie takiej obróbki zachodzą jednocześnie dwa procesy: odkształcenie plastyczne i
rekrystalizacja. W rezultacie nie następuje umocnienie metalu, który miał strukturę
zrekrystalizowaną.

35
JW
3. BUDOWA STOPÓW METALI
3. 1. Rodzaje faz występujących w stopach metali
Stopy to substancje otrzymywane przez stopienie dwóch lub więcej pierwiastków. Jakkolwiek
obecnie stopy można otrzymywać także innymi metodami, np. przez spiekanie lub elektrolizę,
metoda stapiania stosowana jest najczęściej. Oczywiście budowa stopu jest znacznie bardziej
złożona niż czystego pierwiastka i zależy przede wszystkim od rodzaju wzajemnego
oddziaływania na siebie składników stopu podczas krzepnięcia i chłodzenia w stanie stałym. W
zależności od liczba pierwiastków tworzących stop, rozróżnia się stopy dwuskładnikowe, zwane
też podwójnymi, trójskładnikowe (potrójne) itd.
Najprostszymi stopami są stopy dwóch metali lub metalu z niemetalem, czyli stopy
dwuskładnikowe. Pierwiastki tworzące stopy podwójne mogą wykazywać:
a) wzajemną nieograniczoną rozpuszczalność w stanie ciekłym, a w stanie stałym:
• również nieograniczoną rozpuszczalność,
• ograniczoną rozpuszczalność,
• brak rozpuszczalności;
b) rozpuszczalność ograniczoną w stanie ciekłym, a w stanie stałym:
• również ograniczoną rozpuszczalność,
• brak rozpuszczalności;
c) brak rozpuszczalności zarówno w stanie ciekłym, jak i stałym.
Praktyczne znaczenie mają jedynie stopy pierwiastków wykazujących całkowitą wzajemną
rozpuszczalność w stanie ciekłym. W związku z tym, struktura stopów w stanie stałym może
stanowić:
a) jeden roztwór stały, jeśli składniki stopu wykazują nieograniczoną wzajemną
rozpuszczalność;
b) mieszaninę ziarn czystych pierwiastków tworzących stop, jeśli pierwiastki te w stanie stałym
na siebie w ogóle nie oddziałują;
c) mieszaninę kryształów roztworów stałych granicznych, jeśli składniki stopu w stanie stałym
wykazują częściową wzajemną rozpuszczalność;
d) mieszaninę kryształów roztworu stałego oraz kryształów związków chemicznych lub faz
międzymetalicznych, jeśli składniki stopu w stanie stałym nie tylko rozpuszczają się w sobie
wzajemnie, ale również reagują ze sobą chemicznie. W pierwszym przypadku, gdy stop jest
mieszaniną, własności poszczególnych ziarn są takie same jak własności czystych metali
tworzących stop, natomiast własności całego stopu są ich wypadkową, przy czym duży wpływ
wywierają na nie wielkość i kształt ziarn oraz ich wzajemne usytuowanie. Ten przypadek jednak
występuje rzadko, a praktyczne znaczenie mają przede wszystkim stopy zawierające roztwory
stałe i fazy międzymetaliczne.
Roztwory stałe. Roztwory stałe można podzielić na dwa zasadnicze rodzaje:
a) roztwory międzywęzłowe,
b) roztwory różnowęzłowe.
Roztwory międzywęzłowe powstają, gdy atomy pierwiastka stopowego mają małą średnicę i
mogą w sieci krystalicznej metalu podstawowego zajmować pozycje międzywęzłowe.
Pierwiastkami takimi są azot, wodór, węgiel i bor. Wszystkie inne pierwiastki stopowe tworzą
roztwory różnowęzłowe, tzn. atomy pierwiastka stopowego zajmują w sieci krystalicznej
położenie węzłowe, zastępując atomy pierwiastka podstawowego. W obu przypadkach obce
atomy tworzą określone defekty punktowe. Ich ilość i wielkość są funkcjami rodzaju i
zawartości pierwiastka stopowego w stopie.
Niektóre pierwiastki tworzą ze sobą roztwory stałe nieograniczone, tzn. rozpuszczają się w
sobie całkowicie niezależnie od składu stopu, inne rozpuszczają się w stanie stałym tylko
częściowo (np. roztwory międzywęzłowe są z reguły ograniczone). W pierwszym przypadku
stopy mają strukturę jednofazową w całym zakresie składów chemicznych, a maksymalny
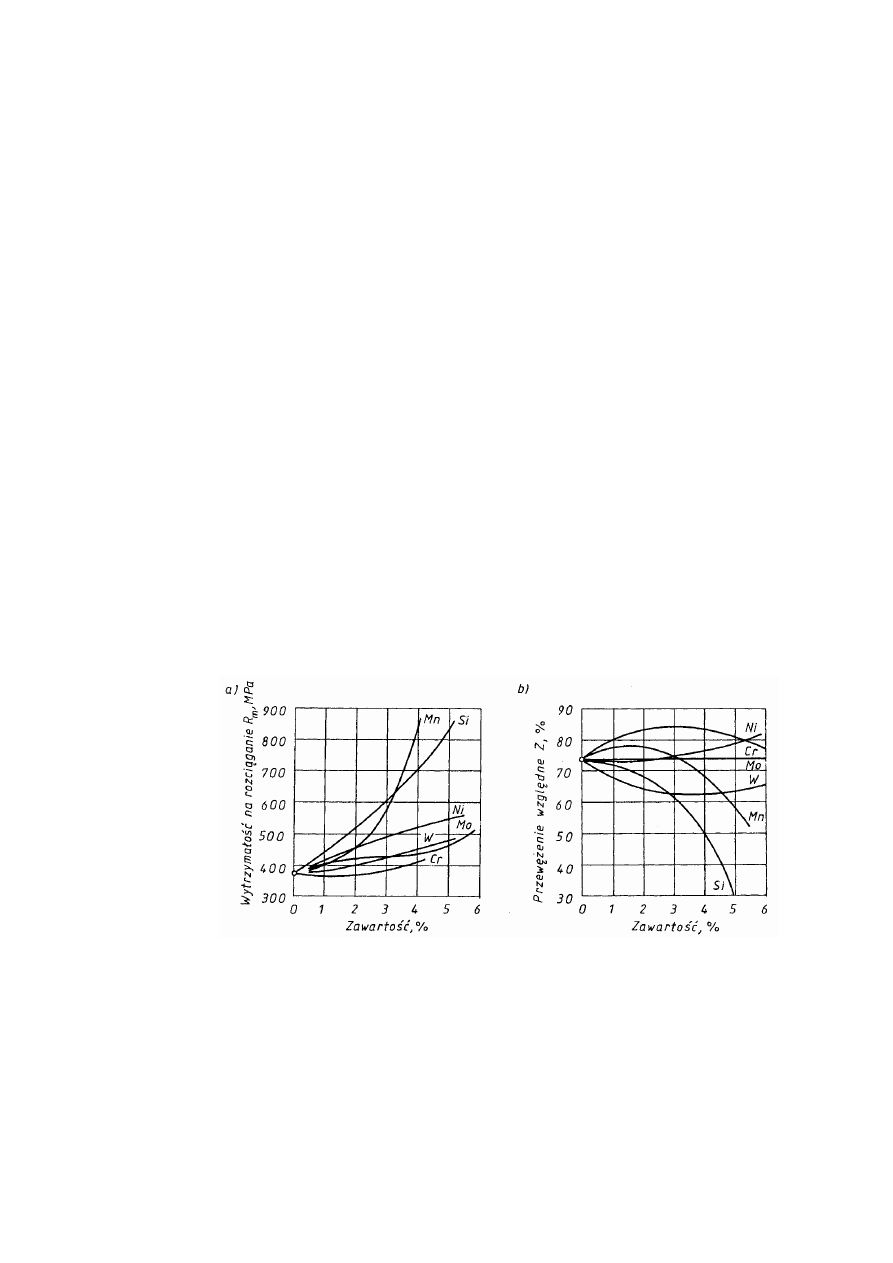
36
JW
wpływ umacniający pierwiastka stopowego występuje przy zawartości około 50%. W
roztworach stałych zostaje zachowana sieć krystaliczna metalu rozpuszczającego.
Roztwory stałe nieograniczone mogą tworzyć ze sobą tylko te pierwiastki, które mają
jednakowy typ sieci krystalicznej, zbliżone wielkości średnic atomowych (różniące się nie
więcej jak o 10-15%), jednakową wartościowość i są do siebie podobne pod względem
charakteru elektrochemicznego. Z warunków tych, zwanych warunkami Hume-Rothery'ego
wynika jednak, że pierwiastki stopowe tworzące z metalem podstawowym roztwory stałe
nieograniczone na własności tych roztworów wywierają wpływ raczej nieznaczny.
Oczywiście nie wszystkie pierwiastki spełniające powyższe warunki tworzą roztwory stałe
nieograniczone, ale strukturę taką wykazuje wiele rzeczywistych stopów podwójnych, m.in.
stopy: złoto-srebro, złoto-platyna, miedź-nikiel, miedź-platyna, kadm-magnez, tytan-molibden,
wolfram-molibden, żelazo-chrom.
Istotny wpływ na własności roztworu stałego wywierają pierwiastki różniące się rodzajem
przestrzennej sieci krystalicznej i średnicą atomową od metalu podstawowego. Jak już
wspomniano, takie pierwiastki mogą tworzyć jedynie roztwory ograniczone, wobec czego w
zależności od składu chemicznego stop może być jednofazowy (jeśli zawartość w nim składnika
stopowego nie przekracza granicznej rozpuszczalności w temperaturze otoczenia) lub
dwufazowy, złożony z mieszaniny kryształów roztworu stałego granicznego składnika B w A i
kryształów roztworu stałego granicznego składnika A w B. W tym drugim przypadku własności
stopu zależą nie tylko o własności roztworów stałych tworzących mieszaninę oraz wielkości i
kształtu ziarn ale także od wzajemnego usytuowania kryształów obu roztworów.
Wśród stopów podwójnych układy z ograniczoną rozpuszczalnością wykazują m.in. stopy:
miedź-srebro, aluminium-krzem, aluminium-cynk, kadm-bizmut, cynk-ołów, ołów-kadm, cynk-
kadm.
Zmianę własności mechanicznych żelaza
α pod wpływem pierwiastków stopowych
tworzących z nim roztwory ograniczone lub nieograniczone pokazano na rys. 3.1. Ja widać,
mangan i krzem wyraźnie podwyższają wytrzymałość żelaza, powoduje jednocześnie znaczny
spadek plastyczności, natomiast nikiel, chrom, wolfram, czy molibden na własności
mechaniczne żelaza oddziałują w niewielkim stopniu
Rys. 3.1. Wpływ niektórych pierwiastków stopowych na własności mechaniczne żelaza
α:
a) na wytrzymałość na rozciąganie, b) na przewężenie
Własności wytrzymałościowe miedzi najbardziej podwyższa krzem, w mniejszym stopniu
cyna, natomiast cynk prawie na nie ma wpływu (rys. 3.2).
W przypadku tytanu wytrzymałość na rozciąganie silnie podwyższają żelazo, mangan, chrom,
molibden i wanad (rys. 3.3).
Fazy międzymetaliczne. W omówionych powyżej typach stopów jako oddzielne występowały
bądź czyste metale, bądź roztwory stałe nieograniczone lub ograniczone. Składniki stopu w
stanie stałym mogą jednak tworzyć też tzw. fazę międzymetaliczną, charakteryzującą się
odmienną siecią krystaliczną niż sieci krystaliczne jej składników.
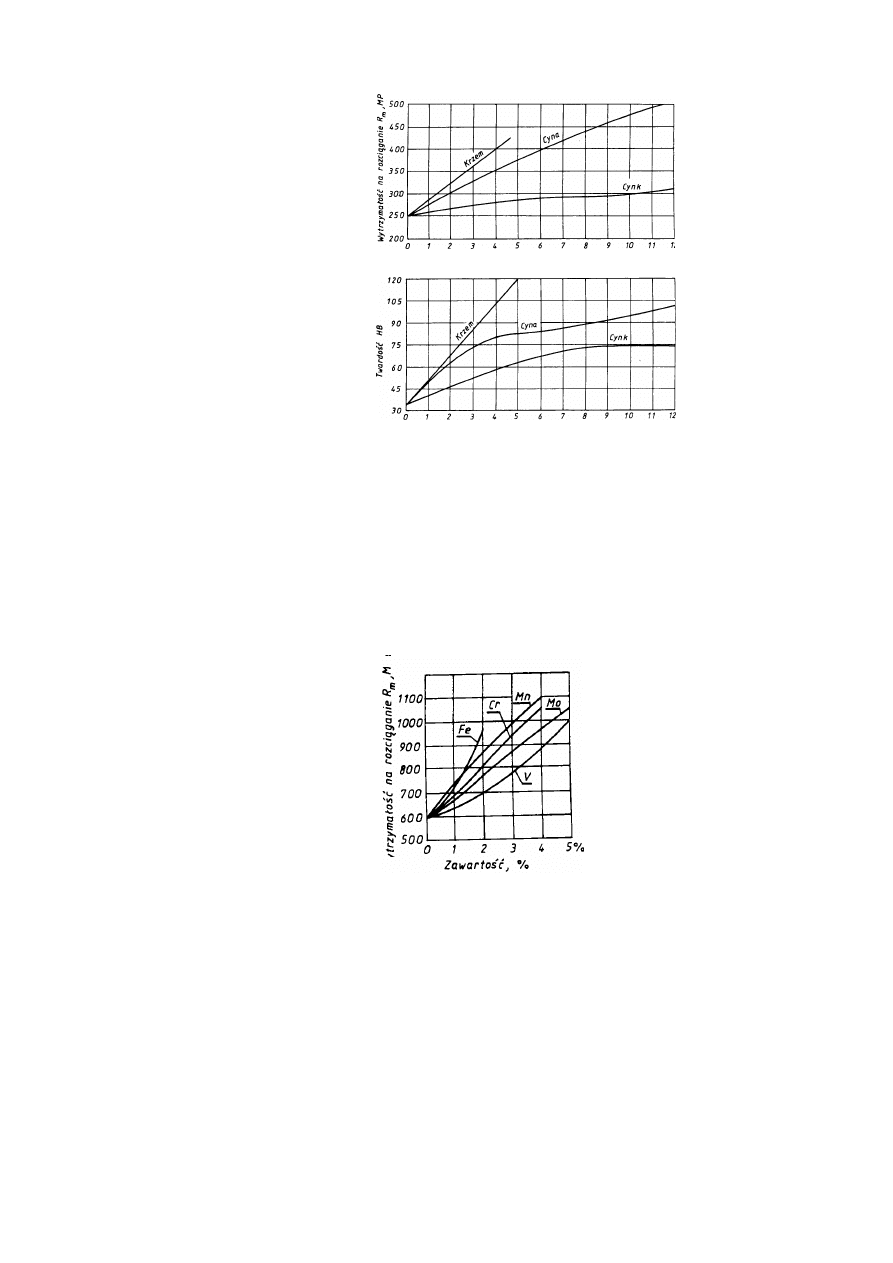
37
JW
Rys. 3.2. Wpływ niektórych pierwiastków stopowych na własności mechaniczne miedzi
Z reguły sieć krystaliczna fazy międzymetalicznej jest bardzo skomplikowana, co powoduje
jej dużą twardość i kruchość.
Ogólnie fazę międzymetaliczną określa się symbolem A
n
B
m
, co oznacza, że składa się ona z n
atomów składnika A i m atomów składnika B. W rzeczywistości tylko niektóre fazy
międzymetaliczne mają stały stosunek składników, większość ich istnieje w szerszym zakresie
stężeń, tzn. występuje również przy nadmiarze jednego ze składników. Takie fazy
międzymetaliczne nazywa się często wtórnymi roztworami stałymi. Mają one sieć krystaliczną
fazy międzymetalicznej
Rys. 3.3. Wpływ niektórych pierwiastków stopowych na wytrzymałość tytanu
Wtórne roztwory stałe, podobnie jak omówione już roztwory stałe, zależnie od pozycji jaką
zajmują atomy składnika będącego w nadmiarze, dzielą się na międzywęzłowe i różnowęzłowe.
Oprócz tego w niektórych przypadkach mogą istnieć charakterystyczne tylko dla faz
międzymetalicznych tzw. roztwory state pustawęzłowe. Powstają one wtedy, gdy pewne węzły
sieci krystalicznej nie są obsadzone przez atomy jednego ze składników. Wywołuje to nadmiar
atomów drugiego składnika w stosunku do teoretycznego składu chemicznego fazy
międzymetalicznej.
Fazy międzymetaliczne klasyfikuje się na podstawie ich struktury sieciowej, z
uwzględnieniem stężenia elektronowego, wielkości promieni atomowych lub jonowych (czyli
czynnika wielkości), lub struktury elektronowej atomów pierwiastków składowych (czyli
czynnika elektrochemicznego).
Fazy elektronowe. Do tej grupy zalicza się fazy międzymetaliczne tworzące się przy
określonych wartościach stężenia elektronowego, tzn. stosunku liczby elektronów walencyjnych

38
JW
do liczby atomów w jednej komórce strukturalnej. Często nazywa się je również fazami Hume-
Rothery'ego od nazwiska ich odkrywcy. Fazy elektronowe występują w stopach, w których
jednym ze składników jest pierwiastek z szeregu Cu, Ag, Au, Mn, Fe, Co, Ni, Rh, Pd, Pt, drugim
- pierwiastek z szeregu Be, Mg, Zn, Cd, Hg, Al, Ga, In, Si, Ge, Sn, Sb. Stwierdzono, że istnieją
trzy rodzaje faz elektronowych, oznaczane literami
β, γ i ε, charakteryzujące się stężeniami
elektronowymi odpowiednio 3/2, 21/13 i 7/4 (oznaczenia pochodzą od faz międzymetalicznych
występujących w stopach Cu-Zn). Fazy
β (np. CuZn, Cu
3
Al, FeAl, NiAl, CoZn
3
, AgZn)
krystalizują bądź w sieci regularnej przestrzennie centrycznej A2, bądź w sieci regularnej
złożonej o 20 atomach, bądź w sieci heksagonalnej zwartej A3. Fazy
γ (np. Cu
5
Zn
8
, Cu
31
Sn
8
,
Fe
5
Zn
21
, Co
5
Zn
21
, Cu
9
Al
4
, Ag
5
Zn
8
) krystalizują w sieci regularnej złożonej o 52 atomach, a fazy
ε (np. CuZn
3,
Cu
3
Sn, Ag
5
Al
3,
AgZn
3
) – w sieci heksagonalnej.zwartej A3.
Przy obliczaniu stężeń elektronowych przyjmuje się dla poszczególnych pierwiastków
następujące liczby elektronów walencyjnych:
Cu, Ag, Au
l
Be, Mg, Zn, Cd, H
2
Al, Ga, In
3
Si, Ge,Sn 4
Sb
5
Mn, Fe, Co, Ni, Rh, Pd, Pt, . . 0
Jak widać, przy obliczaniu stężenia elektronowego pierwiastkom przejściowym przypisuje się
zerową wartościowość, tzn. zakłada się, że pierwiastki te nie wnoszą udziału elektronów do
ogólnego stężenia elektronowego fazy.
Fazy elektronowe istnieją w dość szerokich zakresach stężeń składników, co jest związane z
ich typowym wiązaniem metalicznym.
Fazy Lavesa. Tej grupie faz międzymetalicznych przypisuje się wzór stechiometryczny AB
2
,
ale głównym czynnikiem wpływającym na ich powstanie jest stosunek promieni atomów
składników, tzn. r
A
./r
B
. Stosunek ten teoretycznie wynosi 1,225, w rzeczywistości fazy Lavesa
powstają w zakresie stosunków r
A
./r
B
= 1,05
÷ 1,68. Warunek geometryczny powoduje, że fazy
Lavesa istnieją w wąskim przedziale stężeń składników.
Ogólnie dzielą się na trzy rodzaje, których typowymi przedstawicielami są MgCu
2
, MgZn
2
i
MgNi
2
. Pierwszy rodzaj krystalizuje w sieci regularnej o 24 atomach, pozostałe - w sieci
heksagonalnej o różnym stopniu skomplikowania. W stopach żelaza fazami Lavesa są m.in.
ZrFe
2
, TiFe
2
, WFe
2
i NbFe
2
. Fazy Lavesa cechuje duża twardość i własności typowe dla stanu
metalicznego.
Fazy międzywęzłowe i fazy o strukturach złożonych. Podobnie jak fazy Lavesa, fazy
międzywęzłowe są fazami czynnika wielkości. Są to fazy krystalizujące w sieci Al, A3 i A2
(rzadziej) oraz w sieci heksagonalnej prostej, przy czym węzły sieci są obsadzone atomami
jednego z metali przejściowych (Fe, Cr, Co, Ni, W, i, V, Ta, Nb, Ti), a przestrzenie
międzywęzłowe zajęte są przez atomy jednego z czterech pierwiastków niemetalicznych o
najmniejszych promieniach atomowych (H, C, N, B).
Fazy międzywęzłowe mogą powstawać, jeśli stosunek promieni atomowych pierwiastka
niemetalicznego do metalu nie przekracza 0,59. Można przypisać im wzory stechiometryczne
Me
4
X, Me
2
X, MeX i MeX
2
, gdzie Me jest symbolem metalu, a X - niemetalu. Fazami
międzywęzłowymi są zatem wodorki, azotki oraz niektóre węgliki i borki metali przejściowych.
W stopach technicznych najważniejszymi są azotki (Nb
2
N, TaN, TiN, WN, VN, Fe
4
N, Fe
2
N,
Ni
2
N, Mn
4
N i in.) i węgliki (m.in. Nb
4
C, Nb
2
C, NbC, Ta
2
C, TiC, W
2
C, WC, Mo
2
C, V
2
C).
Fazy międzywęzłowe występują w dość szerokim zakresie stężeń. Wykazują własności
metaliczne, są twarde i kruche, przewodzą prąd elektryczny, mają wysoką temperaturę topnienia
i dość dobrą odporność chemiczną. Izomorficzne węgliki i azotki w stanie stałym wzajemnie się
rozpuszczają.
Fazy o strukturach złożonych powstają, jeśli stosunek promieni atomowych pierwiastka
niemetalicznego do metalu przekracza 0,59. Takimi fazami są węgliki żelaza, chromu, niklu,
manganu i kobaltu, węgliki podwójne żelaza i manganu, żelaza i wolframu oraz żelaza i
39
JW
molibdenu, a także większość borków. Fazy o strukturze złożonej cechuje duża liczba atomów
przypadających na jedną komórkę strukturalną.
Węgliki o złożonej strukturze mają również własności metaliczne, ale występują przy
praktycznie stałym stosunku ilościowym składników, określonym wzorem stechiometrycznym.
W porównaniu do węglików międzywęzłowych mają niższą temperaturę topnienia, mniejszą
twardość i odporność chemiczną, łatwiej przechodzą do roztworu stałego podczas ogrzewania
stali.
W stopach żelaza największe znaczenie ma węglik żelaza Fe
3
C zwany cementytem, o
stosunku promieni atomowych węgla do żelaza wynoszącym 0,63. Może on tworzyć roztwory
stałe różnowęzłowe, w których część atomów żelaza jest zastępowana atomami manganu,
chromu, molibdenu, czy wolframu (cementyt stopowy), a część atomów węgla - atomami azotu
(tzw. węglo-azotki). Krystalizuje w złożonej sieci należącej do układu rombowego, podobnie jak
Mn
3
C i Co
3
C.
Węgliki Cr
23
C
6
i Mn
23
C
6
krystalizują w bardzo złożonej sieci regularnej, której pojedyncza
komórka zawiera 116 atomów, w tym 24 atomy węgla. Węgliki Cr
7
C
3
i Mn
7
C
3
krystalizują w
złożonej sieci heksagonalnej, której komórka zawiera 80 atomów (w tym również 24 atomy
węgla). W stalach wysokostopowych oba rodzaje węglików tworzą również roztwory stałe
różnowęzłowe, przy czym część atomów chromu lub manganu zastępują atomy żelaza,
molibdenu, wolframu i in., podobnie jak podwójne węgliki żelaza i wolframu (Fe
4
W
2
C i
Fe
3
W
3
C), krystalizujące w złożonej sieci regularnej (112 atomów, w tym 16 węgla).
Fazy Sigma. Są to fazy międzymetaliczne krystalizujące w sieci tetragonalnej, której komórka
zawiera 30 atomów. Tworzą się między niektórymi metalami przejściowymi, takimi jak Cr-Fe,
V-Fe, Mo-Fe, Cr-Co, V-Co, Mo-Co, Cr-Mn, V-Ni i in. Fazy Sigma są twarde i kruche.
Największe znaczenie techniczne ma faza Sigma występująca w stopach żelaza z chromem
(stopy odporne na korozję oraz stopy o dużym oporze elektrycznym), o przybliżonym wzorze
stechiometrycznym FeCr, wydzielająca się z roztworu stałego w temperaturze 815°C.
Rys. 3.4. Schematy wzajemnego usytuowania faz w stopach dwufazowych: l — kryształy
granicznego roztwór stałego
α, 2 — kryształy granicznego roztworu stałego β lub
fazy międzymetalicznej
Teoretycznie zawiera 47% atom. chromu, praktycznie istnieje przy zawartościach chromu 43-
50% atom., jest więc roztworem stałym wtórnym. Obecność fazy FeCr w stopach jest bardzo
niekorzystna, gdyż zwiększa ich kruchość i obniża odporność na korozję.

40
JW
Omówione grupy faz międzymetalicznych są najbardziej typowymi i najliczniej
występującymi w stopach technicznych.
W stopach dwu- i wielofazowych, w których tworzą się fazy międzymetaliczne, miękką i
plastyczną osnowę stanowi zwykle roztwór stały o sieci krystalicznej jednego ze składników, a
w nim są rozłożone w określony sposób wydzielenia fazy (lub faz) międzymetalicznej. Każda z
faz ma określone, charakterystyczne własności, natomiast własności stopu są wypadkowe,
podobnie jak w mieszaninie kryształów czystych metali lub mieszaninie kryształów roztworów
granicznych. Trzeba Jednak podkreślić, że własności stopu zależą ponadto w bardzo dużym
stopniu od wzajemnego usytuowania faz. Różne przykłady wzajemnego usytuowania kryształów
roztworu stałego i fazy międzymetalicznej podano na rys. 3.4. Podobne usytuowanie faz
występuje również w przypadku struktur będących mieszaniną kryształów roztworów stałych
granicznych.
3. 2. Analiza termiczna i reguła faz
Analizą termiczną nazywa się badania polegające na określeniu temperatury początku i końca
krzepnięcia (lub topnienia) oraz temperatury przemian zachodzących w stanie stałym podczas
ochładzania (lub ogrzewania) metali i ich stopów. Badania te mają na celu wykreślenie
krzywych przebiegu ogrzewania lub chłodzenia, na których wszelkie efekty cieplne występujące
podczas zmian fazowych uwidaczniają się w postaci przystanków temperaturowych i załamań. te
z kolei służą do zbudowania tzw. wykresów równowagi układów pierwiastków przedstawiają-
cych położenie granic faz w funkcji temperatury i składu chemicznego.
Ogólnie wykresy równowagi obrazują:
a) przemiany w stanie ciekłym (zmiany rozpuszczalności),
b) zmiany stanu skupienia (krzepnięcie, topnienie),
c) przemiany w stanie stałym (zmiany rozpuszczalności, przemiany alotropowe,
eutektoidalne itp.),
d) tworzenie się lub rozpad faz międzymetalicznych w stanie ciekłym, w czasie zmiany stanu
skupienia lub w stanie stałym.
Wykresy równowagi podają więc w zasadzie tylko budowę fazową stopów, tzn. można z nich
wnioskować, z jakich faz stop jest zbudowany, w jakim stosunku ilościowym te fazy pozostają i
jaki jest ich skład chemiczny. Inaczej mówiąc, wykresy równowagi przedstawiają graficznie stan
stopu. Jeśli zachodzi zmiana jego składu chemicznego, temperatury lub ciśnienia, zmienia się
również stan stopu, co znajduje odwzorowanie graficzne na wykresie równowagi.
Wykres równowagi obrazuje stany trwałe stopu, tj. stany, którym w danych warunkach
odpowiada najmniejszy zasób energii swobodnej układu. Również zmiany stanu stopu
odwzorowane na wykresie odpowiadają warunkom równowagi, tzn. nie uwzględniają zjawisk
przegrzania lub przechłodzenia, które w rzeczywistości mają zawsze miejsce (oczywiście w
różnym stopniu). Dlatego wykres równowagi stopu jest wykresem teoretycznym, a posługiwanie
się nim jest uwarunkowane rozpatrywaniem przemian przy małych szybkościach nagrzewania
lub chłodzenia, gdy zjawiska przegrzania lub przechłodzenia występują w minimalnym stopniu i
praktycznie można je pominąć.
Ogólne zasady określające współistnienie trwałych faz odpowiadających teoretycznym
warunkom równowagi wyraża matematycznie tzw. reguła faz, zwana także regułą Gibbsa.
Podaje ona ilościową zależność między liczbą stopni swobody danego stopu czy metalu, a liczbą
jego faz i składników.
Posługując się regułą faz można przewidzieć, w jakich warunkach temperaturowych
przebiegają przemiany fazowe (w stałej temperaturze, czy w zakresie temperatur) i jakie
czynniki mogą ulegać pewnym zmianom bez naruszenia stanu równowagi.
Przy omawianiu reguły faz konieczne jest dokładne zdefiniowanie pojęć: fazy, składnika oraz
liczby stopni swobody.
Faza jest to jednorodna część układu oddzielona od innych jego części (faz) powierzchnią
rozdziału, czyli granicą fazy, po przekroczeniu której własności fizyczne czy też struktura
zmieniają się w sposób nieciągły.

41
JW
Składnikami układu nazywa się substancje tworzące dany układ. Dla przykładu czysty metal
tworzy układ jednoskładnikowy, stop dwóch metali — układ dwuskładnikowy itd. Fazy
międzymetaliczne uważa się także za składniki, jeśli w rozpatrywanym zakresie temperatury nie
rozkładają się na pierwiastki składowe.
Liczba stopni swobody układu jest to liczba zewnętrznych i wewnętrznych czynników
(temperatura, ciśnienie i skład chemiczny), które można zmieniać bez spowodowania zmiany
liczby faz w danym układzie.
Przy założeniu, że w rozpatrywanym układzie wszystkie przemiany zachodzą przy stałym i
niezmiennym ciśnieniu, reguła faz wyraża się wzorem
s = m – f + l
gdzie: s - liczba stopni swobody,
m - liczba składników,
f - liczba faz.
Dla rzeczywistych układów wielofazowych liczba stopni swobody wynosi zwykle 0, l lub 2.
Gdy S = 0, układ jest niezmienny, czyli nie można zmieniać ani temperatury, ani składu
chemicznego bez spowodowania zmiany liczby faz w układzie.
Gdy S = l, układ jest jednozmienny. Oznacza to, że nie zmieniając liczby faz w układzie
można zmienić (w pewnych granicach) temperaturę bądź skład chemiczny.
Gdy S = 2, układ jest dwuzmienny. Oznacza to, że nie zmieniając liczby faz w układzie
można zmienić (w pewnych granicach) temperaturę i skład chemiczny.
3. 3. Budowa stopów podwójnych
Jak już wspomniano, przy badaniu struktury stopów bardzo ważne znaczenie ma znajomość
ich wykresów równowagi fazowej. Jeżeli dwa składniki stopu nie oddziałują na siebie
chemicznie i nie podlegają żadnym przemianom w stanie stałym, to ich wykresy równowagi
podają dla dowolnego składu chemicznego stopu temperaturę początku krzepnięcia (linia
likwidusu) i jego końca (linia solidusu).
Na rysunku 3.5 przedstawiono wykres równowagi typu I, odpowiadający całkowitej
wzajemnej rozpuszczalności składników w stanie ciekłym i jej brakowi w stanie stałym.
Składniki nie tworzą również związków chemicznych. Linia AEB jest linią likwidusu, a linia
DEC linią solidusu. Na linii AE przy chłodzeniu zaczynają się wydzielać z cieczy kryształy
metalu A, na linii EB — kryształy metalu B. Na linii DEC z cieczy o składzie E wydzielają się
jednocześnie bardzo drobne krysztale A i B tworzące mieszaninę, która nazywa się eutektyką
(po grecku łatwo topliwa) a stop o tym składzie - stopem eutektycznym (stopy o składzie
odpowiadającym zakresowi DE są stopami podeutektycznymi, pozostałe - nadeutektycznymi).
Na rysunku podano również schematy struktur stopów podeutektycznych, eutektycznego i
nadeutektycznych. Jak widać, w pierwszym przypadku na tle eutektyki występują kryształy
metalu A, w drugim - sama eutektyką, w trzecim - na tle eutektyki kryształy metalu B.
Oczywiście skrajne odcięte wykresu odpowiadają czystym składnikom A i B. Taki wykres
równowagi tworzą m.in. stopy bizmut-kadm.
Trzeba podkreślić, że zawartość eutektyki w stopie zmienia się liniowo, od 0 dla czystych
składników A i B do 100% dla stopu eutektycznego.
Wykres równowagi typu II, odpowiadający całkowitej rozpuszczalności wzajemnej
składników zarówno w stanie ciekłym, jak i stałym (przy braku faz międzymetalicznych),
podano na rys. 3.6.
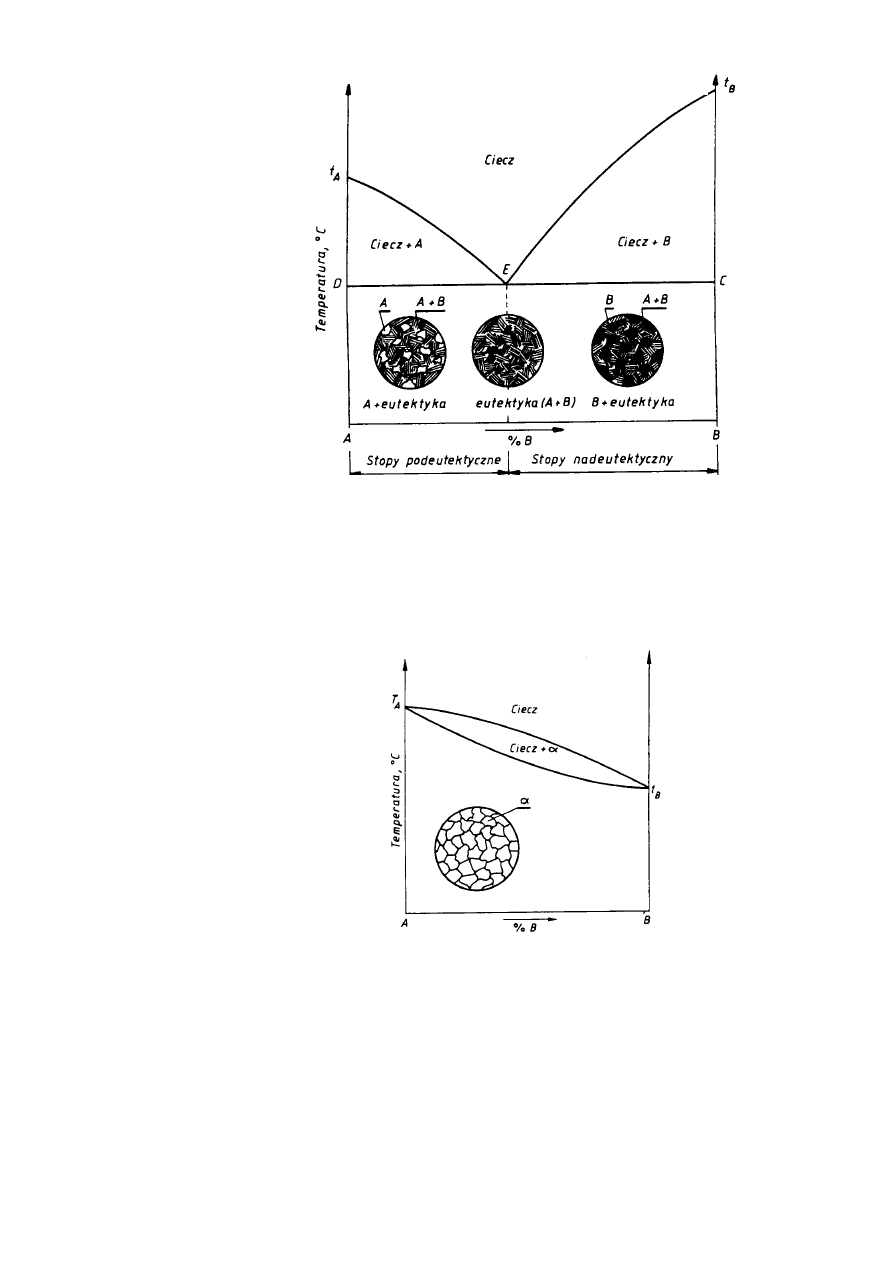
42
JW
Rys. 3.5. Wykres równowagi odpowiadający całkowitej rozpuszczalności wzajemnej
składników w stanie ciekłym i brakowi rozpuszczalności w stanie stałym oraz
schematy struktur poszczególnych grup stopów
W tym przypadku eutektyka nie występuje, a wszystkie stopy w stanie stałym: mają jednakową
strukturę złożoną z kryształów roztworu stałego
α o składzie odpowiadającym składowi stopu.
Rys. 3.6. Wykres równowagi odpowiadający całkowitej rozpuszczalności wzajemnej składników
zarówno w stanie ciekłym, jak i w stanie stałym oraz schemat struktury stopów
Przy rozpuszczalności wzajemnej składników w stanie ciekłym i ograniczonej rozpuszczal-
ności w stanie stałym oraz braku faz międzymetalicznych mogą występować dwie postacie
wykresów równowagi typu III, a mianowicie z eutektyką (rys. 3.7) i perytektyką (rys. 3.8).
W pierwszym przypadku nie występują fazy będące czystymi składnikami, w sąsiedztwie
linii A i B, odpowiadających czystym składnikom, znajdują się obszary roztworów stałych
granicznych
α (składnika B w składniku A) i β (składnika A w składniku B), przy czym
graniczną rozpuszczalność z lewej strony określa linia DF, z prawej — linia CG.
A
B
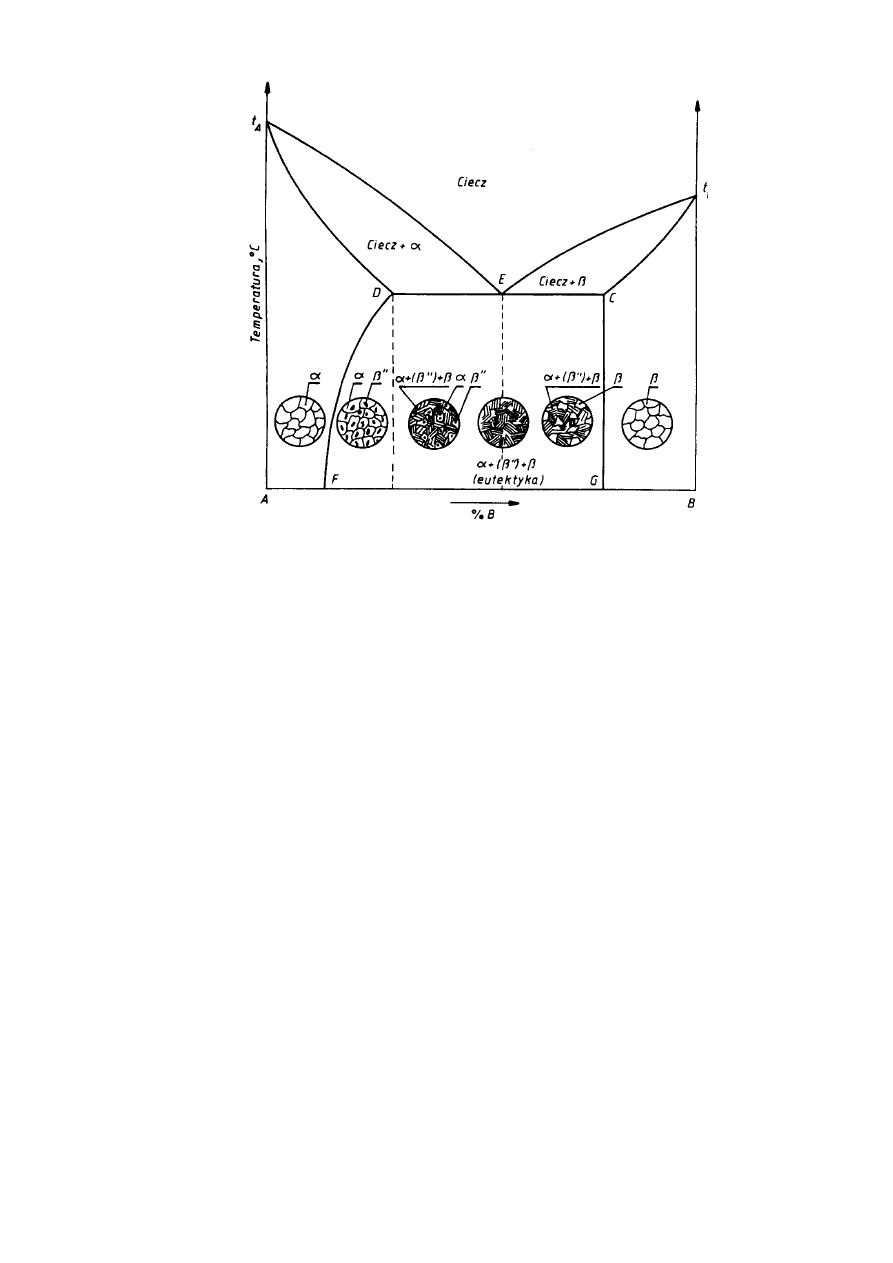
43
JW
Rys. 3.7. Wykres równowagi odpowiadający całkowitej rozpuszczalności wzajemnej
składników w stanie ciekłym i ograniczonej rozpuszczalności w stanie stałym
(z eutektyką) oraz schematy struktur poszczególnych grup stopów
Między tymi liniami znajdują się stopy leżące poza granicami rozpuszczalności i stanowiące
mieszaninę dwóch roztworów stałych granicznych
α + β. Jak widać na wykresie, graniczna
rozpuszczalność składnika B w składniku A stała, lecz zmienia się wraz z temperaturą (linia
DF), co prowadzi do wtórnej krystalizacji i wydzielania się z roztworu stałego
α kryształów β .
Kryształy te noszą nazwę kryształów wtórnych i dla odróżnienia od pierwotnych kryształów
β
oznaczane są zwykle symbolem
β". Kryształów wtórnych nie zawierają jedynie stopy o stężeniu
składnika B mniejszym od stężenia odpowiadającego punktowi F.
W przeciwieństwie do tego, graniczna rozpuszczalność składnika A w składniku B nie zależy
od temperatury (linia CG jest linią pionową), toteż wydzielanie się wtórnych kryształów
α w
tym przypadku nie zachodzi.
Wykres równowagi zbliżony do omówionego (graniczna rozpuszczalność obu składników jest
zmienna) tworzą m.in. stopy srebro-miedź, cyna-ołów, kadm-cynk i aluminium-krzem.
W drugim przypadku, zamiast przemiany eutektycznej, podczas której ciecz krystalizuje tworząc
dwie fazy stałe, występuje przemiana polegająca na reagowaniu cieczy z wcześniej
wydzielonymi kryształami i tworzeniu się nowego rodzaju kryształów. Przemiana ta nosi nazwę
perytektycznej
Jak widać na rys. 3.8 na wykresie poniżej linii solidusu ADPB można wyodrębnić trzy obsza-
ry (dla uproszczenia linie granicznych rozpuszczalności są na rysunku pionowe):
α, α + β i β.
Stopy o stężeniu składnika B mniejszym od stężenia odpowiadającego punktowi F, po
skrzepnięciu składają się z jednorodnych ziarn roztworu stałego
α, które podczas dalszego
chłodzenia nie podlegają żadnym przemianom.
Stopy o stężeniu składnika B odpowiadającym stężeniom zawartym między punktem F i
punktem G zaczynają krzepnąć w temperaturze określonej likwidusem, tak że w temperaturze t
p
składają się z kryształów
α o składzie punktu D i cieczy o składzie punktu C. W tej temperaturze
następuje przemiana perytektyczna, którą ogólnie można przedstawić:
ciecz +
α →β
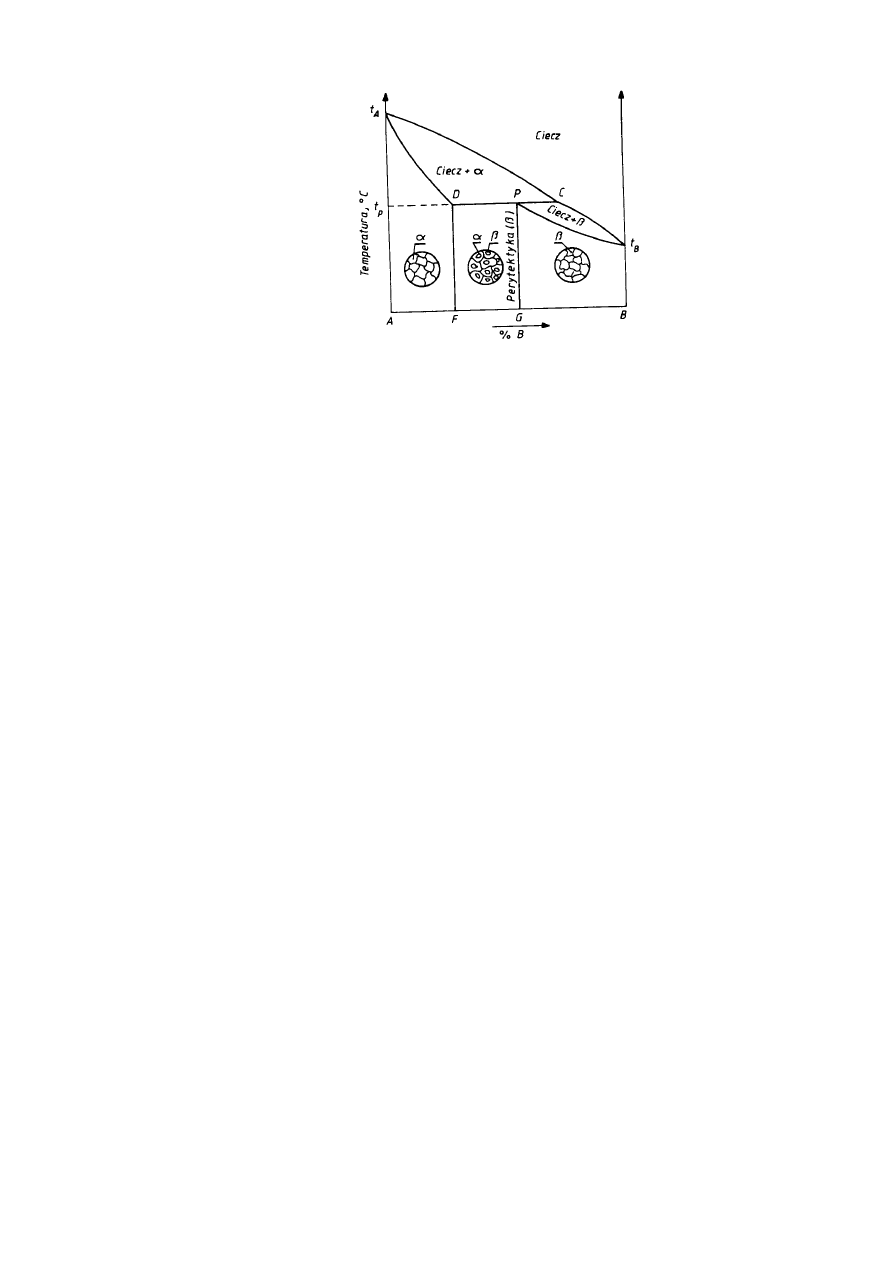
44
JW
Rys. 3.8. Wykres równowagi odpowiadający całkowitej rozpuszczalności wzajemnej
składników w stanie ciekłym i ograniczonej rozpuszczalności w stanie stałym
(z perytektyką) oraz schematy struktur poszczególnych grup stopów
Nowo powstające kryształy
β mają skład punktu P. Ponieważ stopy zawieraj; nadmiar fazy α
w stosunku do ilości potrzebnej do utworzenia kryształów
β, po zakończeniu przemiany składają
się z kryształów
β i pozostałych w nadmiarze kryształów α Oczywiście ilość tych ostatnich
maleje w miarę zbliżania się d punktu P, a stop o składzie tego punktu zawiera wyłącznie
kryształy
β .
Krzepnięcie stopów o stężeniu składnika B większym od stężenia odpowiadającego punktowi
G różni się od poprzednich tym, że w temperaturze perytektycznej istnieje pewien nadmiar
cieczy w stosunku do ilości potrzebnej dla utworzenia kryształów
β o składzie punktu P. Dlatego
przemiana perytektyczna w tym przypadku kończy się z chwilą wyczerpania kryształów
roztworu stałego
α, a pozostała ciecz bezpośrednio krystalizuje jako faza β.
W praktyce rzadko spotyka się układy o stałej granicznej rozpuszczalności. Przeważnie
zmienia się ona wraz z temperaturą i wtedy ma miejsce jeszcze wtórna krystalizacja kryształów
β" z kryształów α, i kryształów α" z kryształów β.
Układ równowagi z perytektyką tworzą m.in. stopy srebro-platyna.
Zawartość perytektyki w stopie, podobnie jak eutektyki, zmienia się liniowo, od 0 dla stopów
o składzie punktów D i C do 100% dla stopu o składzie punktu P.
Wykres równowagi typu IV (rys. 3.9a) tworzą dwa składniki, które w stanie ciekłym
rozpuszczają się wzajemnie bez ograniczenia, natomiast w stanie stałym bądź nie rozpuszczają
się w sobie, bądź tworzą roztwory graniczne, a ponadto tworzą trwałą fazę międzymetaliczną
A
n
B
m
. Taka faza dzieli wykres na dwie nienależne części tak, że w rzeczywistości występują tu
jakby dwa odrębne układy równowagi: metalu A i fazy międzymetalicznej oraz fazy
międzymetalicznej i metalu
B, a każdy z tych układów może należeć do jednego z poprzednio
omówionych typów.
Niektóre fazy międzymetaliczne mogą rozpuszczać w stanie stałym składnik A lub B
względnie oba składniki, tworząc omówione już roztwory stałe wtórne. Wykres równowagi
odpowiadający temu przypadkowi podano na rys. 3.9b.
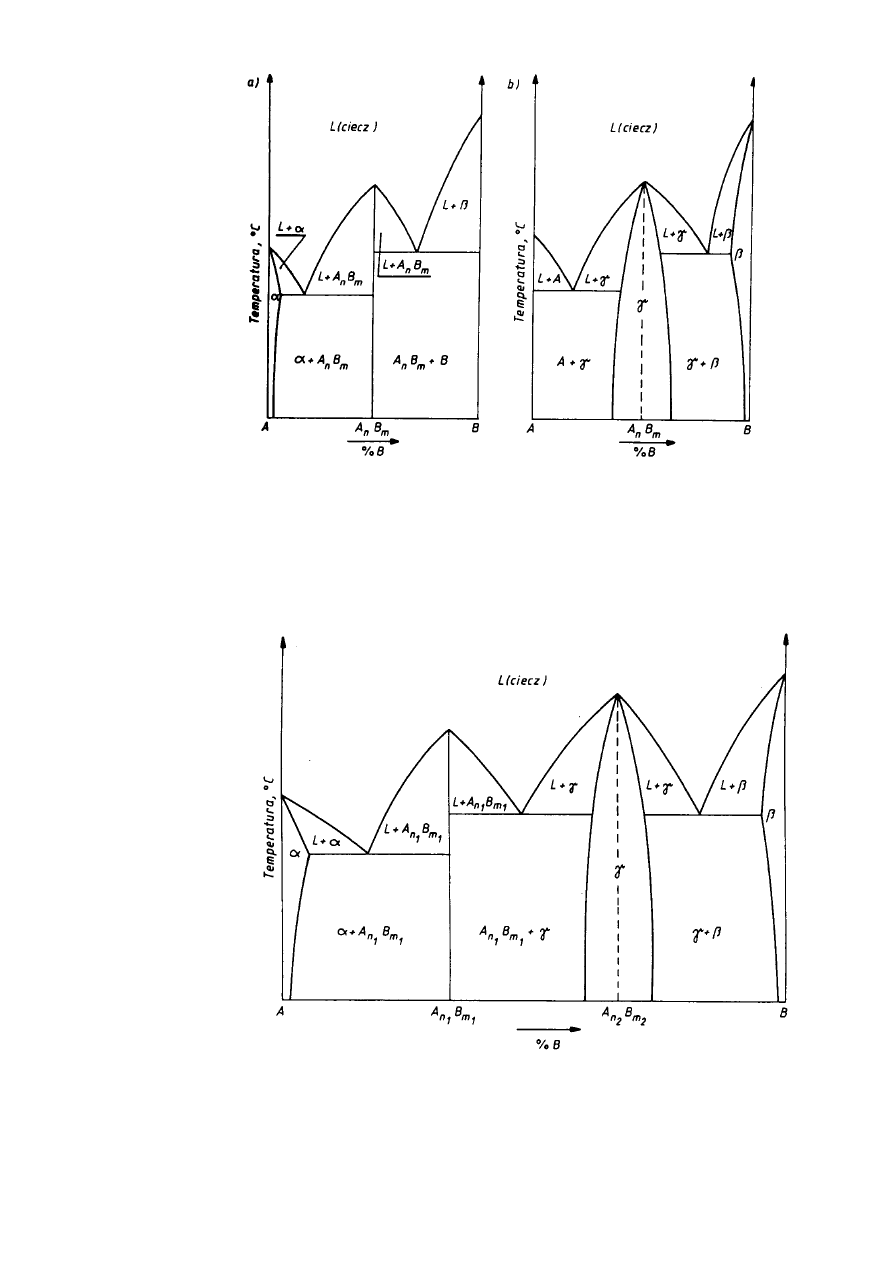
45
JW
Rys.3.9. Wykres równowagi z fazą międzymetaliczną krystalizującą z fazy ciekłej:
a) faza międzymetaliczna o stałym składzie stechiometrycznym,
b) faza międzymetaliczna w postaci wtórnego roztworu stałego
γ
Istnieją układy dwuskładnikowe, w których występują dwie (lub więcej) faz;
międzymetaliczne. Jeśli wszystkie te fazy tworzą się bezpośrednio z fazy ciekłej dzielą one
wykres na trzy, cztery lub więcej części, z których każda może być rozpatrywana samodzielnie
(rys. 3.10).
Rys. 3.10. Wykres równowagi z dwoma fazami międzymetalicznymi krystalizującymi
z fazy ciekłej
Jeśli faza międzymetaliczna krystalizuje poniżej temperatury początku krzepnięcia stopu, to
powstaje ona zwykle w wyniku reakcji perytektycznej. Wykres równowagi typu V
odpowiadający temu przypadkowi, może mieć różną postać w zależności od wzajemnego
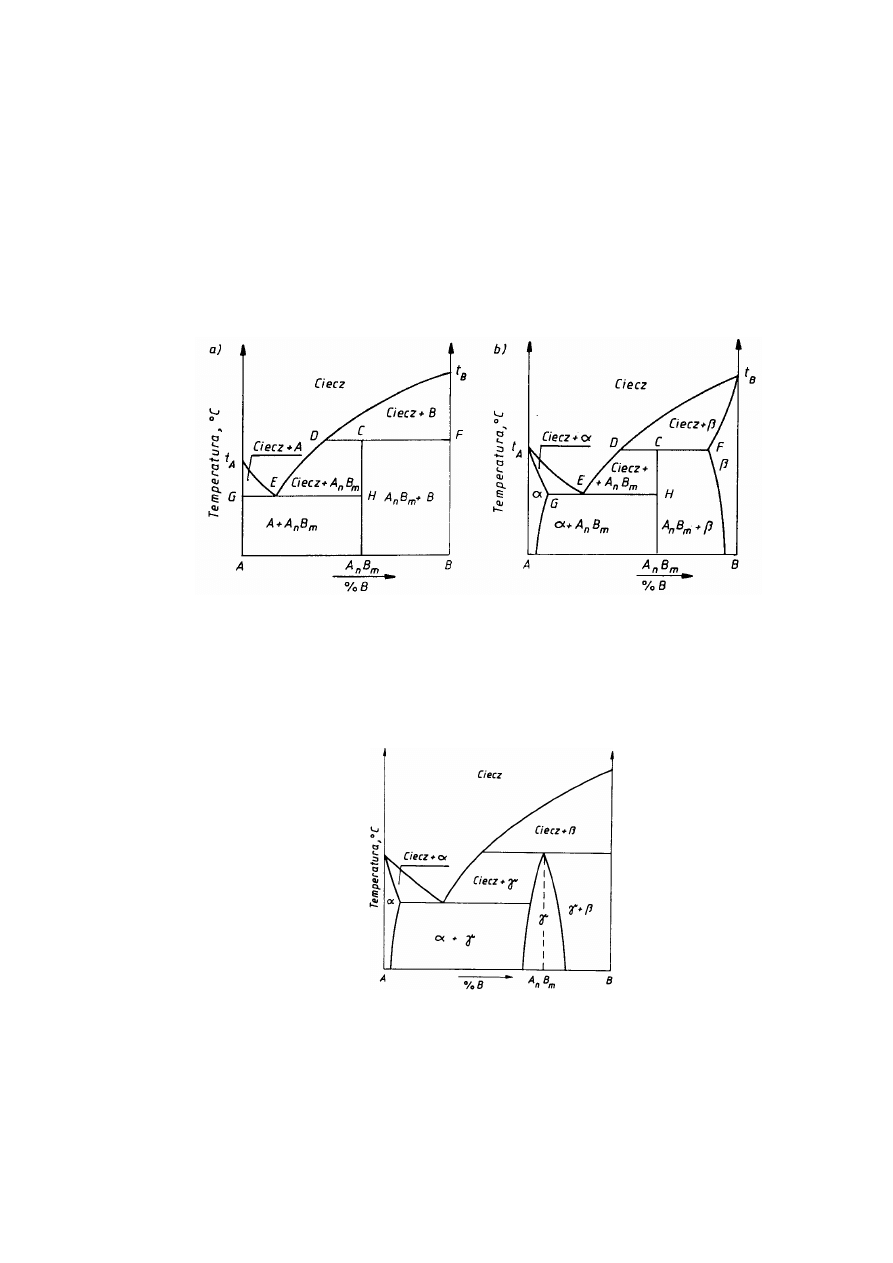
46
JW
oddziaływania składników stopu. Na rys. 3.ll a pokazano wykres równowagi dwóch składników
nie rozpuszczających się wzajemnie w stanie stałym, a na rys. 3.1I b - wykres równowagi dwóch
składników tworzących roztwory graniczne.
W pierwszym przypadku faza międzymetaliczna powstaje w wyniku reakcji perytektycznej
zgodnie ze wzorem
L
D
+B
→A
n
B
m
,
gdzie: L
D
- ciecz o składzie punktu D.
W drugim przypadku reakcja perytektyczna zachodzi wg wzoru
L
D
+
β
F
→
A
n
B
m
gdzie: L
D
- ciecz o składzie punktu D,
β
F
— roztwór stały A w B o składzie punktu F
Rys. 3.11. Wykresy równowagi z fazą międzymetaliczną powstającą w wyniku reakcji
perytektycznej: a) przy braku wzajemnej rozpuszczalności składników w stanie
stałym, b) przy ograniczonej rozpuszczalności składników w stanie stałym
Podobnie jak w przypadku wykresów równowagi typu IV, również na wykresach równowagi
typu V faza międzymetaliczna może występować w postaci roztworu wtórnego
γ (rys. 3.12).
Rys. 3.12. Wykres równowagi z fazą międzymetaliczną w postaci wtórnego roztworu
stałego
γ, powstającą w wyniku reakcji perytektycznej
Omówione
dotychczas
podstawowe wykresy równowagi dotyczyły układów
dwuskładnikowych, w których nie występowały żadne przemiany w stanie stałym. W
rzeczywistych układach przemiany takie występują jednak dość często i są związane z
tworzeniem się fazy międzymetalicznej z innej fazy stałej, bądź z przemianami alotropowymi
jednego lub obu składników stopu.
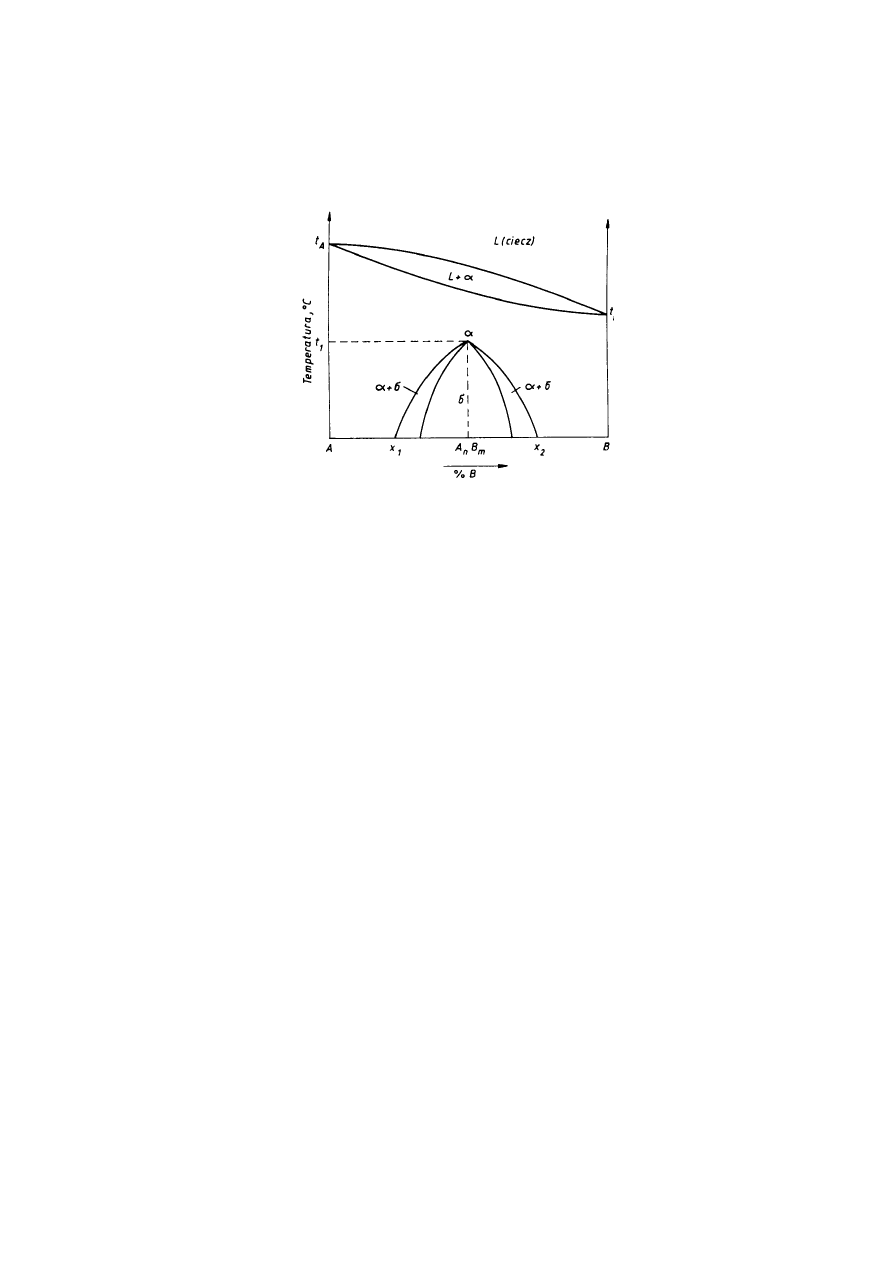
47
JW
Wykres równowagi z fazą międzymetaliczną tworzącą się w stanie stałym przedstawiono na
rys. 3.13. Ja widać, dla pewnego zakresu składów (od x
1
do x
2
) przy obniżaniu temperatury
zachodzi przemiana roztworu stałego
α w fazę międzymetaliczną σ, będącą roztworem stałym
wtórnym obu składników A i B w fazie międzymetalicznej A
n
B
m
. W stopie o składzie A
n
B
m
przemiana ta zachodzi w stałej temperaturze t
1
, w pozostałych stopach podlegających przemianie
— w pewnym zakresie temperatury
Rys.3.13. Wykres równowagi z fazą międzymetaliczną krystalizującą z fazy stałej
Oprócz krystalizacji pierwotnej związanej z krzepnięciem stopu i krystalizacji; wtórnej
związanej bądź ze zmianą rozpuszczalności składników w funkcji temperatury, bądź z
tworzeniem się w stanie stałym fazy międzymetalicznej w niektórych stopach występuje
przekrystalizowanie w stanie stałym, związane z przemianą alotropową przynajmniej jednego ze
składników tworzących stop. Warto przypomnieć, że przemianom alotropowym podlegają m.in.
żelazo, kobalt, mangan, tytan, cyrkon, cyna i inne.
Postać wykresu równowagi zależy w tym przypadku od rodzaju wzajemnego oddziaływania
między poszczególnymi odmianami alotropowymi składników. Najbardziej typowe odmiany
wykresów równowagi z przemianami alotropowymi zostaną omówione poniżej.
l. Przyjmując, że składnik A ma dwie odmiany alotropowe A
α
i A
β
, a składnik B odmian
alotropowych nie ma oraz że odmiana A
α
(istniejąca w niższych temperaturach) i składnik B są
izomorficzne, czyli mogą tworzyć roztwór stały ciągły, otrzymuje się wykres równowagi
przedstawiony na rys. 3.14. Jak widać, wszystkie stopy niezależnie od ich składu chemicznego w
temperaturze otoczenia mają budowę jednofazową
α. Różnią się jedynie sposobem powstania tej
fazy, a mianowicie: stopy o składzie od A do x
1
krzepną jako stopy
β, a faza α powstaje w
wynika przemiany alotropowej w stanie stałym; w stopach o składzie od x
1
do x
2
faza
α
powstaje w wyniku reakcji perytektycznej; w stopach o składzie od x
2
, do B faza
α powstaje
bezpośrednio z fazy ciekłej.
2. Przyjmując, że składnik A ma dwie odmiany alotropowe A
α
i A
β
, a składnik B odmian
alotropowych nie ma oraz że odmiana A
β
i składnik B są izomorficzne. otrzymuje się wykres
przedstawiony na rys. 3.15. W tym przypadku wszystkie stopy bezpośrednio po skrzepnięciu
składają się z roztworu stałego
β. Strukturę tę zachowują do temperatury otoczenia jednak tylko
stopy o składzie od x
2
do B. Pozostałe stopy podlegają całkowicie (o składzie od A do x
1
,) lub
częściowo o składzie od x
1
do x
2
) przemianie alotropowej
β w . Dzięki temu w temperaturze
otoczenia, w zależności od składu chemicznego, można uzyskać jednofazowe: stopy
α,
dwufazowe stopy
α + β bądź jednofazowe stopy β.
3. Przyjmując, że składnik A ma dwie odmiany alotropowe A
α
i A
β
, a składnik B dwie
odmiany alotropowe B
α
i B
β
oraz że odmiany A
α
i B
α
oraz odmiany A
β
i B
β
. są izomorficzne,
otrzymuje się wykres równowagi przedstawiony na rys. 3.16. Jak widać, wszystkie stopy po
skrzepnięciu mają strukturę B, wszystkie też podlegają w niższych temperaturach przemianie
alotropowej
β w α.
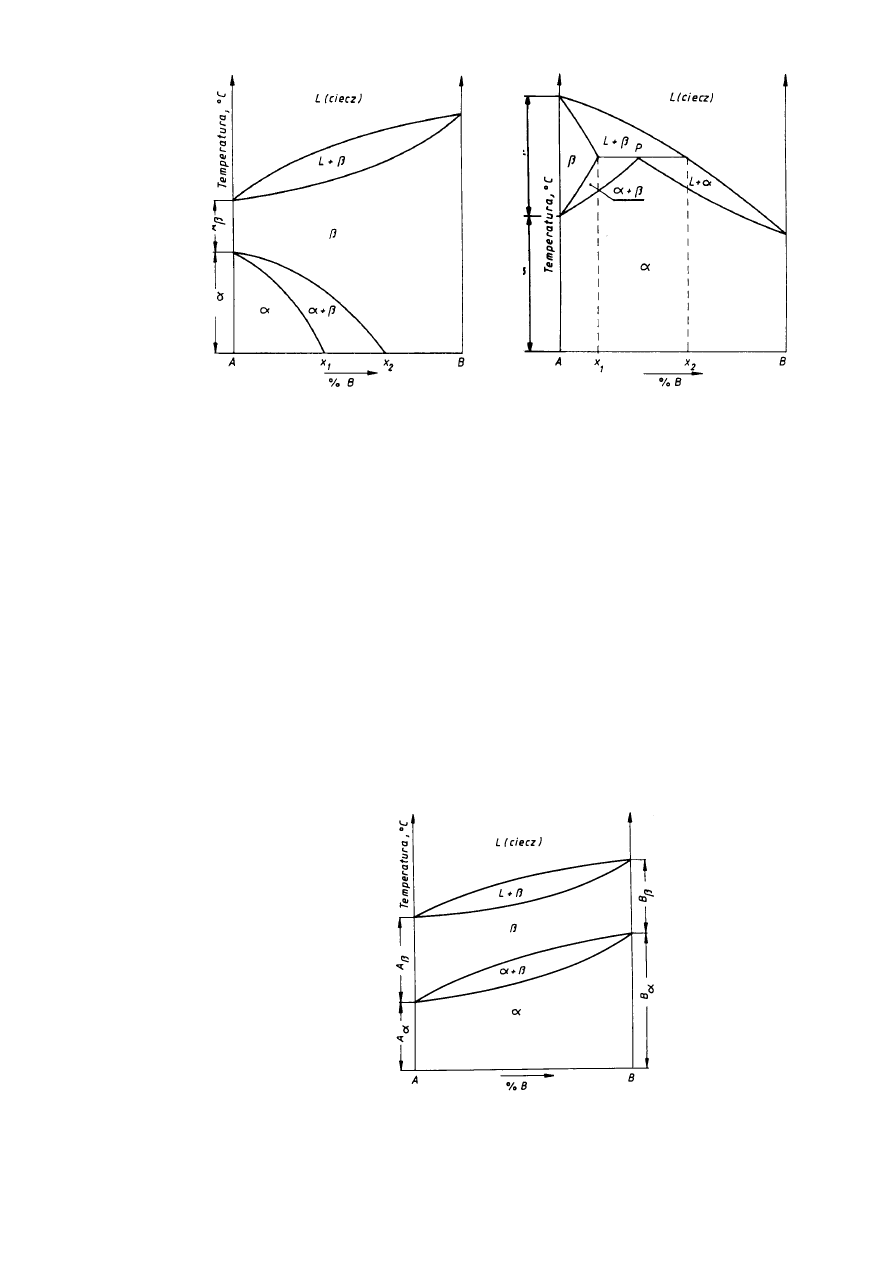
48
JW
Rys. 3.14. Wykres równowagi z przemianą Rys. 3.15. Wykres równowagi z przemianą
alotrotropową składnika A (odmiana A
α
i
alotropową składnika A (odmiana A
β
i
składnik B są
izomorficzne)
składnik B są są izomorficzne
4. Przyjmując, że składnik A ma dwie odmiany alotropowe A
α
i A
β
, a składnik B dwie
odmiany alotropowe B
α
i B
β
oraz że odmiany A
α
i B
β
są izomorficzne, a odmiany A
α
i tworzą
roztwory stałe graniczne, otrzymuje się wykres równowagi przedstawiony na rys. 3.17. W tym
przypadku, bezpośrednio po skrzepnięciu wszystkie stopy mają również strukturę
β, ale w
temperaturze otoczenia ich struktura, zależnie od składu chemicznego, składa się bądź z
kryształów roztworu stałe-
α
1
składnika B
α
w A
α
, bądź z kryształów roztworu stałego
α
2
składnika A
α
w B
α
, bądź z dwufazowej mieszaniny tych kryształów. Temperatury początku
przemiany alotropowej wyznacza linia RES, temperatury końca tej przemiany - linia RMENS. W
temperaturze wyznaczonej linią MEN zachodzi rozkład roztworu stałego
β o składzie punktu E
na dwa roztwory stałe graniczne
α
1
i
α
2
, tworzące mieszaninę zwaną eutektoidem:
β
E
→α
1
+
α
2
Reakcja tego rozkładu nazywa się reakcją eutektoidalną, stała temperatura t
E
, w której
zachodzi - temperaturą eutektoidalną, a stop o składzie e - stopem eutektoidalnym. Wszystkie
stopy o składzie od A do e nazywają się stopami podeutektoidalnymi, a stopy o składzie od e do
B stopami nadeutektoidalnymi
Rys. 3.16. Wykres równowagi z przemianą alotropową obu składników (odmiana A
α
i B
α
oraz A
β
i B
β
są izomorficzne)
Z powyższego wynika, że stopy podeutektoidalne o składzie od A do m i nadeutektoidalne o
składzie od n do B nie podlegają reakcji eutektoidalnej, a roztwory
α
1
i
α
2
tworzą się
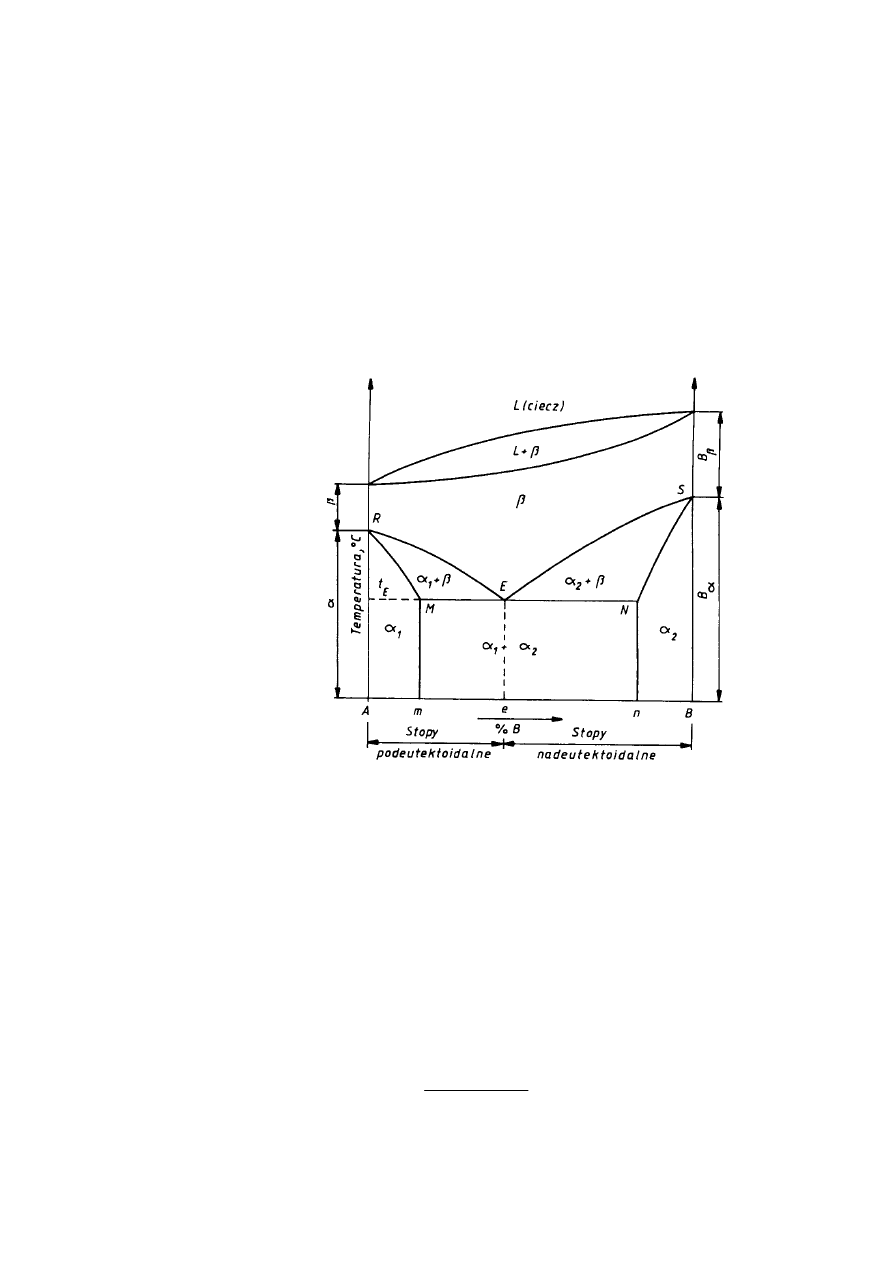
49
JW
bezpośrednio z fazy
β w odpowiednim zakresie temperatury. W stopach o składzie od m do e z
fazy
β wydzielają się najpierw kryształy faz α
1
, bogatej w składnik A (o składzie określonym
linią RM), dzięki czemu faza
β wzbogaca się w składnik B do momentu, gdy osiągnie skład
punktu E. Wtedy reszta fazy
β ulega eutektoidalnemu rozkładowi na mieszaninę bardzo
drobnych kryształów
α
1
i
α
2
. Podobnie w stopach od e do n, z fazy
β wydzielają się najpierw
kryształy fazy
α
2
, bogatej w składnik B (o składzie określonym linią SN) dzięki czemu faza
β
ubożeje w składnik B do momentu, gdy osiągnie skład punktu E i ulegnie eutektoidalnemu
rozkładowi.
Zawartość eutektoidu w stopie zmienia się liniowo, od 0 dla stopów o składzie m i n do 100%
dla stopu eutektoidalnego.
Przedstawiony na rys. 3.17 wykres równowagi dotyczy przypadku stałej rozpuszczalności A
α
w
B
α
i odwrotnie. W rzeczywistości zwykle rozpuszczalność ta zmienia się z temperaturą i wtedy
wykres równowagi przybiera postać pokazaną na rys. 3.18.
Rys. 3.17. Wykres równowagi z przemianą alotropową obu składników (odmiany A
β
i B
β
są izomorficzne, a odmiany A
α
i B
α
tworzą roztwory stałe graniczne o
niezmiennej rozpuszczalności)
Na wykresie tym podano również schematy struktur stopów podeutektoidalnych,
eutektoidalnego i nadeutektoidalnych.
Spotykane w praktyce wykresy równowagi układów dwuskładnikowych rzadko odpowiadają
omówionym typowym układom prostym, z reguły będąc kombinacją dwóch lub więcej typów
układów. Skład procentowy stopów na wykresach równowagi wyraża się zwykle w procentach
wagowych, co w przypadku stopów technicznych jest korzystniejsze. Niekiedy jednak,
zwłaszcza w badaniach strukturalnych, wygodniej jest przedstawić układ chemiczny stopów w
procentach atomowych. Zależność między procentami wagowymi a procentami atomowymi
określają wzory:
%
100
2
2
1
1
1
1
1
G
a
G
a
G
a
C
⋅
+
⋅
⋅
⋅
=
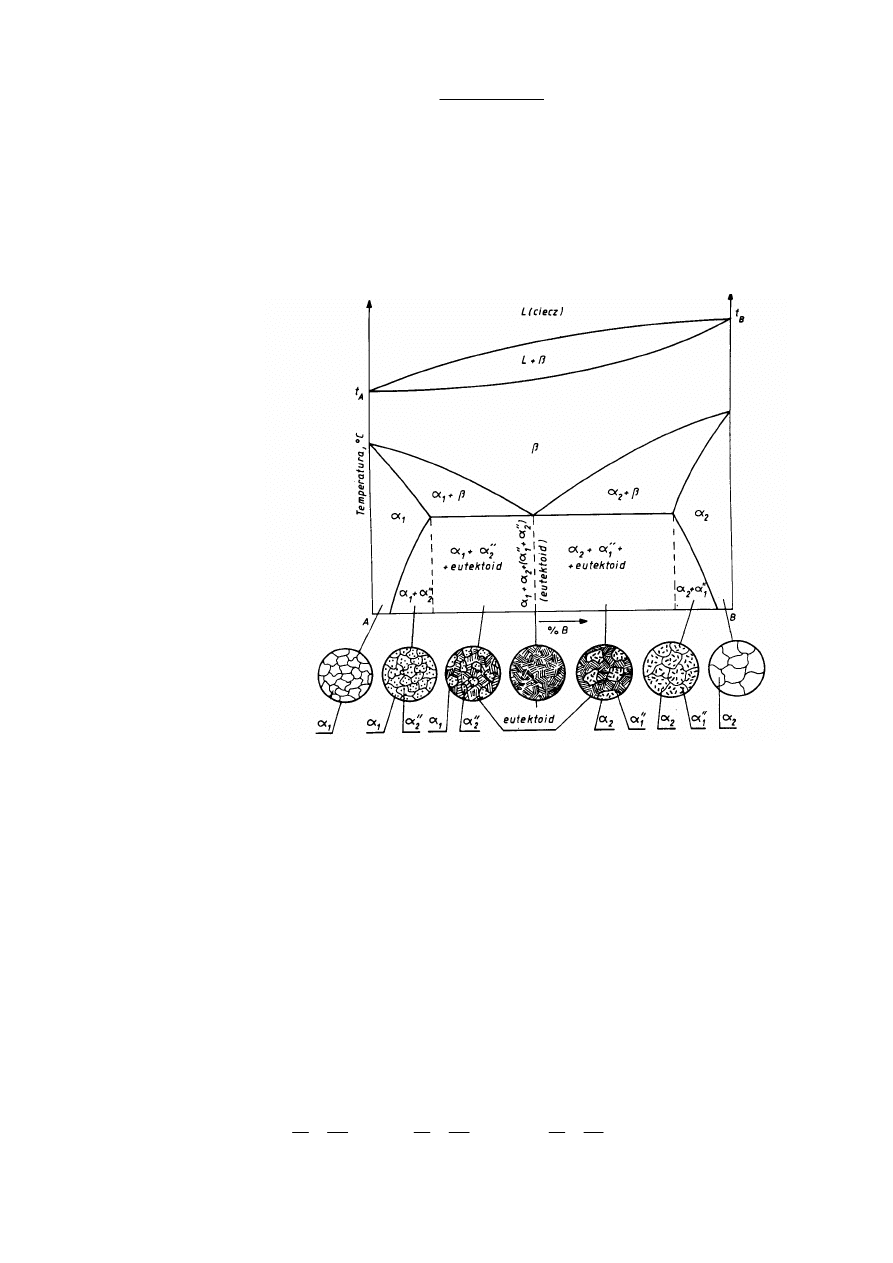
50
JW
gdzie: C
1
- zawartość składnika A w % wagowych,
C
2
- zawartość składnika B w % wagowych,
α
1
, - zawartość składnika A w % atomowych,
α
2
-
zawartość składnika B w% atomowych,
G
1
- ciężar atomowy składnika A,
G
2
- ciężar atomowy składnika B.
Rys. 3.18. Wykres równowagi z przemianą alotropową obu składników (odmiany A
β
i B
β
są izomorficzne, a odmiany A
α
i B
α
tworzą roztwory stale graniczne o zmiennej
rozpuszczalności) oraz schematy struktur poszczególnych grup stopów.
3. 4. Reguła dźwigni
W procesie krystalizacji zarówno pierwotnej, jak i wtórnej (w stanie stałym zmienia się nie
tylko skład poszczególnych faz, ale i ilość każdej fazy. W dowolnym punkcie wykresu
równowagi można w obszarze jednoczesnego występowania) dwóch faz określić ich ilość oraz
ich skład chemiczny. Wykorzystuje się do tego celu tzw. regułę dźwigni.
Przykład zastosowania reguły dźwigni podano na rys. 3.19. Jak widać, aby określić skład
chemiczny współistniejących faz w danej temperaturze t
y
, należy dla tej temperatury
przeprowadzić linię równoległą do osi składów. Rzuty punktów przecięcia tej linii z krzywymi
ograniczającymi obszar dwufazowy na oś składów, określają skład faz. Na rysunku przez x
oznaczono skład stopu, przez x
1
— skład fazy stałej
α, a przez x
2
- skład fazy ciekłej.
Ilościowy stosunek faz określa się za pomocą wzorów:
%
100
2
2
1
1
2
2
2
G
a
G
a
G
a
C
⋅
+
⋅
⋅
⋅
=
bc
ab
r
r
s
c
=
ac
ab
r
r
c
=
ac
bc
r
r
s
=
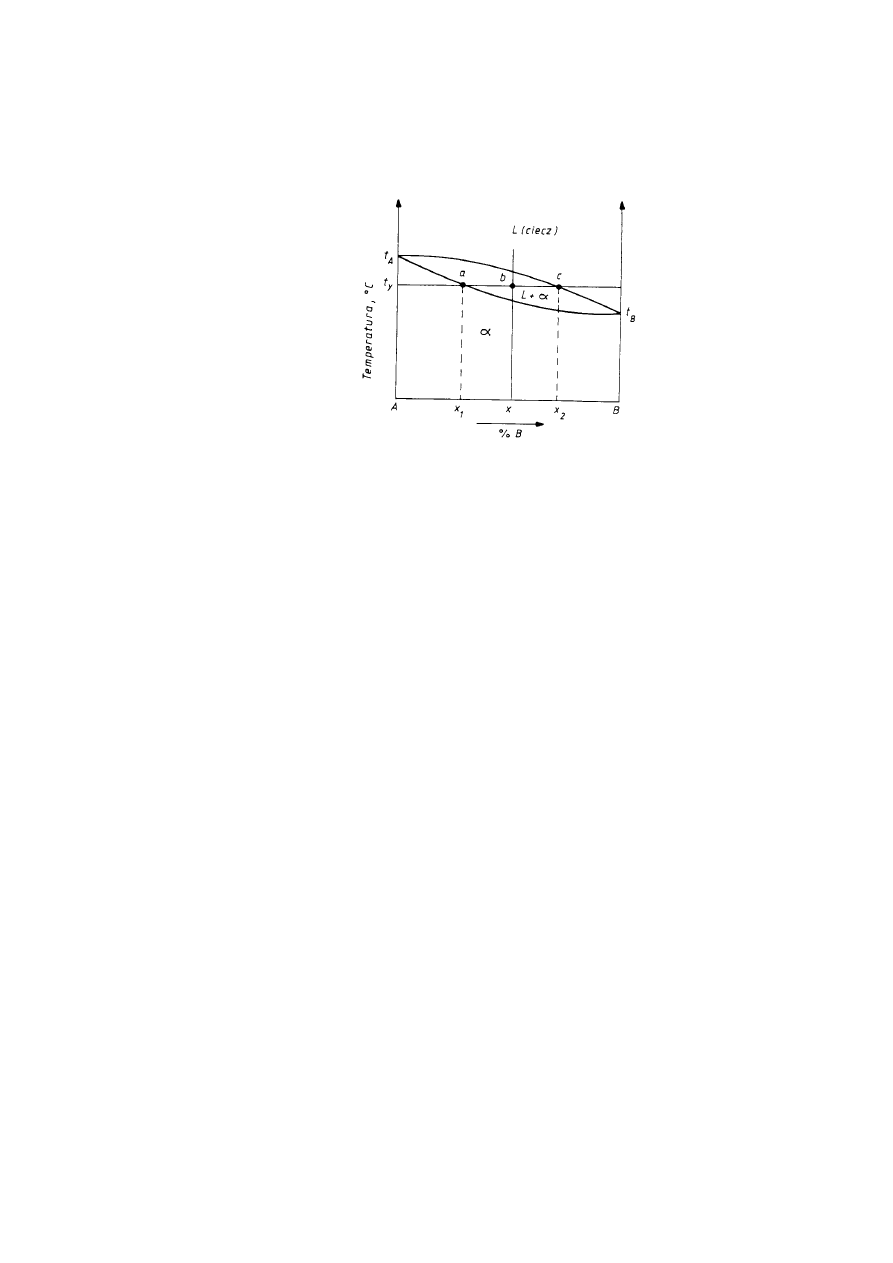
51
JW
gdzie: r
c
— ilość fazy ciekłej,
r
s
- ilość fazy stałej,
r = r
c
+ r
s
— ogólna ilość stopu,
ab, bc i ac — odcinki wyznaczone przez linię t
y
oraz likwidus i solidus
Rys. 3.19. Przykład zastosowania reguły dźwigni w przypadku układu równowagi o
nieograniczonej rozpuszczalności w stanie stałym
3. 5. Budowa stopów potrójnych
Budowa stopów potrójnych jest znacznie bardziej skomplikowana niż stopów podwójnych i
aby graficznie przedstawić zależność struktury od składu chemicznego i temperatury, trzeba
operować modelem przestrzennym albo jego rzutami na płaszczyznę podstawy. Podstawę tego
modelu stanowi trójkąt równoboczny, zwany trójkątem Gibbsa, którego wierzchołki
przedstawiają czyste składniki układu potrójnego A, B i C, boki odpowiadają składom stopów
układów podwójnych A-B, B-C i C-A, a punkty wewnątrz trójkąta reprezentują składy stopów
układu potrójnego A-B-C (rys. 3.20).
Na osiach prostopadłych do podstawy oznaczona jest temperatura. Aby określić skład
dowolnego punktu N podstawy, przeprowadza się z niego równoległe do dwóch boków trójkąta
aż do przecięcia z trzecim; wówczas odcinek CD odpowiada procentowej zawartości składnika
B, odcinek DF — składnika A i odcinek FB -składnika C. Na rysunku 3.20b punkt N
przedstawia stop trzyskładnikowy o składzie: 50% A, 25% B i 25% C.
W układach potrójnych, zamiast linii likwidusu i solidusu, występują powierzchnie likwidusu
i solidusu, a zamiast poziomej linii eutektycznej — płaszczyzna eutektyczna odpowiadająca
potrójnej eutektyce, której temperatura topnienia jest niższa od każdej z trzech eutektyk
dwuskładnikowych. Ponieważ składniki układów potrójnych mogą parami tworzyć rozmaite
przypadki, ilość możliwych typów układów jest bardzo duża.
Wynika stąd, że przestrzenne wykresy równowagi układów trzyskładnikowych są niezbyt
przejrzyste i stąd niewygodne w praktycznym stosowaniu. Dlatego zwykle wykorzystuje się
różne ich rzuty na płaszczyznę trójkąta Gibbsa. Najczęściej stosowany jest tzw. płaski wykres
równowagi, powstający przez przecięcie wykresu przestrzennego płaszczyznami izotermicznymi
(równoległymi do podstawy, odległymi od siebie o stałą liczbę stopni Celsjusza, np. 25 lub 50) i
zrzutowanie otrzymanych izoterm na powierzchnię likwidusu i solidusu, jak również
ewentualnych krzywych i punktów eutektycznych, perytektycznych itp., na płaszczyznę trójkąta
Gibbsa. Dla większej przejrzystości wykresu najczęściej zaznacza się na nim tylko izotermy
likwidusu. Przykładowy płaski wykres równowagi trzyskładnikowy z zaznaczonymi izotermami
likwidusu oraz rzutami eutetyki przedstawiono na rys. 3.20c.
Trzeba jednak podkreślić, że płaskie wykresy trzyskładnikowe podają tylko warunki zmiany
stanu skupienia, tj. warunki topnienia i krzepnięcia stopów, nie odtwarzając przemian fazowych
w stanie stałym. Oczywiście, stopy cztero- i więcej składnikowe mają budowę jeszcze bardziej
złożoną.
52
JW
Rys. 3.20. Wykres równowagi trzech składników: a) przestrzenny, b) na płaszczyźnie z
kładami wykresów równowagi dwuskładnikowych, c) płaski
3.6. Wpływ struktury na własności stopów
Jak wynika z omówionych układów równowagi fazowej, struktura stopów może być
jednofazowa, czyli złożona z ziarn jednego rodzaju, lub może stanowić mieszaninę dwóch lub
więcej faz (w stopach wieloskładnikowych).
W przypadku stopów o budowie jednofazowej głównymi czynnikami wpływającymi na ich
własności są:
a) skład chemiczny ziarn,
b) wielkość i kształt ziarn,
c) budowa granic ziarn,
d) rodzaj, ilość, wielkość, kształt i rozmieszczenie zanieczyszczeń.
W stopach dwu- i wielofazowych oprócz powyższych czynników o własnościach decydują:
• skład chemiczny współistniejących faz,
• własności tych faz,
• udział procentowy poszczególnych faz,
• wzajemne usytuowanie faz.
Rola wymienionych czynników w kształtowaniu poszczególnych własności stopów jest
różna. Na przykład własności fizyczne, a więc m.in. przewodność elektryczna i cieplna, gęstość,
ciepło właściwe i rozszerzalność cieplna, zależą przede wszystkim od składu chemicznego faz i
ich własności oraz udziału procentowego faz w stopie.
Własności mechaniczne są funkcją nie tylko czynników charakteryzujących strukturę, ale
również wielkość ziarn i ich kształtu, sposobu rozłożenia faz itd.

53
JW
Innym czynnikiem istotnie wpływającym na własności mechaniczne stopów jest wielkość
wydzieleń fazy międzymetalicznej. Im są one drobniejsze i bardziej równomiernie rozłożone,
tym bardziej stop jest wytrzymały i twardy. Na przykład twardość stali węglowych w miarę
zmniejszania się grubości płytek cementytu od 2 do 0,5 µm wzrasta od 220 do 350 HB.
W przypadku sferoidalnych wydzieleń cementytu twardość stali węglowych wzrasta od 190 do
400 HB, w miarę zmniejszania się ich średnicy od 2 do 0,2 µm. Podobnie wzrasta wytrzymałość
tych stali, jednocześnie pogarsza się ich plastyczność.
Własności mechaniczne stopu zależą także od kształtu wydzieleń fazy międzymetalicznej.
Wiadomo na przykład, że stop zawierający wydzielenia twardej i kruchej fazy międzymetali-
cznej w postaci sferoidów jest mniej wytrzymały i twardy, ale bardziej plastyczny, niż stop, w
którym ta sama faza występuje w postaci płytek albo siatki na granicach ziarn.
Istotnym zagadnieniem w przypadku stopów dwu- i wielofazowych jest anizotropia
własności, wywołana ukierunkowanym rozłożeniem jednej z faz. Na rysunku 3.21 pokazano
strukturę stali niskowęglowej, w której perlit tworzy wyraźnie pasma ułożone zgodnie z
kierunkiem przeróbki plastycznej. Wytrzymałość na rozciąganie tej stali jest mniejsza w
kierunku zgodnym z ułożeniem pasm perlitu, większa w kierunku prostopadłym. Natomiast
plastyczność zmienia się odwrotnie
Rys. 3.21. Pasmowa struktura stali niskowęglowej, wywołana przeróbką plastyczną.
Widoczne jasne ziarna ferrytu i ciemne perlitu (traw. 5% HN03), 100x
W niektórych przypadkach osnowa stopu wielofazowego jest bardziej twarda i wytrzymała od
fazy tworzącej wydzielenia. Również i wtedy wytrzymałość stopu jest związana z wielkością i
kształtem tych wydzieleń. Przykładem może tu być żeliwo szare, w którym wydzieloną fazę
stanowi grafit. Im drobniejsze i bardziej zbliżone kształtem do kulistych są wydzielenia grafitu,
tym bardziej żeliwo jest wytrzymałe, niezależnie od rodzaju struktury osnowy (ferrytycznej,
ferrytyczno-perlitycznej, czy perlitycznej). Wytrzymałość grafitu w porównaniu z
wytrzymałością osnowy można przyjąć za zerową, toteż jego wydzielenia zmniejszają ogólną
wytrzymałość stopu, gdyż zmniejszają czynny przekrój. Ponadto działają jako koncentratory
naprężeń, tym groźniejsze, im bardziej różnią się kształtem od kuli.
Poważny wpływ na własności mechaniczne metali i stopów wywierają także wszelkie defekty
materiałowe, jak pory gazowe, zażużlenia, wtrącenia niemetaliczne, mikropęknięcia itd.
Stanowią one nieciągłości materiału zmniejszające czynny przekrój elementów konstrukcyjnych,
osłabiając ogólną wytrzymałość, a w wielu przypadkach stanowiące bardzo groźne
koncentratory naprężeń mogące doprowadzić do przedwczesnego zniszczenia części maszyn
przy obciążeniach nie przekraczających dopuszczalnych.
W metalach i stopach obok różnych składników i zanieczyszczeń występują również gazy.
Nawet metale o wysokiej czystości zawierają niewielkie ilości azotu, tlenu i wodoru, a w wielu
przypadkach nawet te niewielkie ilości istotnie wpływają na niektóre własności fizyczne i
mechaniczne metalu. Na przykład, już 0,015% tlenu wyraźnie obniża przewodnictwo
elektryczne i plastyczność miedzi.
Gazy dostają się do metalu zarówno podczas metalurgicznych procesów jego wytwarzania,
obróbki cieplnej w atmosferach utleniających, obróbki chemicznej i elektrochemicznej czy
spawania, jak i podczas eksploatacji w środowiskach agresywnych, zwłaszcza przy
podwyższonych ciśnieniach i temperaturach. Mogą one tworzyć roztwory stałe i związki
54
JW
chemiczne, mogą też wypełniać pęcherze, pory lub inne wewnętrzne wady materiałowe. W stali
w temperaturze otoczenia tlen i azot rozpuszczają się w niewielkim stopniu, tworzą natomiast
liczne tlenki i azotki. Wodór w stali również rozpuszcza się w niewielkim stopniu, a jego
nadmiar powoduje powstawanie pęcherzy.
Gazy rozpuszczone w metalu przeważnie podwyższają jego wytrzymałość, a obniżają
plastyczność. Dla przykładu, atomy azotu rozpuszczonego w żelazie zwykle grupują się wokół
dyslokacji, tworząc tzw. atmosfery Cottrella. Atmosfery te unieruchamiają dyslokacje,
zwiększając tym samym wytrzymałość metalu.
Tlenki i azotki stanową tzw. wtrącenia niemetaliczne. Nazwa ta określa znajdujące się w
każdej stali produkty reakcji fizykochemicznych, zachodzących podczas jej wytapiania (oprócz
tlenków i azotków należą tu siarczki i krzemiany) oraz wtrącenia materiałów ogniotrwałych
używanych do wyłożenia pieców stalowniczych, kadzi itp. Ilość, wielkość, skład chemiczny i
fazowy oraz rozłożenie wtrąceń niemetalicznych wpływają na wiele własności użytkowych stali,
a zależą m.in. od metody wytopu, rodzaju i ilości użytych odtleniaczy, temperatury procesu
stalowniczego, zawartości siarki i fosforu w stali itd.
a)
b)
Rys. 3.22. Złożone wtrącenia niemetaliczne w stali: a) siarczkowo-tlenkowe, b) siarczko-
wo-tlenkowe z wrostkami azotków, c) krzemianowe (nietrawione). Powiększ. 500x
Wtrącenia niemetaliczne bardzo rzadko są prostymi związkami chemicznymi. Najczęściej
stanowią złożone związki lub mieszaniny związków prostych (rys. 3.22). Szczególnie szkodliwe
są wtrącenia twarde i kruche, będące złożonymi spinelami lub zawierające dużo tlenku krzemu.
Te ostatnie często mają kształt zbliżony do kuli (rys. 3.23). Szkodliwość wtrąceń
niemetalicznych wzrasta ponadto przy ich nierównomiernym rozmieszczeniu w metalu i jest tym
większa, im są one większe (rys. 3.24)
Również wodór obecny w stali jest składnikiem zdecydowanie szkodliwym. Jak wspomniano,
tylko niewielkie ilości wodoru rozpuszczają się w żelazie międzywęzłowe, reszta gromadzi się w
postaci gazowej na wszelkiego rodzaju defektach strukturalnych, takich jak granice ziarn,
granice faz, dyslokacje czy skupienia wakansów, wywołując porowatość, powstawanie i rozwój
pęcherzy i pęknięć, rozwarstwianie metalu, pojawianie się lokalnych odkształceń sieci
krystalicznej oraz stref metalu o zmienionym składzie chemicznym, powstawanie płatków
śnieżnych itd. Wszystkie te wady materiałowe mogą powstawać zarówno w procesie
wytwarzania stali i jej przetwórstwa, jak i w czasie jej eksploatacji w środowiskach
agresywnych, zwłaszcza przy podwyższonych ciśnieniach i temperaturach. Prowadzą one do
gwałtownego spadku własności plastycznych stali, co ogólnie określa się terminem kruchości
wodorowej.
Jak widać z powyższego, krótkiego zresztą przeglądu, liczba czynników strukturalnych
kształtujących rzeczywiste własności metali i stopów jest olbrzymia. Poznanie tych czynników i
umiejętne nimi kierowanie jest podstawowym warunkiem uzyskiwania optymalnych dla danego
zastosowania własności materiałów.

55
JW
Rys. 3.23. Globularne wtrącenia złożone (zawierające głównie tlenek krzemu): a)
widoczne na zgładzić metalograficznym, b), c) i d) ujawnione na przełomie
stalowej próbki udarnościowej (SEM - nietrawione. Powiększ. 500x
Rys. 3.24. Wtrącenia niemetaliczne różnej wielkości i o różnym rozłożeniu: a) bardzo
drobne tlenki i siarczki rozłożone mniej więcej równomiernie, b) różnej
wielkości tlenki i siarczki rozłożone nierównomiernie, c) pojedyncze duże
wtrącenia siarczkowe wydłużone w czasie przeróbki plastycznej oraz nieliczne
drobne wtrącenia niemetaliczne rozłożone równomiernie, nie trawione).
Powiększ. 200

56
JW
4. STOPY ŻELAZA Z WĘGLEM
4.1. Charakterystyka żelaza
Żelazo jest pierwiastkiem metalicznym o temperaturze topnienia 1534°C i temperaturze
wrzenia 3070°C. W przyrodzie występuje głównie w postaci tlenków, węglanów,
wodorotlenków i siarczków, jako magnetyt (Fe
3
O
4
), hematyt (Fe
2
O
3
), syderyt (FeCO
3
,), limonit
(2Fe
2
O
3
⋅3H
2
O) i piryt (FeS
2
).
Z rud tlenkowych w redukcyjnym procesie hutniczym w wielkim piecu otrzymuje się tzw.
surówkę, będącą stopem żelaza z węglem, krzemem, manganem, siarką, fosforem, tlenem,
azotem i in. (łącznie do 10%). Surówka podlega dalszej przeróbce w plecach stalowniczych,
podczas której utlenia się znaczna część domieszek, tak że w większości przypadków łączna ich
ilość (nie licząc węgla) nie przekracza 1%. Otrzymany produkt nazywa się stalą węglową.
Żelazo występuje w dwóch odmianach alotropowych:
α i γ.
Żelazo
α, termodynamicznie trwale od niskich temperatur do temperatury 910°C oraz od
temperatury 1390 do 1534°C, ma strukturę krystaliczną o sieci regularnej przestrzennie
centrowanej. Warto wspomnieć, że wysokotemperaturową odmianę żelaza
α często nazywa się
żelazem
δ.
Żelazo
γ, termodynamicznie trwałe w temperaturach 910 do 1390°C, ma strukturę krystaliczną
o sieci regularnej ściennie centrowanej.
Gęstość żelaza
α w temperaturze 20°C wynosi 7,86 g/cm
3
, gęstość żelaza
γ w temperaturze
916°C - 8,05 g/cm
3
.
Przemiany zachodzące w czystym żelazie podczas jego studzenia lub ogrzewania najlepiej
omówić posługując się krzywą studzenia. Jak widać na rys. 4.1, poza przystankiem w
temperaturze 1534°C, związanym z krzepnięciem żelaza, na krzywej występują jeszcze trzy
przystanki temperatury. Pierwszy z nich w temperaturze 1390°C odpowiada przemianie
alotropowej żelaza
α w żelazo γ. Drugi przystanek ma miejsce w temperaturze 910°C i
odpowiada przemianie alotropowej żelaza
γ w żelazo α. Trzeci wreszcie, znacznie krótszy
przystanek w temperaturze 768°C (punkt Curie) związany jest z przemianą magnetyczną żelaza
α (poniżej tej temperatury żelazo jest ferromagnetyczne, powyżej — paramagnetyczne).
Rys. 4.1. Krzywa studzenia żelaza
Przemiany alotropowe są związane z przebudową struktury krystalicznej, co powoduje zmianę
własności fizycznych, chemicznych i mechanicznych. W efekcie powstają inne odmiany tego
samego żelaza, noszące nazwę odmian alotropowych. W przeciwieństwie do tego, przy
przemianie magnetycznej zmieniają się jedynie niektóre własności elektryczne, magnetyczne i
cieplne, tak że jest ona szczególnym rodzajem przemiany, zupełnie różnym od alotropowej.
57
JW
4.2. Układ równowagi żelazo-cementyt
Stopy żelaza z węglem należą do najbardziej rozpowszechnionych stopów w technice. Można
je traktować pod wieloma względami jako stopy dwuskładnikowe, mimo że zawierają one
jeszcze zawsze niewielkie ilości manganu, krzemu. siarki, fosforu i innych pierwiastków
pochodzących z procesu metalurgicznego. W związku z tym struktury tych stopów w stanie
zbliżonym do równowagi (a więc w stanie wyżarzonym zupełnie) można rozpatrywać
korzystając z wykresu równowagi fazowej dwuskładnikowego układu żelazo-węgiel.
Istnieją dwa rodzaje układu żelazo-węgiel: układ stabilny i układ metastabilny (rys. 4.2).
Pierwszy z nich przedstawia równowagę układu żelazo-grafit, drugi — równowagę układu
żelazo-cementyt (węglik żelaza Fe
3
C). Ze względów praktycznych układ metastabilny (z
cementytem) jest rozpatrywany w zakresie zawartości węgla od 0% (czyste żelazo) do 6,6%
(cementyt). Ten układ ma zastosowanie przy analizowaniu przemian fazowych i struktur stali
węglowych.
Rys. 4.2. Wykres równowagi układu żelazo-węgiel; linie ciągłe przedstawiają równowagę
metastabilną układu żelazo-cementyt, linie przerywane — równowagę stabilnego
układu żelazo-grafit (wg Hansena,1958)
Zgodnie z omawianym wykresem, za stale węglowe uważa się wszystkie stopi żelaza z
węglem zawierające 0,02-2,06% C, przy czym górna granica tego zakresu odpowiada
maksymalnej rozpuszczalności węgla w żelazie
γ. Należy wyjaśnić, a stopy zawierające mniej
niż 0,02% C noszą nazwę żelaza technicznego, a stop o zawartości węgla większej od 2,06 -
nazwę żeliw.
Fazy występujące w układzie żelazo-cementyt. Ponieważ żelazo występuje w dwóch
odmianach alotropowych
α i γ, a ponadto tworzy z węglem roztwory stałe i fazę
międzymetaliczną Fe
3
C (cementyt), w układzie równowagi żelazo-cementyt (zależnie od
temperatury i zawartości węgla) istnieją następujące fazy ferryt, austenit, cementyt i ciekły
roztwór węgla w żelazie. Na rysunku 4.2 W poszczególnych polach wykresu oznaczono
następujące fazy (L — roztwór ciekły węgla w żelazie,
α — ferryt, γ — austenit oraz Fe
3
C).
Wykres układu równowagi żelazo-cementyt można podzielić na dwa obszary: a) obszar

58
JW
związany ze zmianą stanu skupienia, ograniczony od góry linią likwidusu ABCD, od dołu - linią
solidusu AHIJECF, b) obszar przemian w stanie stałym — poniżej linii solidusu.
Ferryt jest międzywęzłowym roztworem stałym węgla w żelazie
α. Oznacza się go bądź
symbolem Fe
α
(C), bądź krótko
α. Graniczna zawartość węgla w ferrycie w stanie równowagi
wynosi w temperaturze 20°C zaledwie 0,008% i wzrasta w temperaturze 723°C do 0,02% (punkt
P na wykresie). Natomiast ferryt wysokotemperaturowy może zawierać w temperaturze 1493°C
do 0,1% C.
Własności fizyczne i mechaniczne ferrytu są zbliżone do własności żelaza
α. Na przykład,
twardość ferrytu wynosi ok. 80 HB, R
m
- ok. 300 MPa, A
10
- ok. 40%, KCU2 - ok. 180 J/cm
2
.
Podobnie jak żelazo
α, ferryt jest ferromagnetyczny do temperatury 768°C.
Austenit jest międzywęzłowym roztworem węgla w żelazie
γ i oznaczony jest bądź symbolem
Fe
γ
(C), bądź literą
γ. Graniczna zawartość węgla w austenicie w temperaturze 1147°C wynosi
2,06% (punkt E na wykresie). W stopach żelaza z węglem w stanie równowagi austenit
występuje jedynie w temperaturach wyższych od 723°C. Natomiast w niektórych stalach
stopowych, zawierających np. nikiel lub mangan, austenit w stanie równowagi istnieje również
w temperaturach niższych.
Podobnie jak żelazo
γ, austenit jest paramagnetyczny. Odznacza się przy tym dużą
plastycznością, zwłaszcza przy niższej zawartości węgla. Również gęstość austenitu zależy od
zawartości węgla (średnio wynosi ona 8,1 g/cm3).
Cementyt, czyli węglik żelaza jest fazą międzymetaliczną o złożonej strukturze, krystalizującą
w układzie rombowym. Stosunek liczby atomów żelaza do atomów węgla wynosi 3:1 (Fe
3
C), co
odpowiada wagowej zawartości węgla 6,67%. W temperaturze do 210°C cementyt jest
ferromagnetyczny, powyżej tej temperatury — paramagnetyczny. Gęstość cementytu wynosi 6,9
g/cm3. Jest on fazą bardzo twardą (HB ok. 800) i bardzo kruchą.
Cementyt może tworzyć roztwory stałe różnowęzłowe, przy czym na miejsce atomów węgla
mogą wchodzić do jego sieci atomy azotu, zaś na miejsce atomów żelaza — atomy takich metali,
jak mangan, chrom, wolfram itd. Tak utworzone roztwory stale na osnowie sieci cementytu
noszą nazwę cementytu stopowego.
Warto podkreślić, że zgodnie z układem równowagi żelazo-cementyt, w temperaturze
otoczenia stopy żelaza z węglem do zawartości 0,008% C są jednofazowe (ferryt), natomiast
wszystkie stopy o zawartości węgla od 0,008 do 6,67% składają się z dwóch faz: ferrytu i
cementytu.
Pomijając omówione już przemiany zachodzące w czystym żelazie oraz przemiany w czystym
cementycie, w układzie żelazo-cementyt można wyróżnić następujące trzy podstawowe
przemiany, zachodzące w stałych temperaturach:
przemiana eutektyczna: L
C
→ γ
E
+ Fe
3
C,
przemiana perytektyczna: L
B
+
α
H
→ γ
J
przemiana eutektoidalna:
γ
S
→ α
P
+ Fe
3
C.
Szczególnie duże znaczenie praktyczne ma przemiana eutektoidalna, na której opiera się
obróbka cieplna stali.
Składniki strukturalne występujące w układzie żelazo-cementyt.
Składnikami strukturalnymi nazywa się pojedyncze fazy lub charakterystyczne ugrupowania
kilku faz, tworzące dany stop. Składniki te tworzą strukturę metalograficzną stopu, przy czym
struktura taka składa się z jednego lub więcej składników strukturalnych.
Jak więc widać, określenie struktura jest używane zarówno w odniesieniu do struktury
krystalograficznej, związanej z odpowiednim układem atomów, jak też struktury
metalograficznej, związanej z odpowiednim układem faz. Obecnie coraz częściej w odniesieniu
do struktury metalograficznej używa się pojęcia mikrostruktura. Należy również rozróżniać
określenia: składniki strukturalne i składniki układu. W przypadku układu żelazo-cementyt
składnikami układu są oczywiście żelazo i cementyt.

59
JW
Mikrostruktura metalu lub stopu jest przedmiotem badań metalograficznych, które
przeprowadza się na odpowiednio przygotowanych powierzchniach próbek. Przygotowanie
polega na wyszlifowaniu i wypolerowaniu (mechanicznym lub elektrolitycznym) wybranych
powierzchni próbek, (dzięki czemu otrzymuje się tzw. zgład metalograficzny) i następnie
wytrawieniu tej powierzchni odpowiednio dobranym odczynnikiem, chemicznie lub
elektrolitycznie.
Podczas wytrawiania zgładu odczynnik działa zwykle silniej na granice niż na powierzchnię
ziarn jednej fazy lub różnych faz. Po dłuższym trawieniu poszczególne ziarna danej fazy zostają
zaatakowane w różnym stopniu, zależnie od ich orientacji krystalograficznej i dlatego
przybierają różne zabarwienie.
Na ogół wskutek trawienia na powierzchni zgładu wytwarza się delikatny relief, pozostający
po wypłukaniu produktów reakcji chemicznych zachodzących podczas trawienia.
Do trawienia stali węglowych i żeliw najczęściej stosuje się 1-5% roztwór HNO
3
w alkoholu
etylowym. Stale o większej zawartości węgla oraz żeliwa można wytrawiać również 4%
alkoholowym roztworem kwasu pikrynowego.
Inny rodzaj trawienia, stosowany rzadziej, ma na celu wytworzenie na powierzchni określonej
fazy nalotu umożliwiającego jej identyfikację. Jako przykład można podać trawienie niektórych
stali i żeliw we wrzącym alkalicznym roztworze pikrynianu sodu, podczas którego na
cementycie tworzy się ciemna warstewka, umożliwiająca odróżnienie go od ferrytu.
Oprócz trawienia chemicznego, niekiedy stosuje się również trawienie elektrolityczne.
W stopach układu żelazo-cementyt, zależnie od zawartości węgla i od temperatury, mogą
występować następujące strukturalne składniki jednofazowe: ferryt, austenit i cementyt, oraz
składniki dwufazowe: perlit i ledeburyt.
Na rysunku 4.3 podano wykres równowagi układu żelazo-cementyt z oznaczonymi składnikami
strukturalnymi
.
Ferryt jako składnik strukturalny stopów technicznych na osnowie żelaza ma zwykle budowę
komórkową, której granice ziarn ujawnia się poprzez trawienie (rys. 4.4). Dłuższe trawienie
nadaje poszczególnym ziarnom różne zabarwienie, zależnie od ich orientacji krystalograficznej.
Czasem ferryt może mieć budowę iglastą (np. w strukturach spoin).
Rys. 4.4. Struktura ferrytyczna (żelazo techniczne o zawartości 0,007 %C). Traw. 5%
roztworem alkoholowym HNO
3
. Powiększ. 100x
Austenit w stalach węglowych istnieje w stanie równowagi tylko w wysokich temperaturach.
Może, więc być badany metalograficznie jedynie za pomocą mikroskopu próżniowego,
wyposażonego w urządzenie do obserwacji na gorąco. Zwyczajną techniką mikroskopową bada
się austenit tylko w tych stalach stopowych, w których jest on trwały również w temperaturze
otoczenia. Strukturę austenityczna ujawnia się silnie działającymi odczynnikami, np. roztworem
FeCl
3
i HC1 lub trawieniem elektrolitycznym, np. roztworem kwasu szczawiowego.
Austenit ma również strukturę komórkową, zwykle bardziej regularną niż ferryt
Charakterystyczne dla struktury austenitu jest dość liczne na ogół występowanie
rekrystalizacyjnych kryształów bliźniaczych.
60
JW
Rys. 4.3. Wykres równowagi układu żelazo-cementyt z oznaczonymi składnikami
strukturalnymi
Cementyt jako oddzielny składnik strukturalny występuje w stopach układu Fe-Fe
3
C w
postaci cementytu pierwszorzędowego (pierwotnego), cementytu drugorzędowego (wtórnego)
bądź cementytu trzeciorzędowego.
Cementyt pierwszorzędowy krystalizuje w stopach zawierających ponad 4,3% C, na skutek
zmniejszającej się ze spadkiem temperatury rozpuszczalności węgla w ciekłym żelazie (zgodnie
z linią CD - rys. 4.3). Występuje on w strukturach wysokowęglowych żeliw białych w postaci
grubych igieł, widocznych zwykle już pod niewielkim powiększeniem.
Cementyt wtórny wydziela się z austenitu na skutek zmniejszającej się ze spadkiem
temperatury rozpuszczalności węgla w żelazie
γ (zgodnie z linią ES -rys. 4.3). Jako oddzielny
składnik strukturalny, cementyt wtórny występuje w stalach o zawartości węgla przekraczającej
0,8% i zwykle ma postać siatki otaczającej poszczególne ziarna.
Cementyt trzeciorzędowy wydziela się z ferrytu na skutek zmniejszającej się ze spadkiem
temperatury rozpuszczalności węgla w żelazie
α (zgodnie z linią PQ -rys. 4.3). Jako oddzielny
składnik strukturalny może być wyraźnie zaobserwowany w strukturze stali o niewielkiej
zawartości węgla, zwykle w postaci wydzieleń na granicy ziaren ferrytu.
Perlit jest eutektoidalną mieszaniną dwóch faz: ferrytu i cementytu, zawierającą 0,8% węgla i
tworzącą się w temperaturze 723°C zgodnie z przemianą:
γ
S
→ α
P
+ Fe
3
C. Dla ścisłości należy
dodać, że przy ochładzaniu perlitu od temperatury 723°C do temperatury otoczenia, z ferrytu
zawartego w perlicie wydziela się jeszcze pewna ilość cementytu trzeciorzędowego (zazwyczaj
pomijanego z powodu nieznacznej jego ilości). Perlit obserwowany pod dostatecznie dużym
powiększeniem charakteryzuje się budową pasemkową, gdyż składa się z płytek ferrytu i
cementytu ułożonych na przemian. Odległości między płytkami zmniejszają się ze wzrostem
szybkości chłodzenia i jednocześnie następuje wzrost twardości struktury.
Pod mikroskopem, po wytrawieniu zgładu, ziarno perlitu
1
jest ciemne, jakkolwiek obydwa
składniki perlitu - ferryt i cementyt obserwowane oddzielnie mają jasne zabarwienie. Ciemne
zabarwienie ziarna perlitu wiąże się z jego budową płytkową i sposobem oświetlenia próbki pod
mikroskopem (obserwacja w świetle odbitym). Po wytrawieniu zgładu, bardziej odporne
chemicznie płytki cementytu wystają ponad płytki ferrytu, a strumień świetlny padający na taką
powierzchnię ulega częściowemu rozproszeniu. W wyniku tego oglądane pod mikroskopem

61
JW
ziarno perlitu ma zabarwienie ciemne.
Własności mechaniczne perlitu wynoszą w przybliżeniu: HB = 220
÷ 260, R
m
=700
÷ 800
MPa, A
10
~ 7% i KCU2 = 40 J/cm
Dodatek składników stopowych na ogół przesuwa punkt eutektoidalny w kierunku mniejszych
zawartości węgla, obniża lub podwyższa temperaturę przemiany eutektoidalnej oraz wpływa na
wzrost własności wytrzymałościowych.
Ledeburyt jest eutektyką o zawartości 4,3% C, tworzącą się z roztworu ciekłego L
C
w
temperaturze 1147°C, zgodnie z przemianą: L
C
→ γ
E
+ Fe
3
C. W temperaturze powstania
ledeburyt jest, więc mieszaniną eutektyczną dwóch faz: austenitu (zawierającego 2,06% C) i
cementytu. W miarę obniżania się temperatury do 723°C, z austenitu wydziela się cementyt
wtórny. W temperaturze 723°C austenit przemienia się w perlit i przy dalszym obniżaniu
temperatury z ferrytu zawartego w perlicie wydziela się niewielka ilość cementytu
trzeciorzędowego. W związku z tym, poniżej temperatury 723°C, ledeburyt stanowi już
mieszaninę perlitu i cementytu. Struktura taka nosi nazwę ledeburytu przemienionego.
Ledeburyt przemieniony jest, więc charakterystycznym składnikiem strukturalnym żeliw
białych.
4.3. Struktury stali węglowych
W temperaturze otoczenia, w zależności od zawartości węgla, struktury stali węglowych są
następujące:
Przy zawartości węgla teoretycznie nie przekraczającej 0,008%, występuje struktura
ferrytyczna (rys. 4.4).
Przy zawartości węgla 0,008-0,02%, na granicach ziarn ferrytu pojawiają się wydzielenia
cementytu trzeciorzędowego. Jak już wspomniano, takie stopy nazywane są zwykle
żelazem technicznym.
Stale o zawartości do 0,8% C noszą nazwę stali podeutektoidalnych. Ich struktura składa
się z dwóch składników, a mianowicie ferrytu i perlitu, przy czym w miarę wzrostu
zawartości węgla w stali wzrasta zawartość perlitu w strukturze (rys. 4.5-4.7).
Stal o zawartości 0,8% węgla ma strukturę perlityczną (rys. 4.8) i nosi nazwę stali
eutektoidalnej.
Stale o zawartości 0,8-2,06% węgla nazywają się stalami nadeutektoidalnymi i mają
strukturę składającą się z perlitu i cementytu wtórnego. W miarę wzrostu zawartości
węgla, wzrasta ilość cementytu w strukturze (rys. 4.9 i 4.10). Teoretycznie maksymalna
zawartość cementytu wtórnego występuje w stali o granicznej zawartości węgla 2,06% i
wynosi wtedy około 20%.
Rozpatrując własności mechaniczne stali węglowych można stwierdzić, że najniższą
wytrzymałość i najwyższą plastyczność w temperaturze pokojowej ma stal o strukturze
ferrytycznej. W miarę wzrostu zawartości węgla, a więc również wzrostu zawartości perlitu w
strukturze, rośnie wytrzymałość i twardość stali, przy jednoczesnym obniżaniu się plastyczności.
Maksymalną wytrzymałość (w stanie wyżarzonym) ma stal eutektoidalna (0,8% C). Dalszy
wzrost zawartości węgla powoduje podwyższanie twardości, gdyż w strukturze pojawia się
cementyt wtórny, równocześnie jednak maleje efektywna wytrzymałość stali, ponieważ staje się
ona mało plastyczna.
Struktura stali wykazuje często charakterystyczną pasmowość, która jest wynikiem obróbki
plastycznej na gorąco.
Oprócz omówionych wyżej składników strukturalnych, w każdej stali występują ponadto
różnego typu wtrącenia niemetaliczne, omówione już w rozdz. 3. Pod względem wielkości
wtrącenia te dzieli się na podmikroskopowe, mikroskopowe i makroskopowe.
Oznaczanie wtrąceń niemetalicznych w stali polega na obserwacji mikroskopowej (pod
powiększeniem 90-ll0x) powierzchni odpowiednio reprezentatywnie pobranych i
przygotowanych próbek i określeniu rodzaju, kształtu, ilości, wielkości i rozmieszczenia
1 W przypadku perlitu, za ziarno umownie uważa się nie pojedynczy krystalit jednofazowy, lecz zespół
naprzemianległych płytek cementytu i ferrytu.

62
JW
wtrąceń przez porównanie z ustalonymi wzorcami. Skład chemiczny i skład fazowy wtrąceń
niemetalicznych można określać za pomocą mikrosondy elektronowej.
Rys. 4.5. Struktura ferrytyczna z niewielką Rys. 4.6. Struktura ferrytyczno-perlityczna
ilością perlitu (stal o zawartości 0,05% C), (stal o zawartości 0,21% C). Traw. 5%
Traw. 5% roztworem alkoholowym HNO
3
roztworem alkoholowym HNO
3
.
Powiększ. 100x····· . 100Powiększ. 100x
Rys. 4.7. Struktura perlityczna z siatką ferrytu Rys. 4.8. Struktura perlityczna (stal o
(stal o zawartości 0.52% C). Traw. 5% zawartości 0,8% C). Traw. 5%
roztworem alkoholowym HNO
3
. 100x
roztworem alkoholowym HNO
3
.100x
Rys. 4.9. Struktura perlityczna z siatką ce- Rys. 4.10. Struktura perlityczna z siatką
mentytu (stal o zawartości ok. 1,2% C), i igłami cementytu (stal nawęglona do za-
Traw. 5% roztworem alkoholowym HNC
3
. wartości ok. 1,4%C). Traw. 5% roztworem
Powiększ. 500x
alkoholowym HN03. Powiększ. 100x
Dokładny sposób mikroskopowego oznaczania zanieczyszczenia stali wtrąceniami
niemetalicznymi podaje PN-64/H-04510, która zawiera również tablicę wzorców każdego typu
wtrąceń: tlenków ułożonych łańcuszkowo, tlenków ułożonych punktowo, krzemianów kruchych,
krzemianów plastycznych, krzemianów i tlenków nieodkształcalnych (globulamych), siarczków,
azotków tytanu i azotków aluminium.
Kilka przykładów wtrąceń niemetalicznych występujących w stalach podano na rys. 3.22-3.24
(rozdz. 3).

63
JW
5. OBRÓBKA CIEPLNA STOPÓW ŻELAZA
Związek mikrostruktury z własnościami mechanicznymi stali
Własności wytrzymałościowe i technologiczne stali są związane z jej mikrostrukturą zależną
w zasadniczy sposób od obróbki cieplnej, tj. od różnorodnych zabiegów cieplnych, którym stal
podlegała. Wykorzystując fizykochemiczne zjawiska występujące przy ogrzewaniu i oziębianiu
stali można doprowadzić do wytworzenia się w niej najbardziej pożądanych składników
strukturalnych, nadających je określone własności wytrzymałościowe.
I tak np. w celu wykonania obróbki skrawaniem stal wyżarza się zmiękczająco lub
normalizuje, w wyniku czego powstaje struktura ferrytyczno-perlityczna, odznaczająca się małą
twardością i wytrzymałością, ale dość znaczną ciągliwością. Własności takie ułatwiają
wykonanie obróbki wiórowej, więc w tym przypadku są one pożądane w procesie wytwarzania
elementu konstrukcyjnego.
Natomiast w gotowym wyrobie, podlegającym znacznym naprężeniom, struktura ferrytyczno-
perlityczna często nie zapewnia wystarczającej wytrzymałości i twardości. W celu polepszenia
tych własności, przy jednoczesnym uzyskaniu dobrej ciągliwości i udarności, stosuje się
ulepszanie cieplne, polegające na hartowaniu i odpuszczaniu w odpowiednio wysokiej
temperaturze, w wyniku czego powstaje struktura sorbityczna. Stal w stanie ulepszonym jest
materiałem konstrukcyjnym znacznie bardziej wartościowym niż ta sama stal w stanie
nieulepszonym. Dlatego jest regułą, że wysokojakościową stal konstrukcyjną, zwłaszcza
stopową, należy stosować jedynie w stanie ulepszonym.
Z kolei wyroby podlegające ścieraniu (np. narzędzia) powinny odznaczać się bardzo dużą
twardością. Wykorzystuje się wtedy wysoką twardość jaką odznacza się struktura
martenzytyczna powstająca przy hartowaniu.
Obróbka cieplna zwykła jest to rodzaj obróbki cieplnej, w wyniku której uzyskuje się zmiany
własności metali i stopów będące głównie funkcją temperatury i czasu.
Czasem jednak łączy się również zabiegi obróbki cieplnej z odkształcaniem-plastycznym, z
działaniem pola magnetycznego lub też z działaniem chemicznym środowiska. Mamy wówczas
do czynienia odpowiednio z obróbką cieplno-plastyczną, cieplno-magnetyczną lub cieplno-
chemiczną.
Związek obróbki cieplnej z przemianami fazowymi
Aby do danego stopu można było stosować poszczególne rodzaje obróbki cieplnej, np.
operacje hartowania i odpuszczania lub przesycania i starzenia, powinny się w nim dokonywać
przemiany fazowe, tj. np. podczas nagrzewania stopu powinne zachodzić przemiany alotropowe
lub powinna występować wyraźna zmiana rozpuszczalności pewnych jego składników.
Na podstawie wykresu równowagi fazowej danego układu można ustalić jak; rodzaj obróbki
cieplnej można zastosować do danego stopu i w jakich zakresach temperatury należy tę obróbkę
przeprowadzić.
W związku z tym proces obróbki cieplnej stali należy rozpatrywać, korzystając wykresu
równowagi fazowej układu żelazo-cementyt (rys. 5.1). Temperatury równowagi faz w tym
układzie oraz temperatury przemian (punkty krytyczne) przyjęto powszechnie oznaczać literą A
z odpowiednim wskaźnikiem. Najniższa z tych temperatur A
1
odpowiada równowadze austenitu
z ferrytem i cementytem (linia PSK). Temperatura A
2
jest temperaturą przemiany magnetycznej
ferrytu (linia MO). Temperatura A
3
, wyznaczona przez punkty leżące na linii GS, jest
temperaturą graniczną równowagi austenitu z ferrytem. Temperatura A
cm
(linia SE) to graniczna
temperatura równowagi austenitu z cementytem wtórnym.
Aby odróżnić temperatury początku i końca przemian podczas nagrzewania od tychże
temperatur podczas chłodzenia dodaje się do litery A wskaźnik c w przypadku nagrzewania lub
wskaźnik r w przypadku chłodzenia (np. A
c1
, A
r3
).
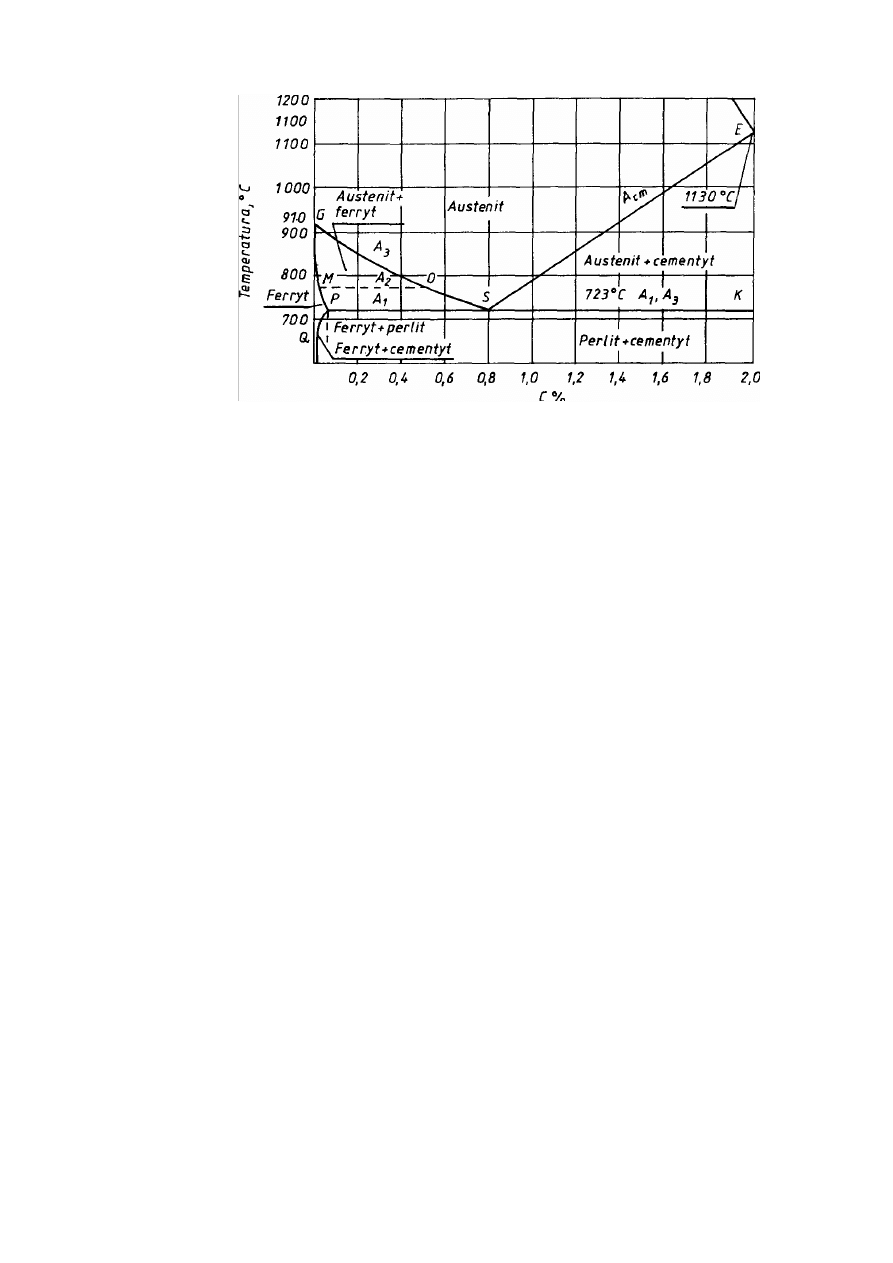
64
JW
Rys. 5.1. Fragment wykresu równowagi fazowej żelazo-cementyt
5.1. Podstawowe przemiany fazowe w stali związane z obróbką cieplną
Przemiany fazowe w stali są wynikiem tego, że wskutek zmiany warunków, np. temperatury,
jeden stan staje się mniej trwały niż drugi. To właśnie jest przyczyną przemian zachodzących w
stali. Należy zaznaczyć, że może w niej występować kilka podstawowych struktur, a istotą
najważniejszych przemian jest właśnie przejście jednej struktury w drugą. Tymi podstawowymi
strukturami są:
W procesach obróbki cieplnej stali występują następujące podstawowe przemiany
I. Przemiana ferrytu w austenit
Fe
α
(C)
→ Fe
γ
(C)
II. Przemiana austenitu w ferryt
Fe
γ
(C)
→ Fe
α
(C)
III. Przemiana perlitu w austenit
(Fe
α
(C) + Fe
3
C)
→ Fe
γ
(C)
IV. Przemiana austenitu w struktury perlityczne (lub bainityczne)
Fe
γ
(C)
→ Fe
α
(C) + Fe
3
C
V. Przemiana austenitu w martenzyt
Fe
γ
(C)
→ Fe
’
α
(C)
VI. Przemiana martenzytu w mieszaninę ferrytu i cementytu
Fe
’
α
(C)
→ Fe
α
(C) + Fe
3
C
ferryt Fe
α
(C)
austenit Fe
γ
(C)
martenzyt Fe
α
'(C)
perlit (Fe
α
(C)+Fe
3
C)
stały roztwór węgla w żelazie
α,
stały roztwór węgla w żelazie
γ
,
stały, przesycony roztwór węgla w żelazie
α
(jest to faza metastabilna),
eutektoidalna mieszanina ferrytu Fe
α
(C) i cementytu Fe
3
C.
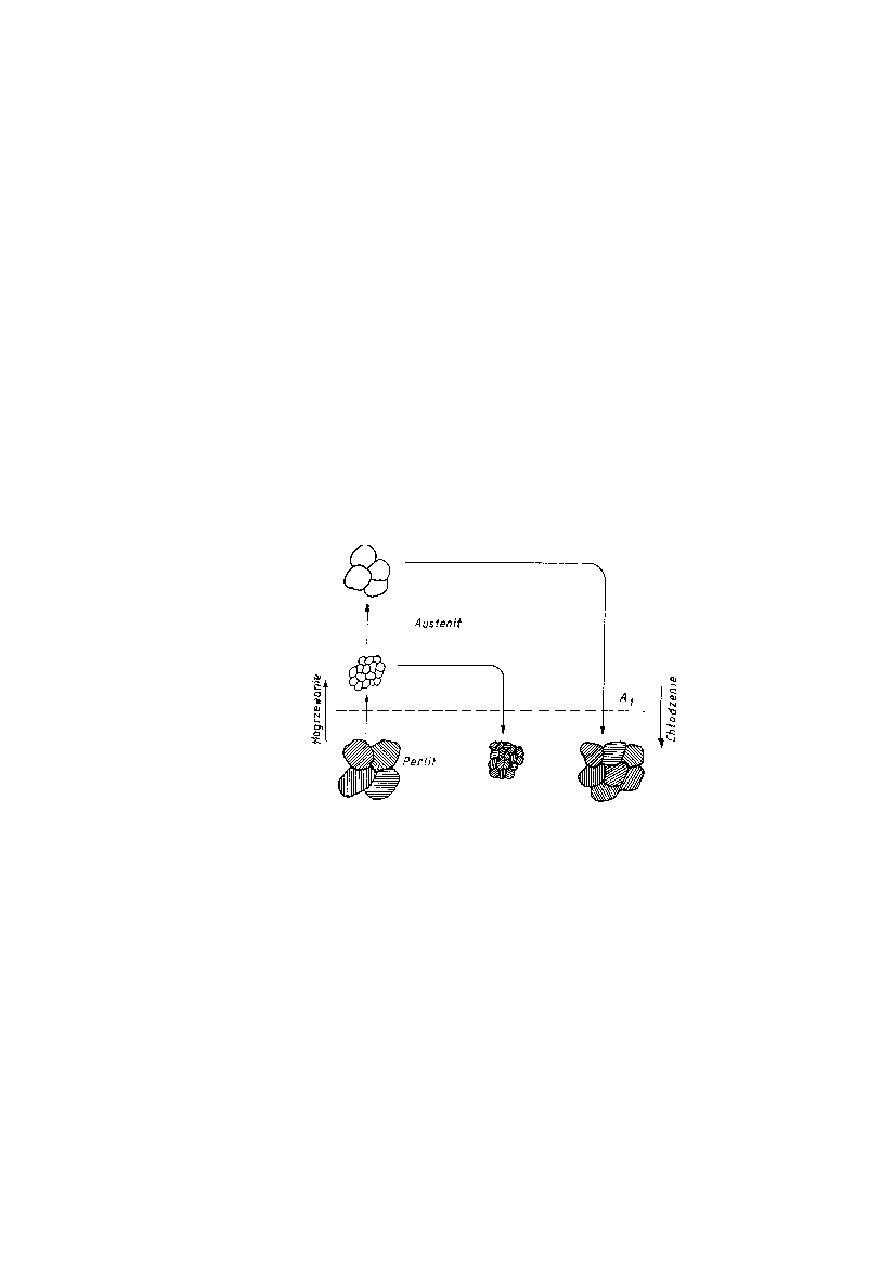
65
JW
5.1.1. Przemiana perlitu w austenit
Przemiana perlitu w austenit przebiega powyżej temperatury równowagi austenitu z ferrytem i
cementytem. Przemiana ta ma charakter dyfuzyjny, tj. zachodzi rozpuszczanie się cementytu i
równomierne rozmieszczanie się węgla w austenicie drogą dyfuzji. W zwykłych warunkach
stosunkowo szybkiego nagrzewania stali występuje opóźnienie przemiany i konieczne jest
podwyższenie temperatury, aby przemiana zaszła w określonym czasie. Perlit przegrzany
powyżej temperatury A
c1
przemienia się w austenit z różną szybkością, zależnie od stopnia
przegrzania. Szybkość przebiegu tej przemiany zależy również w znacznym stopniu od począt-
kowej struktury stali, tj. od stopnia dyspersji cementytu i od jego kształtu. Im drobniejsze są
cząstki cementytu, a tym samym większa ich ogólna powierzchnia, tym szybciej zachodzą
opisane przemiany.
Zabieg cieplny polegający na wygrzewaniu stali w celu wytworzenia struktury austenitu przed
chłodzeniem nazywany jest austenityzowaniem.
4.1.2. Zmiana wielkości ziarna austenitu
Przekroczenie temperatury przemiany A
c1
zaznacza się raptownym zmniejszeniem ziarn to
znaczy nowo powstałe ziarna austenitu są zawsze bardzo drobna i w zasadzie ich wymiary nie
zależą od wielkości ziarn perlitu, z którego utworzył się austenit. Rozdrobnienie ziarna austenitu
w czasie przemiany jest związane z tworzeniem się dużej liczby zarodków nowych ziarn na
olbrzymiej i bardzo rozwiniętej powierzchni granicznej między ferrytem i cementytem.
Dalsze nagrzewanie (lub wygrzewanie) po dokonanej przemianie wywołuje rozrost ziarn
austenitu (rys. 5.2 i 5.3).
Rys. 5.2. Schemat zmiany wielkości ziarna stali eutektoidalnej w czasie nagrzewania
powyżej temperatury A
1
Zjawisko rozrostu jest procesem samorzutnym, gdyż jego następstwem jest zmniejszenie
łącznej powierzchni ziarn (zmniejsza się energia powierzchniowa), wysoka temperatura
zapewnia dostatecznie szybki przebieg tego procesu.
W praktyce rozróżnia się dwa typy stali (rys. 5.3):
stale wykazujące skłonność do rozrostu ziarn austenitu, który zaczyna się po niewielkim
przekroczeniu temperatury A
c1
— stale te nazywamy gruboziarnistymi;
stale nie mające skłonności do rozrostu ziarn austenitu bezpośrednio po przekroczeniu
temperatury A
c1
W stalach tych ziarno zaczyna się rozrastać dopiero po nagrzaniu ich do
temperatury ok. 1000°C.
Zbyt wysokie i długotrwałe wygrzewanie stali podczas austenityzowania powoduje więc
rozrost ziarn austenitu. Z kolei wielkość ziarna perlitu zależy od wyjściowej wielkości ziarna
austenitu, z którego powstał perlit. Im większe są ziarna austenitu, tym większe tworzą się na
ogół ziarna perlitu. Powstanie struktury gruboziarnistej jest niepożądane, gdyż stal taka
charakteryzuje się niższą wytrzymałością i udarnością. Dlatego w czasie austenityzowania stali
skłonnych do rozrostu ziarna należy ściśle przestrzegać określonych temperatury i czasu grzania.
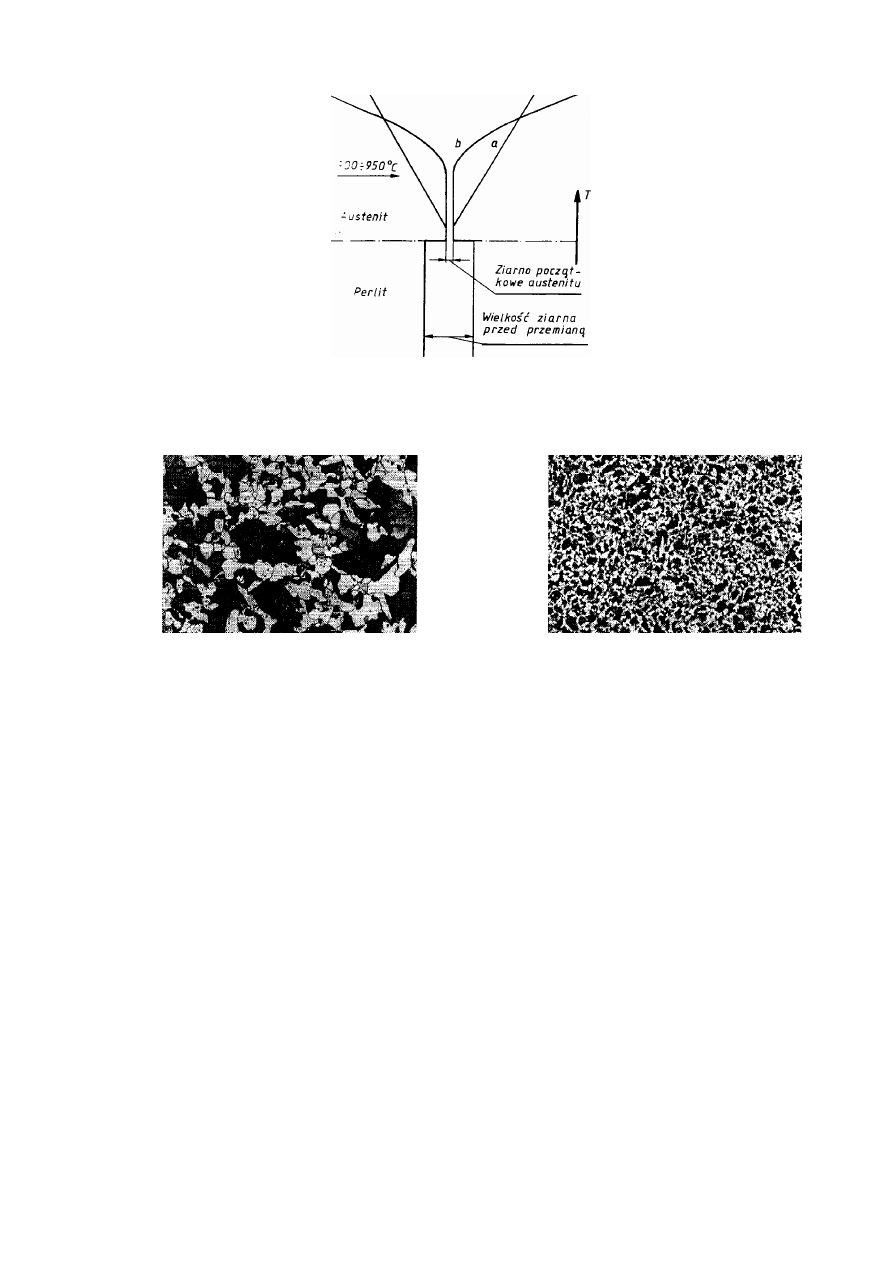
66
JW
Rys. 5.3. Schemat przedstawiający zmianę wielkości ziarna austenitu w czasie
nagrzewania stali gruboziarnistej (krzywa a) i stali drobnoziarnistej (krzywa b)
Rys. 5.4. Stal węglowa o zawartości 0,45%C
Rys. 5.5. Stal węglowa o zawartości 0,45% C
w stanie wyżarzonym o strukturze gruboziar-
w stanie normalizowanym. Struktura drobno-
nistej. Widoczne ciemne pola perlitu i jasne
ziarnista. Traw. 5% Nitalem. Powiększ. 100x
ziarna ferrytu. 5% Nital. Powiększ.100x
Na rysunku 5.4 przedstawiona jest struktura stali węglowej podeutektoidalnej o zawartości
0,45% C w stanie przegrzanym, charakteryzującej się dużym ziarnem. Z kolei na rys. 5.5
widoczna jest struktura drobnoziarnista tej samej stali w stanie normalizowanym, tj. po
nagrzewaniu do temperatury tylko ok. 30
÷ 50
o
C powyżej temperatury A
3
i chłodzeniu na
powietrzu.
5.1.3. Przemiana austenitu w struktury perlityczne
Przemiana austenitu w struktury perlityczne (lub bainityczne) przebiega w temperaturze
niższej niż A
1
. Rozpoczyna się przy pewnym przechłodzeniu, gdy energia swobodna mieszaniny
ferrytu z cementytem (perlitu) stanie się mniejsza od energii swobodnej austenitu.
Im niższa jest temperatura przemiany, tj. im większe przechłodzenie, tym większa jest różnica
swobodnych energii i tym szybciej przebiega przemiana.
Z drugiej strony przemianie austenitu w perlit towarzyszy dyfuzja połączona z
przegrupowaniem węgla, gdyż powstają dwie fazy znacznie różniące się zawartością węgla od
austenitu. Ferryt zawiera bardzo mało węgla (maks. ok. 0,02%). cementyt zaś 6,67% węgla.
Szybkość dyfuzji raptownie zmniejsza się przy obniżaniu temperatury, w związku z tym wzrost
przechłodzenia powoduje zmniejszenie szybkości przebiegu przemiany.
W wyniku łącznego działania obu czynników szybkość przemiany początkowo zwiększa się ze
wzrostem przechłodzenia, osiągając przy pewnej wartości przechłodzenia swe maksimum, a
potem zmniejsza się.
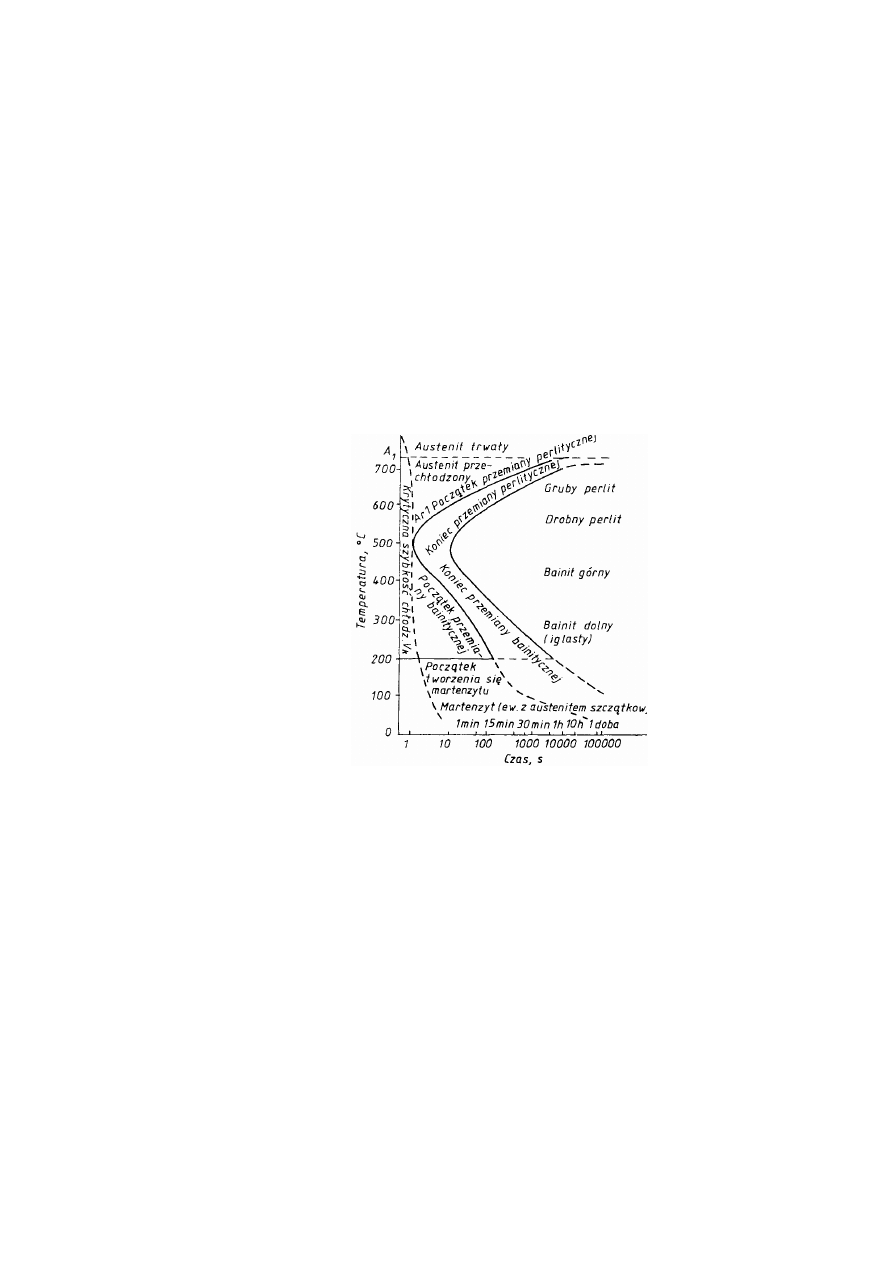
67
JW
Wykresy CTP
Przebieg procesu przemiany przechłodzonego austenitu wygodnie jest rozpatrywać na
podstawie wykresów rozpadu austenitu, zwanych wykresami CTP (czas, temperatura,
przemiana). Na wykresach tych naniesione są linie początku i końca przemian we
współrzędnych logarytm czasu-temperatura, przy czym rozróżnia się wykresy dla przemian
austenitu w warunkach izotermicznych oznaczane CTP
i
oraz wykresy przemian austenitu w
warunkach chłodzenia ciągłego, oznaczane CTP
c
. Na rysunku 4.6 podany jest schematycznie
wykres CTP; dla stali węglowej eutektoidalnej. Trwałość przechłodzonego austenitu zmienia się
w zależności od temperatury. Dla stali eutektoidalnej przy małych przechłodzeniach trwałość
austenitu jest duża, następnie zmniejsza się i minimum występuje w temperaturze ok. 500°C, po
czym znowu trwałość austenitu jest coraz większa aż do temperatury ok. 200°C, poniżej której
przechłodzony austenit przechodzi w martenzyt.
Wykresy CTP
i
, buduje się wykorzystując krzywe kinetyczne przemiany austenitu, dla
określonego stopnia przechłodzenia, wskazujące ilość wytworzonego perlitu w zależności od
czasu jaki upłynął od początku przemiany.
Rys. 5.6. Wykres CTP
i
przedstawiający linie początku i końca przemian austenitu
przechłodzonego w warunkach izotermicznych
Na rysunku 5.7 pokazano kilka krzywych kinetycznych obrazujących przebieg przemiany w
różnych temperaturach, a więc i przy różnych stopniach przechłodzenia. Jak widać, w
początkowym okresie przemiana odbywa się z bardzo małą prędkością, jest to tzw. okres
inkubacyjny. Punkty p
1
, p
2
, p
3
wskazują czas, w którym doświadczalnie stwierdza się początek
przemiany (wytworzone jest już wówczas ok. 0,5% perlitu). W miarę upływu czasu wzrasta
szybkość przemiany (szybkość ta jest maksymalna, gdy przemianie uległo ok. 50% austenitu),
następnie przebiega ona coraz wolniej, aż wreszcie kończy się w punktach k
1
, k
2
, k
3
.
Z
krzywych kinetycznych obrazujących przemianę austenitu w perlit odmierza się czasy od
początku chłodzenia do początków i końców przemiany, a odpowiadające im przy różnych
temperaturach punkty p i k rozmieszcza się w układzie temperatura-log czasu, na prostych
poziomych odpowiadających tym temperaturom. Otrzymuje się w ten sposób wykres CTP
i
, (rys.
5.7 II).
Struktury, jakie otrzymuje się w wyniku przemiany, zależą od stopnia przechłodzenia
austenitu, czyli od temperatury przemiany.
Zakres temperatury od A
1
do ok. 500°C odpowiada przemianie austenitu w perlit. Perlit
utworzony w temperaturze wysokiej przy małym stopniu przechłodzenia jest grubopłytkowy.
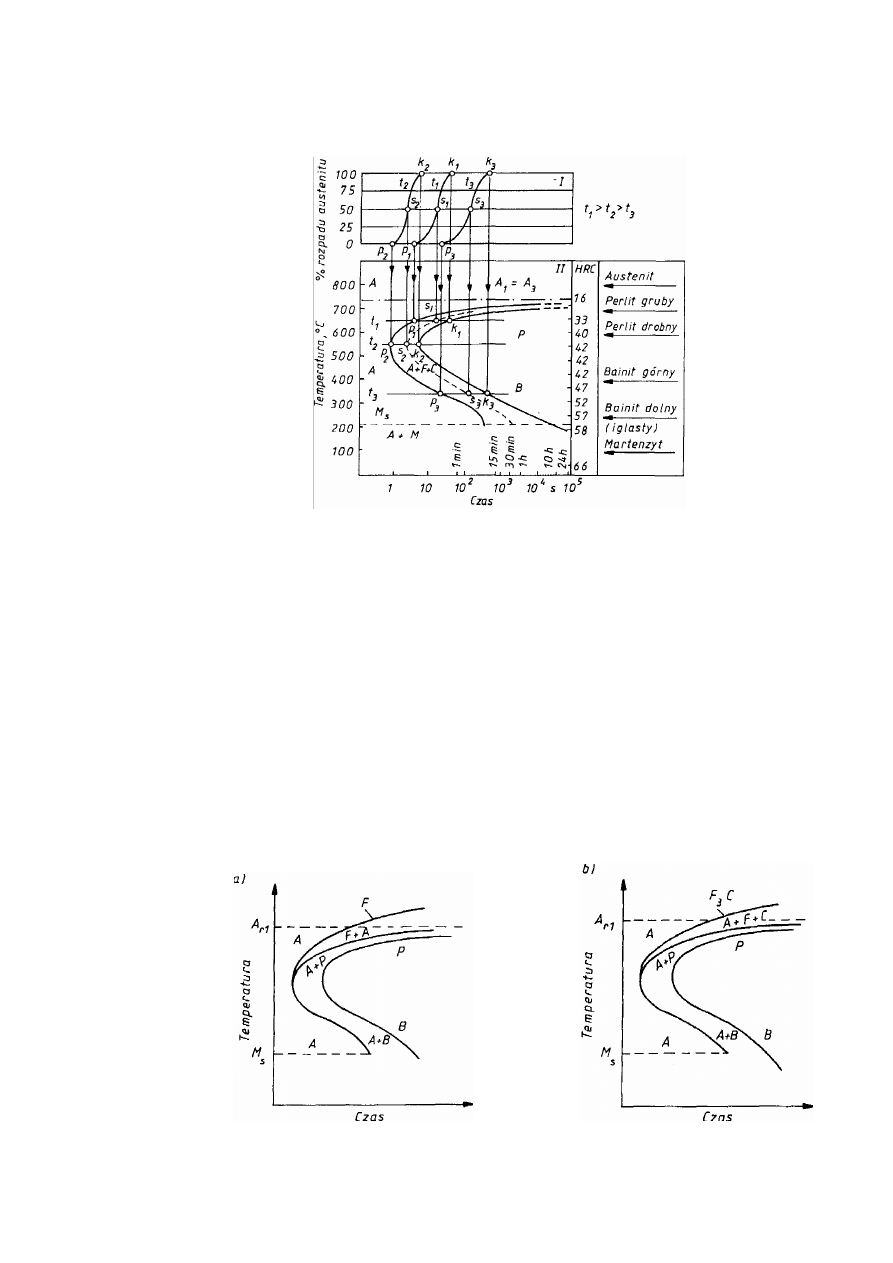
68
JW
Natomiast w miarę obniżania temperatury perlit staje się coraz drobniejszy, jego płytki stają się
coraz cieńsze i powstaje struktura o stosunkowo dużym stopniu dyspersji, która nosi nazwę
perlitu drobnego.
Rys. 5.7. Wykres izotermicznych przemian austenitu dla stali eutektoidalnej;
A - austenit, P - perlit, B - bainit, M –martenzyt
Stopień dyspersji perlitu wpływa na jego własności mechaniczne, tak np. twardość perlitu
grubego w przypadku stali węglowej eutektoidalnej wynosi ok. 15 HRC, a perlitu bardzo
drobnego dochodzi do 40 HRC (rys. 5.7).
W stalach podeutektoidalnych i nadeutektoidalnych przemiana perlityczna poprzedzona jest
innymi przemianami strukturalnymi. W stali podeutektoidalnej z austenitu tworzy się najpierw
ferryt, a w stalach nadeutektoidalnych przed rozpoczęciem przemiany perlitycznej wydziela się z
austenitu cementyt (rys. 5.8). Dalsza przemiana perlityczna przebiega podobnie, jak w stali
eutektoidalnej. Na rysunku 5.9 podany jest schematycznie wykres CTP
i
, dla stali węglowej
podeutektoidalnej, na którym naniesiono dodatkowe linie przemian izotermicznych (dla różnych
temperatur) prowadzących do powstania odpowiednich struktur. Z kolei rys. 5.10 przedstawia
wykres dla tej samej stali ale przy chłodzeniu ciągłym (CTP
c
); zaznaczono również linie
chłodzenia prowadzące do powstania różnych struktur.
Rys. 5.8. Wykres CTP dla stali: a) podeutektoidalnej, b) nadeutektoidalnej
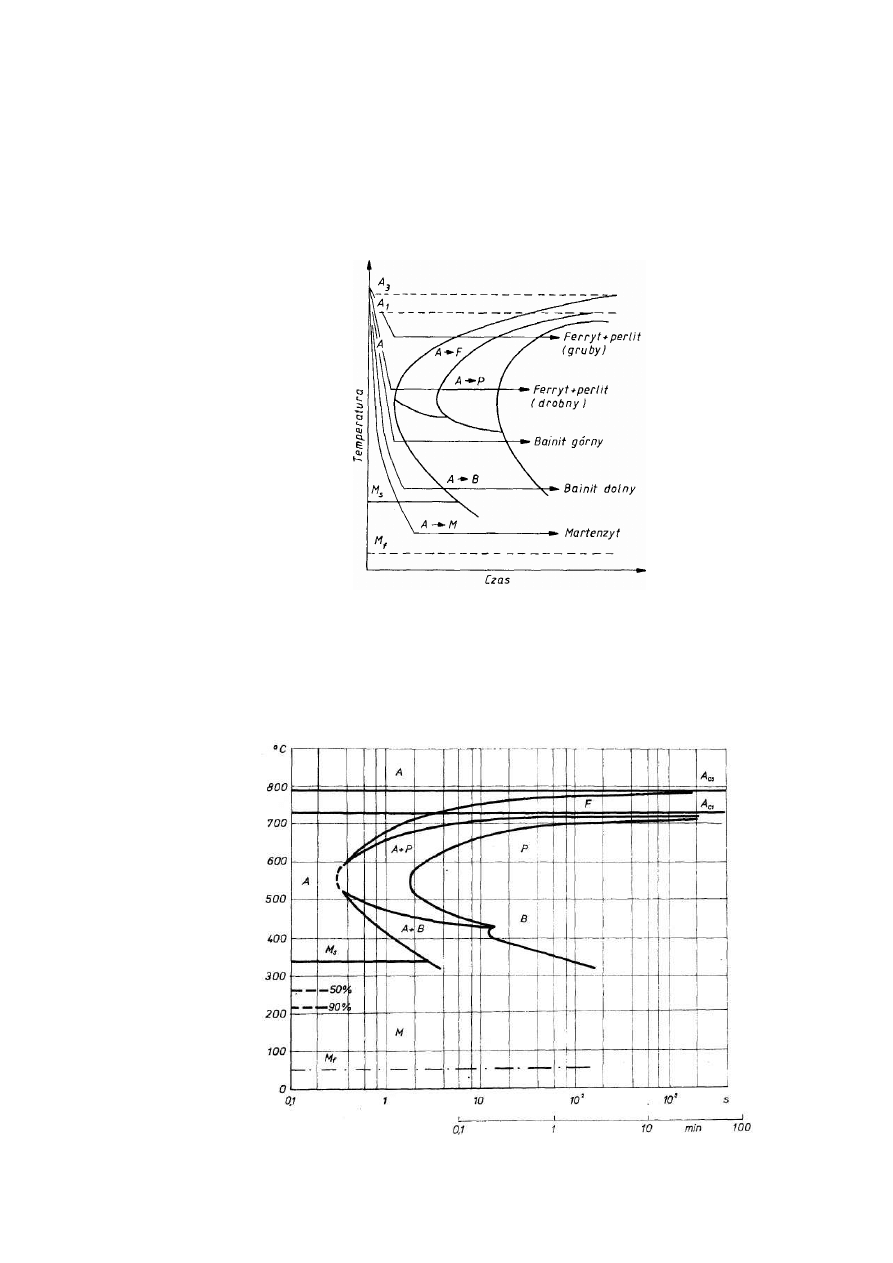
69
JW
Obniżenie temperatury rozpadu austenitu powoduje, że przemiana zachodzi w warunkach
utrudnionej dyfuzji. Struktura produktów rozpadu austenitu w takich linkach nosi nazwę bainitu.
W przypadku stali eutektoidalnych przemiana austenitu w bainit zachodzi w temperaturze ok.
500-200°C (rys. 5.6, 5.7), przy czym rozróżnia się bainit dolny o strukturze drobnoiglastej i
jeszcze większym stopniu dyspersji wydzieleń cementytu. W odróżnieniu od perlitu ferryt w
bainicie zawiera znacznie więcej węgla (tym więcej, im niższa była temperatura przemiany).
Bainit górny wykazuje twardość ok. 45 HRC, natomiast twardość bainitu dolnego wynosi ok. 55
HRC. Tę stosunkowo dużą twardość tłumaczy się znaczną dyspersją struktury oraz
zniekształceniem sieci.
Rys. 5.10. Wykres przemian austenitu w warunkach chłodzenia ciągłego (CTP
c
) dla
stali podeutektoidalnych
Przykładowe wykresy CTP
i
I CTP
c
dla stali 45 (węglowa konstrukcyjna wyższej jakości)
przedstawiono odpowiednio na rysunkach 5.11 i 5.12.
Rys. 5.11. Wykres CTP
i
stali 45
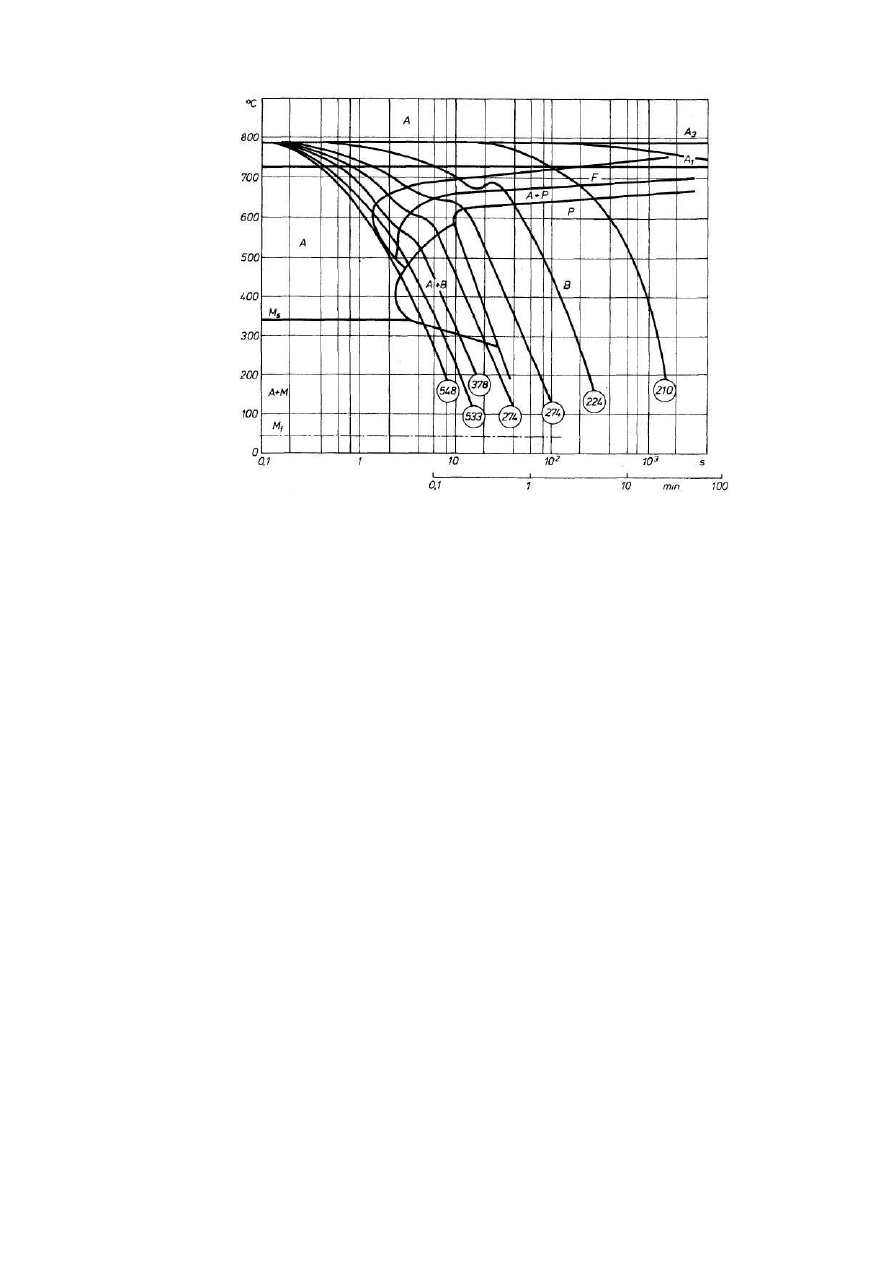
70
JW
Rys. 5.12. Wykres CTP
c
stali 45
Na wykresach CTP
i
izotermy reprezentują temperatury A
c1
, A
c3
lub A
cm
oraz M
s
i M
f
. Krzywe
między izotermami A
c1
i M
s
przedstawiają początek i koniec przemian dyfuzyjnych. Dodatkowa
linia przed krzywą początku przemiany odpowiada początkowi wydzielania ferrytu lub
cementytu (węglików) w stalach odpowiednio przed- lub zaeutektoidalnych. Czasami na
wykresach umieszcza się dodatkowo: krzywa odpowiadającą zaawansowaniu przemian
dyfuzyjnych w 50%, izotermy odpowiadające uzyskaniu 20, 50 lub 90% martenzytu oraz podaje
twardości struktur- produktów przemian. Na wykresach CTP
c
ponadto nanoszone są linie
reprezentujące poszczególne szybkości chłodzenia i odpowiadające im twardości Brinella lub
Vickersa (w kółkach).
5.1.4. Przemiana austenitu w martenzyt
Przemiana austenitu w martenzyt zachodzi poniżej określonej dla danej stali temperatury,
oznaczonej zwykle symbolem M
s
(rys. 5.9, 5.10). Temperatura końca przemiany
martenzytycznej oznaczona jest przez M
f
. W przypadku stali węglowych temperatury M
s
i, M
f
obniżają się wraz ze wzrostem zawartości węgla i składników stopowych, tak że przy większej
ich zawartości temperatura M
f
może być niższa od 0°C (rys. 5.13) i wobec tego przemiana
martenzytyczna zachodzi tylko częściowo W takim przypadku w strukturze pozostaje pewna
ilość tzw. austenitu szczątkowego. Ze względu na niską temperaturę procesu przemiana
martenzytyczna jest przemianą bezdyfuzyjną. W jej wyniku następuje przebudowa sieci
sześciennej zwartej (regularnej ściennie centrowanej) austenitu na sieć sześcienną centrowaną
(regularną przestrzennie centrowaną) żelaza alfa bez dyfuzji umożliwiającej wydzielanie węgla.
Martenzyt w stalach węglowych jest więc przesyconym roztworem stałym węgla w żelazie alfa.
Wtrącony międzyatomowo węgiel zniekształca strukturę żelaza alfa, tak, że po przemianie
martenzyt ma sieć tetragonalną. Stopień tetragonalności zwiększa się proporcjonalnie do
zawartości węgla w martenzycie; przy zawartości ok. 1,2% C stopień tetragonalności, czyli
stosunek parametrów sieci c/a. wynosi ok. 1,05.
Makroskopowo martenzyt ma charakterystyczną strukturę iglastą, co tłumaczy się tym, że
martenzyt składa się z płytek przesyconego węglem ferrytu, które w płaszczyźnie przecięcia
(wykonania szlifu metalograficznego) są podobne do igieł (rys. 5.14).
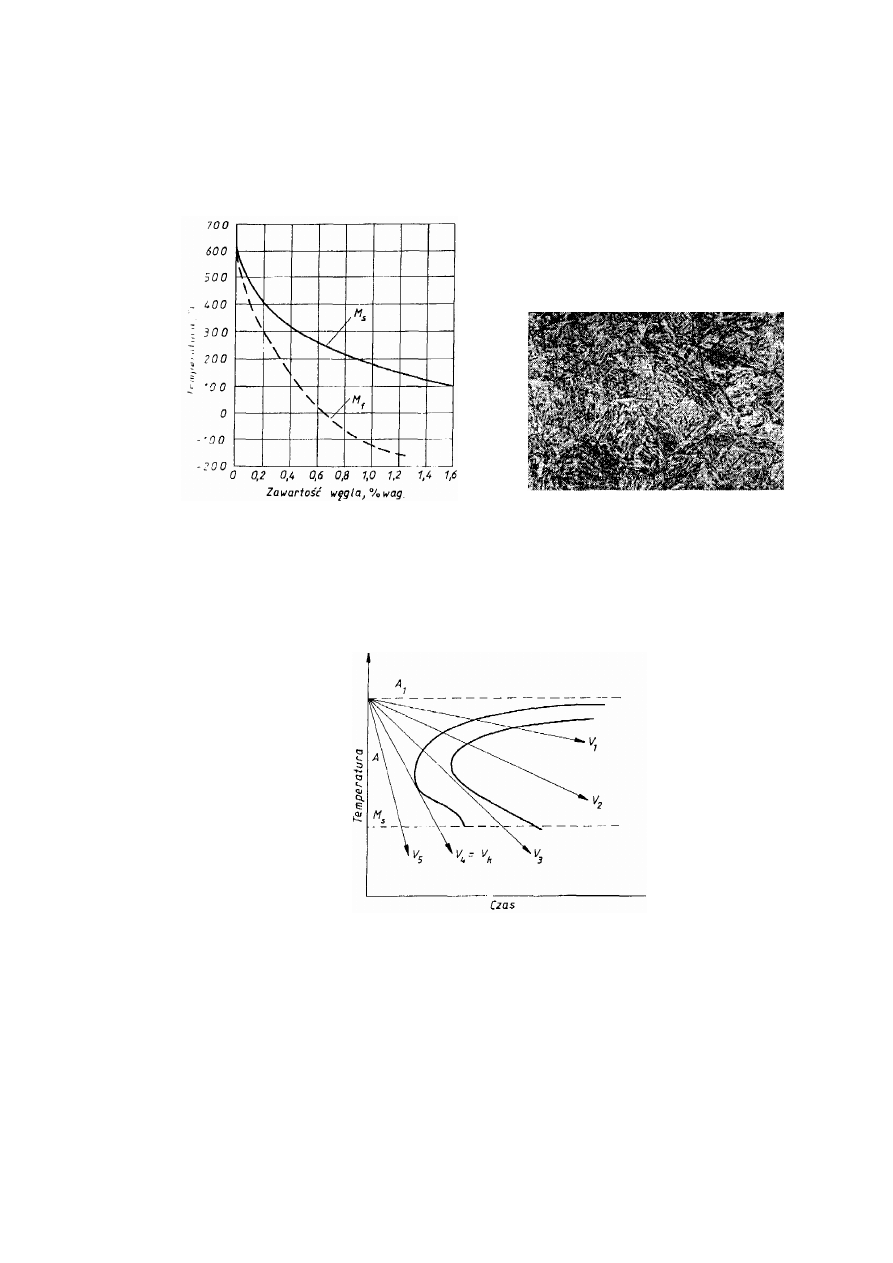
71
JW
Warunkiem przebiegu przemiany martenzytycznej jest ciągłe obniżanie temperatury w
zakresie od M
s
do M
f
.. Przy stałej temperaturze powstawanie martenzytu ustaje. Ponadto
przemiana austenitu w martenzyt może zajść dopiero przy odpowiednio dużej szybkości
chłodzenia. Najmniejsza szybkość chłodzenia, w wyniku której austenit przechodzi wyłącznie w
martenzyt, nazywana jest krytyczną szybkością chłodzenia V
k
(rys. 5.15).
Rys. 5.13. Położenie punktów początku M
s
Rys. 5.14. Typowa struktura martenzytu.
i końca M
f
przemiany martenzytycznej w
Stal 35 zahartowana w wodzie. Traw. 2%
zależności od zawartości węgla
Nitalem,
x500
Rys. 5.15. Przebieg chłodzenia stali z różną szybkością na tle wykresu CTP;
V
k
—szybkość krytyczna chłodzenia
Na kształt krzywych CTP duży wpływ mają składniki stopowe dodawane do stali
konstrukcyjnych. Składniki te powodują przesunięcie linii początku i końca przemiany austenitu
w prawo w stosunku do położenia tych linii dla stali węglowych (rys. 5.16a) lub powodują
jednoczesne przesunięcie i zmianę kształtu tych linii polegającą na ich rozdwojeniu (rys. 5.16b).
Zjawisko to jest korzystne o tyle, że krytyczna szybkość chłodzenia V
k
dla stali stopowych jest
w rezultacie znacznie niższa niż w przypadku stali węglowych.
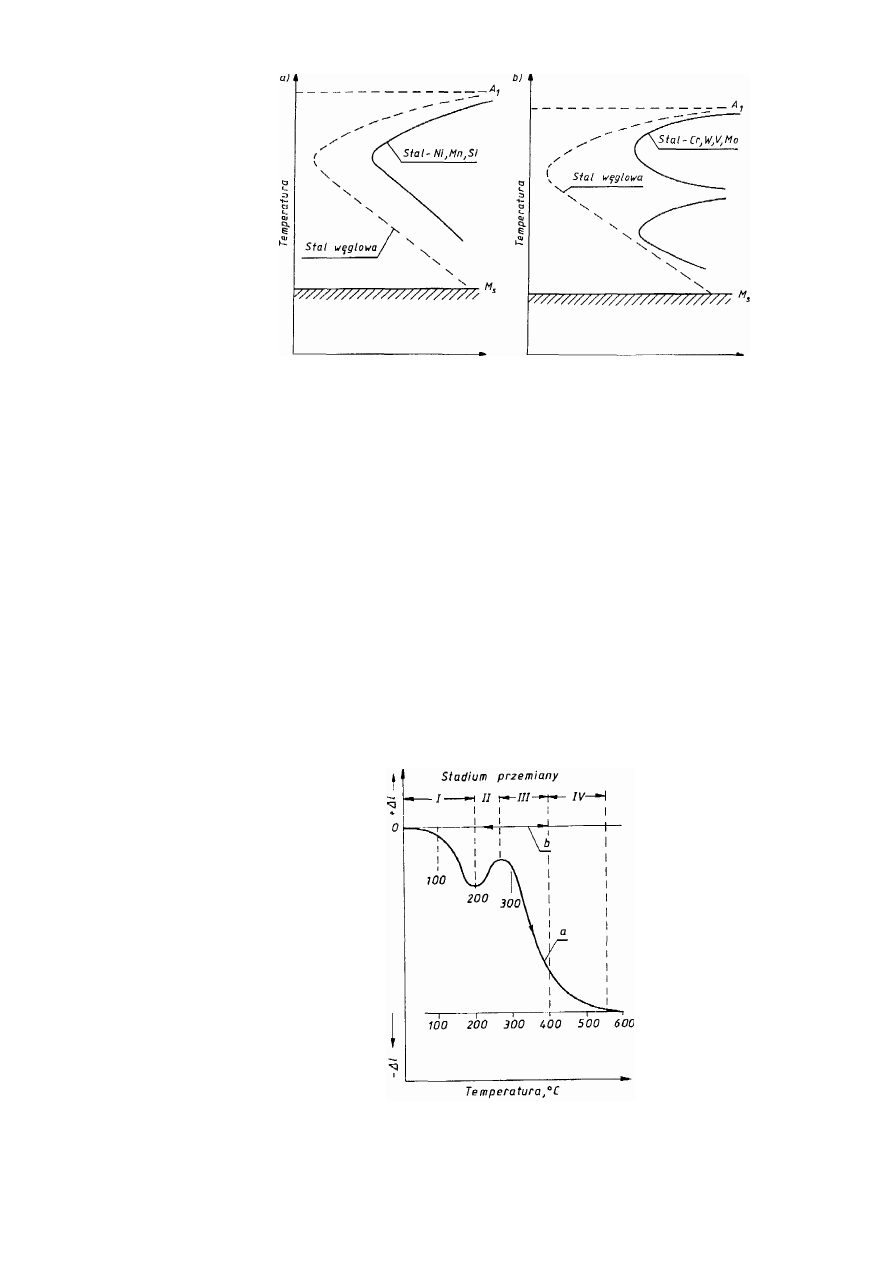
72
JW
Rys. 4.14. Porównanie położenia i kształtu linii początku przemiany przechłodzonego
austenitu na wykresach CTP: a) dla stali węglowej i dla stali stopowych zawierających
pierwiastki nie tworzące węglików, b) dla stali węglowej i dla stali stopowych
zawierających pierwiastki węglikotwórcze
4.1.5. Przemiany zachodzące podczas odpuszczania martenzytu
Martenzyt, który powstaje w wyniku szybkiego chłodzenia austenitu, czyli hartowania, jest
strukturą metastabilną, która już po niewielkim podgrzaniu zaczyna ulegać przemianom.
Operacja obróbki cieplnej, polegająca na wygrzewaniu zahartowanej stali w zakresie temperatur
leżących poniżej temperatury A
1
, i następnie chłodzeniu, nosi ogólnie nazwę odpuszczania.
Podczas odpuszczania martenzytu w stalach węglowych i niskostopowych można
obserwować kolejno, w poszczególnych zakresach temperatur, następujące zmiany:
I. W zakresie temperatury 80-200°C następuje przemiana martenzytu tetragonalnego w
martenzyt regularny - zwany też martenzytem odpuszczonym - co związane jest z
wydzielaniem węgla z martenzytu w postaci węglika
ε. Węglik ten wykazuje zmienny skład
wahający się w granicach Fe
2
C-Fe
3
C. Przemiana w tym zakresie temperatury związana jest ze
skurczem próbki, co można zarejestrować za pomocą badań dylatometrycznych (rys. 5.17).
Rys. 5.17. Krzywa dylatometryczna przedstawiająca różne stadia przemian
zachodzących w czasie odpuszczania stali zahartowanej;
∆l - przyrost długości próbki
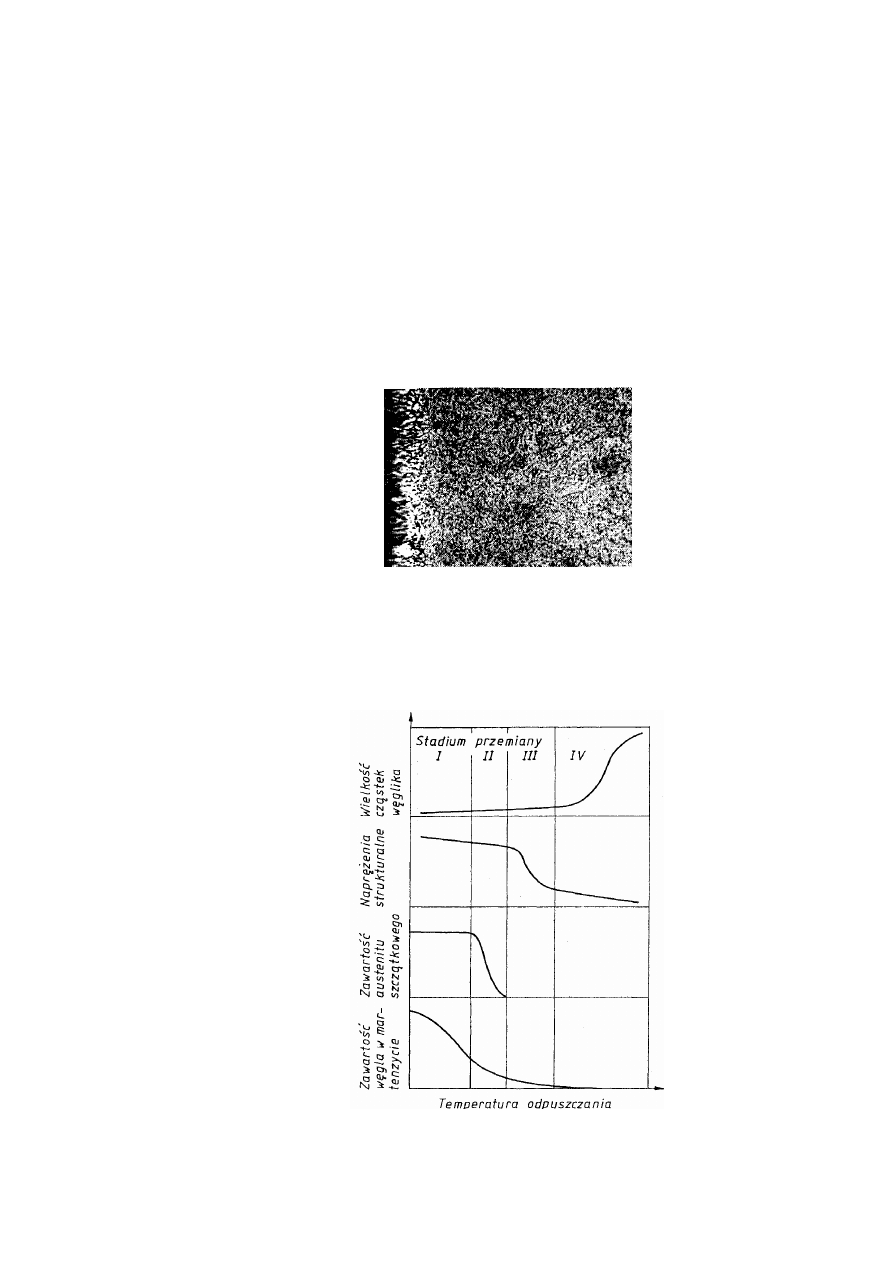
73
JW
II. W zakresie temperatury 200 -300°C zachodzi przemiana austenitu szczątkowego w martenzyt
odpuszczony i węglik żelaza, a także zachodzi w dalszym ciągu wydzielanie węglika z
martenzytu. Ponieważ objętość właściwa austenitu jest mniejsza niż martenzytu w tym stadium
przemiany obserwuje się rozszerzanie się próbki (rys. 5.18).
III. W zakresie temperatury 300-400°C następuje zarodkowanie i wzrost wydzieleń cementytu
Fe
3
C, przy czym węglik
ε rozpuszcza się, a węgiel dyfunduje do rosnących wydzieleń
cementytu Fe
3
C. W tym stadium zachodzi również intensywny proces zanikania naprężeń
własnych. W temperaturze ok. 400°C zawartość węgla w ferrycie osiąga już wartość
odpowiadającą stanowi równowagi. W czasie tych przemian obserwuje się skurcz próbki.
IV. Przy dalszym nagrzewaniu stali zahartowanej powyżej 400°C następuje proces koagulacji
wydzieleń cementytu, a wyjściowe igły martenzytu ulegają zdrowieniu.- i rekrystalizacji. Przy
najwyższych temperaturach odpuszczania procesami dominującymi stają się rozrost ziarna i
sferroidyzacja cementytu. Struktura składa się z ferrytu i cementytu drobnoziarnistego i nosi
nazwę sorbitu (rys. 5.18).
Rys. 5.18. Mikrostruktura stali o zawartości 0,35% C zahartowanej i odpuszczonej w
600°C. Sorbit. Traw. 2% Nitalem. x500
Na rysunku 4.17 przedstawione są schematycznie kolejne stadia procesów zawodzących w
czasie odpuszczania zahartowanej stali.
Rys. 5.19. Schemat przedstawiający procesy zachodzące podczas odpuszczania
zahartowanej stali
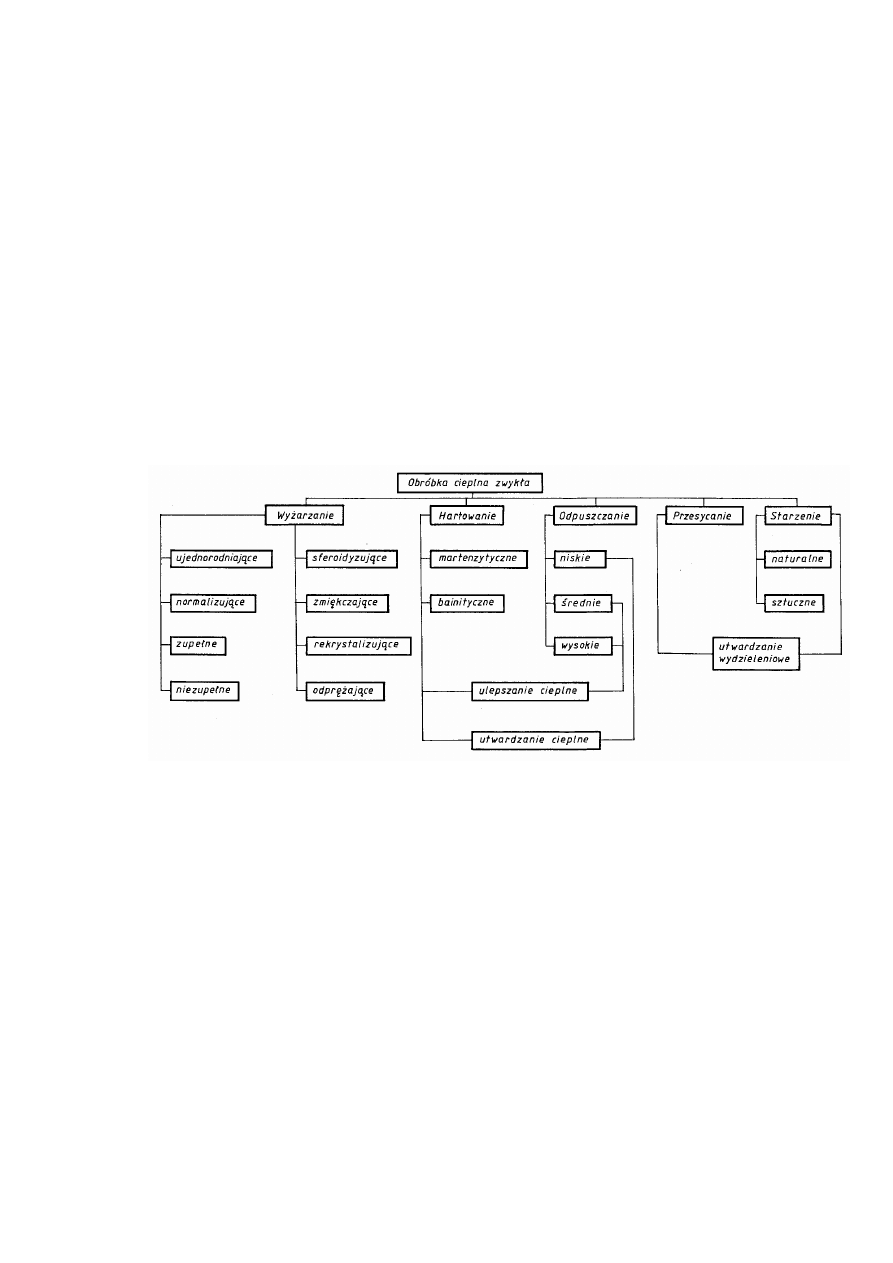
74
JW
5.2. Podstawowe rodzaje obróbki cieplnej stali
Schemat klasyfikacji podstawowych operacji obróbki cieplnej zwykłej stopów żelaza
przedstawiono na rys. 5.20. W obróbce cieplnej stali rozróżnia się trzy główne grupy operacji
cieplnych: operacje wyżarzania, operacje hartowania i odpuszczania, operacje przesycania i
starzenia. Największe znaczenie spośród wymienionych rodzajów obróbki cieplnej stali ma
hartowanie martenzytyczne z następującym po nim odpuszczaniem, czyli tzw. ulepszanie cieplne
lub utwardzanie cieplne. Ulepszanie cieplne polepsza znacznie cały zespół mechanicznych
własności stali i jest obecnie podstawowym rodzajem obróbki cieplnej stali konstrukcyjnych.
5.3. Wyżarzanie
Wyżarzanie jest operacją obróbki cieplnej polegającą na nagrzaniu stali do określonej
temperatury, wygrzaniu w tej temperaturze i powolnym chłodzeniu w celu otrzymania struktury
bardziej zbliżonej do stanu równowagi.
Rozróżnia się kilka rodzajów wyżarzania stali, z których każdy ma na celu osiągnięcie
określonych własności materiału, często bardzo różniących się pomiędzy sobą (rys. 5.20).
Zakresy temperatury, w jakich przeprowadza się poszczególne, ważniejsze rodzaje operacji
wyżarzania, podane są na rys. 5.21.
Rys. 5.20. Podstawowe operacje obróbki cieplnej zwykłej stopów żelaza
Wyżarzanie ujednorodniające (ujednorodnianie, homogenizowanie) polega na nagrzaniu stali
do temperatury niewiele niższej od temperatury solidusu, długotrwałym wygrzaniu w tej
temperaturze i powolnym chłodzeniu w celu zmniejszenia niejedno-rodności składu
chemicznego i struktury. Zakres praktycznie stosowanej temperatur} wynosi ok. 1050-1250°C.
Czas wygrzewania wynosi dla wlewków ok. 12-15 h
Niejednorodność składu chemicznego występuje w stali na skutek likwacji w czasie
krzepnięcia wlewka oraz segregacji dendrytycznej. Podczas wygrzewana pierwiastki
nierównomiernie rozłożone w stali, np. węgiel lub składniki stopowe dyfundują z miejsc
bogatszych do uboższych, przez co wyrównuje się skład chemiczny stali. Przez ujednorodnianie
można zmniejszyć w znacznym stopniu segregację dendrytyczną, nie daje się natomiast usunąć
likwacji strefowej, co może nastąpić dopiero przez obróbkę plastyczną, np. kucie lub
walcowanie.
Wysoka temperatura i długi czas wyżarzania ujednorodniającego powodują silny rozrost
ziarna i dlatego w celu poprawienia struktury przeprowadza się dodatkową obróbkę cieplną —
wyżarzanie normalizujące.
Podczas ujednorodniania zachodzą również takie niekorzystne procesy, jak odwęglenie
powierzchniowe i utlenienie powierzchni, co powoduje straty materiału.
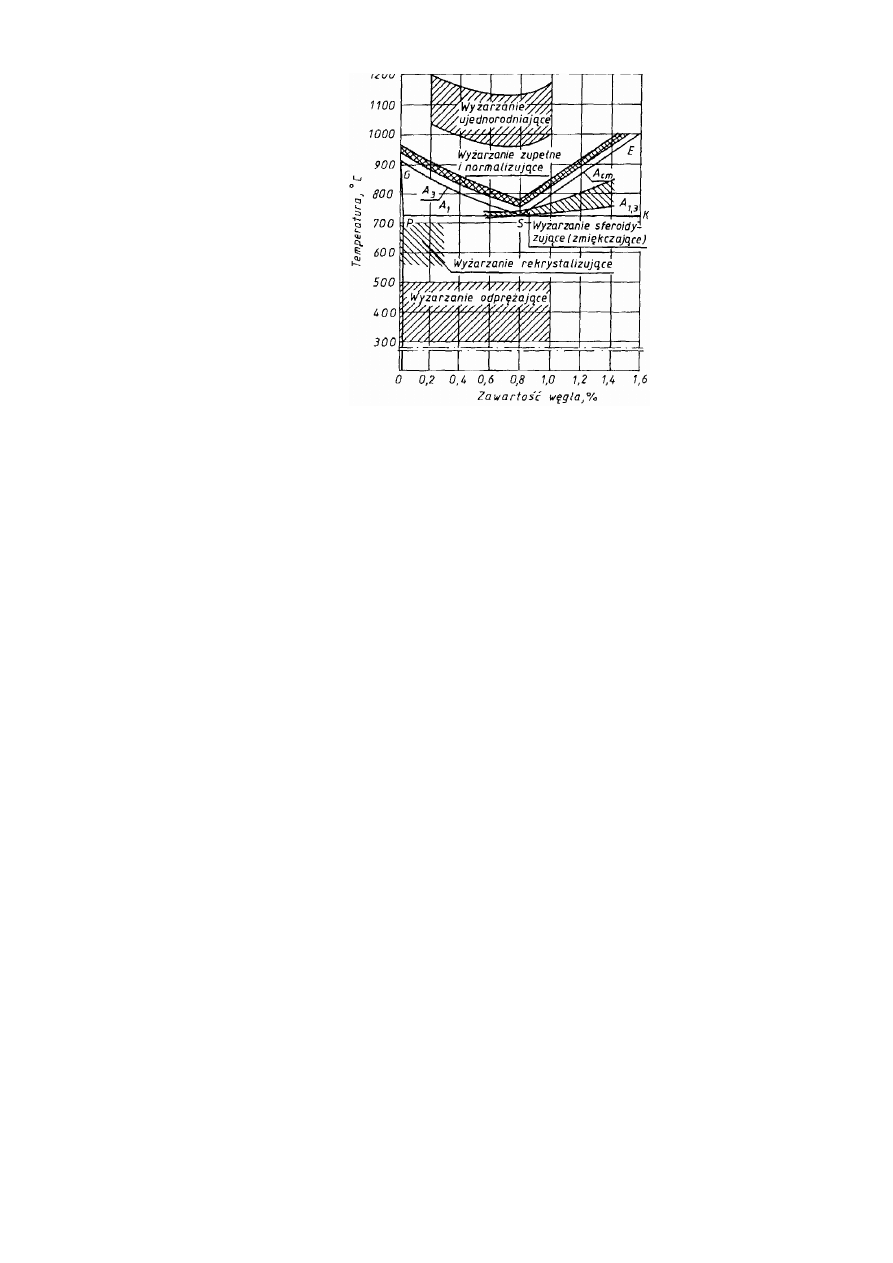
75
JW
Rys. 5.20. Zakresy temperatury wyżarzana stali węglowych na tle wykresu żelazo-cmentyt
Wyżarzanie normalizujące (normalizowanie) polega na nagrzaniu stali ok. 30-50°C
powyżej A
c3
lub A
cm
(w przypadku stali nadeuktoidalnych), wygrzaniu w tej temperaturze i
studzeniu w spokojnym powietrzu w celu uzyskania drobnego ziarna i równomiernego
rozłożenia składników strukturalnych. Stosunkowo krótkie czasy wygrzewania i dość szybkie
chłodzenie w spokojnym powietrzu powoduje uzyskanie korzystnej struktury drobnoziarnistej
(rys. 5.5). Normalizowanie polepsza własności wytrzymałościowe stali i w pewnych
przypadkach poprawia jej przydatność do obróbki mechanicznej przez skrawanie.
Normalizowanie stosuje się też często przed dalszą obróbką cieplną w seryjnej produkcji
celem nadania stali jednakowej struktury wyjściowej, co ma wpływ na własności stali po
obróbce cieplnej.
Wyżarzanie zupełne różni się od normalizowania tylko sposobem studzenia stali.
Polega na nagrzaniu stali powyżej A
c3
i następnie powolnym studzeniu (zwykle z piecem) do
temperatury poniżej A
r1
, w celu zupełnego przekrystalizowania stali. Dalsze studzenie może się
już odbywać na wolnym powietrzu. Wyżarzaniu temu poddaje się szczególnie stale stopowe, w
przypadku których szybkość chłodzenia w spokojnym powietrzu po normalizowaniu jest tak
duża, że może już doprowadzić do zahartowania. Stal po wyżarzaniu zupełnym ma dobrą
plastyczność, małą twardość i dobrą obrabialność.
Wyżarzanie niezupełne jest to wygrzewanie stali podeutektoidalnej w zakresie temperatury
A
c1
÷ A
c3
i studzenie do temperatury poniżej A
r1
w celu częściowego przekrystalizowania stali.
Jako celowa obróbka cieplna ten rodzaj wyżarzania jest rzadziej stosowany.
Wyżarzanie sferoidyzujące (sferoidyzowanie) polega na nagrzaniu do temperaturyty bliskiej A
c1
(nieco wyższej), wygrzewaniu w tej temperaturze i studzeniu w celu sferoidyzacji węglików.
Czas wygrzewania jest stosunkowo długi i może wynosić od kilku do kilkudziesięciu godzin. W
wyniku wyżarzania sferoidyzującego otrzymuje się strukturę ziarnistego cementytu w osnowie
ferrytycznej (rys. 5.21). Cementyt ziarnisty powstaje przez koagulację podczas wygrzewania w
temperaturze bliskiej A
c1
. Zaokrąglenie wydzieleń cementytu następuje w tych warunkach przez
dążność tej fazy do zmniejszenia energii powierzchniowej. Struktura taka charakteryzuje się
małą twardością, co zapewnia optymalną podatność na odkształcenia plastyczne przy obróbce
plastycznej na zimno, a także dobrą skrawalność. W stali eutektoidalnej i nadeutektoidalnej
wyżarzanie sferoidyzujące pozwala otrzymać korzystną strukturę wyjściową do hartowania pod
warunkiem, że cementyt będzie drobny i równomiernie rozłożony w osnowie ferrytu (rys, 5.21).
76
JW
Rys.5.21. Cementyt ziarnisty (sferoidyt) w stali o zawartości ok. 1% C. Traw. 5% nitalem.x500
Wyżarzanie zmiękczające (zmiękczanie) jest to wyżarzanie mające na celu zmniejszenie
twardości. Przeprowadza się je zwykle w temperaturze bliskiej A
c1
Wyżarzanie rekrystalizujące (rekrystalizowanie) jest to wyżarzanie stali utwardzonej
plastycznie na zimno, w temperaturze wyższej od temperatury początku rekrystalizacji, w celu
usunięcia skutków zgniotu i uzyskania określonej wielkości ziarna. W praktyce wyżarzanie
rekrystalizujące stosuje się najczęściej jako zabieg międzyoperacyjny, który usuwa skutki
zgniotu, co umożliwia wykonywanie dalszych operacji obróbki plastycznej na zimno. Zakres
temperatury wyżarzania rekrystalizującego jest podany na rys. 5.20.
Wyżarzanie odprężające ma na celu usunięcie naprężeń możliwie bez wprowadzenia zmian
strukturalnych w stali. Naprężenia występują w odlewach, spoinach materiałach odkształconych
plastycznie oraz hartowanych. Naprężenia te mogą być w pewnych przypadkach tak znaczne, że
powodują powstawanie pęknięć w materiale. Aby temu zapobiec, stosuje się wyżarzanie
odprężające, polegające na nagrzaniu i wygrzaniu przedmiotu w temperaturze poniżej A
c1
i
powolnym studzeniu Zależnie od rodzaju materiału oraz od przyczyn wywołujących naprężenia
stosuje się różną temperaturę (rys. 5.20) i różny czas wygrzewania. Na ogół im wyższa
temperatura, tym krótszy czas wygrzewania (do kilku godzin).
5.4. Hartowanie
Hartowanie polega na nagrzewaniu przedmiotu do temperatury, w której następuje
wytworzenie struktury austenitu, i następnie szybkim chłodzeniu w wodzie lub oleju w celu
otrzymania struktury martenzytycznej. Temperaturę hartowania stali określa się w zależności od
temperatur A
c1
A
c3
A
cm
. Optymalna temperatura hartowania stali podeutektoidalnych jest zwykle
wyższa o 30
÷ 50°C od temperatury A
c3
, a stali eutektoidalnych i nadeutektoidalnych - wyższa o
30
÷ 50°C od A
cm
. Zakres temperatury hartowania stali węglowych podany jest schematycznie
na rys. 5.22 na tle wykresu żelazo-cementyt.
Rys. 5.22. Zakres temperatury hartowania stali węglowych
77
JW
Hartowanie stali podeutektoidalnych od temperatury wyższej od A
c1
lecz niższej A
c3
jest
niekorzystne, ponieważ w strukturze martenzytu występuje również pewna ilość wolnego
ferrytu, który zmniejsza twardość i pogarsza własności mechaniczne po odpuszczeniu.
Natomiast w przypadku stali nadeutektoidalnych zakres temperatury hartowania powyżej A
c1
i
poniżej A
cm
(rys. 5.22) jest korzystny. Nie uzyskuje się wprawdzie pełnego przejścia stali w
austenit, lecz pozostający w strukturze cementyt drugorzędowy jest składnikiem o wysokiej
twardości i nie pogarsza własności mechanicznych. Nagrzewanie zaś powyżej A
cm
jest
niebezpieczne i zbyteczne, ponieważ nie zwiększa twardości stali zahartowanej, lecz przeciwnie
- nawet nieco zmniejsza wskutek zwiększenia ilości austenitu szczątkowego i rozpuszczania się
cementytu. Ponadto podczas nagrzewania powyżej A
cm
rośnie ziarno austenitu i zwiększa się
możliwość powstania dużych naprężeń hartowniczych. Rozrost ziarn austenitu powoduje, że w
stali zahartowanej otrzymuje się strukturę martenzytu o grubych igłach i grubokrystaliczny
przełom, co jest powodem małej ciągliwości i niskiej udarności stali.
Hartowanie zwykłe polega na hartowaniu z ciągłym (nie przerywanym) oziębianiu z
szybkością większą od krytycznej w środowisku o temperaturze niższej od temperatury początku
przemiany martenzytycznej (rys. 5.23a). Stale węglowe hartuje na ogół w wodzie a stale
stopowe w oleju. Przy chłodzeniu w powietrzu nie uzyskuje się szybkości krytycznych
wymaganych dla stali węglowych i niskostopowych. Jedynie stale wysokostopowe o malej
szybkości krytycznej ulegają zahartowaniu
w powietrzu; są to tak zwane stale samohartujące się.
Rys. 5.23. Różne rodzaje hartowania stali. Schemat przebiegu chłodzenia na tle wykresu
CTP: a) hartowanie zwykłe, b) hartowanie stopniowe, c) hartowanie z przemianą
izotermiczną (bainityczne); p — powierzchnia, r — rdzeń przedmiotu
Hartowanie stopniowe. Zwykłe hartowanie martenzytyczne powoduje powstawanie naprężeń
cieplnych i strukturalnych, co jest często przyczyną deformacji i pęknięć elementów obrabianych
cieplnie. Aby tego uniknąć, stosuje się w niektórych przypadkach hartowanie stopniowe. Jest to
hartowanie z pierwszym stopniem oziębiania w kąpieli solnej o temperaturze nieco wyższej od
M
s
, w ciągu czasu niezbędnego do oziębienia całego przekroju przedmiotu do temperatury
kąpieli, i z drugim stopniem oziębiania w powietrzu. Czas przetrzymywania w kąpieli solnej nie
może być dłuższy niż wynosi czas trwałości austenitu w tej temperaturze - rys. 5.23b.
Hartowanie stopniowe jest stosowane w obróbce cieplnej przedmiotów o małych przekrojach i
skomplikowanym kształcie.
Hartowanie bainityczne z przemianą izotermiczną jest zabiegiem cieplnym polegającym na
hartowaniu i oziębianiu w kąpieli solnej o temperaturze bliskiej, lecz nieco wyższej od M
s
i
wytrzymaniu w tej kąpieli w czasie zapewniającym całkowite ukończenie przemiany
bainitycznej i następnie ochłodzeniu na powietrzu (rys. 5.23c). Ten rodzaj hartowania ma
wszystkie dodatnie cechy hartowania stopniowego, a więc zmniejszenie naprężeń cieplnych i
strukturalnych oraz zmniejszenie możliwości powstawania pęknięć i deformacji, a ponadto
zapewnia uzyskanie przez stal dużej udarności, lecz niższej twardości od martenzytu.

78
JW
5.5. Hartowanie powierzchniowe
Hartowanie powierzchniowe polega na szybkim nagrzaniu cienkiej warstwy powierzchniowej
przedmiotu do temperatury powyżej A
c3
(temperatury austenityzacji) i oziębieniu z dużą
szybkością niezbędną do uzyskania struktury martenzytycznej w tej warstwie. Celem hartowania
powierzchniowego jest nadanie warstwie powierzchniowej wysokiej twardości i odporności na
ścieranie, przy zachowaniu ciągliwego rdzenia. Hartowaniu powierzchniowemu poddaje się stale
węglowe o zawartości 0,4-0,6% oraz stale niskostopowe o zawartości 0,3-0,6% C. Elementy, od
których wymaga się większej wytrzymałości, przed hartowaniem powierzchniowym poddaje się
ulepszaniu cieplnemu, tj. hartowaniu i wysokiemu odpuszczaniu. Najczęściej stosowanymi
metodami hartowania powierzchniowego są:
a) hartowanie płomieniowe — polegające na nagrzewaniu powierzchni płomieniem gazowym,
zwykle acetylenowo-tlenowym, za pomocą palnika o dużej wydajności, i na intensywnym
oziębieniu strumieniem wody;
b) hartowanie indukcyjne — polegające na nagrzewaniu warstwy powierzchniowej przedmiotu
prądami wirowymi, wzbudzonymi przez prąd zmienny o wielkiej częstotliwości płynący we
wzbudniku w postaci uzwojenia, i następnie szybkim oziębianiu natryskiem wodnym;
c) hartowanie kąpielowe — polegające na nagrzewaniu powierzchni przez krótkie zanurzenie do
kąpieli solnej lub metalowej i następnie oziębieniu;
d) hartowanie kontaktowe lub oporowe, przy którym powierzchnia przedmiotu nagrzewa się w
miejscu styku elektrody w postaci rolki dociskowej z powierzchni przedmiotu na skutek oporu
omowego;
e) hartowanie elektrolityczne, podczas którego grzanie odbywa się w elektrolicie wskutek
przepływu prądu o dużym natężeniu przez elektrolit, przy czym katodą jest przedmiot
nagrzewany.
Stosowane jest również hartowanie z grzaniem powierzchniowym laserowym, elektronowym
i plazmowym.
Wspólną cechą metod hartowania powierzchniowego jest zapewnienie tak szybkiego
nagrzewania, aby przedmiot osiągnął temperaturę hartowania tylko do pewnej zadanej
głębokości. Temperatura warstwy powierzchniowej przy szybkim nagrzewania przekracza
zwykle znacznie (o ok. 100°C) właściwą temperaturę hartowania, a jednak nie wywiera
ujemnego wpływu na własności stali, gdyż czas nagrzewania jest dużo krótszy niż przy
hartowaniu zwykłym i praktycznie rozrost ziarn nie występuje.
Wszystkie metody hartowania powierzchniowego wymagają bardzo dokładnego opracowania
warunków nagrzewania i ścisłego dostosowania ich do kształtu i żądanej charakterystyki
hartowanej powierzchni.
Wybór jednej z metod hartowania powierzchniowego oraz sposób wykonania zabiegu zależą
m.in. od wielkości i kształtu obrabianych przedmiotów, od ich ilości oraz od żądanej głębokości
utwardzenia.
Hartowanie płomieniowe pozwala na osiągnięcie głębokości zahartowania od około 2 do 8
mm, przy minimalnej średnicy przedmiotu 20 mm. Na wyniki hartowania mają wpływ takie
czynniki, jak: wydajność palnika, kształt jego końcówek, szybkość posuwu palnika lub
przedmiotu, odległość palnika od powierzchni, czas upływający między końcem grzania a
początkiem chłodzenia, intensywność chłodzenia.
Zależnie od kształtu i wielkości hartowanego przedmiotu rozróżnia się dwa sposoby
hartowania: hartowanie jednoczesne obrotowe oraz hartowanie ciągłe posuwowe lub posuwowo-
obrotowe.
Metoda jednoczesnego hartowania polega na nagrzewaniu od razu całej powierzchni
przedmiotu i po osiągnięciu właściwej temperatury na jej szybkim ochłodzeniu. Najczęściej
spotykaną odmianą tego sposobu jest hartowanie obrotowe, w czasie którego palnik jest
nieruchomy, a przedmiot obraca się z określoną prędkością - rys. 5.24. Sposób ten stosowany
jest do przedmiotów okrągłych o niewielkich średnicach.
79
JW
Rys. 5.24. Hartowanie powierzchniowe płomieniowe jednoczesne obrotowe
Metoda hartowania ciągłego polega na postępowym ciągłym nagrzewaniu powierzchni i
postępującym za nim oziębianiu ciągłym za pomocą natryskiwacza znajdującego się za
palnikiem. Metodę tę stosuję się do przedmiotów o dużej powierzchni płaskiej – rys. 5.25, lub
krzywoliniowej, długich przedmiotów walcowych (hartowanie posuwowo-obrotowe – rys. 5.26
oraz przedmiotów o dużej średnicy.
Hartowaniu płomieniowemu poddaje się przedmioty wykonane ze stali węglowych o
zawartości 0,45
÷0,60% C oraz niektóre gatunki stali manganowych chromowych i chromowo-
wanadowych. Największe zastosowanie ta metoda hartowania znalazła przy miejscowym
utwardzaniu dużych części maszyn produkowanych pojedynczo lub w niewielkich seriach.
Stosowana jest również przy hartowaniu kół zębatych o dużych modułach oraz wałów o dużych
średnicach I długości do 10 m.
Hartowanie indukcyjne (rys. 5.27) pozwala na osiągnięcie mniejszych głębokości
zahartowania niż przy hartowaniu płomieniowym (ok. 0,2 - 5 mm).
Głębokość warstwy, w której indukują się prądy wirowe można obliczyć za pomocą
empirycznego wzoru:
gdzie:
ρ - oporność właściwa
µ - przenikalność magnetyczna
f - częstotliwość prądu w Hz
Przenikalność magnetyczna stali węglowej gwałtownie maleje w temperaturze
przemiany magnetycznej (punkt Curie) i przy dalszym nagrzewaniu prawie nie
ulega zmianie. Głębokość przenikania prądów wirowych wynosi dla:
f
C
d
⋅
µ
ρ
=
]
mm
[
f
600
d
:
austenitu
]
mm
[
f
17
d
:
ferrytu
Fe
Fe
=
=
γ
α
80
JW
Głębokość warstwy zahartowanej zależy od trzech czynników: częstotliwości prądu, mocy
właściwej urządzenia (mocy we wzbudniku przypadającej na jednostkę powierzchni
nagrzewanego przedmiotu) oraz czasu nagrzewania. Ze względu na konieczność szybkiego
nagrzewania powierzchni przedmiotu w grzejnictwie indukcyjnym stosowane są częstotliwości
prądu w granicach 1-5000 kHz.. Dla przykładu można podać, że przy częstotliwości f = 1000 Hz
głębokość hartowania d = 6 mm, natomiast przy f = 450 000 Hz - d = 0,9 mm.
Wielkość nagrzewanej powierzchni zależy od mocy generatora. Orientacyjne
zapotrzebowanie mocy niezbędnej do nagrzania l cm
2
wynosi 0,3-3,0 kW. Rozrzut ten jest
spowodowany zróżnicowaną konstrukcją wzbudników, których kształt uzależniony jest od
hartowanej powierzchni. Dużą rolę odgrywa też szczelina pomiędzy wzbudnikiem a
powierzchnią nagrzewaną. W praktyce szczelina ta powinna zawierać się w granicach 1-3 mm.
Rys.5.27. Schemat grzania indukcyjnego; l — induktor - wzbudnik, .1 -pręt nagrzewany,
3 - linie pola magnetycznego, I
w
— prąd we wzbudniku, I
p
— prąd w przedmiocie
Czas grzania, niezbędny do osiągnięcia temperatury austenityzacji, zależny jest częstotliwości
prądu i mocy generatora. Teoretycznie dla bardzo małych powierzchni i małych głębokości czas
grzania może wynosić ułamek sekundy, w praktyce zawiera się w granicach 2
÷ 20 s.
Ze względu na bardzo duży koszt urządzeń, hartowanie indukcyjne stosuje się w produkcji
wielkoseryjnej i masowej. Dla każdego typu przedmiotu wykonuje się specjalny wzbudnik,
Rys. 5.25. Hartowanie powierzchniowe Rys. 5.26. Hartowanie powierzchnie
płomieniowe ciągle
posuwowe
płomieniowe ciągle posuwowo-obrotowe
81
JW
ściśle dostosowany do kształtu i wymiarów przedmiotu. Podobnie jak przy hartowaniu
płomieniowym, również przy hartowaniu indukcyjnym rozróżnia się dwie podstawowe metody
hartowania: hartowanie jednoczesne obrotowe oraz hartowanie ciągłe posuwowe lub posuwowo-
obrotowe – rys. 5.28
Rys. 5.28. Schemat hartowania indukcyjnego ciągłego, posuwowo-obrotowego;
l - wzbudnik dwuzwojowy, 2 - natryskiwacz, 3 - przedmiot
Ogólna zasada hartowania indukcyjnego jest podobna do hartowania płomieniowego z tą
różnicą, że w miejscu palników umieszczony jest wzbudnik, który bardzo często spełnia rolę
natryskiwacza.
W przemyśle największe zastosowanie znalazło hartowanie indukcyjne, a następnie
płomieniowe. Inne metody hartowania powierzchniowego jak: kąpielowe, kontaktowe czy
elektrolityczne, stosowane są sporadycznie.
5.6. Hartowność i utwardzalność stali
Cechami charakteryzującymi stal zahartowaną są utwardzalność i hartowność. Pojęcia te są
zbieżne, gdyż określają własności stali zahartowanej, które są ściśle od siebie uzależnione.
Przez utwardzalność rozumie się zdolność stali do utwardzania się przy hartowaniu, a określa ją
maksymalna twardość mierzona na powierzchni stali, którą uzyskano przy optymalnych
parametrach hartowania. Twardość po hartowaniu jest zależna od zawartości węgla w stali.
Wyższa zawartość węgla w martenzycie zwiększa twardość stali, ale tylko do zawartości ok.
0,9% C. W stalach nadeutektoidalnych, dla których optymalną temperaturą hartowania jest A
c1
+
30°C, zawartość węgla w martenzycie po hartowaniu jest stała, zmienia się natomiast ilość
cementytu, który jednak nie wpływa w sposób istotny na zmianę twardości.
Z kolei przez hartowność rozumie się głębokość na jaką stal da się zahartować. Miarą
hartowności jest więc grubość strefy zahartowanej.
Przy hartowaniu przedmiotów stalowych nie następuje zwykle zahartowanie na wskroś, gdyż
szybkość chłodzenia jest większa na powierzchni, a mniejsza w rdzeniu. Rozkład szybkości
chłodzenia na przekroju okrągłego pręta podczas hartowania przedstawiono w przybliżeniu linią
ciągłą na rys. 5.28a. Jeżeli szybkość hartowania w środkowej części pręta będzie mniejsza od
krytycznej szybkości hartowania V
kr
to pręt nie zahartuje się na wskroś, jego struktura w rdzeniu
będzie się składała z perlitu i bainitu, a głębokość strefy zahartowanej będzie równa tylko
grubości warstwy zakreskowanej.

82
JW
Strefa nie zahartowana
Rys. 5.29. Hartowanie pręta stalowego V
kr
- krytyczna szybkość chłodzenia. V
p
-
szybkość chłodzenia powierzchni. V
r
- szybkość chłodzenia rdzenia
Na rysunku 5.29 przedstawiono również wykres CTP, na którym naniesiono linie szybkości
chłodzenia: powierzchni – V
p
i rdzenia - V
r
próbki oraz zaznaczono szybkość krytyczną V
kr
. Jest
oczywiste, że ze zmniejszeniem krytycznej szybkości hartowania wzrasta głębokość warstwy
zahartowanej. Tak więc, im mniejsza jest V
kr
dla danej stali, tym większa jest jej hartowność.
Wartość V
kr
jest ściśle związana z szybkością przemiany austenitu w struktury perlityczne, a
zatem z położeniem krzywej początku przemiany na wykresie CTP, które z kolei zależne jest od
gatunku stali.
Głębokość warstwy zahartowanej zmienia się także zależnie od użytego środka oziębiającego.
Jeżeli środek oziębiający szybciej będzie odbierał ciepło ze stali, to na większej głębokości od
powierzchni stal będzie chłodzona z szybkością większą od krytycznej. Na przykład warstwy
zahartowane w przedmiotach chłodzonych w wodzie są grubsze niż warstwy po hartowaniu w
oleju.
Jako głębokość warstwy zahartowanej przyjmuje się zwykle umownie odległość od
powierzchni do początku warstwy o strukturze półmartenzytycznej, czyli do tej warstwy
przekroju, w której struktura składa się w 50% z martenzytu i w 50% ze struktur
niemartenzytycznych. Największa średnica pręta okrągłego, przy której zachodzi zahartowanie
na wskroś (tj. w środku pręta będzie 50% martenzytu), nazywana jest średnicą krytyczną D
o
.
Strefę półmartenzytyczną można łatwo określić na podstawie badań mikrostruktury lub, co jest
łatwiejsze, na podstawie pomiarów twardości. Należy zaznaczyć, że twardość strefy
półmartenzytycznej, podobnie jak twardości martenzytu, zależy od zawartości węgla i dla
różnych gatunków stali będzie inna.
Metody określania hartowności
Metoda krzywych
U. Hartowność i średnicę krytyczną dla danego gatunku stali można
określić metodą pomiaru twardości na przekroju poprzecznym zahartowanej próbki. W tym celu
poddaje się hartowaniu w tych samych warunkach kilka próbek różnych średnicach, następnie
przecina się je i dokonuje pomiaru twardości wzdłuż średnicy próbki. Wyniki pomiarów nanosi
się na wykres, który wyglądem przypomina literę U (rys . 5.30). Stąd metoda ta nosi nazwę
krzywych U. W celu pełnego scharakteryzowania hartowności badanej stali, wyniki pomiarów
twardości dla wszystkich próbek nanosi się na jeden zbiorczy wykres. Na rysunku 5.31
przedstawione są dwa wykresy zbiorcze dla stali węglowej o zawartości 0,4% C i stali
chromowej o zawartości 0,45% C i 1,0% Cr. Twardość struktury półmartenzycznej dla stali o tej
zawartości węgla wynosi ok. 45 HRC. Jak widać na wykresie, stal stopowa ma większą
hartowność, gdyż próbka o średnicy 50 mm zahartowała się na wskroś (D
o
=
50 mm), natomiast
83
JW
w przypadku stali węglowej na wskroś zahartowała się jedynie próbka o średnicy ok. 15 mm (D
o
=
15 mm).
Rys. 5.30. Badanie hartowności stali metodą krzywych U. Rozkład twardości na
przekroju próbki
Rys. 5.31. Krzywe U dla prętów o różnej średnicy: a) stal węglowa o zawartości 0,45%
C, b) stal stopowa o zawartości 0,40% C i 1,0% Cr
Metoda Jominy'ego.
Metoda krzywych U jest dość kłopotliwa, gdyż wymaga wykonania i
przebadania wielu próbek. Z tego względu obecnie najczęściej stosowaną metodą oznaczania
hartowności stali jest metoda hartowania od czoła (Jminy'ego). Próba ta jest znormalizowana i
opisana w normie PN-79/H-04402 polega ona na nagrzaniu próbki o znormalizowanych
wymiarach (
φ 25 mm, długość 100 mm) do temperatury austenityzacji i następnie oziębieniu jej
od czoła strumieniem wody. Następnie po obu stronach próbki wzdłuż tworzącej dokonuje się
pomiarów twardości metodą Rockwella lub Vickersa. Średnie arytmetyczne kolejnych pomiarów
z obu stron próbki nanosi się na wykres przedstawiający zmianę twardości w funkcji odległości
od czoła (rys. 5.31). Korzystając z tego wykresu oraz znając twardość struktury
półmartenzytycznej dla danej stali, można określić, w jakiej odległości od czoła otrzymamy
strukturę półmartenzytyczną. Następnie na podstawie odpowiednich nomogramów
uwzględniających ośrodek chłodzący można określić średnicę krytyczną Dp badanej stali.
Wykonując szereg prób hartowności dla różnych wytopów tego samego gatunku stali i nanosząc
wyniki pomiarów twardości na ten sam wykres, otrzymuje się tzw. pasmo hartowności (rys.
5.32
84
JW
Rys. 5.32 Kształt i wymiary próbki do badania hartowności metodą Jominy'ego; na tle
próbki krzywa rozkładu twardości wzdłuż tworzącej
Rys.5.33. Pasmo hartowności dla stali węglowej o zaw. ok. 0,50% C
5.7. Odpuszczanie
Hartowanie martenzytyczne jest pierwszym etapem obróbki cieplnej stali konstrukcyjnych.
Mała plastyczność i duże naprężenia własne uniemożliwiają bezpośrednie stosowanie stali
konstrukcyjnej w takim stanie, jaki otrzymuje się po hartowaniu. Niezbędny jest następny zabieg
cieplny - odpuszczanie, który zwiększa plastyczność i ciągliwość, a zmniejsza naprężenia
własne. Odpuszczanie jest więc końcowym zabiegiem obróbki cieplnej (ulepszania cieplnego)
stali konstrukcyjnej, ustalającym ostatecznie jej własności. Wyjątek stanowi tu jedynie
hartowanie bainityczne, po którym odpuszczanie nie jest wymagane.
Odpuszczanie polega na nagrzaniu uprzednio zahartowanej stali do temperatury niższej od
temperatury przemiany eutektoidalnej i chłodzeniu do temperatury otoczenia.
Zależnie od stosowanej temperatury rozróżnia się odpuszczanie niskie, średnie i wysokie.
Odpuszczanie niskie przeprowadza się w zakresie temperatury 150-250°C celem
usunięcia naprężeń hartowniczych, przy zachowaniu dużej twardości i odporności na
ścieranie. Stosuje się głównie do stali narzędziowych.
Odpuszczanie średnie przeprowadza się w zakresie temperatury 250-500°C w celu
uzyskania przez stal dużej wytrzymałości i sprężystości. Twardość ulega przy tym dość
znacznemu obniżeniu. Tego rodzaju odpuszczaniu poddaje się sprężyny, resory, matryce,
części silników, samochodów itp.
Odpuszczanie wysokie przeprowadza się w zakresie temperatury powyżej 500°C i poniżej
A
cl
. Ma ono na celu m.in. uzyskanie możliwie najwyższej udarności dla danej stali, przy
jednoczesnym zwiększeniu stosunku R
e
do R
m
. Stal konstrukcyjna odpuszczona wysoko po
hartowaniu uzyskuje strukturę sorbityczną i odznacza się z reguły wyższą granicą
plastyczności i wyższym wydłużeniem i przewężeniem niż ta sama stal o strukturze
perlitycznej. Podczas wysokiego odpuszczania, poza zmianami strukturalnymi, zachodzi
85
JW
jednocześnie prawie całkowite usunięcie naprężeń powstałych podczas hartowania.
Odpuszczanie wysokie stosuje się do większości stali konstrukcyjnych.
Temperaturę i czas odpuszczania dobiera się w zależności od własności, jakie mają być
otrzymane.
Schemat zmian własności mechanicznych stali konstrukcyjnych w zależności od temperatury
odpuszczania przedstawiony jest na rys. 5.34.
Rys. 5.34. Zmiana własności mechanicznych stali konstrukcyjnej w zależności od
temperatury odpuszczania
Kruchość odpuszczania
. Temperatura odpuszczania i szybkość chłodzenia przy
odpuszczaniu mają znaczny wpływ na udarność konstrukcyjnej stali stopowej. W przypadku
powolnego chłodzenia stali po odpuszczaniu
krzywa charakteryzująca jej udarność ma dwa
minima: dla około 300°C i około 500 ÷ 600°C. Jest to zjawisko tzw. kruchości odpuszczania
pierwszego i drugiego rodzaju.
Rys. 5.35. Wpływ temperatury odpuszczania i szybkości chłodzenia po odpuszczaniu
na udarność stali
Kruchość odpuszczania pierwszego rodzaju powstaje podczas odpuszczania w
temperaturze około 300°C niezależnie od składu chemicznego stali i szybkości chłodzenia po
odpuszczaniu (rys. 5.35 i 5.36). Z tego względu należy unikać odpuszczania w tym zakresie
temperatury.
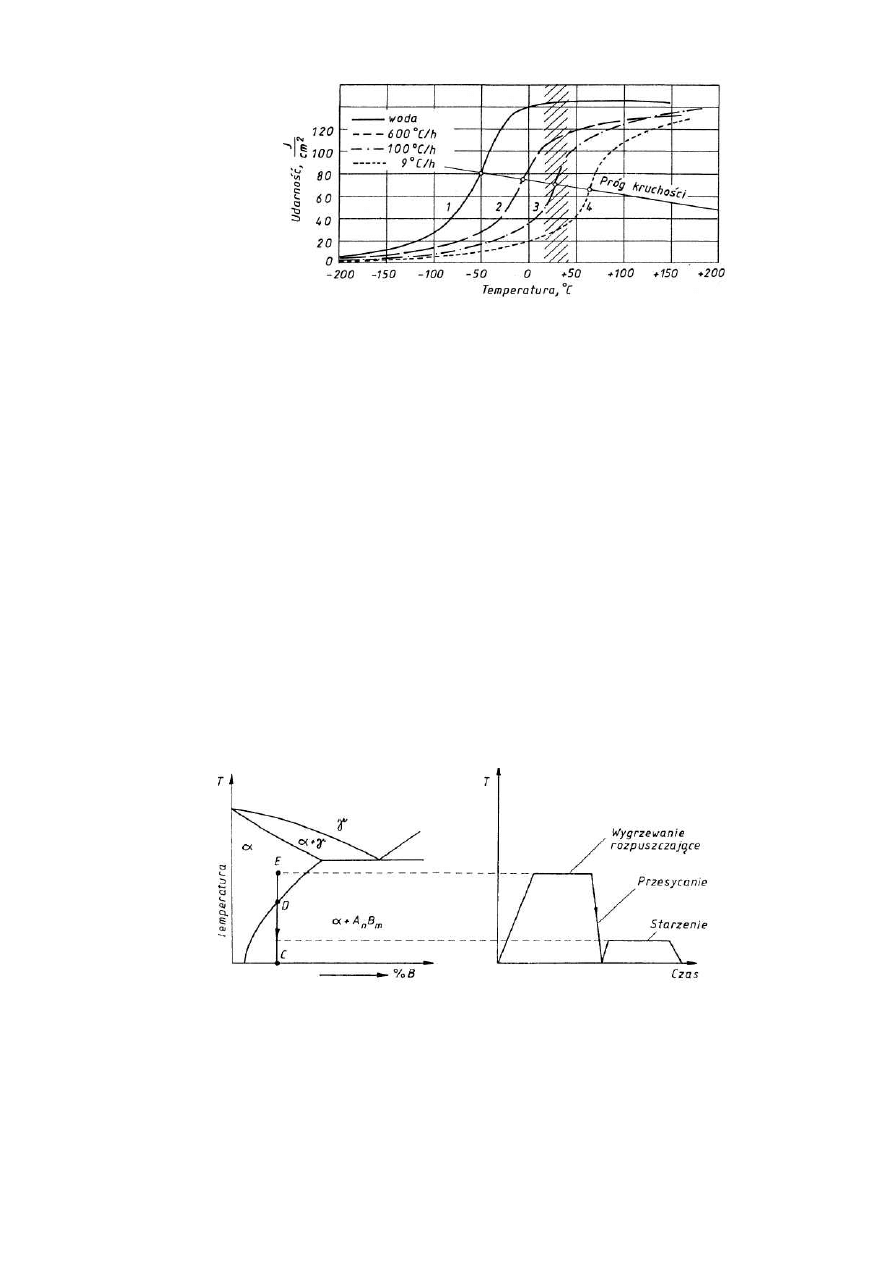
86
JW
Rys. 5.36. Udarność stali w różnych temperaturach w zależności od szybkości chłodzenia
po odpuszczaniu
Kruchość odpuszczania drugiego rodzaju
ujawnia się po odpuszczaniu w temperaturze
powyżej 500°C w przypadku, gdy po odpuszczaniu przedmiot jest chłodzony powoli,
natomiast w razie szybkiego chłodzenia udarność nie zmniejsza się, wzrasta monotonicznie z
podwyższaniem temperatury odpuszczania (rys. 5.35.). Wzrost szybkości chłodzenia po
odpuszczaniu powoduje również przesunięcie progu kruchości w kierunku niższych
temperatur (rys. 5.36). Skłonność do kruchości odpuszczania drugiego rodzaju wykazują tylko
niektóre konstrukcyjne stale stopowe np. chromowo-manganowe, chromowo-niklowe lub
chromowo-wanadowe, natomiast nie są do niej skłonne np. stale węglowe i stale stopowe z
dodatkiem Mo.
5.8. Przesycanie i starzenie stopów żelaza
Przesycaniem nazywa się operację cieplną polegającą na: 1) nagrzaniu stali do temperatury, w
której wydzielona faza przechodzi do roztworu stałego, tj. powyżej temperatury granicznej
rozpuszczalności, 2) wygrzaniu w tej temperaturze, 3) oziębieniu w celu zatrzymania
rozpuszczonego składnika w roztworze przesyconym (rys. 4.37). Stan przesycony jest
nietrwały i stop dąży do przejścia w stan równowagi, co może nastąpić stosunkowo łatwo
np. po podgrzaniu. W stanie przesyconym stop ma większą plastyczność, natomiast twardość i
wytrzymałość ulegają zmniejszeniu.
Przesycanie stosowane jest np. do stali chromowo-niklowej o strukturze austenitycznej
(stale kwasoodporne) lub o dużej zawartości manganu. Stale te nagrzewa się do temperatury
ok. 1100°C i następnie oziębia się w wodzie. Celem tego zabiegu jest rozpuszczenie
węglików i uzyskanie jednorodnej struktury austenitycznej. Obróbka taka zwiększa przede
wszystkim odporność na korozję międzykrystaliczną stali typu 18-8 (18% Cr, 8% Ni).
Przesycanie stosuje się również w przypadku wysokostopowych stali żarowytrzymałych i
stali o specjalnych własnościach magnetycznych. Zabieg ten ponadto jest szeroko
stosowany do wielu stopów metali nieżelaznych.

87
JW
Starzenie polega na nagrzaniu i wytrzymaniu uprzednio przesyconego roztworu w
temperaturze znacznie niższej od temperatury granicznej rozpuszczalności w celu wydzielenia
o odpowiednim stopniu dyspersji składnika lub składników znajdujących się w nadmiarze
w przesyconym roztworze stałym. W przypadku niektórych stopów procesy starzenia
zachodzą już w temperaturze otoczenia, co nosi nazwę starzenia naturalnego
(samorzutnego). W czasie starzenia zachodzą zmiany strukturalne zbliżające skład stopu do
stanu równowagi. Wydzielanie się w czasie starzenia składnika (znajdującego się w
przesyconym roztworze stałym) w postaci skupień lubfaz o dużej dyspersji powoduje
utwardzanie stopu. Z tego względu połączenie zabiegów przesycenia i starzenia nosi nazwę
utwardzania wydzieleniowego.
W stalach niskowęglowych przeznaczonych do głębokiego tłoczenia, a także w
stalach kotłowych starzenie jest niekorzystne, gdyż obniża plastyczność i powoduje
kruchość. Zjawisko to występuje silniej w stalach nieuspokojonych, gdyż oprócz węgla w
ferrycie rozpuszczony jest także azot, który tworzy z żelazem fazę międzywęzłową Fe
4
N.
Szybkie chłodzenie np. od temperatury walcowania powoduje zatrzymanie prawie całej ilości
rozpuszczonych składników w ferrycie, które następnie wydzielają się podczas starzenia,
zwłaszcza na granicach ziarn. Starzenie może zachodzić już w temperaturze otoczenia,
zwłaszcza w ciągu dłuższych okresów czasu, i powoduje pogorszenie własności plastycznych
stali.

90
JW
6. Obróbka cieplno-chemiczna stali
6.1. Wiadomości ogólne
Obróbkę cieplno-chemiczną, podobnie jak omówione już hartowanie powierzchniowe, stosuje
się w celu uzyskania wysokiej twardości warstwy powierzchniowe przedmiotu, przy zachowaniu
ciągliwego rdzenia. Zapewnia to dużą odporność ni ścieranie i wysoką wytrzymałość na
obciążenia dynamiczne, a w niektórych przepadkach zabezpiecza stal przed korozją.
W stosunku do hartowania powierzchniowego obróbka cieplno-chemiczną jest procesem
mniej wydajnym, ale za to zapewnia większe różnice między własnościami rdzenia i warstwy
powierzchniowej, gdyż są one wynikiem nie tylko różnic struktury, ale także składu
chemicznego. Dodatkową jej zaletą jest możliwość stosowania do dowolnych przedmiotów,
niezależnie od ich kształtu (hartowanie powierzchniowe jest niemożliwe przy zbyt
skomplikowanych kształtach).
Obróbka cieplno-chemiczną polega na dyfuzyjnym wprowadzeniu do przypowierzchniowej
warstwy przedmiotu obcego pierwiastka, celem spowodowania odpowiednich zmian jej
własności (w niektórych przypadkach właściwy efekt uzyskuje się dopiero po dodatkowej
obróbce cieplnej). Ogólnie dzieli się na:
• dyfuzyjne nasycanie niemetalami (nawęglanie, azotowanie, utlenianie, siarkowanie,
borowanie, krzemowanie),
• dyfuzyjne nasycanie metalami (aluminiowanie, chromowanie, cynkowanie,
tytanowanie),
• dyfuzyjne nasycanie wieloskładnikowe (węgloazotowanie, węglotytanowanie
siarkowęgloazotowanie itd.).
Obróbkę cieplno-chemiczną przeprowadza się w środowisku bogatym w składnik
dyfundujący do stali. W większości przypadków stosuje się środowisko gazowe i wówczas w
czasie obróbki zachodzą trzy podstawowe procesy:
a) dysocjacja — polegająca na rozkładzie cząsteczek gazu i utworzeniu aktywnych atomów
pierwiastka dyfundującego, np.
2CO
→ CO
2
+ C,
NH
3
→ 3H + N;
b) adsorpcja — polegająca na wchłanianiu (rozpuszczaniu) wolnych atomów przez
powierzchnię metalu (zachodzi tylko wtedy, gdy pierwiastek wprowadzany rozpuszcza
się w obrabianym metalu),
c) dyfuzja — polegająca na przemieszczaniu się obcych atomów w sieci przestrzennej
obrabianego metalu.
W wyniku tych trzech procesów powstaje warstwa dyfuzyjna, w której stężenie
dyfundującego pierwiastka osiąga maksimum na powierzchni i maleje w miarę oddalania się od
niej.
Przemieszczanie dyfuzyjne atomów uwarunkowane jest następującymi czynnikami:
• wzajemną rozpuszczalnością metalu nasycanego i pierwiastka nasycającego,
• dążeniem układu do wyrównywania składu chemicznego w całej objętości,
• ruchami cieplnymi atomów.
Szybkość przemieszczania się atomów, czyli szybkość dyfuzji w dużej mierze zależy od
temperatury i wzrasta z jej podwyższeniem.
W zależności od przebiegu dyfuzji rozróżnia się dyfuzję atomową i dyfuzję reakcyjną.
Dyfuzja atomowa polega na przemieszczaniu się atomów jednego pierwiastka do sieci
elementarnej pierwiastka drugiego, przy czym powstaje roztwór stały o sieci elementarnej
pierwiastka rozpuszczającego. Tworzenie się nowych faz o budowie różnej od budowy
pierwiastka rozpuszczającego jest niemożliwe i maksymalne stężenie pierwiastka dyfundującego
nie przekracza granicznej rozpuszczalni w temperaturze dyfuzji. Widać więc, że przy dyfuzji
atomowej zachodzi jedynie zmiana stężenia składnika rozpuszczanego w sieci elementarnej

91
JW
składnika rozpuszczającego, tworzącego w wyniku tego roztwór stały. Zmiana ta powoduje, że
różnice we własnościach warstwy powierzchniowej metalu rozpuszczającego, czemu towarzyszy
zazwyczaj zmiana mikrostruktury. Zdarzają się jednak przypadki, że mikrostruktura powstałego
roztworu stałego nie różni się wyraźnie od mikrostruktury metalu rozpuszczającego i ujawnienie
granicy między nimi jest trudne.
Mechanizm dyfuzji atomowej jest następujący: Jak wiadomo, temperatura określa wielkość
energii układu, która rozdzielona jest między poszczególne atomy nierównomiernie. W związku
z tym w sieci elementarnej znajduje się pewna ilość atomów, których energia jest znacznie
większa niż atomów pozostałych. Energia ta przejawia się w drganiach i atom mający taką
zwiększoną ilość energii wychodzi ze swego normalnego położenia w węźle sieci elementarnej,
zajmując położenie nienormalne - międzywęzłowe. W sieci elementarnej pojawia się puste
miejsce (wakans).
Istnienie wolnych miejsc w sieci elementarnej umożliwia powstawanie na drodze dyfuzji tzw.
roztworów stałych różnowęzłowych, tzn. roztworów, w których część atomów w węzłach sieci
metalu rozpuszczającego jest zastąpiona atomami pierwiastka rozpuszczonego.
Nieco inaczej przebiega proces powstawania na drodze dyfuzji roztworów stałych
międzywęzłowych. W tym przypadku atomy pierwiastka rozpuszczanego wnikają w przestrzenie
międzywęzłowe sieci elementarnej metalu rozpuszczającego. Taki proces zachodzi przede
wszystkim w przypadku dyfuzji pierwiastków o małych średnicach atomowych, jak wodór,
węgiel, azot, czy bor
.
Energię, która jest konieczna do przesunięcia atomu z jednego położenia w sieci elementarnej
w drugie, nazywa się energią aktywacji. Oczywiście energia ta podczas tworzenia się stałych
roztworów międzywęzłowych jest znacznie mniejsza niż podczas tworzenia się roztworów
różnowęzłowych, gdyż w pierwszym przypadku odpada konieczność przesuwania atomów
metalu rozpuszczającego z węzłów sieci w położenie nienormalne.
Drugim rodzajem dyfuzji jest dyfuzja reakcyjna, której wynikiem jest powstanie nowej fazy
międzymetalicznej, zgodnie z wykresem równowagi między pierwiastkiem rozpuszczanym i
metalem rozpuszczającym. Proces dyfuzji reakcyjnej można podzielić na dwa etapy:
a) powstanie nowej fazy na powierzchni metalu na skutek zachodzącej reakcji chemicznej,
b) rozrost nowej fazy na skutek zachodzącej dyfuzji.
W wielu przypadkach z wykresu równowagi wynika, że pierwiastek rozpuszczany może
tworzyć z metalem rozpuszczającym zarówno graniczne roztwory stałe, jak i fazy
międzymetaliczne. Powstaje więc pytanie, która dyfuzja (atomowa czy reakcyjna) zachodzi
wcześniej. Przeważa opinia, że faza międzymetaliczna powstaje w drugiej kolejności, tzn. po
granicznym nasyceniu metalu rozpuszczającego pierwiastkiem rozpuszczanym.
Budowa faz międzymetalicznych i ich skład chemiczny zależą od takich czynników, jak
budowa krystaliczna reagujących pierwiastków, stan ich powierzchni, temperatura itp.
Wielkością charakterystyczną dla procesów dyfuzyjnych jest tzw. współczynnik dyfuzji D,
wyrażany równaniem
D = D
o
e
-Q/RT
gdzie: D
o
- stały współczynnik dla danej sieci krystalicznej,
e - podstawa logarytmów naturalnych,
Q - energia aktywacji (dla wytrącenia atomu z położenia równowagi),
R - stała gazowa,
T - temperatura bezwzględna.
Jak widać, współczynnik ten zależy przede wszystkim od temperatury i wzrasta wraz z jej
wzrostem (powiększa się ilość wolnych miejsc w węzłach sieci).
Grubość warstwy dyfuzyjnej w zależności od czasu (przy ustaleniu pozostałych parametrów
procesu, takich jak temperatura, ciśnienie itd.), określa równanie
Y
2
= k
τ
gdzie: y - grubość warstwy dyfuzyjnej,
k - stała zależna od współczynnika dyfuzji,
τ
-
czas procesu.

92
JW
Dyfuzja w metalach może zachodzić bądź równolegle do powierzchni, bądź w głąb ziarn,
bądź też wzdłuż ich granic. Doświadczalnie ustalono, że największa wartość ma współczynnik
dyfuzji na powierzchni metalu, mniejszą na granicy ziarn, a najmniejszą w samych ziarnach.
Ustalono także, że szybkość dyfuzji w metalach o elementarnej sieci regularnej (żelazo,
aluminium, nikiel, kobalt, miedź, molibden: praktycznie nie zależy od kierunku osi
krystalograficznych. Natomiast metale o innych sieciach wykazują pewną anizotropowość
dyfuzji. Spośród różnych procesów obróbki cieplno-chemicznej najczęściej są stosowane
nawęglanie, azotowanie i węgloazotowanie.
6.2. Węgloutwardzanie cieplne
Węgloutwardzaniem cieplnym nazywa się zespól operacji polegających na kolejnym
dyfuzyjnym nasycaniu węglem przypowierzchniowych stref przedmiotu (nawęglaniu),
hartowaniu i niskim odpuszczaniu. W wyniku tego procesu uzyskuje się twardą (60-64 HRC),
odporną na ścierania warstwę powierzchniową przy zachowaniu ciągliwego rdzenia. W
niektórych przypadkach stosuje się wzbogacanie węglem do określonego stężenia warstw
powierzchniowych przedmiotu, odwęglonych w poprzedzających operacjach technologicznych.
Taki proces nazywa się dowęglaniem.
Rozróżnia się dwa rodzaje nawęglania: w proszkach i gazowe.
Nawęglanie w proszkach przeprowadza się w szczelnych skrzynkach wykonanych
ze stali żaroodpornych lub stopów niklowo-chromowych, wypełnionych najczęściej mieszaniną
węgla drzewnego (w postaci granulek o średnicy kilku mm) intensyfikatorów, czyli środków
przyspieszających nawęglanie. Mieszaninę tę nazywa się proszkiem do nawęglania lub
karburyzatorem. Jako intensyfikatory zwykle stosuje się węglan baru (BaCO
3
) oraz węglan sodu
(Na
2
CO
3
,), których zawartość w świeżym proszku do nawęglania wynosi 10-30%.
W temperaturze nawęglania (900-950°C) tlen zawarty w powietrzu znajdującym się między
granulkami łączy się z węglem, tworząc z powodu małej ilości tlenek węgla CO. Tlenek ten w
zetknięciu z żelazem rozkłada się wg reakcji
2CO
→ CO
2
+ C,
tworząc węgiel atomowy (in statu nascendi), wchłaniany przez powierzchnię przedmiotu.
Jednocześnie zachodzą reakcje:
BaC0
3
+ C
→ BaO + 2CO,
2CO
→ CO
2
+ C,
które uaktywniają proces nawęglania.
Proces nawęglania w proszkach jest długotrwały (czasem trwa do kilkudziesięciu godzin) i nie
można go kontrolować, dlatego stosuje się go jedynie w produkcji jednostkowej i małoseryjnej.
Natomiast w produkcji masowej powszechnie stosuje nawęglanie gazowe, polegające na
wygrzewaniu obrabianych przedmiotów w komorze pieca, przez którą z określoną szybkością
przepływa gaz nawęglający.
Jako gazy nawęglające najczęściej stosuje się gaz ziemny, gaz świetlny, gaz generatorowy i
gaz koksowniczy, gazy pochodzące z rozkładu benzolu i ropy naftowej, a także specjalnie
wytwarzane mieszaniny tych gazów z węglowodorami.
Najtańszym i najsilniej działającym jest gaz ziemny, zawierający 92-96% metanu CH
4
. Nie
można go jednak stosować w czystej postaci, gdyż powstaje wtedy dużo sadzy, która pokrywa
obrabiane przedmioty, hamując ich nawęglanie. Dlatego zwykle używa się go po połączeniu z
produktami niezupełnego spalania gazu ziemnego. W wyniku otrzymuje się mieszaninę głównie
CH
4
, CO i N
2
, która charakteryzuje się dobrą aktywnością, a nie wytwarza sadzy. W
temperaturze nawęglania (900-950°C) zachodzą reakcje
:
CH
4
→ 2H
2
+ C,
2CO
→ CO
2
+ C
,
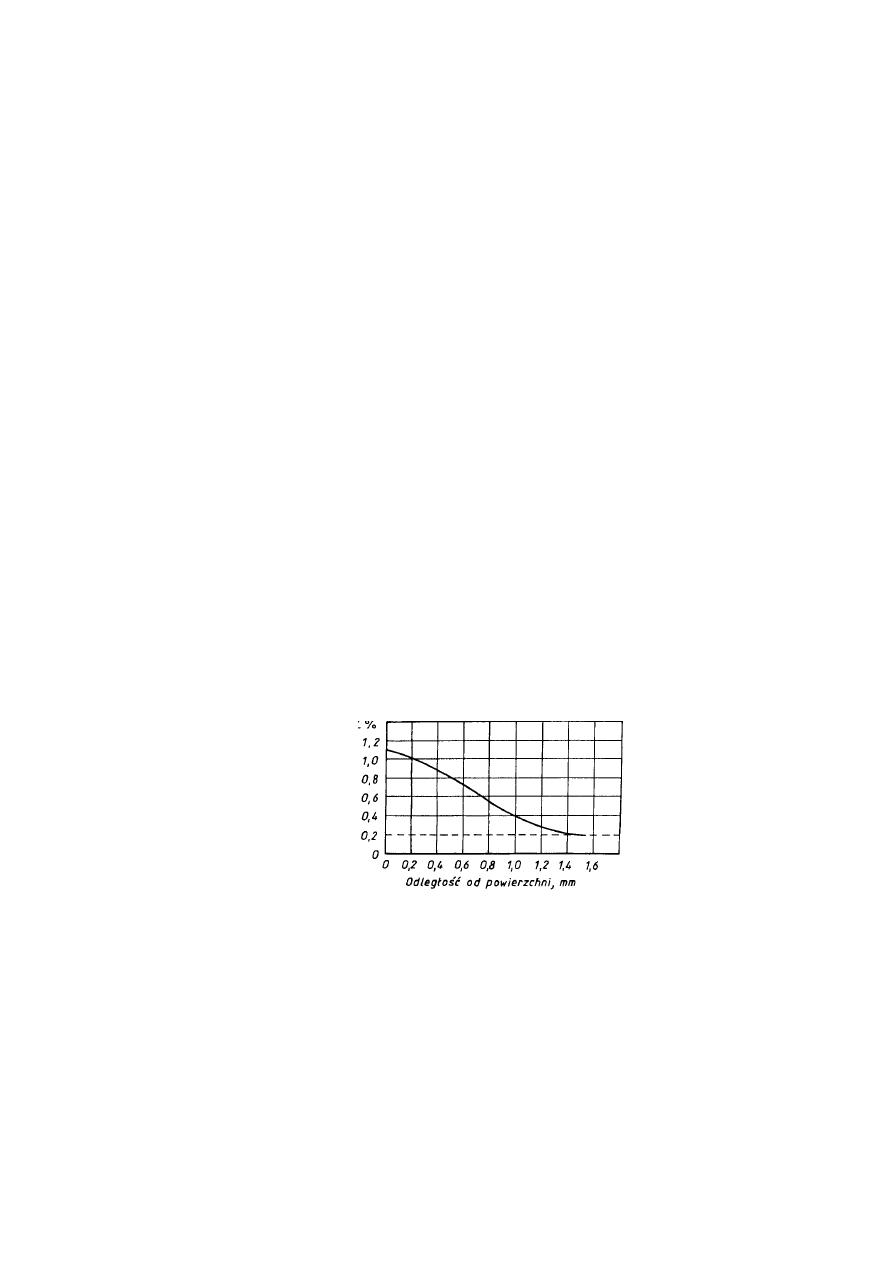
93
JW
w wyniku których powstaje węgiel „in statu nascendi", dyfundujący do stali.
Główną zaletą nawęglania gazowego jest możliwość regulowania stężenia węgla w warstwie
powierzchniowej zarówno przez dobór składu gazu nawęglającego, jak i zmiany szybkości jego
przepływu przez komorę pieca. Proces trwa krócej niż nawęglanie w proszkach (unika się
nagrzewania skrzynki i karburyzatora), jest prostszy (unika się czynności związanych z
umieszczaniem przedmiotów obrabianych w skrzynkach i ich wyjmowaniem po nawęglaniu) i
łatwiejszy do automatyzacji. Dodatkową zaletą jest możliwość hartowania przedmiotów
bezpośrednio po nawęgleniu.
Nawęglaniu poddaje się wyroby ze stali niestopowych niskowęglowych (np gatunku 10, 15,
20 wg PN-93/H-84019) lub ze stali stopowych konstrukcyjnych (np. gatunku 15H, 20H, 16HG,
15HGM, 17HGN wg PN-89/H-84030/02), o zawartości węgla 0,07-0,24%.
Głębokość warstwy nawęglonej dla danych warunków nawęglania zależy od temperatury i
czasu trwania procesu. Im wyższa temperatura (praktycznie wynosi 900-950°C), tym szybkość
nawęglania jest większa, ale niezależnie od temperatura proces nawęglania najintensywniej
zachodzi w pierwszym okresie, a potem stopniowo szybkość jego się zmniejsza. W praktyce
grubość warstwy nawęglonej zawiera się w granicach 0,6-2,0 mm.
Zawartość węgla na powierzchni nawęglonej jest określona graniczną rozpuszczalnością
węgla w austenicie w temperaturze procesu. Na przykład dla temperatury nawęglania 900°C,
zgodnie z wykresem równowagi Fe-Fe
3
C, wynosi ona 1,2-1,3%. W miarę oddalania się od
powierzchni zawartość węgla stopniowo maleje, aż do zawartości odpowiadającej stali
nienawęglonej. W związku z tym warstwa nawęglona po powolnym chłodzeniu od temperatury
nawęglania składa się z:
a) przypowierzchniowej warstwy nawęglonej do zawartości węgla powyżej 0,8%; jest to strefa
nadeutektoidalna o strukturze złożonej z perlitu otoczonego cienką siatką cementytu
;
b) warstwy o zawartości węgla około 0,8%; jest to strefa eutektoidalna o strukturze perlitu;
c) warstwy o zawartości węgla poniżej 0,8%; jest to strefa podeutektoidalna
o strukturze perlityczno-ferrytycznej, przechodząca stopniowo w ferrytyczno-perlityczną
strukturę rdzenia.
Rozkład stężenia węgla w warstwie nawęglonej pokazano na rys. 6.1, a strukturę warstwy -
na rys. 6.
2.
6.1. Rozkład stężenia węgla w warstwie nawęglonej (linią przerywaną zaznaczono
stężenie węgla w rdzeniu
Maksymalna zawartość węgla w strefie nadeutektoidalnej nie powinna przekraczać 1,1-1,2% C.
W przeciwnym przypadku podczas chłodzenia tworzy się duża ilość cementytu wtórnego, który
zwiększa jej kruchość.
W wielu przypadkach niektóre powierzchnie przedmiotów muszą być chronione przed
nawęglaniem. Zabezpiecza się je bądź przez pokrycie odpowiednimi pastami (złożonymi z
glinki, piasku, boraksu, szkła wodnego itd.), bądź przez galwaniczne miedziowanie (warstwa
grubości kilku setnych mm). Jak już wspomniano, celem węgloutwardzania jest wytworzenie
twardej i odpornej na ścieranie warstwy powierzchniowej, przy zachowaniu ciągliwego rdzenia.
Samo nawęglanie wytwarza jedynie korzystny rozkład węgla na przekroju przedmiotu, a
ostateczny efekt uzyskuje się dopiero przez odpowiednią obróbkę cieplną. Mniej
odpowiedzialne przedmioty można hartować bezpośrednio z temperatury nawęglania. W tym

94
JW
jednak przypadku z ziarn austenitu rozrośniętych na skutek długotrwałego wygrzewania w
wysokiej temperaturze otrzymuje się gruboiglasty martenzyt i gruboziarnistą strukturę rdzenia.
W warstwie nawęglonej występuje ponadto większa ilość austenitu szczątkowego (wyższa
temperatura hartowania), co oczywiście zmniejsza jej twardość. Przedmioty bardziej
odpowiedzialne po nawęgleniu chłodzi się w powietrzu, a następnie hartuje z temperatury 850-
900°C, zależnie od gatunku stali. Najbardziej właściwą obróbką cieplną przedmiotów
nawęglonych, zapewniającą szczególnie wysokie własności, jest hartowanie dwustopniowe.
Pierwsze przeprowadza się z temperatury 850-900°C i jego głównym celem jest rozdrobnienie
struktury rdzenia oraz rozpuszczenie siatki cementytu w warstwie powierzchniowej (jeśli siatka
taka utworzyła się podczas nawęglania). W związku z tym w przypadku stali stopowych może
być zastąpione normalizowaniem. Powtórne hartowanie przeprowadza się z temperatury 760-
800°C, dzięki czemu w warstwie powierzchniowej powstaje drobnoiglasty martenzyt z
ewentualnymi wtrąceniami cementytu drugorzędowego. Ta temperatura jest jednak za niska do
zupełnego zahartowania rdzenia, toteż nawet w przypadku stali o dużej hartowności będzie on
miał strukturę złożoną z ferrytu i martenzytu niskowęglowego (stale o małej hartowności
zachowują w rdzeniu strukturę ferrytyczno-perlityczną
).
Rys. .2. Struktura nawęglonej stali 15 (ok. 0,15% C) nie poddanej obróbce cieplnej:
a) 100x; b), c), d), i e) 500x. Traw. 5% alkoholowym roztworem HN03
Końcową operacją jest w każdym przypadku odpuszczanie w temperaturze 150-200°C,
usuwające naprężenia własne.
Po prawidłowej obróbce cieplnej przedmioty nawęglone wykazują na powierzchni twardość
60-64 HRC, przy twardości rdzenia 25-35 HRC.
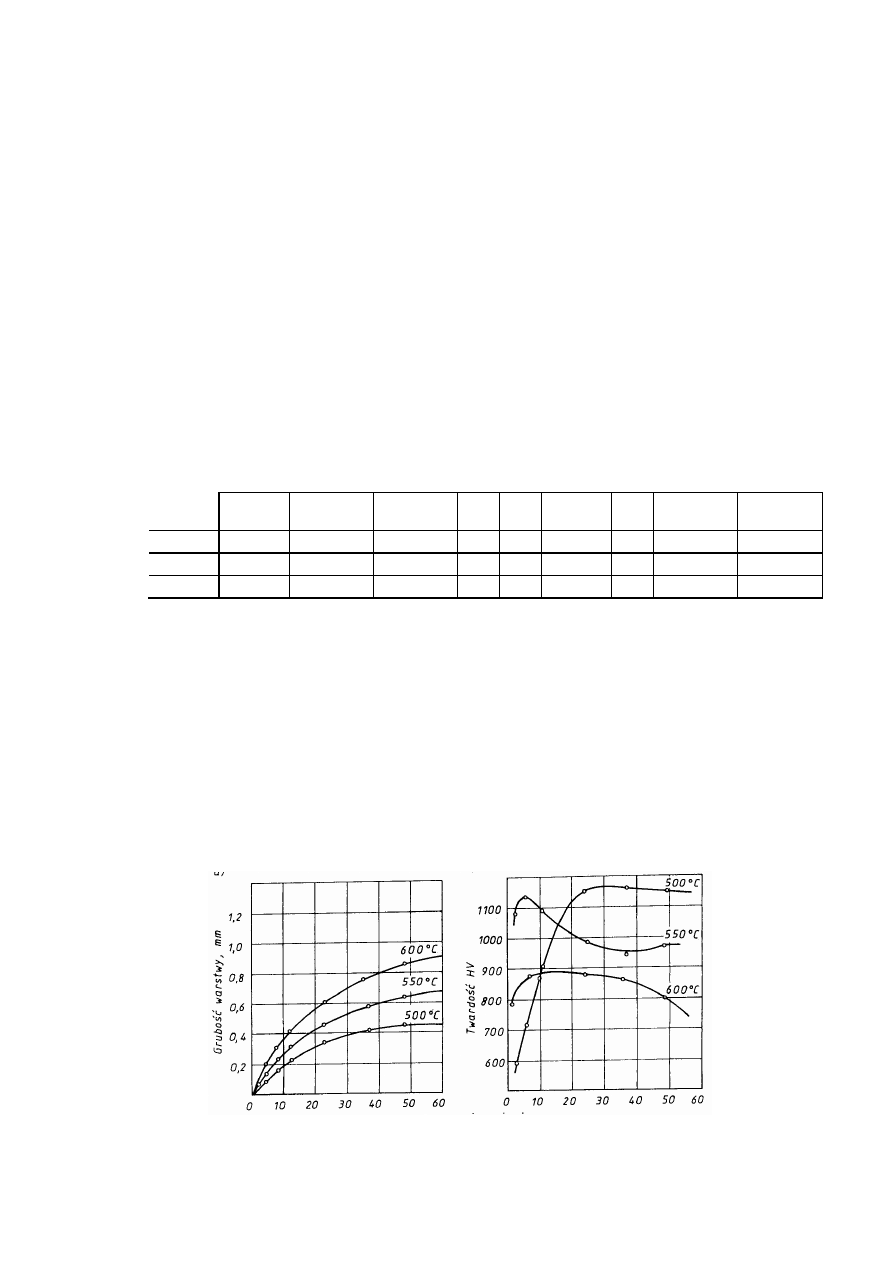
95
JW
6.3. Azotowanie
Azotowaniem nazywa się dyfuzyjne nasycanie azotem przypowierzchniowych
stref przedmiotu. Zależnie od celu azotowania i sposobu jego przeprowadzania rozróżnia się:
a) azotowanie utwardzające,
b) azotowanie przeciwkorozyjne.
W przeciwieństwie do nawęglania, azotowanie powoduje utwardzenie stref
przypowierzchniowych bez dodatkowej obróbki cieplnej. Dlatego azotowaniu poddaje się stale
uprzednio ulepszone cieplnie (hartowane i wysokoodpuszczone), przy czym temperatura ich
odpuszczania jest nieco wyższa od temperatury azotowania utwardzającego.
Azotowaniu utwardzającemu poddaje się specjalne stale stopowe, zawierające pierwiastki
tworzące trwałe azotki o wysokiej dyspersji (AlN, CrN, MoN), które zapewniają zachowanie
bardzo wysokiej twardości stali aż do temperatury ok. 500°C.
Skład chemiczny znormalizowanych w Polsce stali stopowych do azotowania PN-89/H-
84030/03) podano w tabl. 6.1. Stale te stosuje się głównie na części silników spalinowych, jak
np. wały korbowe, korbowody, sworznie tłokowe, koła zębate, wałki rozrządowe, pompy
paliwowe itd. Na przykład, twardość warstwy naazotwanej stali 38HMJ wynosi 900-1200 HV.
Skład chemiczny stali stopowych konstrukcyjnych do azotowania
(wg PN-89/H-84030/03) Tablica 5.1
Znak
Skład chemiczny, %
gatunku
stali
C Mn Si
P
max
S
max
Cr Ni
max
Mo Inne
38HMJ 0,35-0,42
0,30+0,60 0,17+0,37
0,025 0,025 1,35+1,65 0,25
0,15+0,25
0,70+1,10AI
33H3MF 0,29-0,36 0,50+0,80 0,17+0,37 0,035 0,035 2,40+2,80 0,30 0,35+0,45 0,20+0,30
V
25H3M 0,20+0,30
0,40+0,65 0,17+0,37
0,035 0,035 2,90+3,50 0,40 0,40+0,55
-
Azotowanie innych stali stopowych, a także stali węglowych nie zapewnia uzyskania tak
wysokich twardości warstwy powierzchniowej i stosowane jest jedynie w celu wytworzenia
warstwy odpornej na korozję lub w niektórych przypadkach (stale konstrukcyjne chromowo-
niklowe) — zwiększenia wytrzymałości na zmęczenie.
Azotowanie utwardzające przeprowadza się w temperaturze 480-600°C (najczęściej 500-
520°C), w czasie 10-100 h. Uzyskuje się twardą trudno ścieralną warstwę o grubości 0,1-0,6 mm
(najczęściej 0,2-0,3 mm), wyraźnie odcinającą się od nie naazotowanego rdzenia. Warstwa ta
zachowuje swą twardość do temperatury ok. 500°C (po nawęgleniu tylko do ok. 200°C). Wpływ
czasu azotowania i temperatury na grubość i twardość warstwy naazotowanej podano na rys. 5.3.
Maksymalna twardość warstwy naazotowanej występuje na głębokości około 0,05 mm od
powierzchni. Aby ją ujawnić, należy powierzchnię przeszlifować na tę głębokość.
a)
b)
Czas azotowania, h
Rys. 5.3. Wpływ czasu i temperatury azotowania stali 38HMJ na: a) głębokość warstwy
naazotowanej, b) twardość warstwy naazotowanej

96
JW
Azotowanie przeciwkorozyjne przeprowadza się w temperaturze 600-700°C w czasie 0,5-1 h
(maks. do kilku godzin). Uzyskuje się warstwę o grubość 0,02-0,04 mm, niezbyt twardą, ale
bardzo szczelną i odporną na korodujące działanie pary wodnej, wody wodociągowej i
atmosfery.
Azotowanie przeprowadza się w szczelnie zamkniętej mufli lub komorze pieca, do której z
określoną szybkością dopływa amoniak. W temperaturze azotowania zachodzi dysocjacja
amoniaku zgodnie z reakcją
NH
3
→ 3H + N,
przy czym obecność żelaza działa na tę reakcję katalitycznie. Powstający azot atomowy jest
adsorbowany przez powierzchnię stali, a następnie dyfunduje w głąb Stopień dysocjacji
amoniaku zależy od temperatury, ciśnienia, szybkości przepływu gazu przez komorę pieca i
wielkości wsadu (działającego jako katalizator).
6.4. Azotonawęglanie i węgloazotowanie
Obróbka cieplno-chemiczna polegająca na jednoczesnym dyfuzyjnym nasycaniu węglem i
azotem przypowierzchniowych stref przedmiotu dzieli się na:
• azotonawęglanie przeprowadzane zwykle w zakresie temperatury 800-880
o
C w którym
dominuje dyfuzja węgla,
• węgloazotowanie przeprowadzane zwykle w zakresie temperatury 500-600
o
C w którym
dominuje dyfuzja azotu.
Zależnie od sposobu przeprowadzania procesu rozróżnia się azotonawęglanie gazowe, w
przypadku stosowania atmosfery gazowej, oraz azotonawęglanie kąpielowe w przypadku
stosowania kąpieli cyjanowych. Azotonawęglanie stosuje się do różnego rodzaju części maszyn i
urządzeń (np. maszyn do pisania, broni, silników motocyklowych itd.) ze stali o małej i średniej
zawartości węgla zarówno węglowych, jak i stopowych. Ostateczne własności warstwy
przypowierzchniowej, podobnie jak po nawęglaniu, uzyskuje się dopiero po hartowaniu i niskim
odpuszczaniu. Łączny proces azotonawęglania, hartowania i niskiego odpuszczania nazywa się
azotowęgloutwardzaniem.
Węgloazotowanie (zwykle kąpielowe) stosuje się do narzędzi ze stali szybkotnących jako
końcowy zabieg (po obróbce cieplnej) mający na celu podwyższenie powierzchniowej
twardości, odporności na ścieranie, a także żaroodporności. Warstwa azotonawęglona w
stosunku do warstwy nawęglonej wykazuje większą twardość i odporność na ścieranie oraz
większą odporność na korozję. Po azotonawęglaniu kąpielowym przedmioty mają ładną, matową
powierzchnię, toteż obróbkę stosuje się również w celu nadania im estetycznego wyglądu.
Azotonawęglanie, podobnie jak azotowanie, podwyższa wytrzymałość zmęczeniową stali (w
warstwie powierzchniowej występują naprężenia ściskające). Hartowanie można przeprowadzać
bezpośrednio po azotonawęglaniu, gdyż niższa temperatura i krótszy czas procesu nie wywołują
rozrostu ziarna, jak to ma miejsce podczas nawęglania.
Najczęściej stosowanymi w praktyce procesami są azotonawęglanie i węgloazotowanie
kąpielowe w roztopionych solach, zawierających zwykle cyjanek sodu NaCN, cyjanek potasu
KCN, chlorek sodu NaCl, chlorek baru BaCl
2
i węglan sodu Na
2
CO
3
.
Cyjanki stanowią aktywną część kąpieli i ilość ich waha się w granicach 7 – 70%, w
zależności od wymaganej aktywności kąpieli. Zwiększenie zawartości cyjanków sprzyja
zwiększeniu zawartości węgla i azotu w warstwie nasycanej, ale nie zwiększa jej głębokości,
która zależy od temperatury i czasu nasycania.
W temperaturze procesu NaCN reaguje z tlenem, wskutek czego powstają azot i węgiel w
stanie atomowym (in statu nascendi).
Azotonawęglanie i węgloazotowanie kąpielowe prowadzi się w czasie od kilkunastu minut do
kilku godzin. Węgloazotowanie pozwala na otrzymanie warstw nasyconych o głębokości 0,02-
0,15 mm, azotonawęglanie - o głębokości kilku dziesiątych milimetra.
Istotną wadą procesu kąpielowego jest trujące działanie cyjanków. Dlatego musi
- odbywać w specjalnie wydzielonych pomieszczeniach, przy ścisłym przy przestrzeganiu

97
JW
przepisów bezpieczeństwa pracy.
Azotonawęglanie gazowe przeprowadza się w temperaturze 700-800°C w mieszaninie gazów
nawęglających i azotujących, np. w gazie świetlnym i amoniaku (20
÷ 30 %).
Struktura warstwy nasyconej węglem i azotem zależy głównie od temperatury procesu. W
przypadku obróbki wysokotemperaturowej i następnie hartowania warstwa nasycona składa się z
martenzytu węglowego i azotowego, węglików, azotków, faz węglikowo-azotkowych typu
Fe
2
(NC) lub Fe
3
(C,N) oraz pewnej ilości austenitu szczątkowego. W przypadku obróbki
niskotemperaturowej warstwa nasycona ma strukturę podobną do struktury warstwy
naazotowanej.
6.5. Metalizowanie dyfuzyjne
Metalizowanie dyfuzyjne polega na nasycaniu przypowierzchniowych warstw przedmiotów
stalowych różnymi metalami, najczęściej chromem lub aluminium, rzadziej cynkiem lub
tytanem. Niekiedy stosuje się jednoczesne nasycanie dwoma różnymi metalami, np. chromem i
aluminium (chromoaluminiowanie), czy chromem i wolframem (chromowolframowanie).
Uzyskane w ten sposób warstwy cechuje bardzo wysoka twardość i odporność na ścieranie (w
przypadku chromowania stali wysokowęglowej) podwyższona odporność na korozję (w
przypadku chromowania stali niskowęglowej) oraz dobra żaroodpomość, tj. odporność na
utlenianie w wysokich temperaturach (w przypadku aluminiowania). W stosunku do
analogicznych pokryć galwanicznych, metaliczne warstwy dyfuzyjne są mniej porowate i lepiej
zespolone ze stalowym podłożem.
Metalizowanie dyfuzyjne można przeprowadzać zarówno w środowisku stałym jak i ciekłym
lub gazowym. W pierwszym przypadku metalizowanie odbywa się w sproszkowanym
żelazostopie (np. żelazochromie, czy żelazoaluminium), zmieszanym z pewną ilością salmiaku
NH
4
C1: w wysokiej temperaturze metal nasycająca zawarty w żelazostopie reaguje z HC1 lub
Cl
2
tworząc lotny chlorek (odpowiednie CrCl
3
, AlCl
3
itd.), który rozkłada się przy zetknięciu z
metaliczną powierzchnią nasycanego przedmiotu. W ten sposób powstają wolne, aktywne atomy
metalu nasycającego, które wnikają następnie do stali, tworząc odpowiednią warstwę dyfuzyjną.
Metalizowanie kąpielowe przeprowadza się przez zanurzenie obrabianego przedmiotu w
roztopionym metalu nasycającym. Ta metoda obróbki jest stosowana przede wszystkim w celu
aluminiowania stali.
Metalizowanie gazowe odbywa się w środowisku gazowym, utworzonym z chlorków
odpowiednich metali.
Dyfuzja metali w żelazie przebiega znacznie wolniej niż dyfuzja węgla lub azotu, gdyż metale
tworzą z nim roztwory stałe różnowęzłowe (węgiel i azot — międzywęzłowe). Dlatego proces
metalizowania dyfuzyjnego wymaga długotrwałego wygrzewania w wysokiej temperaturze
(1000-1200°C), co oczywiście wpływa na jego koszt.
Struktury warstw metalizowanych dyfuzyjnie są zgodne z odpowiednimi układami
równowagi żelazo-metal nasycający. Na przykład, struktura warstwy aluminiowej składa się (od
powierzchni) z: prawie czystego aluminium, kruchych faz pośrednich i fazy
α będącej
roztworem stałym aluminium w żelazie.

96
JW
7. Klasyfikacja stali*
Klasyfikacji gatunków stali dokonuje się zgodnie z PN-EN 10020:1996 według składu
chemicznego oraz wg ich zastosowania i własności mechanicznych lub fizycznych.
Klasyfikacja stali według składu chemicznego
- stale niestopowe (węglowe),
- stale stopowe.
Do stali niestopowych zalicza się te gatunki stali, w których zawartość pierwiastków jest
mniejsza od zawartości granicznych podanych w tabl. 7.1.
Do stali stopowych zalicza się gatunki stali, w których zawartość przynajmniej jednego
pierwiastka jest równa lub większa od zawartości granicznej podanej w tabl. 7.1.
Tablica 7.1
Granica między stalami niestopowymi i stopowymi (wg PN-EN 10020:1996)
Nazwa i symbol
chemiczny pierwiastka
Zawartość graniczna
(% wagowy)
Aluminium, Al
0,10
Bor, B
0,0008
Bizmut, Bi
0,10
Chrom, Cr*
0,30
Cyrkon, Zr*
0,05
Kobalt, Co
0,10
Krzem, Si
0,50
Lantanowce, każdy
0,05
Mangan, Mn
1.65**
Miedź, Cu*
0,40
Molibden, Mo*
0,08
Nikiel, Ni*
0,30
Niob, Nb*
0,06
Ołów.Pb
0,40
Selen, Se
0,10
Tellur, Te
0,10
Tytan, Ti*
0,05
Wanad, V*
0,10
Wolfram, W
0,10
Inne (każdy oprócz 0,05
fosforu, siarki i azotu),
* Jeżeli te pierwiastki określa się dla stali w kombinacji dwu, trzech lub czterech, a ich zawartości są
mniejsze niż podane w tablicy, to przy kwalifikacji stali należy dodatkowo uwzględnić zawartość
graniczną wynoszącą 70% sumy poszczególnych zawartości granicznych tych dwu, trzech lub
czterech pierwiastków
** Jeżeli jest określona tylko maksymalna zawartość manganu, jego graniczna zawartość wynosi
1,80% i nie stosuje się zasady 70%.
Klasyfikacja stali według zastosowania i własności mechanicznych lub fizycznych
A. Klasy jakości stali niestopowych
• stale niestopowe podstawowe,
• stale niestopowe jakościowe,
• stale niestopowe specjalne.
Stale niestopowe podstawowe
Stale podstawowe to gatunki stali o takich wymaganiach jakościowych, jakie można
osiągnąć w ogólnie stosowanym procesie stalowniczym, bez dodatkowych zabiegów
technologicznych.
* Oznaczanie stali wg PN-EN 10027-1 na stronie 146

97
JW
Wyroby z tych stali nie są przeznaczone do obróbki cieplnej (z wyjątkiem wyżarzania
odprężającego, zmiękczającego i normalizowania).
Z wyjątkiem manganu i krzemu (oraz granicznych zawartości C, P, S), zawartość innych
pierwiastków stopowych nie jest wymagana.
Nie określa się dodatkowych wymagań jakościowych dotyczących np. głębokiego tłoczenia,
ciągnienia, kształtowania na zimno itp.
Własności w stanie walcowanym na gorąco lub wyżarzonym odprężające, zmiękczająco albo
normalizowanym powinny odpowiadać następującym wartościom granicznym dla wyrobów o
grubości do 16 mm:
minimalna wytrzymałość na rozciąganie (R
m
) < 690 MPa,
minimalna granica plastyczności (R
e
)
< 360 MPa,
minimalne wydłużenie (A) . < 26%,
minimalna praca łamania w temp. 20°C na próbkach
wzdłużnych ISO < 27 J,
minimalna średnica trzpienia w próbie zginania
(e oznacza grubość próbki) >1 e
maksymalna zawartość węgla > 0,10%,
maksymalna zawartość fosforu > 0,045%,
maksymalna zawartość siarki > 0,045%.
Przykłady stali należących do tej klasy:
• stale miękkie niskowęglowe na taśmy i blachy walcowane na gorąco lub na zimno
ogólnego zastosowania,
• stale konstrukcyjne walcowane na gorąco ogólnego zastosowania,
• stale do wyrobu walcówki do ciągnienia (drutu).
Stale niestopowe jakościowe
Stale niestopowe jakościowe to gatunki stali, których własności w stanie obrobionym cieplnie
w zasadzie się nie określa, nie określa się również czystości metalurgicznej wyrażonej stopniem
zanieczyszczenia wtrąceniami niemetalicznymi.
Ze względu na warunki stosowania wyrobów ze stali jakościowych, wymagania dotyczące np.
wrażliwości na kruche pękanie, regulowanej wielkości ziarna czy podatności na kształtowanie,
są wyższe niż dla stali podstawowych, co wymusza większą staranność podczas produkcji.
Przykłady stali należących do tej klasy:
• stale na wyroby płaskie do kształtowania na zimno;
• stale konstrukcyjne o zawartości P
max
i S
max
poniżej 0,045%, np.:
stale o podwyższonej wytrzymałości,
stale do budowy statków,
stale na wyroby ocynkowane ogniowo,
stale na butle gazowe,
stale na kotły i zbiorniki ciśnieniowe;
• stale z wymaganą podatnością na odkształcenie plastyczne;
• stale konstrukcyjne z wymaganą minimalną zawartością Cu;
• stale do zbrojenia betonu;
• stale szynowe;
• stale automatowe;
• stale do ciągnienia drutu;
• stale do spęczania na zimno;
• stale sprężynowe;
• stale z wymaganymi własnościami magnetycznymi lub elektrycznymi;
• stale do produkcji blach cienkich, ocynowanych (na opakowania);
• stale do produkcji elektrod otulonych lub drutu spawalniczego o zawartości P
max
, i S
max
większej niż 0,02%.

98
JW
Stale niestopowe specjalne
Stale niestopowe specjalne charakteryzują się wyższym niż stale jakościowe stopniem
czystości metalurgicznej, szczególnie w zakresie zawartości wtrąceń niemetalicznych. Są one
przeważnie przeznaczone do ulepszania cieplnego lub hartowania powierzchniowego.
Dzięki dokładnemu doborowi składu chemicznego oraz przestrzeganiu specjalnych warunków
produkcji stali i kontroli przebiegu procesów technologicznych uzyskuje się różnorodne
własności przetwórcze i użytkowe stali. Często otrzymuje się równocześnie i w zawężonych
granicach np. wysoką wytrzymałość lub hartowność z równocześnie dobrą ciągliwością,
podatnością na kształtowanie, spawanie itp.
Stale niestopowe specjalne spełniają jeden lub więcej z niżej wymienionych warunków:
a) określona udarność w stanie ulepszonym cieplnie;
b) określona hartowność lub twardość powierzchniowa w stanie hartowanym i odpuszczonym
lub utwardzonym powierzchniowo;
c) określona mała zawartość wtrąceń niemetalicznych;
d) określona maksymalna zawartość fosforu i siarki (każdy z nich):
< 0,020% według analizy wytopowej,
< 0,025% według analizy chemicznej wyrobu (np. walcówka przeznaczona do
produkcji mocno obciążonych sprężyn, elektrod, drutu do zbrojenia opon
Przykłady stali należących do tej klasy:
• stale konstrukcyjne o określonej minimalnej pracy łamania próbek wzdłużnych ISO z
karbem V, większej niż 27 J w temperaturze -50°C;
• stale konstrukcyjne przeznaczone do produkcji reaktorów jądrowych, o ograniczonej
zawartości następujących pierwiastków: miedź < 0,10%, kobalt < 0,05%, wanad <
0,05%;
• stale do ulepszania cieplnego;
• stale do nawęglania;
stale utwardzalne wydzieleniowo o wymaganej zawartości węgla minimum 0,25% lub
większej (w analizie wytopowej) i strukturze ferrytyczno-perlitycznej: zawierające jeden
lub więcej mikrododatków stopowych, takich jak niob albo wanad, jednak ich zawartość
powinna być niższa niż wartość graniczna dla stali stopowych; utwardzanie
wydzieleniowe uzyskuje się zwykle przez kontrolowane chłodzenie z temperatury
przeróbki plastycznej na gorąco;
• stale do sprężania betonu;
• stale do ciągnienia (drutu);
• stale do spęczania na zimno;
• stale sprężynowe;
• stale narzędziowe;
• stale o określonej przewodności elektrycznej większej niż 9 Sm/mm;
• stale do produkcji elektrod otulonych lub na drut spawalniczy o zawartości P
max
i S
max
mniejszej niż 0,02%.
B. Klasy jakości stali stopowych
- stale stopowe jakościowe,
- stale stopowe specjalne.
Stale stopowe jakościowe
Stale stopowe jakościowe mają podobne zastosowanie jak stale niestopowe jakościowe, lecz
wymagane własności powodują konieczność zwiększenia w nich zawartości pierwiastków
stopowych powyżej wartości granicznych podanych w tabl. 7.1.
Stale te zwykle nie są przeznaczone do ulepszania cieplnego lub utwardzania
powierzchniowego.
Do grupy stali stopowych jakościowych należą:
stale konstrukcyjne drobnoziarniste spawalne, w tym stale przeznaczone do produkcji

99
JW
zbiorników i rurociągów pracujących pod ciśnieniem, spełniające następujące warunki:
a) wymagana minimalna granica plastyczności dotycząca wyrobów o grubości do 16 mm -
poniżej 380 N/mm,
b) zawartości pierwiastków stopowych powinny być niższe niż wartości graniczne według
tabl. 6.la,
c) wymagana praca łamania próbek wzdłużnych ISO z karbem V w temperaturze -50°C - do
27 J;
stale elektrotechniczne zawierające jako pierwiastki stopowe tylko krzem lub krzem i
aluminium w celu uzyskania wymaganych własności w zakresie stratności magnetycznej,
minimalnej wartości indukcji magnetycznej, polaryzacji lub przenikalności magnetycznej;
stale stopowe przeznaczone do produkcji szyn i grodzic oraz kształtowników na obudowy
górnicze;
stale stopowe przeznaczone do produkcji wyrobów płaskich walcowanych na gorąco lub na
zimno do dalszej trudniejszej przeróbki plastycznej na zimno (wyłączając stale przeznaczone
do produkcji zbiorników ciśnieniowych lub rur), zawierające pierwiastki rozdrabniające
ziarno, takie jak B, Ti, Nb, V i/lub Zr, -albo „stale dwufazowe" (struktura wyrobów płaskich
ze stali dwufazowych składa się z ferrytu i 10
÷ 35% martenzytu wysepkowego);
stale, w których miedź jest jedynym wymaganym pierwiastkiem stopowym.
Tablica 7.1a
Stale stopowe drobnoziarniste spawalne. Granica składu chemicznego między stalami
stopowymi jakościowymi i specjalnymi
Pierwiastek
Zawartość graniczna
(% wagowy)
Cr Chrom*
0,50
Cu Miedź*
0,50
La Lantanowce
0,06
Mn Mangan
1,80
Mo Molibden*
0,10
Nb Niob*
0,08
Ni Nikiel*
0,50
Ti Tytan*
0,12
V Wan*
0,12
Zr Cyrkon*
0,12
Inne nie wymienione
patrz tablica 6.1
pierwiastki (każdy)
*Jeżeli te pierwiastki występują w stali w kombinacji dwu, trzech lub czterech, a ich zawartości są
mniejsze niż podane w tablicy 7.1, to przy klasyfikacji stali należy dodatkowo uwzględnić wartość
graniczną, która stanowi 70% sumy poszczególnych zawartości granicznych tych dwu, trzech lub
czterech pierwiastków.
Stale stopowe specjalne
Stale stopowe specjalne dzięki precyzyjnie określonemu składowi chemicznemu
odpowiednim warunkom wytwarzania i kontroli procesów produkcyjnych maję różnorodne
własności przetwórcze i użytkowe często uzupełniające się i utrzymywane w zawężonych
granicach.
Ta klasa obejmuje następujące grupy stali:
• stale odporne na korozję,
• stale żaroodporne i żarowytrzymałe,
• stale przeznaczone do produkcji łożysk tocznych,
• stale narzędziowe,
• stale maszynowe,
• stale do nawęglania,

100
JW
• specjalne stale konstrukcyjne (spawalne drobnoziarniste stale konstrukcyjne, stale
odporne na korozję atmosferyczną),
• stale o specjalnych własnościach fizycznych (niemagnetyczne, magnetyczne lub o
wymaganym współczynniku rozszerzalności cieplnej).
Skład chemiczny stali stopowych specjalnych stanowi podstawę ich podziału na następujące
główne kategorie:
l) stale odporne na korozję o zawartości węgla < 1,20% i chromu > 10,50%, które pod
względem zawartości niklu dzieli się na:
a) poniżej 2,50% Ni,
b) nie mniej niż 2,50% Ni;
2) stale szybkotnące zawierające (wraz z innymi składnikami lub bez nich):
- co najmniej dwa z trzech następujących pierwiastków: Mo, W lub V łącznie nie mniej niż
7% wagowych,
- 0,60% lub więcej węgla,
- i 3
÷ 6% wagowych chromu;
3) inne stale stopowe specjalne.
7.2. Stale niestopowe (węglowe)
7. 2.1. Wpływ węgla na własności stali
Węgiel bardzo silnie wpływa na własności stali nawet przy nieznacznej zmianie jego
zawartości i z tego względu jest bardzo ważnym składnikiem stali.
Zwiększenie zawartości węgla powoduje, jak już poprzednio wspomniano, zmianę struktury
stali. Jeżeli stal zawiera mniej niż 0,8% C, to jej struktura składa się ferrytu i perlitu. Struktura
stali zawierającej 0,8% C składa się tylko z perlitu, natomiast w stali o zawartości powyżej 0,8%
C oprócz perlitu występuje również cementyt wtórny. Zmiana struktury stali spowodowana
różną zawartością węgla wiąże się ściśle ze zmianą własności mechanicznych. Na rysunku 7.1
przedstawiono wpływ węgla na własności mechaniczne stali walcowanej na gorąco.
7.1. Wpływ węgla na własności mechaniczne stali
Jak widać zwiększenie zawartości węgla zwiększa wytrzymałość na rozciąganie R
m
i zmniej-
sza plastyczność stali. Maksymalną wytrzymałość osiąga stal przy zawartości ok. 0,85% węgla.
Przy większej zawartości węgla wytrzymałość zmniejsza się na skutek pojawiania się coraz
większej ilości cementu wtórnego, który wydziela się na granicach ziarn.
Zwiększenie zawartości węgla, oprócz obniżenia własności plastycznych, pogarsza również
własności technologiczne stali węglowej; szczególne znaczenie ma pogorszenie spawalności.

101
JW
7.2.2. Domieszki zwykłe w stali
Za domieszki zwykłe stali uważa się mangan, krzem, fosfor, siarkę oraz wodór, azot i tlen,
ponieważ te pierwiastki występują zawsze w mniejszej lub większej ilości w przemysłowych
gatunkach stali. Zawartość tych pierwiastków w stalach węglowych nie przekracza zwykle
następujących granic: Mn do 0,8% (w niektórych gatunkach stali granica ta jest rozszerzona do
1,5%), Si do 0,5%, P do 0,05% (z wyjątkiem stali automatowych), S do 0,05% (z wyjątkiem stali
automatowych).
Mangan wprowadza się do wszystkich stali w procesie stalowniczym w celu ich odtlenienia,
tj. usunięcia szkodliwego tlenku żelazawego lub związania siarki w MnS, przez co zapobiega się
powstaniu FeS powodującemu powstanie kruchości stali na gorąco. W ilościach (1,0 ÷ 1,5)%
Mn rozpuszczając się zarówno w ferrycie, jak i w cementycie umacnia roztworowo stal,
zmniejsza wielkość ziarna ferrytu w wyrobach walcowanych na gorąco oraz zwiększa
hartowność. Ponieważ jednak wszystkie stale węglowe mają zazwyczaj mniej więcej taką samą
zawartość manganu, to jego wpływ na własności różnych gatunków tych stali jest jednakowy.
Krzem w ilościach do 0,5% jest dodawany do stali podczas jej wytapiania w celu odtlenienia.
W ilościach (0,5 ÷ 1,0)% jest dodawany w celu umocnienia ferrytu. W większych ilościach (0,5 ÷
4,5)% powoduje zwiększenie oporu elektrycznego oraz zmniejszenie stratności stali magnetycznie
miękkich. Zwiększa również żaroodporność stali. Krzem stabilizuje bardzo mocno ferryt, dlatego
stale zawierające więcej niż 3% Si zachowują strukturę ferrytyczną od temperatury otoczenia do
temperatury solidusu.Wpływ krzemu, który rozpuszcza się w ferrycie, jest podobny do wpływu
manganu.
Fosfor dostaje się do stali z rud żelaza, które zawierają różne jego ilości. Podczas wytapiania
stali fosfor zostaje z niej usunięty w mniejszym lub większym stopniu, zależnie od rodzaju
procesu stalowniczego. Fosfor rozpuszczony w ferrycie (graniczna rozpuszczalność w
temperaturze pokojowej wynosi ok. 1,2%) zmniejsza bardzo znacznie jego plastyczność i
podwyższa temperaturę, w której stal staje się krucha, wywołując tzw. kruchość na zimno. Ten
wpływ fosforu jest bardzo wyraźny wówczas, gdy jego zawartość w stali jest większa niż 0,1%.
Jednak w stalach przeznaczonych na odpowiedzialne wyroby zawartość nawet 0,05% P jest
niebezpieczna i należy jej unikać, ponieważ w czasie krystalizacji stali zachodzi silna segregacja
fosforu, wskutek czego w pewnych miejscach zawartość fosforu będzie dość znaczna i będzie
powodować kruchość.
W zależności od przeznaczenia stali ustala się ostrzejsze wymagania dotyczące zawartości
fosforu (np. max 0,025%).
Należy zaznaczyć, że w niektórych wyjątkowych przypadkach zawartość fosforu w stali może
być pożyteczna. Na przykład w stalach automatowych dodatek ok. 0,1% P polepsza
skrawalność, zaś do ok. 0,35% - zwiększa odporność na ścieranie. Przy jednoczesnej zawartości
miedzi fosfor zwiększa odporność stali na korozję atmosferyczną.
Siarka podobnie jak fosfor dostaje się do stali z rud żelaza, a ponadto z gazów piecowych,
tzn. z produktów spalania paliwa zawierających dwutlenek siarki (SO
2
). Siarkę można w
znacznej mierze usunąć ze stali, jeżeli stosuje się podczas wytapiana zasadowy proces
martenowski lub zasadowy proces elektryczny. W stalach wysokojakościowych zawartość siarki
ogranicza się zazwyczaj do 0,02
÷ 0,03%.
W stali zwykłej jakości dopuszcza się większą zawartość siarki (do 0,05%).
Siarka nie rozpuszcza się w żelazie, lecz tworzy siarczek żelazawy FeS, który jest
składnikiem eutektyki Fe + FeS o temperaturze topnienia 985°C. Występowanie w stalach tej
łatwo topliwej i kruchej eutektyki, rozmieszczonej przeważnie a granicach ziarn, powoduje
kruchość stali nagrzanych do temperatury 800°C i powyżej. Zjawisko to nosi nazwę kruchości
na gorąco. Wskutek tej wady stal zawierająca większy procent siarki nie nadaje się do przeróbki
plastycznej na gorąco. W stali pojawiają się naderwania i pęknięcia, m.in. dlatego, że podczas

102
JW
nagrzewania poczynając od temperatury 985°C, zachodzi nadtapianie otoczek z siarczku
żelazawego wokół ziarn. Z tego powodu należy uważać siarkę za szkodliwą domieszkę stali.
Dodatek manganu do stali zmniejsza szkodliwe działanie siarki, gdyż wówczas w ciekłej stali
następuje reakcja, w wyniku której tworzy się siarczek manganawy MnS. Siarczek ten topi się w
1620°C, a więc w temperaturze o wiele wyższej niż temperatura przeróbki plastycznej na gorąco
(800
÷ 1200°C). Siarczki w temperaturze przeróbki plastycznej na gorąco są plastyczne i ulegają
odkształceniu, tworząc wydłużone wtrącenia. Pogarszają one wytrzymałość na zmęczenie i
obciążenia dynamiczne stali. Siarka pogarsza również spawalność stali.
Natomiast siarka, podobnie jak fosfor, polepsza skrawalność stali i w ilości 0,15-0,30% jest
wprowadzana celowo do stali automatowych.
Wodór, azot i tlen występują w stali w niedużych ilościach, a ich zawartość zależy w dużym
stopniu od sposobu wytapiania.
W stali będącej w stanie stałym, gazy mogą występować w kilku postaciach:
w stanie wolnym, skupiając się w różnych nieciągłościach wewnątrz metalu najczęściej
tworząc tzw. pęcherze);
mogą być rozpuszczone w żelazie;
mogą tworzyć związki (azotki, tlenki) występujące w stali jako tzw. wtrącenia
niemetaliczne.
Wpływ wodoru na własności stali jest zdecydowanie ujemny. Rozpuszcza się on stosunkowo
łatwo w żelazie i to w całym zakresie temperatury, szczególnie zaś przy przejściu fazy
α w γ
oraz w stanie ciekłym. Zmniejsza on w znacznym stopniu własności plastyczne i technologiczne
stali oraz powoduje występowanie wielu wad materiałowych, jak np. tzw. płatków śnieżnych (tj.
wewnętrznych pęknięć o jasnej powierzchni), odwęglania, skłonności do tworzenia pęcherzy
przy trawieniu itp.
Azot powoduje zwiększenie wytrzymałości i zmniejszenie plastyczności stali, co objawiać
się może jako tzw. kruchość na niebiesko. Niekorzystne działanie azotu przejawia się także
zwiększeniem skłonności stali do starzenia, powodowanym wydzielaniem się azotków z
przesyconego roztworu. Zjawisko to jest szczególnie niekorzystne w stalach w stanie
zgniecionym, gdyż wówczas występuje już w temperaturze otoczenia.
W niektórych stalach stopowych azot jest stosowany jako korzystny dodatek stopowy
stabilizujący austenit, zastępując drogi nikiel.
Tlen występuje w stali głównie w postaci związanej, najczęściej tlenków FeO, SiO
2
, Al
2
O
3
i
in. Tlen powoduje pogorszenie prawie wszystkich własności mechanicznych i dlatego dąży się
przez odpowiednie prowadzenie procesu metalurgicznego do obniżenia jego zawartości w stali.
Odtlenianie stali przeprowadza się za pomocą stopów krzemu, manganu i aluminium. Sposób
odtleniania wywiera także duży wpływ na wielkość ziarna stali węglowej. Stale odtleniane
żelazomanganem wykazują skłonności do intensywnego rozrostu ziarn przy nagrzaniu już nieco
powyżej temperatury A
c3
. W przeciwieństwie do tego stale odtlenione aluminium, a także
żelazokrzemem wykazują wyraźny wzrost ziarn dopiero w temperaturze 150-200°C powyżej
A
c3
, co praktycznie wystarczy, aby przeciwdziałać zjawisku przegrzania stali.
Bardzo skutecznym sposobem zmniejszania ilości wodoru, azotu i tlenu oraz wtrąceń
niemetalicznych w stali jest wytapianie lub odlewanie jej w próżni. Można w ten sposób
otrzymać stal o lepszych własnościach dzięki większej czystości i prawie zupełnemu brakowi
rozpuszczonych w metalu gazów.
7.2.3. Stale niestopowe (węglowe) podstawowe konstrukcyjne ogólnego zastosowania
Stale niestopowe podstawowe konstrukcyjne są stosowane zazwyczaj w stanie surowym lub
rzadziej w stanie normalizowanym.
Według PN-88/H-84020 rozróżnia się 6 podstawowych gatunków stali w tej grupie. w
zależności od składu chemicznego i wymaganych własności mechanicznych. Znak gatunku stali
składa się z liter St oraz liczby porządkowej 0, 3, 4, 5, 6 lub 7.
103
JW
Gatunki stali przeznaczone na konstrukcje spawane o liczbie porządkowej 0, 3 i 4 oznacza się
dodatkowo literą S (np. St0S, St3S, St4S) oraz w przypadku określonej zawartości miedzi (z
wyjątkiem St0S) dodatkowo literami Cu (np. St3SCu. St4SCu). Gatunki o liczbie porządkowej 3
i 4 o podwyższonych wymaganiach jakościowych (o obniżonej zawartości C oraz P i S) oznacza
się dodatkowo literą V lub W (np. St3V, St4W).
Znak gatunku stali St5, St6 i St7 w przypadku określonej dodatkowo zawartości węgla, manganu
i krzemu uzupełnia się na początku literą M (np. MSt5).
Gatunki stali o liczbie porządkowej 3 i 4 z literą S lub V mogą być dodatkowe oznaczane
literą X w przypadku stali nieuspokojonej (np. St3SX, St3VX, St3SCuXC lub literą Y w
przypadku stali półuspokojonej (np. StSCuY, St4SY, St4W).
Skład chemiczny i własności mechaniczne tych stali podane są w tabl. 7.2.
Tablica 7.2
Skład chemiczny i własności mechaniczne stali węglowych konstrukcyjnych
ogólnego zastosowania (PN-88/H-84020)
Skład chemiczny, %
R
e
*
i
R
m
**
A
5
***
Znak
stali
C
Mn
Si
P max S max
MPa
MPa
%
StOS 0,23 max 1,30 0,40 max 0,070
0,065
185
300-540 W 20
P 18
St3S 0,22
max 1,10 0,10
0,35
0,050 0,050 225 360-490 W
26
P 24
St3W 0,17
max 1,30 0,10
0,35
0,040 0,040 225 360-490 W
26
P 24
St4S 0,24
max 1,10 0,10
0,35
0,050 0,050 265 420-550 W
22
P 20
St4W 0,20
max 1,30 0,10-
0,35
0,040 0,040 265 420-550 W
22
P 20
MSt5 0,26+0,37 0,80 0,35 max 0,050
0,050
285
470-640 W 20
P 18
MSt6 0,38+0,49 0,80 0,35 max 0,050
0,050
325
570-740 W 15
P 13
MSt7
0,50-0,62
0,80 0,35
max 0,050 0,050
355 670-840
W11
P9
* Dla wyrobów o grubości lub średnicy powyżej 16 ÷ 40 mm.
** Dla wyrobów o grubości lub średnicy powyżej 3 ÷ 100 mm.
*** Dla wyrobów o grubości lub średnicy powyżej 3 ÷ 40 mm.
Kierunek osi próbki: W - wzdłużny, P - poprzeczny (w stosunku do kierunku walcowania).
Gatunki stali o liczbie porządkowej 3 i 4 mogą mieć dodatkowo określoną wymaganą
udarność w temperaturze +20°C, 0°C i -20°C. Szczegółowe wymagania odnośnie do tych
odmian stali i ich oznaczenia podane są w PN-88/H-84020. Znaki gatunków tych stali uzupełnia
się na końcu znakiem odmiany plastyczności B, C, D lub U, M, J (np. St3SYU, St4WD).
7.2.4. Stale niestopowe specjalne do ulepszania cieplnego i utwardzania powierzchniowego
Stale te należą do grupy stali o wyższych wymaganiach w porównaniu do stali jakościowych
i charakteryzują się wyższym stopniem czystości. Zawartość fosforu i siarki nie może w nich
przekraczać po 0,040%. Są przeznaczone do wyrobu maszyn i urządzeń i stosuje się je w stanie
ulepszonym cieplnie, normalizowanym, hartowanym powierzchniowo lub po nawęglaniu.
Dzięki dokładnemu doborowi składu chemicznego oraz przez zastosowanie specjalnych
warunków wytwarzania uzyskuje się wymagane właściwości technologiczne i użytkowe często
w kombinacji z wysoką lub wąsko ograniczoną wytrzymałością lub hartownością.
Znak tych stali wg PN-93/H-84019 składa się z liczb dwucyfrowych, które mogą być
uzupełnione literami. Liczby te określają przybliżone średnie zawartości węgla w setnych
częściach procentu (np. 10, 15, 20, 25, 30 itd). Litery po liczbach oznaczają:
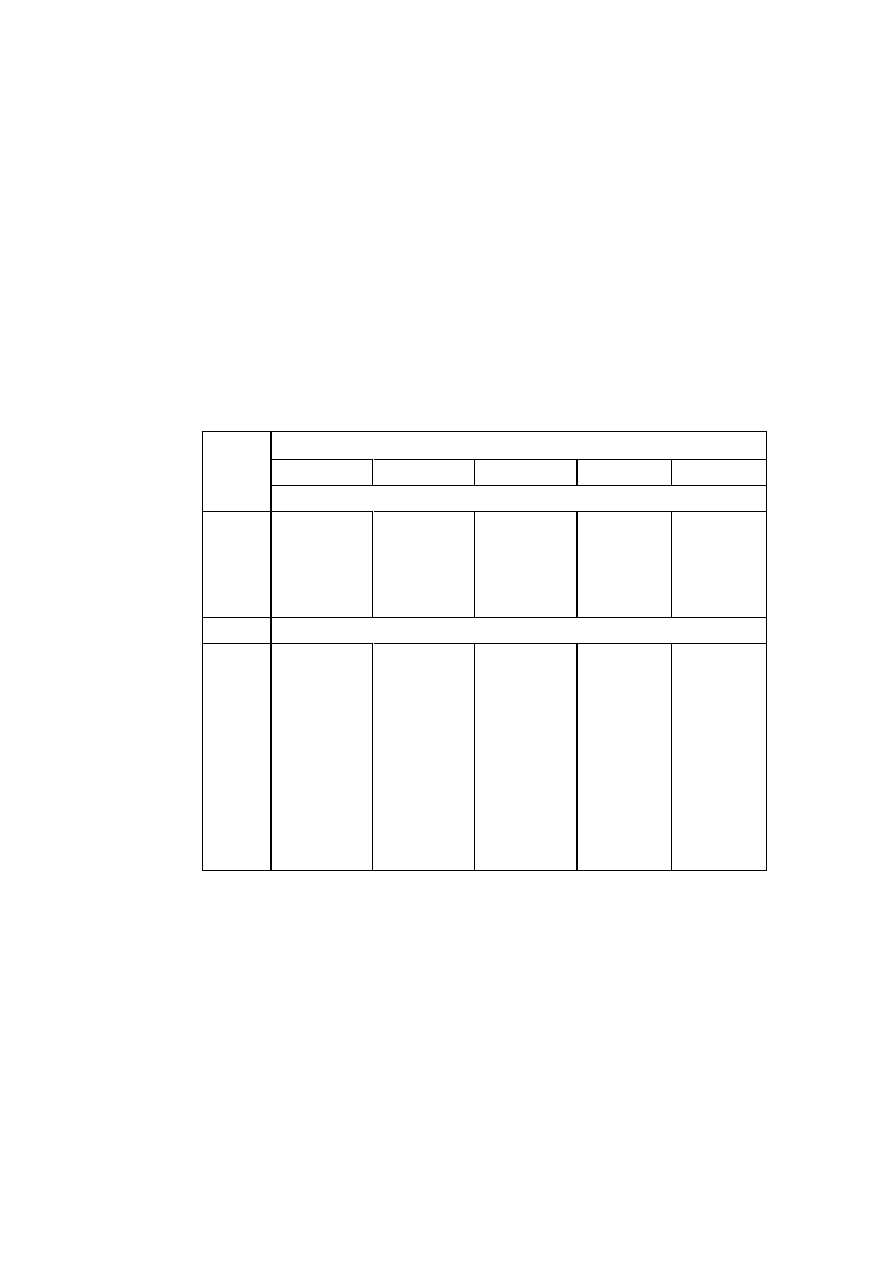
104
JW
G - stal o podwyższonej zawartości manganu,
A - stal o podwyższonej czystości w zakresie fosforu i siarki,
AA - stal o zaostrzonych wymaganiach w zakresie składu chemicznego (np. dotyczących
zawartości węgla, obniżonej zawartości fosforu i siarki ograniczonej sumie zawartości
Cr+Mo+Ni, itp.),
rs - stal o regulowanej zawartości siarki,
h - stal o wymaganej hartowności,
H - stal o podwyższonej dolnej granicy twardości w stosunku do wymaganego pasma
hartowności,
L - stal o obniżonej granicy twardości w stosunku do wymaganego pasma hartowności, przy
czym cyfry (np. 4, 5, 15) po literach hH i hL oznaczają odległości od czoła próbki w
milimetrach (4 mm, 5 mm, 15 mm). Skład chemiczny niektórych stali niestopowych do
nawęglania oraz normalizowania, ulepszania cieplnego i hartowania powierzchniowego
podano w tabl. 7.3
Tablica 7.3.
Skład chemiczny niektórych gatunków stali niestopowej specjalnej do nawęglania oraz
normalizowania, ulepszania cieplnego i hartowania powierzchniowego (wg PN-93/H-840191
Znak Skład chemiczny, % wag.
gatunku C
Mn
Si P
max S
stali
Stale do nawęglania
10 0,07-0,14 0,35+0,65 0,15-0,40 0,040 max
0,040
15 0,12-0,19 0,35-0,65 0,15-0,40 0,040 max
0,040
14A 0,12-0,18 0,30+0,60
0,15+0,40
0,035 max
0,035
20 0,17-0,24 0,35+0,65
0,15+0,40 0,040 max
0,040
20G 0,17+0,24 0,70+1,00
0,15+0,40
0,040 max
0,040
Stale do normalizowania, ulepszania cieplnego i hartowania powierzchniowego
25 0,22-0,29 0,40+0,70
0,10+0,40 0,040 max
0,040
26A
0,22+0,29
0,40+0,70
0,10+0,40
0,035
max 0,035
30 0,27-0,34 0,50+0,80
0,10+0,40
0,040 max
0,040
35
0,32-0,39
0,50+0,80
0,10+0,40
0,040
max 0,040
40 0,37+0,44 0,50+0,80
0,10+0,40
0,040 max
0,040
45
0,42-0,50
0,50+0,80
0,10-0,40
0,040
max 0,040
46A
0,42+0,50
0,50+0,80
0,10-0,40
0,035
max 0,035
46rs 0,42-0,50 0,50+0,80
0,10+0,40
0,035 0,020+0,040
45G
0,42+0,50
0,70+1,00
0,10+0,40
0,040
max 0,040
50
0,47+0,55
0,60+0,90
0,10+0,40
0,040
max 0,040
55
0,52+0,60
0,60+0,90
0,10+0,40
0,040
max 0,040
60 0,57-0,65 0,60+0,90
0,10+0,40
0,040 max
0,040
65 0,62+0,70 0,50+0,80
0,10+0,40
0,040 max
0,040
Własności mechaniczne w stanie normalizowanym i dla porównania w stanie ulepszonym
cieplnie (po hartowaniu i odpuszczaniu w temperaturze 550
÷
660°C) niektórych stali podano w
tabl. 7.4. Należy zwrócić uwagę, że wytrzymałość na rozciąganie R
m
granica plastyczności R
e
i
udarność KCU2 są znacznie wyższe w stanie ulepszonym cieplnie, w porównaniu ze stanem
normalizowanym, a dla stali o większej zawartości węgla (gatunku 55, 60) większe jest również
wydłużenie.
7.2.5 Stale niestopowe jakościowe i specjalne o określonym zastosowaniu
W
przemyśle, oprócz omówionych wyżej stali węglowych konstrukcyjnych ogólnego
zastosowania, stosuje się również wiele gatunków stali węglowych o określonym z góry
zastosowaniu. Stale te z uwagi na konieczność zapewnienia szczególnych własności użytkowych
lub technologicznych mają skład chemiczny różniący się od składu stali węglowych ogólnego
zastosowania i to zarówno w odniesieniu do składników zasadniczych, jak i przypadkowych lub
zanieczyszczeń. Poza tym w niektórych przypadkach stale te wykazują wyższe lub niższe
105
JW
własności mechaniczne, w porównaniu do odpowiednich stali ogólnego zastosowania o
zbliżonym składzie chemicznym, jednakże zapewniają żądane własności technologiczne i
użytkowe.
Tablica 7.4
Własności mechaniczne niektórych gatunków stali niestopowej specjalnej w stanie
normalizowanym oraz ulepszanym cieplnie wg PN-93/H-84019 (dla wyrobów o średnicy lub
grubości do 16 mm*)
Znak gatunku
stali
Stan obróbki
cieplnej
R
m
MPa
R
e
(R
eH
,R
0,2
)
MPa, min
A
5
, %
min
KCU 2, J/cm
2
min
N
**)
min
470 275
22
60
25
T
***)
550 ÷ 700
370
19
90
N min
510 295 20
60
30
T
600 ÷ 750
400
18
80
N min
550 315 18
50
35
T
630 ÷ 780
430
17
70
N min
580 335 16
50
40
T
650 ÷ 800
460
16
60
N min
620 355 14
40
45
T
700 ÷ 850
490
14
50
N min
680 380 11
-
55
T
800 ÷ 950
550
12
-
N min
710 400 10
-
60
T
850 ÷ 1000
580
11
-
* Dla większych wartości grubości wyrobów własności wytrzymałościowe są odpowiednio niższe.
** N - normalizowanie.
*** T - ulepszanie cieplne (hartowanie i odpuszczanie wysokie).
Wśród stali węglowych konstrukcyjnych o określonym zastosowaniu można wyodrębnić
następujące ważniejsze grupy gatunków:
stale do wyrobu drutu do patentowania, na liny, na sprężyny, do konstrukcji sprężanych,
drutu ogólnego przeznaczenia i dla przemysłu włókienniczego (PN 91/H-84028);
stale, dla kolejnictwa (PN-84/H-84027, PN-91/H-84027/03, PN-88/H-84027/04-05);
stale do wyrobu rur (PN-89/H-84023/07);
stale do wyrobu nitów (PN-89/H-84023/04-05);
stale na blachy kotłowe (PN-81/H-92123);
stale do budowy mostów (PN-89/H-84023/04);
stale na blachy grube i uniwersalne do budowy statków (PN-85/H-92147);
stale na blachy karoseryjne (PN-89/H-84023/03);
stale do wyrobu ogniw łańcuchów technicznych i okrętowych (PN-89/H-84023/08);
stale automatowe (łatwo obrabialne mechanicznie) (PN-73/H-84026);
stale magnetycznie miękkie (PN-89/H-84023/02).
Stale niestopowe przeznaczone na walcówkę do produkcji drutu są wysokiej czystości.
Zawartość węgla w tych stalach zawiera się w granicach 0,33
÷
0,98%. W stalach o najwyższej
czystości do wyrobu drutu na liny zawartość fosforu i siarki nie może przekroczyć po 0,020%,
ale łącznie zawartość P+S nie może być wyższa niż 0,035%.
Stale automatowe (oznaczone wg PN-73/H-84026 znakami A10X, A10XN, A11. A35, A45,
A35G2), a także stal do wyrobu nakrętek prasowanych (10P) są stalami o podwyższonej
zawartości fosforu i siarki (np. stal automatowa A10 zawiera 0,04
÷
0,08% P i 0,24
÷
0,34% S, a
stal do wyrobu nakrętek 10P - 0,20
÷
0,35% P i 0,06% S). Duża zawartość tych pierwiastków
zapewnia dobrą skrawalność stali, które dzięki temu nadają się szczególnie dobrze do obróbki
106
JW
wiórowej na automatach i szybkobieżnych obrabiarkach do nacinania gwintów, gdyż obecność
dużej ilości wtrąceń niemetalicznych (siarczków i fosforków) ułatwia łamanie się wióra podczas
skrawania. Skład chemiczny i własności mechaniczne stali automatowych podano w tabl. 7.5.
Stale węglowe magnetycznie miękkie są to stale o bardzo małej zawartości węgla (max
0,04%). Stale te odznaczają się małą koercją i dużą przenikalnością magnetyczną. Stosuje się je
najczęściej na rdzenie elektromagnesów. Własności magnetyczne materiałów magnetycznie
miękkich pogarszają się ze wzrostem ilości zanieczyszczeń, zwłaszcza C, S, P, O i N. Dlatego
wymaga się, aby w tych stalach ich ilość była jak najmniejsza.
Szczegółowe wymagania, dotyczące wymienionych wyżej grup stali węglowych o
określonym przeznaczeniu i o szczególnych własnościach, podają Polskie Normy.
Tablica 7.5
Skład chemiczny i własności mechaniczne stali automatowych (wg PN-73/H-84026)
Średnia zawartość, %
Znak
stali
Stan**
R
m
***
MPa
R
e
***
min
MPa
A
5
***
min
%
C
Mn
Si
P
S
min
min
A10X 0,12 1,10 0,05 0,06 0,29 W 380
÷
510
- —
max
max
C
490
÷
740
390
8
A11 0,10 0,70 0,27 0,06 0,20 W 380
÷
510
— —
max
C 490
÷
740
390 8
T
440
÷
740
260 14
A35 0,35 0,70 0,27
0,06
0,20
W
490
÷
660
—
max
C
540
÷
740
310 8
TC
620
÷
770
500 12
A45 0,45 0,70 0,27
0,06
0,20
W
590
÷
770
max
C
640
÷
830
370 7
TC
700
÷
890
580 10
A35G2 0,35 1,60 0,27 0,035 0,14 WN min
690 410
13
max
C — - —
T
780
÷
930
590 12
* Wytwarzany jest również gatunek z azotem A10XN zawierający średnio ok. 0,013% N.
**Własności mechaniczne podano dla grubości wyrobów powyżej 16 ÷ 40 mm; dla grubości
mniejszej własności wytrzymałościowe są nieco wyższe, a plastyczne nieco niższe, natomiast
dla grubości większej własności wytrzymałościowe są nieco niższe, a plastyczne wyższe.
***W - walcowanie na gorąco, WN - walcowanie i normalizowanie, T – ulepszanie cieplne, TC -
ciągnienie po ulepszaniu cieplnym, C - ciągnienie po walcowaniu.
7.3. Stale niestopowe (węglowe) narzędziowe
Stale narzędziowe służą w głównej mierze do wyrobu wszelkiego rodzaju narzędzi w tym
skrawających, na odpowiedzialne części przyrządów mierniczych, uchwytów itd. Zasadnicze
cechy, których wymaga się od stali narzędziowych, to: twardość po zahartowaniu, odporność na
ścieranie i zużycie, ciągliwość, niewrażliwość na przegrzanie, mała odkształcalność przy
hartowaniu - przy czym nie zawsze wszystkie cechy są wymagane jednocześnie.
Podstawowym wymaganiem stawianym narzędziom skrawającym jest trwałość ostrza, która
stępia się i zużywa podczas skrawania. Im bardziej stal jest odporna na zużycie i ścieranie, tym
lepiej nadaje się na narzędzia skrawające. Aby stal była odporna na ścieranie, powinna mieć
dużą twardość, zazwyczaj powyżej 60 HRC.
Największą twardość po hartowaniu uzyskują stale o większej zawartości węgla i z tego względu
stale narzędziowe są z reguły stalami wysokowęglowymi.
Zawartość węgla w stalach węglowych narzędziowych objętych Polską Normą PN-84/H-
85020 wynosi 0,5
÷ 1,24. Stale te w porównaniu ze stalami węglowymi konstrukcyjnymi
charakteryzują się większą czystością (mniejszą zawartością fosforu i siarki), mniejszą
zawartością manganu oraz drobnoziarnistością.
Charakterystyczną zaletą stali narzędziowych węglowych jest mała głębokość hartowania, tzn.
że hartuje się tylko warstwa wierzchnia narzędzia, a rdzeń pozostaje bardziej miękki i ciągliwy.
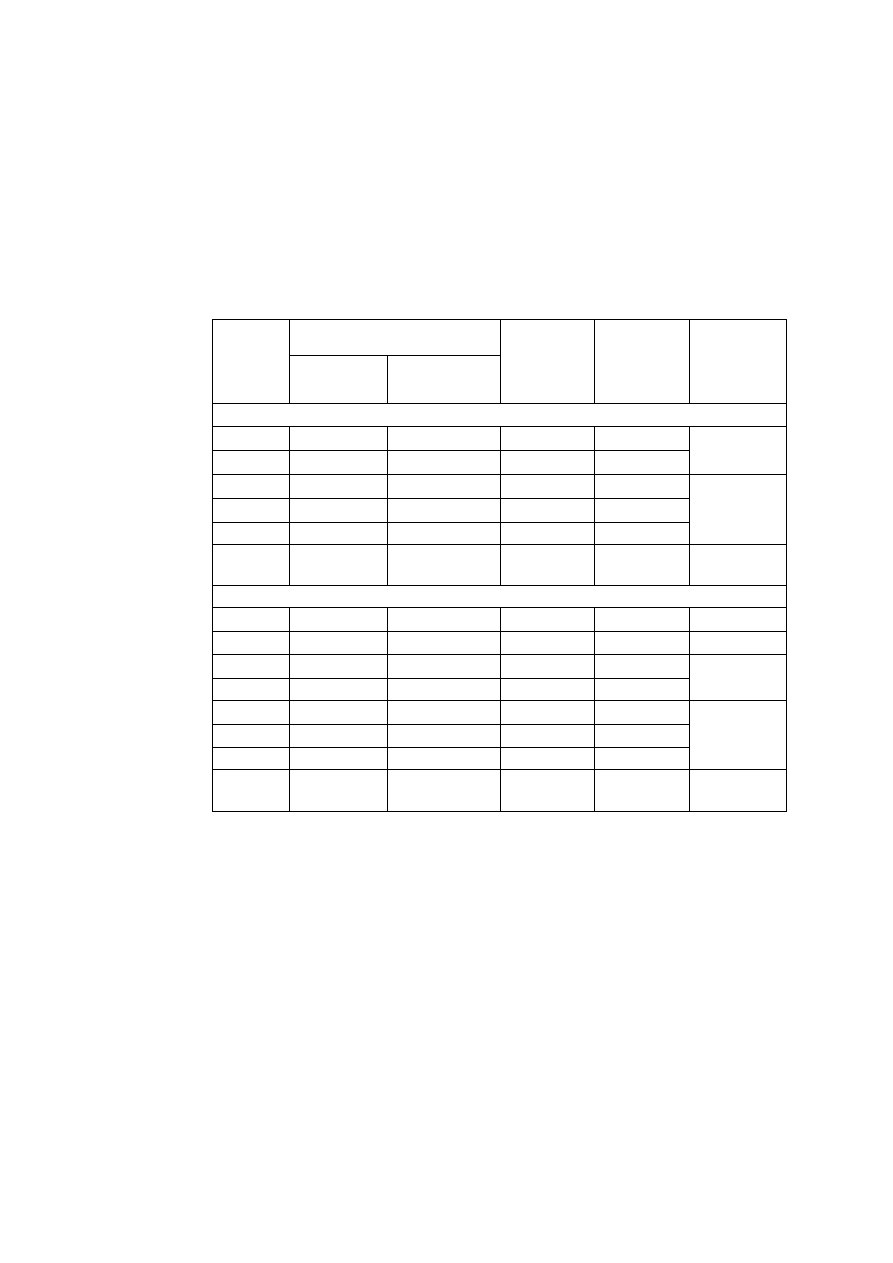
107
JW
Daje to możliwość uzyskania narzędzia twardego i odpornego na ścieranie, a jednocześnie
mającego dostateczną odporność na uderzenia.
Według Polskich Norm PN-84/H-85020 stale węglowe narzędziowe dzielą się na dwie grupy:
- stale hartujące się płytko,
- stale hartujące się głęboko.
W tablicy 7.6. podano skład chemiczny tych stali oraz ich twardość w stanie zmiękczonym i
po hartowaniu. Stale hartujące się płytko oznaczone są literą N (oznaczają stal narzędziową),
liczbą oznaczającą w przybliżeniu średnią zawartość węgla w dziesiętnych częściach procentu
oraz na końcu literą E. Stale hartujące się głęboko są oznaczone analogicznie, ale bez litery E.
Tablica 7.6
Skład chemiczny i twardość w stanie zmiękczonym i po hartowaniu stali
węglowych narzędziowych (wg PN-84/H-85020)
Skład chemiczny, %
Znak stali
C
inne pierwiastki
Twardość w
stanie
zmiękczony
m HB, max
Temp.**)
hartowania,
o
C
Twardość w
stanie harto-
wanym HRC,
min
Stale hartujące się płytko
N7E 0,65
÷
0,74 Mn
0,15
÷
0,30
187 790
÷
810
N8E 0,75
÷
0,84 Si
0,15
÷
0,30 187 780
÷
800
61
N9E 0,85
÷
0,94 P
max
0,025
197
770
÷
790
N10E 0,95
÷
1,04 S
max
0,025
197
770
÷
790
N11E 1,05
÷
1,14 Cr
max
0,15
207
770
÷
790
62
1,15
÷
1,24 Ni
max
0,20
Cu
max
0,20
207 760
÷
780 63
Stale hartujące się głęboko
N5 0,50
÷
0,60 Mn
0,40
÷
0,60
*
183 790
÷
810 58
N6 0,61
÷
0,70 Mn
0,30
÷
0,50
*
183 790
÷
810 61
N7 0,65
÷
0,74 Mn
0,15
÷
0,35
187 790
÷
810
N8 0,75
÷
0,84 Si
0,15
÷
0,35 187 790
÷
800
61
N9 0,85
÷
0,94 P
max
0,030
197
770
÷
790
N10 0,95
÷
1,04 S
max
0,030
197
770
÷
790
N11 1,05
÷
1,14 Cr
max
0,20
207
770
÷
790
62
N12
1,15
÷
1,24
Ni
max
0,25 Cu
max
0,25
207 760
÷
780
63
* Pozostałe pierwiastki dla stali N5 i N6; Si max 0,15%, P max 0,035%, S max 0,035%, Cr, C, i Ni nie
określa się.
* Hartowanie w wodzie czystej lub słonej.
Stale płytko i głęboko się hartujące, które mają taką samą zawartość węgla, różnią się tylko
zawartością domieszek pochodzących z wytopu, które jednak wpływają na ich hartowność.
Stale hartujące się płytko są stalami o małej hartowności (głębokość zahartowania wynosi 2
÷ 5 mm w zależności od temperatury hartowania), wykazują małą wrażliwość na przegrzanie i
ze względu na małą zawartość zanieczyszczeń należą do stali najwyższej jakości.
Stale hartujące się głęboko są bardziej wrażliwe na przegrzanie, tzn. że hartowane z wyższej
temperatury wykazują większą gruboziarnistość i większą skłonność do rys i pęknięć. Stale te
odznaczają się nieco większą hartownością (głębokość zahartowania wynosi 5
÷12 mm, w
zależności od temperatury hartowania) i mają nieco większą dopuszczalną zawartość
zanieczyszczeń (fosforu i siarki) i innych domieszek, co powoduje, że są stalami niższej klasy
niż stale hartujące się płytko.
Stale hartujące się płytko są stosowane w zasadzie do wyrobu narzędzi, których grubość nie
przekracza 20 mm, natomiast stale głęboko hartujące się - do wyrobu narzędzi, których grubość
lub średnica jest większa niż 20 mm.

108
JW
Obróbka cieplna stali narzędziowych węglowych polega na hartowaniu i niskim opuszczaniu
(ok. 180°C). Typowa struktura wysokowęglowej stali narzędziowej przedstawiona jest na rys.
7.2. Nagrzewanie zahartowanych stali węglowych powyżej temperatury 180°C zaczyna
powodować odpuszczanie martenzytu i obniżanie twardości. Wrażliwość na podwyższoną
temperaturę jest główną wadą stali węglowych narzędziowych, które z tego powodu są
zakwalifikowane jako stale do pracy na zimno i do obróbki materiałów przy niewielkiej
szybkości skrawania.
Rys. 7..2. Mikrostruktura stali węglowej narzędziowej N11E po hartowaniu i niskim
odpuszczaniu (180°C). Widoczne jasne wydzielenia cementytu na tle drobnoiglastego
martenzytu. Traw. 2% nitalem. Powiększ. 630x
Rys.7.3. Stal narzędziowa węglowa w stanie zmiękczonym. Widoczny cementyt kulkowy
(sferiodyt) na tle osnowy ferrytycznej. Traw. 5% nitalem. Powiększ. 500x
Stal narzędziowa jest dostarczana z huty w stanie zmiękczonym i aby ułatwić dalszą jej
przeróbkę lub obróbkę skrawaniem, wyżarzana w celu uzyskania struktury cementytu
kulkowego (rys. 7.3), gdyż stal mająca strukturę perlitu płytkowego trudniej poddaje się
obróbce. Strukturę taką otrzymuje się najprościej przez wyżarzanie sferoidyzujące w
temperaturze nieco wyższej od A
c1
7.4. Stale stopowe
Stalą stopową nazywa się stal, do której celowo wprowadzono pierwiastki stopowe, aby
nadać jej wymagane własności.
Według Polskich Norm do stali stopowych zalicza się gatunki stali, w których
najmniejsza wymagana zawartość chociażby jednego z pierwiastków jest równa lub
większa niż podano w tabl. 7.1.
Wprowadzenie do stali dodatków stopowych może mieć na celu:
• uzyskanie określonych własności wytrzymałościowych,
• wywołanie pożądanych zmian strukturalnych,
• uzyskanie specjalnych własności chemicznych lub fizycznych,
• podwyższenie hartowności,
• ułatwienie technologii i polepszenie efektów obróbki cieplnej.

109
JW
Najczęściej stosowanymi dodatkami stopowymi są: mangan, krzem, chrom, nikiel, molibden,
wanad, wolfram. Nieco rzadziej stosuje się aluminium, kobalt, tytan i niob. Ponadto coraz
częściej jako celowe dodatki stopowe zyskują na znaczeniu bor i azot.
7.4.1. Wpływ pierwiastków stopowych na strukturę i własności stali
Pierwiastki stopowe dodawane do stali w procesie metalurgicznym w przeważającej
ilości przechodzą do roztworu ciekłego. Po skrzepnięciu stali pierwiastki stopowe mogą
wystąpić w następujących fazach:
• w roztworach stałych: ferrycie i austenicie;
• w związkach z węglem i azotem: węglikach, azotkach i węgliko-azotkach;
• w związkach międzymetalicznych;
• w postaci wolnej (czystego pierwiastka).
Ze względu na różnice potencjału chemicznego pierwiastków w poszczególnych fazach,
składniki stopowe nie są równomiernie rozłożone we wszystkich składnikach strukturalnych
stopu, ale wykazują tendencję do skupiania się w poszczególnych fazach.
Węgliki są w stalach tworzone przez metale położone w układzie okresowym na lewo
od żelaza (Mn, Cr, V, Ti, Mo, Nb, Zr, W, Ta, Hf). Pierwiastki te należą podobnie jak
żelazo, do metali przejściowych. Im dalej na lewo od żelaza znajduje się w układzie
okresowym pierwiastek węglikotwórczy, tym aktywniej łączy się z węglem i trwałość
utworzonych węglików jest większa. Według wzrastającej skłonności do tworzenia w
stali węglików, pierwiastki węglikotwórcze można uszeregować w następującej
kolejności: Fe, Mn, Cr, W, Mo, V, Ti, Zr, Nb.
W stalach powstają najczęściej następujące węgliki:
węgliki grupy I - Fe
3
C, Mn
3
C, Cr
23
C
6
, Cr
7
C
3
, Fe
3
Mo
3
C, Fe
3
W
3
C;
węgliki grupy II - VC, TiC, NbC, ZrC, WC, W
2
C, Mo
2
C, TaC, Ta
2
C.
Węgliki grupy I mają złożoną sieć krystaliczną i charakteryzują się tym, że łatwo się
rozpuszczają w austenicie podczas nagrzewania.
Węgliki grupy II mają prostą sieć krystaliczną (regularną lub heksagonalną) znacznie trudniej
rozpuszczają się w austenicie, tak że przy nagrzewaniu nawet do wysokich temperatur mogą nie
przejść do roztworu stałego.
W stalach jednak węgliki z reguły nie występują w postaci czystej. Zawierają zwykle
rozpuszczone żelazo, a gdy w skład stali stopowej wchodzi kilka pierwiastków, to węgliki
zawierają również te pierwiastki w roztworze. Na przykład w stali chromowo-manganowej
tworzy się nie czysty węglik chromu Cr
23
C
6
, lecz węglik (Cr, Mn, Fe)
23
C
6
, zawierający w
roztworze żelazo i mangan.
Dodatki stopowe rozpuszczające się w żelazie wpływają silnie na zmianę temperatury
przemian alotropowych A
3
i A
4
. Niektóre z pierwiastków w pewnym zakresie stężeń albo
podwyższają temperaturę A
3
i obniżają temperaturę A
4
, wskutek czego ulega rozszerzeniu
obszar istnienia odmiany alotropowej
γ np. (Ni, Mn), albo obniżają temperaturę A
4
a
podwyższają temperaturę A
3
, zwężając obszar istnienia odmiany
γ (np. Cr, Si, W, Mo, V, Ti),
względnie mogą podwyższać (Co) lub obniżać obie te temperatury jednocześnie (Cr).
W wyniku oddziaływania pierwiastków stopowych na temperatury przemian alotropowych
żelaza oraz punkty krytyczne układu Fe-Fe
3
C, struktura stali stopowych może różnić się
zasadniczo od występującej w stalach węglowych przy tych równoważnych zawartościach
węgla. Duże znaczenie ma również wpływ pierwiastków stopowych na przemiany austenitu
przechłodzonego, w szczególności na krytyczną szybkość chłodzenia oraz temperaturę
przemiany martenzytycznej M
s
.
Pierwiastki, które rozpuszczają się jedynie w ferrycie lub cementycie, jak np. Mn, Ni, Si, Al,
Cu, wpływają na przemianę austenitu tylko ilościowo, opóźniając ją i przesuwając krzywą
początku rozkładu austenitu (na wykresie CTP) w kierunku większych wartości czasu (rys. 7.4)
w stosunku do stali węglowej (wyjątkiem jest jedynie Co, który przyspiesza przemianę).

110
JW
Natomiast pierwiastki węglikotwórcze wywołują w kinetyce przemiany izotermicznej
austenitu zmiany nie tylko ilościowe, ale i jakościowe. Krzywe początku przemiany ulegają nie
tylko przesunięciu, lecz również zmienia się ich kształt (rys. 7.4d). Obszary przemian
perlitycznej oraz bainitycznej zostają w tych stalach przedzielone zakresem o zwiększonej
trwałości przechłodzonego austenitu
Rys.7. 4. Schemat krzywych izotermicznych przemian austenitu przechłodzonego dla stali
stopowych: a) stal węglowa (0,45% C), b) stal manganowa (0,45% C, 0,2% Mn), c) stal
chromowo-wanadowa (0,5%C, 1,0% Cr, 0,1% V), d) stal chromowo-niklowo-molibdenowa
(0,30% C, 1,5% Cr, 2,0% Ni, 0,35 Mo)
Najważniejszy dla praktyki wpływ pierwiastków stopowych polega na zmniejszeniu
szybkości rozkładu austenitu w zakresie jego przemiany w struktury perlityczne. Zapewnia to
większą hartowność stali, a przechłodzenie austenitu do zakresu przemiany martenzytycznej
można osiągnąć stosując powolniejsze chłodzenie, np. podczas chłodzenia w oleju lub w
powietrzu.
Zwiększenie hartowności jest szczególnie duże, gdy stal zawiera jednocześnie kilka
pierwiastków stopowych, np. nikiel, chrom i molibden itp.
Stwierdzono również, że bardzo małe dodatki niektórych pierwiastków zwiększają bardzo
wyraźnie hartowność stali, natomiast większa ich zawartość nie wywołuje tak skutecznego
działania. Do takich pierwiastków należy przede wszystkim bor (B). Optymalna zawartość boru
w stali, zapewniająca największą hartowność wynosi zaledwie 0,001
÷0,003%. W razie większej
ilości boru jego stężenie na granicach ziarn austenitu przekracza maksymalną rozpuszczalność,
wskutek czego powstają odrębne fazy zawierające bor (borki), które jako ośrodki krystalizacji
ułatwiają wykrystalizowanie struktur perlitycznych i hartowność zmniejsza się.
Wpływ pierwiastków stopowych na wykresy CTP stali zaznacza się nie tylko zmianą
położenia i kształtu krzywych przemian, lecz również przesunięciem punktu przemiany
martenzytycznej M
s
. Większość pierwiastków obniża punkt M
s
, zwiększając tym samym
zawartość austenitu szczątkowego po zahartowaniu. Odwrotne działanie wywierają jedynie Al i
Co.
7.4.2. Klasyfikacja stali wg struktury po wyżarzaniu i po chłodzeniu na powietrzu
Przyjmując zasadę podziału wg struktury w stanie wyżarzonym, można wyróżnić następujące
grupy stali stopowych:
• podeutektoidalne, w których strukturze obok perlitu występuje wolny ferryt;
• eutektoidalne, o strukturze perlitycznej;
• nadeutektoidalne, zawierające w strukturze wydzielone z austenitu węgliki wtórne
• ledeburytyczne, w których strukturze występuje eutektyka - ledeburyt, zawierająca
węgliki pierwotne wydzielone z ciekłej stali;
• ferrytyczne, ewentualnie z wydzieleniami węglików;
• austenityczne, mogące również zawierać wydzielone węgliki.

111
JW
Zgodnie z wykresem Fe-Fe
3
C stale węglowe podeutektoidalne zawierają mnie niż 0,8% C,
eutektoidalne ok. 0,8% C, nadeutektoidalne 0,8
÷2,0% C, ledeburytyt natomiast pojawia się
powyżej ok. 2% C. Ponieważ jednak większość pierwiastków stopowych przesuwa punkty S i E
wykresu Fe-Fe
3
C w lewo, tj. w kierunku mniejszych zawartości węgla, więc granica między
stalami podeutektoidalnymi i nadeutektoidalnymi oraz nadeutektoidalnymi i ledeburytycznymi
odpowiada w stalach stopowych mniejszym zawartościom węgla niż w stalach węglowych.
Stale ferrytyczna i austenityczna są to najczęściej stale o dużej zawartości dodatków
stopowych i niskiej zawartości węgla.
Podział stali stopowych ze względu na strukturę przeprowadza się również w zależności od
tego, jaką strukturę otrzymuje się po ochłodzeniu w spokojnym powietrzu próbek o niedużym
przekroju. Struktura ta może się zasadniczo różnić od struktury uzyskanej po wyżarzaniu. W
tym przypadku można rozróżnić trzy podstawowe klasy stali:
• perlityczną,
• martenzytyczną,
• austenityczną
(mogą także występować klasy pośrednie). Klasę perlityczną cechuje dość mała zawartość
pierwiastków stopowych, stale klasy martenzytycznej zawierają więcej, a klasy austenitycznej
- najwięcej tych pierwiastków.
Wytworzenie się jednej z tych trzech struktur stali następuje wskutek tego, że w miarę
zwiększania się zawartości pierwiastków stopowych wzrasta trwałość przechłodzonego austenitu
(krzywe C na wykresie CTP przesuwają się w prawo), zaś początek przemiany martenzytycznej
obniża się w kierunku niższych temperatur.
Należy podkreślić, że podana klasyfikacja jest umowna i ma znaczenie w przypadku
chłodzenia w powietrzu próbek o dość małych wymiarach. Zmieniając warunki chłodzenia,
można oczywiście otrzymać w tej samej stali różne struktury.
7.4.3. Oznaczanie stali stopowych konstrukcyjnych i maszynowych
Sposób oznaczania różnych gatunków stali stopowych konstrukcyjnych został opracowany i
ujęty przez Polską Normę PN-89/H-84030/01.
Stale stopowe konstrukcyjne oznaczane są za pomocą znaku składającego się z: cyfr i liter.
Pierwsze dwie cyfry określają średnią zawartość węgla w setnych procenta. Litery oznaczają
pierwiastki stopowe:
G — mangan,
S — krzem,
H — chrom,
N — nikiel,
M — molibden,
T - tytan,
F - wanad (także V),
J — aluminium.
Liczby występujące za literami oznaczają zaokrąglone do liczby całkowitej średnie zawartości
składnika w stali w przypadku, gdy jego średnia zawartość przekracza 1,5% (w przypadku stali
niskostopowych, gdy średnia zawartość składnika przekracza 1%).
Stale o wyższych wymaganiach co do składu chemicznego (np. co do zawartości fosforu i
siarki) oznacza się na końcu znaku literą A.
Stale przetapiane elektrożużlowo oznacza się przez dodanie na końcu znaku stali Ż. Stale
modyfikowane związkami chemicznymi litu, sodu lub wapnia i innymi oznacza się literą D.
Według takich samych zasad, jak stale stopowe konstrukcyjne, oznacza się stale odporne na
korozję i stale żaroodporne. Natomiast stale stopowe narzędziowe oznacza się w odrębny sposób
wg dawnych cech hutniczych (patrz rozdz. 7.5).

112
JW
7.4.4. Stale niskostopowe o podwyższonej wytrzymałości
W wyniku dążenia do obniżania ciężaru konstrukcji, zwłaszcza budowlanych. i poprawy
wskaźników użytkowych opracowanych zostało szereg gatunków stali niskostopowych, które
bez dodatkowej obróbki cieplnej odznaczają się lepszymi własnościami mechanicznymi niż stale
węglowe. Są to stale zawierające niewielkie dodatki składników stopowych i wykazujące w
stanie dostawy podwyższone własności wytrzymałościowe i strukturę ferrytyczno-perlityczną.
Stale te są stosowane głównie na konstrukcje budowlane, mosty, siatki i pręty do zbrojenia
betonu, na zbiorniki i rury ciśnieniowe. Od materiałów tych, oprócz odpowiednio dużych war-
tości R
e
i R
m
, wymaga się odpowiedniej plastyczności, niskiej wartości temperatury progu
kruchości, dobrej spawalności oraz niskiej ceny.
Duże znaczenie przy opracowywaniu nowych gatunków stali o podwyższone wytrzymałości
miały osiągnięcia w zakresie fizyki metali, a w szczególności poznanie mechanizmów
umocnienia metali i stopów. Stwierdzono, że obok utwardzenia roztworu stałego i udziału perlitu
w strukturze, często znacznie większy wpływ na podwyższenie wytrzymałości stali wywierają
inne czynniki, w tym głównie wielkość ziarna i obecność w strukturze dyspersyjnych wydzieleń
węglików i azotków lub innych faz.
Szczególne znaczenie w produkcji stali o podwyższonej wytrzymałości ma tzw. regulowane
walcowanie, polegające na obniżeniu temperatury nagrzewania wsadu, na niewielkich, lecz
licznych zgniotach, a przede wszystkim na obniżeniu temperatury końca walcowania i
przyspieszeniu chłodzenia wyrobów po walcowaniu. W efekcie ulega zahamowaniu
rekrystalizacja zgniecionego austenitu, a uzyskane w wyniku jego przemiany drobne ziarno
ferrytu zapewnia odpowiednio wysokie własności wytrzymałościowe i plastyczne. Zasadnicze
znaczenie ma również obecność w stali mikrododatków Al, V, Ti, Nb i Zr, tworzących trudno
rozpuszczalne dyspersyjne wydzielenia, które wpływają na opóźnienie rekrystalizacji i rozrostu
ziarn austenitu, oddziałując tym samym na wzrost umocnienia i obniżenie progu kruchości.
Jedną z grup stali spawalnych o podwyższonej wytrzymałości stanowią stale niskostopowe o
strukturze ferrytyczno-perlitycznej zawierające maksymalnie 0,20% C dodatek manganu max do
ok. 1,8% oraz mikrododatki Al, V, Ti, Nb i N, tworzące dyspersyjne wydzielenia węglików i
azotków. Zawartości tych pierwiastków na ogół nie przekraczają 0,02% Al, 0,15% V, 0,05% Nb
oraz do ok. 0,025% N. Stale te stosowane po regulowanym walcowaniu lub normalizowaniu
zapewniają uzyskanie granicy plastyczności R
e
305
÷ 460 MPa (dla wyrobów o grubości 3 ÷ 16
mm).
Polska Norma PN-86/H-84018 obejmuje 11 gatunków stali niskostopowych podwyższonej
wytrzymałości oznaczonych znakami:
09G2
18G2A
09G2Cu
18G2ACu
15GA
18G2ANb
15G2ANb 18G2AV
15G2ANNb 18G2AVCu
18G2
Stale te, w zależności od wymaganych własności wytrzymałościowych na rozciągnie i
technologicznych na zginanie, dzielą się na 7 kategorii oznaczonych symbolami E305, E325,
E355, E390, E420, E440, E460. Trzycyfrowa liczba po literze E oznacza w przybliżeniu granicę
plastyczności R
e
w MPa. Granica ta wykazuje pewne niewielkie różnice w zależności od
grubości wyrobu (3
÷ 70 mm). Należy określić, że stale te mają znacznie wyższą (o 50 ÷ 80%)
granicę plastyczności porównaniu ze stalami węglowymi zwykłej jakości przeznaczonymi do
spawania, co stwarza możliwość uzyskania znacznych oszczędności materiałowych.
W zależności od wymaganej udarności w temperaturze od +20 do -60°C stale te dzielą się na
odmiany.
Jak wspomniano już na wstępie, omawiana grupa stali musi charakteryzować się dobrą
spawalnością. Muszą to być zatem stale o ograniczonej hartowności, tj. możliwie niskim
ekwiwalencie węgla C
E
, który można wyliczyć z zależności;
113
JW
Stale niskostopowe o podwyższonej wytrzymałości ujęte w PN-86/H-84018 mają ekwiwalent
węgla C
E
nie przekraczający 0,44
÷ 0,52.
7.4.5. Stale stopowe konstrukcyjne i maszynowe do ulepszania cieplnego
W przypadkach nie pozwalających na użycie stali węglowych ze względu na małą
hartowność lub też zbyt niskie własności wytrzymałościowe, stosuje się stale stopowe
konstrukcyjne i maszynowe do ulepszania cieplnego. Wykonuje się z nich głównie wysoko
obciążone i ważne elementy konstrukcyjne maszyn, silników, pojazdów mechanicznych itp.,
zwłaszcza o dużych przekrojach.
Grupa stali konstrukcyjnych stopowych do ulepszania cieplnego obejmuje znaczą ilość
gatunków o bardzo zróżnicowanym składzie chemicznym. Polskie Normy wyszczególniają 35
gatunków stali stopowych konstrukcyjnych do ulepszania cieplnego (PN-89/H-84030/04) oraz
ponadto 9 gatunków stali o większej zawartości pierwiastków stopowych, przeznaczonych do
wyrobu sprzętu szczególnie obciążonego PN-72/H-84035), np. sprzętu lotniczego, części
silników spalinowych itp. Skład chemiczny tych dwóch grup stali oraz ich własności
mechaniczne podano w tabl. 7.7
÷ 7.10.
Tablica 7.7
Skład chemiczny niektórych stali stopowych konstrukcyjnych do ulepszania cieplnego (wg
PN-89/H-84030/04)
Grupa stali
Średnia zawartość, %
Znak stali
C Mn Si Cr Ni Mo Inne
Mn 30G2
4502
0,30
0,45
1,60
1,60
0,27
0,27
Mn-Si
35SG
0,35
1,25
1,25
Cr
30H
40H
45H
50H
0,30
0,40
0,45
0,50
0,65
0,65
0,65
0,65
0,27
0,27
0,27
0,27
0,95
0,95
0,95
0,95
Cr-Si 37HS
0,37
0,45
1,15
1,45
Cr-Mn-Si
20HGS
30HGS
35HGS
0,20
0,30
0,35
0,95
0,95
0,95
1,05
1,05
0,15
0,95
0,95
1,25
Cr-Mo
25HM
30HM
35HM
40HM
0,25
0,30
0,35
0,40
0,55
0,55
0,55
0,55
0,27
0,27
0,27
0,27
0,95
0,95
1,05
0,95
0,20
0,20
0,20
0,20
Cr-Mo-V
40H2MF 0,40
0,65
0,27
1,75 0,35
V
-
0,20
Cr-Ni 45HN
0,45
0,65
0,27
0,60
1,2
Cr-Mn-Ni-Mo
37HGN
0,37 0,95 0,27 0,55 0,55 0,20
Cr-Ni-Mo
36HNM
34HNM
40HNM
0,36
0,36
0,40
0,65
0,55
0,65
0,27
0,27
0,27
1,05
1,50
0,75
1,05
1,50
1,45
0,20
0,20
0,20
Cr-Ni-Mo-V
45HNMF 0,45 0,65 0,27 0,95 1,55 0,20 V-0,15
Zawartość fosforu i siarki max po 0,025-0,035%.
Obróbka cieplna stali stopowych konstrukcyjnych polega na hartowaniu w oleju z
temperatury 820-950°C oraz odpuszczaniu najczęściej w zakresie 500-650°C. Uzyskuje się
wówczas sorbit złożony z ferrytu stopowego oraz bardzo drobnych węglików (rys. 7.5).
15
Cu
Ni
5
V
Mo
Cr
6
Mn
C
C
E
+
+
+
+
+
+
=
114
JW
Własności mechaniczne zależą od zawartości węgla i pierwiastków stopowych oraz od
temperatury odpuszczania. Niższa temperatura odpuszczania pozwala uzyskiwać wysokie
własności wytrzymałościowe przy gorszych plastycznych i odwrotnie, zależnie od stawianych
wymagań (rys. 7.6).
Hartowność stali stopowych.
Najistotniejszym kryterium stosowania poszczególnych gatunków
stali stopowych konstrukcyjnych jest hartowność. W tablicy 7.8. podano dla poszczególnych
gatunków stali wielkości średnic krytycznych, tj. największych średnic wyrobów hartujących się
na wskroś z utworzeniem w rdzeniu struktury zawierającej 50% martenzytu oraz 50% struktur
perlityczno-bainitycznych.
Rys. 7..5. Mikrostruktura stali 30HGSA po ulepszaniu cieplnym. Sorbit. Traw. 3% nitalem. 300x
Tablica 7.8
Własności mechaniczne w stanie ulepszonym cieplnie oraz hartowność (średnica
krytyczna) niektórych stali stopowych konstrukcyjnych
Własności wytrzymałościowe
Udarność
Znak stali
Średnica kry-
tyczna (50%
martenzytu)
hartów. w
oleju, min
R
m
MPa,
min
R
e
, MPa, min
A
5
, %
Z,%
KCU2,
J/cm
2
30G2
20 780 540 14 50 80
45G2 25 880
690
10
40 -
35SG 30 880
690
15
40 60
30H
30
880
740
12
45
70
40H 40 980
780
10
45 60
38HA 40 930
780
12
50 90
45H 40 1030
830
9
45 50
50H 45 1080
930
8
40 40
37HS 80 930
740
12
50 70
20HGS 40 780
640
12
45 70
30HGS 65 1080
830
10
45 45
35HGS 90 1620
1280
9
40 40
25HM 50 740
590
15
55 100
30HM
55
930
740
11
45
80
35HM 55 980
780
12
45 80
40HM
65
1030
880
10
45
70
40H2MF
250
1230
1030
9
40
50
45HN 50 1030
830
10
45 70
37HGNM
60 930
780
13
50 80
36HNM
110 980
780
11
50 80
34HNM
160 1080
880
10
45 70
40HMNA
165 1080
930
12
50 90
45HNMF
180 1470 1320 7 45 40
Na rysunkach 7.7 i 7.8 przedstawiono przykładowo pasma hartowności dla prób hartowania od
czoła dwóch gatunków stali o małej (40H) i bardzo dużej hartowności (40HNMA). Spośród stali
o jednakowej hartowności należy zawsze stosować najekonomiczniejszą, oczywiście o ile
dodatkowe wymagania (np. udarność) nie uzasadniają stosowania stali droższej, wyżej stopowej.
115
JW
Stale manganowe (30G2, 45G2), krzemowo-manganowe (35SG) oraz chromowe (30H, 40H,
45H, 50H) charakteryzują się stosunkowo niedużą hartownością w porównaniu z innymi
gatunkami stali stopowych. Znacznie wyższą hartowność wykazują stale chromowo-
manganowo-krzemowe (30HGS, 35HGS). Zastępują one w wielu przypadkach drogie stale
zawierające Ni, Mo, W i V.
Największą hartowność oraz najkorzystniejszy zespół własności wytrzymałościowych po
ulepszaniu cieplnym wykazują stale chromowo-niklowo-molibdenowe, ewentualnie z dodatkiem
wanadu lub wolframu, a także manganu i krzemu (40HNMA, 36HNM, 45HNMF, 30H2N2M,
30HGSNA, 25H2N4W, 30HN2MFA i inne). Stale te są używane na części maszyn o
największych wymaganiach wytrzymałościowych, jak wały korbowe silników lotniczych, wały
napędowe, na części turbin o dużych przekrojach, na koła zębate i inne części, gdzie występują
największe i zmienne obciążenia.
Tablica 7.9
Skład chemiczny stali stopowych konstrukcyjnych do ulepszania cieplnego
przeznaczonych do wyrobu sprzętu szczególnie obciążonego (wg PN-72/H-84035)
Średnia zawartość, %
Znak stali
C Mn Si Cr Ni Mo inne
25HGS 0,25 0,95 1,05 0,95
-
-
-
30HGSNA 0,30 1,15
1,05
1,05
1,60
- -
20HN3A
0,20
0,45
0,27
0,75
3,00
-
-
30HN3A 0,30 0,45
0,27
0,75
3,00
- -
37HN3A
0,37
0,40
0,27
1,40
3,25
-
-
25H2NWA 0,25 0,40
0,27
1,50
4,20
- W-1,00
30H2N2M 0,30 0,45
0,27
1,95
1,05
0,30
30HN2MFA
0,30
0,45
0,27
0,60
2,00
0,20
V - 0,22
65S2WA 0,65 0,85 1,75 - 0,40
max - W-1,00
Tablica 7.10
Własności mechaniczne stali stopowych konstrukcyjnych w stanie ulepszonym cieplnie,
przeznaczonych do wyrobu sprzętu szczególnie obciążonego (wg PN-72/H-84035)
Znak stali
Obróbka cieplna
hart.
o
C/odp. °C
R
m
MPa
min
R
e
MPa
min
A
5
, %
Z,%
KCU,
J/cm
2
25HGS 880/480
1080
830
10
40
60
30HGSNA 900/200-300
1620
1370
9
45 60
20HN3A 820/500
930
780
12
55
100
30HN3A 820/530
1080
880
10
50
80
37HN3A 820/520
1130
980
10
50
60
25H2N4W 850/560
1080
930
11
45 90
30H2N2M A.
830/600
980
830
13
50 80
B. 830/530
1230
1030
9
40
50
30HN2MFA 860/680
880
780
10
40 90
65S2WA 850/420
1860
1670
5
20 -
Stale stopowe konstrukcyjne o zawartości węgla 0,4
÷ 0,6% mogą być także poddawane
hartowaniu powierzchniowemu, co w wielu przypadkach jest korzystne, gdyż unika się w ten
sposób długotrwałego i bardziej kłopotliwego procesu nawęglania.
116
JW
Rys. 7.7. Pasmo hartowności dla próby hartowania od czoła stali 40H
Rys. 7.6. Wpływ temperatury odpuszczania oraz grubości wyrobu na własności
mechaniczne: a) stali węglowej 40, b) stali stopowej 40HNMA
Rys. 7.8. Pasmo hartowności dla próby hartowania od czoła stali 40HNMA

117
JW
7.4.6. Stale stopowe konstrukcyjne do nawęglania
Nawęglanie ma na celu uzyskanie twardej i odpornej na ścieranie warstwy wierzchniej
elementu konstrukcyjnego, przy zachowaniu wysokiej udarności i ciągliwości rdzenia.
Własności te uzyskuje się przez odpowiednią obróbkę cieplną. Dużą twardość osiąga się przez
wzbogacenie warstwy powierzchniowej w węgiel i następnie zahartowanie. Drugim
zagadnieniem jest sprawa wytrzymałości rdzenia nawęglonego przedmiotu. Na ogół wymaga się
od rdzenia dużej udarności i ciągliwości, aby skompensować niebezpieczeństwo, które
przedstawia warstwa powierzchniowa o dużej twardości i kruchości. Z tego względu zawartość
węgla w stalach do nawęglania jest niska i wynosi zazwyczaj 0,10
÷0,25%, natomiast wyższą
wytrzymałość rdzenia uzyskuje się dzięki obecności pierwiastków stopowych.
W porównaniu ze stalami węglowymi stale stopowe do nawęglania mają wyższą wytrzymałość
na rozciąganie zarówno w stanie zmiękczonym, jak i zahartowanym, a dzięki większej
hartowności wysoką wytrzymałość można uzyskać w elementach o większych przekrojach przy
jednocześnie dużej udarności, dużym przewężeniu i wydłużeniu. Przedmioty wykonane ze stali
stopowej charakteryzuje więc po nawęgleniu i zahartowaniu duża wytrzymałość rdzenia, której
nie można uzyskać przy użyciu stali węglowych. Z tego względu stal stopową do nawęglania
stosuje się wyłącznie na wysoko obciążone, ważne elementy konstrukcyjne silników, pojazdów
mechanicznych i samolotów oraz na inne odpowiedzialne części maszyn.
Aby spełnić zasadniczy postulat uzyskania najwyższej twardości powierzchniowej, należy
warunki hartowania dostosować do składu chemicznego warstwy nawęglonej, dla której
właściwa temperatura hartowania jest znacznie niższa niż temperatura hartowania właściwa dla
rdzenia. Poza tym temperatura odpuszczania po hartowaniu musi być niska, gdyż już przy 150°C
twardość warstwy nawęglanej zaczyna się zmniejszać. Wobec tego, że własności stali do
nawęglania nie można zmieniać przez odpuszczanie, skład chemiczny stali jest zasadniczym
czynnikiem rozstrzygającym o własnościach wytrzymałościowych rdzenia.
Wynika stąd, że dobrawszy odpowiednio zawartość pierwiastków stopowych można uzyskać
jednocześnie potrzebną wytrzymałość rdzenia w wymaganym przekroju i pożądaną twardość
powierzchniową po nawęgleniu. Ponieważ jednak każdy gatunek stali pozwala na osiągnięcie
tylko wąskiego zakresu wytrzymałości rdzenia, aby uzyskać szeroki zakres wytrzymałości R
m
700
÷ 1500 MPa i spełnić różnorodne wymagania dotyczące twardości powierzchniowej, należy
mieć do dyspozycji dość dużo gatunków stali do nawęglania.
Polskie Normy obejmują łącznie 20 gatunków stali stopowych do nawęglania. W grupie stali
stopowych konstrukcyjnych (PN-89/H-84030/02) Polskie Normy wyszczególniają 16 gatunków
stali do nawęglania: 15H, 20H, 16HG, 20HG, 18HGT, 15HGM, 15HGMA, 18HGM, 17HGN,
15HGN, 15HN, 15HNA, 20HNM, 22HNM, 17HNM, 18H2N2 a w grupie stali stopowych
konstrukcyjnych przeznaczonych do wyrobu sprzętu szczególnie obciążonego (PN-72/H-84035)
- 4 gatunki stali do nawęglania: 12HN3A, 12H2N4A, 20H2N4A, 18H2N4WA. Stale te
odznaczają się niską zawartością węgla (średnio 0,12-0,22%), zawierają prawie zawsze 0,5
÷ 2%
Cr oraz zależnie od gatunku również Mn, Ni, Mo oraz rzadziej Ti i W.
Najniższe własności mechaniczne rdzenia uzyskuje się w przypadku stali chromowych i
chromowo-manganowych (15H, 20H, 16HG, 20HG). Mangan w omawianych stalach sprzyja
niekorzystnemu rozrostowi ziarn. Przeciwdziała się temu przez dodatek Ti, np. w stali 18HGT.
Wobec mniejszej skłonności do rozrostu ziarn, stal może być nawęglana w szerokim zakresie
temperatury. Wytrzymałość rdzenia na rozciąganie w tych stalach może dochodzić do ponad
1200 MPa.
Stale chromowo-niklowe (15HN, 17HNM, 18H2N2) uzyskują znacznie lepsze własności, ze
względu jednak na drogi dodatek niklu zastępowane są coraz częściej stalami chromowo-
manganowo-molibdenowymi (15HGM, 18HGM, 19HM) również wykazującymi wysokie
własności mechaniczne i dużą hartowność.
Elementy maszyn wymagające wysokich własności plastycznych rdzenia i jednocześnie
bardzo wysokiej wytrzymałości (Rm = 1200
÷ 1400 MPa), jak np. części silników lotniczych,
wykonuje się ze stali chromowo-niklowych wyższej jakości: większej zawartości chromu (ok.

118
JW
1,5%) i niklu (3
÷ 4,5%) z dodatkiem Mo (0,2 ÷ 0,3) lub W (ok. 1%) (np. stali 12HN3A,
12H2N4A, 20H2N4A, 18H2N4WA).
7 4.7. Stale do azotowania
Dzięki zawartości niektórych pierwiastków stopowych, a w szczególności aluminium, chromu
i molibdenu stale stopowe do azotowania pozwalają na uzyskanie po azotowaniu największej
twardości i odporności na ścieranie warstwy wierzchniej, bez potrzeby stosowania dodatkowej
obróbki cieplnej. Twardość warstwy naazotowanej nie tylko nie zmniejsza się po nagrzaniu do
temperatury dochodzącej do 500°C, lecz także pozostaje nie zmieniona podczas dłuższego
wygrzewania w tym zakresie temperatury.
W związku z tym stale do azotowania znajdują duże zastosowanie na cylindry, wały, sworznie
tłokowe i inne części silników spalinowych, na części turbin, armaturę do pary przegrzanej,
wrzeciona zaworów, sprawdziany itp.
Czynnikiem rozstrzygającym o wysokiej twardości naazotowanej warstwy powierzchniowej jest
niemal wyłącznie skład chemiczny stali, a mianowicie zawartość pierwiastków tworzących
trwałe azotki (Al, Cr, Mo i V).
Polska Norma PN-89/H-84030/03 przewiduje 3 gatunki konstrukcyjnych stali stopowych do
azotowania: 38HMJ, 33H3MF i 25H3M.
Oprócz specjalnych gatunków do azotowania, również niektóre stale chromowo-
-molibdenowe i zawierające wanad (40HMF, 40HGM, 35HM) mogą być stosowane do tego
celu, nie pozwalając jednak na uzyskanie maksymalnej twardości powierzchniowej. Przed
azotowaniem stale ulepsza się cieplnie, stosując hartowanie w wodzie lub oleju i wysokie
odpuszczanie, aby uzyskać możliwie wysokie własności wytrzymałościowe rdzenia. Stale te
dzięki większej zawartości węgla i pierwiastków stopowych odznaczają się dużą hartownością.
7.4.8. Stale sprężynowe
Stale konstrukcyjne przeznaczone do wyrobu sprężyn i resorów powinny się charakteryzować
wysoką granicą sprężystości i plastyczności oraz dużą wytrzymałością na zmęczenie.
Jednocześnie jednak stale te muszą mieć pewne minimalne własności plastyczne, aby w razie
przekroczenia granicy sprężystości raczej nastąpiło odkształcenie, a nie pęknięcie. Duża ilość
różnorodnych sprężyn i metod ich wytwarzania wymaga stosowania różnych materiałów i
różnych gatunków stali. Typowe stale sprężynowe cechuje zwiększona zawartość węgla,
wynosząca zazwyczaj 0,5
÷ 0,7%. Stale te zawierają również dodatki manganu, krzemu i chromu
oraz wanadu. Wysoką granicę sprężystości tych stali osiąga się przez hartowanie (przeważnie w
oleju) i odpuszczanie w temperaturze 380
÷ 520°C. Ten zakres temperatury odpuszczania
zapewnia najkorzystniejszy stosunek granicy sprężystości R
sp
(lub granicy plastyczności R
e
,)
do
wytrzymałości na rozciąganie R
m
.
Stale sprężynowe są znormalizowane. W tablicy 7.11 podano średnią zawartość głównych
dodatków stopowych oraz własności mechaniczne w stanie ulepszonym cieplnie wg PN-74/H-
84032. Pierwsze trzy gatunki są stalami niestopowymi o zawartości węgla 0,65
÷ 0,85%
(±0,05%), podlegającym hartowaniu w oleju i odpuszczaniu. Sprężyny mniej odpowiedzialne
wykonuje się ze stali węglowej również w stanie surowym z taśm walcowanych na zimno lub
drutu ciągnionego. Sprężyny bardziej odpowiedzialne wykonuje się ze stali stopowych
zawierających 0,4
÷ 2,0% Si z ewentualnym dodatkiem Mn, Cr i V. Krzem jest pierwiastkiem
stopowym, który najintensywniej zwiększa R
sp
, R
e
, i R
m
i dlatego jest składnikiem większości
gatunków stali sprężynowych.
Stale sprężynowe krzemowe (45S, 50S, 40S2, 50S2, 55S2, 60S2, 60S2A) wykazują
stosunkowo małą hartowność, co ma jednak mniejsze znaczenie, gdyż sprężyny mają zwykle
małe przekroje. W przypadku większych przekrojów zaleca się stale zawierające chrom oraz Si,
Mn lub V zapewniające większą hartowność. Do wyrobu sprężyn o szczególnie ważnym
przeznaczeniu stosuje się stal chromowo-wanadową 50HF, która charakteryzuje się bardzo
119
JW
drobnym ziarnem oraz wykazuje mniejszą skłonność do odwęglania powierzchniowego niż stale
krzemowe
Tablica 7.11
Skład chemiczny i własności mechaniczne w stanie ulepszonym cieplnie
stali sprężynowych (wg PN-74/H-84032)
Temp. hart.,
°C
Grupa
stali
Znak
stali'
1
Średnia za-
wartość
składników Temp. odp.,
°C
R
m
MPa
min
R
e
MPa
min
A
5
%
min
Z,% min
65
0,65%
C
840/480
980
780
10
35
C 75
0,75%
C
820/480
1080
880
9 30
85
0,85%
C
820/480
1030
980
8
30
Mn 65G
1,1%
Mn
830/480
980 780 8 30
45S
1,15%Si
830/420
1180
980
6
-
50S
0,45% Si
800/380
1080
930
5
-
40S2
1,70%Si
840/430
1370
- 6 -
50S2
1,65%Si
870/460
1280
1080
6 30
Si 55S2
1,65%Si
870/460
1320
1180 6 30
60S2
1,65%Si
870/460
1370
1180 5 25
60S2A
1,80%Si
870/420
1520
1180 5 20
Mn-Si 60SG 0,95% Mn
860/460
1570
1370
6
25
1,55%Si
1,0%
Mn
Si-Mn-Cr
60SGH 1,15%Si 850/480 1370 1230 7
-
0,50% Cr
Cr-Mn
50HG
1,05%Cr
840/440
1370
1180
7
35
0,95% Mn
Cr-Si 50HS 1,05%Cr 850/520 1320 1180 6 30
1,00%Si
Cr-V 50HF 0,95%
Cr
850/500
1280
1080 8 35
0,15% V
Wiele sprężyn wykonuje się również z innych stali, np. ze stali narzędziowych węglowych
lub stopowych, a do pracy w podwyższonych temperaturach ze stali narzędziowych
szybkotnących. Natomiast sprężyny pracujące w środowiskach korozyjnych wykonywane są ze
stali nierdzewnych hartowanych i odpuszczonych lub utwardzonych przez zgniot.
7.
4.9. Stal na łożyska toczne
Stal do wyrobu łożysk tocznych (pierścieni łożyskowych, kulek, wałeczków itp.) powinna się
odznaczać wysoką twardością i odpornością na ścieranie, a także dużą wytrzymałość na
ściskanie i zginanie. W tym celu stosuje się stale wysokowęglowe (ok. 1% C) z dodatkiem
chromu (ok. 1,5% Cr) i ewentualnie manganu i krzemu, głównie w celu zwiększenia
hartowności. Ze względu na warunki pracy oraz metody produkcji stalom tym stawia się
szczególne wymagania pod względem czystości i struktury. Dopuszczalna zawartość fosforu i
siarki jest w nich bardzo ograniczona i wynosi wg PN-74/H-84041 max 0,027% P i max 0,020%
S. Ponadto w stalach tych kontroluje się ściśle stopień zanieczyszczenia wtrąceniami
niemetalicznymi, pasmowość ułożenia węglików (segregację) oraz przeprowadza inne
szczegółowe badania mikroskopowe i makroskopowe.
W kraju stosuje się dwa gatunki stali łożyskowych (PN-74/H-84041):
ŁH15 (1,0% C, 0,3% Mn, 0,25% Si, 1,50% Cr),
ŁH15SG (1,0% C, 1,1% Mn, 0,55% Si, 1,5% Cr).
Stal ŁH15SG ze względu na wyższą zawartość manganu i krzemu ma większą hartowność i
jest stosowana do wyrobu pierścieni łożyskowych o większej grubości (powyżej 30 mm).

120
JW
Obróbka cieplna stali łożyskowych polega na hartowaniu w oleju od temperatur 815
÷ 860°C
(zależnie od grubości wyrobu) i niskim odpuszczaniu w temperaturze ok. 160°C. Po obróbce
cieplnej stal powinna mieć twardość co najmniej 61HRC.
Struktura stali łożyskowych w stanie obrobionym cieplnie składa się z drobnoziarnistego
odpuszczonego martenzytu i drobnych wtrąceń równomiernie rozłożonych węglików chromu
(rys. 7.9).
Rys. 7.9. Mikrostruktura stali łożyskowej ŁH15 po hartowaniu i niskim odpuszczaniu. Traw.
3% nitalem Powiększ. 630x
Na łożyska toczne pracujące w środowiskach powodujących korozję stosuje się najczęściej
stal H 18 zawierającą ok. 1% C i 18% Cr. Duża zawartość chromu w tej stali jest niezbędna, by
nadać jej znaczną odporność na korozję. Obróbka cieplna tej stali polega na hartowaniu w oleju
od temperatury 1050°C, obróbce podzerowej w 70°C i odpuszczaniu w temp. ok. 160°C.
Twardość stali po takiej obróbce wynosi 60
÷ 61 HRC.
7.5. Stale narzędziowe stopowe
Zależnie od warunków pracy, od stali narzędziowych wymaga się wysokie twardości i
hartowności, odporności na ścieranie, odpowiedniej wytrzymałość i ciągliwości (zwłaszcza w
przypadku obciążeń udarowych), odporności na odpuszczające działanie ciepła oraz twardości i
wytrzymałości w podwyższonych temperaturach. Własności te w decydującej mierze zależą od
składu chemicznego tych stali rodzaju i ilości dodatków stopowych), a także od przeróbki
plastycznej i obróbki cieplnej, które w istotny sposób wpływają na strukturę i własności stali.
Twardość stali zahartowanej zależy przede wszystkim od zawartości węgla, przy czym
maksymalną twardość ok. 66 HRC osiąga martenzyt przy zawartości węgla ok. 0,8%.
Pierwiastki stopowe nie powiększają twardości w sposób istotny, ale głównie zwiększają
hartowność i tworzą twarde węgliki odporne na ścieranie.
Większa hartowność jest wymagana w odniesieniu do stali narzędziowych, szczególnie w
tych przypadkach, gdy podczas pracy narzędzia występują znaczne naciski. Wówczas warstwa
zahartowana na martenzyt musi być odpowiednio grubsza i potrzebna jest większa wytrzymałość
rdzenia. Osiąga się to przez stosowanie stali narzędziowych stopowych. Przy bardzo dużych
naciskach konieczne jest stosowanie stali hartujących się na wskroś. Narzędzia wykonane ze
stali stopowych hartuje się w oleju. Łagodniejsze chłodzenie (w porównaniu ze stalami
narzędziowymi węglowymi, które hartuje się w wodzie) zmniejsza niebezpieczeństwo pęknięć i
odkształceń, co jest bardzo istotne w przypadku narzędzi o złożonych kształtach.
Wysoka odporność na ścieranie narzędzi, zwłaszcza stosowanych do obróbki w produkcji
seryjnej lub jako narzędzia pomiarowe, staje się parametrem decydującym. Odporność na
ścieranie stali narzędziowych osiąga się przez zwiększenie ilości twardych węglików stopowych
w strukturze zahartowanej stali, a zwłaszcza węglików chromu (typu M
23
C
6
) i wolframu (M
6
C).
Klasyfikacja stali narzędziowych stopowych opiera się głównie na ich zastosowaniu.
W szczególności można wyróżnić następujące grupy objęte normami:
- stale narzędziowe stopowe do pracy na zimno (PN-86/H-85023),
- stale narzędziowe stopowe do pracy na gorąco (PN-86/H-85021),
- stale szybkotnące (PN-86/H-85022).

121
JW
Przyjęty przez polskie normy sposób oznaczania stali narzędziowych stopowych różni się od
oznaczeń stali stopowych konstrukcyjnych. Znak stali składa się z liter liczb, przy czym
pierwsza litera oznacza zawsze grupę stali narzędziowych:
N — stale narzędziowe stopowe do pracy na zimno,
W — stale narzędziowe stopowe do pracy na gorąco,
S — stale szybkotnące.
Następna litera lub kilka liter określają składniki stopowe lub grupę składników, przy czym
symbolika jest tu nieco inna niż w przypadku stali konstrukcyjnych specjalnych, a mianowicie:
M - mangan, W - wolfram,
S - krzem, K - kobalt,
C - chrom, B - bor,
N - nikiel, P - chrom + nikiel + wanad,
L - molibden, Z - krzem + chrom + wolfram.
V - wanad,
Liczba znajdująca się na końcu lub w środku znaku służy do odróżnienia poszczególnych
gatunków stali zawierających te same składniki stopowe. W stalach szybkotnących liczby te
oznaczają średnią zawartość głównego składnika stopowego w procentach.
7.5.1. Stale narzędziowe stopowe do pracy na zimno
Ze stali narzędziowych stopowych do pracy na zimno wykonuje się narzędzia służące do
obróbki materiałów w temperaturze otoczenia. Stale te w porównaniu ze stalami narzędziowymi
węglowymi mają większą hartowność, wyższą wytrzymałość i ciągliwość oraz lepszą odporność
na ścieranie.
Polska Norma PN-86/H-85023 obejmuje 18 gatunków stali narzędziowych stopowych do
pracy na zimno:
NV (V 0,22)
NW1 (W 1,25)
NWV (Mn 1,9, V 0,15)
NZ2 (Si 0,95, Cr 1,05, W 1,85, V 0,22)
NCV1 (Cr 0.55, V 0,22)
NZ3 (Si 0,95, Cr 1,05, W 1,85, V 0,22)
NCMS (Cr 1,45, Mn 1,1)
NW1 (W 1,25)
NC5 (Cr 0,55)
NWC (W 1,4, Cr 1,05)
NC6 (Cr 1,45, V 0,20)
NPW (Cr 1,35, Ni 3, V 0,5)
NC4 (Cr 1,45)
NMWV (Mn 1,2, W 0,6, V 0,2)
NC10 (Cr 12, C 1,65)
NCLV (CR 5, V 0,4, Mo 1,0)
NC11 (Cr 12, C 1,95)
NW9 (Cr 4,3, W 9, V 2).
NC11LV (Cr 11, Mo 0,85, V 0,75)
Obok znaku stali w nawiasie podano średnią zawartość pierwiastków stopowych w
procentach (liczby za symbolami pierwiastków).
Gatunki NZ2, NZ3 i NPW są stalami średniowęglowymi o zawartości 0,40
÷ 0,55% C.
Pozostałe gatunki są stalami wysokowęglowymi zawierającymi 0,75
÷ 2,10% C. Stale
średniowęglowe znalazły zastosowanie na narzędzia, od których wymagana jest większa
plastyczność i odporność na obciążenia dynamiczne, jak np. matryce, stemple, tłoczniki, rolki do
prasowania, wybijaki itp. Stale wysoko-węglowe stosowane są głównie do wyrobu narzędzi
skrawających.
Podstawowymi dodatkami stopowymi w stalach narzędziowych do pracy na zimno są: Cr, W,
V oraz w stali NPW - Ni. Dodatki te nadają stali dużą hartowność i drobnoziarnistą strukturę,
zapewniają wysokie własności wytrzymałościowe, a w szczególności wysoką odporność na
ścieranie wskutek tworzenia się węglików stopowych o dużej twardości i dyspersji. Węgliki te
nie ulegają całkowitemu rozpuszczeniu w czasie nagrzewania do hartowania, dzięki czemu
przeciwdziałają rozrostowi ziarn austenitu, zapewniając tym samym drobnoziarnistość stali.
Zależnie od gatunku stale hartuje się w wodzie lub oleju z temperatur) 780
÷ 1020°C, a stale
wysokochromowe (NC10, NC11 i NC11LV) z temperatur) 970
÷ 1020°C. Po hartowaniu stosuje

122
JW
się w zasadzie niskie odpuszczanie w temperaturze 150
÷ 350°C. Stale wysokochromowe
odpuszcza się w nieco wyższych temperaturach w zakresie 220
÷ 450°C. Stal NW9 hartuje się z
temperatury 1200°C i odpuszcza w temperaturze 500
÷ 560°C.
Twardość stali wysokowęglowych w stanie zahartowanym wynosi 60
÷ 68 HRC, natomiast
stale średniowęglowe mają po hartowaniu twardość 50
÷ 57 HRC.
Należy podkreślić, że własności stali narzędziowych stopowych po hartowaniu i
odpuszczaniu zależą w dużej mierze od temperatury austenityzowania, z którą ściśle wiąże się
stopień nasycenia roztworu stałego (austenitu) dodatkami stopowymi i późniejsze wydzielanie
się węglików wtórnych podczas odpuszczania.
7.5.2. Stale narzędziowe stopowe do pracy na gorąco
Ze stali narzędziowych do pracy na gorąco wytwarza się narzędzia służące do przeróbki
plastycznej materiałów uprzednio nagrzanych do wysokich temperatur oraz formy do odlewania
metali pod ciśnieniem. W stanie nagrzanym przerabiane metale są plastyczne, więc stale
narzędziowe do pracy na gorąco nie muszą mieć tak dużej twardości w temperaturze otoczenia
jak stale do pracy na zimno. Wymagania stawiane stalom do pracy na gorąco to przede
wszystkim wysoka wytrzymałość i twardość przy wyższych temperaturach, wysoka udarność,
stabilność struktury, odpowiednio wysoka hartowność oraz możliwie mała skłonność do
zmęczenia cieplnego i pęknięć ogniowych. Odporność na ścieranie i erozję, którą powoduje
odkształcony plastycznie metal, jest również ważną cechą, ale główną przyczyną zużywania
się narzędzi są pęknięcia ogniowe.
Odpowiednie własności osiąga się przez stosowanie stali o stosunkowo niskiej zawartości
węgla 0,25
÷ 0,60%, zawierających jako podstawowe dodatki stopowe wolfram, molibden,
wanad i chrom, a także czasem krzem i nikiel. Wolfram, molibden i wanad są pierwiastkami
silnie węglikotwórczymi, które po rozpuszczeniu podczas austenityzacji i po zahartowaniu dają
podczas odpuszczania twardość wtórną lub znacznie hamują spadek twardości stali. Chrom
silnie zwiększa hartowność, a przy wyższych zawartościach również odporność na utlenianie.
Polska Norma PN-86/H-85021 obejmuje 12 gatunków stali narzędziowych stopowych do
pracy na gorąco, a mianowicie:
WNLV (Cr 1,2, Ni 1,75, Mo 0,6, V 0,1),
WLV (Cr 3,0, Mo 2,75, V 0,55),
WLK (Cr 2,75, Mo 2,75, V 0,5, Co 3,0),
WCLV (Cr 5,25, Mo 1,35, V 1,0),
WCL (Cr 5,0, Mo 1,35, V 0,4),
WLB (Cr 2,4, Mo 0,4, B 0,003),
WNLB (Cr 1,1, Mo 0,3, B 0,003, Ti 0,03, A1 0,03),
WNL (Cr 0,7, Ni 1,6, Mo 0,22),
WNL1 (jak WNL + V 0,1),
WWS1 (Cr 2,5, V 0,5, W 4,5, Si 1,0),
WWV (Cr 2,7, W 9,0, V 0,3),
WWN1 (jak WWV + Ni 1,4).
Obok znaku stali w nawiasach podano średnią zawartość dodatków stopowych liczby za
symbolami pierwiastków). Zawartość węgla w tych stalach wynosi średnio 0,3
÷ 0,6%.
Obróbka cieplna stali do pracy na gorąco polega na hartowaniu w oleju lub na powietrzu z
temperatury w zakresie 840
÷
1160°C (zależnie od gatunku stali) i następnym odpuszczaniu w
zakresie 400
÷
600°C, tj. w temperaturach wyższych niż stali do pracy na zimno. Zapewnia to
dobrą udarność, niezbędną ze względu na charakter pracy oraz twardość min 43
÷
50 HRC.
Temperatura odpuszczania stali powinna być wyższa od temperatury pracy, aby zapewnić
stabilność struktury i własności.
Ze względu na zastosowanie, stale do pracy na gorąco można podzielić na trzy grupy. Do
pierwszej należą stale, z których wykonuje się matryce do pras i formy dla odlewów pod
ciśnieniem. Wspólną cechą tych zastosowań jest stosunkowo długi kontakt gorącego materiału z
narzędziem i związane z tym silniejsze nagrzewanie się powierzchni pracującej. Są to stale
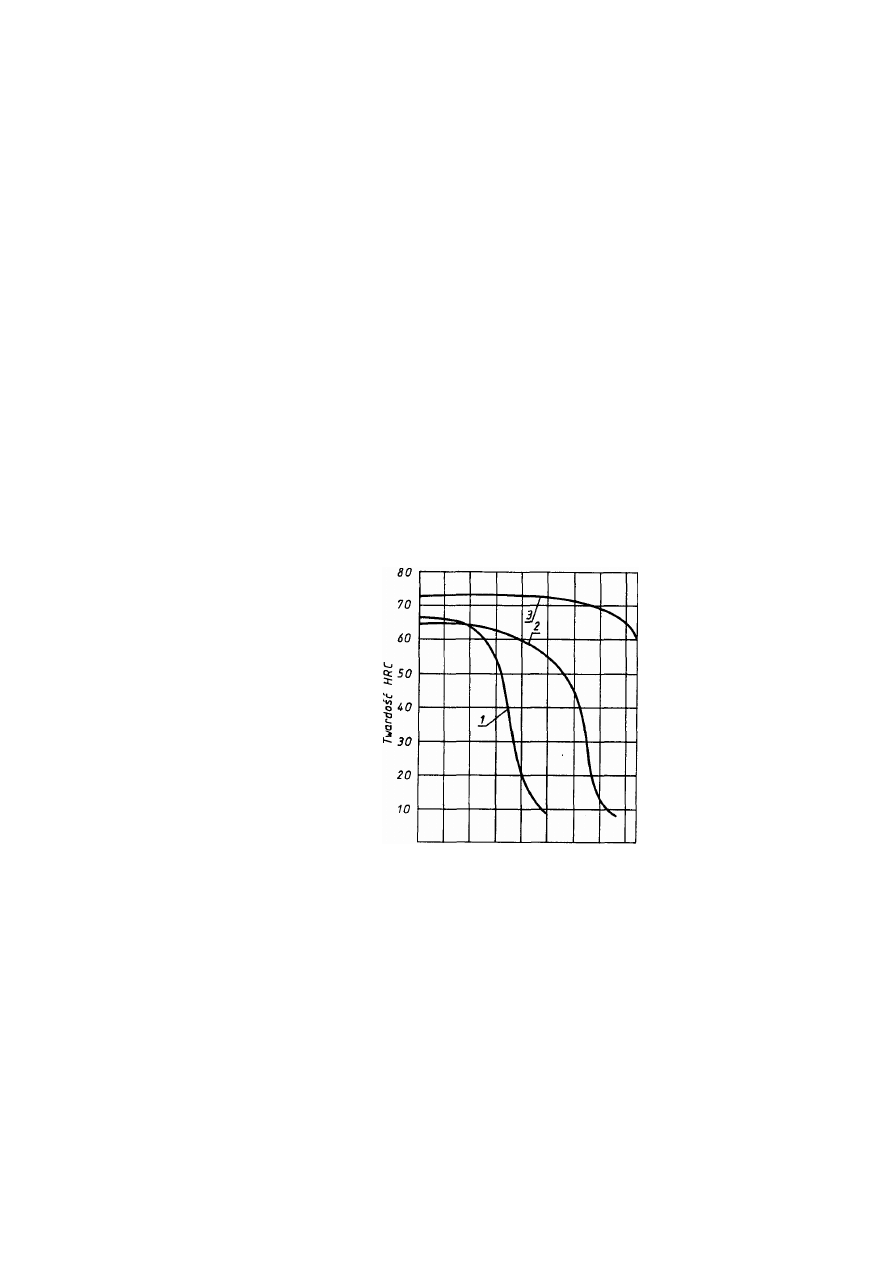
123
JW
wolframowe (WWS1, WWV) i chromowo-molibdenowa (WCL). Zawartość węgla w tych
stalach jest stosunkowo niska (0,3
÷ 0,4%), co zapewnia ciągliwość. Temperatura
austenityzowania tych stali jest wysoka (950
÷ 1120°C) ze względu na konieczność
rozpuszczania węglików zawierających W, Mo, V, Cr.
Drugą grupę stanowią stale używane przede wszystkim na matryce kuzienne i kowadła do
młotów (WNL, WNLV). Matryce mają stosunkowo krótki kontakt z gorącą odkuwką, natomiast
w czasie pracy występują duże naciski i uderzenia związane z kuciem, co wymaga materiału
twardego, ale równocześnie bardzo ciągliwego. W stalach tych bardzo ważna jest hartowność,
ponieważ wymiary matryc często są znaczne.
Do trzeciej grupy zalicza się stale używane na walce do walcowania na gorąco oraz na
wkładki matrycowe do pras i kuźniarek, oraz stemple do wyciskania i spęczania wyrobów ze
stopów miedzi i aluminium.
7.5.3. Stale szybkotnące
Nazwa „stale szybkotnące" pochodzi stąd, że służą one do wyrobu narzędzi skrawających,
pracujących przy dużych prędkościach skrawania lub przy dużych przekrojach wióra. Praca w
takich warunkach jest przyczyną bardzo silnego rozgrzewania się narzędzia, nawet do
temperatury czerwonego żaru, wskutek tarcia o skrawany materiał. Stale węglowe i
niskostopowe w tych warunkach szybko tracą twardość, a narzędzia tępią się, natomiast stale
szybkotnące zachowują wysoką twardość do znacznie wyższych temperatur (rys. 7.10). Wysoką
twardość „na gorąco" oraz odporność na ścieranie nadaje stalom szybkotnącym twarda i nie
mięknąca pod wpływem odpuszczania osnowa, w której rozmieszczone są twarde węgliki.
Rys. 7.10. Twardość na gorąco różnych materiałów narzędziowych: l — stal narzędziowa
węglowa, 2 — stal szybkotnąca, 3 — węgliki spiekane
Podstawowymi składnikami stopowymi stali szybkotnących są pierwiastki węglikotwórcze:
wolfram, wanad, chrom i molibden. Stale o najlepszych własnościach zawierają również znaczne
dodatki kobaltu.
Skład chemiczny stali szybkotnących produkowanych w kraju, podany jest w tabl. 7.12 (wg
PN-86/H-85022).
Stale szybkotnące przyjęto zaliczać do tzw. stali ledeburytycznych, ponieważ w stanie odlanym
w ich strukturze występuje częściowo eutektyka, na skutek nie osiągania stanu równowagi
podczas krzepnięcia. Po przekuciu i wyżarzeniu struktura stali szybkotnących składa się z
ferrytu stopowego i mniej lub więcej równomiernie rozmieszczonych węglików.
O 100 200 300 400 500 600 700 800
Temperatura,
o
C
124
JW
Tablica 7.12
Stale szybkotnące (wg PN-86/H-85022)
Średnia zawartość, %
Średnia tem-
peratura,
o
C
Twardość w
stanie harto
wania i odpu
Znak
stali
C
Cr
W
Mo
V
Co
hart.
odp.
szczania HRC,
min
SW18 0,8 4,0 18,0 — 1,3 1250
560
64
SW7M 0,87 4,0 6,5 5,0 2,0 - 1210
560
65
SW12 1,1 4,0 12,0 - 2,5 - 1190
560
64
SK5M 0,92 4,0 6,3
4,7
1,9
5,0
1200
560
65
SK8M 1,1 4,0 1,6 9,5 1,2 8,0 1190
560
66
SK5 1,1 4,0 12,0 - 2,3 5,0
1200
560
65
SK5V 1,38 4,0 12,8 1,0 4,5 5,5 1270
560
65
SK10V 1,22 4,0 10,0 3,3 3,0 10,0 1220
560
66
SW2M5 0,95 4,0 1,75 5,0 1,3 - 1160
560
64
SK5MC
1,1 4,0 7,0 4,0 1,9 5,0 1200
560
66
Obróbka cieplna narzędzi ze stali szybkotnących polega na hartowaniu i odpuszczaniu. Do
hartowania stale nagrzewa się do wysokiej temperatury ok. 1160
÷ 1270°C (tabl. 7.12), aby
zapewnić rozpuszczenie się dostatecznie dużej ilości węgla i składników stosowanych w
austenicie, które tym samym, po hartowaniu zostaną w martenzycie. Ze względu na małe
przewodnictwo cieplne oraz wysoką temperaturę hartowania, nagrzewanie stali szybkotnącej
przy hartowaniu prowadzi się stopniowo wg schematu przedstawionego na rys. 7.11.
Odpuszczanie po hartowaniu przeprowadza się w temperaturze ok. 560°C, przy czym zabieg
ten powtarza się dwu- albo trzykrotnie (rys. 7.11); drugie i ewentualne trzecie odpuszczanie
przeprowadza się w temperaturze niższej o 20
÷ 30°C od pierwszego odpuszczania. Podczas
odpuszczania zachodzi wydzielanie się węglików wtórnych z martenzytu oraz austenitu
szczątkowego, który ubożeje w dodatki stopowe i podczas studzenia od temperatury
odpuszczania przemienia się w martenzyt.
Czas
Rys. 7.11. Schemat obróbki cieplnej stali szybkotnącej
W wyniku odpuszczania w temperaturze 550-570°C pojawia się w tych stalach efekt
twardości wtórnej, tj. wzrost twardości na skutek wydzielania się z przesyconego roztworu
metastabilnych węglików typu M
3
C i MC. Strukturę o najkorzystniejszych własnościach
użytkowych, tzn. o wysokiej twardości, bez austenitu szczątkowego i o odpowiedniej
żarowytrzymałości i ciągliwości, uzyskuje się po wielokrotnym odpuszczaniu w ciągu ok. 2
godzin (rys. 7.11). Struktura stali szybkotnącej po hartowaniu i odpuszczaniu pokazana jest na
rys. 7.12.
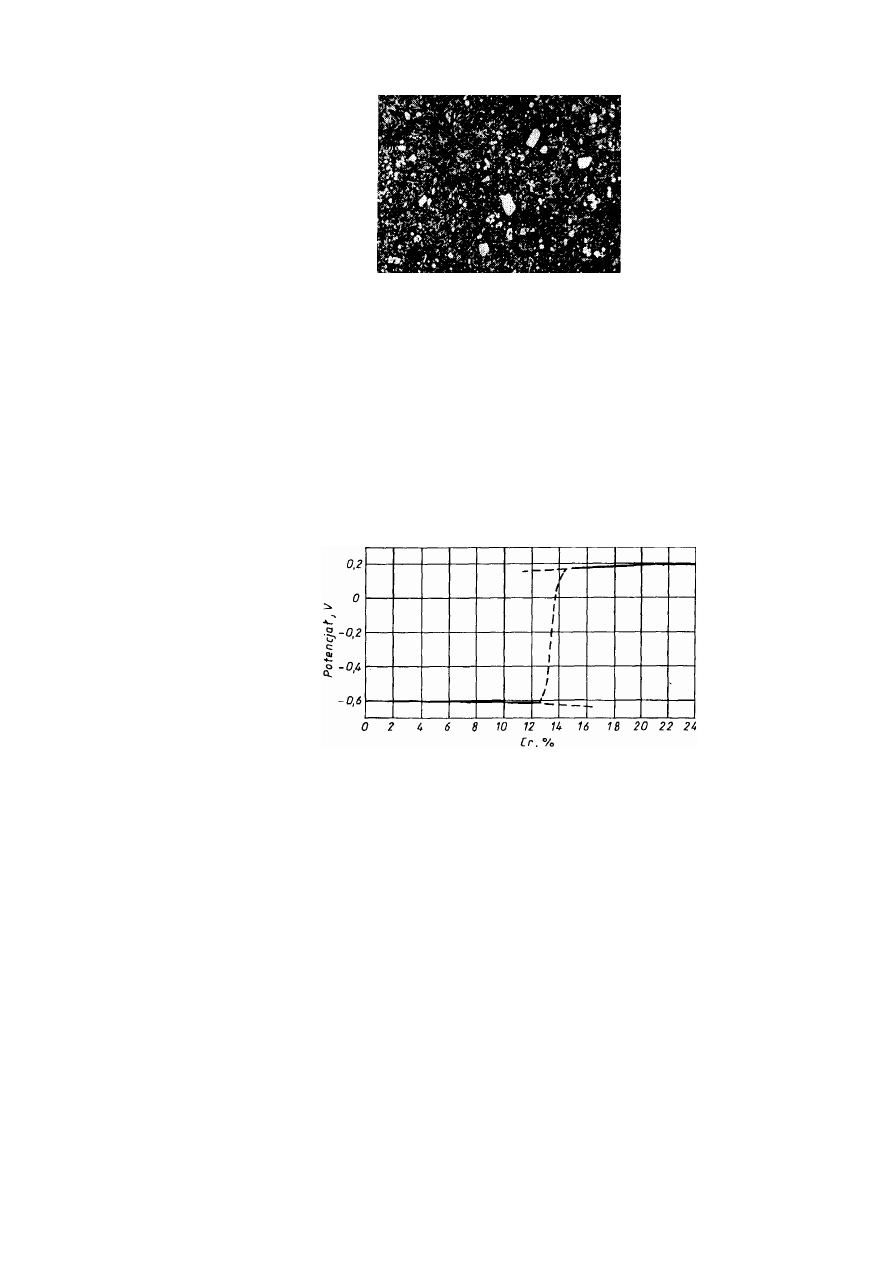
125
JW
Rys. 7.12. Mikrostruktura stali szybkotnącej SW7M hartowanej i odpuszczonej. W osnowie
drobnoiglastego martenzytu widoczne jasne węgliki. Traw. 5% nitalem. Powiększ. 650x
7.6. Stale stopowe odporne na korozję, żaroodporne, żarowytrzymałe i o specjalnych
własnościach fizycznych
7.6.1. Stale odporne na korozję (nierdzewne i kwasoodporne)
Odporność stali nierdzewnych na korozję związana jest przede wszystkim z działaniem
chromu, który powiększa zdolność tzw. pasywacji stopów żelaza. Przejście w stan pasywny
zaznacza się skokową zmianą potencjału elektrochemiczego metalu lub stopu na bardziej
dodatni (rys. 7.13).
Rys. 7.13. Potencjał elektrochemiczny stopów żelaza z chromem
Zjawisko pasywowania się metali polega na pokrywaniu się ich powierzchni bardzo cienką,
szczelnie przylegającą i odporną warstewką tlenków, która chroni metal przed korozją.
Pasywacja jest zjawiskiem zależnym od składu chemicznego stopu i od zdolności utleniania
jaką mają różne środowiska. Żelazo i miękka stal pasywują się np. w stężonym kwasie
azotowym i w roztworach związków silnie utleniających. Pasywacja żelaza jest jednak bardzo
nietrwała. Natomiast niektóre metale o większym powinowactwie do tlenu pasywują się łatwiej,
a ich stan pasywny jest znacznie trwalszy. Do takich metali należy chrom, którego odporność na
korozję związana jest właśnie z łatwością pasywowania się.
Chrom ma tę własność, że przenosi skłonność do pasywacji również na stopy z innymi
metalami. Stopy żelaza z chromem przy zawartości powyżej 13
÷ 14% Cr pasywują się pod
wpływem tlenu zawartego w powietrzu, co zapewnia im odporność chemiczną.
Podstawowym składnikiem wszystkich stali nierdzewnych jest więc chrom, przy czym jego
zawartość winna wynosić co najmniej 12% (rys. 7.13). Oprócz chromu w skład stali odpornych
na korozję często wchodzi nikiel jako drugi składnik podstawowy. Na podstawie składu
chemicznego można najogólniej podzielić stale odporne na korozję na: chromowe i chromowo-
niklowe
. Jednak częściej stosuje się klasyfikację tych stali według struktury i rozróżnia się stale
ferrytyczne, martenzytyczne i austenityczne
.
126
JW
Stale chromowe ferrytyczne i martenzytyczne (nierdzewne).
Stale chromowe odporne na korozję, zależnie od zawartości chromu i węgla, mogą być
ferrytyczne lub martenzytyczne.
W przypadku stali martenzytycznych występuje w czasie nagrzewania całkowita przemiana
ferrytu w austenit, dzięki czemu możliwe jest hartowanie i powstawanie struktury
martenzytycznej. Stale te hartują się już w czasie chłodzenia na powietrzu i właśnie z tego
powodu nazywane są martenzytycznymi.
W przypadku stali ferrytycznych, ferryt jest fazą trwałą od temperatury pokojowej aż do
temperatury topnienia i przemiany fazowe nie zachodzą. Z tego względy stali ferrytycznych nie
można utwardzać przez obróbkę cieplną (hartowanie).
W tablicy 7.13 podano skład chemiczny częściej stosowanych stali ferrytycznych i
martenzytycznych (wg PN-71/H-86020).
W stalach ferrytycznych (w temperaturze otoczenia) ferryt stanowi osnowę, ale oprócz niego
występują również często niewielkie ilości węglików, które rozpuszczając się w wyższych
temperaturach powodują tworzenie się pewnej ilości austenitu, a szybkie chłodzenie może
spowodować przemianę tego austenitu w martenzyt.
Mikrostruktura stali ferrytycznej 0H13 z niewielką ilością węglików widoczna jest na rys.
7.14. Taki sam wpływ mają również azot i nikiel, których małe ilości zawsze spotyka się w tych
stalach. Martenzyt, który powstaje po szybkim chłodzeniu od wysokiej temperatury, np. podczas
spawania, jest przyczyną kruchości i pęknięć w strefie wpływu cieplnego spoiny.
Tym niepożądanym zjawiskom przeciwdziała dodatek tytanu w ilości wystarczającej do
związania węgla i azotu, jak to ma miejsce np. w stali 0H17T (tabl. 7.13), albo dodatek
pierwiastka stabilizującego ferryt, jakim jest aluminium, np. w stali 0H13J (tabl. 7.13).
Tablica 7.1.
Stale chromowe odporne na korozję: ferrytyczne i martenzytyczne (wg PN-71/H-08620)
Znak stali
Średnia zawartość, %*
Średnia temp,
o
C Struktura
po
C
Cr
inne
składn
hartów.
odp.
obróbce
cieplnej
Stale ferrytyczne
OH13
OH13J
H17
OH17T
0,08 max
0,08 max
0,10 max
0,08 max
13,0
13,0
17,0
17,0
AI0.2
Ti5XC
1025
750
ferryt
ferryt
ferryt
ferryt
Stale martenzytyczne
1H13
2H13
0,12 0,20
13,0
13,0
-
1000
1020
750
720
ferryt i perlit
sorbit
3H13
0,30
13,0
-
980
250
350
martenzyt
sorbit
650
4H13
0,40
13,0 - 1030 150
250
martenzyt
sorbit
600
H18
H17N2
2H17N2
1,0 0,14
0,20
18,0
17,0
17,0
Ni 2,0
Ni 2,0
1020
1000
1080
250 310 700
martenzyt
sorbit i ferryt
sorbit
*
Zawartość P
max
≤ 0,040, S
max
≤ 0,030.

127
JW
Na rysunku 7.15 pokazana jest mikrostruktura stali ferrytycznej 0H17T, z wyraźnie
widocznymi węglikami i azotkami tytanu.
Rys. 7.14. Mikrostruktura stali chromowej
0H13. Widoczne ziarna ferrytu i wydzielenia
wielkiej ilości węglików. Traw. elektrol. w
kwasie szczawiowym. Powiększ. 150x
Rys. 7.15. Mikrostruktura stali chromowe
OH17T. Widoczne ziarna ferrytu i
wydzielenie węglików i azotków tytanu.
Traw. elektrol. w kwasie szczawiowym.
Powiększ. 150x
Największą odporność na korozję i największą ciągliwość wykazują stale ferrytczne w stanie
wyżarzonym w ok. 800°C. Ich odporność chemiczna jest lepsza niż stali martenzytycznych i
wzrasta z zawartością chromu, dlatego stale o zawartości 17% Cr są bardziej odporne na korozję
niż stale o zawartości 13% Cr.
Stale ferrytyczne są odporne na korozję atmosferyczną, z wyjątkiem warunków szczególnie
agresywnych, jak np. zanieczyszczona atmosfera przemysłowa. Są dość odporne na działanie
kwasu azotowego i środowisk utleniających, słabych kwasów organicznych i różnych produktów
żywnościowych.
Stal 0H17T i H17 w stanie wyżarzonym (ok. 800°C) jest stosunkowo miękka i ciągliwa,
nadaje się do tłoczenia na zimno, w związku z czym gatunki te są szeroko stosowane na
naczynia kuchenne, aparaty w przemyśle spożywczym, elementy karoserii samochodowych itp.
Do najbardziej rozpowszechnionych stali martenzytycznych, tj. takich, które można hartować
na martenzyt, należą stale o zawartości 13% Cr i 0,1-0,45% węgla, stale o zawartości ok. 17%
Cr, 2% Ni i ok. 0,2% węgla oraz stal zawierająca 18% Cr i ok. 1% węgla (tabl. 7.13). Stale
1H13, 2H13, 3H13 hartuje się od temperatury 950-1050°C. Temperatura odpuszczania wynosi
zwykle 600-700°C.
Stale 4H13 i H18 są stosowane w stanie hartowanym i odpuszczanym, ale przy stosunkowo
niskiej temperaturze ok. 200°C, ponieważ chodzi o możliwie dużą twardość.
Mikrostruktura stali H18 w stanie zahartowanym pokazana jest na rys. 7.16. Widoczne są
skupienia dużych pierwotnych węglików chromu oraz rozsiane małe węgliki w osnowie
drobnego martenzytu.
Stale H17N2 i 2H17N2 ze względu na wyższą zawartość chromu mają lepszą odporność
chemiczną niż stale z zawartością 13% Cr. Dodatek niklu w tych stalach rozszerza zakres
występowania austenitu i umożliwia osiągnięcie jednofazowej struktury podczas
austenityzowania.
Stale 1H13, 2H13 i 3H13 są używane w stanie ulepszonym cieplnie na silnie obciążone
części maszyn, które muszą być odporne na korozję, oraz na przedmioty gospodarstwa
domowego. Stal 2H17N2 jest dość odporna na działanie wody morskiej i ma zastosowanie w
budowie okrętów. Stal 4H13 jest używana na noże. sprężyny i narzędzia, a stal H18 na łożyska
kulkowe odporne na korozję, na elementy maszyn odporne na ścieranie pracujące np. w
środowisku wodnym, na noże chirurgiczne, narzędzia skrawające, przyrządy pomiarowe itp.
128
JW
Rys. 7.16. Mikrostruktura stali H18 (1%
C, 18% Cr). Widoczne skupienia dużych
pierwotnych węglików chromu oraz
drobne węgliki na tle osnowy
martenzytycznej. Traw. elektrol. w kwasie
szczawiowym. Powiększ. 500x
Rys. 7.17. Mikrostruktura austenitycznej
stali kwasoodpomej OH18N9 w stanie
przesyconym. Widoczne ziarna austenitu z
bliźniakami rekrystalizacji. Traw. elektrol.
w kwasie szczawiowym. Powiększ. 150x
Stale martenzytyczne są odporne na działanie kwasu azotowego, szeregu kwasów
organicznych i produktów spożywczych. Na korozję atmosferyczną są odporne poć warunkiem
braku agresywnych zanieczyszczeń w powietrzu. Odporność chemiczne tych stali zależy
ponadto od gładkości powierzchni.
Austenityczne stale chromowo-niklowe (kwasoodporne).
Stale austenityczne odporne na korozję są w zasadzie stalami chromowo-niklowymi o niskiej
zawartości węgla. Dodatek niklu w ilości ok. 8% do niskowęglowych stali chromowych zawie-
rających ok. 18% Cr zwiększa ich odporność na korozję i na działanie kwasów podwyższa
wytrzymałość i udarność. Duży dodatek niklu powoduje, że stale te mają strukturę
austenityczną. Mikrostruktura tego typu stali pokazana jest na rys 7.17. Ze względu na dobrą
odporność na działanie wielu kwasów stale te są również nazywane kwasoodpornymi.
Stale zawierające 18% Cr i 8% Ni oznaczane popularnie znakiem 18/8, zyskały ogromne
znaczenie praktyczne. Większość dziś stosowanych gatunków stali kwasoodpornych stanowi
modyfikację tego podstawowego składu.
Ze względu na niebezpieczeństwo korozji
międzykrystalicznej, zawartość węgla w stalach 18/8
powinna być jak najniższa. Na rysunku 7.18 przedstawiony
jest przekrój przez układ Fe-Cr-Ni-C dla stałej zawartości
18% Cr i 8% Ni, przy zmiennej zawartości węgla,
ilustrujący zakres jednorodnego roztworu i granice
rozpuszczalności węglików M
23
C
6
. Przy powolnym chło-
dzeniu, np. w stali o zawartości 0,1% C, część węgla
pozostaje w roztworze stałym w austenicie, część zaś
wydziela się w postaci węglików (Cr, Fe)
23
C
6
bogatych w
chrom, zgodnie z krzywą rozpuszczalności.
Stal o jednofazowej strukturze austenitycznej, bez
wydzielonych węglików, można otrzymać przez
przesycanie od temperatury 1050
÷ 1100°C z chłodzeniem
w wodzie. W temperaturze 1050
÷1100°C węgliki roz-
puszczają się w austenicie, natomiast szybkie chłodzenie
zapobiega ich wydzielaniu. Po takiej obróbce cieplnej stal
18/8 jest najbardziej odporna na korozję, gdyż stanowi
materiał jednofazowy. Natomiast podczas wygrzewania
stali 18/8 w temperaturze 500
÷ 800°C na granicach ziarn
następuje wydzielanie się węglików chromu typu M
23
C
6
,
zawierających zwykle co najmniej 60% Cr. Jest to
szczególnie niekorzystne, gdyż w wyniku tego procesu stal staje się skłonna do korozji
Rys. 7.18. Fragment układu
równowagi Fe-Cr-Ni-C.
Przekrój dla zawartości 18% Cr
i 8% Ni

129
JW
międzykrystalicznej.
W środowisku korozyjnym atakowane są szczególnie granice ziarn.
Ilustruje to rys. 7.19 przedstawiający skorodowaną powierzchnię blachy ze stali 18/8. Ten rodzaj
korozji jest bardzo niebezpieczny, gdyż niszczy materiał w głąb, nie pozostawiając wyraźnych
śladów na powierzchni. Stal traci wytrzymałość i plastyczność i nie daje metalicznego dźwięku
przy uderzeniu.
Zjawisko korozji międzykrystalicznej można wytłumaczyć zubożeniem granic ziarn w
chrom na skutek wydzieleń węglików chromu, które zarodkują prawie wyłącznie na granicach
ziarn. W temperaturze powyżej 500°C szybkość dyfuzji węgla jest większa od szybkości dyfuzji
chromu, w związku z czym węgiel potrzebny dla tworzących się węglików chromu na granicach
ziarn pochodzi z całego ziarna, podczas gdy chrom - tylko z zewnętrznej warstewki w pobliżu
granic ziarn. Zawartość chromu w pobliżu granic ziarn może więc spaść poniżej 12%, co
stanowi minimum konieczne dla pasywacji (rys. 7.20). W tych warunkach rozpuszczanie
zubożałej w chrom warstewki może postępować bardzo szybko nawet w roztworach, na które
stal 18/8 normalnie jest odporna.
Stale typu 18/8 stosuje się najczęściej w stanie przesyconym (od temp. 1050
÷ 1100°C) i w
tym stanie niebezpieczeństwo korozji międzykrystalicznej nie istnieje. Jednak w budowie
aparatów chemicznych, zbiorników, rurociągów itp. stosuje się powszechnie spawanie. Sama
spoina i jej najbliższe sąsiedztwo stygną szybko od wysokich temperatur. W strefie wpływu
cieplnego spoiny znajdują się jednak zawsze obszary, które podczas spawania nagrzewają się
tylko do niebezpiecznego zakresu temperatur, tj. do 500
÷ 800°C. W tych obszarach może
następować wydzielanie węglików chromu na granicach ziarn i możliwe jest występowanie
korozji międzykrystalicznej
Rys. 7.19. Korozja międzykrystaliczna auste-
nitycznej stali chromowo-niklowej. Próbka
nietrawiona. Powiększ. 600x
Rys. 7.20. Schemat zmian koncentracji
chromu w pobliżu granicy ziarna,
spowodowany wydzielaniem się węglików
chromu typu M
23
C
6
. Materiał w takim stanie
jest wrażliwy na korozję międzykrystaliczna
Obszary te leżą zwykle w odległości kilku do kilkunastu milimetrów od spoiny. Zjawisko to
stanowiło początkowo poważną trudność w stosowaniu stali 18/8. Obecnie istnieje szereg
sposobów opracowanych w celu zapobiegania korozji międzykrystalicznej. Najważniejsze z nich
to:
a) przesycanie,
b) stabilizacja,
c) zmniejszenie zawartości węgla.
Pierwszy z tych sposobów, o którym już wspomniano, jest najprostszy i polega na
zastosowaniu przesycania od temperatury 1050-1100°C spawanego przedmiotu. Sposób ten
jednak jest ograniczony do przedmiotów o małych rozmiarach.
Drugi sposób polega na wprowadzeniu do stali dodatków tytanu lub niobu, w ilości
wystarczającej do związania węgla w postaci węglików. Ti i Nb wykazują silniejsze
powinowactwo do węgla niż chrom i tworzą bardzo trwałe węgliki TiC i NbC, które nie
rozpuszczają się w austenicie w normalnie stosowanych temperaturach przesycania, co utrudnia
tworzenie się węglików chromu. Stale takie nazywa się stabilizowanymi. Normy przewidują
zawartość tytanu równą co najmniej 5-krotnej zawartości węgla, a niobu co najmniej 10-krotnej

130
JW
(tabl. 7.14), Są to ilości większe niż potrzeba do związania węgla, ale tytan i niob wiążą również
azot, którego zawartość w tych stalach wynosi zwykle 0,01
÷ 0,02% i dlatego dodatki tych
pierwiastków oblicza się z pewną rezerwą na związanie azotu.
Tablica 7.14
Stale austenityczne odporne na korozję wg PN-71/H-86020
Znak stali
Skład chemiczny, %
C Cr
Ni
inne
składniki
00H18N10 max
0,03 17,0+19,0 10,0+12,5
—
0H18N9 max
0,07 17,0-19,0 9,0+11,0
-
1H18N9 max
0,12 17,0+19,0 8,0-10,0
-
0H18N10T max
0,08 17,0+19,0 9,0-11,0 Ti5xC
÷0,7
1H18N9T max
0,10
17,0+19,0
8,0+10,0
Ti5xC
÷0,8
1H18N12T max
0,10 17,0-19,0 11,0+13,0 Ti5xC
÷0,8
0H18N12Nb max
0,08
17,0-19,0
10,0+13,0
Nb10xC
÷1,1
00H17N14M2 max
0,3 16,0---18,0 12,0+15,0 Mo 2,0
÷2,5
H17N13M2T max
0,08 16,0+18,0 11,0+14,0 Ti 5xC0
÷,7
H18N10MT
max 0,10
17,0+11,0
9,0+11,0
Mo 1,5
÷2,2
Ti5xC
÷0,8
Mo 3,0
÷4,0
0H17N16M3T
max 0,08
16,0+18,0
14,0+16,0
Ti 0,3
÷0,6
Mn 7,0-9,0
0H17N4G8 max
0,07 16,0+18,0 4,0+5,0 N 0,12
÷0,25
1H17N4G9 max
0,12 16,0+18,0 3,5+4,5 Mn 8,0
÷10,5
N 0,15
÷0,25
00H18N5M3S max
0,03 17,0+19,0
4,5+5,5 Mo 3,0
÷4,0**
Si 1,0
÷2,0
*' Zawartość Mn max 2,0 z wyjątkiem stali 0H17N4G8 i 1H17N4G9, zawartość Si max 0,8,
zwartość P max 0,045, zawartość S max 0,030.
** Stal OOH18N5M3S nie jest ujęta w normie PN-71/H-86020.
Trzecim sposobem (najskuteczniejszym) zapobiegania korozji międzykrystalicznej jest
obniżenie zawartości węgla do 0,02
÷ 0,03%. Zastosowanie tego sposobu stało się możliwe
dopiero po opanowaniu metod wytapiania stali o tak niskiej zwartości węgla. Obniżenie
zawartości węgla wywołuje niestabilność austenitu pojawienie się ferrytu. Aby tego uniknąć,
powiększa się zawartość niklu z 8
÷ 9% do 10 ÷ 13%. To samo dotyczy stali stabilizowanych
tytanem lub niobem (tabl. 7.14).
Oprócz korozji międzykrystalicznej stale austenityczne 18/8 ulegają również korozji wżerowej
i naprężeniowej.
Korozję wżerową
wywołują głównie jony chloru, bromu, jodu, fluoru i inne. Powodują one
lokalną depasywację powierzchni, na skutek czego miejsca pozbawione warstwy ochronnej stają
się anodą wobec pasywnej powierzchni i w miejscach tych rozpuszczanie się metalu zachodzi
bardzo szybko. Korozji wżerowej zapobiega w pewnym stopniu dodatek molibdenu. Stosuje się
dodatki w ilości 2
÷ 4% Mo. Mikrostruktura stali z dodatkiem Mo widoczna jest na rys. 7.21. Ze
względu na ferrytyzujący wpływ molibdenu (dodatek Mo sprzyja powstawaniu struktury
ferrytycznej) powiększa się w tych stalach zawartość niklu. Na przykład gdy dodatek Mo wynosi
3-4%, zawartość niklu dochodzi do 16% (stal OH17N16M3T, tabl. 7.14).
Stale austenityczne są również wrażliwe na korozję naprężeniową. Warunkiem jej
wystąpienia jest równoczesne działanie naprężeń rozciągających i środowiska korozyjnego,
głównie roztworów chlorków magnezu, wapnia, sodu, roztworów alkalicznych i in.
131
JW
Większą odporność na korozję naprężeniową wykazują stale austenityczne o znacznie
zwiększonej zawartości niklu oraz stale o strukturze częściowo ferrytycznej, co z kolei wymaga
obniżenia zawartości niklu. Przykładem jest stal 00H18N5M3S (tabl. 7.14) o strukturze
austenityczno-ferrytycznej (rys. 7.22).
Rys. 7.21. Mikrostruktura austenitycznej stali
chromowo-niklowej z dodatkiem molibdenu i
tytanu (H17N12M2T). Widoczne ziarna
austenitu z bliźniakami rekrystalizacji oraz
wydzieleniami węglików Mo i Ti oraz azotków
Ti. Traw. elektrol. w kwasie szczawiowym.
Powiększ. 150x
Rys. 7.22. Austenityczno-ferrytyczna struktura
stali 00H18N5M3S o zwiększonej odporności
na korozję naprężeniową. Traw. elektrol.
w kwasie szczawiowym. Powiększ. 150x
Najskuteczniejsze są jednak takie środki, które pozwalają na uniknięcie naprężeń lub obróbka
powierzchni wywołująca naprężenia ściskające w warstwie wierzchniej.
Najważniejsze gatunki stali austenitycznych odpornych na korozję zestawiono w tabl. 7.14.
Stale 0H17N4G8 i 1H17N4G9 są gatunkami oszczędnościowymi, w których nikiel zastąpiono
częściowo manganem i azotem. Odporność tych stali na korozję międzykrystaliczną jest
porównywalna ze stalami 0H18N9 i 1H18N9. Nadają się one do głębokiego tłoczenia i są
stosowane głównie w przemyśle spożywczym, w architekturze, do wyrobu przedmiotów
gospodarstwa domowego itp.
Szczegółowe dane dotyczące odporności na korozję stali austenitycznych w różnych
środowiskach podane są w specjalnych tablicach, wykazach i normach PN-71/H-86020). W
praktyce jednak odporność tych stali wymaga skrupulatnego sprawdzenia przed ich
zastosowaniem, zwłaszcza w środowiskach szczególnie agresywnych.
Stale austenityczne w stanie przesyconym (od temp. 1050
÷ 1100°C, chłodzenie w wodzie)
są stosunkowo miękkie i bardzo ciągliwe, R
m
wynosi ok. 500
÷ 700 MPa, a wydłużenie A
powyżej 40%. Umowna granica plastyczności R
02
wynosi ok. 200
÷ 250 MPa. Obniżenie
zawartości węgla poniżej 0,03% powoduje obniżenie R
m
do 450
÷ 650 MPa i R
0,2
do 180 MPa.
Ta stosunkowo niska wytrzymałość powoduje często konieczność stosowania grubszych
ścianek w elementach konstrukcyjnych. Zwiększenie
wytrzymałości tych stali można uzyskać przez
zwiększenie zawartości azotu bez szkodliwego wpływu
na odporność chemiczną. Inną możliwością
powiększenia granicy plastyczności i wytrzymałości stali
austenitycznych jest zgniot (rys. 7.23). Sposób ten
stosuje się zwłaszcza do cienkich blach i taśm
walcowanych na zimno. Na tej drodze istnieje
możliwość uzyskania wytrzymałości na rozciąganie ok.
1200 MPa, przy zachowaniu wystarczającej ciągliwości.
Rys. 7.23. Zmiana własności mechanicznych stali austenitycznej (18% Cr, 8% Ni, 0,2% C)
pod wpływem zgniotu)
132
JW
7.6.2. Stale żaroodporne i żarowytrzymałe
Stale przeznaczone do pracy w podwyższonych temperaturach powinny się odznaczać
odpornością na korozyjne działania gazów, zwłaszcza utleniających, czyli powinny być
żaroodporne. Od stali tych wymaga się również, aby były żarowytrzymałe tj. aby wykazywały
znacznie wyższe własności wytrzymałościowe w wysokich temperaturach w porównaniu z
innymi stalami.
Szybkość utleniania żelaza i stali niskostopowych wzrasta gwałtownie powyżej ok. 560
o
C na
skutek tworzenia się tlenku FeO (wustytu), który umożliwia szybką dyfuzję tlenu do żelaza i
dalsze jego utlenianie.
Podstawowymi pierwiastkami stopowymi, które chronią stal przed utlenianiem są: Cr, Al i Si.
Pierwiastki te mają większe powinowactwo do tlenu aniżeli żelazo i tworzą szczelne warstewki
tlenków Cr
2
O
3
, Al
2
O
3
SiO
2
, które utrudniają dyfuzję tlenu w głąb metalu. Im wyższa
temperatura pracy danego elementu, tym większa jest potrzebna zawartość pierwiastka
stopowego dla zapewnienia żaroodporności.
Na rysunku 7.24 przedstawiono wpływ zawartości chromu na żaroodporność stali. Jak
widać, wpływ na żaroodporność ma również struktura stali. Przy tej samej zawartości chromu
stale austenityczne są nieco bardziej żaroodporne niż stale ferrytyczne.
Rys.7.24. Wpływ zawartości chromu na żaroodporność stali;
l - stale austenityczne, 2 - stale ferrytyczne
Wzrost żarowytrzymalości, która jest związana przede wszystkim z wysoką odpornością na
pełzanie, powodują dodatki stopowe podwyższające temperaturę topnienia i rekrystalizacji stali,
a więc: Mo, W, C, Co oraz Ti, Cr i Si. Również bardziej korzystna jest struktura austenityczna
stali, co wynika m.in. z wyższej temperatury rekrystalizacji austenitu. Ponadto na podwyższenie
żarowytrzymałości znacznie wpływa wzrost wielkości ziarna i wydzielanie faz o dużej dyspersji
(utwardzanie dyspersyjne).
Ze względu na zastosowanie i strukturę wśród stali przeznaczonych do pracy w
podwyższonych temperaturach można wyróżnić kilka grup. Zasady znakowania tych stali są
analogiczne, jak stali stopowych konstrukcyjnych i stali odpornych na korozję.
Stale żarowytrzymałe stosowane do budowy kotłów i turbin parowych.
Są to na ogół stale ulepszane cieplnie, które po chłodzeniu na powietrzu mogą mieć
strukturę ferrytyczno-perlityczną, perlityczno-bainityczną, martenzytyczną, martenzytyczno-
ferrytyczną itp. W większości są to stale niskostopowe (z wyjątkiem martenzytycznych, które
zawierają ok. 12% Cr) o małej i średniej zawartości węgla (0,10
÷ 0,35%). Po ulepszaniu
cieplnym, które kończy się wysokim odpuszczaniem struktura większości tych stali składa się z
ferrytu i węglików stopowych. Polska norma PN-75/H-84024 obejmuje 26 gatunków stali
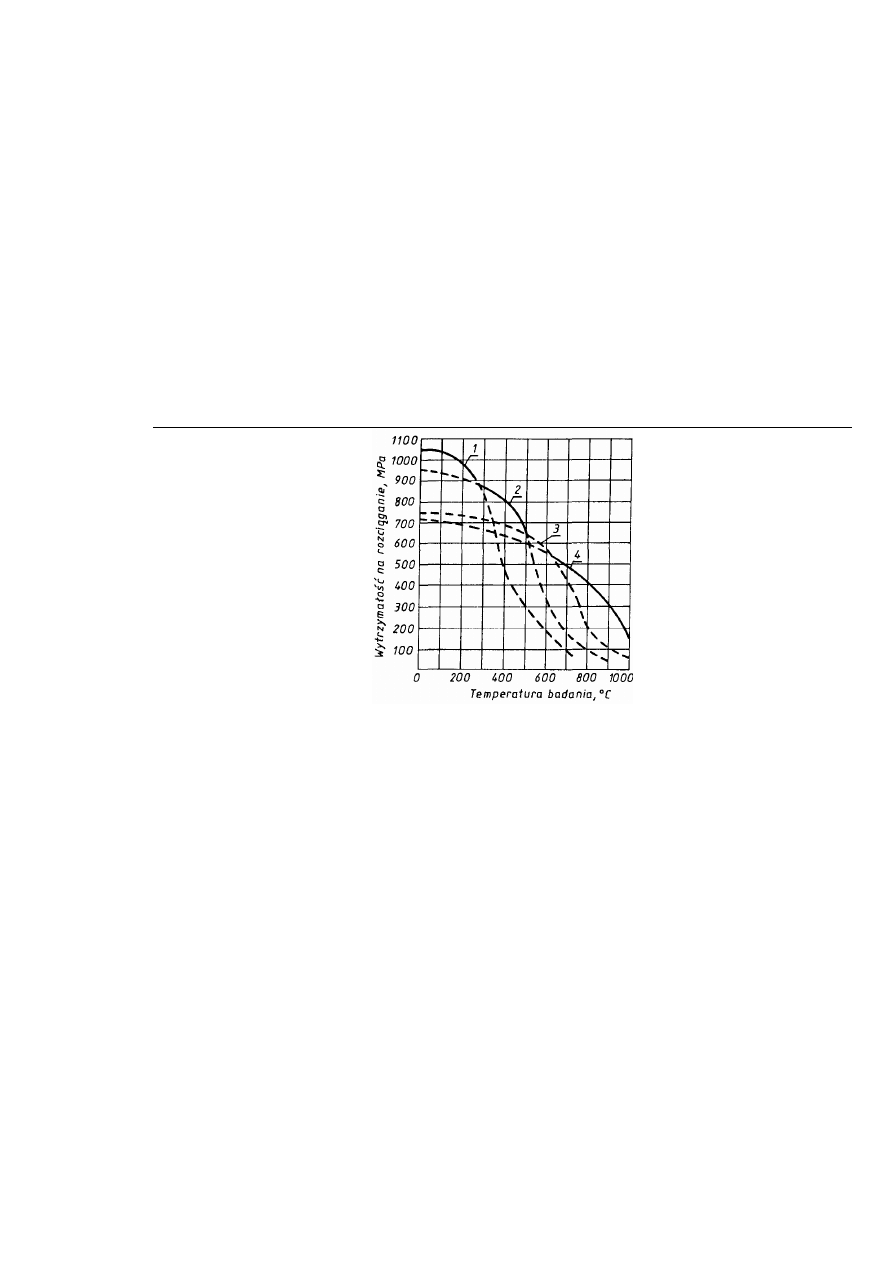
133
JW
stopowych przeznaczonych do pracy w podwyższonych temperaturach, stosowanych w budowie
kotłów parowych i wodnych, zbiorników ciśnieniowych, turbin, rurociągów pary i wody oraz
innych urządzeń energetycznych. Stale te mają następujące oznaczenia, z których wynika ich
orientacyjny skład chemiczny: 19G2, 16M, 20M, 15HM, 20HM, 10H2M 12HMF, 13HMF,
15HMF, 20MF, 21HMF, 20HMFTB, 15HCuMNb, 23H2MF 24H2MF, 26H2MF, 30H2MF,
22H2NM, 33H2NMJ, 20H3MWF, 32HN3M 34HN3M, 15H11MF, 15H12WMF, 20H12M1F,
23H12MWF.
Wszystkie te stale, z wyjątkiem czterech ostatnich zawierających po ok. 12% Cr (stale
martenzytyczne), są stalami niskostopowymi. Dobre własności mechaniczne w podwyższonych
temperaturach uzyskuje się przede wszystkim dzięki zawartości molibdenu i wanadu, i
utwardzaniu wydzieleniowemu węglikami. Temperatura długotrwałej pracy tych stali wynosi,
zależnie od gatunku 400
÷ 600°C. Górna granica ich zastosowania nie może przekraczać 600°C,
ponieważ powyżej tej temperatury szybkość dyfuzji węgla i pierwiastków stopowych wzrasta,
następuje koagulacja węglików i zanik utwardzenia, co powoduje obniżenie naprężeń
wywołujących odkształcenia plastyczne, i wzrost szybkości pełzania. Ponadto temperatura
600°C jest dla tych stali (z wyjątkiem wysokochromowych) krytyczna ze względu na
gwałtowny wzrost szybkości utleniania.
Rys. 7.25. Wytrzymałość na rozciąganie różnych stopów żarowytrzymałych w zależności od
temperatury badania; l - stal konstrukcyjna niskostopowa chromowo-niklowa, 2 -
stal żarowytrzymała ferrytyczna, 3 - stal żarowytrzymała austenityczna, 4 -
żarowytrzymały stop na osnowie niklu
Z tego względu do pracy w wyższych temperaturach lub w warunkach powodujących
intensywną korozję gazową stosuje się stale wysokostopowe ferrytyczne dużej zawartości
chromu lub austenityczne chromowo-niklowe, wykazujące wyższą odporność na utlenianie.
Stale wysokochromowe ferrytyczne.
Stale te charakteryzują się wysoką żaroodpornością, którą zapewnia duża zawartość chromu oraz
dodatki Al i Si (tabl. 7.15). Natomiast żarowytrzymałość tych stali jest stosunkowo niska i z tego
względu są one stosowane na nisko obciążone elementy pracujące w wysokich temperaturach,
jak np. części żaroodporne kotłów parowych, pojemniki do wyżarzania, szyny, kołpaki i rury do
pieców przemysłowych, części aparatury do destylacji siarki, części gazogeneratorów itp.
Stale austenityczne chromowo-niklowe.
Odznaczają się również wysoką żaroodpornością jak
stale ferrytyczne wysokochromowe, natomiast są bardziej żarowytrzymałe i dlatego mogą być
stosowane na części obciążone mechanicznie, pracujące w wysokich temperaturach (rys. 7.25).
Skład chemiczny tych stali podano w tabl. 7.15. Wysoką odporność na utlenianie zapewnia
znaczna zawartość chromu (16
÷ 26%) i dodatek krzemu (1 ÷ 2,5%). Na żaroodporność wpływa
dodatnio także nikiel, który nadaje stali strukturę austenityczną. Obróbka cieplna stali
austenitycznych polega na przesycaniu od temperatur 1050
÷ 1150°C w wodzie lub w powietrzu
134
JW
(elementy o małym przekroju o grubości do 2 mm). Stale o większej zawartości niklu (20
÷36%)
są stosowane na części aparatury i urządzeń pracujących pod bardzo silnym obciążeniem
mechanicznym w wysokich temperaturach.
Stale zaworowe.
Zawory w silnikach spalinowych pracują w bardzo trudnych warunkach.
Narażone są na działanie wysokich temperatur dochodzących w przypadku zaworów
wylotowych do 900°C, zaworów wlotowych - do 500°C, a jednocześnie są silnie obciążone
mechanicznie na skutek uderzenia o gniazda zaworów i narażone na ścieranie w prowadnicach i
w miejscach styku z popychaczami. Ponadto na zawory działają spaliny często zawierające
tlenki ołowiu, które powodują silną korozję.
W tablicy 7.15 podano gatunki stali zaworowych produkowane w kraju. Dwa pierwsze H9S2 i
H10S2M są to tzw. silchromy, czyli stale chromowo-krzemowe, które można hartować na
martenzyt. Temperatura austenityzowania wynosi dla tych stali ok. 1050°C. Hartowanie
przeprowadza się w oleju, a następnie odpuszcza się w zakresie 750
÷ 850°C. Silchromy
wykazują dość wysoką żarowytrzymałość do ok. 700°C. Powyżej tej temperatury wytrzymałość
zaczyna jednak dość szybko spadać. Stale te są stosowane głównie na wylotowe oraz wlotowe
zawory silników spalinowych samochodowych i motocyklowych.
Stale żaroodporne i żarowytrzymałe (wg PN-71/H-86022)
Tablica 7.15
Średnia zawartość, %
Żaroodpo Struktura
po
Znak stali
rność w
obróbce
Inne
powietrzu
cieplnej
C
Si
Cr
Ni
składnik do temp. °C
H5M
0,15* 0,50* 5,0 0,5* Mo0,5
650
ferryt i perlit
H6S2 0,15* 1,75
6,0
0,6*
800 ferryt i perlit
2H17 0,15* 1,2* 17,0 0,6*
850 ferryt
H13JS 0,12* 1,15 13,0 0,5*
AI0.9
950 ferryt
H18JS 0,12* 0,9
18,0 0,5*
AI1,0
1050 ferryt
H24JS 0,12* 1,45 24,0 0,5*
Al 1,4
1200 ferryt
H25T 0,15* 1,0* 25,0 0,6* Ti(4xC)
1100 ferryt
H26N4 0,20*
2,5*
26,0
4,5 1100
ferryt
i
austenit
H18N9S
0,15
1,5* 18,0
9,5
850
austenit
H23N13 0,20* 1,0* 23,0 13,5
1050 austenit
H20N12S2 0,20* 2,2
20,0 12,0
1050 austenit
H23N18 0,20* 1,0* 23,0 18,5
1050 austenit
H25N20S2 0,20* 2,5
25,0 19,5
1150 austenit
H18N25S2
0,35
2,5
18,0 25,0
1100
austenit
H16N36S2 0,15* 1,7 16,0 36,0
1100 austenit
Stale zaworowe
H9S2 0,40
2,5
9,0
0,60*
850 sorbit
H10S2M
0,40 2,2
10,0 0,50* Mo0,8
900
sorbit
4H14N14W 0,45 0,8*
14,0 14,0
W 2,5
900
austenit i węgliki
Mo 0,35
50H21G9N 0,50 0,5* 21,0 4,0 N0,45 900
austenit
i
węgliki
* Zawartość maksymalna
Dwa następne gatunki (4H14N14W2M i 50H21G9N4) są to stale austenityczne, które w
zakresie temperatury 800
÷ 900°C wykazują jeszcze wystarczającą wytrzymałość i są stosowane
w trudniejszych warunkach pracy: stal 4H14N14WZM na najbardziej obciążone zawory
wylotowe i wlotowe silników lotniczych, a stal 50H21G9N4, która wykazuje dobrą odporność
na korozję powodowaną przez tlenki ołowiu - na najbardziej obciążone zawory wylotowe
silników samochodowych. Struktura obu tych stali składa się z austenitu i węglików. Można je
utwardzać wydzieleniowo, stosując przesycanie od temperatury 1100
÷ 1150°C i starzenie w
temperaturze 700
÷ 750°C przez kilkanaście godzin. Jednak stal 4H14N14WZM ze względu na

135
JW
niebezpieczeństwo rozrostu ziarn w czasie nagrzewania do przesycania i niekorzystne
wydzielanie się węglików na granicach ziarn w czasie starzenia, częściej jest stosowana tylko po
kuciu w zakresie temperatury 1150
÷ 900°C i wyżarzaniu zmiękczającym w temperaturze ok.
850°C.
7.6.3. Stale stopowe o szczególnych własnościach fizycznych
Do tej grupy stali można zaliczyć stale o specjalnych własnościach magnetycznych (są
omówione w rozdziale dotyczącym materiałów magnetycznych), stale oporowe, stopy żelaza o
specjalnej rozszerzalności cieplnej i inne.
Jako materiały o dużym oporze elektrycznym używane są stale chromowo-niklowe i
chromowo-aluminiowe. Stosuje się je głównie na oporniki oraz elementy grzejne pieców
przemysłowych i laboratoryjnych i innych urządzeń.
Stale oporowe Cr-Ni zawierają ok. 20% Ni i 20% Cr, 60% Fe oraz dodatki Mn i Si (1
÷ 2%).
Temperatura pracy wynosi maksymalnie ok. 1000°C.
Stale oporowe Cr-Al są to stale ferrytyczne zawierające 12-27% Cr i 3-7% Al. W kraju
produkowane są z następującymi oznaczeniami: H13J4, H17J5, H25J5, 0H25J5. Ze wzrostem
zawartości chromu odporność na utlenianie zwiększa się coraz bardziej. Górne temperatury
stosowania tych stali wynoszą odpowiednio 850-1200°C. Wadą tych stali jest to, że po pewnym
okresie pracy stają się kruche i pękają przy próbach zginania; z tego względu elementy grzejne
umieszcza się w specjalnych obudowach o kształtach dostosowanych do określonych wymagań.
Jako stopy o specjalnej rozszerzalności cieplnej znalazły zastosowanie głównie stopy Fe-Ni.
Zależnie od zawartości niklu wartość współczynnika rozszerzalności cieplej a tych stopów
zmienia się w granicach l ,2
÷ 20,0 ⋅10
-6
°C
-1
. Najniższy współczynnik z tej grupy stopów ma
stop o zawartości ok. 36% Ni noszący nazwę inwaru.
Stopy Fe-Ni z dodatkiem 6
÷ 12% Cr odznaczają się tym, że ich moduł sprężystości podczas
nagrzewania zmienia się bardzo mało, czyli mają stałą sprężystość nie zmieniającą się z
temperaturą. Jednym ze stopów tego typu jest tzw. elinwar stosowany na sprężyny do
dokładnych przyrządów pomiarowych.
Współczynnik rozszerzalności cieplnej podobny do współczynnika niektórych szkieł ma stop
o zawartości 54% Fe, 28% Ni i 18% Co noszący nazwę femico, który jest stosowany do
wtapiania przejść elektrycznych w różnych przyrządach i lampach.
Istnieje również szereg odmian tych stopów o podobnych własnościach.
7.7. Staliwa węglowe i stopowe
Staliwo jest to stop żelaza z węglem i innymi pierwiastkami, zawierający do około 2,0%
węgla, otrzymywany w procesach stalowniczych w stanie ciekłym odlewany do form
odlewniczych. Odlewy takie mogą być używane bezpośrednio po zakrzepnięciu bez obróbki
cieplnej lub mogą być obrabiane cieplnie, względnie poddawane obróbce cieplno-chemicznej.
Jako materiał konstrukcyjny staliwo wykazuje wiele zalet, ma lepsze własności
wytrzymałościowe i plastyczne w porównaniu z żeliwem, a także dobrą spawalność zwłaszcza
niskowęglowe i niskostopowe). Wykazuje jednak gorsze własności odlewnicze ze względu na
skurcz dochodzący do 2% i wysoką temperaturę topnienia dochodzącą do 1600°C.
7.7.1. Staliwa węglowe konstrukcyjne ogólnego przeznaczenia
Polska norma PN-ISO 3755:1994 wymienia 8 gatunków staliw węglowych konstrukcyjnych
ogólnego przeznaczenia. Gatunki te oznacza się dwiema liczbami trzycyfrowymi lub dwiema
liczbami trzycyfrowymi i literą W: 200-400, 200-400W, 230-450, 230-450W, 270-480, 270-
480W, 340-550, 340-550W. Pierwsza liczba oznacza wymaganą minimalną wartość R
e
lub Rg,
w MPa, a druga - minimalną wytrzymałość na rozciąganie R
m
również w MPa. Gatunki
zawierające na końcu literę W mają dodatkowo określoną maksymalną zawartość
poszczególnych pierwiastków (czyli tzw. ograniczony skład chemiczny), w celu zapewnienia
dobrej (jednolitej) spawalności.
Staliwa, których oznaczenie nie zawiera litery W, nie mają obowiązującego składu
chemicznego poza fosforem (max 0,035%) i siarką (max 0,035%). Natomiast gatunki z literą W

136
JW
mają max 0,25% C i zróżnicowaną w zależności od gatunku zawartość Mn od max 1,00% do
max 1,50%, oraz określoną maksymalną zawartość pozostałych pierwiastków (jednakowa dla
tych gatunków):
≤ 0,60% Si, ≤ 0,035% P, ≤ 0,035% S, ≤ 0,40% Ni, ≤ 0,35% Cr, ≤ 0,40% Cu, ^ ≤
0,15% Mo i
≤ 0,05% V.
Wytrzymałość na rozciąganie R
m
zależy od gatunku staliwa i zawiera się w granicach od 400
÷ 550 MPa do 550 ÷ 700 MPa, a wydłużenie A
min
odpowiednio - od 25% do 15%.
Staliwa węglowe konstrukcyjne ogólnego przeznaczenia mogą być obrabiane cieplnie. Zwykle
poddaje się je normalizowaniu, wyżarzaniu zupełnemu lub wyżarzaniu odprężającemu.
7.7.2. Staliwa stopowe
Staliwa stopowe, podobnie jak stale, zawierają specjalnie wprowadzone dodatki stopowe,
które nadają im określone własności. Sposób znakowania gatunków staliw stopowych jest
analogiczny, jak stali stopowych konstrukcyjnych, z tą różnicą, że w przypadku staliw na
początku znaku znajduje się litera L. Za literą L znajdują się cyfry określające średnią zawartość
węgla w setnych procentu, następnie litery symbole) analogiczne jak w przypadku stali
stopowych konstrukcyjnych rozdz. 7.4.3), które określają pierwiastki stopowe, i cyfry, które
podają średnią zawartość danego pierwiastka w procentach. Jeżeli zawartość pierwiastka
stopowego nie przekracza średnio 2%, to podaje się tylko litery stanowiące symbole tego
pierwiastka.
Staliwa stopowe ze względu na zastosowanie dzielą się na:
Staliwa stopowe konstrukcyjne
— Polska Norma PN-H/83156:1997 obejmuje 23 gatunki
staliw tej grupy o następujących oznaczeniach: L20G, L35G, L15GM, L30GS, L35GM, L35GN,
L30H, L40H, L17HM, L25HM, L25HN, L35HM, L40HF, L30HMF, L30HGNM, L35HGS,
L35HNM, L20HN3M, L30H2N2M, 35H2MF, L12H13, L12H13N4M, L0H13N4M. W normie
podany jest skład chemiczny poszczególnych gatunków i ich własności mechaniczne.
Wytrzymałość na rozciąganie R
m
powyższych staliw w stanie normalizowanym zawiera się w
graniach od 450 do 800 MPa, a w stanie ulepszonym cieplnie po normalizowaniu – od 450 do
1200 MPa.
Staliwa do pracy w podwyższonych temperaturach —
PN-89/H-83157 (9 gatunków: L20,
L16M, L20M, L20HM, L18H2M, L15HMF, L18HM, L21HMF, L17HMF). Staliwa te
charakteryzują się określonymi własnościami mechanicznymi określoną granicą pełzania w
zakresie temperatury do 600°C.
Staliwa stopowe odporne na korozję (nierdzewne i kwasoodporne) charakteryzujące się
zwiększoną odpornością na działanie korozyjne atmosfery, kwasów oraz niektórych ośrodków
korozyjnych - PN-86/H-83158 (14 gatunków). Ze względu na zawartość pierwiastków
stopowych i struktury osnowy rozróżnia się następujące staliwa odporne na korozję:
- chromowe martenzytyczne (LOH13, LH14, LH14N),
- chromowo-niklowe austenityczne (LH18N9, LH18N9T, LH16N5G6),
- chromowo-niklowo-molibdenowe austenityczne (LH18N10M2, L0H18N10M2, L0H18N9M,
LH18N10M2T),
- chromowo-niklowe austenityczno-ferrytyczne (L0H12N4M, LH21N5, LH12N5M, LH21N5T).
Wszystkie gatunki staliwa odpornego na korozję mogą być spawane. Zastosowanie tych
staliw jest podobne jak stali nierdzewnych i kwasoodpomych o podobnym składzie chemicznym.
Staliwa żaroodporne i żarowytrzymałe
- PN-90/H-83159 (9 gatunków). Staliwo żaroodporne
charakteryzuje się odpornością na bezpośrednie działanie płomienia lub spalin w wysokich
temperaturach. Staliwo żarowytrzymałe wykazuje w wysokich temperaturach wyższe własności
wytrzymałościowe niż inne staliwa pracujące w tych temperaturach.
Gatunki LH18S2, LH26, LH29S2G, LH26N4S2 są wysokochromowymi staliwami
żaroodpornymi przeznaczonymi do pracy przy małych obciążeniach. Zawartość węgla jest
wysoka (1,3
÷ 1,5% C, z wyjątkiem LH26 - 0,5% C). Struktura tych staliw składa się z perlitu i
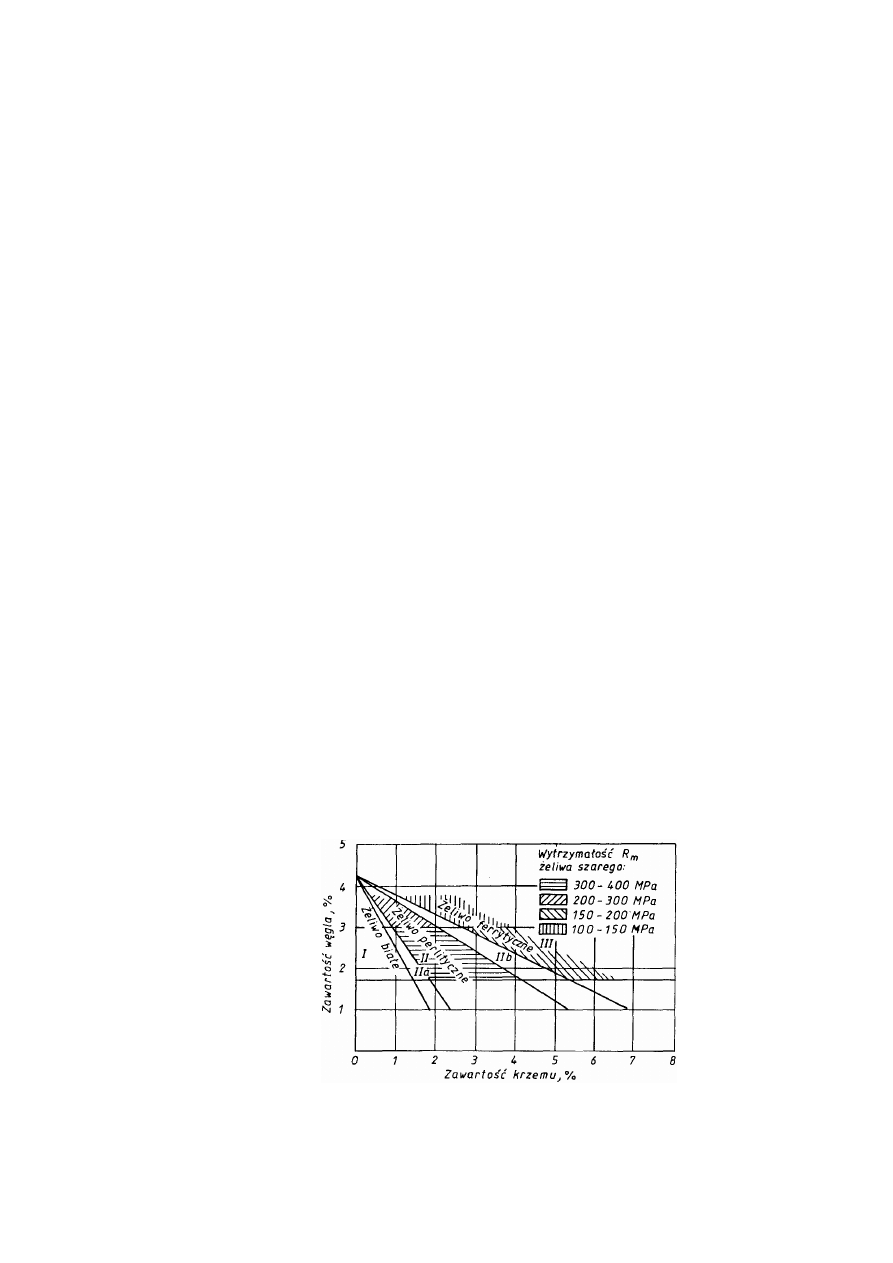
137
JW
węglików lub ferrytu i węglików. Staliwo LH29S2G są ponadto bardzo odporne na ścieranie w
wysokich temperaturach.
Gatunki LH17N8G, LH19N14G, LH23N18G, LH25H19S2, LH17N37S2G są chromowo-
niklowymi staliwami żarowytrzymałymi i żaroodpornymi, o strukturze austenitycznej. Obróbka
cieplna i zastosowanie tych staliw są analogiczne jak austenitycznych stali żarowytrzymałych.
Stosowane są one również jako kwasoodporne w podwyższonych temperaturach.
Staliwa odporne na ścieranie -
PN-88/H-83160 (12 gatunków: L20HGSNM, L25SHNM,
L30HGN2M, L35GSM, L40GM, L40H3T, L100AGM, L40HM, [20G13, L120G13H,
L120G13T oraz L30GS wg PN-87/H-83156). Stosowane są wyjątkiem L120G13, L120G13T i
L120G13H) w stanie normalizowanym i ulepszonym na: korpusy sprzęgieł, elementy
czerpaków, koparek, koła zębate, części maszyn budowlanych, ogniwa gąsienicowe, płyty
pancerne, szczęki do kruszarek koła jezdne do suwnic itp. Staliwa L120G13, L120G13H i
L120G13T (wysokowęglowe i wysokomanganowe) mają strukturę austenityczną i są
szczególnie odporne na zużycie. Stosowane są w stanie przesyconym najczęściej na rozjazdy
kolejowe, gąsienice traktorowe i części łamaczy i kruszarek.
Staliwa narzędziowe
- PN-90/H-83161. Norma obejmuje 18 gatunków staliw do pracy na
zimno i gorąco: L150HSM, L155HNM, L180HNM, L200HNM. L200HSNM, L70H2GNM,
L90HMF, L120H21NM, L180H20F, L35H17N2M. L40H5MF, L45HN2MF, L65HNM,
L75HMF, L100H2M, L120HWMF. L120HNMF, L210H21S. Staliwa te stosuje się w stanie
obrobionym cieplnie, aby zapewnić odpowiednią twardość.
7.8. Żeliwa węglowe
Żeliwami węglowymi nazywa się odlewnicze stopy żelaza z węglem, zawierające
teoretycznie powyżej 2,06% C, a praktycznie 2,5
÷ 4,5% C. Poza tym żeliwa te podobnie jak
stale, zawsze zawierają pewne ilości krzemu, manganu, fosforu i siarki pochodzenia
metalurgicznego. W przeciwieństwie do stali, większość żeliw odznaczają się niską
plastycznością.
Węgiel w żeliwach może występować w dwóch postaciach: bądź w stanie wolnym jako grafit,
bądź w postaci związanej w cementycie. W zależności od tego rozróżnia się żeliwa szare, które
niezależnie od struktury osnowy (ferrytycznej, perlitycznej lub ferrytyczno-perlitycznej)
zawierają wydzielenia grafitu, oraz żeliwa białe, w których węgiel występuje prawie wyłącznie
w postaci cementytu. Nazwy te związane z kolorem ich przełomów. Niekiedy spotyka się żeliwa
połowiczne, które miejscami mają budowę żeliw szarych, a miejscami - białych.
Struktura żeliw zależy zarówno od ich składu chemicznego (rys. 7.26), jak szybkości
krystalizacji metalu, co jest związane z grubością ścianek odlewu.
Rys. 7.26. Wpływ zawartości węgla i krzemu na strukturę żeliw (odlewy piaskowe o grubości
ścianek około 30 mm)
Krzem, którego zawartość w żeliwach waha się od 0,3 do 5%, sprzyja tworzeniu grafitu.
Zmieniając zawartość krzemu można otrzymać różne rodzaje żeliw, całkowicie odmienne

138
JW
zarówno pod względem struktury, jak i własności, od żeliwa białego do ferrytycznego szarego.
Proces grafityzacji ułatwiają również takie pierwiastki, jak miedź i nikiel. Mangan utrudnia
proces grafityzacji, sprzyjając tworzeniu się cementytu. Podobnie działa siarka, której zawartość
w żeliwach nie może przekraczać 0,08
÷ 0,12% (w zależności od wielkości odlewów), ponieważ
pogarsza ona własności odlewnicze i zwiększa kruchość. Poza tym do pierwiastków
przeciwdziałających grafityzacji należą między innymi chrom, wolfram, molibden i wanad.
Ważnym składnikiem żeliw jest fosfor, który zwiększa ich rzadkopłynność dzięki tworzeniu
eutektyki fosforowej, nie oddziałując w wyraźnym stopniu na proces grafityzacji.
7.8.l. Żeliwa szare
Największe zastosowanie przemysłowe mają jak dotąd żeliwa szare. W żeliwach i grafit
występuje w postaci nieregularnych płatków różnej wielkości, tworząc nieciągłości w osnowie
metalicznej (rys. 7.27). Wytrzymałość grafitu w porównaniu z wytrzymałością tej osnowy
można przyjąć za równą zeru, stąd też żeliwa szare odznaczają się niską wytrzymałością na
rozciąganie i zginanie, przy dość dobrej wytrzymałości na ściskanie. Również wytrzymałość
zmęczeniowa żeliw jest niewielka, ze względu na istnienie wspomnianych karbów naturalnych.
Z tego samego powodu żeliwa szare są mało wrażliwe na działanie wad powierzchniowych,
wszelkiego rodzaju karbów konstrukcyjnych itp.
Rys. 7.27, Struktura żeliwa szarego nie
trawionego. Widoczne płatki grafitu.
Powiększ. 100x
Rys. 7.28. Struktura żeliwa szarego
ferrytyczno-perlitycznego z wyraźnie
widoczną eutektyką fosforową (jasne,
kropkowane obszar. Traw. 5% roztworem
alkoholowym HN03. Powiększ. 500x
Główną zaletą żeliwa szarego są przede wszystkim dobre własności odlewnicze przejawiające
się wysoką rzadkopłynnością, dobrym wypełnianiem form, mały skurczem odlewniczym (-1%)
itd. Inne zalety związane z obecnością wydzielę-grafitu to: dobre własności przeciwcierne i
zdolność tłumienia drgań. Dodatkowi zaletą tych żeliw jest niska cena.
Grafit, będący jednym z głównych składników strukturalnych żeliw szarych, jest
rozmieszczony w osnowie ferrytycznej, ferrytyczno-perlitycznej lub perlityczne przy czym ferryt
jest tu nie tylko roztworem stałym węgla w żelazie a, lecz także roztworem krzemu oraz
ewentualnie innych pierwiastków w żelazie a i dlatego nosi nazwę krzemoferrytu. Oczywiście
perlit jest w przypadku żeliw mieszaniną krzemoferrytu i cementytu.
Charakterystycznym składnikiem strukturalnym żeliw szarych jest potrójna eutektyka
fosforowa, zwana niekiedy steadytem. W temperaturze powstawania składa się ona z cementytu
(Fe
3
C), fosforku żelaza (Fe
3
P) i austenitu, w temperatura pokojowej - z cementytu, fosforku
żelaza oraz produktów przemiany eutektoidalnej austenitu (perlitu, cementytu wtórnego i
trzeciorzędowego).
Przy większej zawartości siarki w strukturze żeliw szarych uwidaczniają się również siarczki
manganu w postaci lekko niebieskawych wieloboków.
Strukturę żeliwa szarego o osnowie ferrytyczno-perlitycznej pokazano na rys. 7.28. Oprócz
grafitu płatkowego i składników osnowy wyraźnie widać potrójną eutektykę fosforową w
postaci jasnych, kropkowanych obszarów o charakterystycznych kształtach.
139
JW
Żeliwa szare zgodnie z PN-92/H-83101 dzielą się na gatunki, przy czym podstawą podziału
jest wytrzymałość na rozciąganie, określana na próbkach o średnicy pomiarowej 20 mm,
wytoczonych z oddzielnie odlewanych wlewków próbnych.
Polska Norma podaje 6 gatunków żeliwa szarego z określoną minimalną wytrzymania na
rozciąganie, a mianowicie: 100, 150, 200, 250, 300 i 350 (trzycyfrowa liczba oznacza min. R
m
w
MPa). Niższe wartości odnoszą się do żeliw o strukturze ferrytycznej, wyższe - do żeliw o
strukturze perlitycznej. Wytrzymałość i twardość zesłane na próbkach wykonanych z wlewków
próbnych różnią się od wytrzymali i twardości odlewów, gdyż własności te w istotny sposób
zależą od grubości ścianek odlewów, zmniejszając się z jej wzrostem. Na przykład żeliwo szare
gatunek 100, ze zwiększaniem grubości ścianek odlewu, wykazuje wytrzymałość na rozciąganie
120
÷ 90 MPa, a żeliwo gatunku 350 – 315 ÷270 MPa.
W przypadku wymaganej dobrej obrabialności i odporności odlewów na ścieranie żeliwa
szare klasyfikuje się na podstawie twardości, przy czym ustala się 6 klas twardości
oznaczających przewidywaną średnią twardość HB w określonym miejscu odlewu (tabl. 7.16).
Przewidywane zakresy twardości HB dla różnych grubości ścianki odlewu podano w tabl. 7.17.
.
Klasy twardości żeliw szarych (wg PN-92/H-83101) Tablica 7.16
Klasa twardości Zakres
twardości odlewu
HB
H 145
max 170
H 175
150-200
H 195
170-200
H 215
190-240
H 235
210-260
H 255
230-280
Dla żeliw szarych istnieją ustalone empirycznie zależności między twardością i
wytrzymałością na rozciąganie w przypadkach, gdy:
R
m
≥ 196 MPa, wówczas HB = RH (100 + 0,438) R
m
R
m
< 196 MPa, wówczas HB = RH (100 + 0,724) R
m
Czynnik RH, czyli tzw. twardość względna, zmienia się w granicach 0,8
÷1,2 w zależności od
materiału wyjściowego, procesu topienia i rzeczywistego procesu metalurgicznego. W
poszczególnych odlewniach można ustalić wartość czynnika RH na prawie stałym poziomie i w
takich przypadkach, mierząc twardość HB na powierzchni odlewu, można określić jego
wytrzymałość na rozciąganie wykorzystując podane zależności.
Tablica 7.17
Orientacyjne zakresy twardości żeliw szarych dla różnych grubości ścianki
odlewu (wg PN-92/H-83101)
Twardość HB
Gatunek
2,5
÷ 5
5
÷ 10
Grubość
ścianki, mm
20
÷ 40
40
÷ 80
350
280-200
260-185
300
280-200
255-180
240-165
250
280-200
250-180
235-160
220-145
200
<280
260-170
230-150
210-135 190-120
150
260-170
225-140
205-125
185-110 170-100
100 <210 <185 <175 <160 <150

140
JW
Dzięki swym zaletom, żeliwa szare są materiałem konstrukcyjnym powszechnie stosowanym
w przemyśle maszynowym, kolejowym, samochodowym i in. (np. na korpusy maszyn, płyty
fundamentowe, pierścienie tłokowe, bębny hamulcowe, tuleje cylindrowe, armaturę).
Odmianą żeliw szarych są żeliwa modyfikowane, zawierające bardzo drobny grafit płatkowy.
To rozdrobnienie grafitu uzyskuje się przez dodanie do żeliwa przed odlaniem tzw. modyfikato-
ra, najczęściej w postaci sproszkowanego żelazokrzemu. Żeliwa modyfikowane mają wyższą
wytrzymałość niż żeliwa zwykłe.
7.8.2. Żeliwa sferoidalne
Żeliwami sferoidalnymi nazywa się żeliwa, w których grafit wydziela się podczas krzepnięcia
w postaci kulek.
Otrzymuje się je w wyniku procesu modyfikacji, który polega na wprowadzeniu do metalu -
bezpośrednio przed jego odlewaniem - niewielkiego dodatku magnezu (w stopie z niklem lub
miedzią).
Struktura osnowy żeliw sferoidalnych, podobnie jak struktura osnowy zwykłych żeliw
szarych, może być ferrytyczna (rys. 7.29), ferrytyczno-perlityczna, perlityczno-ferrytyczna lub
perlityczna (rys. 7.30).
Żeliwa sferoidalne są w Polsce znormalizowane (PN-92/H-83123), przy czym podstawą
klasyfikacji są ich własności mechaniczne. Polska Norma podaje dwie odrębne klasyfikacje
żeliw sferoidalnych. Pierwsza - opiera się na własnościach mechanicznych określanych na
próbkach wyciętych z wlewków próbnych oddzielnie odlewanych. Według tej klasyfikacji
rozróżnia się 9 gatunków żeliw (tabl. 7.18). Oznaczenie poszczególnych gatunków składa się z
liczby określające minimalną wytrzymałość na rozciąganie w MPa oraz liczby określającej
minimalne wydłużenie w procentach. Na przykład oznaczenie 400-15 oznacza żeliwo
sferoidalne o R
m
min. 400 MPa i wydłużeniu A
5
min. 15%.
Rys. 7.29. Struktura żeliwa sferoidalnego
ferrytycznego. Widoczne kuliste wydzielenia
grafitu na tle ferrytycznej osnowy. Traw. 5%
roztworem alkoholowym HNO
3
. Powiększ.
200x
Rys. 7.30. Struktura żeliwa sferoidalnego
perlitycznego. Widoczne kuliste wydzielenia
grafitu w otoczce ferrytycznej na tle
perlitycznej osnowy. Traw. 5% roztworem
alkoholowym HNO
3
. Powiększ. 200x
Druga klasyfikacja opiera się na własnościach mechanicznych określanych na próbkach
wykonanych z wlewków próbnych tzw. przylanych (odlewanych razem z odlewem). W tym
przypadku, w oznaczeniu gatunku za liczbą określającą minimalne wydłużenie podaje się literę
A, np. 400-15A. Ta klasyfikacja zawiera 6 garnków żeliw o wytrzymałości na rozciąganie 320-
700 MPa, granicy plastyczności 210
÷ 400 MPa, wydłużeniu 15 ÷ 2% i twardości HB 130 ÷ 320.
Dodatkowa klasyfikacja (również zawarta w PN) oparta na twardości mierzonej na
samych
odlewach rozróżnia 9 gatunków oznaczanych literą H i podaje średnią twardość HB danego
gatunku, np. H330, H150 itd.
Niezależnie od przyjętej klasyfikacji, wyższa wytrzymałość i twardość odpowiada perlitycznej
strukturze osnowy, wyższa plastyczność - strukturze ferrytycznej.

141
JW
Gatunki i własności mechaniczne żeliw sferoidalnych (wg PN-92/H-83123) Tablica 7.18
Gatunek
żeliwa
R
m
min
R
0,2
min
A
5
min
Twardość
HB
Struktura osnowy
900-2
900
600
2
280-360 bainit lub martenzyt odpuszczony
800-2 800 480 2 245-335
perlit
lub
struktura.odpuszczona
700-2 700 420 2 225-305
perlit
600-3
600
370
3
190-270
perlit + ferryt
500-7
500
320
7
170-230
perlit + ferryt
450-10 450 310 10 160-210
ferryt
400-15 400 250 15 130-180
ferryt
400-18 400 250 18 130-180
ferryt
350-22 350 220 22 <150
ferryt
Żeliwo sferoidalne zastępuje z powodzeniem nie tylko staliwo, lecz również niektóre
odkuwki stalowe. Wytwarza się z niego takie części silników samochodowych, jak wały
wykorbione, wałki rozrządcze, cylindry i pierścienie tłokowe. W budowie obrabiarek żeliwo
sferoidalne wykorzystuje się na koła zębate, wrzeciona, korpusy itd.
7.8.3. Żeliwa białe
Żeliwa białe ze względu na zawartość węgla dzielą się na: podeutektyczne, eutektyczne i
nadeutektyczne.
Struktura żeliwa podeutektycznego (o zawartości węgla poniżej 4,3%), zgodnie z układem
równowagi żelazo-cementyt (p. rozdz. 3), składa się w temperaturze 1147°C z austenitu i
ledeburytu. W miarę obniżania temperatury z austenitu wydziela się cementyt wtórny. W
temperaturze 723°C następuje przemiana austenitu w perlit, a ledeburytu - w ledeburyt
przemieniony. Przy dalszym ochładzaniu stopu do temperatury pokojowej, w miarę
zmniejszania się rozpuszczalności węgla w żelazie a, wydziela się cementyt trzeciorzędowy (w
bardzo małej ilości). W rezultacie, w temperaturze pokojowej struktura żeliwa białego
podeutektycznego składa się z perlitu, cementytu i ledeburytu przemienionego (rys. 7.31).
Struktura żeliwa eutektycznego (zawierającego 4,3% C) w temperaturze 1147°C składa się z
ledeburytu, a w temperaturze pokojowej - z ledeburytu przemienionego.
Struktura żeliwa białego nadeutektycznego (zawierającego ponad 4,3% węgla) składa się w
temperaturze 1147°C z ledeburytu i cementytu pierwszorzędowego (pierwotnego)
krystalizującego w postaci grubych igieł, w temperaturze pokojowej — z ledeburytu
przemienionego i cementytu pierwotnego (rys. 7.32).
Rys. 7.31. Struktura żeliwa białego podeutek-
tycznego. Na tle przemienionego ledeburytu
widoczne ciemne kryształy perlitu z wydzie-
lonym cementytem wtórnym. Traw. 5% roz-
tworem alkoholowym HNO
3
. Powiększ. 100x
Rys. 7.32. Struktura żeliwa białego nadeutek-
tycznego. Na tle przemienionego ledeburytu
widoczne jasne, iglaste kryształy cementytu
pierwszorzędowego. Traw. 5% roztworem al-
koholowym HNO
3
. Powiększ. 100x

142
JW
Żeliwa białe, jako materiał konstrukcyjny, prawie nie mają bezpośredniego zastosowania
technicznego, natomiast powierzchniowa warstwa żeliwa białego na żeliwie szarym, powstająca
przez tzw. zabielenie (tj. szybkie lokalne ochłodzenie odlewu), jest często stosowana w celu
zwiększenia odporności materiału na ścieranie. Taką twardą warstwę w żeliwie otrzymuje się
umieszczając w formie tzw. ochładzalniki, czyli odpowiednie wkładki metaliczne szybko
odprowadzające ciepło. Zabielenie żeliwa szarego stosuje się czasem w przypadku mniej
odpowiedzialnych prowadnic korpusów maszyn, bieżni kół wagoników roboczych itp.
Żeliwo białe jest materiałem wyjściowym przy wytwarzaniu przedmiotów z żeliwa ciągłego.
7.8.4. Żeliwa ciągliwe
Żeliwami ciągliwymi nazywa się żeliwa białe, które wskutek długotrwałego (rzędu
kilkudziesięciu godz.) wyżarzania w wysokiej temperaturze (ok. 1000°C) ulegają określonemu
uplastycznieniu, dzięki odwęgleniu lub grafityzacji lub obu tym procesom łącznie. W zależności
od sposobu przeprowadzania tej obróbki otrzymuje się:
- Żeliwa ciągliwe białe, przez wyżarzanie żeliw białych w środowisku utleniającym, np. w
rudzie żelaza. Podczas wyżarzania znaczna część węgla zawartego w żeliwie utlenia się, a w
warstwie powierzchniowej grubości 1,5
÷ 2 mm zachodzi zupełne odwęglenie. Przy ochładzaniu
zazwyczaj nie wygrzewa się żeliwa w temperaturze poniżej temperatury przemiany, w wyniku
czego w metalicznej osnowie rdzenia zachowuje się znaczna ilość perlitu. Przy powierzchni
odlewu żeliwo to wykazuje matowobiałą barwę przełomu (ferryt) przechodzącą łagodnie w
srebrzystą bliżej środka ścianki odlewu (perlit).
- Żeliwa ciągliwe czarne, przez wyżarzanie żeliw białych w środowisku obojętnym. W czasie
tego wyżarzania cementyt zawarty w żeliwie rozpada się, a wydzielający się z niego węgiel w
postaci grafitu tworzy skupienia zwane węglem żarzenia. Struktura żeliwa w temperaturze
wyżarzania składa się więc z austenitu i węgla żarzenia. Kolejnym zabiegiem jest bardzo wolne
chłodzenie, warunkujące zachodzenie przemian fazowych zgodnie ze stabilnym układem
równowagi żelazo-grafit (z austenitu zamiast cementytu wydziela się grafit). W efekcie, w
temperaturze pokojowej otrzymuje się żeliwo, którego struktura składa się ze skupień grafitu
(węgla żarzenia) rozmieszczonych w ferrytycznej osnowie (rys. 7.33). Duża ilość wydzieleń
grafitu wywołuje ciemną barwę przełomu.
- Żeliwa ciągliwe perlityczne, przez wyżarzanie żeliw białych w środowisku obojętnym, lecz bez
doprowadzania do końca procesu grafityzacji (szybsze chłodzenie poniżej temperatury
przemiany, dzięki czemu w strukturze zachowuje się część cementytu). W wyniku uzyskuje się
żeliwo o osnowie perlitycznej lub perlityczno-ferrytycznej i srebrzystej barwie przełomu.
W procesie produkcji żeliwa ciągliwego bardzo ważnym czynnikiem jest uzyskanie w odlewie
żeliwa całkowicie białego, ponieważ częściowa grafityzacja podczas krzepnięcia i utworzenie
się w żeliwie płatków grafitu zakłócają zachodzące w czasie wyżarzania grafityzującego
powstawanie zwartych skupień grafitu. W związku z tym zawartość pierwiastków wchodzących
w skład żeliwa ciągliwego musi mieścić się w stosunkowo wąskich granicach.
Zazwyczaj skład chemiczny żeliwa ciągliwego jest następujący: 2,4
÷ 2,8 % węgla, 0,8 ÷
1,4% krzemu, do 1% manganu, do 0,1% siarki i do 0,2 % fosforu.
Rys. 7.33. Struktura żeliwa ciągliwego czarnego. Na tle ferrytu widoczne wydzielenia węgla
żarzenia. Traw. 5% roztworem alkoholowym HN03. Powiększ. 100x
143
JW
Żeliwa ciągliwe są w Polsce znormalizowane (PN-92/H-83221), przy czym norma rozróżnia 4
gatunki żeliwa ciągliwego białego (tabl. 7.19), 3 - żeliwa ciągliwego czarnego i 7 - żeliwa
ciągliwego perlitycznego (tabl. 7.20). Oznaczenie poszczególnych gatunków składa się z liter i
cyfr. Litery oznaczają: W - żeliwo ciągliwe białe, B - żeliwo ciągliwe czarne, P - żeliwo ciągliwe
perlityczne. Po literze oddzielonej odstępem podawane są dwie cyfry oznaczające minimalną
wytrzymałość na rozciąganie w MPa próbki o średnicy 12 mm podzieloną przez 10, a następnie,
oddzielone znakiem pauzy, dwie cyfry oznaczające minimalne wydłużenie A
3
wyrażone w %.
Jeśli wartość wydłużenia jest mniejsza niż 10%, pierwszą cyfrą jest 0. Przykładowe oznaczenia
żeliw ciągliwych: W 35—04, B 32—10, P 65—02.
Żeliwo ciągliwe odznacza się dobrą skrawalnością, dużą odpornością na działanie dymu i
kwaśnej wody kopalnianej. Wykonuje się z niego odlewy o dużej wytrzymałości, dobrej
plastyczności, obrabialności i odporności na uderzenia, gdyż łączy w sobie dobre własności
odlewnicze żeliwa z dobrymi własnościami mechanicznymi staliwa
Tablica 7.19
Własności mechaniczne i twardość żeliw ciągliwych białych (wg PN-92/H-83221)
Oznaczenie
gatunku
Średnica
próbki mm
R
min
MPa
Rmin
MPa
A
5
min
Twardość HB
max
9
340
5
W 35-04
12
350
-
4
230
15
360
-
3
9
320
170
15
W 38-12
12
380
200
12
200 ;
15
400
210
8
9
360
200
8
W 40-05
12
400
220
5
220
15
420
230
4
9
400
230
10
W 45-07
12
450
260
7
220
15
480
290
4
Jest szeroko stosowane w przemyśle maszyn rolniczych, samochodowym, obrabiarkowym, w
kolejnictwie itp
Tablica 7.20
Własności mechaniczne i twardość żeliw ciągliwych czarnych i perlitycznych
(wg PN-92/H-83221)
Oznaczenie
gatunku
R
m
min
R
0,2
min MPa
A
5
min
Twardość HB
B 30-06
300
-
6
max 150
B 32-12
320
190
12
max 150
B 35-10
350
200
10
max 150
P 45-06
450
270
6
150-200
P 50-05
500
300
5
160-220
P 55-04
550
340
4
180-230
P 60-03
600
390
3
200-250
P 65-02
650
430
2
210-260
P 70-02
700
530
2
240-290
P 80-01*
800
600
1
270-310
*
Hartowanie w oleju, a następnie odpuszczanie.

144
JW
7.9. Żeliwa stopowe
Żeliwami stopowymi nazywa się żeliwa zawierające dodatkowo pierwiastki takie jak nikiel,
chrom, molibden, aluminium, tytan, wanad, miedź, wolfram, bor lub zwiększone ilości krzemu i
manganu. Dobór ww. składników oraz ich wzajemne stosunki ilościowe decydują o
własnościach wytrzymałościowych żeliw stopowych, ich odporności na ścieranie i działanie
środowisk korozyjnych oraz na oddziaływane utleniających atmosfer w wysokich temperaturach.
Polska Norma PN-88/H-83144 podaje 48 gatunków żeliw stopowych dzielących się, w
zależności od własności i zastosowania, na 3 grupy: żaroodporne, odporne na korozję i odporne
na ścieranie.
Żeliwo stopowe oznacza się znakiem gatunku, który zawiera: litery Zl dla żeliwa stopowego
szarego i połowicznego, litery Zb dla żeliwa stopowego białego, litery Zs dla żeliwa stopowego
sferoidalnego, symbole chemiczne pierwiastków stopowych wg malejącej procentowej
zawartości składnika, oraz liczby określające średnią procentową zawartość pierwiastka
stopowego, jeżeli jest ona równa lub większa od 0,8%.
Żeliwa stopowe żaroodporne.
Jest to grupa żeliw wykazujących odporność na korozyjne
działanie gazów utleniających w wysokich temperaturach dzięki zawartości takich dodatków
stopowych, jak krzem (do 6%), chrom (do 34%) i aluminium (do 8%). Graniczna temperatura
pracy tych żeliw zależy od zawartości i wzajemnego stosunku ilościowego ww. dodatków
stopowych i w zależności od gatunku żeliwa wynosi 550
÷1100°C. Oprócz żaroodporności, ta
grupa żeliw charakteryzuje się również dobrą odpornością na ścieranie i twardością, a także
odpornością na korozyjne oddziaływanie różnych środowisk chemicznych. Polska Norma podaje
10 ganków żeliw stopowych żaroodpornych.
Żeliwa stopowe odporne na korozję.
Żeliwa węglowe zwykłe są stosunkowo mało odporne na
działanie czynników chemicznych. Wprowadzenie do tych żeliw dodatków stopowych, takich
jak krzem, nikiel, chrom i miedź znakomicie podwyższa ich odporność na korozyjne i erozyjne
oddziaływanie różnorodnych środowisk chemicznych. Polska Norma podaje 8 gatunków żeliw
stopowych odpornych na korozję, w tym: l gatunek żeliwa wysokokrzemowego (14
÷16% Si), 5
gatunków żeliw wysokoniklowych (13,5
÷32% Ni) o podwyższonej zawartości krzemu, chromu
i miedzi oraz 2 gatunki żeliw wysokochromowych (25
÷34% Cr), wykazujących również bardzo
dobrą odporność na ścieranie i doskonałą żaroodporność.
Żeliwa stopowe odporne na ścieranie.
Jest to najliczniejsza grupa żeliw stopowych,
obejmująca zgodnie z Polską Normą 33 gatunki. W większości są to żeliwa wysokostopowe
zawierające: 0,5
÷ 3,1% Si, 0,5 ÷1,2% Mn (tylko l gatunek zawiera do 12% Mn), 0,15 ÷ 2,4% Cr
(tylko 2 gatunki żeliw mają wysoką zawartość chromu: jeden do 19%, drugi - do 30% Cr), 0,13
÷ 5% Ni, 0,5 ÷ 2,0% Cu. Ponadto w 7 gatunkach występują niewielkie zawartości molibdenu,
tytanu, wanadu bądź boru. Skład chemiczny tych żeliw jest tak dobrany, że wykazują dobre
własności przeciwcierne oraz wytrzymałościowe przy zadowalającej odporności korozyjnej w
określonych ośrodkach chemicznych; niektóre gatunki zachowują te własności również w
podwyższonych temperaturach.
7.10. Oznaczanie stali wg: PN-EN 10027-1 Systemy oznaczania stali.
Znaki stali, symbole
główne.
EN 10027-1:1992 jest zalecana przez CEN (Europejski Komitet Normalizacyjny) do
stosowania przez krajowe komitety normalizacyjne bez jakichkolwiek zmian. PN-EN 10027-1
jest identyczna z EN 10027-1:1992 i została ustanowiona przez Polski Komitet Normalizacyjny
15.12.1994 r.
W tej klasyfikacji oznaczeń stali wyróżnia się dwie główne grupy znaków:
• znaki zawierające symbole wskazujące na skład chemiczny stali,
• znaki zawierające symbole wskazujące na zastosowanie oraz mechaniczne lub fizyczne
własności stali.

145
JW
W obu grupach znaków po symbolach głównych mogą być podawane symbole dodatkowe.
Poniżej podano jedynie, z jakich symboli głównych składa się znak stali. W przypadku staliwa znak
gatunku zawierający symbole wskazujące na skład chemiczny poprzedza litera G.
Oznaczanie stali wg składu chemicznego
W znakach stali wg składu chemicznego wyróżnia się cztery podgrupy;
• stale niestopowe (bez stali automatowych) o średniej zawartości manganu <1%. Znak
tych stali składa się z następujących symboli głównych, umieszczonych kolejno po sobie:
litery C i liczby będącej 100-krotną średnią wymaganą zawartością węgla;
• stale niestopowe o średniej zawartości manganu ≥ 1 %, niestopowe stale automatowe i
stale stopowe
(bez stali szybkotnących) o zawartości każdego pierwiastka stopowego <5%.
Znak tych stali składa się z: liczby będącej 100-krotną wymaganą średnią zawartością węgla,
symboli pierwiastków chemicznych składników stopowych stali w kolejności malejącej zawartości
pierwiastków oraz liczb oznaczających zawartości poszczególnych pierwiastków stopowych w
stali. Każda liczba oznacza odpowiednio, średni procent zawartości pierwiastka pomnożony
przez współczynnik wg tabl. 7.21 i zaokrąglony do najbliższej liczby całkowitej. Liczby
oznaczające zawartości poszczególnych pierwiastków stopowych należy oddzielić poziomą
kreską.
TABLICA 7.21.
Współczynnik do ustalania symboli liczbowych pierwiastków stopowych przy oznaczaniu stali
stopowych (bez stali szybkotnących) o zawartości każdego pierwiastka stopowego <5% (PN-
EN 10027-1)
Pierwiastek
Współczynnik
Cr, Co, Mn, Ni, Si, W
4
Al, Be, Cu, Mo, Nb, Pb, Ta, Ti, V,
10
Ce, N, P, S
100
B
1000
Na przykład 55NiCrMoV6-2-2 jest znakiem stali o średnim składzie: 0,55% C, l,5%Ni, 0,6%
Cr, 0,2% Mo i poniżej 0,1 % V (jest to stal narzędziowa do pracy na gorąco);
• stale stopowe (bez stali szybkotnących) zawierające przynajmniej jeden pierwiastek stopowy
w ilości ≥ 5%.
Znak tych stali składa się z: litery X, liczby będącej 100-krotną wymaganą
średnią zawartością węgla, symboli chemicznych składników stopowych stali w kolejności
malejącej zawartości oraz liczb (zaokrąglonych do najbliższej liczby całkowitej) oznaczających
średni procent zawartości poszczególnych pierwiastków. Na przykład X5CrNiMol7-12-2 jest
znakiem stali o składzie: maks. 0,07% C, 17,5% Cr, 11,6% Ni, 2,25% Mo. Stal ta wg polskiej
normy (PN) miałaby oznaczenie 0H17N12M2;
• stale szybkotnące. Znak tych stali składa się z następujących symboli literowych i
liczbowych: liter HS oraz liczb oznaczających procentowe zawartości (zaokrąglone do
najbliższych liczb całkowitych) pierwiastków stopowych w następującej kolejności; wolfram,
molibden, wanad, kobalt; np. HS18-0-1 jest znakiem stali oznaczanej wg PN SW18; średnia
zawartość pierwiastków w tej stali wynosi: 0,80% C, 18,0% W, 1,25% V. Zawartość Cr w
stalach szybkotnących nie jest podawana, gdyż jest we wszystkich gatunkach tych stali taka sama i
wynosi od 3,5 do 4,5 %.
Oznaczanie stali wg zastosowania i własności
Znak stali oznaczanych wg ich zastosowania i własności mechanicznych lub fizycznych
zawiera następujące główne symbole:
a) S - stale konstrukcyjne,
P - stale pracujące pod ciśnieniem,
L - stale na rury przewodowe,
E - stale maszynowe,
146
JW
za którymi umieszcza się liczbę będącą minimalną granicą plastyczności w MPa;
b) B - stale do zbrojenia betonu,
za którym umieszcza się liczbę będącą charakterystyczną granicą plastyczności;
c) Y - stale do betonu sprężonego,
R - stale na szyny lub w postaci szyn,
za którymi umieszcza się liczbę będącą wymaganą minimalną wytrzymałością ni rozciąganie;
d) H - wyroby płaskie walcowane na zimno ze stali o podwyższone wytrzymałości przeznaczone do
kształtowania na zimno, za którym umieszcza się liczbę będącą wymaganą minimalną granicą plastyczności
albo jeżeli jest wymagana tylko wytrzymałość na rozciąganie, wtedy umieszcza się literę T, za którą
podaje się wymaganą minimalną wytrzymałość na rozciąganie;
e) D - wyroby płaskie ze stali miękkich przeznaczonych do kształtowania na zimno, za którym umieszcza
się jedną z następujących liter:
1) C - dla wyrobów walcowanych na zimno,
2) D - dla wyrobów walcowanych na gorąco przeznaczonych do kształtowania na zimno,
3) X - dla wyrobów bez charakterystyki walcowania (na zimno lub na gorąco);
oraz dwa symbole cyfrowe lub literowe charakteryzujące stal;
f) T - wyroby walcowni blachy ocynowanej, za którym umieszcza się:
1) dla wyrobów o jednokrotnie redukowanej grubości - literę H, za którą podaje się liczbę będącą
wymaganą nominalną twardością wg HR 30Tm;
2) dla wyrobów o dwukrotnie redukowanej grubości - liczbę będącą wymaganą nominalną
granicą plastyczności;
g) M - stale elektrotechniczne, za którym umieszcza się:
1) liczbę będącą 100-krotną wymaganą maksymalną stratnością w W·kg
-1
,
2) liczbę będącą 100-krotną nominalną grubością wyrobu w mm,
3) liczbę oznaczającą rodzaj blachy lub taśmy elektrotechnicznej, tj.:
A - o niezorientowanym ziarnie,
D - ze stali niestopowych, nie wyżarzonych końcowo,
E - ze stali stopowych, nie wyżarzonych końcowo,
N - o normalnie zorientowanym ziarnie,
S - o zorientowanym ziarnie i zmniejszonej stratności,
P - o zorientowanym ziarnie i dużej przenikalności magnetycznej.
Tablica 7.22
Skład chemiczny i własności wytrzymałościowe niektórych stali konstrukcyjnych
Rodzaj stali
Znak stali wg
PN-EN
Średni skład stali, %
R
e
MPa
R
m
MPa
C
Si
Mn
Cr
Inne
Stal niestopowa
C10
C35
C60
0,10
0,35
0,61
≤0,40
≤0,40
≤0,40
0,45
0,65
0,75
—
—
—
-
280
390
540
480
680
950
Stal
niskostopowa o
podwyższonej
S355NL
S460N
≤0,18
≤0,20
≤0,50
≤O,6O
1,28
1,35
—
—
0,05 ≥ Nb; 0,12 ≥ V; 0,03 ≥ Ti
0,05 ≥ Nb; 0,20 ≥ V; 0,03 ≥
Ti
355
460
550
640
Stal do
nawęglania
C15R
16MnCrB5
20NiCrMoS2-2
0,15
0,16
0,20
≤0,40
≤0,40
≤S0,40
0,45
1,15
0,80
—
0,95
0,53
0,03 S
0,035 ≥ S; 0,0029 B
0,55 Ni; 0,20 Mo; 0,03 S
320
—
—
550
—
—
Stal do ulepszania
cieplnego
C45E
25CrMoS4
42CrMoS4
34CrNiMo6
0,45
0,25
0,42
0,34
≤0,40
≤0,40
≤0,40
≤0,40
0,65
0,75
0,75
0,65
—
1,05
1,05
1,50
—
0,23 Mo; 0,03 S
0,23 Mo; 0,03 S
1,50 Ni; 0,23 Mo
620
650
880
950
900
920
1100
1100
Stal sprężynowa
51CrV4
60Si7
0,51
0,60
≤0,40
1,65
0,90
0,75
1,05
—
0,18 V
—
1100
1200
1300
1400
147
JW
Tablica 7.23.
Składy chemiczne i twardości wybranych stali narzędziowych
Tablica 7.24.
Skład chemiczny wybranych stali o szczególnych własnościach
Rodzaj stali
Znak stali
wg PN-EN ISO 4957
Średni skład stali, %
Twardość
HV min-
C
Si
Mn
Ci
Inne
Stal nicslupowa do pracy
na zimno
C45U
C70U
C90U
C12OIJ
0,45
0,70
0,90
1,20
0,27
0,20
0,20
0,20
0,70
0,25
0.25
0,25
−
−
−
−
−
−
−
−
54
57
60
62
Stal słupowa do
pracy na zimno
102Cr6
60WCW8
X2IOCrWI2
X153C>MoV12
1,02
0,60
2,15
1,53
0,25
0,85
0,25
0,35
0,35
0,30
0,45
0,40
1,50
1,05
12,00
12,00
−
1,95 W; 0,15 V
0,7 W
0,85 Mo; 0,85V
60
58
62
61
Stal stopowa do
pracy na gorącu
55NiCrMuV7
32CrMoVI2-2R
X40CrMnV5-l
X30WOV9-3
0,55
0,32
0,40
0,30
0,25
0,25
1,00
0,25
0,75
0,30
0,38
0,30
1,00
2,95
5,15
2,85
1,65 Ni; 0.45 Mo; 0.10 V
2,75 Mo; 0,55 V
1,35 Mo; 1,00 V
9,00 W; 0,40 V
42
46
50
4ff
Stal szybkotnąca
HSl8-0-1
HS2-9-2
HS6-5-4
HS2-9-1-8
0.78
1,00
1,32
1,10
≤0,45
≤0,70
≤0,45
≤0,70
≤0,40
≤0,40
Ą0.40
Ą0.40
4.15
4,00
4,15
4,00
17.95 W; 1,10 V
8,70 Mo; 1.95 V; 1.80 W
5,60 W; 4,60 Mo; 3,95 V
9,50 Mo; K,00 Co; 1,55 W; 1,10 V
63
64
64
66
Rodzaj stali
Znak wg PN-EN
Średni skład stali, %
C
Si
Mn
Cr
Inne
Stal odporna na korozję;
ferryty czna
X6Crl3
X6Crl7
X6CrMol7-l
≤0,08
≤0,08
≤0,08
≤1,00
≤1,00
≤1,00
≤1,00
≤1,00
≤1,00
13,00
17,00
17,00
−
−
1,15 Mo
Stal odporna na korozję;
martenzytyczna
X30Crl3
X17CrNil6-2
X90CrMoV18
0,30
0,17
0,90
≤1,00
≤1,00
≤1,00
≤1,00
≤1,00
≤1,00
13,00
16,00
18,00
-
2,00 Ni
1,10 Mo; 0,10 V
Stal odporna na korozję;
austenityczna
X2CrNil9-ll
X2CrNiMol7-12-2
X2CrNiMoN17-13-5 X1
CrNiMoCuN25-25-5
≤0,03
≤0,03
≤0,03
≤0,02
≤1,00
≤1,00
≤1,00
≤0,70
≤2,00
≤2,00
≤2,00
≤2,00
19,00
17,50
17,50
25,00
11,00 Ni
11,50 Ni; 2,25 Mo
13,50 Ni; 4,50 Mo; 0,17 N
25,50 Ni; 5,20 Mo; 1,50 Cu; 0,21 N
Stal odporna na korozję;
ferrytyczno-austenityczna
X3CrNiMoN27-5-2
X2CrNiMoCuN25-6-3
≤0,05
≤0,03
≤1,00
≤0,70
≤2,00
≤2,00
26,50
25,00
5,50 Ni; 1,65 Mo; 0,125 N
6,50 Ni; 3,35 Mo; 1,75 Cu; 0,225 N
Stal żaroodporna;
ferrytyczna
X10CrAlSil3
X10CrAlSi25
≤0,12
≤0,12
1,05
1,05
≤1,00
≤1,00
13,00
24,50
0,95 Al.
1,45 Al
Stal żaroodporna;
austenityczna
X15CrNiSi20-12
X6CrNiSiNCel9-10
≤0,12
0,06
2,00
1,50
≤2,00
≤1,00
20,00
19,00
12,00 Ni
10,00 Ni; 0,16 N; 0,055 Ce
Oznaczenia i składy stali odpornych na korozję zaczerpnięto z PN-EN 10088-1, natomiast stali żaroodpornych z PN-EN 10095.

148
JW
8. Miedź i stopy miedzi
Miedź jest metalem barwy czerwonawej, o gęstości 8,96 g/cm
3
i temperaturze topnienia
1083°C. Można ją przerabiać plastycznie na zimno i na gorąco, ale w przypadku przeróbki na
zimno następuje utwardzenie metalu (w wyniku zgniotu), które usuwa się przez wyżarzenie
rekrystalizujące (w temp. 400-600°C). Przeróbkę plastyczną na gorąco przeprowadza się w
temp. 650-800°C. Cennymi własnościami miedzi są wysoka przewodność elektryczna i cieplna
oraz odporność na korozję.
8.1. Miedź technicznie czysta
Zawiera 0,01-1,0% zanieczyszczeń, zależnie od sposobu wytwarzania i oczyszczania. Dzieli
się na miedź surową (konwertorową lub anodową), rafinowaną oraz przetopioną (beztlenową,
tlenową i odtlenioną). Gatunki miedzi rafinowanej i przetopionej są w Polsce znormalizowane.
Oprócz tlenu wszystkie rodzaje miedzi technicznie czystej zawierają drobne ilości innych
pierwiastków (Bi, Pb, Sb, As, Fe, Ni, Sn, Zn, S i Ag), które również uważane są za
zanieczyszczenia (wyjątkiem jest srebro).
Miedź beztlenowa (zawierająca max 0,003% O) stosowana jest na elementy konstrukcyjne
lamp elektronowych, aparatury próżniowej, przewody elektrotechniczne itd. Pozostałe rodzaje
miedzi, zależnie od czystości, są stosowane do wyrobu różnych elementów konstrukcyjnych
oraz przerabianych plastycznie i odlewniczych stopów miedzi. Duże ilości miedzi zużywa się do
wytwarzania powłok galwanicznych na stali, zwykle jako podkładu pod powłoki niklowe lub
niklowo-chromowe.
8.2. Stopy miedzi
Stopami miedzi nazywa się stopy, w których metalem podstawowym (głównym
składnikiem) jest miedź, z wyjątkiem stopów zawierających złoto lub srebro, które uważa się za
stopy złota lub srebra, jeśli zawartość tych metali wynosi conajmniej 10%.
Ogólnie stopy miedzi, będące obecnie najbardziej rozpowszechnionymi materiałami
konstrukcyjnymi po stopach żelaza i stopach aluminium, dzielą się na:
a) stopy wstępne miedzi,
b) miedź stopową,
c) mosiądze,
d) miedzionikle,
e) brązy,
f) stopy oporowe miedzi.
W zależności od przeznaczenia stopy miedzi dzielą się na odlewnicze i do przeróbki
plastycznej.
Stopy wstępne miedzi są pomocniczymi, dwu- lub trzyskładnikowymi stopami,
wytwarzanymi w celu ułatwienia wprowadzenia dodatków stopowych lub technologicznych
(odtlenianie). Na przykład, stop zawierający 50% Al stosowany jest jako dodatek stopowy przy
produkcji brązów i mosiądzów aluminiowych, stop zawierający 12% P — jako dodatek stopowy
lub jako odtleniacz itd.
Miedź stopowa jest ogólną nazwą stopów do przeróbki plastycznej, zawierających nie więcej
niż 2% głównego dodatku stopowego. Znormalizowane gatunki obejmują miedź arsenową,
chromową, cynową, kadmową, manganową, niklową, siarkową, srebrową, tellurową i
cyrkonową. Miedź arsenowa, zawierająca 0,3
÷ 0,5% As, jest stosowana na elementy aparatury
chemicznej, miedź chromowa (0,4
÷ 1,2% Cr) - na elektrody zgrzewarek, miedź srebrowa (0,045
÷ 2% Ag) - na uzwojenia silników elektrycznych, luty, elektrody do spawania, druty wspierające
siatki lamp elektrycznych itd. (PN-79/H-87053).
Mosiądze są stopami miedzi, w których głównym składnikiem stopowym jest cynk w ilości
powyżej 2%. Dzielą się na mosiądze odlewnicze (tabl. 8.1) i do przeróbki plastycznej. Te
ostatnie, zgodnie z PN-92/H-87025, dzielą się na dwuskładnikowe, zawierające 0,4
÷ 40,5%
149
JW
cynku (gatunki M95, M90, M85, M80, M75, M70, M67, M65, M63 i M60, w symbolu M
oznacza mosiądz, a liczba - nominalną zawartość miedzi w %), i wieloskładnikowe. Mosiądze
wieloskładnikowe dzielą się z kolei na ołowiowe (tabl. 8.2) i bezołowiowe, zwane też
mosiądzami specjalnymi (tabl. 8.3).
Tablica 8.1
Skład chemiczny i własności mechaniczne odlewów piaskowych z mosiądzów odlewniczych (wg
PN-91/H-87026)
Gatunek mosiądzu Skład chemiczny, % (reszta cynk)
znak
cecha
Cu
Mn
Fe
inne
zanie-
czysz-
czenia,
max
R
m
MPa
A
5
%
CuZn43Mn4Pb3Fe MM47 48-50
3,0-4,0 0,5-1,2 2,0-3,5
Pb
1,0
360
10
CuZn37Mn4Fe1Sn1 MM54 54,5-57 3,0-4,2 0,7-1,6 0,6-1,5Sn
2,0 390
12
CuZn50Mn3Fe MM55 53-58
3,0-4,0 0,5-1,5
- 1,2
450
15
CuZn38Mn2Pb2 MM58 57-60
1,5-2,5
-
1,5-2,5Pb
1,8
250
15
CuZn38AI12Mn1Fe MA58 56-60
1,0-2,0 0,5-1,5 1,5-2,5AI
1,2 400
12
2,5-3,5 1,0-2,5 5,0-6,5 Al
CuZn26AI6Mn3Fe2Ni1,5
MA62
61-63,5
1,0-2,0
Ni
1,0
600
5
CuZn39Pb2 M059
57-60
-
-
1,0-2,5
Pb
1,8
250
12
CuZn38Pb2 M060
56-62
-
-
1,0-3,OPb
2,2
250
10
CuZn16Si4 MK80 79-81
-
-
3,0-4,5
Si
2,0
300
15
Tablica 8.2
Skład chemiczny i gęstość mosiądzów ołowiowych do przeróbki plastycznej
(wg PN-92/H-87025)
Gatunek mosiądzu
Skład chemiczny, %
(reszta cynk)
znak cecha Cu Pb
Gęstość, g/cm
3
CuZn37PbO,5
M063
62,0 ÷ 64,0
0,3 ÷ 0,7
8,5
CuZn36Pb1,5
M062
62,0 ÷ 64,0
0,7 ÷ 2,5
8,5
CuZn36Pb3
M061
60,0 ÷ 62,0
2,5 ÷ 3,5
8,5
CuZn38Pb1,5
M060
59,5 ÷ 61,5
1,0 ÷ 2,0
8,4
CuZn39Pb2
M059
58,5 ÷ 60,0
1,5 ÷ 2,5
8,4
CuZn40Pb2
M058
56,0 ÷ 60,0
1,0 ÷ 3,5
8,5
CuZn39Pb3
M058A
57,0 ÷ 59,0
2,5 ÷ 3,5
8,5
CuZn40Pb2
M058B
57,0 ÷ 59,0
1,5 ÷ 2,5
8,5
Grupę mosiądzów do przeróbki plastycznej stanowią mosiądze wysokoniklowe, zawierające
11
÷19,5% niklu. Osobną grupę znormalizowanych mosiądzów do przeróbki plastycznej (PN-
93/H-87027) stanowią mosiądze wysokoniklowe, zwane często (od zabarwienia) nowym
srebrem (tabl. 8.4).
Mosiądze odlewnicze cechuje rzadkopłynność i dobre wypełnianie form, tak że nadają się
one na odlewy piaskowe, kokilowe i pod ciśnieniem (temperatura odlewania waha się od 950 do
1100°C). Ich wadą jest skłonność cynku do parowania (temperatura wrzenia cynku wynosi
907°C) i wiążące się z tym duże straty tego pierwiastka. Dlatego mosiądz należy topić pod
przykryciem i w miarę możliwości bez przegrzewania. Inną wadą mosiądzów jest duży skurcz
odlewniczy (1,8
÷ 2%). Mosiądze stosowane są na wszelkiego rodzaju części maszyn, armatury,
silników itd. Z mosiądzu MM55 odlewa się m.in. śruby okrętowe, mosiądz MA58 jest
wykorzystywany przez przemysł lotniczy i okrętowy.
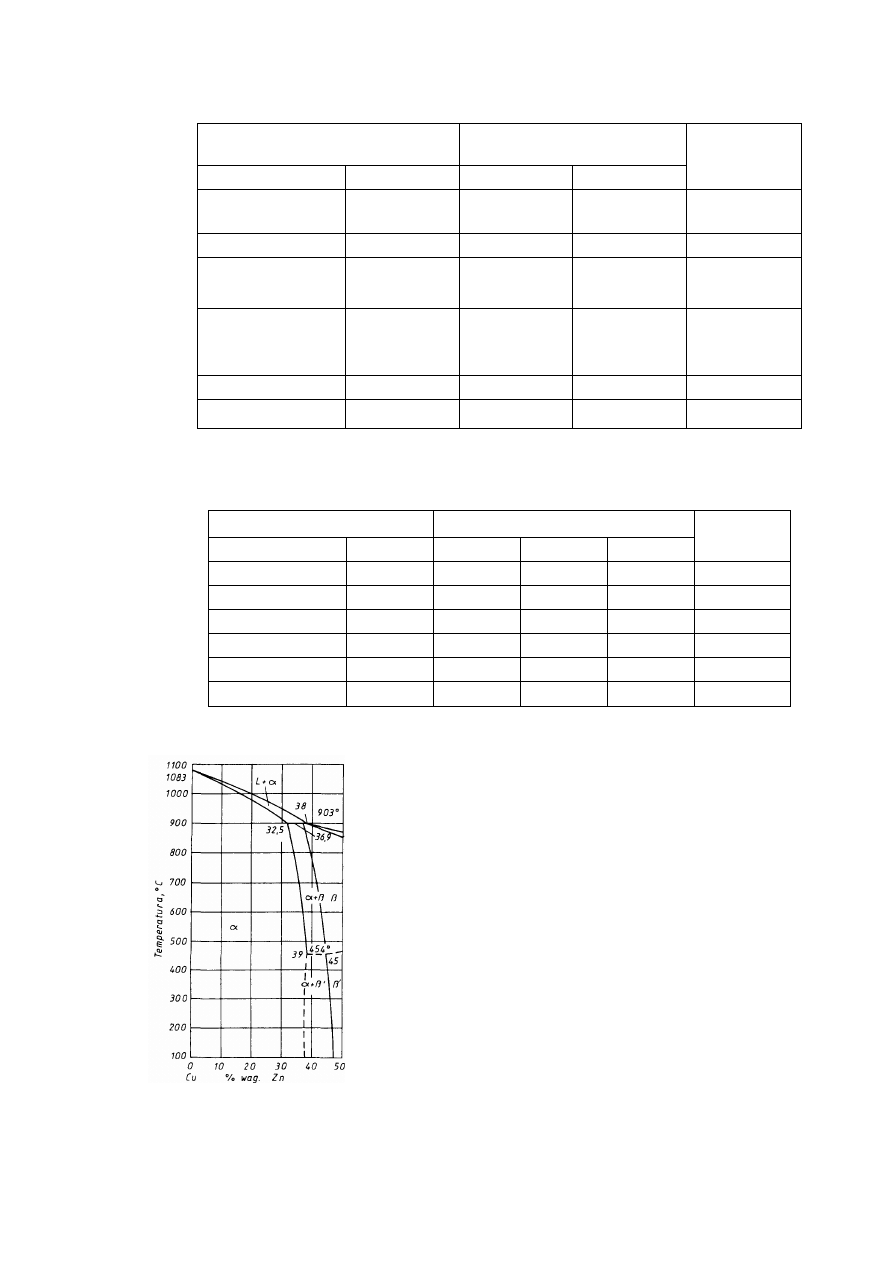
150
JW
Tablica 8.3
Skład chemiczny i gęstość mosiądzów specjalnych do przeróbki plastycznej (wg PN-92/H-87025)
Gatunek mosiądzu
Skład chemiczny, %
(reszta cynk)
znak cecha
Cu inne
Gęstość g/cm
3
CuZn28Sn1 MC70
70,0
÷
72,5 0,02
÷
0,06 As
0,9
÷
1,3 Sn
8,5
CuZn38Sn1 MC62
59,0
÷
62,0
0,5
÷
1,0 Sn
8,4
CuZn20AI2
MA77
76,0
÷
79,0
0,02
÷
0,06 As
1,8
÷
2,3Al
8,4
CuZn39AI1Fe1Mn1
MA58
56,0
÷
61,0
0,2
÷
1,5 Fe
0,2
÷
1,5 AI
0,2
÷
2,0 Mn
8,3
CuZn4Z0Mn1,5 MM58 57,0
÷
59,0
1,0
÷
2,0Mn
8,3
CuZn31Si1 MK68
66,0
÷
70,0 0,7
÷
1,3 Si
8,4
Tablica 8.4
Skład chemiczny i gęstość wysokoniklowych mosiądzów do przeróbki plastycznej
(wg PN-93/H-87027)
Gatunek mosiądzu Skład chemiczny, % (reszta cynk)
znak cecha
Cu Ni Pb
Gęstość
g/cm
3
CuNi18Zn27 MZN18
53,5
÷
56,5 17,0
÷
19,0
- 8,7
CuNi18Zn20 MZ20N18
60,0
÷
63,0 17,0
÷
19,0
- 8,8
CuNi15Zn21 MZN15
63,0
÷
66,0 14,0
÷
16,0
- 8,7
CuNi12Zn24 MZN12
63,0
÷
66,0 11,0
÷
13,0
- 8,7
CuNi18Zn19Pb11 MZN181 59,0
÷
63,0 17,0
÷
18,0
0,5
÷
1,5
8,8
CuNi10Zn28Pb1 MZN101 59,0
÷
63,0 9,0
÷
11,0
1,0
÷
2,0
8,6
Mosiądze dwuskładnikowe, czyli stopy miedzi z cynkiem, są najczęściej stosowanymi
stopami miedzi. Jak wynika z układu równowagi miedź-cynk
(rys. 8.1). stopy zawierające do 39% Zn mają strukturę
roztworu stałego
α cynku w miedzi, powyżej tej zawartości -
strukturę dwufazową, będącą mieszaniną roztworu stałego
α i
roztworu stałego
β (β' - uporządkowany roztwór stały β na
osnowie fazy międzymetalicznej CuZn).
Roztwór stały
α odznacza się dobrymi własnościami
wytrzymałościowymi, łatwo poddaje się przeróbce plastycznej
na zimno i jest odporny na działanie wielu ośrodków
korozyjnych. Roztwór
β jest bardziej twardy od roztworu
stałego
α, mniej jednak ciągliwy i mniej odporny na korozję.
W zasadzie cynk zwiększa wytrzymałość i plastyczność
stopu, ale maksymalną plastyczność ma stop zawierający około
30% Zn. Przekroczenie granicy obszaru jednofazowego
powoduje gwałtowne pogorszenie plastyczności. Z tego
powodu do przeróbki plastycznej na zimno (cienkie blachy i
druty) stosuje się raczej mosiądze o maksymalnej
plastyczności w temperaturze pokojowej, tj. mosiądze jedno-
fazowe
α zawierające około 30% Zn (rys. 8.2). Natomiast do
przeróbki plastycznej na gorąco lepiej nadają się mosiądze
zawierające więcej niż 32% Zn, gdyż w wysokiej temperaturze struktura takich stopów składa
Rys. 8.1. Część układu
równowagi miedź-cynk od
strony miedzi

151
JW
się z kryształów
α+ β (roztwór stały β w temp. 300 ÷ 700°C jest mniej wytrzymały i bardziej
plastyczny niż roztwór stały
α). Mikrostrukturę mosiądzu dwufazowego pokazano na rys. 8.3.
Mosiądze do przeróbki plastycznej są stosowane przeważnie w stanie utwardzonym przez
zgniot, dzięki czemu uzyskuje się znaczne podwyższenie ich wytrzymałości, przy pewnym
jednak pogorszeniu własności plastycznych. Z mosiądzów dwuskładnikowych wykonuje się
rurki włoskowate i chłodnicowe, wężownice, membrany manometrów, łuski amunicyjne, części
tłoczne i kute.
Mosiądze ołowiowe są przeznaczone na części obrabiane skrawaniem i dla przemysłu
zegarowego, mosiądze specjalne, zależnie od składu chemicznego - na rury wymienników ciepła
(MC70 i MA77), elementy aparatury, elementy ślizgowe (MA58 i MK68) itp.
Mosiądze wysokoniklowe są przeznaczone do wyrobów przedmiotów artystycznych, naczyń
stołowych, widelców, łyżek (jako imitacja srebra), części sprężynujących aparatów, elementów
głębokotłocznych. Gatunki zawierające ołów są przeznaczone na elementy obrabiane
skrawaniem, szczególnie dla mechaniki precyzyjnej i optyki.
Rys.8.2. Mikrostruktura mosiądzu
jednofazowego (30% Zn). Widoczne
kryształy roztworu stałego
α, częściowo
bliźniacze. Traw. roztworem NF
4
OH + H
2
O
2
;.
Powiększ. 200x
Rys. 8.3. Mikrostruktura mosiądzu dwufazo-
wego (40% Zn) po przeróbce plastycznej na
gorąco. Widoczne jasne kryształy roztworu
stałego
α i ciemne kryształy roztworu stałego
β'. Traw. odczynnikiem chromowym, 150x
Miedzionikle są przerabialnymi plastycznie stopami miedzi, w których głównym -
składnikiem stopowym jest nikiel w ilości powyżej 2% (tabl. 8.5).
Tablica 8.5
Skład chemiczny miedzionikli (wg PN-92/H-87052)
Gatunek miedzioniklu
Skład chemiczny, % (reszta miedź)
znak cecha Ni Mn Inne
CuNi25 MN25 24,0
÷
26,0
0,10
÷
0,50
-
CuNi9Sn2 MNC92 8,5
÷
10,5
-
1,8
÷
2,8 Sn
CuNi10FelMn MNŻ101
9,0
÷
11,0
0,5
÷
1,0 1,0
÷
2,0 Fe
CuNi30Mn1Fe MNM301 30,0
÷
32,0
0,5
÷
1,5 0,4
÷
1,0 Fe
CuNi30Fe2Mn
MNŻM3022
29,0
÷
32,0
1,5
÷
2,5 1,5
÷
2,5 Fe
CuNi44Mn1 MNM441 43,0
÷
45,0 0,5
÷
2,5
-
Miedzionikle cechuje bardzo dobra odporność na korozję i ścieranie oraz dobra plastyczność,
która umożliwia wytwarzanie z nich blach, taśm, prętów, rur i drutów. W szczególności
miedzionikiel MN25 przeznaczony jest na monety, MNC92 – na elementy sprężynujące,
połączenia wtykowe i przełączniki, MNŻ101, MNM301 i MNŻM – na rury wymienników ciepła
zwłaszcza w urządzeniach okrętowych, elementy aparatury i urządzeń klimatyzacyjnych.
MNM441 - na oporniki urządzeń pomiarowych i elementy elektroniczne. Gęstość wszystkich
miedzionikli wynosi 8,9 g/cm
3
.
Brązy są stopami miedzi, w których głównym składnikiem stopowym (ponad 2% jest cyna,
aluminium, krzem, beryl, ołów i inne, z wyjątkiem cynku i niklu. W zależności od głównego
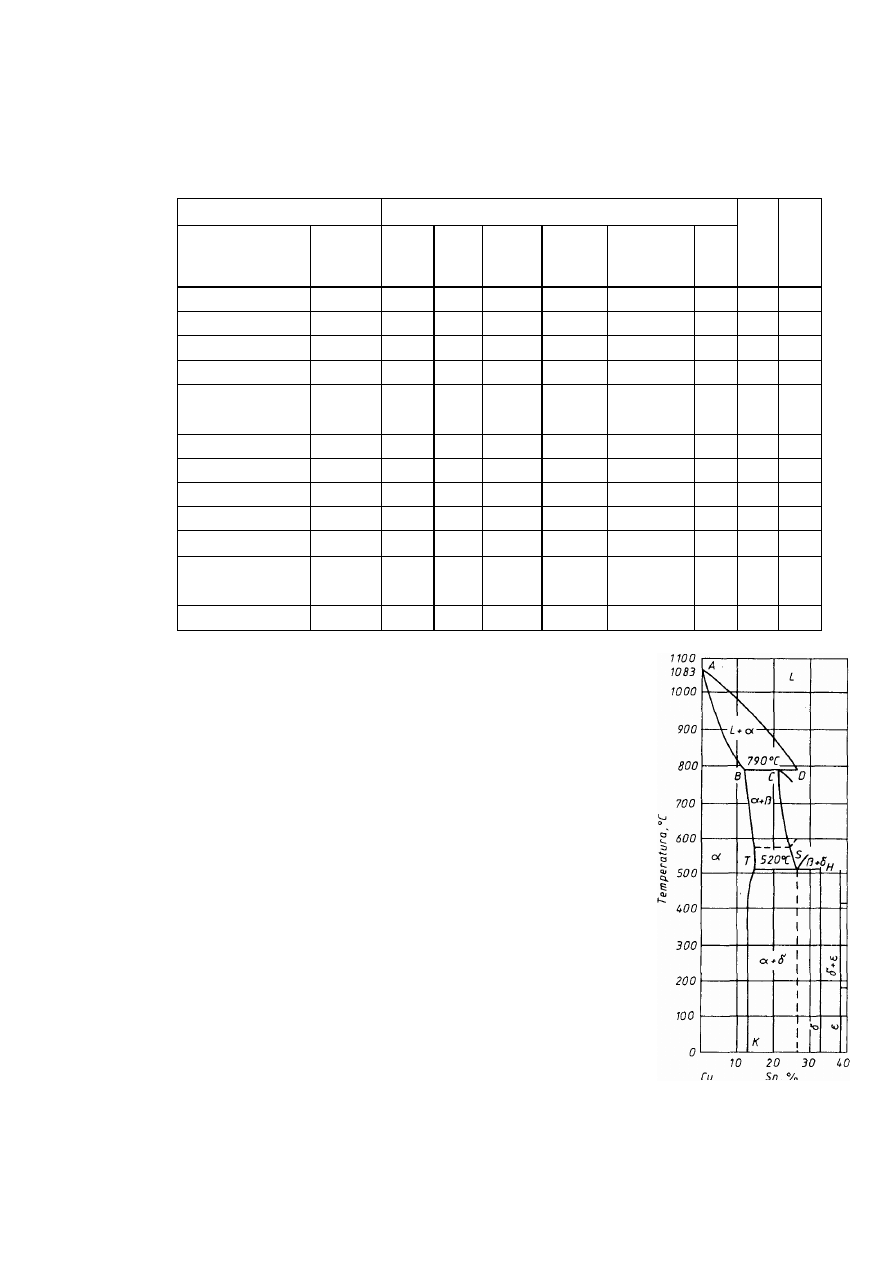
152
JW
składnika stopowego (aluminium, beryl, cyna, krzem. kobalt, ołów, antymon, mangan, tytan)
noszą nazwę brązów aluminiowych, berylowych itd. Podobnie jak mosiądze, dzielą się na
odlewnicze (tabl. 8.6) i do przeróbki plastycznej.
Tablica 8.6
Skład chemiczny i własności mechaniczne odlewów piaskowych z brązów
odlewniczych (wg PN-91/H-87026)
Gatunek brązu Skład chemiczny, % (reszta miedź)
znak
cecha
Sn
Zn
Fe
Mn
inne
Zanie-
czysz-
czenia
max
R
m
MPa
A
5
%
CuSn10 B10
9
÷
11
- - -
- 1,0
240
12
CuSn10P B101
9
÷
11
- - -
0,5
÷
1,0 P 0,8 220 3
CuSn10Zn2 B102
9
÷
11 1
÷
3
- - - 1,0
240
10
CuSn10Pb10 B1010
9
÷
11
- - -
8,5
÷
11 Pb 0,8 180 7
CuSn8Pb15Ni
B815
7,3
÷
9
-
-
-
13,5
÷
17 Pb
0,5
÷
1,5 Ni
1,2
150
7
CuSn5Zn5Pb5 B555 4
÷
6
4
÷
6
- -
4
÷
6 Pb
1,0 200 13
CuSn4Zn7Pb6 B476 3
÷
5
6
÷
8
- -
5
÷
7 Pb
1,0 200 15
CuSn5Pb20 B520
4
÷
6
- - -
18
÷
23Pb 1,2 150 5
CuAI9Fe3 BA93 - -
2
÷
4
-
8
÷
10AI
1,0 500 13
CuAI10Fe3Mn2 BA1032
-
- 2
÷
4 1
÷
2 8,5
÷
10,5AI 0,8 500 15
CuAI10Fe4Ni4
BA1044
-
-
3,6
÷
5,7
-
9
÷
11,2 AI
3,5
÷
5,5 Ni
1,5
590
5
CuSi3Zn3Mn BK331 - 3
÷
5 0,5
÷
1,2 0,5
÷
1,5
3
÷
4 Si
1,0 280 8
Brązy cynowe należą do najstarszych znanych stopów i już w
starożytności stosowane były do wyrobu mieczów, ozdób,
naczyń i przedmiotów codziennego użytku.
Na rysunku 8.4 przedstawiono część układu równowagi
miedź-cyna. Jak widać w stopach zawierających do około 14% Sn
występuje roztwór stały
α cyny w miedzi, powyżej tej zawartości
- mieszanina roztworu stałego
α i fazy δ (faza elektronowa).
Praktycznie jednak struktura lanych stopów miedzi z cyną ze
względu na wzmożoną likwację znacznie odbiega od stanu
równowagi. Przy zawartości 5
÷ 6% Sn składa się ona z
niejednorodnego roztworu stałego
α, mającego jak każdy metal
lany budowę dendrytyczną. Przy większej zawartości cyny na tle
niejednorodnego roztworu występuje eutektoid (
α + δ ) mający
niejednorodną budowę (rys. 8.5 i 8.6). Obecność kruchej fazy
δ
wyklucza możliwość walcowania, dlatego brązy o większej
zawartości cyny stosuje się wyłącznie na odlewy.
Brązy cynowe wykazują wyjątkowo mały skurcz odlewniczy,
co umożliwia wykonywanie z nich odlewów o skomplikowanych
kształtach (np. pomników). Jednak wskutek znacznej różnicy
temperatur początku i końca krzepnięcia, brązy te mają małą
rzadkopłynność i nie tworzą skupionej jamy usadowej. Rzadko
więc można uzyskać odlew o dobrej ścisłości (bez rzadzizn i
porów).
Dzięki dużej odporności chemicznej, zwłaszcza na działanie
czynników atmosferycznych, dobrej wytrzymałości i odporności na ścieranie, z cynowych
Rys. 8.4. Część układu
równowagi miedź-cyna od
strony miedzi
153
JW
brązów odlewniczych wytwarza się wszelkiego rodzaju armaturę wodną i parową, panewki do
łożysk ślizgowych, odlewy artystyczne i inne o skomplikowanym kształcie (tabl. 8.7). Trzeba
wspomnieć, że obecność wtrąceń twardego eutektoidu zapewnia dużą odporność na ścieranie i
dlatego brąz zawierający ponad 10% Sn jest jednym z najlepszych materiałów przeciwciernych,
znajdują zastosowanie jako stop łożyskowy. Brązy cynowe przerabialne plastycznie (tabl. 8.8)
mają także dobrą wytrzymałość, są sprężyste oraz odporne na korozję i ścieranie (ze wzrostem
zawartości cyny w brązie następuje wzrost tych własności. Wszystkie gatunki są dobrze
skrawalne, podatne lutowanie i spawanie oraz przeróbkę plastyczną na zimno.
Tablica 8.7
Przykłady zastosowania brązów odlewniczych (wg PN-91/H-87026)
Cecha Przykłady zastosowania
B10
silnie obciążone części maszyn, jak łożyska, panewki i napędy oraz osprzęt
parowy, wodny; odporny na działanie niektórych kwasów
B101
wysoko obciążone, szybkoobrotowe, źle smarowane i narażone na korozję
łożyska, części maszyn oraz armatura chemiczna
B102
wysoko obciążone i narażone na korozję części maszyn w przemyśle
B1010
łożyska i części trące maszyn pracujących przy dużych naciskach i
B815
panwie ślizgowe pracujące przy znacznych naciskach, pierścienie
B555
części maszyn, osprzętu aparatury pojazdów, silników i traktorów
podlegające korozji w środowisku wodnym, ścieranie wytrzymujące
ciśnienie do 2,5 MPa
B476
części maszyn, tuleje i łożyska pracujące przy obciążeniach statycznych i
normalnej temperaturze, armatura wodna wytrzymująca ciśnienie 2,5 MPa
B520
łożyska i części maszyn narażone na ścieranie przy dużej szybkości i
małych naciskach
BA1032
BA93
BA1044
silnie obciążone części maszyn, silników oraz osprzętu i aparatury
narażone na korozję i ścieranie przy równoczesnym obciążeniu
mechanicznym w przemyśle komunikacyjnym, okrętowym, lotniczym,
chemicznym itp.
BK331 części maszyn i osprzętu (łożyska, elementy napędów, pompy) narażone na
korozję, zmienne obciążenia i złe smarowanie
Tablica 8.8
Skład chemiczny i gęstość przerabialnych plastycznie brązów cynowych (wg PN-92/H-87050)
Gatunek brązu Skład chemiczny, % (reszta miedź)
znak
cecha
Sn Zn Pb P
Gęstość
g/cm
3
CuSn2 B2
1,0
÷ 2,5
- -
0,01
÷ 0,35
8,9
CuSn4 B4
3,5
÷ 4,5
- -
0,01
÷ 0,35
8,8
CuSn6 B6
5,5
÷ 7,0
-
-
0,01
÷ 0,35
8,8
CuSn8 B8
7,5
÷ 8,5
- -
0,01
÷ 0,35
8,8
CuSn4Pb4Zn3 B443 3,5
÷ 4,5 1,5 ÷ 4,5 3,5 ÷ 4,5 0,01 ÷ 0,50
8,8
Z brązu B2 wytwarza się śruby i giętkie węże, z brązu B4 - śruby, sprężyny manometryczne,
elementy przyrządów kontrolno-pomiarowych i połączenia wtykowe z brązów B6 i B8 -
sprężyny, membrany, sita papiernicze, rurki manometryczne elementy przyrządów, z brązu B443
- elementy ślizgowe. Brązy o zawartości 4
÷6% Sn ze względu na dobre własności plastyczne i
piękne zabarwienie znalazły zastosowanie m.in. do wyrobu monet i medali. Pod wpływem
przeróbki plastycznej na zimno wzrasta bardzo ich twardość, co wpływa korzystnie na
zwiększenie odporności na ścieranie. Brąz o zawartości 10% Sn jest stosowany do wyrobu kół
zębatych.
154
JW
Z brązów cynowych wieloskładnikowych trzeba wymienić stopy z cynkiem (5
÷ 10% Sn,
2
÷ 6% Zn), zwane dawniej spiżami. Mają one nieco mniejszą wytrzymałość i odporność na
korozję niż brązy dwuskładnikowe, ale lepsze własności odlewnicze, co umożliwia
wykonywanie z nich skomplikowanych odlewów cienkościennych (części maszyn, armatura,
okucia budowlane, wyroby artystyczne).
Rys. 8.5. Mikrostruktura brązu cynowego
(10% Sn) w postaci lanej. Widoczna budowa
dendrytyczna. Traw. roztworem NH
4
OH +
H
2
O
2
. Powiększ. 100x
Rys. 8.6. Mikrostruktura brązu cynowego(10%
Sn) w postaci lanej. Widoczna faza a w postaci
dendrytów (bogate w miedź środki dendrytów
są ciemne, bogate w cynę brzegi tych
dendrytów są jasne) i szare, kropkowane
wydzielenia eutektoidu a + 8 . Traw. roztwo-
rem NH
4
OH + H
2
O
2
. Powiększ. 500x
Brązy aluminiowe produkowane są zarówno jako odlewnicze (tabl. 8.6), jak przerabialne
plastycznie (tabl. 8.9). Dzielą się na dwuskładnikowe, zawierające 4
÷ 8% Al, i wieloskładniko-
we, zawierające zwykle żelazo i mangan, żelazo i nikiel i inne dodatki. Główne ich cechy to
wysoka wytrzymałość i plastyczność zarówno w temperaturze otoczenia, jak i w temperaturach
podwyższonych, oraz dobra odporność na ścieranie i korozję (m.in. wody morskiej).
Tablica 8.9
Skład chemiczny i gęstość przerabialnych plastycznie brązów aluminiowych (wg PN-92/H-87051)
Gatunek brązu aluminiowego
Skład chemiczny, % (reszta miedź)
znak cecha
Al Fe Inne
Gęstość
g/cm
3
CuAl5As BA5
4,0
÷
6,0
-
0,1
÷
0,4 As
8,2
CuAl 8
BA8
7,5
÷
9,0
- - 7,8
CuAl8Fe3 BA83
6,5
÷
8,5
1,5
÷
3,3
- 7,7
CuAl10Fe3Mn2 BA1032 8,5
÷
11,0
2,0
÷
4,0
1,5
÷
3,5Mn
7,6
CuAl10Ni5Fe4 BA1054
8,5
÷
11,0 2,0
÷
5,0 4,0
÷
6,0 Ni
7,6
W postaci lanej brązy aluminiowe stosuje się na silnie obciążone części maszyn, silników
oraz części osprzętu i aparatury, narażone na korozję i ścieranie przy równoczesnym obciążeniu
mechanicznym. Orientacyjne własności i przykładowe zastosowanie brązów aluminiowych
przerabialnych plastycznie podano w tabl. 8.10. Brązy aluminiowe podlegają ulepszaniu
cieplnemu (hartowanie z temp. ok. 900°C, odpuszczanie w temp. 300
÷ 450°C).
Mikrostrukturę brązu aluminiowego w postaci lanej pokazano na rys. 8.7.
Z pozostałych brązów znormalizowane są: odlewniczy brąz krzemowy BK331 (tabl. 8.6)
oraz specjalne stopy miedzi do przeróbki plastycznej, w tym brązy krzemowe i berylowe (tabl.
8.11). Orientacyjne własności tych stopów i ich zastosowanie podano w tabl. 8.12.
Brązy berylowe podlegają obróbce cieplnej (umocnieniu wydzieleniowemu), złożonej z
przesycania z temperatury 800°C i starzenia w temperaturze 350°C. Wadą ich jest stosunkowo
wysoki koszt berylu.
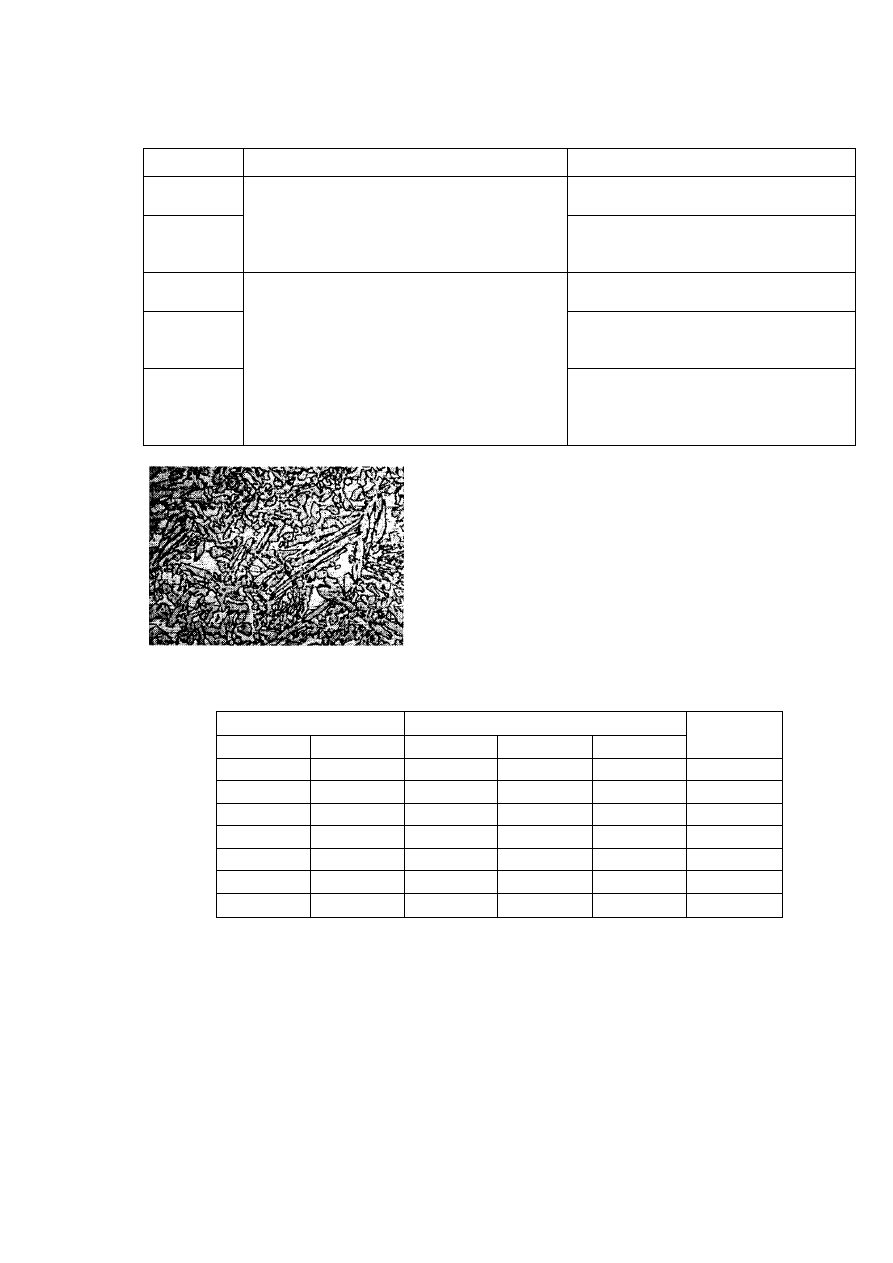
155
JW
Tablica 8.10
Orientacyjne własności i przykłady zastosowania brązów aluminiowych do przeróbki plastycznej
(wg PN-92/H-87051
)
Cecha brązu
Orientacyjne własności
Przykłady zastosowania
BA5
elementy pracujące w wodzie morskiej,
części aparatury chemicznej
BA8
duża odporność na korozję, dobra podatność
na obróbkę plastyczną na zimno; BA5 jest
szczególnie odporny na działanie gorących
roztworów soli, BA8- na działanie kwasu
siarkowego i octowego
części aparatury chemicznej
BA83
dna sitowe wymienników ciepła, części
aparatury chemicznej
BA 1032
części aparatury kontrolno-pomiarowej i
chemicznej, wały, śruby, części narażone
na ścieranie
BA1054
wysokie własności wytrzymałościowe
(również w temperaturach podwyższonych),
dobra odporność na korozję szczególnie w
roztworach kwaśnych, wysoka odporność na
erozję i kawitację wysoka odporność na
obciążenia zmienne, dobra odporność
na ścieranie, dobra podatność na obróbkę
plastyczną na zimno
dna sitowe wymienników ciepła, wały,
śruby części narażone na ścieranie, części
urządzeń hydraulicznych, gniazda
zaworów, koła zębate
Rys. 8.7. Mikrostruktura brązu aluminiowego (9% Al w
postaci lanej (szybko chłodzonego). Widoczne jasne
kryształy roztworu stałego i nieco ciemniejsze iglaste
kryształy roztworu stałego
β. Traw. roztworem NH
4
OH +
H
2
O
2
. Powiększ. 200x
Tablica 8.11
Skład chemiczny i gęstość specjalnych stopów miedzi
do przeróbki plastycznej
(wg PN-92/H-87060
)
Gatunek Skład chemiczny, % (reszta Cu)
znak cecha Si Be inne
Gęstość
g/cm
3
CuSi1 BK1
0,8-2,0 -
-
8,5
CuSi3Mn1 BK31
2,7-3,5
-
1,0-1,5
Mn
8,5
CuBe1,7 BB1,7
-
1,6-1,8
-
8,4
CuBe2 BB2
-
1,8-2,1
-
8,3
CuBe2Pb BB21
-
1,8-2,1 0,2-0,6
Pb
8,3
CuCo2Be BC2
-
0,4-0,7 2,0-2,8
Co
8,8
CuNi2Si BN2
0,5-0,8
-
1,6-2,5
Ni 8,8
Brązy ołowiowe zawierają do 26% ołowiu oraz najczęściej mniejsze dodatki cyny, niklu,
manganu itd. Odznaczają się dobrą odpornością na korozję dobrą obrabialnością, a przede
wszystkim dobrą odpornością na ścieranie, w związku z czym wykonuje się z nich tulejki i
panewki do silnie obciążonych maszyn. Ze względu na brak rozpuszczalności ołowiu w miedzi,
w stanie ciekłym brązy ołowiowe maja skłonność do likwacji składników. Warunkiem dobrych
własności przeciwciernych stopu jest równomierne rozmieszczenie ziarn ołowiu i miedzi.
Brązy manganowe są odporne na działanie wysokich temperatur, w których zachowują dużą
twardość i ciągliwość. Znalazły zastosowanie w budowie maszyn parowych, turbin i silników
spalinowych, przemyśle elektrotechnicznym (sprężyny, kontakty, szczotki) itd. Stop o
zawartości 85% Cu, 12% Mn i 3% Ni nosi nazwę manganinu. Cechuje go wysoki opór
elektryczny.
156
JW
Tablica 8.12
Orientacyjne własności i przykłady zastosowania specjalnych stopów miedzi do
przeróbki plastycznej (wg PN-92/H-87060)
Cecha Orientacyjne
własności Przykłady zastosowania
BK1
śruby, szczególnie w
środowisku morskim
BK31
wysokie własności wytrzymałościowe, duża
odporność na korozję, dobra podatność na
przeróbkę plastyczną na zimno; BK31 - duża
podatność na spawanie
elementy konstrukcji
spawanych
BB1,7
BB2
BB21
BC2
bardzo wysokie własności wytrzymałościowe i
sprężyste, bardzo duża odporność na ścieranie i
korozję, brak skłonności do iskrzenia, średnie
przewodnictwo elektryczne, podatność na
przeróbkę plastyczną na zimno, szczególnie w
stanie przesyconym; BB21 - podwyższona
skrawalność
sprężyny, elementy
sprężynujące i narażone
na ścieranie, narzędzia
nieiskrzące
BN2
wysokie własności wytrzymałościowe, średnie
przewodnictwo elektryczne, podatność na
przeróbkę plastyczną na zimno
śruby, osprzęt
Stopy oporowe miedzi są stopami z niklem (do 41%), cynkiem (do 28%), manganem (do
13%), aluminium (do 3,6%) i żelazem (do 1,5%). Charakteryzują się stosukowo wysokim
oporem elektrycznym (rezystywnością) i małym współczynnikiem cieplnym oporu oraz
stabilnością obu tych własności, dzięki czemu są stosowane do wyrobu elektrycznych oporników
pomiarowych i rozruszników. Stopy te mają strukturę jednofazową. Najbardziej znane, to
omówione wyżej konstantan, nikielina, manganin i nowe srebro (27% Zn, 18% Ni) oraz inmet
albo nowokonstantan (12% Mn, 3% Al, 1% Fe).
9. Aluminium i stopy aluminium
Aluminium jest pierwiastkiem metalicznym, krystalizującym w układzie regularnym
płaskocentrycznym Al, o gęstości 2,7 g/cm
3
, temperaturze topnienia 660°C i temperaturze
wrzenia 2450°C. Cechuje go dobra przewodność cieplna i elektryczna (ta ostatnia wynosi 66%
przewodności elektrycznej miedzi), duży współczynnik rozszerzalności cieplnej (23,6 •10
-6
1/°C) i dość dobra odporność na korozję atmosferyczną (aluminium samorzutnie tworzy na
powierzchni cienką, ale bardzo szczelną i ściśle przylegającą warstewkę tlenku aluminium, która
zabezpiecza go przed dalszym utlenianiem) oraz na działanie wody, niektórych kwasów
organicznych. dwutlenku siarki i wielu innych związków chemicznych.
Zwiększenie odporności korozyjnej aluminium (a także jego stopów) uzyskuje się przez
sztuczne wytwarzanie powłoki tlenkowej bądź chemicznie (alodynowanie) bądź elektrochemi-
cznie (eloksalacja). Obecnie proces eloksalacji jest powszechnie stosowany w budownictwie
(blachy osłonowe, ramy okienne i drzwiowe), w przemyśle samochodowym, przy wyrobie
naczyń i sprzętu gospodarstwa domowego Warstewka tlenków Al
2
O
3
ma grubość 5
÷30 µm, a
jej porowatość umożliwia barwienie na dowolny kolor.
9.1. Aluminium technicznie czyste
Zawiera 0,01-1,0% zanieczyszczeń (głównie żelazo, krzem, miedź, cynk i tytan. w
mniejszych ilościach Mg, Mn, Cr, V, Pb i Ni), zależnie od sposobu oczyszczania. W Polsce,
zgodnie z PN-79/H-82160, produkowane są dwa rodzaje aluminium technicznie czystego:
rafinowane, o zawartości 99,995, 99,9 oraz 99,95% Al, i hutnicze, o zawartości 99,8, 99,7, 99,5 i
99,0% Al. Aluminium rafinowane stosuje się przede wszystkim do budowy specjalnej aparatury
chemicznej oraz na wyroby dla elektrotechniki i elektroniki, aluminium hutnicze - do produkcji
kabli i przewodów elektrycznych, do platerowania, budowy aparatury chemicznej, farb i
produkcji stopów aluminium. Ostatni gatunek aluminium hutniczego służy ponadto do wyrobu
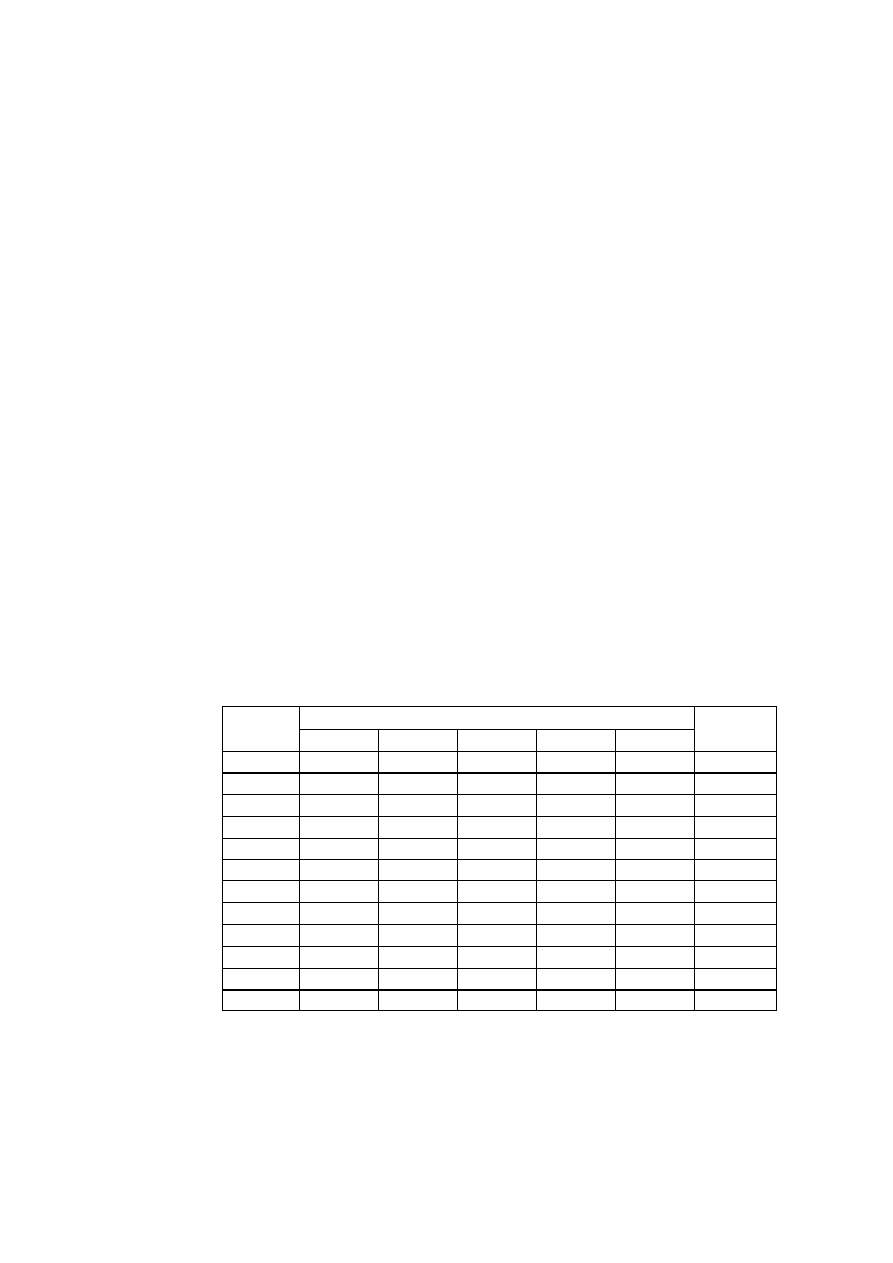
157
JW
naczyń kuchennych i przedmiotów codziennego użytku. Przykłady oznaczania gatunków
aluminium technicznego: Al 99,99 R (rafinowane), Al 99,8 H (hutnicze), 99,7 HE (hutnicze dla
elektrotechniki).
Aluminium technicznie czyste jest metalem bardzo plastycznym, ale ma niewielką
wytrzymałość, w związku z czym jego zastosowanie w budowie maszyn jest bardzo
ograniczone.
9.2. Stopy aluminium
Stopy aluminium są obecnie po stopach żelaza najbardziej rozpowszechnionymi materiałami
konstrukcyjnymi, znajdującymi zastosowanie we wszystkich gałęziach przemysłu. Szczególnie
ważnym tworzywem są w budowie samolotów i statków ulicznych, przede wszystkim dzięki
wysokim wskaźnikom własności wytrzymałościowych odniesionych do gęstości (wytrzymałości
właściwej). Na przykład w samolocie „Caravelle" różne stopy aluminium stanowią 70%
materiałów konstrukcyjnych, stale - 26%, a inne tworzywa tylko 4%.
Ogólnie stopy aluminium dzielą się na stopy odlewnicze i stopy do przeróbki plastycznej.
Obie grupy są w Polsce znormalizowane (tabl. 9.1 i 9.2).
9.2.1. Stopy aluminium odlewnicze
Ta grupa stopów obejmuje 12 znormalizowanych gatunków. Cecha każdego stopu składa się
z litery A (stop aluminium), z litery K, G lub M (odpowiednio krzemowy, magnezowy lub
miedziowy) oraz liczby określającej zawartość procentową głównego lub dwóch głównych
składników stopowych. Wśród odlewniczych stopów aluminium można wyróżnić stopy
dwuskładnikowe (Al-Si, Al-Cu i Al-Mg) oraz wieloskładnikowe (Al-Si-Cu, Al-Si-Ms,, Al-Si-
Cu-Mg-Ni, Al-Cu-Ni i Al-Cu-Ni-Mg).
Stopy aluminium z krzemem jako głównym składnikiem stopowym noszą nazwę siluminów.
Pod względem zawartości krzemu siluminy dzielą się na podeutektyczne 10% Si), eutektyczne
(10
÷13% Si) i nadeutektyczne (17 ÷ 30% Si).
Tablica 9.1
Skład chemiczny i gęstość odlewniczych stopów aluminium (wg PN-76/H-88027)
Skład chemiczny, % (reszta aluminium)
Cecha
stopu
Si Cu Mg Mn
inne
Gęstość
g/cm
3
AK20 20,0-23,0
1,1-1,5 0,5-0,9 0,1-0,3
0,8-1,1
Ni 2,60
AK12 11,5-13,0
0,8-1,5 0,8-1,5
- 0,8-1,3
Ni 2,72
AK11
10,0-13,0
- - - -
2,65
AK9 8,5-10,5 - 0,2-0,4 0,25-0,5 -
2,65
AK7 6,0-8,0 -
0,2-0,4
0,1-0,5
-
2,68
AK64 5,0-7,0 3,0-5,0 - 0,3-0,6 -
2,77
AK52 4,0-6,0 1,5-3,5 0,2-0,8 0,2-0,8
-
2,70
AK51 4,5-5,5 1,0-1,5 0,35-0,6 0,2+0,5 -
2,67
AG10 - - 9,0-11,0 - - 2,55
AG51
0,8-1,3 - 4,0-6,0
0,1-0,4 - 2,60
AM5
-
4,0-5,0
- - -
2,80
AM4 — 4,2-5,0
0,15-0,4
-
0,15-0,30 2,80
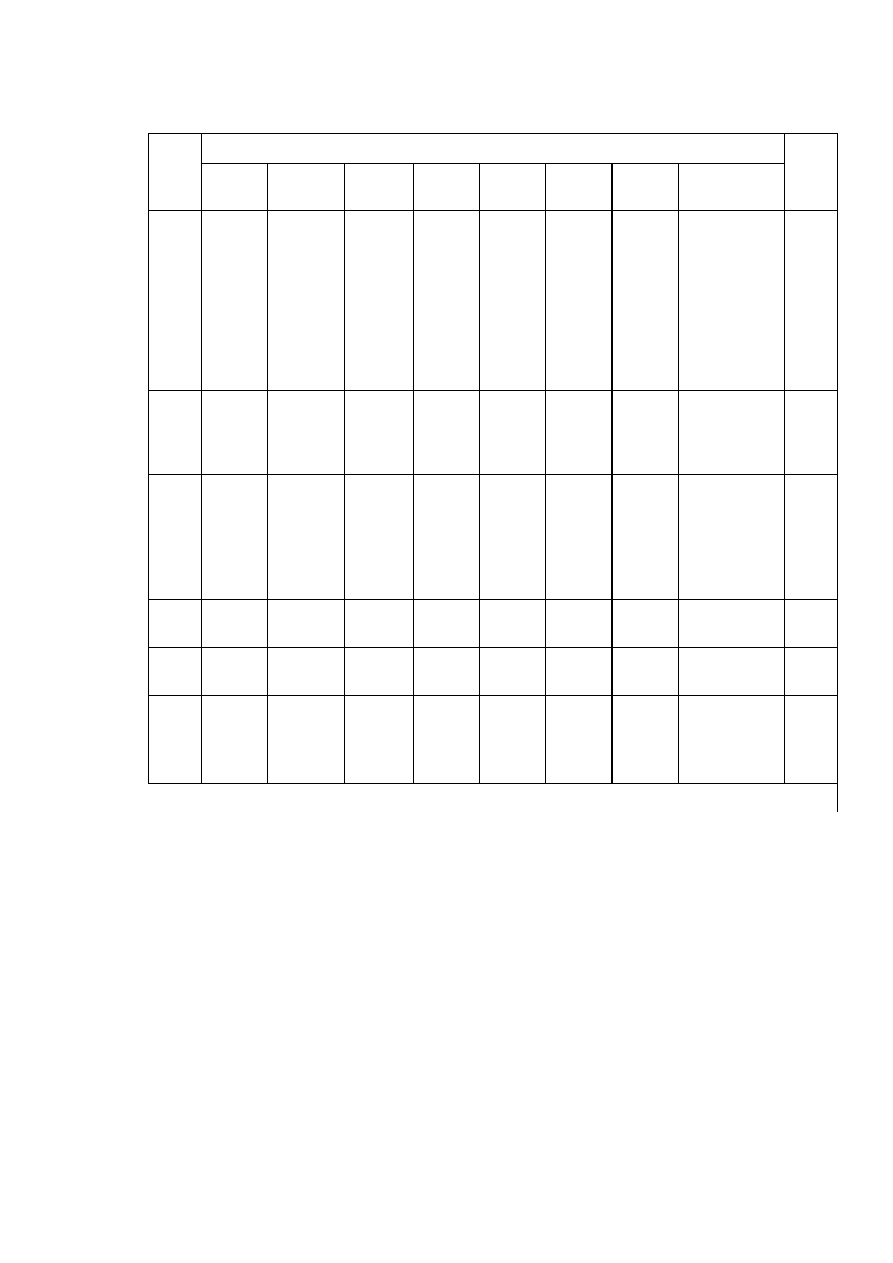
158
JW
Tablica 9.2
Skład chemiczny i gęstość stopów aluminium do przeróbki plastycznej (wg PN-79/H-88026)
Cecha Skład chemiczny, % (reszta aluminium)
*
Gęs
stopu
tość
Cu Mg Mn Si Ni Fe Zn inne g/cm
3
PA43 — 0,7
÷1,2
—
- - - 2,69
PA15 - 0,9
÷1,3
0,4
÷0,7
-
-
0,4+0,7
-
-
2,74
PA16 - 0,2
÷0,8 0,3÷0,8
-
-
-
- -
2,72
PA2 - 1,7
÷2,6
do 0,6
-
-
-
- -
2,68
PA11 - 2,7
÷3,6
do 0,6
-
-
-
- -
2,66
PA13 - 4,0
÷4,9 0,4÷1,0
-
-
-
-
0,05
÷0,25 Cr
2,66
PA20 - 4,3
÷5,8 0,2÷0,6
-
-
-
- -
2,64
PA5 - 0,8
÷1,5 1,0÷1,5
-
-
-
- -
2,70
PA1 -
- 1,0
÷1,5
- - - -
- 2,73
PA38 - 0,4
÷0,9
-
0,3
÷0,7
- - -
- 2,69
PA4 - 0,7
÷1,5 0,2-1,0 0,7÷1,5
-
-
- -
2,70
PA45 0,15
÷0,4 0,8÷1,2
-
0,4
÷0,8
-
-
-
0,15
÷0,35 Cr
2,71
PA10 0,1
÷0,5 0,45÷0,9 0,15÷0,3 0,5÷1,2
- - -
- 2,72
PA6 3,8
÷4,8 0,4÷1,0 0,4÷1,0
- - - -
- 2,80
PA7 3,8
÷4,9
1,2
÷1,8
0,4
÷0,9
-
-
-
-
-
2,77
PA21 3,8
÷4,5 0,4÷0,8 0,4÷0,8
-
-
-
- -
2,80
PA23 3,8
÷4,5 1,2÷1,6 0,3÷0,7
-
-
-
- -
2,77
PA24 2,0
÷3,0 0,2÷0,5
-
-
-
-
- -
2,75
PA25 3,9
÷4,5 0,15÷0,3 0,3÷0,5
- - - -
- 2,7-7
PA29 1,9
÷2,5 1,4÷1,8
—
0,5
÷1,2 0,8÷1,3 0,8÷1,3
- -
2,80
PA30 1,9
÷2,7 1,2÷1,8
- -
0,8
÷1,4 0,8÷1,4
-
0,02
÷0,10 Ti
2,80
PA31 1,8
÷2,6 0,4÷0,8 0,4÷0,8 0,7÷1,2
— — -
- 2,80
PA33 3,9
÷4,8 0,4÷1,0 0,4÷1,0 0,6÷1,2
- - -
- 2,80
PA9 1,4
÷2,0 1,8÷2,8 0,2÷0,6
— — —
5,0
÷7,0 0,1÷0,25 Cr
2,80
PA47 - 1,15
÷1,4 0,15÷0,4
-
-
-
4,3
÷5,0 0,1÷0,25 Cr
2,75
0,1
÷0,2 Zr
0,10
÷0,13 Ti
*
Maksymalna ilość zanieczyszczeń — 0,15%.
Podstawą tego podziału jest struktura stopów wynikająca z układu równowagi Al-Si (rys. 9.1).Niektóre siluminy
oprócz krzemu zawierają niewielkie ilości miedzi i magnezu oraz niekiedy niklu, manganu i tytanu.
Stopy aluminium-krzem tworzą eutektykę o zawartości 11,6% Si, złożoną z kryształów
roztworu stałego granicznego a krzemu w aluminium i roztworu stałego granicznego
β
aluminium w krzemie. W temperaturze eutektycznej (577°C) rozpuszczalność krzemu w
aluminium wynosi 1,65%, w temperaturze 300°C ok. 0,5%. Natomiast rozpuszczalność
aluminium w krzemie nawet w temperaturze eutektycznej jest tak mała, że się jej nie określa, a
w wielu publikacjach fazę
β traktuje się jako czysty krzem.
Siluminy charakteryzują się doskonałymi własnościami odlewniczymi (mały skurcz liniowy,
dobra lejność, mała skłonność do pękania na gorąco) i stosunkowo dobrymi własnościami
mechanicznymi oraz dostateczną odpornością na korozję. Z tego względu są one szeroko
stosowane na odlewy tłoków silników spalinowych AK 12), głowic cylindrów silników
spalinowych (AK51, AK52), części maszyn (AK7, AK9, AK11, AK51, AK52 i AK64),
armatury okrętowej (AK11) itd.
Siluminy praktycznie nie podlegają obróbce cieplnej, a ich własności mechaniczne polepsza
się przez specjalne zabiegi w stanie ciekłym, zwane modyfikowaniem.
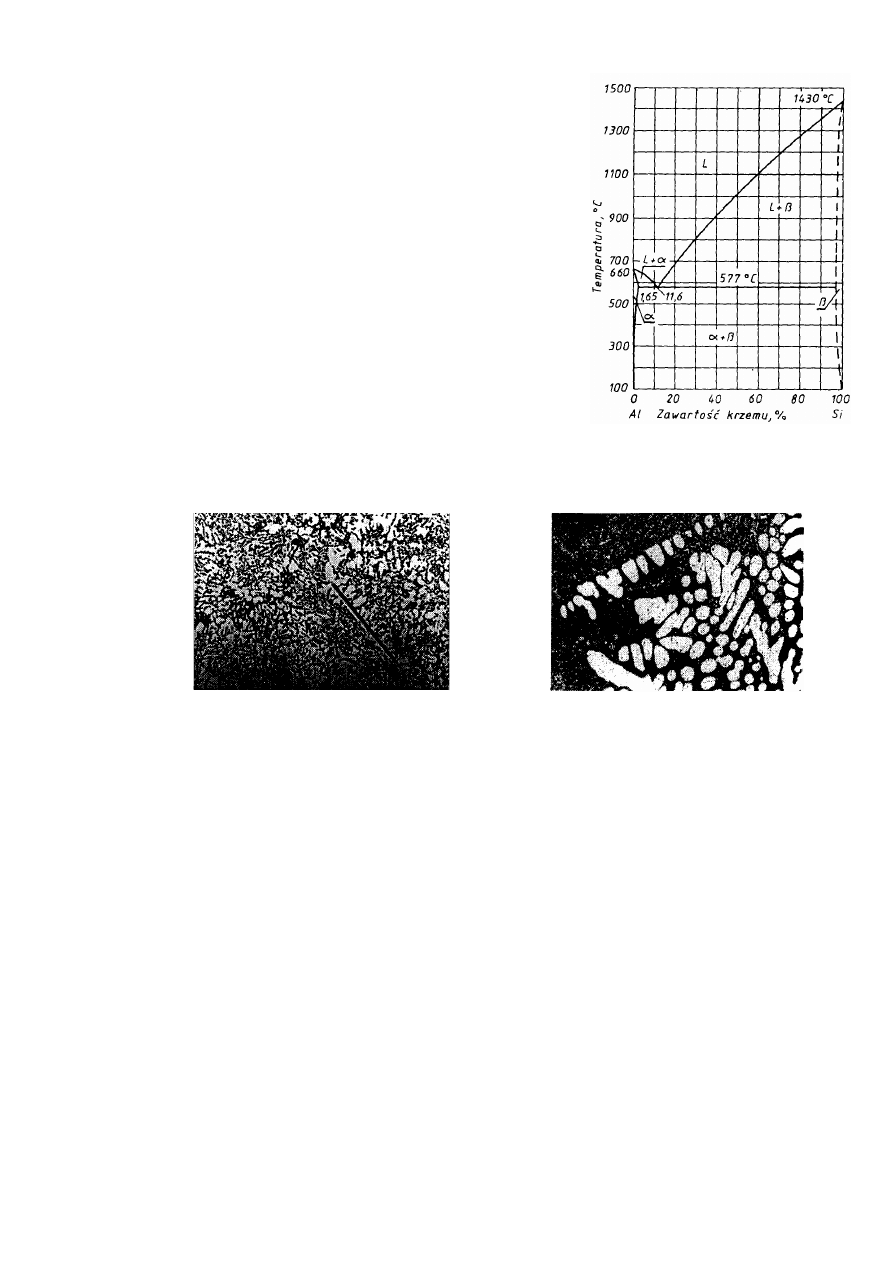
159
JW
Celem modyfikacji jest z jednej strony
rozdrobnienie ziarn, z drugiej - zmiana ich kształtu. Na
przykład, przy zawartości 11,6% Si siluminy krzepną
jako stopy eutekyczne, przy czym ich struktura składa
się z grubych, iglastych lub pierzastych kryształów
roztworu stałego
β na tle kryształów roztworu stałego α
(rys. 9.2). Taka gruboziarnista struktura ujemnie wpływa
na własności mechaniczne stopu. Przez dodanie w stanie
ciekłym pewnej ilości sodu metalicznego lub soli sodu (z
których na skutek reakcji wydziela się sód) uzyskuje się
dużą liczbę aktywnych zarodków krystalizacji.
Jednocześnie wywołuje się jakby przesunięcie punktu
eutektycznego w kierunku wyższych zawartości krzemu,
z jednoczesnym obniżeniem temperatury eutektycznej do
564°C. Dzięki temu silumin o składzie ściśle
eutekycznym zachowuje się podczas krzepnięcia jak stop
podeutektyczny i jego struktura składa się z
dendrytycznych kryształów roztworu stałego
α oraz
drobnoziarniste eutektyki, w której kryształy
β mają
kształt zaokrąglony (rys. 9.3).
Rys. 9.2. Mikrostruktura siluminu
eutektyczne-go przed modyfikacją. Na tle
roztworu stałego a widoczne ciemne
kształty fazy P. Traw. 0,5% roztworem
wodnym HF (40%). Powiększ. 100x
Rys. 9.3. Mikrostruktura siluminu
eutektyczne go po modyfikacji. Na tle
ciemnej, drobnoziarnistej eutektyki
widoczne dendrytyczne kryształy roztworu
stałego a. Traw. 0,5% roztworem wodnym
HF (40%). Powiększ. 100x
Dzięki opisanym zmianom strukturalnym wzrasta zarówno wytrzymałość, jak i
plastyczność stopów. Na przykład stop niemodyfikowany o zawartości 13% Si ma R
m
= 140
MPa i A
5
= 3%. Taki sam stop po modyfikacji ma R
m
= 175 MPa i A
5
= 8%.
W procesie modyfikacji siluminów nadeutektycznych rolę modyfikatora spełnił fosfor,
który tworzy z aluminium związek A1P. Związek ten charakteryzuje się dużym pokrewieństwem
do krzemu pod względem struktury sieciowej i dzięki temu wytwarza aktywne zarodki
krystalizacji. Praktycznie modyfikację przeprowadza się bądź czystym fosforem, bądź
pięciochlorkiem fosforu, bądź też jego stopami z miedzią. W wyniku takiej modyfikacji
otrzymuje się strukturę podobną do pierwotne przed modyfikacją (rys. 9.4), ale kryształy
roztworu
β są znacznie drobniejsze i bardziej równomiernie rozłożone w eutektyce (rys. 9.5).
Rozdrobnienie kryształów roztworu stałego
β, z jednej strony polepsza własności
mechaniczne stopu, z drugiej umożliwia obróbkę skrawaniem. Przed modyfikacją pojedyncze
kryształy
β osiągają wymiary nawet kilku milimetrów. Jako twarde i bardzo kruche utrudniają, a
nawet uniemożliwiają obróbkę skrawaniem odlewów, powodując bardzo szybkie niszczenie
narzędzi. Niemożliwe jest także uzyskanie gładkiej powierzchni obrabianego przedmiotu z
powodu łatwego wykruszania się dużych kryształów.
Rys.9.1. Układ równowagi
aluminium-krzem

160
JW
Rys. 9.4. Mikrostruktura siluminu
nadeutektycznego (20% Si) przed
modyfikacją. Na tle eutektyki widoczne duże
kryształy fazy
β. Traw. 0,5% roztworem
wodnym HF. Powiększ. 100x
Rys. 9.5. Mikrostruktura siluminu nadeutek-
tycznego (20% Si) po modyfikacji. Na tle
eutektyki widoczne drobne kryształy fazy
β.
Traw. 0,5% roztworem wodnym HF. Po-
większ. 100x
Dwuskładnikowe stopy Al-Cu charakteryzują się dobrą lejnością i stosunkowo dobrą
plastycznością, ale niską wytrzymałością. Toteż ich zastosowanie z reguły ogranicza się do
wytwarzania galanterii stołowej i innych odlewów, od których wymaga się dobrej plastyczności.
Główne zastosowanie przemysłowe mają stopy wieloskładnikowe, z których wytwarza się m.in.
odlewy części samochodowych maszynowych średnio i wysoko obciążonych.
Stopy Al-Cu podlegają obróbce cieplnej, powodującej znaczny wzrost wytrzymałości, ale
spadek plastyczności.
Stopy Al-Mg charakteryzują się wysoką odpornością na korozję, dość dobrą wytrzymałością i
plastycznością. Podobnie jak stopy Al-Cu, podlegają przesycaniu i starzeniu. Stopy te są
szczególnie odporne na obciążenia dynamiczne, mają ładny połysk i są stosowane na części
aparatury chemicznej, a także w budowie okrętów i samolotów.
9.2.2. Stopy aluminium do przeróbki plastycznej
Stopy te można podzielić na dwie podgrupy:
a) stopy stosowane bez obróbki cieplnej,
b) stopy stosowane w stanie utwardzonym dyspersyjnie.
Pierwsza podgrupę tworzą stopy aluminium-mangan, aluminium-magnez i aluminium-
magnez-mangan.
Stopy aluminium-mangan umacnia się jedynie przez obróbkę plastyczną na zimno (zgniot).
Wykazują one dużą plastyczność, dzięki czemu dobrze się tłoczą, ale ich wytrzymałość
niewiele przewyższa wytrzymałość czystego aluminium. Cenną zaletą jest duża odporność na
korozję atmosferyczną, na działanie wody morskiej, olejów, materiałów napędowych i in. (w
odróżnieniu od innych pierwiastków stopowych mangan podwyższa odporność aluminium na
korozję). Są stopami spawalnymi. W lotnictwie stosuje się je m.in. na zbiorniki, przewody i
elementy łączne instalacji paliwowej i olejowej, owiewki, pływaki i pokrycia kadłubów
hydroplanów.
Stopy aluminium-magnez można obrabiać cieplnie, ale efekt tej obróbki jest niewielki, toteż
praktycznie umacnia się je również tylko przez obróbkę plastyczną a zimno. Własności
mechaniczne stopów aluminium-magnez zbliżone są do własności stopów aluminium-mangan,
przy mniejszej jednak ich gęstości (2,6 g/cm
3
). Wadami są gorsza obrabialność skrawaniem i
gorsza odporność na korozję, zwłaszcza przy większych zawartościach magnezu. Do stopów
tego typu często wprowadza się dodatkowo mangan (kilka dziesiątych procentu), który
podwyższa własności mechaniczne i polepsza odporność na korozję. Zastosowanie stopów
aluminium-magnez i aluminium-magnez-mangan w lotnictwie jest podobne jak stopów
alumiium-mangan.
Orientacyjne własności mechaniczne omówionych stopów podano w tabl. 9.3.
161
JW
Tablica 9.3
Orientacyjne własności mechaniczne niektórych stopów aluminium do przeróbki
plastycznej
Własności mechaniczne
Cecha
stopu
Typ stopu
Stan stopu
R
m
, MPa
R
0,2
MPa
A
10
,%
wyżarzony
150
- 21
PA1
AI-Mn
zgnieciony
190
- 4
PA43 wyżarzony
120
50 27
wyżarzony
190
80 23
PA2
półzgnieciony
250
210 6
PA11
AI-Mg
wyżarzony
240
100 20
PA20 AI-Mg-Mn
wyżarzony 300 160
17
Znacznie liczniejszą podgrupę stanowią stopy aluminium przerabialne plastycznie,
stosowane po umacniającej obróbce cieplnej. Należą tu stopy Al-Mg-Si, Al-Cu-Mg, Al-Zn-Mg,
Al-Zn-Mg-Cu, Al-Cu-Mn, Al-Cu-Mg-Mn, Al-Cu-Mg-Mn-Si wiele innych stopów
wieloskładnikowych.
Niezależnie od składu chemicznego struktura tych stopów w stanie zbliżonym do równowagi
składa się ze stosunkowo miękkiego i plastycznego roztworu stałego pierwiastków stopowych
(ew. domieszek pochodzących z przerobu hutniczego) w aluminium i określonych faz
międzymetalicznych utworzonych bądź przez aluminium i pierwiastki stopowe lub domieszki
(np. Al
2
Cu, Al
2
CuMg, Al
2
Mg
3
Zn
3
Al
3
Mg
2
Al
4
Si
2
Fe i Al
3
Fe), bądź przez pierwiastki stopowe
między sobą (Mg
2
Si, MgZn
2
i in). Wszystkie te fazy międzymetaliczne są twarde i kruche i
spełniają w stopach rolę składnika utwardzającego. Oczywiście stopień utwardzenia stopu o
danym składzie chemicznym i fazowym jest zależny od wielkości, kształtu i rozmieszczenia
kryształów tych faz. Obróbka cieplna omawianych stopów polega więc na:
a) wprowadzeniu do roztworu stałego wydzielonych faz międzymetalicznych i uzyskaniu
jednorodnego roztworu stałego składników stopowych w aluminium (w temperaturze
otoczenia będzie to oczywiście roztwór przesycony, stąd nazwa obróbki - przesycanie),
b) wydzieleniu z przesyconego roztworu stałego faz międzymetalicznych (czyli tzw.
starzeniu). Wynika z tego, że podstawowym warunkiem tej obróbki cieplnej, zwanej
utwardzaniem wydzieleniowym, jest zmienna rozpuszczalność składników stopowych w
aluminium, wzrastająca w miarę podwyższania temperatury aż do temperatury przemiany
eutektycznej lub eutektoidalnej.
Najważniejszym składnikiem stopowym tej podgrupy stopów aluminium jest miedź,
podwyższająca wytrzymałość i twardość.
Z aluminium miedź tworzy eutektykę o zawartości 33% Cu (rys. 9.6), złożoną z kryształów
roztworu stałego granicznego
ω
miedzi w aluminium i kryształów roztworu stałego granicznego
θ aluminium w fazie międzymetalicznej Al
2
Cu. W temperaturze eutektycznej (548°C)
rozpuszczalność miedzi w aluminium wynosi 5,7%, w temperaturze otoczenia zaledwie 0,5%
(wg niektórych danych rozpuszczalność miedzi w aluminium w temperaturze otoczenia jest
mniejsza od 0,1%). Wynika z tego, że stopy zawierające do 0,5% Cu są stopami jednofazowymi
ω, stopy zawierające 0,5-5,7% Cu są stopami dwufazowymi, składającymi się z roz tworu
stałego
ω i wydzielonych wtórnych kryształów fazy θ. Stopy te można jednk przez nagrzanie do
odpowiedniej temperatury przekształcić w stopy jednofazowe, czyli można je obrabiać cieplnie.
Przy zawartości miedzi przekraczającej 7% w strukturze stopów pojawia się eutektyka, której
ilość jest oczywiście proporcjonalna do zawartości miedzi w stopie. Stopy te również można
obrabiać cieplnie, ale efekt obróbki będzie mniejszy, gdyż pierwotne kryształy fazy
θ wchodzące
skład eutektyki nie uczestniczą w procesie dyspersyjnego utwardzania, a ponadto za
θ jest
składnikiem kruchym i w większych ilościach w stopach niepożądanym dlatego zawartość
miedzi w stopach do przeróbki plastycznej nie przekracza 5,5% (w stopach krajowych 4,8%).
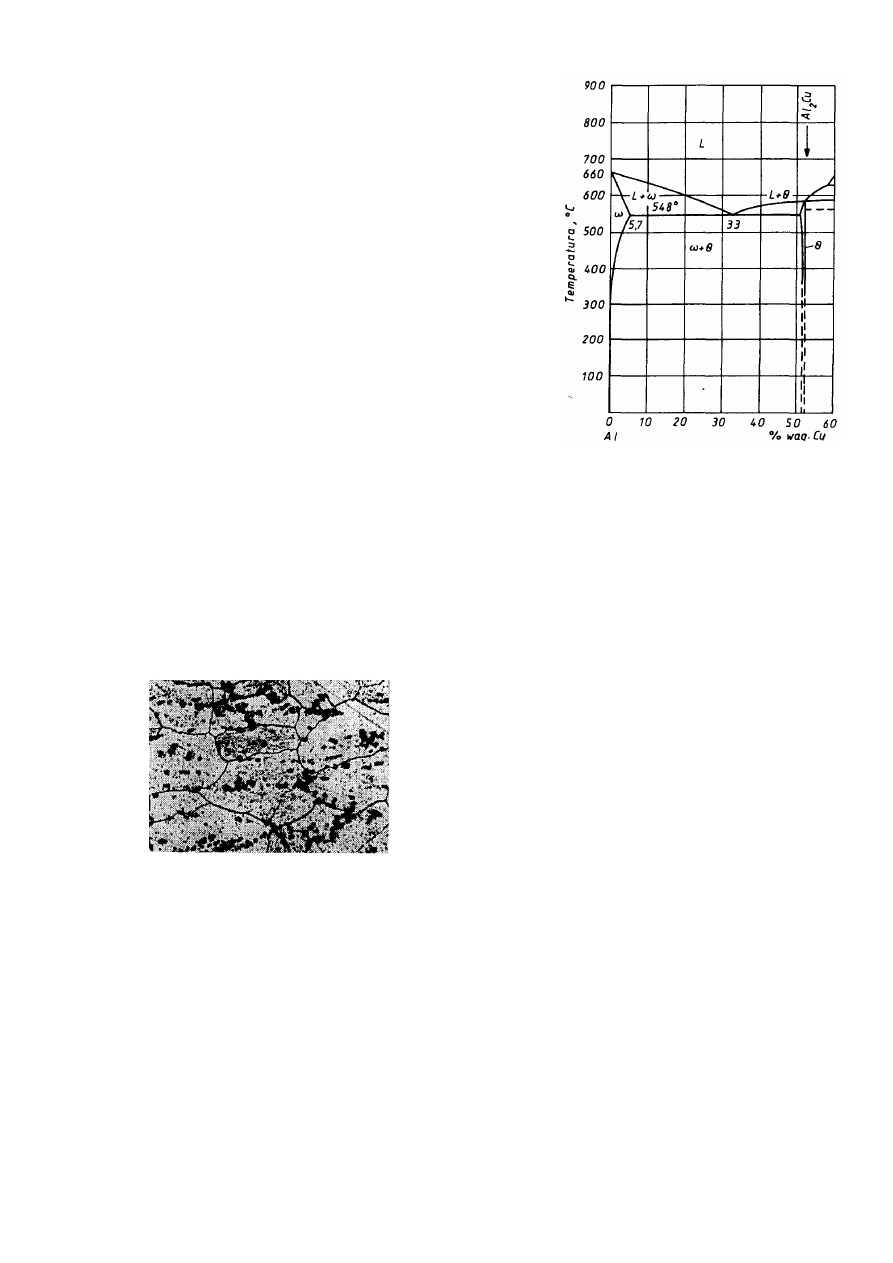
162
JW
W stopach wieloskładnikowych, a takimi są
przerabialne plastycznie stopy aluminium utwardzane
wydzieleniowo, jak już wspomniano, tworzą się
określone fazy międzymetaliczne, których skład
chemiczny i ilość są funkcją składu chemicznego
stopu, a które również wykazują zmienną
rozpuszczalność w tworzącym osnowę stopu
roztworze stałym.
Najstarszymi stopami aluminium, mającymi
zresztą do dziś szerokie zastosowanie przede
wszystkim w lotnictwie, są durale (nazwa
duraluminium lub krótko dural oznacza „twarde
aluminium", z francuskiego dur — twardy). Rozróżnia
się dwa rodzaje durali: bezcynkowe, których skład
chemiczny zawiera się w granicach:1
÷5,2% Cu, 0,4
÷1,8 Mg, 0,3 ÷1,0% Mn, max 0,7% Si, max 0,5% Fe
max 0,5% Zn, oraz durale zawierające cynk, o
składzie: 1,4
÷ 2,0% Cu, 5 ÷ 2,8% Mg, 0,2 ÷ 0,9%
Mn, 4,0
÷ 8,0% Zn, max 0,5% Si, max 0,5% Fe,
ewentualnie kilka dziesiątych procentu chromu. Do
pierwszej grupy należą stopy PA6, PA7, do drugiej - stop PA9. Do durali bezcynkowych należą
również stopy PA21, P23, PA24 i PA25.
W duralach bezcynkowych głównymi dodatkami stopowymi umacniającymi są miedź i
magnez. Mangan dodawany jest w celu polepszenia odporności na korozję, pozostałe pierwiastki
są nieuchronnymi zanieczyszczeniami.
W stanie wyżarzonym, tj. w stanie zbliżonym do równowagi fazowej, struktura durali
składa się z roztworu stałego i wydzieleń różnych faz międzymetalicznych (rys. 9.7), w stanie
przesyconym - z roztworu stałego na osnowie aluminium i nie rozpuszczonych związków żelaza.
Rys. 9.7. Mikrostruktura duralu (PA29) w stanie
wyżarzonym. Widoczne duże kryształy roztworu stałego
bogatego w aluminium oraz ciemne wydzielenia
międzymetalicznych faz umacniających (Al
2
Cu, Al.CuMg,
Al
2
CuMg, Mg
5
Cu i in.). Traw. odczynnikiem o składzie: l
ml HF (30%) + 2,5 ml HNO + l,5 ml HCl + 95 ml H
2
O
Powiększ. 200x
Durale zawierające cynk są najbardziej wytrzymałymi stopami aluminium (po utwardzeniu
dyspersyjnym R
m
osiąga wartość do 600 MPa), wykazują jednak mniejszą podatność do
przeróbki plastycznej i nieco obniżoną odporność na korozję naprężeniową.
Blachy zabezpiecza się przed korozją za pomocą platerowania specjalnym stopem (Al+Zn),
co jednak powoduje zmniejszenie ogólnej ich wytrzymałości, tym większe, im większy procent
przekroju blachy stanowi warstwa platerowana (o stosunkowo małej wytrzymałości).
Platerowanie jako ochronę przed działaniem środowisk korodujących stosuje się zresztą również
często i dla durali bezcynkowych. W tym przypadku platerowanie wykonuje się czystym
aluminium, przy czym grubość warstwy ochronnej wynosi 4
÷ 8% grubości blachy (odkuwki,
pręty, rury, druty i kształtowniki zabezpiecza się przed korozją innymi metodami).
Charakterystykę i zastosowanie znormalizowanych stopów aluminium do przeróbki
plastycznej podano w tabl. 9.4.
Rys. 8.6. Część układu równowagi
miedź-cyna od strony miedzi
163
JW
Tablica 9.4
Charakterystyka i zastosowanie stopów aluminium do przeróbki plastycznej (wg PN-79/H-88026)
Własności technologiczne**
podatność
Cecha
stopu
Wyrob
y*
do
przeróbki
plasty-
cznej
do
polero-
wania
do wytwa
rzania an-
odowych
powłok
tlenkowych
Odporn
-ość na
korozję
spa-
wal-
ność
Zastosowanie
PA43
B, T, D
Pr, R,
K, Ok
5 5
5
4 4
w przemyśle chemicznym i spożywczym,
elementy dekoracyjne, części głęboko tłoczone,
odkuwki matrycowe
PA2
B, Pr,
R, Rk,
D, K
5
5
5
5
4
średnio obciążone elementy konstrukcji
lotniczych, okrętowych i in., przemysł
spożywczy i chemiczny, konstrukcje budowlane
PA11
B, Pr,
R, D, K
5 5 3 5
4
elementy konstrukcyjne i nadbudówki okrętów,
elementy konstrukcji lotniczych, przemysł
spożywczy i chemiczny
PA13 B, Pr,
R, D, K
4 3 3 5
4
PA20 Pf, R,
K, D
4 3 3 4
4
obciążone konstrukcje okrętowe, transport,
przemysł chemiczny
PA1
B, T, D
Pr, R,K
5 - 4
5
w przemyśle spożywczym i chemicznym,
spawane zbiorniki do cieczy i gazów
PA38
Pr, R,
D, K
5 5 3 4
-
elementy dekoracyjne w budownictwie i
meblarstwie
PA4
B, Pr,
R, D,
K, Ok
5
5
3
4***
4
średnio obciążone elementy konstrukcji lotnicz-
ych i pojazdów mechanicznych, meble, ozdoby,
części głęboko tłoczone, odkuwki matrycowe
PA45
B, Pr,
R, D, K
5 5 5 4***
4
budownictwo, elementy dekoracyjne i
konstrukcyjne
PA10
B, Pr,K
R,D,Ok
5
5
5
4***
4 jak stopu PA4
PA6
B, Bpl,
Pr, R,
4 - -
w transporcie konstrukcje lotnicze, pojazdy
mechaniczne konstrukcje budowlane
PA7
B, Bpl,
Pr, R,
D, K,
Ok
4 - - 3***
-
silnie obciążone elementy konstrukcji
lotniczych i pojazdów mechanicznych, w
transporcie, części maszyn, konstrukcje
budowlane
PA21
PA23
PA24
PA25
D 4
-
-
-
-
nity lotnicze
PA29
PA30 Pr,0k 4
konstrukcje lotnicze, części pracujące w
temperaturze200 do 300°C
PA31 Pr,0k
4
-
-
-
-
konstrukcje lotnicze, odkuwki o
skomplikowanych kształtach
PA33 Ok
4
-
-
-
- konstrukcje
lotnicze, odkuwki matrycowe
PA9
Bpl, Pr,
K, Ok
4 - - 3
-
bardzo silnie obciążone elementy konstrukcji
lotniczych, środków transportu i maszyn
PA47
B, Pr,
K
3 - 3 4
4
silnie obciążone spawane konstrukcje nośne,
przemysł okrętowy, pojazdy mechaniczne,
pawilony wystawowe, sprzęt sportowy
PA15
B, T
5
-
-
4
4
elementy pojazdów mechanicznych, urządzenia
przemysłu spożywczego i chemicznego,
elementy konstrukcji budowlanych
B - blachy, Bpl - blachy platerowane (stopy PA6 i PA7 - aluminium, stop PA9 stopem AIZn1) K - kształtowniki,
Ok –odkuwki ** 5 - bardzo dobra, 4 - dobra, 3 – dostateczna *** Po utwardzaniu dyspersyjnym
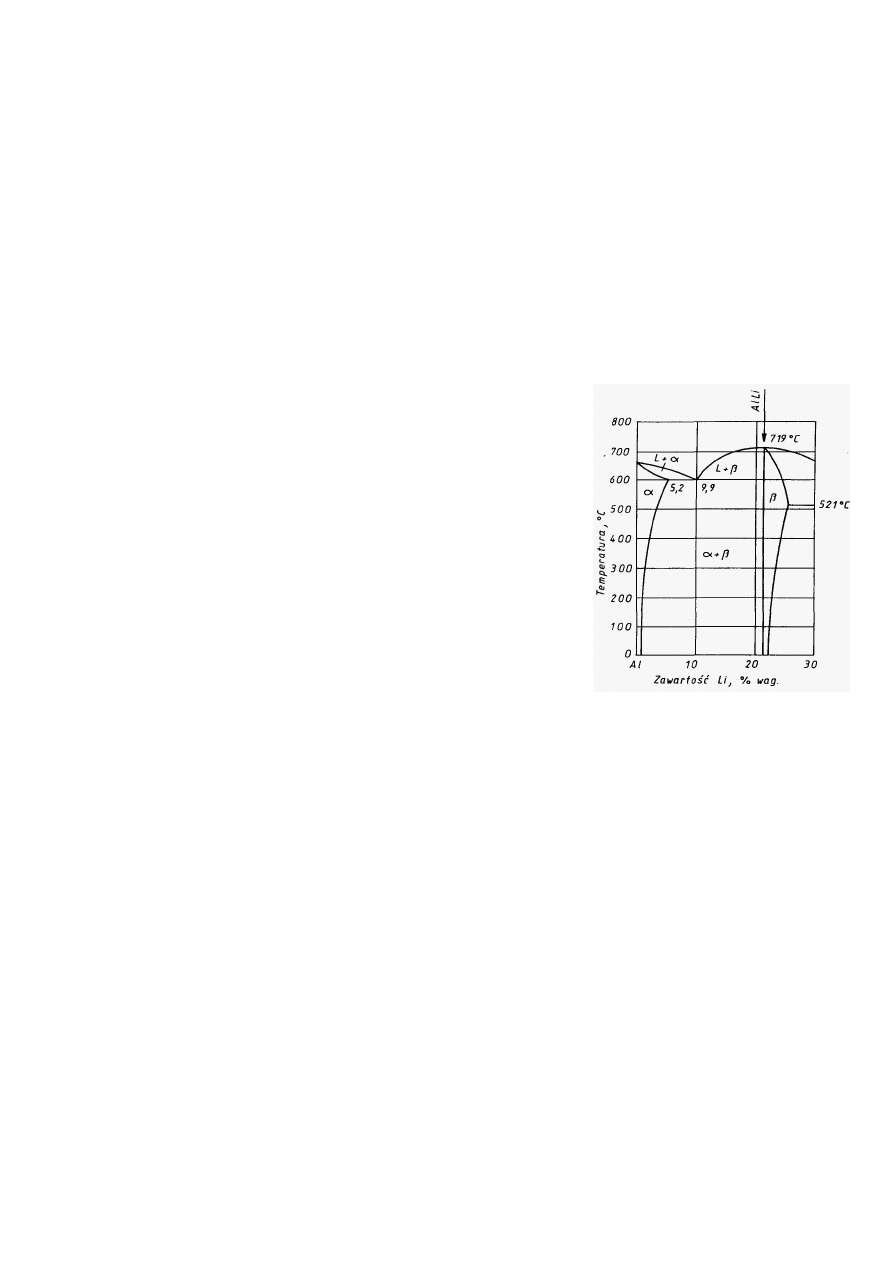
164
JW
Stopy aluminium-lit
Najnowszą generacją stopów aluminium są stopy z litem, jako głównym składnikiem
stopowym. Wykorzystanie litu do tego celu od dawna przyciągało uwagę metaloznawców,
głównie jako możliwość uzyskania stopów o gęstości znacznie mniejszej niż gęstość metalu-
bazy. Sukces osiągnięto w ostatnich latach.
Lit; jest najlżejszym metalem. Jego gęstość w temperaturze 20°C wynosi 0,536 g/cm
3
. Każdy
procent litu wprowadzony do aluminium obniża gęstość stopu o ok. 0,l g/cm
3
, co pozwala na
uzyskanie stopów o dość wysokim stosunku wytrzymałości do gęstości. Ponadto stopy Al-Li
cechuje wyższy moduł sztywności, niż konwencjonalne. Te właściwości powodują, że
zainteresowanie stopami aluminum-lit stale rośnie.
Optymalne połączenie wytrzymałości i plastyczności mają stopy podwójne zawierające 2,0-
2,5% Li, po obróbce cieplnej składającej się z przesycania z temperatury 580°C i starzenia w
temperaturze 130°C przez 48 godzin. Ich wytrzymałość na rozciąganie wynosi wówczas około
160 MPa, granica plastyczności 100 MPa, a wydłużenie 14%.
Zastosowanie obróbki plastycznej na zimno po prze-
sycaniu, a przed starzeniem, powoduje wzrost wskaźni-
ków wytrzymałościowych, ale spadek plastyczności.
Podobnie dzieje się przy zwiększaniu zawartości litu.
Zgodnie z układem równowagi (rys. 9.8), struktura
stopów podwójnych aluminium-lit do zawartości 5,2% Li
składa się z kryształów
α roztworu stałego granicznego
litu w aluminium i wtórnych kryształów
β roztworu na
osnowie fazy międzymetalicznej AlLi.
Jak stwierdzono, zawartość litu do 5,2% nie wpływa
praktycznie na odporność korozyjną stopów. Większa
zawartość litu powoduje jednak spadek tej odporności, co
wiąże się z pojawieniem się w strukturze eutektyki
α + β.
Szczególnie interesujące są stopy zawierające 2-3% Li i do
5% Mg. Ich granica plastyczności po obróbce cieplnej
osiąga 400 MPa. Wadą, podobnie jak wszystkich stopów
aluminium-lit, jest wrażliwość na naprężenia zmienne.
Przewiduje się, że stopy aluminium z litem znajdą
zastosowanie w budowie samolotów, przede wszystkim w
postaci cienkich blach na powłoki skrzydeł i kadłuba.
9.3. Obróbka cieplna stopów aluminium
9.3.1. Przesycanie i starzenie stopów Al
Obróbka cieplna stopów aluminium, mająca na celu przede wszystkim podwyższenie ich
wytrzymałości, polega na utwardzaniu dyspersyjnym, tj. na kolejnym przeprowadzeniu operacji
przesycania roztworu stałego i starzenia.
Podstawowym warunkiem, na którym opiera się proces utwardzania wydzieleniowego
stopów, jest zmniejszanie się granicznej rozpuszczalności składników stopowych w stanie
stałym wraz z obniżaniem się temperatury.
Typowym przykładem układu równowagi faz, który może służyć jako model u wyjaśnienia
procesów zachodzących podczas obróbki cieplnej stopów Al, jest ukłd Al-Cu, którego fragment
widoczny jest na rys. 9.9. Układ Al-Cu, a właściwie jest część odpowiadająca układowi
równowagi Al i fazy międzymetalicznej
θ o składzie bardzo bliskim Al
2
Cu, przedstawiono na
rys. 9.6.
Na rysunku 9.9 widać, że maksymalna rozpuszczalność miedzi w temperaturze
548°C wynosi około 5,7%, natomiast w temperaturze pokojowej jest znikoma.
Rozpatrzmy na przykład stop o składzie C (rys. 9.9) o zawartości ok. 4% Cu. W stanie
równowagi w temperaturze pokojowej składa się on z dwóch faz: kryształów roztworu stałego
(
ω
stanowiącego osnowę, i kryształów fazy międzymetalicznej
θ. Nagrzanie tego stopu do
Rys. 9.8. Fragment układu równo-
wagi alumnium-lit od strony
aluminium
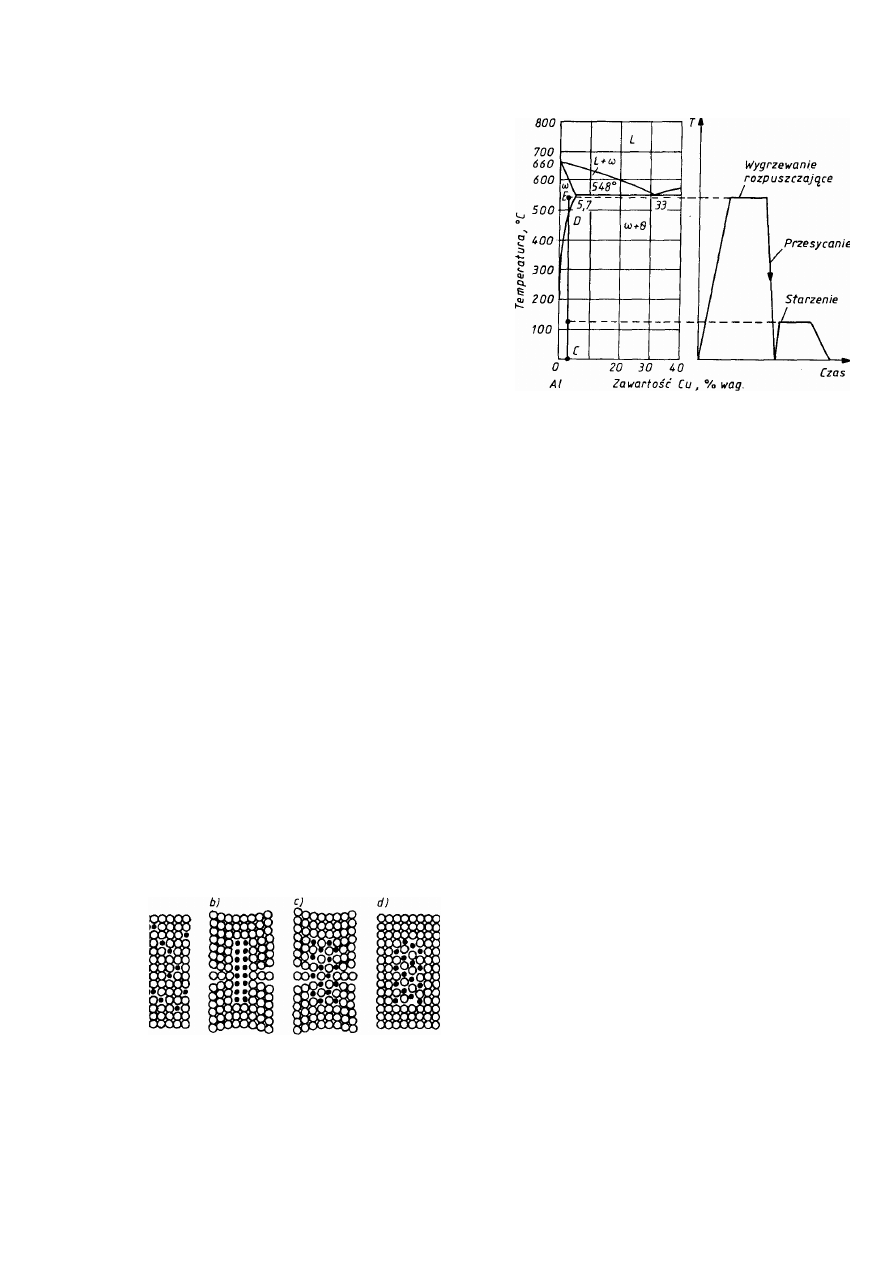
165
JW
temperatury E (powyżej punktu D) spowoduje, że będzie on jednorodnym roztworem stałym
ω,
gdyż kryształy fazy
θ ulegną rozpuszczeniu.
Jeżeli stop ten zostanie z kolei szybko
ochłodzony od tej temperatury, wówczas faza
θ nie zdąży się wydzielić i otrzymamy roztwór
stały przesycony. Stan taki jest nietrwały i jeżeli
stop będzie starzony, czyli wygrzewany w nieco
podwyższonej temperaturze (rys. 9.9), to zaczną
w nim zachodzić zmiany, które poprzez szereg
studiów pośrednich doprowadzą w końcowym
wyniku do wydzielenia się fazy
θ, czyli do
ustalenia się stanu równowagi.
Jednak jeżeli temperatura starzenia nie jest
dostatecznie wysoka, a czas starzenia nie jest
zbyt długi, zmiany zachodzące w przesyconym
stopie nie przebiegają do końca, a proces
starzenia ulega zatrzymaniu na pewnym
stadium pośrednim i nie dochodzi do
wydzielenia się fazy
θ.
Starzenie może zachodzić już w temperaturze
pokojowej i wówczas nosi nazwę starzenia naturalnego, jeśli zaś odbywa się wskutek nagrzania
stopu do określonej temperatury, nosi nazwę starzenia przyspieszonego.
9.3.2. Procesy zachodzące podczas starzenia
W początkowym okresie procesu starzenia, nazywanym pierwszym stadium starzenia, atomy
rozpuszczonego składnika (np. miedzi) rozmieszczone przypadkowo w przesyconym roztworze
stałym (rys. 9.10a) skupiają się w określonych miejscach sieci krystalicznej (rys. 9. l0b). W
wyniku tego procesu powstają wewnątrz kryształu submikroskopowe strefy o dużej dyspersji o
zwiększonej zawartości rozpuszczonego składnika, zwane strefami Guiniera-Prestona lub w
skrócie - strefami G-P (rys. 9.10).
W stopach Al-Cu strefy G-P są skupieniami atomów miedzi o kształcie podobnym do płytek,
które są ułożone wzdłuż płaszczyzn {100}. Grubość tych płytek jest rzędu zaledwie kilku
odstępów międzyatomowych, a średnica ok. 100 A. Ich obecność można wykryć metodą
małokątowego rozpraszania promieni X lub za pomocą mikroskopu elektronowego. Tworzenie
stref G-P powoduje powstawanie m.in. dużych naprężeń własnych w krysztale oraz
rozdrobnienie bloków mozaiki. Obecność stref G-P o dużej dyspersji oraz związane z nimi
zniekształcenia sieci krystalicznej, cznie utrudniają ruch dyskolacji, co w efekcie objawia się
wzrostem twardości wytrzymałości stopu.
Rys. 9.10. Schemat zmian zachodzących w sieci
przesyconego stopu AlCu4: a) rozmieszczenie
atomów Cu (czarne kółka) po przesycaniu, b)
powstawanie stref G-P, c) tworzenie się koherentnych
wydzieleń
θ'' i θ', d) wydzielenia fazy θ (Al
2
Cu)
Następne stadium stanowią koherentne
1)
wydzielenia pośrednie oznaczane symbolem
θ" (rys.
9.10c).
Wydzielenia te o maksymalnej grubości ok. 100 A i średnicy ok. 1500 A mają strukturę
tetragonalną, której parametry a i b są zgodne z parametrem komórki elementarnej Al, natomiast
parametr c jest znacznie większy. Wydzielenia te również
powodują umocnienie starzonego
stopu.
Rys. 9.9. Fragment układu równowagi Al-
Cu oraz schemat przebiegu obróbki cieplnej
polegającej na przesycaniu i starzeniu
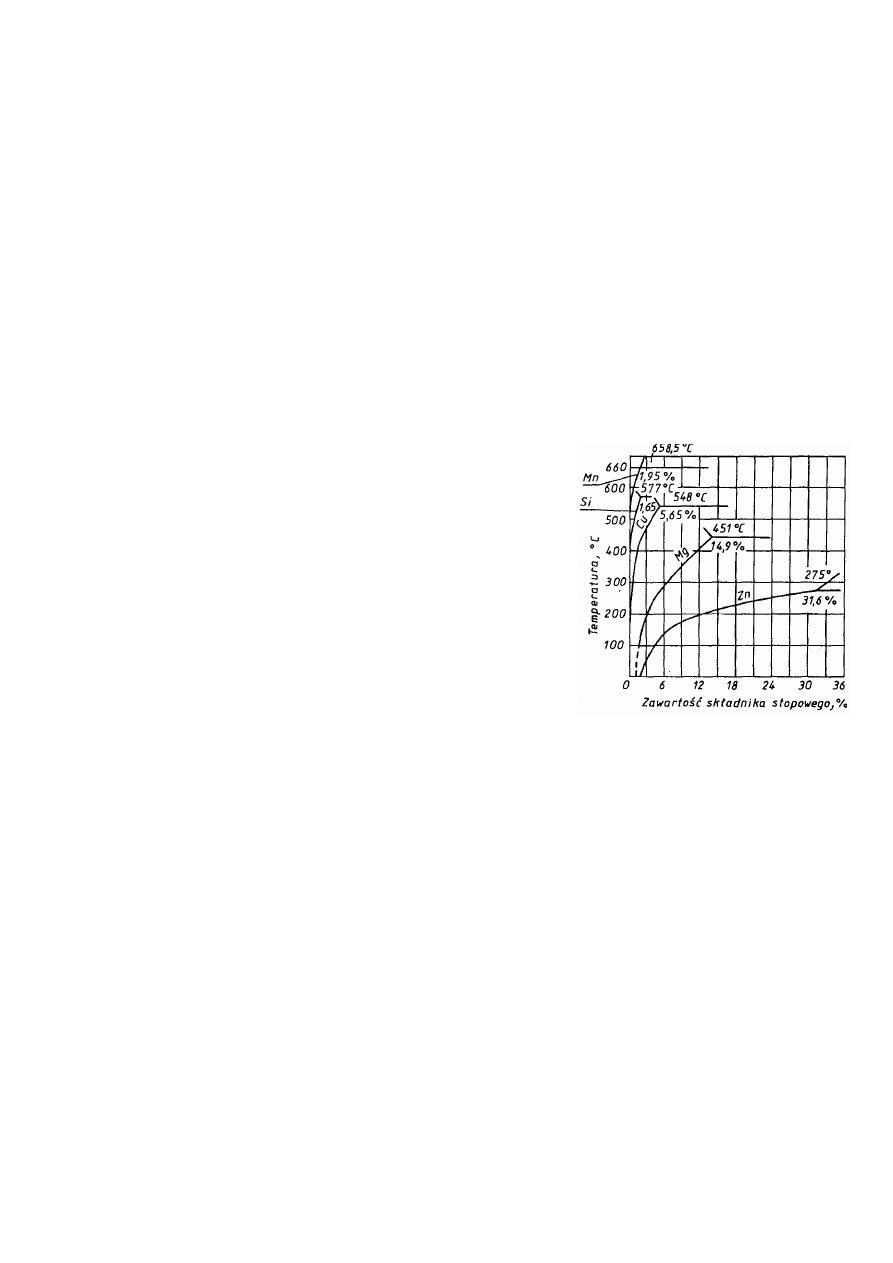
166
JW
Kolejna faza pośrednia
θ' jest także tetragonalną, ale o innym parametrze c aniżeli faza θ".
Tworzenie się wydzieleń
θ' powoduje już spadek twardości stopu. Faza θ (Al.
2
Cu) ma również
strukturę tetragonalną, ale nie jest już koherentna z siecią osnowy. Jej tworzenie się zawsze
prowadzi do spadku twardości stopu, poważ zanikają naprężenia związane z koherencją (rys.
9.10d). Kolejne przeobrażenia struktury wydzieleń w stopach Al-Cu zachodzące w czasie
starzenia można więc przedstawić następującym szeregiem:
(strefy G-P)
→ θ" → θ' → θ (AL
2
Cu)
Wszystkie powyższe stadia występują wówczas, gdy zawartość miedzi w stopie
stosunkowo duża (ok. 4,5%), a temperatura starzenia niezbyt wysoka (do ok. 190°C). Jeżeli
starzenie odbywa się w wyższych temperaturach (np. ok. 190°C) niektóre stadia pośrednie mogą
nie wystąpić, co uwidacznia się w przebiegu zmiany twardości podczas starzenia.
W stopach Al-Cu starzonych naturalnie, tj. w temperaturze pokojowej, występuje tylko
pierwsze i drugie stadium starzenia, tj. utworzenie się stref G-P oraz koherentnej fazy
θ". Dalsze
stadia starzenia zachodzą w temperaturze wyższej od temperatury otoczenia.
Stopień utwardzenia stopu jest związany z krytyczną dyspersją stref G-P i koherentnych
wydzieleń. Jeżeli w danej temperaturze proces starzenia
ulegnie zbytniemu przedłużeniu, następuje koagulacja i
wzrost wielkości wydzieleń. Małe wydzielenia ulegają
wtórnemu rozpuszczeniu, a ich kosztem rosną
wydzielenia większe, których dyspersja jest mniejsza.
Powoduje to zmniejszenie twardości i spadek
umocnienia stopu, o którym mówimy wówczas, że jest
przestarzony.
Składnikami konstrukcyjnych stopów aluminium,
które mają techniczne znaczenie, są, jak już
wspomniano poprzednio, takie pierwiastki jak Cu, Si,
Mg, Mn, Zn. Metale te tworzą graniczne roztwory stałe
w Al, charakteryzujące się spadkiem rozpuszczalności
w stanie stałym wraz z obniżaniem się temperatury (rys.
9.11). Stopy te można więc umacniać, poddając je
obróbce cieplnej polegającej na przesycaniu i starzeniu.
Oprócz stopów podwójnych również stopy potrójne i
poczwórne na bazie Al można umacniać dyspersyjnie,
przy czym obróbka cieplna takich stopów wielo-
składnikowych jest z reguły bardziej skuteczna, aniżeli stopów podwójnych. Jako przykład
można tu wymienić stopy: Al-Mg-Si, Al-Cu-Mg, Al-Zn-Mg.
W przypadku wieloskładnikowych stopów Al zamiast fazy
θ (Al.
2
Cu) tworzą się inne fazy
międzymetaliczne, które spełniają analogiczną rolę. W stopach układu Al-Mg-Si tworzy się np.
faza
β (Mg
2
Si), w stopach Al-Cu-Mg — faza
δ (Al
2
CuMg), a w stopach Al-Zn-Mg — faza M
(Mg
2
Zn).
9.3.3. Zmiany własności mechanicznych stopów Al zachodzące pod wpływem obróbki
cieplnej
W stanie wyżarzonym stop aluminium zawierający ok. 4% Cu ma wytrzymałość na
rozciąganie R
m
≅ 200 MPa. Bezpośrednio po przesycaniu, tj. gdy zaraz po tej operacji następuje
próba rozciągania, wytrzymałość jest nieco większa i w przybliżeniu wynosi 250 MPa (rys.
9.12). Szybkie chłodzenie po wygrzewaniu rozpuszczającym powoduje pewne niewielkie
zmiany własności mechanicznych, przede wszystkim na skutek tego, że atomy składnika
rozpuszczonego (tj. miedzi) oraz defekty punktowe znajdują się w osnowie w nadmiarze w
stosunku do stanu równowagi w temperaturze pokojowej.
Rys. 9.11. Krzywe granicznej
rozpuszczalności poszczególnych
składników stopowych w aluminium
w stanie stałym
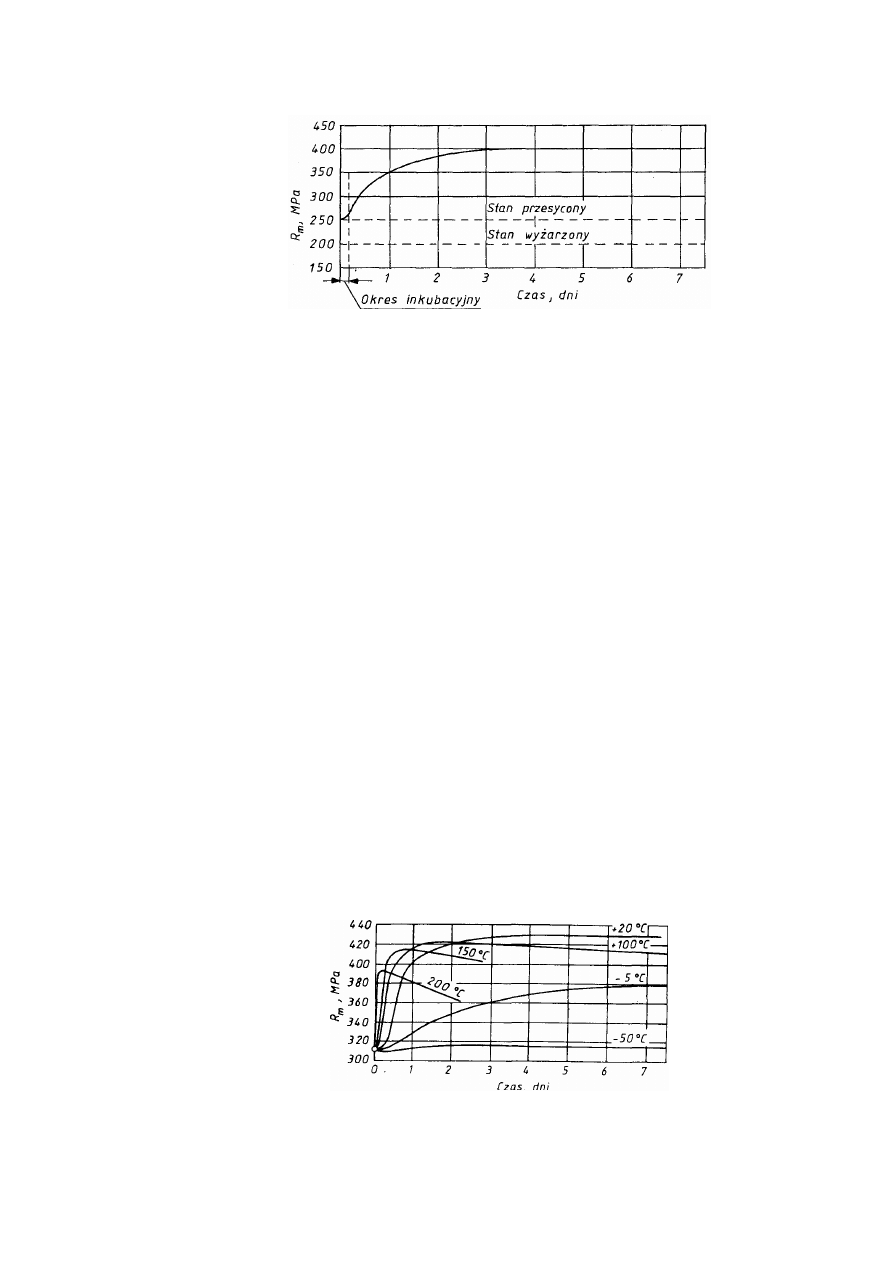
167
JW
Rys. 9.12. Zmiana wytrzymałości stopu A1Cu4 podczas starzenia naturalnego
Natomiast starzenie przesyconego stopu powoduje znaczne zmiany własności mechanicznych.
Wytrzymałość na rozciąganie znacznie się zwiększa i dla stopu AlCu4 osiąga wartość ok. 400
MPa, a więc wzrasta prawie dwukrotnie, natomiast własności plastyczne (wydłużenie i
przewężenie) oraz udarność maleją. Maksymalna wytrzymałość stop ten uzyskuje po starzeniu
naturalnym (tj. w temperaturze 3°C) po upływie 4
÷5 dni od chwili przesycania.
Typowy przebieg krzywej obrazującej zmianę wytrzymałości stopu Al-Cu podczas starzenia
naturalnego pokazano na rys. 9.12.
W początkowym stadium starzenia istnieje okres inkubacyjny, w którym nie stwierdza się
jeszcze wzrostu wytrzymałości. Dla procesów technologicznych okres ten ma duże znaczenie,
ponieważ stop wykazuje w tym okresie dużą plastyczność, co umożliwia poddawanie
przedmiotów przesycanych różnym operacjom technologicznym połączonym z odkształcaniem
(zakuwanie nitów, gięcie, tłoczenie itp.). Długość okresu inkubacyjnego jest różna dla stopów
aluminium o różnym składzie chemicznym i zależy od temperatury, w której stop jest starzony.
Dla stopów Al-Cu kres ten w temperaturze pokojowej wynosi ok. 2
÷3 godzin.
Szybkość starzenia i umocnienie stopów zależy w dużym stopniu od temperatury. Wykres
widoczny na rys. 9.13 przedstawia zależność wytrzymałości na rozciąganie duralu, tj. stopu Al-
Mg-Cu (o zawartości około 4% Cu i 1% Mg), od czasu starzania w różnych temperaturach w
zakresie 50
÷ 200°C.
W niskich temperaturach (-5°, -50°C) zbyt małe strefy G-P i zbyt mała ich ilość nie daje
dostatecznego umocnienia stopu.
W temperaturze zbyt wysokiej (+200°C) powstają już wydzielenia fazy
θ, a po dłuższym
przetrzymywaniu w tej temperaturze następuje ich koagulacja i wytrzymałość stopu spada. Na
rysunku 9.13 widać, że umocnienie stopu do 420 MPa można osiągnąć po około 24 godzinach
starzenia w temperaturze 100°C, stosując jednak starzenie naturalne można po dłuższym okresie
czasu uzyskać większe umocnienie.
Rys. 9.13. Krzywe starzenia duralu w różnych temperaturach

168
JW
Stan stopu osiągnięty w wyniku starzenia naturalnego nie jest trwały. Jeśli stop tak
umocniony zostanie nagrzany do temperatury 200
÷ 250°C i wytrzymany przez krótki okres
czasu (2
÷ 3 min) w tej temperaturze, to umocnienie zaniknie własności stopu będą odpowiadały
tym, jakie stop miał w stanie świeżo przesyconym, przy czy czym stop zyskuje ponownie
zdolność do starzenia naturalnego. Zjawisko to nazywa się nawrotem. Przyczyną nawrotu jest
rozpuszczanie się nietrwałych stref G-P o małych rozmiarach i powrót do struktury pierwotnie
przesycnego roztworu stałego o równomiernym rozłożeniu atomów rozpuszczonych. Po
ostudzeniu stop może być powtórnie starzony i będzie ulegał umocnieniu.
9.3. 4. Wyżarzanie stopów aluminium
Stopy aluminium można poddawać następującym rodzajom wyżarzania:
• wyżarzaniu ujednorodniającemu,
• wyżarzaniu zmiękczającemu,
• wyżarzaniu rekrystalizującemu,
• wyżarzaniu odprężającemu.
Wyżarzanie ujednorodniające przeprowadza się głównie w celu ujednorodnienia struktury,
zwłaszcza odlewów. Polega ono na nagrzaniu stopu do temperatury, w której ma on strukturę
roztworu stałego, wygrzaniu w tej temperaturze przez dłuższy okres czasu (2
÷ 12 godzin) i
następnie powolnym chłodzeniu.
Wyżarzanie zmiękczające ma na celu zmniejszenie twardości i polepszenie plastyczności
stopu poprzez koagulację wydzielonych faz. Przeprowadza się je w zakresie temperatur leżących
poniżej krzywej granicznej rozpuszczalności. W praktyce stopy aluminium w zależności od
składu wyżarza się w temperaturze 320
÷ 400°C przez 2 ÷ 3 godziny. Stopy wyżarzone
zmiękczająco mają niższą twardość i wytrzymałość niż stopy przesycone. Wysoka plastyczność
stopów uzyskana w wyniku wyżarzania ułatwia ich walcowanie, kucie i inne rodzaje przeróbki
plastycznej na zimno.
Wyżarzanie rekrystalizujące przeprowadza się w celu usunięcia niektórych skutków zgniotu
zwykle w temperaturze nieco wyższej od temperatury rekrystalizacji (300
÷ 400°C). Wyżarzanie
to przeprowadza się jako zabieg międzyoperacyjny w czasie obróbki plastycznej na zimno lub
jako zabieg końcowy, należy jednak pamiętać, że w niektórych przypadkach może ono
spowodować nadmierny rozrost ziarn, np. gdy nastąpił zgniot krytyczny lub gdy temperatura
wyżarzania była zbyt wysoka, względnie gdy czas wyżarzania był zbyt długi.
Wyżarzanie odprężające ma na celu usunięcie naprężeń własnych, zwłaszcza w odlewach
kokilowych. Temperatura wyżarzania wynosi, zależnie od gatunku stopu, 200
÷ 300°C. Po
wyżarzaniu stosowane jest powolne chłodzenie.
10. Magnez i jego stopy
Ze względu na swoją gęstość (1,74 g/cm
3
) magnez jest zaliczany do najlżejszych metali.
Temperatura topnienia czystego magnezu wynosi 650°C, temperatura topnienia stopów magnezu
460 ÷ 650°C, w zależności od ilości i rodzaju składników stopowych.
Magnez jest metalem bardzo aktywnym chemicznie i podobnie jak aluminium, łatwo łączy się z
tlenem, tworząc na powierzchni warstewkę tlenku MgO. Warstewka ta jest jednak mało szczelna
i nie chroni metalu przed korozją. Z tego powodu magnez i jego stopy są na ogół nieodporne na
korozję (wyjątek stanowi atmosfera suchego powietrza). W temperaturze 600
÷ 650°C magnez
zapala się i płonie oślepiająco białym płomieniem, co wywołuje konieczność stosowania
specjalnych środków zabezpieczających przy jego topieniu i odlewaniu.
Czysty magnez ma niewielką wytrzymałość i plastyczność, np. w postaci lanej R
m
= 78
÷
120 MPa, A
5
= 4
÷ 6 w postaci walcowanej R
m
= 160
÷ 180 MPa, A
5
= 5
÷ 6%. W związku z
tym magnez nie znajduje zastosowania jako materiał konstrukcyjny. Wykorzystywany jest on
natomiast w pirotechnice (do produkcji rakiet sygnalizacyjnych i lotniczych bomb zapalających),
w przemyśle chemicznym, w energetyce jądrowej (jako ciekły nośnik ciepła w niektórych

169
JW
typach reaktorów) oraz w metalurgii jako odtleniacz. W postaci stopów z miedzią i niklem
używany jest także jako modyfikator żeliw.
W Polsce magnez otrzymuje się przez redukcję termiczną tlenku magnezu dolomitu. Zgodnie
z PN-79/H-82161 produkowane są dwa gatunki magnezu: Mg 99,95 (zawierający 99,95% Mg,
reszta to Al, Zn, Fe, Si, Cu i inne) i Mg 99,9 (zawierający 99,9% Mg). Pierwszy jest
przeznaczony dla przemysłu chemicznego i celów specjalnych, drugi - do produkcji stopów
magnezu i stopów z magnezem.
Znacznie szersze zastosowanie przemysłowe znajdują stopy magnezu, które często osiągają
wytrzymałość R
m
= 300
÷ 340 MPa. Głównymi składnikami tych stopów obok magnezu są:
a) aluminium (do 11%), które podwyższa własności wytrzymałościowe i twardość, a w
stopach odlewniczych polepsza lejność i zmniejsza skurcz; wzrost zawartości aluminium w
stopie wywołuje jednak zwiększenie kruchości na gorąco;
b) cynk (do 7%) polepszający zarówno własności wytrzymałościowe, jak i plastyczne;
c) mangan zwiększający odporność na korozję i wywołujący rozdrobnienie ziarna; w stopach
nie zawierających aluminium zawartość manganu dochodzi do 5%, w stopach z aluminium,
które zmniejsza rozpuszczalność manganu w magnezie, wynosi kilka dziesiętnych procentu;
d) cyrkon (do 1%) polepszający własności mechaniczne i obrabialność stopów wywołuje
rozdrobnienie ziarna);
e) cer, tor i metale ziem rzadkich (lantan, neodym, prazeodym) polepszające własności w
temperaturach podwyższonych. Spotyka się również stopy magnezu zawierające takie dodatki
stopowe, jak: krzem, wapń, kadm i nikiel, przy czym zawartość ich zwykle nie przekracza 1%.
Inne pierwiastki występują w stopach magnezu w nieznacznych ilościach i poza berylem
dodawanym w celu zmniejszenia skłonności magnezu do zapalania się podczas odlewania,
pochodzenie ich jest przypadkowe.
Osobną, najmłodszą grupę stopów magnezu stanowią stopy z litem (zawierające do
kilkunastu % Li), których gęstość (1,35
÷ 1,62 g/cm
3
) jest znacznie mniejsza niż pozostałych
stopów magnezu (ok. 1,80 g/cm
3
).
Ogólnie stopy magnezu dzielą się na odlewnicze i do przeróbki plastycznej. W obu tych
grupach podstawowymi typami są podwójne stopy magnez-mangan oraz wieloskładnikowe
stopy magnez-aluminium-cynk-mangan i magnez-cynk-cyrkon. W krajach wysoko
uprzemysłowionych (WNP, USA) na bazie tych podstawowch typów stopów opracowano i
wprowadzono do przemysłu wiele stopów pochodnych, zawierających dodatkowo cer, tor,
lantan, neodym i inne, a więc pierwiastki powodujące wyraźny wzrost własności mechanicznych
w temperaturach podwyższonych. Skład chemiczny krajowych stopów magnezu podano w tabl.
9.1.
Stopy magnezu, podobnie jak większość stopów aluminium, można obrabiać cieplnie
(przesycać i starzyć), gdyż rozpuszczalność głównych składników stopowych (aluminium,
cynku i manganu) w magnezie jest ograniczona i zmniejsza się z obniżeniem temperatury.
Obróbka ta jednak tylko w niewielkim stopniu polepsza własności mechaniczne stopów i rzadko
jest stosowana. Wyjątkiem są stopy odlewnicze, zawierające powyżej 6% aluminium, które po
obróbce cieplnej mają wytrzymałość o 40
÷ 50% wyższą.
Na przykład, stop GA8 w stanie surowym ma wytrzymałość na rozciąganie 150 MPa. Po
przesyceniu w temperaturze w temperaturze 415°C (w czasie 20h, chłodzenie na powietrzu) w
starzeniu w temperaturze 175°C (w czasie 16 h) jego wytrzymałość wzrasta do 230 MPa.
Z reguły natomiast odlewy ze stopów magnezu poddaje się wyżarzaniu odprężającemu w
temperaturze 200
÷ 250°C.
Zastosowanie stopów magnezu zależy od ich składu chemicznego i własności. Na przykład
stopy odlewnicze przeznaczone są na: GA3 - korpusy pomp i armatury, GA6 - odlewy części
lotniczych i samochodowych, obudowy przyrządów aparatów, GA8 - silnie obciążone części
lotnicze, części aparatów fotograficznych maszyn do pisania, GRE3 - skomplikowane odlewy
pracujące w temp. do 250°C; stopy przerabialne plastycznie; GA6 - na obciążone elementy
konstrukcji lotniczych, poszycia samolotów i śmigłowców itd. Dokładne własności i główne i
zastosowania wszystkich krajowych stopów magnezu podają odpowiednie Polskie Normy.
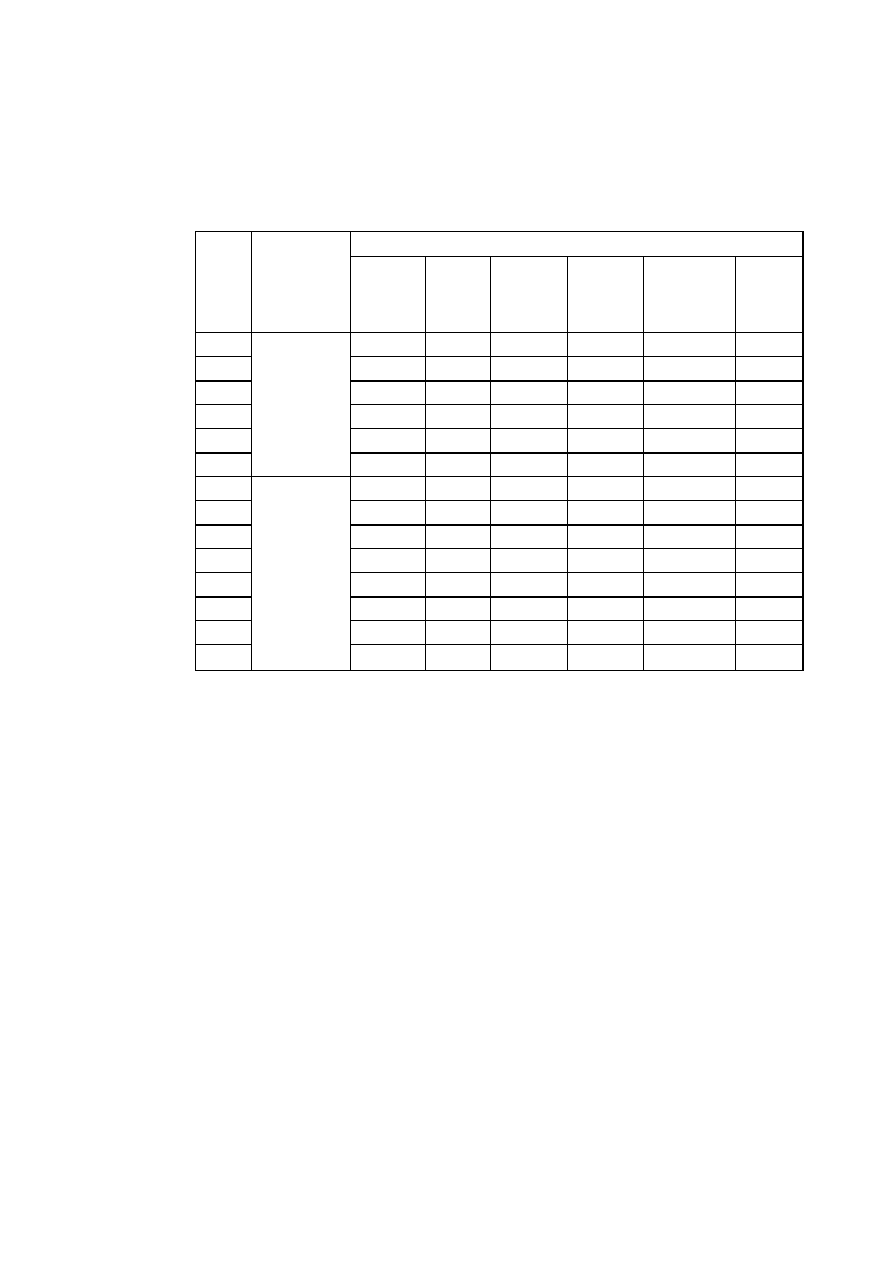
170
JW
Warto dodać, że zakres stosowania stopów magnezu jako tworzywa konstrukcyjno w
lotnictwie, kosmonautyce, budowie rakiet i energetyce jądrowej, w przemyśle światowym stale
wzrasta. Coraz szerzej stopy magnezu stosuje się w przemyśle elektronicznym i
elektrotechnicznym, poligraficznym, samochodowym, transporcie kolejowym itp.
Tablica 9.1
Skład chemiczny krajowych stopów magnezu
Skład chemiczny, % (reszta magnez)
Cecha
stopu
Rodzaj stopu
Al
Zn
Mn
Zr
inne
zanieczy-
szczenia
ogółem,
max
GA8*
7,5
÷
9,0 0,2
÷
0,8 0,15
÷
0,5
- -
0,5
GA10
9,0
÷
10,2 0,6
÷
1,2 0,1
÷
0,5
- -
0,5
GZ5 -
4,0
÷
5,0
-
0,6
÷
1,1
- 0,2
GZ6 -
5,5
÷
6,6
-
0,7
÷
1,1 0,2
÷
0,8Cd
0,2
GN3 -
0,1
÷
0,7
-
0,4
÷
1,0 2,2
÷
2,8Nd
0,2
GRE3
odlewniczy
-
0,2
÷
0,7
-
0,4
÷
1,0 2,5
÷
4,0RE*
0,2
GM -
-
1,3
÷
2,5
- -
0,2
GA3
3,0
÷
4,0 0,2
÷
0,8 0,15
÷
0,5
- -
0,5
GA6
5,5
÷
7,0 0,5
÷
1,5 0,15
÷
0,5
— - 0,7
GA5
5,0
÷
7,0 2,0
÷
3,0 0,2
÷
0,5
- -
0,7
GA8
7,8
÷
9,2 0,2
÷
0,8 0,15
÷
0,5
- -
0,7
GZ3 -
2,5
÷
4,0
-
0,5
÷
0,9
- 0,5
GZ5 -
4,0
÷
5,5
-
0,5
÷
0,9
- 0,5
GME
do przeróbki
plastycznej
- -
1,5
÷
2,5
-
0,15
÷
0,35C
1,0
* Norma zawiera jeszcze stop GA8A różniący się od stopu GA8 tylko dopuszczalną zawartością
zanieczyszczeń, wynoszącą 0,13%.
** RE — mieszanina pierwiastków ziem rzadkich, zawierająca min. 45% ceru.
11. Tytan i jego stopy
Tytan jest metalem o dużej wytrzymałości, zarówno w temperaturze otoczenia, jak i
temperaturach podwyższonych, stosunkowo małej gęstości i dużej odporności na korozję w
powietrzu, wodzie morskiej i wielu środowiskach agresywnych.
Tytan występuje w dwóch odmianach alotropowych
α i β. Odmiana α (Ti
α
) istniejąca do
temperatury 882°C krystalizuje w sieci heksagonalnej zwartej, natomiast odmiana
β (Ti
β
)
istniejąca powyżej temperatury 882°C aż do temperatury topnienia (1668°C) krystalizuje w sieci
regularnej przestrzennie centrowanej.
W temperaturze otoczenia czysty tytan ma kolor srebrzysty i przypomina wyglądem stal
nierdzewną lub nikiel. Gęstość tytanu a w temperaturze 20°C wynosi 4,507 g/cm
3
, tytanu
β w
temperaturze 900°C - 4,32 g/cm
3
. Tytan jest metalem paramagnetycznym.
Własności mechaniczne tytanu zależą przede wszystkim od jego czystości, a ta z kolei
zarówno od rodzaju procesu metalurgicznego przerobu rudy tytanowej (proces jodkowy, proces
Krolla, elektroliza), jak i od metody przerobu otrzymanych m procesie półwyrobów (topienie
gąbki tytanowej, spiekanie proszku). Zwiększenie ilości zanieczyszczeń w tytanie zawsze
prowadzi do podwyższenia jego wytrzymałości i twardości, a obniżenia własności plastycznych,
przy czym bardzo poważny wpływ wywierają nawet setne części procentu zanieczyszczeń.
W przemyśle praktycznie wykorzystuje się głównie tytan produkowana metodą Krolla,
zawierający 99,8
÷ 98,8% Ti. Taki tytan nosi nazwę tytanu technicznego.

171
JW
Szczególnie cenną własnością tytanu jest jego wielka odporność na korozję chemiczną,
dorównująca, a w wielu przypadkach przewyższająca odporność korozyjną austenitycznych stali
chromowo-niklowych.
Istotną również cechą tytanu jest jego silne powinowactwo w stanie nagrzanym i ciekłym do
gazów atmosferycznych (tlenu, azotu i wodoru), co powoduje, że we wszystkich prawie
procesach technologicznych, w których tytan zostaje ogrzany do temperatury umożliwiającej
dyfuzję wymienionych gazów, należy stosować atmosfery ochronne lub próżnię. Praktycznie
tytan jest odporny na działanie atmosfery tlenowej tylko do temperatury 120°C, powyżej tej
temperatury na powierzchni metalu tworzą się tlenki. Absorpcja i dyfuzja wodoru zaczynają się
w temperaturze powyżej 150°C. Z powietrzem tytan reaguje w temperaturze powyżej 500°C,
przy czym jego powierzchnia pokrywa się szczelną warstewką tlenków i azotków. Trzeba jednak
podkreślić, że w miarę wzrostu temperatury chemiczna aktywność tytanu silnie wzrasta i w
powietrzu tytan zapala się płomieniem w temperaturze 1200°C w czystym tlenie - już w
temperaturze 610°C.
11.1. Tytan techniczny
Jak już wspomniano, tytan techniczny zależnie od gatunku zawiera 0,2-1,2% zanieczyszczeń,
na które składają się przede wszystkim tlen, azot, węgiel, żelazo, wodór i krzem.
Zanieczyszczenia te powodują istotne zmiany własności mechanicznych, wyrażające się we
wzroście wytrzymałości na rozciąganie, granicy plastyczności oraz twardości, a zmniejszeniu
wskaźników własności plastycznych. Na przykład, wytrzymałość na rozciąganie tytanu
technicznego zawierającego 0,8% zanieczyszczeń wynosi ok. 400 MPa, a tytanu zawierającego
1% zanieczyszczeń — ok. 550 MPa.
Tytan techniczny jest produkowany w skali przemysłowej w postaci odlewów, blach cienkich
i grubych, taśm, prętów prasowanych wypływowo i kutych, rur, części tłoczonych i kutych.
Podlega obróbce plastycznej na zimno i na gorące (w temp. 1000-750°C) oraz obróbce
skrawaniem (ostre narzędzia, obfite chłodzenie), nie podlega natomiast obróbce cieplnej, a
umacnia się go jedynie przez zgniot. Można go spawać łukowo w osłonie gazów szlachetnych
(argonu lub helu) i elektrożużlowo, poza tym zgrzewać punktowo, liniowo i doczołowo oraz
lutować lutami miękkimi i twardymi.
Tytan techniczny jest stosowany przede wszystkim w przemyśle lotniczym, zarówno na
elementy silników, jak i kadłubów samolotów. Wykorzystuje się go także w przemyśle
okrętowym (części silników, armatura, pompy do wody morskiej), chemicznym (aparatura), w
protetyce stomatologicznej i w chirurgii kostnej (nie jest toksyczny dla organizmu ludzkiego)
itd. Maksymalna temperatura pracy nie może przekraczać 300
÷ 350°C.
11.2. Stopy tytanu
Wpływ pierwiastków stopowych na temperaturę przemiany alotropowej tytanu jest różny.
Aluminium, tlen, azot i węgiel podwyższają temperaturę przemiany tym samym zwiększają
obszar istnienia tytanu
α. Stąd często noszą one nazwę stabilizatorów fazy α. Większość
pozostałych pierwiastków stopowych (np. moliben, wanad, niob, tantal, chrom, mangan, żelazo,
wodór) obniża temperaturę przemiany i rozszerza obszar istnienia tytanu
β. Te pierwiastki noszą
nazwę stabilizatorów fazy
β. Osobną grupę stanowią pierwiastki, których wpływ na temperaturę
przemiany alotropowej jest nieznaczny. Należą tu cyna, cyrkon, tor, hafn i inne. Te pierwiastki
nazywa się zwykle neutralnymi.
Dwuskładnikowe układy równowagi faz tytanu z pierwiastkami wchodzącymi w skład
stopów można podzielić na trzy główne typy, w zależności od wpływu pierwiastka stopowego na
strukturę stopu w stanie równowagi.
Na rysunku 11.1 pokazano układ równowagi typu I, w którym pierwiastek stopowy rozszerza
zakres istnienia roztworu stałego
α (międzywęzłowego w przypadku tlenu, azotu i węgla,
różnowęzłowego w przypadku aluminium), stabilizując fazę
α w strukturze stopów. Jak widać,
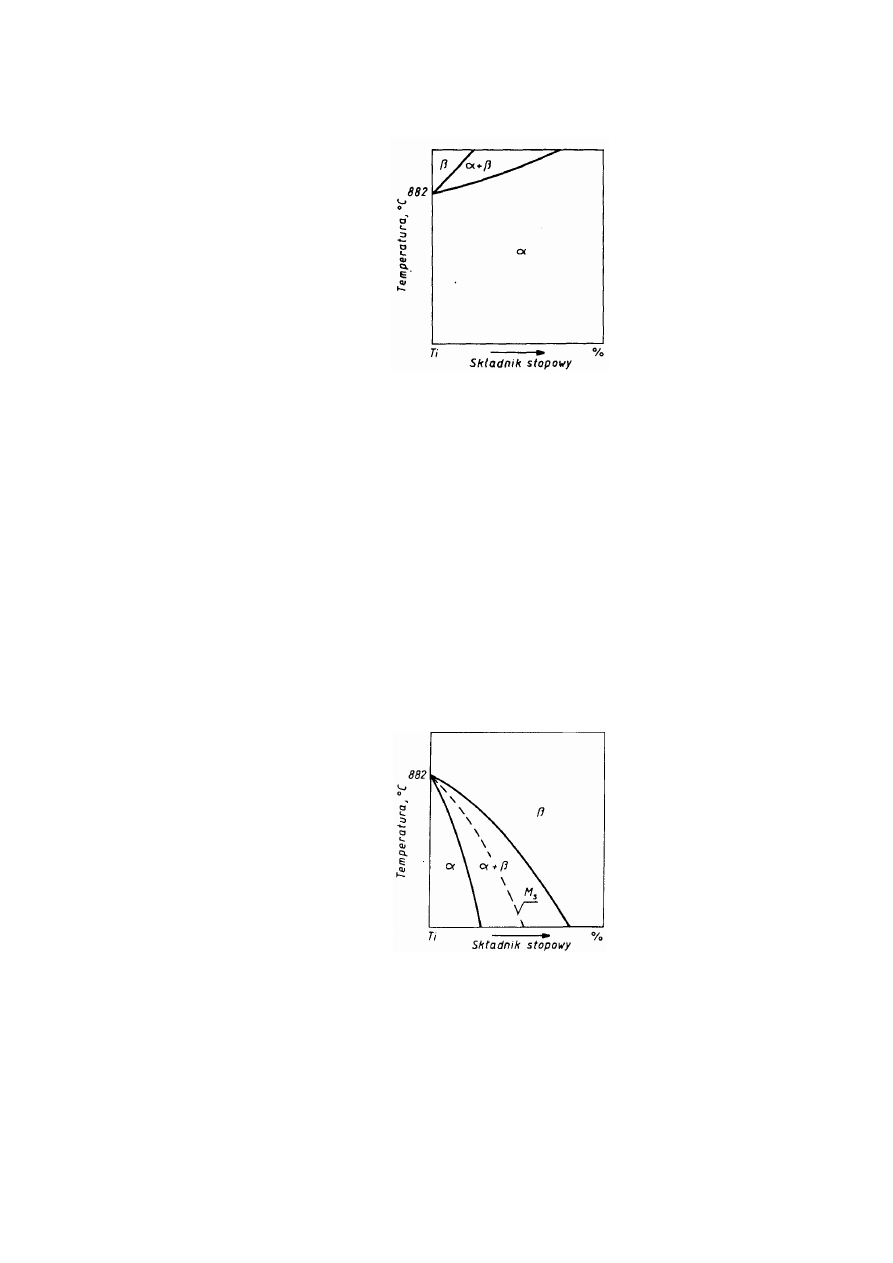
172
JW
ze wzrostem zawartości pierwiastka stopowego granice obszaru dwufazowego
α + β przesuwają
się w kierunku wyższych temperatur.
Rys. 11.1. Typ I układu równowagi tytan-pierwiastek stopowy
(pierwiastek stopowy podwyższa temperaturę przemiany alotropowej)
Na rysunku 11.2 przedstawiono układ równowagi typu II, w którym pierwiastek stopowy
rozszerza zakres istnienia roztworu stałego
β, stabilizując w strukturze stopów fazę β. Tego typu
układy równowagi występują dla molibdenu, wanadu, niobu i tantalu, które znacznie lepiej
rozpuszczają się w tytanie
β, niż w tytanie α, tworząc roztwory stałe różnowęzłowe. Przy bardzo
małej zawartości tych pierwiastków w stopie, strukturą równowagi w temperaturze pokojowej
będzie faza
α, przy dużej - faza β, przy zawartościach pośrednich - mieszanina faz α+ β. W tym
ostatnim przypadku istnieje możliwość otrzymania w temperaturze pokojowej jednofazowej
struktury
β przez szybkie przechłodzenie stopu z temperatury istnienia obszaru trwałej fazy β,
ale możliwość ta jest ograniczona występowaniem bezdyfuzyjnej przemiany typu
martenzytycznego. W wyniku tej przemiany z przechłodzonej fazy
β powstaje przesycona faza
α, oznaczana na ogół jako faza α' i mająca budowę iglastą, podobną do martenzytu w stali, ale w
przeciwieństwie do niego miękka i ciągliwa. Stanowi ona modyfikację fazy
α i krystalizuje
również w sieci heksagonalnej zwartej, tylko o nieco innych parametrach.
Rys. 11.2. Typ II układu równowagi tytan-pierwiastek stopowy (pierwiastek
stopowy obniża temperaturę przemian alotropowej)
Temperaturę początku przemiany bezdyfuzyjnej dla różnych stężeń pierwiastka stopowego
określa na rys. 11.2 kreskowa krzywa M
s
. Jak widać, temperatura ta dla określonego stężenia
pierwiastka stopowego (zw. stężeniem krytycznym) staje się niższa od pokojowej. Warunkiem
więc uzyskania jednorodnej fazy
β w temperaturze pokojowej przez przechłodzenie stopu z
obszaru stabilnej fazy
β jest zawartość pierwiastka stopowego przekraczająca stężenie
krytyczne. Trzeba jednak podkreślić, że tak uzyskana faza
β nie jest fazą stabilną i w
temperaturach podwyższonych wykazuje skłonność do rozkładu (starzenia).
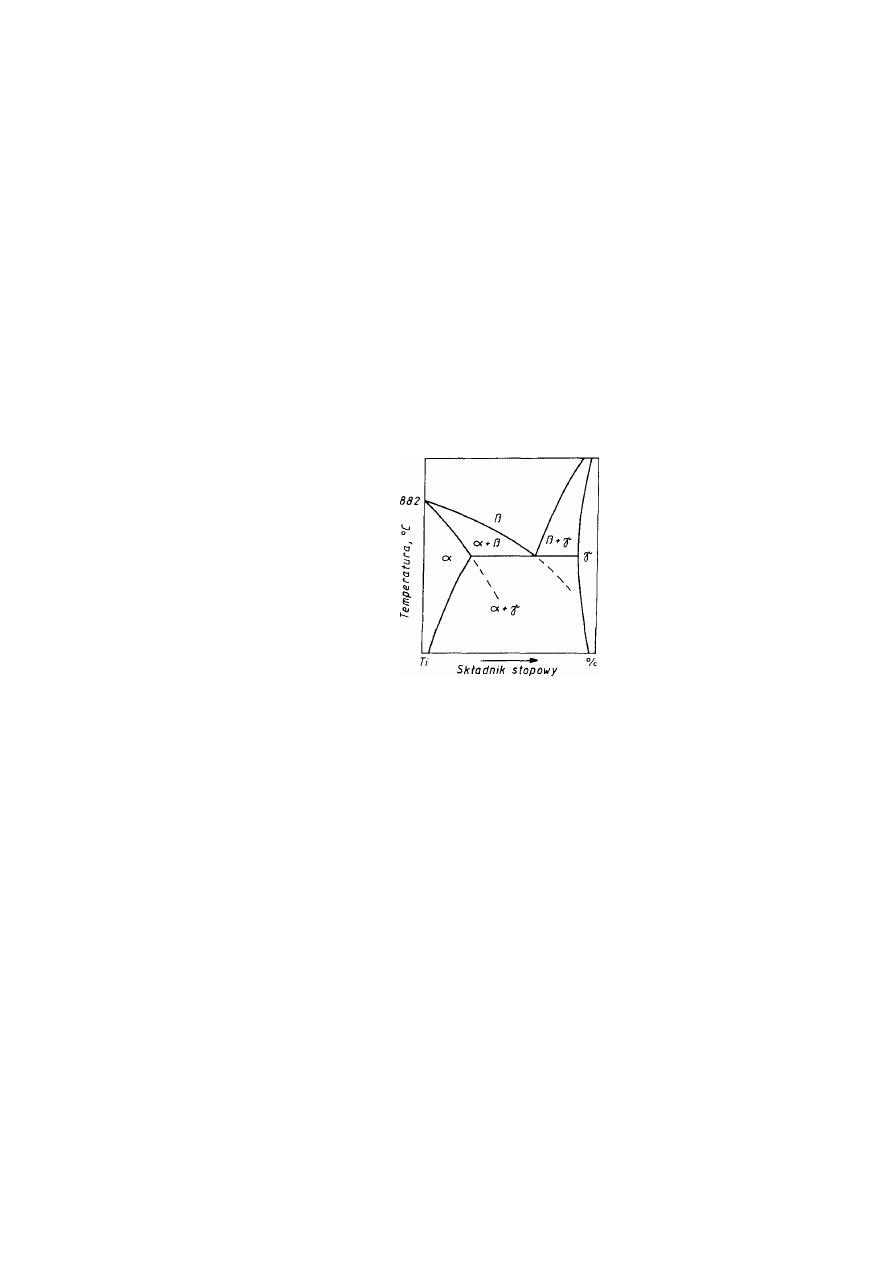
173
JW
W niektórych stopach tytanu (m.in. z Mo, V, Nb, Ta, W i Re) może pojawić się faza
martenzytyczna
α", będąca także przesyconym roztworem stałym pierwiastka stopowego w
tytanie, ale krystalizująca w układzie rombowym. Powstaje ona przy dużych zawartościach
składników stopowych, jest drobniejsza niż faza
α' i bardziej plastyczna. Może współistnieć z
fazą
α i metastabilną fazą β, nie występuje obok fazy α'. Faz α' i α" często się nie rozróżnia,
traktując je jako jedną fazę typu martenzytycznego.
Układem dwuskładnikowym tytan-pierwiastek stopowy III typu jest układ z przemianą
eutektoidalną (rys. 11.3), podczas której następuje rozkład roztworu stałego pierwiastka
stopowego w tytanie
β. Zgodnie z wykresem równowagi produktem przemiany eutektoidalnej
powinna być mieszanina faz
α + γ (faza międzymetaliczna). Okazuje się jednak, że w stopach
tytanu z niektórymi metalami (tzw. przejściowymi), przy ich ochładzaniu z obszaru istnienia
trwałej fazy
β, dla pewnego zakresu stężeń przemiana eutektoidalną jak gdyby nie zachodzi i
poniżej temperatury eutektoidu utrwala się mieszanina faz
α + β (linie kreskowe na rys. 11.3).
Taki nieprawidłowy przebieg przemiany eutektoidalnej wykazują przede wszystkim podwójne
stopy tytanu z chromem, manganem, kobaltem lub żelazem, na skutek bardzo małej prędkości
reakcji rozkładu eutektoidalnego, toteż przy odpowiedim stężeniu pierwiastka stopowego i
określonej prędkości chłodzenia łatwo można w nich uzyskać dwufazową strukturę
α + β.
Rys. 11.3. Typ III układu równowagi tytan-pierwiastek stopowy
(pierwiastek stopowy wywołuje przemianę eutektoidalna)
Jak więc z powyższych rozważań wynika, stopy tytanu w zależności od struktury
występującej w temperaturze pokojowej (uzyskanej przez odpowiedni dobór składników
stopowych oraz ewentualną obróbkę cieplną) można podzielić na trzy główne grupy:
• jednofazowe stopy α,
• dwufazowe stopy α + β,
• jednofazowe stopy β.
Każda z tych grup wykazuje charakterystyczne połączenie własności mechanicznych i
technologicznych, decydujące o ich przeznaczeniu. Wszystkie stopy tytanu stosowane są przede
wszystkim w przemyśle lotniczym i chemicznym.
Skład chemiczny ważniejszych przemysłowych stopów tytanu podano tabl. 11.1.
Stopy
α. Głównym składnikiem stopowym w stopach α jest aluminium, które podwyższa
wytrzymałość i zmniejsza gęstość, ale pogarsza plastyczność, dlatego, jego zawartość ogranicza
się zwykle do 8%. Również cyna podwyższa wytrzymałość stopów, nie zmniejszając jednak ich
plastyczności i zdolności do odkształceń plastycznych w wysokich temperaturach. Jej zawartość
w stopach
α nie przekracza 6%. Podobne własności wykazuje cyrkon.
Niektóre stopy
α obok aluminium zawierają małe ilości (1-2%) niektórych pierwiastków
stabilizujących fazę
β (Nb, Ta, V, Mo). Dodatek tych pierwiastków z jednej strony podwyższa
wytrzymałość stopów, z drugiej - polepsza ich zdolność do obróbki plastycznej na gorąco, co
jest szczególnie ważne w przypadku stopów zawierających większą ilość aluminium.
Jednocześnie wysoka zawartość aluminium równoważy ich wpływ na strukturę, tak że stopy
zachowują jednofazową strukturę
α.
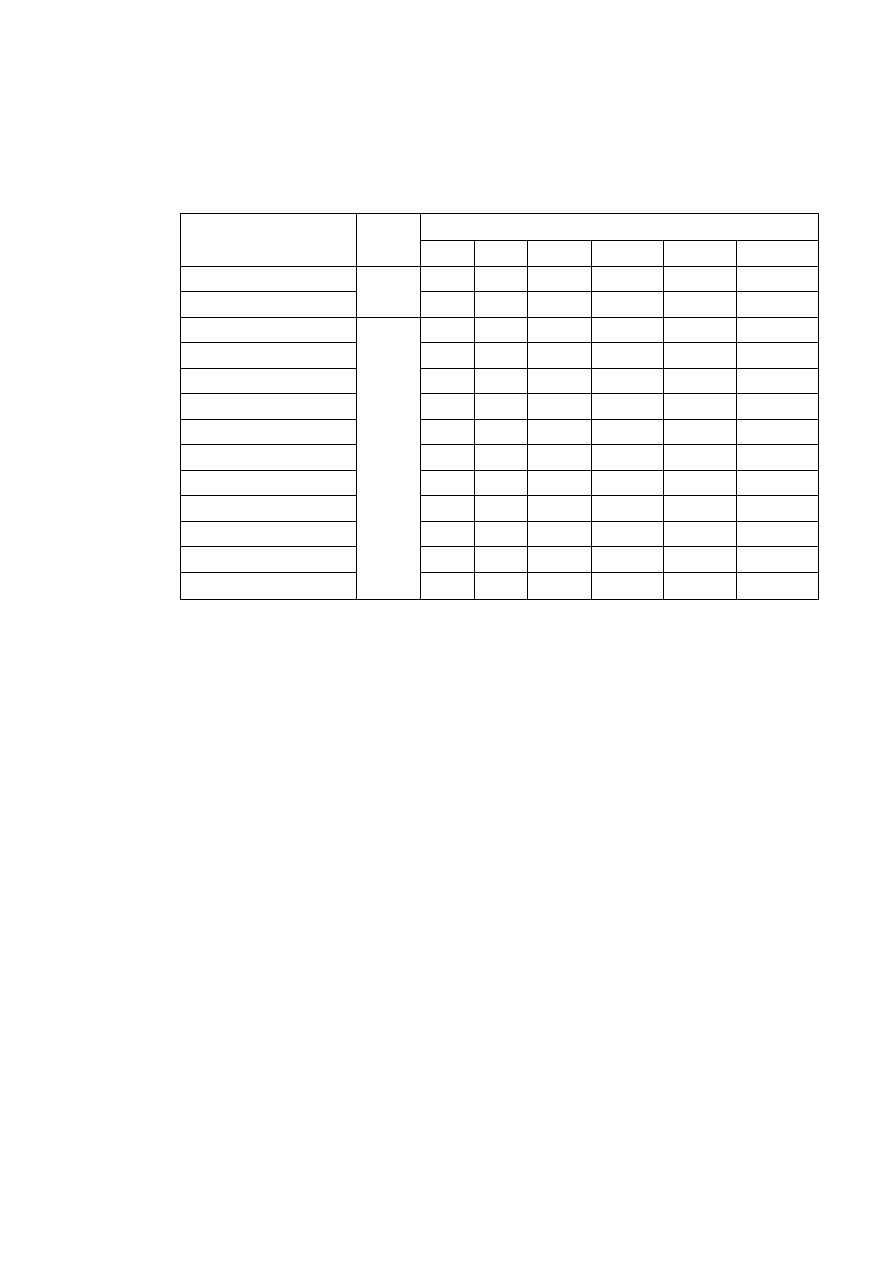
174
JW
Wszystkie stopy
α cechuje dobra spawalność i żarowytrzymałość. Pierwsza własność jest
wynikiem jednofazowej struktury, druga - obecności aluminium. Stopy a nie podlegają obróbce
cieplnej poza wyżarzaniem rekrystalizującym i wyżarzaniem odprężającym, stosowanymi
oczywiście w razie potrzeby. Umacnia się je jedynie przez zgniot, podobnie jak tytan techniczny.
Tablica 11.1
Skład chemiczny ważniejszych stopów tytanu
Skład chemiczny, % (reszta tytanu)
Oznaczenie stopu
Typ
stopu
Al Mo Sn Si
V
inne
Ti-5Al-2,5Sn, BT5-1
*
5 - 2,5 - -
-
RMI 5621
α
5 1 6 -
- 2
Zr
RMI 3A1-2,5V
3
-
-
-
2,5
-
Ti.4Al-3Mo.lV 4
3
-
-
1
-
Ti-6Al-2Sn-4Zr-2Mo 6
2
2
-
-
4
Zr
Ti-6Al-4V, BT6*
6
-
-
-
4
-
Ti-6Al-6V-2Sn 6
-
2
-
6
-
Ti-7Al-4Mo 7
4
-
-
-
-
BT3-1
*
5,5
2
-
0,2
-
2 Cr, l Fe
BT4
*
4 - - - - 1,5
Mn
BT8* 6,5
3,5
-
0,2
-
-
BT9* 6,5
3,5
-
0,2
-
2
Zr
BT20*
α + β
6 1 - -
1 2
Zr
*
Stopy rosyjskie, pozostałe amerykańskie.
Stopy
α + β. Warunkiem uzyskania dwufazowej struktury α + β jest obecność w stopie
odpowiedniej ilości pierwiastków stabilizujących fazę
β. Najbardziej odpowiednimi zarówno ze
względu na własności ich roztworów w tytanie, jak i cenę są mangan, wanad, molibden, chrom i
żelazo. Wszystkie te pierwiastki rozpuszczają się bardzo dobrze w tytanie
β i bardzo słabo w
tytanie
α, w związku z czym ich wpływ na własności mechaniczne występuje przede wszystkim
w fazie
β. Własności mechaniczne stopów tej grupy zależą więc od ilości i własności fazy β.
Większość jednak stopów
α + β oprócz wymienionych pierwiastków zawiera jeszcze
aluminium, które dobrze rozpuszcza się zarówno w tytanie
α, jak i w tytanie β. W takim
przypadku własności stopu są wypadkową własności obu faz.
Ogólnie więc stopy
α + β można podzielić na dwie podgrupy:
a) stopy zawierające tylko pierwiastki stabilizujące fazę
β,
b) stopy zawierające pierwiastki stabilizujące fazę
β i aluminium.
Stopy
α + β zawierające aluminium cechują wysokie wskaźniki własności mechanicznych.
Na rys. 11.4a, b i c pokazano zakresy wytrzymałości na rozciąganie w podwyższonych
temperaturach dla poszczególnych typów stopów tytanu, a na rys. 11.4d - krzywe reprezentujące
średnie wartości tej wytrzymałości. Wyraźnie widać, że stopy
α + β zawierające aluminium są
stopami najbardziej wytrzymałymi i w temperaturze pokojowej i w temperaturach
podwyższonych. Natomiast pozostałe stopy
α + β i stopy α do temperatury około 370°C mają
wytrzymałość zbliżoną, powyżej tej temperatury bardziej wytrzymałe są stopy
α (wpływ
aluminium).
Wytrzymałość zmęczeniowa i udarność stopów
α + β zawierających aluminium są mniej
więcej takie same, jak stopów bez aluminium, wytrzymałość na pełzanie nieco wyższa. Ponadto
stopy
α + β zawierające aluminium cechuje mniejsza gęstość, lepsza obrabialność skrawaniem i
niższa temperatura przemiany martenzytycznej. Przykładową mikrostrukturę stopu
α + β (BT3-
1) po przeróbce plastycznej okazano na rys. 11.5.

175
JW
Rys. 11.4. Wytrzymałość na rozciąganie w stanie wyżarzonym: a) stopów
α, b) stopów α + β
nie zawierających aluminium, c) stopów
α + β zawierających aluminium; d) średnia
wytrzymałość na rozciąganie: l — stopów
α, 2 — stopów α + β nie zawierających
aluminium, 3 - stopów
α + β zawierających aluminium
Rys.11.5. Mikrostruktura stopu tytanu
α + β (BT3-1) po obróbce plastycznej. Na tle ciemnych
kryształów
β widoczne jasne, iglaste kryształy α. Traw. odczynnikiem Krolla. Powiększ. 250x
Wytrzymałość większości stopów
α + β może być dodatkowo podwyższona przez
odpowiednią obróbkę cieplną, składającą się z przechłodzenia i starzenia. Pierwszy proces
polega na nagrzaniu do temperatury istnienia stabilnej fazy
β lub nieco poniżej (tzn. do obszaru
dwufazowego
α + β, ale w pobliżu jego górnej granicy), wygrzaniu w tej temperaturze i
następnie szybkim ochłodzeniu. W wyniku otrzymuje się bądź fazę
β w stanie nierównowagi,
bądź mieszaninę faz
α + β, w której faza β jest także w stanie nierównowagi. W żadnym
przypadku nie wolno jednak dopuścić do przemiany martenzytycznej i wydzielenia się fazy
α'.
Proces starzenia polega na nagrzaniu do temperatury 450
÷ 600°C, zależnie od składu
chemicznego obrabianego stopu. Czas wygrzewania i sposób chłodzenia (powietrze, woda)
również zależą od składników stopu. W czasie starzenia następuje częściowy rozkład nietrwałej
fazy
β na α + β. Bez względu na pierwotną strukturę stopu podlegającego starzeniu (β czy α +
β), własności mechaniczne po starzeniu zależą od postaci wydzieleń fazy α powstającej w
wyniku rozkładu fazy
β. oraz od ilościowego stosunku faz α i β .
Przechłodzenie i starzenie zwykle powodują spadek wskaźników własności plastycznych,
natomiast wytrzymałość wzrasta o około 35% w stosunku do wytrzymałości stopów w stanie
wyżarzonym.
Stopy
α + β podlegają również wyżarzaniu rekrystalizującemu i odprężającemu. podobnie jak
stopy
α.
Spawalność stopów
α + β jest zależna przede wszystkim od procentowej zawartości
pierwiastków stabilizujących fazę
β. Przy zawartości do 3% stopy α + β są mniej czułe na
szybkość chłodzenia po spawaniu i wykonane z nich złącza spawane mają zadowalające
własności mechaniczne. Jeśli jednak zawartość pierwiastków stopowych (bez aluminium)
przekracza 3%, złącza bezpośrednio po spawaniu są kruche i wymagają odpowiedniej obróbki
cieplnej.

176
JW
Stopy
β. Trzecią grupę stopów tytanu stanowią jednofazowe stopy β, które można uzyskać bądź
przez odpowiednią zawartość pierwiastków stabilizujących fazę
β, bądź przez przechładzanie z
obszaru stabilnej fazy
β w wyższych temperaturach, przy stężeniach składnika stopowego
niższych od stanu równowagi. Praktycznie wykorzystuje się drugą metodę, otrzymując jednak
stopy
β o strukturze niestabilnej.
Obecnie znanych jest kilka seryjnie produkowanych stopów tytanu o strukturze
β
(niestabilnej): amerykańskie Ti-13V-llCr-3Al, Beta 3 (11,5% Mo, 4,5% Sn, 6% Zr) i RMI lAl-
8V-5Fe oraz rosyjskie BT14 (4% Al, 3% Mo, 1% V), BT15 (3% Al. 8% Mo, 11% Cr) i BT16
(2,5% Al, 7,5% Mo).
Stopy
β cechuje bardzo wysoka wytrzymałość, zwłaszcza po obróbce cieplnej. Na przykład,
stop Ti-13V-llCr-3Al w stanie wyżarzonym wykazuje wytrzymałość na rozciąganie R
m
= 930
MPa, w stanie przechłodzonym i starzonym R
m
= 1275 MPa, a po walcowaniu na zimno i
starzeniu R
m
= 1750 MPa, co czyni go metalem o najwyższej wytrzymałości właściwej ze
wszystkich tworzyw konstrukcyjnych (gęstość stopu wynosi 4,85 g/cm
3
).
Stopy
β są spawalne zarówno w stanie wyżarzonym, jak i starzonym. Również ich obróbka
skrawaniem nie przedstawia większych trudności.
12. Stopy łożyskowe
Stopy łożyskowe stanowią specjalną grupę materiałów stosowanych do wytwarzania i
wylewania panewek łożysk ślizgowych. Ze względu na specyficzne warunki pracy tych łożysk,
materiał na panewki musi spełniać następujące warunki:
• współczynnik tarcia między powierzchnią czopu wału a panewką powinien być
możliwie mały,
• materiał panewki powinien być odporny na ścieranie,
• materiał panewki powinien mieć dostateczną wytrzymałość w temperaturze -200°C.
Panadto stopy łożyskowe powinny być dostatecznie odporne na korozję oraz nie wykazywać
przy odlewaniu skłonności do likwacji składników. Dlatego stopy łożyskowe powinny
wykazywać własności twardych materiałów w celu zapewnienia dostatecznej wytrzymałości i
uzyskania małego współczynnika tarcia, oraz miękkich materiałów w celu umożliwienia
panewce dostosowania się kształtu czopu wału. Takie skojarzenie przeciwnych sobie własności
można uzyskać jedynie w stopach złożonych z dwóch lub więcej faz o różnych własnościach.
Struktura takich stopów powinna składać się z miękkiego podłoża i możliwie równomiernie
rozłożonych w nim twardych kryształów. W czasie pracy twarde kryształy przejmują obciążenie
i przekazują je na całą panewkę. Jednocześnie ich ilość powoduje wytworzenie między
powierzchnią wału i powierzchnią panewki pewnej przestrzeni, w której umieszcza się smar. W
przypadku, gdy poszczególne części panewki zostaną przeciążone, twarde kryształy wgniatają
się w tych miejscach w miękkie podłoże i następuje wyrównanie obciążenia.
Jako stopy łożyskowe w praktyce przemysłowej stosuje się żeliwa, brązy oraz łatwo topliwe
stopy na osnowie cyny, ołowiu, cynku i aluminium. Panewki żeliwne wytwarza się z szarego
żeliwa perlitycznego, które jest materia najtańszym i może przenosić dość duże naciski
jednostkowe, ale ze względu na stosunkowo duży współczynnik tarcia nie nadaje się do pracy
przy dużej liczbie obrotów.
Do wyrobu panewek brązowych wykorzystuje się omówione już (rozdz. 8) brązy cynowe,
ołowiowe, krzemowe itd. Do tego celu stosuje się także niektóre mosiądze zawierający 3,0
÷
4,5% Si i 2,5
÷ 4,0% Pb). Materiały te mają dość dobrą wytrzymałość, toteż panewki z nich
wykonane mogą pracować przy dużych naciskach jednostkowych i dużej liczbie obrotów.
Mikrostrukturę panewki łożyskowej wylanej brązem ołowiowym pokazano na rys. 12.1.
Zgodnie z Polską Normą PN-82/H-87111 (tabl. 12.1), stopy łożyskowe na osnowie cyny
(zwane także babitami cynkowymi) zawierają 7
÷12% antymonu i 2,5 ÷ 6,5 % miedzi. Struktura
tych stopów (rys. 12.2) składa się z kryształów roztworu stałego
α antymonu w cynie
(tworzących miękkie podłoże) oraz twardych kryształów fazy międzymetalicznej SnSb
(krzepnących w postaci regularnych sześcianów) i twardych kryształów fazy międzymetalicznej
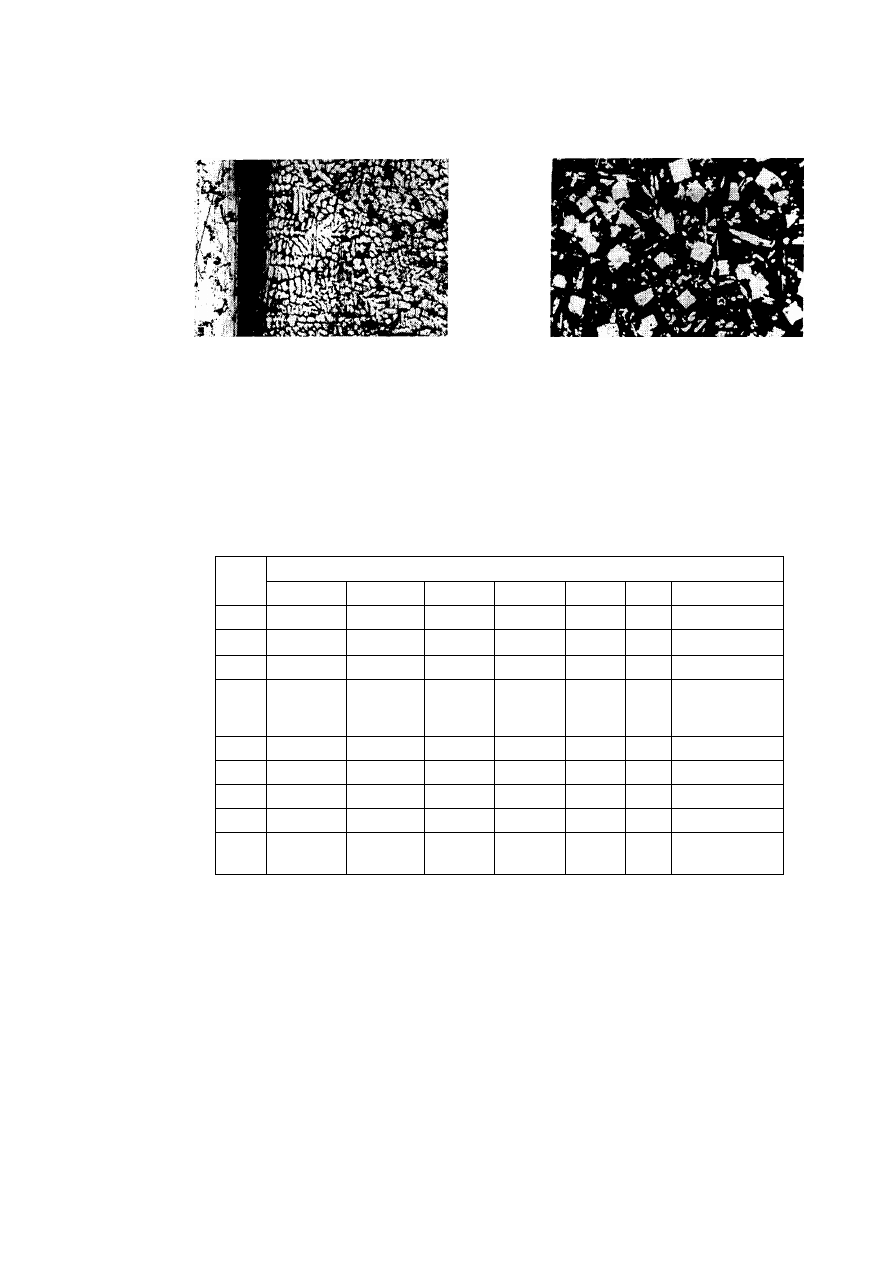
177
JW
Cu
3
Sn (krzepnących w połaci igieł). Te ostatnie, charakteryzując się największą temperaturą
topnienia, krzepną pierwsze, tworząc jak gdyby rodzaj szkieletu, który utrudnia przesuwanie się
krzepnących kryształów SnSb i zapewnia ich równomierne rozmieszczenie w roztworze
α.
Rys. 12.1. Panewka łożyska. Od lewej: stal,
ciemna warstewka stopu Sn-Pb oraz brąz
ołowiowy składający się z jasnych kryszta-
łów miedzi i ciemnych kryształów ołowiu
(osnowa). Nie trawione. Powiększ. 100x
Rys. 12.2. Mikrostruktura stopu łożyskowce na
osnowie cyny (Ł83), zawierającego 11% Sb i 6%
Cu. Na ciemnym tle roztworu stałego antymonu w
cynie widać jasne regularna kryształy fazy
międzymetalicznej SnSb oraz iglaste kryształy fazy
międzymetaliczne Cu
3
Sn. Traw. 5% roztworem
HNO
3
. Powiększ. 100x
Tablica 12.1
Skład chemiczny łożyskowych stopów cyny i ołowiu (wg PN-82/H-87111)
oraz stopów cynku (wg PN-80/H-87101)
Skład chemiczny, %
Cecha
stopu
Sn Sb Cu
As
Pb
2n inne
Ł89 reszta
7,25-8,25
2,5-3,5 - - -
-
Ł83 reszta
10,0-12,0
5,5-6,5 - - -
-
Ł83Te
reszta
10,0-12,0
5,5-6,5
-
max 1,5
-
0,2-0,5 Te
Ł808
reszta
11,0-13,0
5,0-6,5
0,2-0,5
-
-
1,0-1,5Cd
0,3-0,6 Ni
0,03-0,2 Cr
Ł16 15,0-17,0
15,0-17,0
1,5-2,0 - reszta -
-
Ł10As 9,0-11,0 13,0-15,0
1,0-2,0 0,5-0,9 reszta -
-
Ł6 5,0-7,0
5,5-7,5 -
- reszta
-
-
Z105 -
- 4,5-5,8 - -
reszta
9,0-11,5
Al
Z284
-
- 3,0-5,4 - -
reszta
26,0-30,0 Al
0,02-0,05 Mg
Łożyskowe stopy na osnowie cyny mają bardzo dobre własności, w związku z czym
wykonane z nich panewki mogą pracować zarówno przy obciążeniach statycznych, jak i
dynamicznych. Ze względu jednak na wysoką cenę i deficytowość cyny, w wielu przypadkach
stosuje się zastępcze stopy na osnowie ołowiu, w których zawartość cyny jest ograniczona do
kilku lub kilkunastu procent, a nawet stopy bezcynowe, zawierające wapń, sód, lit, aluminium i
inne metale.
Krajowe stopy łożyskowe na osnowie ołowiu zawierają antymon, cynę, miedź, czasem arsen
lub tellur (tabl. 12.1). W stopach tych miękką osnowę stanowią roztwory stałe pierwiastków
stopowych w ołowiu lub eutektyki, twarde wtrącenia – odpowiednie fazy międzymetaliczne, np.
SnSb, Cu
3
Sn, SnAs
2
itd. – rys.12.3.
Łożyskowe stopy na osnowie cynku (PN-80/H-87101) zawierają głównie aluminium i miedź.
Orientacyjne warunki pracy i zastosowanie znormalizowanych w Polsce stopów łożyskowych
na osnowie cyny, ołowiu i cynku podano w tabl. 12.2.

178
JW
Spośród stopów aluminium na panewki łożysk ślizgowych stosuje się stopy z antymonem i
magnezem, z niklem, a także z miedzią i krzemem. Ich znaczenie jest jednak niewielkie.
Tablica 12.2.
Orientacyjne warunki pracy i zastosowanie stopów łożyskowych (wg PN-82/H-87111 i PN-
80/H-87102)
Cecha
stopu
Orientacyjne warunki pracy
Zastosowanie
Ł89
odlewane odśrodkowo taśmy bimetalowe na
panewki łożysk ślizgowych mocno obciążonych
Ł83
obciążenia statyczne i dynamiczne: naciski
do 10 MPa, prędkość obwodowa powyżej 5
m/s, iloczyn nacisku i prędkości poniżej 50
MPa • m/s
wylewane panewki łożysk ślizgowych mocno
obciążonych
Ł83Te
obciążenia statyczne i dynamiczne: naciski
do 10 MPa, prędkość obwodowa powyżej 3
m/s, iloczyn nacisku i prędkości 15
÷50 MPa
-m/s
panewki łożysk ślizgowych mocno obciążonych
Ł80S
obciążenia statyczne i dynamiczne: naciski
do 19 MPa, prędkość obwodowa do 20 m/s,
iloczyn nacisku i prędkości do 38 MPa •m/s
panewki łożysk turbin parowych oraz wysoko
obciążonych przekładni zębatych
Ł16
obciążenia statyczne: naciski do 10 MPa,
prędkość obwodowa powyżej 1,5 m/s,
iloczyn nacisku i prędkości do 15 MPa
·m/s
panewki łożysk średnio obciążonych
Ł10As
obciążenia statyczne: naciski do 10 MPa,
prędkość obwodowa powyżej 1,5 m/s,
iloczyn nacisku i prędkości do 30 Mpa
·m/s
panewki łożysk średnio obciążonych
Ł6 obciążenia uderzeniowe
taśmy bimetalowe na panewki łożysk
samochodowych
Z105
małe i średnie naciski, małe i średnie
prędkości obwodowe
w warunkach pracy niekorozyjnej zastępuje
brąz B555, a nawet stop Ł 10As
Z284 naciski do 20 MPa, maks. temperatura pracy
100°C
w warunkach pracy niekorozyjnej zastępuje
brązy B10, B101 i B555
Rys. 12.3. Mikrostruktura stopu łożyskowego na osnowie ołowiu, zawierającego 16% Sb i 6%
Sn. Na tle eutektyki ołów-antymon-cyna widoczne jasne kryształy fazy międzymetalicznej SnSb
i ciemne kryształy ołowiu. Traw. 5% roztworem HN03. Powiększ. 200x

179
JW
13. Stopy żarowytrzymałe
Stopami żarowytrzymałymi nazywa się stopy wykazujące:
a) dużą wytrzymałość doraźną w temperaturze otoczenia i temperaturach wysokich,
b) odporność na długotrwałe działanie obciążeń stałych w wysokich temperaturach
(wytrzymałość na pełzanie),
c) odporność na długotrwałe działanie obciążeń zmiennych w wysokich temperaturach
(wytrzymałość zmęczeniowa),
d) odporność na wielokrotne zmiany temperatury związane lub nie związane z zmianą
obciążeń (wytrzymałość na zmęczenie cieplne),
e) odporność na korozyjne działanie gazów w wysokich temperaturach (żaroodporność).
Oczywiście poszczególne stopy żarowytrzymałe spełniają powyższe warunki w różnym
stopniu.
Zasadniczym czynnikiem określającym przydatność stopu żarowytrzymałego do danego
zastosowania jest jego optymalna temperatura pracy. Temperatura ta zależy przede wszystkim
od składu chemicznego stopu, ale również od wielkości i rodzaju losowanych obciążeń,
dopuszczalnych odkształceń i założonego czasu pracy (np. czas pracy elementów turbin
lotniczych wynosi około 1000 h, czas pracy turbin stacjonarnych - 10000 do 100000 h).
Najważniejsze grupy stopów żarowytrzymałych to stopy niklu, stopy kobaltu stopy żelazowo-
niklowe, które łącznie nazywane są często nadstopami lub superstopami. Perspektywicznymi
materiałami żarowytrzymałymi są stopy metali trudno topliwych (molibdenu, wolframu, niobu,
tantalu, wanadu), a także stopy berylu.
13.1. Żarowytrzymałe stopy niklu
Do tej grupy materiałów należą stopy niklu z chromem, molibdenem, kobaltem, wolframem, tytanem,
aluminium, borem, żelazem i inne, charakteryzujące się wysoką żaroodpornością i żarowytrzymałością, a
przeznaczone głównie do budowy turbin gazowych i silników odrzutowych, na elementy pracujące w
warunkach w wysokich naprężeń i temperaturze 550
÷1030°C. Na rynkach światowych stopy te znane
pod różnymi nazwami (np. Hastelloy, Inconel, MAR, Nimocast, Nimonic, Rene, Udimet itd.), przy czym
jeśli pod jedną nazwą produkowanych jest kilka stopów, różniących się składem chemicznym i
własnościami, nazwa ta jest uzupełnia dodatkowym oznaczeniem liczbowym lub literowym (tabl. 13.1).
Dzielą się stopy odlewnicze i do przeróbki plastycznej.
Rys. 13.1. Mikrostruktura żarowytrzymałego stopu niklu do przeróbki plastycznej w stanie
wyżarzonym. Widoczne jasne kryształy roztworu stałego
γ i drobne ciemne wydzielenia faz
międzymetalicznych. Traw. elektrolitycznie w 10% roztworze kwasu szczawiowego. Powiększ.
500x
Większość żarowytrzymałych stopów niklu podlega obróbce cieplnej złożonej z przesycania i
starzenia (utwardzanie dyspersyjne). Po takiej obróbce struktura stopów składa się z jednoro-
dnych ziarn roztworu stałego pierwiastków stopowych w niklu i równomiernie rozłożonych,
bardzo drobnych wydzieleń faz umacniających np. Ni
3
Ti, Ni
3
Al, Ni
3
(Al,Ti) (rys. 13.1).
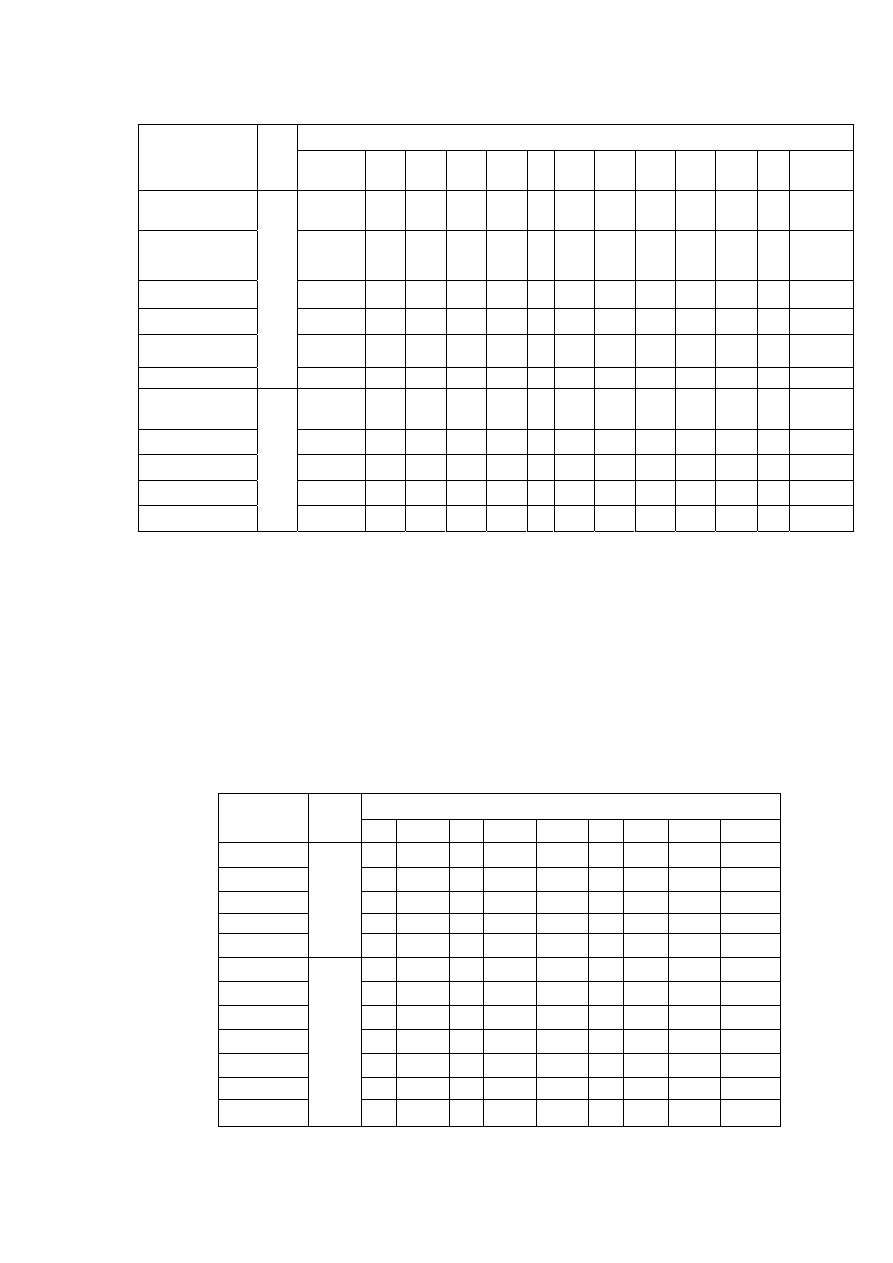
180
JW
Tabl.13.1.
Skład chemiczny niektórych żarowytrzymatych stopów niklu
Skład chemiczny, % (reszta nikiel)
Nazwa stopu
Rod
zaj
stop
C
Mn
max
Si
max
Cr Mo Nb Co W AI
Ti
Zr
Fe inne
Nimonic 90*
max 0,13 1,0 1,0 19,5
-
-
18
-
1,5 2,5 0,15 1,5
max
0,02 B
Nimonic 105*
max 0,12
1,0
1,0
14,8
5
-
20
-
4,7
1,2
0,15
1,0
0,003-
0,010 B
Nimonic 115*
0,16
1,0 1,0 14,2 3,2 -
15
-
4,8 3,7
-
1,0 0,2 Cu
Inconel X-750
0,04
0,5 0,25 15,5
- 1,0
-
-
0,7 2,5
-
7,0
-
Udimet 700
0,15
-
-
15 5,2 - 18,5
-
4,25 3,5
-
0,5 0,05 B
Renę 85
prze-
rabia
-lny
pla-
sty-
czni
e
0,27 -
-
9,3 3,25 -
15 5,35 5,25 3,25 0,03
-
0,015 B
MAR-M246 0,15
0,5
0,5
9
10
-
10
10
5,5
1,5
0,05
-
0,015
B
1,5 Ta
WAZ-20
0,15 - - - - - -
18,5 6,2 - 1,5
- -
IN-6201
0,03 - - 20 0,5 1,0 20 2,3 2,4 3,6 0,05
- 1,5
Ta
TAZ-8B
0,125 - - 6 4
1,5
5 4 6,0 - 1,0 - 8,0
Ta
Nimocast 258*
odle-
wni-
czy
0,2 0,3
0,4
10 - -
20 - 4,8
3,7 2,0
-
* Stopy angielskie, pozostałe amerykańskie.
13.2. Żarowytrzymałe stopy kobaltu
Stopy kobaltu stanowią dużą grupę stopów przeznaczonych do pracy w wysokich
temperaturach. Wytrzymałość ich w wysokich temperaturach (860
÷1090°C) jest jednak niższa
niż stopów niklu, co w pewnym stopniu ogranicza ich zastosowanie. Poważną natomiast zaletą
stopów kobaltu jest tańsza technologia produkcji (nie wymagają topienia próżniowego) i duża
odporność na zmęczenie cieplne. Ta ostatnia cecha powoduje, że znalazły one zastosowanie na
łopatki kierujące w dyszach inne części silników turboodrzutowych.
Tablica 13.2
Skład chemiczny niektórych żarowytrzymałych stopów kobaltu produkcji USA
Skład chemiczny, % (reszta kobalt)
Nazwa
stopu
Rodzaj
stopu C Mn Si Cr Ni Mo W Fe inne
HS-25 0,10
1,5
0,4
20
10
-
15
3
-
S-816 0,38
1,2
0,4
20
20
4
4
4
4,0
Nb
V-36 0,27
1,0
0,4
25
20
4
2
3
2,0
Nb
Haynes 188
0,10 1,25
0,4
22
22
-
14 3
0,03 La
Stellite 6
prze-
rabia-
ny
plasty-
cznie
1,0
1,4 0,7
30 2,5 1,5
4 3
-
HS-21 0,25
0,6
0,6
27
3
5
-
1
-
HS-31 0,50
0,5
0,5
25
10
-
7,5
1,5
-
X-45 0,25
1,0
-
25,5
10,5
-
7
2
0,01
B
Stellite 12
1,4
1,0
2,0
30
3
1
8,3
3
-
Sellite F
1,75
1,0
2,0
25
22
1 12,3
3
-
Sellite 1
2,4
1,0
2,0
31
3
1 12,5
3
-
Stellite 190
odle-
wni-
czy
3,3
1,0 2,0
26 3 1 14,5 3
-
Wszystkie przemysłowe stopy kobaltu zawierają chrom, który podwyższa ich odporność na
korozję, a ponadto - zależnie od gatunku - różne ilości wolframu, niklu, niobu, tantalu,
181
JW
molibdenu, aluminium i in. (tabl. 13.2)..
W obecności dostatecznej ilości węgla niektóre z tych pierwiastków tworzą trudno topliwe
węgliki (np. V, Mo, Ta, Nb), inne wpływają na własności osnowy.
Dzielą się na stopy do przeróbki plastycznej i odlewnicze. Te ostatnie wykazują bardzo dużą
odporność na ścieranie i pod nazwą stellitów są wykorzystywane także jako materiały
narzędziowe oraz do napawania powierzchni części maszyn. Stopy kobaltu są stosowane bądź w
stanie surowym (niektóre odlewy), bądź obrobionym cieplnie (przesycanie i starzenie).
13.3. Żarowytrzymałe stopy żelazowo-niklowe
Stopy żelaza z niklem i chromem oraz - zależnie od gatunku - z molibdenem. wolframem,
niobem, kobaltem, tytanem, aluminium, borem i in. (tabl. 13.3) charakteryzują się wysoką
żarowytrzymałością i żaroodpornością, niższą jednak niż omówione wyżej stopy niklu i kobaltu.
Są natomiast od nich o wiele tańsze (dzięki znacznej zawartości żelaza).
Stopy żelazowo-niklowe są stosowane zarówno w postaci lanej, jak i przerobionej
plastycznie, zwykle po obróbce cieplnej (przesycanie i starzenie).
Tablica 13.5
Skład chemiczny niektórych żarowytrzymałych stopów żelazowo-niklowych produkcji
USA
Skład chemiczny, % (reszta żelazo)
Nazwa stopu
Rodzaj
stopu
C Mn Si Cr Ni Mo W Ti
inne
Discaloy
0,04 0,9 0,8 13,5
26 2,75
- 1,75
0,1
AI
Incoloy 800
0,05 0,75
0,5
21
32,5
-
-
0,38
0,38 AI
Incoloy
901
0,05
0,45
0,4 13,5
42,7 6,2 - 2,5
0,25
AI,
0,015
B
S-590
0,46 1,25
0,4
20,5
20
4
4
-
20 Co, 4Nb
Refractaloy 26
przera-
bialny
plastycz-
nie
0,03
0,8
1
18
38
3,2
-
2,6
0,2 AI, 20 Co
CRM 60
1,05
5
0,5
22
5
1
1
-
1 Nb, 0,003 B
Duraloy 0,50
0,8
1
25,5
45,5
3,25 3,25
-
3,25
Co
lllium PD
odlewniczy
0,08 - - 27 5 2 - -
7
Co
13.4. Molibden i jego stopy
Molibden jest metalem o temperaturze topnienia 2610°C i gęstości 10,2 g/cm
3
. Cechują go:
•
wysoki moduł sprężystości,
•
dobra odporność na gwałtowne zmiany temperatury (dzięki małemu współczynnikowi
rozszerzalności cieplnej i wysokiej przewodności cieplnej),
•
dobra przewodność elektryczna (około 33% przewodności Cu),
•
stosunkowo mały przekrój czynny pochłaniania neutronów.
Do jego zalet należy również dość szerokie rozpowszechnienie w przyrodzie i dobrze
opracowaną technologię wytwarzania.
Zasadniczą natomiast wadą molibdenu i stopów na jego osnowie jest brak odporności w
podwyższonych temperaturach (powyżej 650°C) na korodujące działanie gazów
atmosferycznych, a szczególnie tlenu, tak że stosowanie w wysokich temperaturach jest
uwarunkowane specjalnymi ochronnymi pokryciami ceramicznymi.
Jako materiały konstrukcyjne wykorzystuje się obecnie molibden techniczny zawierający
około 0,02% C), stop molibden-tytan (zawierający 0,04% C i 0,5% Ti), stop molibden-wolfram
(30% W), stop molibden-ren (41% Re), stop TZC 1,2% Hf i 0,05% C) i stop TZM (0,015% C,
0,5% Ti i 0,08% Zr). Ten ostatni w temperaturze 1315°C ma R
m
= 310 MPa.
Molibden i jego stopy są stosowane w lotnictwie i kosmonautyce na dysze rakiet, części
silników, przednie części skrzydeł itd.

182
JW
13.5. Wolfram i jego stopy
Szczególnymi zaletami wolframu są bardzo wysoka temperatura topnienia (3415
o
C) i
wyjątkowa wytrzymałość w wysokich temperaturach, ujemnymi cechami - duża gęstość (19,3
g/cm
3
) i kruchość w niskich temperaturach. Poza tym wolfram, jak większość metali trudno
topliwych, łatwo utlenia się w wysokich temperaturach, co powoduje konieczność stosowania
pokryć ochronnych. Te same własności cechują stopy wolframu z tlenkiem toru (l lub 2% ThO
2
),
wolframu z renem (4% lub 25% Re) i molibdenem (15% Mo).
Wolfram i jego stopy stosowane są doświadczalnie w konstrukcjach lotniczych i
kosmonautycznych.
13.6. Niob i jego stopy
Niob i jego stopy z molibdenem, wolframem, tantalem, cyrkonem, hafnem, tytanem,
wanadem i in. są zaliczane do najcenniejszych tworzyw żarowytrzymałych, głównie dzięki
wysokiej temperaturze topnienia niobu (2468°C), jego małej gęstości (8,57 g/cm
3
) i małemu
przekrojowi czynnemu pochłaniania neutronów.
Inne cenne własności niobu to plastyczność w temperaturach obniżonych i obrabialność,
lepsze niż molibdenu i wolframu. W podwyższonych temperaturach niob staje się miękki i
plastyczny, ale za pomocą pierwiastków stopowych można jego wytrzymałość podwyższyć do
tego stopnia, że stopy niobu z powodzeniem mogą konkurować z innymi metalami
żarowytrzymałymi do temperatury 1815°C. Poważną wadą niobu i jego stopów jest mała
odporność na utlenianie w wysokich temperaturach i związana z tym konieczność stosowania
specjalnych pokryć ochronnych.
Stopy niobu są stosowane na elementy konstrukcyjne sztucznych satelitów, osłony i
elementy przegrzewaczy reaktorów jądrowych, zbiorniki i rurociągi na ciekłe metale, dysze
silników rakietowych, elementy komór spalania i części poszycia samolotów naddźwiękowych,
np. C 103 (10% Hf, 1% Ti, 0,7% Zr), B 66 (5% Mo,5%V, 1% Zr), C 129Y (10% W, 10% Hf,
0,1% Y), B 99 (22% W, 2% Hf, 0,07% C), Cb 132M (20% Ta, 15% W, 5% Mo, 1,5% Zr, 0,1%
C), F-48 (15% W, 5% Mo, 1% Zr, 0,05% C).
13.7. Tantal l jego stopy
Tantal cechuje bardzo wysoka temperatura topnienia (2996°C) doskonała obrabialność i
plastyczność, także w temperaturze poniżej -255°C, oraz dobra spawalność. Wadą tego
pierwiastka jest duża gęstość (16,6 g/cm
3
), mała odporność na utlenianie w wysokich
temperaturach (powyżej 650°C) i co najważniejsze niewielkie zapasy w skorupie ziemskiej (ok.
1,5% znanych zapasów niobu).
Stopy tantalu oprócz wymienionych własności cechuje wysoka żarowytrzymałość.
Stosowane są na elementy konstrukcyjne pojazdów kosmicznych i dysze silników rakietowych,
np. FS 61 (7,5% W), PS 63 (2,5% W, 0,15% Nb), T-lll (8% W, 2% Hf), KBI 41 (37,5% Nb,
2,5% W, 2% Mo).
13.8. Beryl
Bardzo ciekawym i perspektywicznym materiałem dla lotnictwa i techniki rakietowej jest
metaliczny beryl. Charakteryzuje się on bardzo małą gęstością (1,85 g/cm
3
), dość wysoką
temperaturą topnienia (1282°C), wysokim modułem sprężystości, wysoką wartością stosunku
wytrzymałości do gęstości oraz wysoką pojemnością i przewodnością cieplną. Wady berylu to
toksyczność, ograniczona plastyczność w niskich temperaturach i stosunkowo wysoka cena. Jak
dotąd, przemysłowe zastosowanie znalazł beryl technicznie czysty o kontrolowanej zawartości
tlenu (w postaci tlenku BeO). W postaci kutej w temperaturze otoczenia materiał ten ma R
m
ok.
700 MPa, w temperaturze 600°C - R
m
= 330 MPa.

JW
Pytania egzaminacyjne
1. Omówić budowę atomu.
2. Co to jest masa atomowa?
3. Omówić budowę układu okresowego pierwiastków.
4. Wyjaśnić strukturę elektronową atomów na przykładzie kilku pierwiastków.
5. Co to są metale przejściowe?
6. Co to są elektrony walencyjne?
7. Co to są pierwiastki elektrododatnie i elektroujemne?
8. Wymienić wiązania międzyatomowe.
9. Scharakteryzować wiązanie jonowe.
10. Scharakteryzować wiązanie atomowe.
11. Scharakteryzować wiązanie metaliczne.
12. Podać główne cechy metali.
13. Scharakteryzować wiązanie siłami van der Waalsa.
14. Omówić elementarne komórki sieci krystalicznych (A l, A2, A3 i tetragonalną).
15. Omówić wpływ budowy krystalicznej na własności metali.
16, Co to jest anizotropia i izotropia własności?
17. Co to są defekty punktowe struktur krystalicznych (podać przykłady)?
18. Co to jest dyslokacja krawędziowa?
19. Podać własności dynamiczne dyslokacji krawędziowej.
20. Co to jest dyslokacja śrubowa?
21. Omówić budowę granic ziarn.
22. Co to jest struktura mozaikowa?
23. Omówić zależność między wytrzymałością metalu a liczbą defektów jego sieci krystalicznej.
24. Co to są kryształy włoskowe?
25. Wyjaśnić powstawanie dyslokacji ze źródła Franka-Reada.
26. Omówić wpływ ilości dyslokacji na własności metali i stopów.
27. Co to jest stopień przechłodzenia ciekłego metalu?
28. Omówić zarodkowanie homogeniczne i heterogeniczne w procesie krystalizacji.
29. Co mówi reguła Bravais'go?
30. Co to są dendryty?
31. Jaka jest struktura wlewka stalowego?
32. Jakie skutki powoduje odkształcenie plastyczne metalu?
33. W jaki sposób można usunąć zmiany w metalu spowodowane odkształceniem plastycznym?
34. Jakie procesy zachodzą w metalu podczas zdrowienia?
35. Na czym polega rekrystalizacja metalu?
36. Jak można wyznaczyć temperaturę rekrystalizacji?
37. Od czego zależy wielkość ziarna po rekrystalizacji?
38. Co to jest krytyczne odkształcenie plastyczne?
39. Dlaczego w czasie nagrzewania metalu następuje rozrost ziarna?
40. Omówić techniczne znaczenie rekrystalizacji.
41. Jakie fazy występują w stopach metali?
42. Co to jest roztwór stały międzywęzłowy i różnowęzłowy?
43. Co to jest roztwór stały ciągły i ograniczony?
44. Co to są fazy międzymetaliczne?
45. Podać główne grupy faz międzymetalicznych (przykłady).
46. Jakie może być wzajemne usytuowanie faz w stopie?
47. Co to jest analiza termiczna?
48. Co to jest wykres równowagi fazowej?
49. Co nazywa się fazą, a co składnikiem układu?
50. Podać i wyjaśnić regułę faz.

184
51. Narysować i wyjaśnić wykres równowagi z brakiem rozpuszczalności wzajemnej
składników w stanie stałym
52. Narysować i wyjaśnić wykres równowagi z ograniczoną rozpuszczalnością składników w
stanie stałym i eutektyką.
53. Narysować i wyjaśnić wykres równowagi z ograniczoną rozpuszczalnością składników
stanie stałym i perytektyką.
54. Narysować i wyjaśnić wykres równowagi z nieograniczoną rozpuszczalnością składników
stanie stałym.
55. Narysować i wyjaśnić wykres równowagi z fazą międzymetaliczną krystalizującą z fazy
ciekłej.
56. Narysować i wyjaśnić wykres równowagi z fazą międzymetaliczną tworzącą się podczas
reakcji perytektycznej.
57. Narysować i wyjaśnić wykres równowagi z fazą międzymetaliczną krystalizującą z fazy
stałej.
58. Narysować i wyjaśnić przykładowy wykres równowagi z przemianą alotropową jednego lub
obu składników stopu.
59. Wyjaśnić regułę dźwigni.
60. Wyjaśnić budowę stopów potrójnych.
61. Wyjaśnić wpływ struktury na własności stopów jednofazowych.
62. Wyjaśnić wpływ struktury na własności stopów dwu- i wielofazowych.
63. Wyjaśnić wpływ defektów materiałowych na własności mechaniczne stopów.
64. Wyjaśnić rolę gazów atmosferycznych w metalach i stopach.
65. Co to są wtrącenia niemetaliczne?
66. Scharakteryzować żelazo.
67. Narysować układ żelazo-cementyt z zaznaczonymi fazami. Zdefiniować te fazy.
68. Narysować układ żelazo-cementyt z zaznaczonymi składnikami strukturalnymi. Zdefiniować
te składniki.
69. Podać ogólny podział stopów żelaza.
70. Co to jest stal węglowa i jakie zawiera pierwiastki?
71. Podać i wyjaśnić przemiany zachodzące w stopach żelaza z węglem w stanie stałym.
72. Omówić struktury stali węglowych w stanie równowagi w temperaturze otoczenia i w
temperaturach podwyższonych.
73. W jakim celu przeprowadza się obróbkę cieplną stali?
74. Co stanowi podstawę do ustalenia rodzaju obróbki cieplnej danego stopu?
75. Jakie podstawowe przemiany fazowe są związane z obróbką cieplną stali?
76. Jak zmienia się wielkość ziarna w stali przy nagrzewaniu i chłodzeniu?
77. Co to są wykresy CTP i w jaki sposób się je otrzymuje?
78. Jak zmienia się trwałość przechłodzonego austenitu w zależności od przechłodzenia?
79. W jaki sposób można otrzymać stal o strukturze bainitycznej?
80. Scharakteryzować przemianę martenzytyczną stali.
81. Co to jest szybkość krytyczna chłodzenia stali?
82. Jakie przemiany zachodzą w stali podczas odpuszczania martenzytu?
83. Jakie główne grupy operacji cieplnych można wyróżnić w obróbce cieplnej stali?
84. Scharakteryzować poszczególne rodzaje wyżarzania stali.
85. Jakie są optymalne temperatury hartowania stali? Podać uzasadnienie.
86. Jakie rodzaje hartowania stali są stosowane, jakie są ich zalety i w jakich przypadkach
są stosowane?
87. Na czym polega hartowanie powierzchniowe i jakie metody hartowania
powierzchniowego są najczęściej stosowane?
88. Od czego zależy głębokość strefy zahartowanej w przypadku hartowania indukcyjnego?
Co to jest hartowność stali i czym się różni od utwardzalności?
89. W jaki sposób można określić hartowność stali?
90. Na czym polega odpuszczanie stali? Jakie procesy zachodzą w czasie odpuszczania?
91. Jak zmieniają się własności mechaniczne stali w zależności od temperatura odpuszczania?

185
92. Co to jest kruchość odpuszczania i jak można jej zapobiec?
93. Scharakteryzować proces przesycania i starzenia. Podać przykłady zastosowania tego
rodzaju obróbki cieplnej do stopów żelaza.
94. Co to jest obróbka cieplno-chemiczna stali?
95. Wyjaśnić pojęcie dyfuzji atomowej i reakcyjnej.
96. Co to jest nawęglanie stali?
97. Jaki jest cel obróbki cieplnej po nawęglaniu?
98. Co to jest azotowanie stali i w jakim celu jest przeprowadzane?
99. Co to jest węgloazotowanie stali i w jakim celu jest przeprowadzane?
100. Scharakteryzować procesy metalizowania dyfuzyjnego?
101. Podać zasady klasyfikacji stali. Jakie mogą być stany kwalifikacyjne stali?
102. Jaki wpływ wywiera węgiel na własności stali?
103. Które pierwiastki i w jakiej ilości stanowią zwykłe domieszki w stali?
104. Jaki jest wpływ siarki i fosforu na własności technologiczne stali?
105. Porównać stale konstrukcyjne niestopowe podstawowe ze stalami niestopowymi
jakościowymi. Podać sposób oznaczania tych stali.
106. Jaki wpływ wywiera ulepszanie cieplne na własności mechaniczne stali niestopowych
jakościowych?
107. Jakie grupy gatunków można wyodrębnić wśród stali węglowych konstrukcyjnych o
szczególnych własnościach?
108. Co to są stale automatowe, jaki jest ich skład, własności mechaniczne i technologiczne?
109. Jakie wymagania muszą spełniać stale narzędziowe?
110. Jak dzielą się stale węglowe narzędziowe i jakie mają własności?
111. Na czym polega obróbka cieplna stali narzędziowych węglowych?
112. W jakim celu wprowadza się dodatki stopowe do stali?
113. Jakie dodatki stopowe i w jakiej ilości wprowadza się do stali stopowych konstrukcyjnych?
W jakich fazach mogą występować te dodatki?
114. Które pierwiastki tworzą węgliki w stalach?
115. Porównać węgliki grupy pierwszej i drugiej i ich zachowanie się w stalach?
116. Jaki jest wpływ dodatków stopowych rozpuszczających się w żelazie na punkty krytyczne i
strukturę stali?
117. Scharakteryzować wpływ dodatków stopowych na krzywe CTP stali stopowych.
118. Podać klasyfikację stali stopowych wg struktury po wyżarzaniu oraz po chłodzeniu na
powietrzu.
119. Jakie są zasady oznaczania stali stopowych konstrukcyjnych wg Polskich Norm?
120. Jakie czynniki wywierają istotny wpływ na własności mechaniczne stali niskostopowych o
podwyższonej wytrzymałości? Jakie dodatki stopowe zawierają te stale?
121. Scharakteryzować poszczególne grupy gatunków stali stopowych konstrukcyjnych do
ulepszania cieplnego.
122. Jakie jest najistotniejsze kryterium stosowalności poszczególnych gatunków stali
stopowych konstrukcyjnych?
123. Na czym polega obróbka cieplna stali stopowych konstrukcyjnych?
124. Uzasadnić skład chemiczny stali stopowych konstrukcyjnych do nawęglania.
125. Jakie gatunki stali do nawęglania są stosowane do wyrobu sprzętu szczególnie
obciążonego?
126. Co decyduje o wysokiej twardości stali po azotowaniu?
127. Podać główne dodatki stopowe i ich średnią zawartość w stalach sprężynowych.
128. Na czym polega obróbka cieplna stali sprężynowych?
129. Scharakteryzować stale na łożyska toczne. Jakie gatunki stali łożyskowych są produkowane
w kraju?
130. Jak dzielą się stale stopowe narzędziowe ze względu na zastosowanie? Jak oznacza się stale
stopowe narzędziowe wg PN?
131. Porównać stale narzędziowe stopowe do pracy na zimno ze stalami narzędziowymi
węglowymi.

186
132. Podać wymagania stawiane stalom narzędziowym stopowym do pracy na gorąco. Jakie
dodatki stopowe zawierają te stale?
133. Jakie zastosowanie mają poszczególne grupy gatunków stali narzędziowych do pracy na
gorąco?
134. Jakie główne dodatki stopowe zawierają stale szybkotnące i jakie mają własności?
135. W jaki sposób przeprowadza się obróbkę cieplną stali szybkotnących? Jakie główne dodatki
stopowe zawierają stale odporne na korozję i jaki jest ich wpływ na własności i strukturę
tych stali?
136. Jakie główne zastosowanie mają stale nierdzewne chromowe w zależności od zawartości
węgla?
137. Na czym polega obróbka cieplna stali kwasoodpornych chromowo-niklowych o strukturze
austenitycznej?
138. Jaki jest mechanizm korozji międzykrystalicznej stali austenitycznych i w jaki sposób
można jej zapobiec?
139. Jakie czynniki wpływają na żaroodporność i na żarowytrzymałość stali?
140. Scharakteryzować gatunki stali do pracy w podwyższonych temperaturach. Podać
zastosowanie tych stali.
141. Jakie wymagania muszą spełniać stale zaworowe?
142. Wymienić gatunki stali zaworowych produkowanych w kraju, podać ich obróbkę cieplną i
zastosowanie.
143. Wyszczególnić gatunki stali o szczególnych własnościach fizycznych, podać ich
orientacyjne składy chemiczne i zastosowanie.
144. Podać charakterystyczne cechy staliw.
145. Jakie rodzaje staliw można wyodrębnić w zależności od ich składu chemicznego,
zastosowania i własności?
146. Co to są żeliwa węglowe?
147. Scharakteryzować żeliwa szare.
148. Co to są żeliwa sferoidalne?
149. Co to są żeliwa białe?
150. Scharakteryzować żeliwa ciągliwe.
151. Scharakteryzować główne grupy żeliw stopowych.
152. Scharakteryzować własności miedzi.
153. Co to jest miedź technicznie czysta?
154. Podać ogólny podział stopów miedzi.
155. Co to jest miedź stopowa?
156. Scharakteryzować własności i zastosowanie mosiądzów.
157. Co to są miedzionikle?
158. Scharakteryzować własności i zastosowanie brązów.
159. Wymienić i scharakteryzować stopy oporowe miedzi.
160. Scharakteryzować własności aluminium.
161. Co to jest aluminium techniczne?
162. Scharakteryzować odlewnicze stopy aluminium.
163. Scharakteryzować stopy aluminium do przeróbki plastycznej nie obrabialne cieplnie.
164. Scharakteryzować durale.
165. Na czym polega obróbka cieplna stopów Al prowadząca do ich umocnienia?
166. Jakie warunki musi spełniać stop, by mógł być utwardzany dyspersyjnie?
167. Na przykładzie stopu AlCu4 podać, jakie procesy zachodzą w stopach podczas ich
starzenia.
168. Co to jest nawrót i jakie są jego przyczyny?
169. W jakim celu i w jakich zakresach temperatury przeprowadza się poszczególne rodzaje
wyżarzania stopów aluminium.
170. Scharakteryzować własności magnezu.
171. Scharakteryzować stopy magnezu i podać ich zastosowanie.
172. Scharakteryzować własności tytanu.

187
173. Co to jest tytan techniczny?
174. Scharakteryzować własności tytanu technicznego.
175. Scharakteryzować stopy tytanu
α.
176. Scharakteryzować stopy tytanu
α + β.
177. Scharakteryzować stopy tytanu
β .
178. Scharakteryzować ogólnie stopy żarowytrzymałe.
179. Scharakteryzować żarowytrzymałe stopy niklu.
180. Scharakteryzować żarowytrzymałe stopy kobaltu.
181. Scharakteryzować żarowytrzymałe stopy żelazowo-niklowe.
182. Podać własności i zastosowanie molibdenu i jego stopów.
183. Podać własności i zastosowanie wolframu i jego stopów.
184. Podać własności i zastosowanie niobu i jego stopów.
185. Podać własności i zastosowanie tantalu i jego stopów.
186. Podać własności berylu.
Wyszukiwarka
Podobne podstrony:
referaty na materia oznawstwo www.przeklej.pl, Rok II, laborki z termy
miernictwo mojeodp www.przeklej.pl, 1 STUDIA - Informatyka Politechnika Koszalińska, muniol, I rok,
referaty na materia oznawstwo www.przeklej.pl, Rok II, laborki z termy
prognozowanie popytu www.przeklej.pl www.przeklej.pl, Szkoła materiały, Logistyka, Prognozowanie
materia y www przeklej pl
wytrzymalosc materialow sciaga www przeklej pl
materialy www przeklej pl
obr bka cieplna by dr zek www.przeklej.pl, AGH, Semestr II, Podstawy Nauk o materiałach[Kot,Dymek,
konspekt prezentacji czapor www.przeklej.pl, studia, nano, 2rok, 3sem, polimery i materiały funkcjon
podstawy elektrotechniki materialy www przeklej pl[1]
phmetria www przeklej pl
inventor modelowanie zespolow www przeklej pl
prob wki www.przeklej.pl, Ratownictwo Medyczne
rozw j teorii literatury wyk zag do egz www przeklej pl
pytania www przeklej pl
hih wyniki kolokwium 21012010 www przeklej pl
micros atmel www przeklej pl
więcej podobnych podstron