
Dziedziczny rak jelita grubego
Józef Kładny, Jan Lubiński
Szacuje się, że dziedziczne predyspozycje są przyczyną 10-20% wszystkich raków je-
lita grubego (RJG) 1, 2, 3). Opisano szereg postaci dziedzicznego RJG (4, 5) Do dobrze
znanych chorób genetycznych w przebiegu których dochodzi do rozwoju RJG należą dziedzi-
czące się zgodnie z prawami Mendla takie zespoły jak: dziedziczny nie związany z polipowa-
tością rak jelita grubego (HNPCC, zespół Lyncha), polipowatość rodzinna w tym zespół
Gardnera, zespoły Zankasa, Turcota, Peutz-Jeghersa i polipowatości młodzieńczej.
Zespół Lyncha (HNPCC)
Opisany przez Lyncha (6) w latach sześćdziesiątych zespół dziedzicznego nie związa-
nego z polipowatością RJG stanowi około 5% wszystkich RJG. Wykazano, że HNPCC po-
wstaje w wyniku mutacji jednego z kilku genów takich jak MSH2, MLH1, MSH6, PMS1,
PMS2. Mutacje w obrębie dwóch pierwszych z nich są najczęstszą przyczyną zespołów Lyn-
cha (7, 8, 9, 10) Do charakterystycznych cech klinicznych zespołu Lyncha należą:
- wczesny wiek zachorowania (średnio ok. 45 r.ż.)
- częstsza prawostronna lokalizacja guza
-
2 i więcej przypadków RJG wśród krewnych I1
- wiele synchronicznych i metachronicznych ognisk RJG
-
występowanie choroby w kolejnych pokoleniach (transmisja pionowa)
-
zwiększona częstość występowania wśród krewnych raków trzonu macicy, jelita cienkiego
i dróg moczowych.
Według międzynarodowej grupy ekspertów (International Collaborative Group on
HNPCC - ICG-HNPCC) zespół Lyncha można jednoznacznie rozpoznawać wówczas, gdy
wykryta zostanie mutacja konstytucyjna w jednym z genów związanych z HNPCC jak np.
MSH2 czy MLH1 lub też na podstawie danych rodowodowo-klinicznych, wówczas gdy spe-
łnione są kryteria przedstawione w Tabeli 1. (11, 14)
64
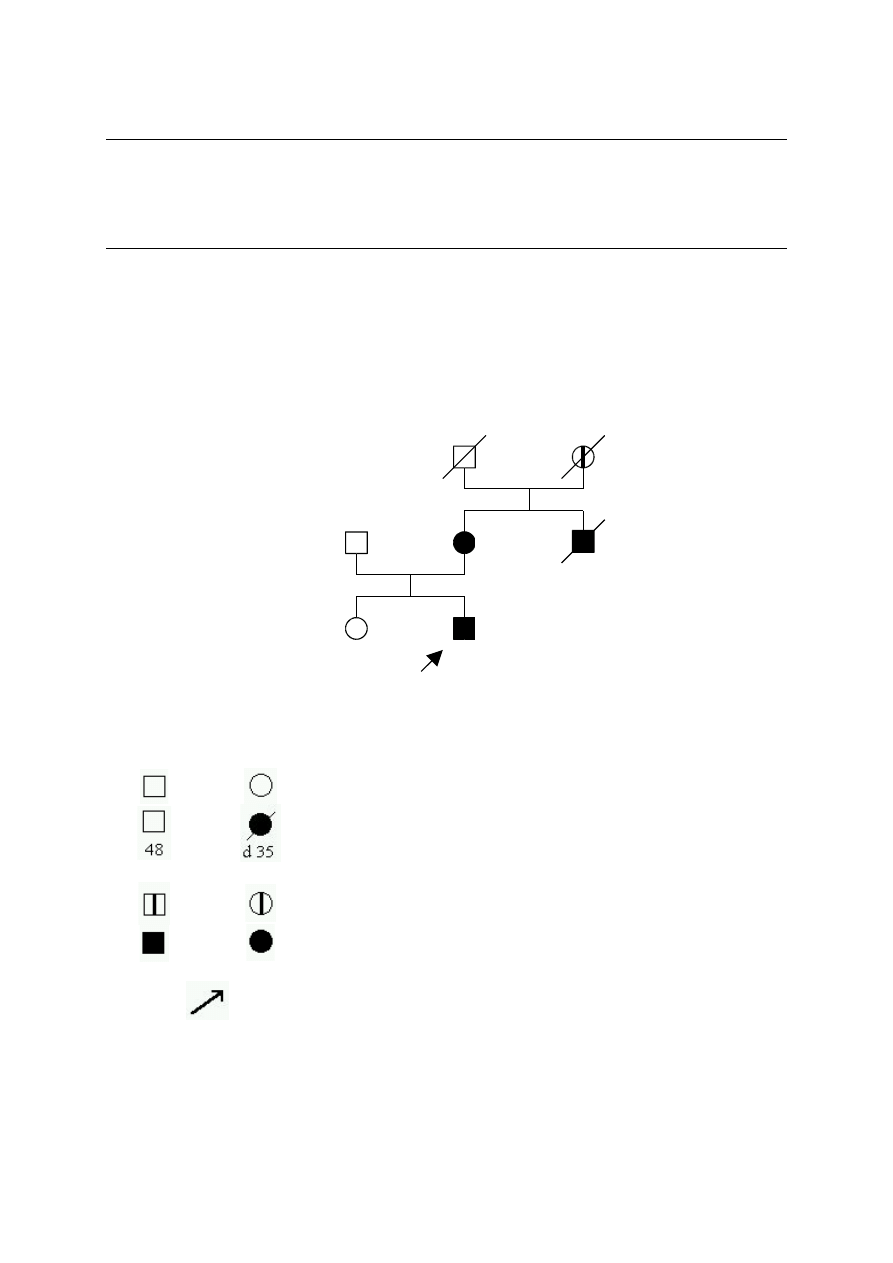
Tabela 1. Kryteria diagnostyczne HNPCC wg. ICG-HNPCC (14).
1.
u co najmniej 3 członków danej rodziny wykryto zweryfikowanego histopatologicznie
RJG lub raka trzonu macicy, jelita cienkiego, lub dróg moczowych.; jeden z nich jest
krewnym I° dwóch pozostałych; wykluczono polipowatość rodzinną
*
,
2. co najmniej 2 z tych osób to krewni I° w dwóch różnych pokoleniach,
3. przynajmniej u 1 spośród tych osób zdiagnozowano raka przed 50 r.ż.
Wszystkie pozostałe parametry (prawostronna lokalizacja, syn- lub metachroniczne guzy) we-
dług tych badaczy powinny być traktowane jako cechy dodatkowe.
* wykluczono występowanie mnogich polipów jelita grubego, wrodzonego przerostu nabłon-
ka barwnikowego siatkówki, torbieli i kostniaków kości twarzoczaszki oraz desmoidów
Na ryc.1 przedstawiono rodzinę spełniającą kryteria definitywnego HNPCC wg ICG-HNPCC.
(14)
Ryc. 1. Rodowód rodziny z cechami HNPCC wg ICG-HNPCC. (14)
Legenda do ryc. 1-3:
zdrowy mężczyzna lub kobieta
aktualny wiek lub wiek zgonu
Col 34
LU 62
lokalizacja raka i wiek zachorowania
rak w wywiadzie
rak zweryfikowany histopatologicznie
probant
Lokalizacja raka:
Bl – pęcherz moczowy
Col - jelito grube
CSU - lokalizacja ogniska pierwotnego nieznana
End – trzon macicy
Ov - jajnik
65
CSU
d49
Col 48
d52
End 43
50
Col 35
36
Pan - trzustka
SB – jelito cienkie
Ze względu na niepełną penetrację genów typową dla chorób mendlowskich dominu-
jących, zgony z powodu różnych chorób, czy też trudności w uzyskaniu pełnych informacji o
wszystkich członkach rodzin, znaczna część - być może większość rodzin w rzeczywistości
obciążonych HNPCC, nie może być rozpoznana w oparciu o kryteria amsterdamskie.
Dlatego też szereg autorów proponuje stosowanie innego typu kryteriów, spełnienie
których nie upoważnia do jednoznacznego rozpoznania HNPCC, jednak jest pomocne w wy-
krywaniu rodzin ze zwiększonym ryzykiem(12, 13, 14, 15,) Naszym zdaniem w identyfikacji
przypadków podejrzanych o HNPCC szczególnie użyteczne są kryteria zestawione w tab. 2.
Tabela 2. Kryteria rozpoznawania rodzin podejrzanych o HNPCC (16)
1 - U pacjenta z RJG wśród krewnych Iº stwierdzono zachorowanie na RJG, raka trzonu
macicy, jelita cienkiego lub dróg moczowych.
2 - Co najmniej jeden z tych nowotworów rozpoznano poniżej 50 r.ż.
3 - Wykluczono polipowatość rodzinną
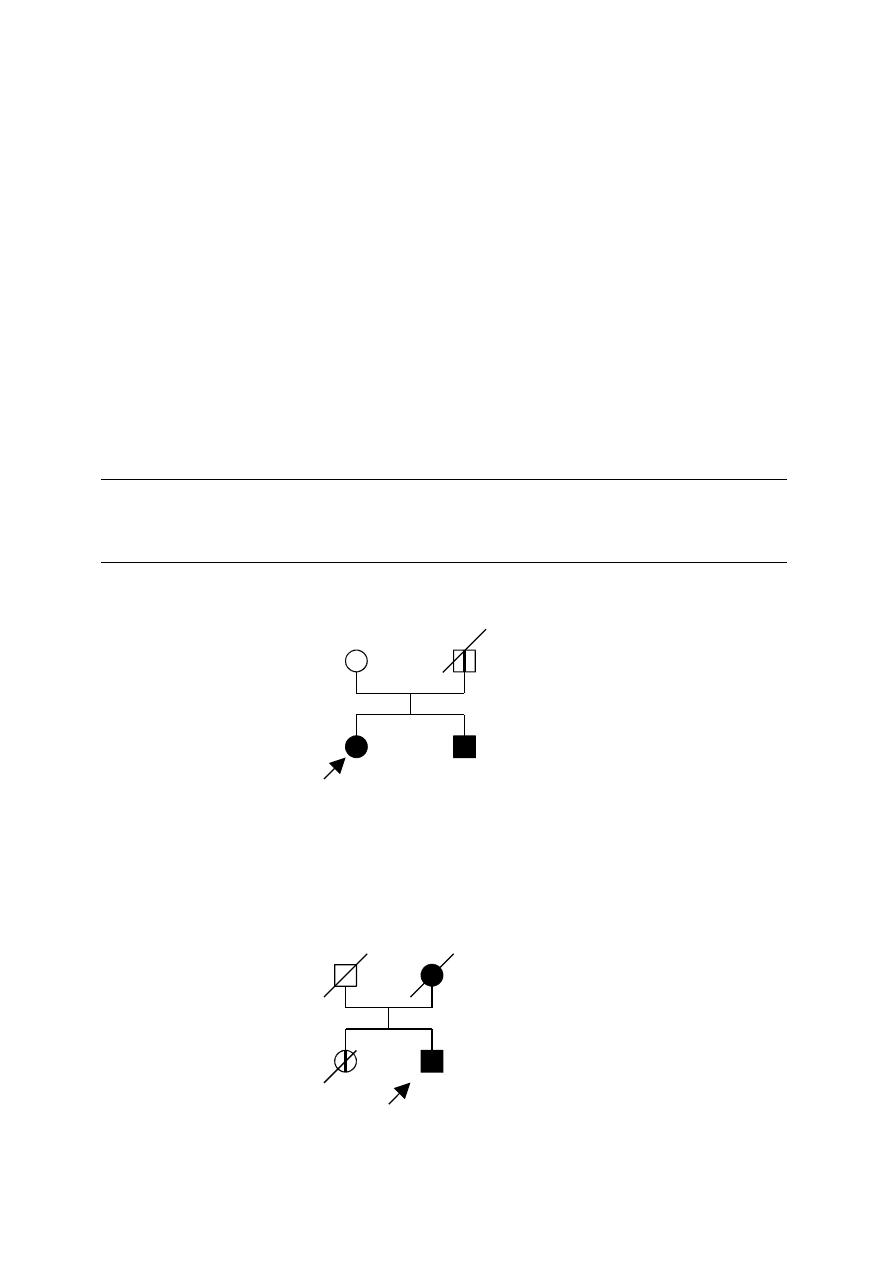
Przykłady rodzin spełniających kryteria „podejrzenia HNPCC” przedstawiono na ryc. 2-3.
Ryc. 2. Rodowód rodziny „podejrzanej o HNPCC”
Legenda – patrz Ryc.1
Ryc. 3. Rodowód rodziny „podejrzanej o HNPCC”
66
Pan
d56
SB 45
46
Col
52
Bl 49
53
Ov
d45
Col 55
d56

Legenda – patrz Ryc.1
67

Diagnostyka molekularna nosicielstwa mutacji w genach związanych z rozwojem
HNPCC
Diagnostyka ta została opisana we wcześniejszym rozdziale – test MSH2/MLH1.
Wykrycie mutacji markerowej dla rodziny z HNPCC ma bardzo istotne znaczenie kliniczne
ponieważ: A) umożliwia wykluczenie ok. 50% członków rodziny z grupy wysokiego ryzyka.
B) ułatwia podjęcie decyzji o radykalności zabiegów chirurgicznych np. kolektomii zamiast
klasycznej resekcji odcinkowej oraz profilaktycznej histerektomii i owariektomii w sytuacji
gdy zabiegi te wykonywane są u zdiagnozowanych nosicieli mutacji.
Prowadzenie rodzin z HNPCC
Posiadana obecnie wiedza na temat tego zespołu wskazuje na konieczność zastosowa-
nia specjalnej formy opieki profilaktyczno-diagnostycznej i leczniczej. W poszczególnych
ośrodkach obowiązują różne programy (4, 17, 18) W świetle najnowszych danych u członków
rodzin z zespołami Lynch uzasadnione wydaje się przyjęcie następujących zasad opieki me-
dycznej:
1. Optymalizacja diety
U osób z wysokim ryzykiem rozwoju RJG zalecana jest dieta ubogotłuszczowa z
ograniczeniem mięsa czerwonego, z dodatkiem otrąb pszennych i bogata w błonnik. (19)
2. Prewencja farmakologiczna
Istnieją doniesienia, że do leków hamujących karcinogenezę RJG należą: aspiryna,
sulindac, piroxicam, wapń, witamina C. Przydatność tych preparatów w zapobieganiu
HNPCC jest prawdopodobna, jednak jak dotąd nie udowodniona.(18, 20, 21)
3. Kolonoskopia
Pełna kolonoskopia zalecana jest co 1-2 lata poczynając od 20-25 r.ż. W rodzinach w
których RJG wystąpił w młodszym wieku kolonoskopie należy rozpocząć wykonywać o 5 lat
wcześniej od wieku najmłodszej osoby z RJG. W przypadkach, w których w trakcie endo-
skopii nie można było dokładnie ocenić całego jelita wskazane jest wykonanie wlewu kon-
trastowego.(18, 22)
68

4. Diagnostyka guzów pozaokrężniczych
W związku ze zwiększoną częstością występowania w rodzinach z HNPCC nowotwo-
rów narządu rodnego, u kobiet zalecane są coroczne szczegółowe badania ginekologiczne z
dopochwowym USG i histopatologicznym badaniem wyskrobin z jamy macicy włącznie.
Ponadto w części przypadków wskazane są badania ukierunkowane na wykrywanie innych
nowotworów częściej występujących w danej rodzinie (np. żołądek, układ moczowy, pierś)
(5, 18).
5. Chirurgia
Polipektomia endoskopowa zalecana jest w przypadku polipów łagodnych, nienaw-
rotowych. Natomiast u pacjentów z gruczolakami: mnogimi i/lub - nawrotowymi i/lub - o
dużym stopniu dysplazji i/lub - kosmkowymi, należy rozważyć profilaktyczną kolektomię
(22). Przeważa pogląd, że nie jest wskazana profilaktyczna chirurgia u pacjentów bez zmian
patologicznych jelita grubego nawet wówczas, gdy osoby badane są nosicielami zmutowanych
genów dla HNPCC (18). Stwierdzenie wysokiego odsetka guzów synchronicznych (u ponad
15% chorych w chwili pierwotnej diagnozy) oraz metachronicznych (ok. 45% pojawia się w
ciągu 10 lat od usunięcia pierwotnego guza) zdecydowało, iż zarówno w profilaktycznej
chirurgii jak i u chorych z rodzin z HNPCC z rozpoznanym histopatologicznie RJG polecane
są następujące rodzaje zabiegów chirurgicznych .(18, 22):
1. proktokolektomia z ileostomią;
2. kolektomia z zespoleniem ileo-rektalnym;
3. proktokolektomia z ileoanalnym "pouchem" - S, J, W lub H.
Pierwszy z proponowanych zabiegów jest wprawdzie najbardziej radykalny, pozbywa-
my się bowiem w całości śluzówki jelita grubego, a zatem i ryzyka nawrotu, ale zabieg ten
oznacza równocześnie ciężkie okaleczenie często połączone z zaburzeniami w oddawaniu
moczu i dysfunkcją seksualną.
Kolektomia z zespoleniem ileo-rektalnym chroni od tego rodzaju powikłań wymaga
jednak częstej kontroli pozostawionego odcinka odbytnicy z uwagi na ryzyko nawrotu.
Proktokolektomia z ileoanalnym "pouchem" S, J, W lub H jest metodą mającą dopiero
historię kilkunastoletnią stąd trudno o jej jednoznaczną ocenę.
U kobiet z zespołem Lyncha operowanych z powodu RJG, ze względu na zwiększone
ryzyko rozwoju raków trzonu macicy i jajnika zalecane jest poszerzenie zabiegu o wycięcie
macicy wraz z przydatkami.(18, 25).
69

Wszystkie te zabiegi są bardziej rozległe od zabiegów klasycznych stosowanych w
leczeniu RJG i cechuje je zwiększona częstość w/w powikłań. Mimo to są one zalecane w
leczeniu HNPCC, ponieważ nadrzędnym problemem u chorych z zespołem Lyncha jest wy-
sokie ryzyko rozwoju drugiego pierwotnego ogniska RJG.
Wykazano już, że zastosowanie odpowiednich programów opieki medycznej w rodzi-
nach z HNPCC zapewnia zwiększoną wykrywalność wczesnych, bezobjawowych RJG.
Ponadto badania prospektywne potwierdziły założenie, iż dzięki odpowiedniemu postępowa-
niu zmniejsza się również zachorowalność na RJG – ryzyko spada z ok. 80 do 30% oraz więk-
szy jest odsetek wyleczeń i czas przeżycia chorych z RJG w rodzinach z zespołem Lyncha.
Odpowiednio prowadzony nosiciel mutacji MSH2/MLH1 nie powinien umrzeć z powodu
RJG (26).
Piśmiennictwo
1. Lovett E.; Family studies in cancer of the colon and rectum. Br. J. Surg. 1976, 63, 13.
2.
Lynch H.T., Lynch J., Lynch P.; Management and control of familial cancer. In:
MulvillJ.J., Miller R.W., Fraumeni J.F., eds. Genetics of Human Cancer. New York:
Raven , 1977, 235-255.
3.
Ponz de Leon M., Sassatelli R., Sacchetti C., Zanghieri G., Scalmati A., Roncucci L.:
"Familial aggregation of tumors in the three-year experience of a Population-based
Colorectal Cancer Registry; Cancer Research. 1989, 49: 4344.
4. Lynch H.T.,Smyrk T, Watson P., Lanspa St., Boman B., Lynch P., Lynch J., Cavalieri
J.; Hereditary colorectal cancer ; Seminars in Oncology ;1991, 18, No 4 ; 337.
5. Vasen H. Inherited forms of colorectal, breast, and ovarian cancer. Surgical Oncology
Clin. N-Am. 1994 Vol. 3 No 3 , 501.
6. Lynch H.T., Krush A.J.: Cancer family "G" revisited: 1895 - 1970 : Cancer 1971, 27 :
1505.
7. Fishel R., Lescoe M.K., Rao M.R.S., Copeland N.G., Jenkins N.A., Garber J., Kane
M., Kolodner R.: The human mutator gene homolog MSH2 and its association with
hereditary nonpolyposis colon cancer. Cell, 1993, 75, 1027.
8. Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomaki
P., Sistonen P., Aaltonen L.A., Nystrom-Lahti M., Guan X.Y., Zhang J., Meltzer P.S.,
Yu J-W., Kao F-T., Chen D.J., Cerosaletti K.M., Fournier R.E.K., Todd S., Lewis T.,
Leach R.J., Naylor S.L., Weissenbach J., Mecklin J.P., Järvinen H., Peterson G.M.,
Hamilton S.R., Green J., Jass J., Watson P., Lynch H.T., Trent J.M., De la Chapelle
70

A., Kinzler K.W., Vogelstein B.: Mutations of a mutS homolog in hereditary non-
polyposis colorectal cancer. Cell, 1993, 75, 1215.
9.
Nicolaides N.C., Papadopoulos N., Liu B., Wei Y-F., Carter K.C., Ruben S.M., Rosen
C.A., Haseltine W.A., Fleischmann R.D., Fraser C.M., Adams M.D., Venter J.C.,
Dunlop M.G., Hamilton S.R., Petersen G.M., De la Chapelle A., Vogelstein B., Kin-
zler K.W.: Mutations of two PMS homologues in hereditary nonpolyposis colon can-
cer; Nature, 1994, 371, 75.
10.
Papadopoulos N., Nicolaides N.C., Wei Y-F., Ruben S.M., Carter K.C., Rosen C.A.,
Haseltine W.A., Fleischmann R.D., Fraser C.M., Adams M.D., Venter J.C., Hamilton
S.R., Petersen G.M., Watson P., Lynch H.T., Peltomaki P., Mecklin J.P., De la Cha-
pelle A., Kinzler K.W., Vogelstein B.: Mutation of a mutL homolog in hereditary co-
lon cancer. Science,1994, 263, 1625.
11.
Vasen H.F.A., Mecklin J.-P., Meera Khan P., Lynch H.T.: The International Colla-
borative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis
Colon Rectum 1991, 34 , 424.
12.
Rodriques-Bigas MA, Boland CR, Hamilton SR, et al. A National Cancer Institute
Workshop on Hereditary on-polyposis Colorectal Cancer Syndrome: meeteng highli-
ghts and Bethesda Guidelines. J. Nat. Cancer Ist 1997; 89: 1758-62.
13.
Park JG, Vasen FA, Park KJ, Peltomaki P et al. Suspected Hereditary Nonpolyposis
Colorectal Cancer. Dis Colon Rectum 1999;42: 710-716.
14.
Vasen HFA, Watson P, Mecklin JP, Lynch H. New Clinical Criteria for Hereditary
Nonpolyposis Colorectal Cancer (HNPCC, Lynch syndrome) Proposed by the Inter-
national Collaborative Group on HNPCC. Gastroenterology 1999; 116: 1453-1456.
15.
Park JG, Vasen FA, Park KJ, Peltomaki P et al.Suspected HNPCC and Amsterdam
criteria II: evaluation of mutation detection rate, an international collaborative study.
Int. J. Colorectal Dis. 2002; 17: 109-114.
16.
Kladny J, Möslein G, Myrhøj T, Kurzawski G, Jakubowska A, Debniak T, Petriczko
W, Kozlowski M, Al-Amawi T, Brzosko M, Flicinski J, Jawien A, Banaszkiewicz A,
Rychter P, Lubinski J; Nuclear pedigree criteria of suspected HNPCC. Hereditary
Cancers in Clinical Praxis 2002 (in press).
17.
Vasen H.F. , Mecklin J.P., Watson P., Utsunomiya J., Bertario L., Lynch P., Svend-
sen L., Cristofaro G., Muller HJ., Meera Khan P., Lynch H.T.: Surveillance in Heredi-
71

tary Nonpolyposis Colorectal Cancer: an international cooperative study of 165 fa-
milies.: Dis. Colon Rectum 1993, 36, 1.
18. Lynch H, Lynch J. Lynch syndrome: Natural history, Genetic Counseling and Preven-
tion. J. Clin. Oncol. 2000; 18: 19-31.
19. Willet W., Stampfer M., Colditz G., Rosner B., Speizer F., Relation of meat, fat, and
fiber intake to the risk of colon cancer in a prospective study among women. N.Engl.
J. Med. 1990; 323, 1664.
20.
Bralow S.P. Primary and secondary chemoprevention of colorectal cancer: Hereditary
colorectal cancer; Springer Verlag Tokyo 1990, 231.
21.
Muscat J.E., Stellman S.D.,Wynder E.L., Nonsteroidal antiiflamatory drugs and colo-
rectal cancer ; Cancer 1994, 74, 1847.
22. Vasen H.F.A., Nagengast F.M., Meera Khan P. Interval cancers in hereditary non-po-
lyposis colorectal cancer (Lynch syndrome); Lancet 1995, 345, 1183.
23.
Kurzawski G, Suchy J, Kladny J, Safranow K, Jakubowska A, Elsakov P, Kucinskas
V, Gardovski J, Irmejs A, Sibul H, Huzarski T, Byrski T, Debniak T, Cybulski C,
Gronwald J, Oszurek O, Clark J, Gozdz S, Niepsuj S, Slomski R, Plawski A, Lacka-
Wojciechowska A, Rozmiarek A, Fiszer-Maliszewska L, Bebenek M, Sorokin D, Sta-
wicka M, Godlewski D, Richter P, Brozek I, Wysocka B, Jawien A, Banaszkiewicz Z,
Kowalczyk J, Czudowska D, Goretzki PE, Moeslein G, Lubinski J.: Germline MSH2
and MLH1 mutational spectrum in HNPCC families from Poland and the Baltic Sta-
tes. J Med Genet. 2002 Oct;39(10):E65.
24.
Dębniak T, Kurzawski G, Górski B, Kładny J, Domagał W, Lubiński J. Value of
pedigree/clinical data, immunochemistry and microsatellite instability analyses in
reducing the cost of determining hMLH1 and hMSH2 gene mutations in patients with
colorectal cancer; Eur J. Cancer 2000, 36, 49-54.
25.
Watson P.,Vasen H.F.A., Mecklin J.P., Järvinen H., Lynch H.T.; The risk of endome-
trial cancer in hereditary nonpolyposis colorectal cancer; Am.J.Med 1994, 96, 516.
26.
Jarvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year Trial on screening for
colorectal cancer in families with hereditary nonpolyposis colorectal cancer.
Gastroenterology 2000; 118: 829-834.
72

Skorowidz
Aspiryna – 67
Charakterystyczne cechy kliniczne zespołu Lyncha – 64
Diagnostyka molekularna mutacji HNPCC – 67
Dieta – 67
dopochwowe USG – 68
Dziedziczny rak jelita grubego – 64
HNPCC- 64 , 68
ICG-HNPCC – 65
kolektomia – 68
Kolonoskopia – 67
Kryteria diagnostyczne HNPCC – 65
Kryteria podejrzanych o HNPCC – 66
MLH1- 64
MSH2 – 64
MSH6 – 64
Piroxicam – 67
PMS1 – 64
PMS2 – 64
polipektomia – 68
polipowatość rodzinna – 64
profilaktyczna chirurgia - 68
Sulindac – 67
Wapń – 67
Witamina C – 67
Wlew kontrastowy – 67
zespół Gardnera -64
zespół Lyncha – 64
zespół Peutz-Jeghersa –64
zespół polipowatości młodzieńczej – 64
zespół Turcota - 64
zespół Zankasa -64
73
Wyszukiwarka
Podobne podstrony:
Lynch Burg Lemonade
Lynch 2
Jennifer Lynch Sekretny dziennik Laury Palmer
jennifer lynch sekretny dziennik laury palmer
Lynch Sekretny dziennik Laury Palmer
Lynch Jennifer Sekretny dziennik Laury Palmer
Scott Lynch Red Seas Under Red
Of Mistletoe and Mating a Claw Jessica Lynch
Foucault s Critical Ethics Lynch, Richard A ;
Duchowość i sztuka Lynch David
Art of the Ridiculous Sublime On David Lynch s Lost Highway Slavoj Zizek
Lynch Sarah Kate Dom córek
Tory Richards [Wild Marauders MC 01] Lynch (pdf)
Wywiad z Reginą Lynch prowadzony przez Marię Lozano
więcej podobnych podstron