Forum: Therapeutic Applications of Reactive Oxygen and Nitrogen Species
in Human Disease
REACTIVE OXYGEN SPECIES, CELL SIGNALING, AND CELL INJURY
K
ENNETH
H
ENSLEY
, K
ENT
A. R
OBINSON
, S. P
RASAD
G
ABBITA
, S
COTT
S
ALSMAN
,
and
R
OBERT
A. F
LOYD
Free Radical Biology and Aging Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, USA
(Received 23 December 1999; Accepted 4 January 2000)
Abstract—Oxidative stress has traditionally been viewed as a stochastic process of cell damage resulting from aerobic
metabolism, and antioxidants have been viewed simply as free radical scavengers. Only recently has it been recognized
that reactive oxygen species (ROS) are widely used as second messengers to propagate proinflammatory or growth-
stimulatory signals. With this knowledge has come the corollary realization that oxidative stress and chronic inflam-
mation are related, perhaps inseparable phenomena. New pharmacological strategies aimed at supplementing antioxidant
defense systems while antagonizing redox-sensitive signal transduction may allow improved clinical management of
chronic inflammatory or degenerative conditions, including Alzheimer’s disease. Introduction of antioxidant therapies
into mainstream medicine is possible and promising, but will require significant advances in basic cell biology,
pharmacology, and clinical bioanalysis.
© 2000 Elsevier Science Inc.
Keywords—Inflammation, Antioxidant, Phosphatase, Nitric oxide, Nitrone, Free radical
INTRODUCTION
During the past 10 –15 years, the field of “free radical
research” has risen from relative obscurity to become a
mainstream element of biomedical science, and for good
reason. Since Commoner’s first detection of free radicals
in a biological system (germinating barley seeds) in 1954
[1], free radical biology had mostly been the proprietary
domain of physical chemists. The chemical entities stud-
ied by these scientists were ephemeral, almost to the
point of abstraction. Very few techniques existed for the
detection or manipulation of free radicals in vitro, let
alone in vivo. Moreover, the techniques brought to bear
on free radical chemistry were esoteric, largely limited to
spin trapping methods, and required expensive and often
inaccessible instrumentation. Most importantly, the
pathophysiological sequelae of oxidative stress have
been notoriously difficult to quantify. Despite these im-
pediments, the medical significance of oxidative stress
has become increasingly recognized to the point that it is
now considered to be a component of virtually every
disease process. The ascendancy of free radical biology
is attributable to several major factors. First, new tech-
niques have been invented (and are still being invented)
to quantify oxidative stress in vivo, although the existing
technology is poorly suited for routine clinical applica-
tions. Second, the inseparable relationship of oxidative
stress to inflammation has become incontrovertible along
with the recognition that certain reactive ROS function
as messenger molecules to propagate inflammatory sig-
nals. Third, the discovery of nitric oxide (NO) as a
vasodilator and immune mediator has stimulated the
interest of mainstream biologists and clinicians to an
almost unprecedented degree. As free radical/oxidative
stress research enters the 21st century, we face the chal-
lenge of transferring our nascent (but burgeoning)
Kenneth Hensley holds a Ph.D. in Physical Chemistry from the
University of Kentucky. He has served as a research scientist at the
Oklahoma Medical Research Foundation for the past four years. His
current research investigates the relationship between oxidative stress
and neuroinflammation in the aging human brain, with special empha-
sis on basic mechanisms of neurodegeneration in Alzheimer’s disease.
Dr. Robinson, Dr. Gabbita, and Mr. Salsman currently pursue studies of
oxidative injury at the Oklahoma Medical Research Foundation with
special emphasis on Alzheimer’s disease. Dr. Floyd is head of the Free
Radical Biology and Aging Research Program at the Oklahoma Med-
ical Research Foundation. His current research interests center on the
biology of aging and the role of nitric oxide in age-related pathologies
of the central nervous system.
Address correspondence to: Kenneth Hensley, Ph.D., Free Radical
Biology and Aging Research Program, Oklahoma Medical Research
Foundation, Oklahoma City, OK 73104, USA; Tel: (405) 271-7569;
Fax: (405) 271-1795; E-Mail: kenneth-hensley@omrf.ouhsc.edu.
Free Radical Biology & Medicine, Vol. 28, No. 10, pp. 1456 –1462, 2000
Copyright © 2000 Elsevier Science Inc.
Printed in the USA. All rights reserved
0891-5849/00/$–see front matter
PII S0891-5849(00)00252-5
1456

knowledge of oxidative pathology from the laboratory
into the clinic and the pharmacy. New therapeutic strat-
egies can, and will be developed which rationally incor-
porate antioxidants into the management of chronic dis-
ease. The purpose of this review is to highlight promising
new developments in antioxidant therapy, particularly
with respect to strategies aimed at uncoupling oxidative
stress from redox-sensitive signal transduction. A final
section of the review summarizes current challenges in
the practical assessment of oxidative stress, which must
be overcome before antioxidant therapy can achieve its
clinical potential.
ROS AS TOXINS: ANTIOXIDANTS AS SCAVENGERS OF
REACTIVE INTERMEDIATES
Until relatively recently, oxidative stress was consid-
ered purely from the toxicological perspective. A rela-
tively small number of free radicals such as the super-
oxide anion (O
2
•
⫺
) and the hydroxyl radical (HO
•
) were
recognized as minor by-products of oxidative phosphor-
ylation. By 1973, Britton Chance and colleagues [2] had
determined that approximately 2% of the oxygen re-
duced by the mitochondrion forms O
2
•
⫺
or the dismuta-
tion product H
2
O
2
. This estimate has been confirmed
repeatedly [3]. Superoxide and peroxide react with metal
ions to promote additional radical generation, with the
release of the particularly reactive hydroxyl [4]. Hy-
droxyl radicals react at nearly diffusion-limited rates
with any component of the cell, including lipids, DNA
and proteins. The net result of this nonspecific free
radical attack is a loss of cell integrity, enzyme function,
and genomic stability [5– 8]. Consequently, numerous
detoxification mechanisms have evolved to deal with
oxyradical stress. Superoxide dismutase (SOD) converts
O
2
•
⫺
to H
2
O
2
, which is subsequently reduced to water by
catalase or otherwise decomposed by glutathione-depen-
dent peroxidases. Small-molecule reducing agents such
as glutathione thereby buffer the intracellular environ-
ment against ROS. In synergy with the aqueous defense
mechanisms, lipid-phase antioxidants exist naturally to
scavenge radical intermediates.
〈-tocopherol (
␣-toc, vi-
tamin E) is the principle lipid-phase antioxidant [9 –11].
Hydroxyl (or alkoxyl) radical attack on tocopherol forms
a stabilized phenolic radical which is reduced back to the
phenol by ascorbate and NADH/NADPH-dependent re-
ductase enzymes [9]. Over the past decade, the menag-
erie of ROS has been expanded to include reactive ni-
trogen species (RNS) derived from NO reaction with
superoxide or peroxide [12,13]. Specific defense mech-
anisms evolved to counteract RNS stress will probably
be identified in coming years.
Given the extreme reactivity of most oxyradicals and
the number of defense mechanisms evolved to counteract
oxidative stress, it seems reasonable that dietary or phar-
macological practices that bolster the ROS scavenging
capacity should somehow improve health. Considerable
epidemiological and clinical data, and huge amounts of
animal data, corroborate this hypothesis. While a com-
plete review is outside the scope of this discussion, it is
worth noting that natural variation in antioxidant levels
correlate negatively with certain pathologies, particularly
of the cardiovascular system. Most famously, plasma
␣-tocopherol correlates negatively with risk of ischemic
heart disease in several large, cross-sectional studies
[14 –16]. The usual explanation for this phenomenon is
that
␣-toc inhibits low density lipoprotein oxidation, an
etiological factor in atherosclerotic plaque development
[reviewed in 17]. Clinical studies designed to supplement
antioxidant defenses, particularly by dietary administra-
tion of
␣-toc (50–1000 mg/day) have shown some mar-
ginal benefit but not as much as might be expected based
on epidemiological statistics. For instance, a 40% in-
crease in plasma
␣-toc is epidemiologically correlated
with a 60 – 80% reduced risk of ischemic heart disease
[14]. Paradoxically, clinical augmentation of plasma
␣-tocopherol by the same amount confers only small
cardiovascular benefit in heart disease patients [18] with
no effect, or even a marginal increase, in all-cause mor-
tality [19]. Even more disconcerting, supplementation
with the lipophilic antioxidant
-carotene actually exac-
erbates cancer risk among smokers [20]. Thus, while
antioxidant levels are clearly important in promoting
health, the supplementation of antioxidant defenses in
human subjects will prove much more complicated than
the simple, casual administration of presumptively ben-
eficial free radical scavengers.
The main problem faced by clinicians and basic sci-
entists is that “antioxidant” function is much more com-
plex than simple free radical scavenging, and dietary
supplementation with a particular antioxidant is likely to
perturb the natural balance of other antioxidants. As a
case in point, dietary supplementation with
␣-toc causes
a profound and immediate decrease in plasma concen-
tration of
␥-tocopherol (␥-toc), a minor unmethylated
tocopherol [21–23].
␥-Tocopherol has been virtually un-
studied, but some reports indicate that
␣-toc may scav-
enge reactive nitrogen species (RNS) in a way that
␣-toc
cannot, forming the nitration product 5-nitro-
␣-tocoph-
erol as a reaction product [21,22]. A very recent cardio-
vascular study reports that dietary
␥-toc is much more
efficacious than
␣-toc at decreasing susceptibility to oc-
clusive thrombus, with plasma concentration-normalized
efficacy of
␥-toc exceeding that of ␣-toc by a factor of 20
or more [24]. Clearly, much more basic research is
needed to understand the interplay among natural anti-
oxidant systems and the synergies inherent to these sys-
tems.
1457
ROS and cell signaling

ROS AS SIGNALING MOLECULES: POTENTIAL
TARGETS FOR ANTI-INFLAMMATORY THERAPEUTICS
As discussed previously, oxidative stress has long
been considered an “accident” of aerobic metabolism; a
stochastic process of free radical production and nonspe-
cific tissue damage which is fundamentally unregulated
aside from the normal phalanx of antioxidant defense
mechanisms. In recent years, a paradigm shift has been
occurring wherein certain ROS and RNS have become
appreciated as signaling molecules whose production
may be regulated as a part of routine cellular signal
transduction [reviewed in 25]. The seminal work by
Baeurle and colleagues first showed that certain tran-
scription factors of the NF
B/rel family can be activated
not only by receptor-targeted ligands but also by direct
application of oxidizing agents (particularly H
2
O
2
) or
ionizing radiation [26,27]. Subsequently, several other
protein kinase cascades and transcription factors have
been discovered to possess redox-sensitive elements. The
common paradigm in all redox-sensitive signal transduc-
tion pathways is the presence of intermediate protein
kinases which are activated by phosphorylation of spe-
cific regulatory domains. For example, NF-
B is acti-
vated upon phosphorylation of an inhibitory subunit
(I
B). Conveniently, specific antibodies are now avail-
able against the phosphorylated activation sites of many
protein kinases so that activation of a particular enzyme
can be assessed by standard immunoblot techniques.
Figure 1 illustrates the phosphoactivation of several ma-
jor protein kinase pathways in cultured primary rat as-
trocytes exposed to low concentrations of exogenous
H
2
O
2
.
Work from our group and others indicates that H
2
O
2
may be synthesized endogenously in certain cell types as
a response to activation by specific cytokines or growth
factors [28 –30]. This endogenous H
2
O
2
then acts as a
second messenger to stimulate protein kinase cascades
coupled to inflammatory gene expression, or in control of
the cell cycle. The earliest convincing studies that impli-
cated H
2
O
2
as an endogenous messenger were performed
by Sunderesan et al. [31] using, as a model system,
vascular smooth muscle cells (VSMCs) stimulated with
Fig. 1. Western blots demonstrating synchronous phospho-activation of four distinct protein kinase cascades in primary rat astrocytes
initiated by addition of exogenous H
2
O
2
. Stat-3
⫽ Signal Transducer and Activator of Transcription-3 (target residue: pSer
727
); JNK
⫽
c-Jun amino terminal kinase (target residues: pThr
183
-pro
184
-pTyr
185
); AKT
⫽ protein kinase B or RAC (target residue : pSer
183
);
p38
⫽ p38
MAPK
(target residues: pThr
180
-Gly
181
-pTyr
182
). Antibodies recognize phosphorylated residues and other epitopic compo-
nents near the phosphorylation sites. Cells were stimulated with the indicated bolus of peroxide for 5 min, lysed, electrophoresed on
12% polyacrylamide gels, and probed with the appropriate phosphorylation-state specific primary antibody (New England Biolabs,
Beverly, MA, USA). Blots were developed using horseradish peroxidase-conjugated secondary antibodies and chemiluminescent
substrates.
1458
K. H
ENSLEY
et al.

platelet-derived growth factor (PDGF). PDGF receptor
binding caused peroxide formation which could be in-
hibited by intracellular expression of catalase. Catalase
expression inhibited PDGF signal transduction by sup-
pressing protein tyrosine phosphorylation [31]. Antioxi-
dants,
particularly
thiol-reducing
agents
such
as
N-acetyl-cysteine, could mimic the inhibitory effects of
catalase and prevent redox activation of ligand-coupled
protein kinase cascades [31].
Subsequent studies by a number of groups, particu-
larly that of Sue Goo Rhee and colleagues [29], have led
to the hypothesis that H
2
O
2
acts through the transient
oxidative inactivation of protein tyrosine phosphatases
(PTPs) which contain a nucleophilic cysteine as a cata-
lytic element of the active site. Rhee has shown that
epidermal growth factor (EGF) binding to epidermoid
cells induces rapid loss of PTP reactivity that can be
restored by glutathione-dependent reductive pathways
[29]. As in the case of PDGF, EGF receptor binding
causes intracellular production of H
2
O
2
within the time-
frame of PTP inactivation [28,29]. In separate but con-
temporaneous work, Denu and Tanner demonstrated that
H
2
O
2
reacts with PTPs in vitro to convert the active-site
cysteine into a metastable sulfinic acid [32]. Subsequent
reduction by glutathione restores the enzyme to its active
form. Alternatively, phosphatase reaction with oxidized
glutathione could transiently inactivate a PTP during a
redox signaling event [33].
We have observed strong evidence for peroxide-me-
diated, phosphatase-dependent signal transduction using
a cytokine stimulus directed against primary rat astro-
cytes [30,34]. We find that both interleukin-1
 (IL1)
and H
2
O
2
will promote phospho-activation of the p38-
mitogen activated protein kinase (p38
MAPK
) in a manner
that can be antagonized with submillimolar quantities of
NAC or the nitrone-based antioxidant phenyl-N-tert-bu-
tylnitrone (PBN) [30]. Interestingly, PBN has been found
efficacious in preventing ischemia/reperfusion injury,
septic shock, and other trauma, though the mechanism of
action has been indeterminate [reviewed in 35]. In IL1
-
treated astrocytes, total phosphatase activity decreases
simultaneously with p38
MAPK
phospho-activation, and
returns to baseline as p38
MAPK
becomes dephosphory-
lated (inactivated). Both NAC and PBN maintain phos-
phatase activity at or above baseline values [30] while
promoting global protein dephosphorylation [34]. Fi-
nally, we were able to measure H
2
O
2
biosynthesis in
IL1
-treated astrocytes and found this to be inhibited by
1 mM PBN [30]. Thus, several lines of evidence argue
that H
2
O
2
is used as a ubiquitous messenger substance to
inactivate regulatory phosphatase enzymes and promote
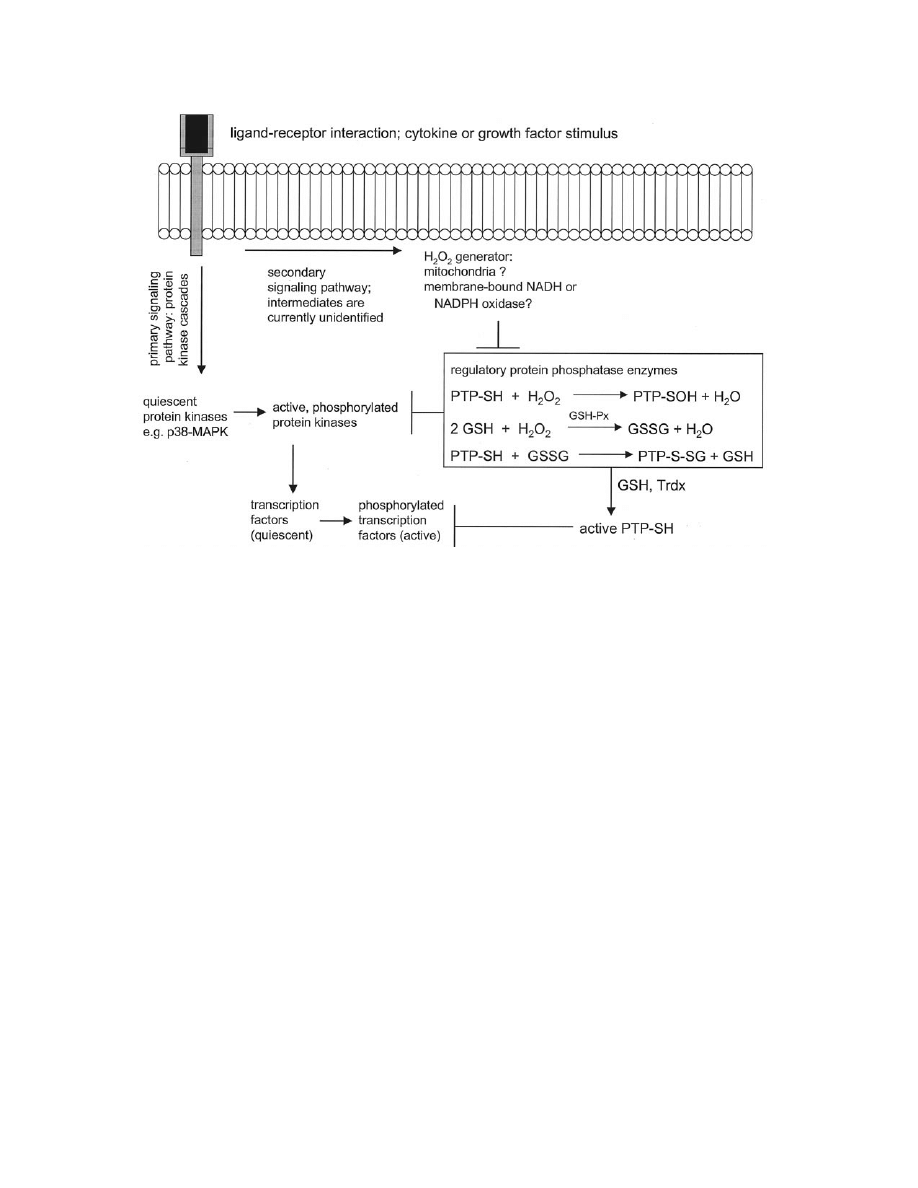
inflammatory signal transduction. Figure 2 schematically
summarizes the probable function of H
2
O
2
as a signal
transducer, and illustrates possible targets for pharmaco-
logical antagonism.
The p38
MAPK
pathway is a particularly relevant target
for antioxidant antagonism in chronic inflammatory dis-
ease. p38
MAPK
regulates expression of inflammatory cy-
tokines including IL1
 [36] and largely regulates expres-
sion of iNOS and COX-II [37,38]. We have observed
p38
MAPK
hyperphosphorylation in Alzheimer’s diseased
(AD) brain tissue, in plaques and neurons where protein
nitration is also evident [39,40]. In separate work, Wal-
ton and colleagues [41] have observed similar p38
MAPK
phosphorylation in microglia of postischemic rodent
brain, where protein oxidation and nitration are salient
pathological phenomena [42,43]. Brain-accessible anti-
oxidants and antagonists of redox signaling may, there-
fore, have wide utility in the therapeutic interdiction of
neuroinflammatory events occurring in AD, stroke, and
other neurodegenerative disease.
The recognition that ROS may stimulate inflamma-
tory signaling pathways comes with considerable clinical
ramifications. Once we can identify the sources and
targets of “second-messenger” ROS, new avenues will
be open for the development of novel pharmacophores
that function both as antioxidants and nonsteroidal anti-
inflammatory agents. PBN, for instance, decreases brain
protein oxidation during ischemia/reperfusion injury or
normal aging [44,45]. Additionally, PBN can protect
animals from systemic inflammation induced by bacte-
rial endotoxin [46]. We have shown that inflammatory
gene transcription and iNOS expression are simulta-
neously suppressed by the nitrone within the same ani-
mal models [47– 49]. Moreover, the transcription of pro-
apoptotic elements such as caspase 3 and Fas antigen are
suppressed by PBN in rats subjected to experimental
septic shock [49]. These diverse actions can be explained
by nitrone antagonism of redox-sensitive signal trans-
duction pathways including, but not limited to, the
p38
MAPK
cascade. Unfortunately, the precise site of ac-
tion of PBN has not been elucidated. Future research will
need to identify the exact source of second-messenger
ROS, better pinpoint the targets of this ROS, and identify
regulatory elements against which novel pharmaco-
phores may be designed.
MONITORING OXIDATIVE STRESS: A BIOANALYTICAL
CHALLENGE AND A BIOMEDICAL NECESSITY
Despite widespread scientific and public perception
that antioxidants are “good,” and the incontrovertible
evidence that oxidative damage is deleterious in chronic
disease, serious barriers exist to the introduction of an-
tioxidant therapies into clinical medicine. The greatest of
these barriers is the fact that we cannot currently deter-
mine which individuals might benefit from which anti-
1459
ROS and cell signaling
oxidant therapy. The optimum daily dose of even com-
mon antioxidants such as
␣-tocopherol and vitamin C are
subject to some debate, while no guidelines have ever
been considered for less-common, but possibly no less
significant antioxidants such as
␣-tocopherol. While we
have a poor idea of the biological effects inherent to
supplementation with natural antioxidants, we have no
idea whatsoever of the effects of synthetic antioxidants
in the human subject. As alluded to previously, certain
subgroups might even react negatively to antioxidants, as
evidenced by the apparent exacerbation of lung cancer
among patients taking
-carotene [20]. Beyond the de-
termination of therapeutic strategy, a clinician should
have some means of determining the responsiveness of
his patient to the prescribed treatment. How can one
monitor antioxidant status in a clinical setting? Cur-
rently, there is no satisfying answer to such a question.
The onus is upon free radical researchers to develop
sensitive, facile, and accurate assays for oxidative stress
that predict the type of antioxidant supplementation that
might be appropriate to a specific individual. Moreover,
such bioanalytical tools must allow a clinician to monitor
a patient’s response to treatment, in much the same way
as the physician would monitor cholesterol or blood
glucose or any other clinically-relevant parameter. Our
group has been active in the development of high per-
formance liquid chromatography with electrochemical
detection (HPLC-ECD) as a tool for the routine assess-
ment of oxidative stress [50 –52]. Specific, discreet ana-
lytes can be selectively measured by HPLC-ECD, and
these analytes may indicate something of the nature of an
oxidative insult. For example, HPLC-ECD can detect
nitrated tyrosines (3-nitrotyrosine) and 5-nitro-
␥-tocoph-
erol as indicators of NO involvement in a disease process
Fig. 2. Postulated mechanism of peroxide-mediated redox signaling. Arrows indicate stimulatory pathways;
indicate inhibitory
pressures. Signaling is initiated by specific ligand-receptor interactions. Typically, a series of protein kinase intermediates propagate
the signal toward nuclear transcription factors. Other signaling pathways must exist to facilitate the H
2
O
2
production observed by
several labs [e.g., references 28,30]. The sites of intracellular peroxide generation are currently subject to some debate; however,
mitochondria and plasma membrane-bound oxidoreductase enzymes have been postulated to serve this function. Endogenously-
generated H
2
O
2
causes transient inactivation of sensitive protein tyrosine phosphatases (PTP-SH); this reaction may occur directly
through a sulfenic acid intermediate (PTP-SOH) or indirectly via formation of a mixed glutathione intermediate (PTP-S-SG).
Glutathione oxidation by peroxide is readily catalyzed by glutathione peroxidase (GSH-Px). Removal of phosphatase inhibition will
allow maximal signal output through the protein kinase cascade. The oxidized, inactive protein phosphatase can be regenerated into
the active form by further reduction by GSH in a reaction catalyzed by thioredoxin (Trdx). Reactivated phosphatase activity will cause
dephosphorylation of intermediate protein kinases and transcription factors, thereby terminating the redox-sensitive signal. Potential
sites of pharmacological action would include the putative peroxide-generator, as well as various intermediate kinase enzymes such
as p38
MAPK
. Agents that maintain phosphatase activity in the face of an oxidative challenge would, in general, be expected to
antagonize the redox signaling process.
1460
K. H
ENSLEY
et al.

[50]. Nonspecific oxidation might be indicated by in-
creases in the hydroxyl reaction products o-tyrosine or
m-tyrosine or by tyrosine dimers; or, alternatively, by
increased conversion of
␣-toc to the corresponding p-
quinone [50 –52].
Other researchers have successfully indexed oxidative
stress by gas chromatography in combination with mass
spectrometry (GC-MS). GC-MS analysis of low molec-
ular weight hydrocarbons in breath [53], or specific ara-
chidonic acid peroxidation products (isoprostanes) in
fluids [54,55], may prove amenable to clinical medicine.
Morrow, Montine and colleagues [55], for instance, have
measured increased F1-isoprostanes in cerebrospinal
fluid of patients with Alzheimer’s disease. AD is one
illness with a clear oxidative stress component wherein
antioxidant supplementation (specifically, with
␣-to-
copherol) confers a small, but significant clinical benefit
manifest by delays in primary outcome indicators (e.g.,
time of entry into a nursing home or loss of ability to
perform routine daily function) [56]. Before antioxidant
therapy becomes accepted, detailed longitudinal studies
will need to be conducted which evaluate panels of
oxidative biomarkers along with traditional clinical end-
points in patients undergoing treatment for diverse
chronic illnesses. The publication of such studies will
usher in a new and exciting period in the history of
oxidative stress research and will signal the final matu-
ration of the discipline.
Acknowledgements — This work was supported in part by the National
Institutes of Health (NS35747) and the Oklahoma Center for the
Advancement of Science and Technology (OCAST H67-097).
REFERENCES
[1] Commoner, B.; Townsend, J.; Pake, G. E. Free radicals in bio-
logical materials. Nature 174:689 – 691; 1954.
[2] Boveris, A.; Chance, B. The mitochondrial generation of hydro-
gen peroxide: general properties and effect of hyperbaric oxygen.
Biochem. J. 134:707–716; 1973.
[3] Hensley, K.; Pye, Q. N.; Maidt, M. L.; Stewart, C. A.; Robinson,
K. A.; Jaffrey, F.; Floyd, R. A. Interaction of
␣-phenyl-N-tert-
butyl nitrone and alternative electron acceptors with complex I
indicates a substrate reduction site upstream from the rotenone
binding site. J. Neurochem. 71:2549 –2557; 1998.
[4] Stadtman, E. R. Metal ion-catalyzed oxidation of proteins: bio-
chemical mechanism and biological consequences. Free Radic.
Biol. Med. 9:315–325; 1990.
[5] Stadtman, E. R.; Berlett, B. S. Fenton chemistry. Amino acid
oxidation. J. Biol. Chem. 266:17201–17211; 1991.
[6] Floyd, R. A. The role of 8-hydroxyguanine in carcinogenesis.
Carcinogenesis 11:1447–1450; 1990.
[7] Gille, J. J.; van Berkel, C. G.; Joenje, H. Mutagenicity of meta-
bolic oxygen radicals in mammalian cell cultures. Carcinogenesis
15:2695–2699; 1994.
[8] Halliwell, B. Oxygen and nitrogen are pro-carcinogens. Damage
to DNA by reactive oxygen, chlorine and nitrogen species: mea-
surement, mechanism and the effects of nutrition. Mutat. Res.
443:37–52; 1999.
[9] Buettner, G. R. The pecking order of free radicals and antioxi-
dants: lipid peroxidation, alpha tocopherol and ascorbate. Arch.
Biochem. Biophys. 300:535–543; 1993.
[10] Burton, G. W.; Joyce, A.; Ingold, K. U. First proof that vitamin E
is major lipid-soluble, chain-breaking antioxidant in human blood
plasma. Lancet 2:27; 1982.
[11] Ingold, K. U.; Webb, A. C.; Witter, D.; Burton, G. W.; Metcalfe,
T. A.; Muller, D. P. Vitamin E remains the major lipid-soluble,
chain-breaking antioxidant in human plasma even in individuals
suffering severe vitamin E deficiency. Arch. Biochem. Biophys.
259:224 –225; 1987.
[12] Squadrito, G. L.; Pryor, W. A. Oxidative chemistry of nitric
oxide: the roles of superoxide, peroxynitrite, and carbon dioxide.
Free Radic. Biol. Med. 25:392– 403; 1998.
[13] Koppenol, W. H. The basic chemistry of nitrogen monoxide and
peroxynitrite. Free Radic. Biol. Med. 25:385–391; 1998
[14] Gey, K. F.; Puska, P.; Moser, U. K. Inverse correlation between
plasma vitamin E and mortality from ischemic heart disease in
cross-cultural epidemiology. Am. J. Clin. Nutr. 53(Suppl. 1):
326S–334S; 1991.
[15] Stampfer, M. J.; Hennekens, C. H.; Manson, J. E.; Coldizt, G. A.;
Rosner, B.; Willett, W. C. Vitamin E consumption and the risk of
coronary artery disease in women. N. Engl. J. Med. 328:1444 –
1449; 1993.
[16] Rimm, E. B.; Stampfer, M. J.; Ascherio, A.; Giovannucci, E.;
Colditz, G. A.; Willett, W. C. Vitamin E consumption and the risk
of coronary heart disease in men. N. Engl. J. Med. 328:1450 –
1456; 1993.
[17] Esterbauer, H.; Gebicki, J.; Puhl, H.; Jurgens, G. The role of lipid
peroxidation and antioxidants in modification of LDL. Free
Radic. Biol. Med. 13:341–390; 1992.
[18] Stephens, N. G.; Parsons, A.; Schofield, P. M.; Kelly, F.; Cheese-
man, K.; Mitchinson, M. J. Randomised controlled trial of vitamin
E in patients with coronary disease: Cambridge Heart Antioxidant
Study (CHAOS). Lancet 347:781–786; 1996.
[19] Rapola, J. M.; Virtamo, J.; Ripatti, S.; Huttumen, J. K.; Albanes,
D.; Taylor, P. R.; Heinonen, O. P. Randomised trial of alpha
tocopherol and beta carotene supplements on incidence of major
coronary events in men with previous myocardial infarction.
Lancet 349:1715–1720; 1997.
[20] The Alpha-Tocopherol Beta Carotene Cancer Prevention Study
Group. The effect of vitamin E and beta carotene on the incidence
of lung cancer and other cancers in male smokers. N. Engl.
J. Med. 330:1029 –1035; 1994.
[21] Cooney, R. V.; Franke, A. A.; Harwood, P. J.; Hatch-Pigott, V.;
Custer, L. J.; Mordan, L. J.
␣-Tocopherol detoxification of nitro-
gen dioxide: superiority to
␣-tocopherol. Proc. Natl. Acad. Sci.
USA 90:1771–1775; 1993.
[22] Christen, S.; Woodall, A. A.; Shigenaga, M. K.; Southwell-Keely,
P. T.; Duncan, M. W.; Ames, B. N.
␣-Tocopherol traps mutagenic
electrophiles such as NOx and complements
␣-tocopherol: phys-
iological implications. Proc. Natl. Acad. Sci. USA 94:3217–3222;
1997.
[23] Goss, S. P. A.; Hogg, N.; Kalyanaraman, B. The effect of
␣-to-
copherol on the nitration of
␣-tocopherol by peroxynitrite. Arch.
Biochem. Biophys. 363:333–340; 1999.
[24] Saldeen, T.; Li, D.; Mehta, J. L. Differential effects of alpha- and
gamma-tocopherol on low-density lipoprotein oxidation, super-
oxide activity, platelet aggregation and arterial thrombogenesis.
J. Am. Coll. Cardiol. 34:1208 –1215; 1999.
[25] Suzuki, Y. J.; Forman, H. J.; Sevanian, A. Oxidants as stimulators
of signal transduction. Free Radic. Biol. Med. 22:269 –285; 1997.
[26] Schreck, R.; Rieber, P.; Baeuerle, P. A. Reactive oxygen inter-
mediates as apparently widely used messengers in the activation
of the NF-kappa B transcription factor and HIV-1. EMBO J.
10:2247–2258; 1991.
[27] Schreck, R.; Albermann, K.; Baeuerle, P. A. Nuclear factor kappa
B: an oxidative stress-responsive transcription factor of eukary-
otic cells (a review). Free Radic. Res. Commun. 17:221–237;
1992.
[28] Bae, Y. S.; Kang, S. W.; Seo, M. S.; Baines, I. C.; Tekle, E.;
Chock, P. B.; Rhee, S. G. Epidermal growth factor (EGF)-induced
generation of hydrogen peroxide. Role in EGF receptor-mediated
tyrosine phosphorylation. J. Biol. Chem. 272:217–221; 1997.
1461
ROS and cell signaling

[29] Lee, S. R.; Kwon, K. S.; Kim, S. R.; Rhee, S. G. Reversible
inactivation of protein-tyrosine phosphatase 1B in A431 cells
stimulated with epidermal growth factor. J. Biol. Chem. 273:
15366 –15372; 1998.
[30] Robinson, K.; Stewart, C. A.; Pye, Q. N.; Nguyen, X.; Kenney,
L.; Salsman, S.; Floyd, R. A.; Hensley, K. Redox sensitive protein
phosphatase activity regulates the phosphorylation state of p38
protein kinase in primary astrocyte culture. J. Neurosci Res.
55:724 –732; 1999.
[31] Sundaresan, M.; Yu, Z. X.; Ferrans, V. J.; Irani, K.; Finkel, T.
Requirement for generation of H
2
O
2
for platelet-derived growth
factor signal transduction. Science 270:296 –299; 1995.
[32] Denu, J. M.; Tanner, K. G. Specific and reversible inactivation of
protein tyrosine phosphatases by hydrogen peroxide: evidence for
a sulfenic acid intermediate and implications for redox regulation.
Biochemistry 37:5633–5642; 1998.
[33] Barrett, W. C.; DeGnore, J. P.; Konig, S.; Fales, H. M.; Keng,
Y. F.; Zhang, Z. Y.; Yim, M. B.; Chock, P. B. Regulation of
PTP1B via glutathionylation of the active site cysteine 215. Bio-
chemistry 18:6699 – 6705; 1999.
[34] Robinson, K. A.; Stewart, C. A.; Pye, Q. N.; Floyd, R. A.;
Hensley, K. Basal protein phosphorylation is decreased and phos-
phatase activity increased by an antioxidant and a free radical trap
in primary rat glia. Arch. Biochem. Biophys. 365:211–215; 1999.
[35] Hensley, K.; Carney, J. M.; Stewart, C. A.; Tabatabaie, T.; Pye,
Q. N.; Floyd, R. A. Nitrone-based free radical traps as neuropro-
tective agents in cerebral ischemia and other pathologies. Int. Rev.
Neurobiol. 40:299 –317; 1997.
[36] Baldassare, J. J.; Bi, Y.; Bellone, C. J. The role of p38 mitogen-
activated protein kinase in IL-1 beta transcription. J. Immunol.
162:5367–5673; 1999.
[37] Bhat, N. R.; Zhang, P.; Lee, J. C.; Hogan, E. L. Extracellular
signal-regulated kinase and p38 subgroups of mitogen-activated
protein kinases regulate inducible nitric oxide synthase and tumor
necrosis factor
␣ gene expression in endotoxin-stiumulated pri-
mary glial cultures. J. Neurosci. 18:1633–1641; 1998.
[38] Da Silva, J.; Pierrat, B.; Mary, J. L.; Lesslauer, W. Blockade of
p38 mitogen-activated protein kinase pathway inhibits inducible
nitric oxide synthase expression in mouse astrocytes. J. Biol.
Chem. 272:28373–28380; 1997.
[39] Hensley, K.; Floyd, R. A.; Zheng, N.-Y.; Nael, R.; Robinson,
K. A.; Nguyen, X.; Pye, Q. N.; Stewart, C. A.; Geddes, J.;
Markesbery, W. R.; Patel, E.; Johnson, G. V. W.; Bing, G. p38
kinase is activated in Alzheimer disease brain. J. Neurochem.
72:2053–2058; 1999.
[40] Smith, M. A.; Harris, P. L. R.; Sayre, L. M.; Beckman, J. S.;
Perry, G. Widespread peroxynitrite-mediated damage in Alzhei-
mer’s disease. J. Neurosci. 17:2653–2657; 1997.
[41] Walton, K. M.; DiRocco, R.; Bartlett, B. A.; Koury, E.; Marcy,
V. R.; Jarvis, B.; Shaefer, E. M.; Bhat, R. V. Activation of p38
MAPK in microglia after ischemia. J. Neurochem. 70:1764 –
1767; 1998.
[42] Tanaka, K.; Shirai, T.; Nagata, E.; Dembo, T.; Fukuuchi, Y.
Immunohistochemical detection of nitrotyrosine in postischemic
cortex in gerbil. Neurosci. Lett. 235:85– 88; 1997.
[43] Eliasson, M. J.; Huang, Z.; Ferrante, R. J.; Sasamata, M.; Mol-
liver, M. E.; Snyder, S. H.; Moskowitz, M. A. Neuronal nitric
oxide synthase activation and peroxynitrite formation in ischemic
stroke linked to neural damage. J. Neurosci. 19:5910 –5918;
1999.
[44] Oliver, C. N.; Starke-Reed, P. E.; Stadtman, E. R.; Liu, G. J.;
Carney, J. M.; Floyd, R. A. Oxidative damage to brain proteins,
loss of glutamine synthetase activity, and production of free
radicals during ischemia/reperfusion-induced injury to gerbil
brain. Proc. Natl. Acad. Sci. USA 87:5144 –5147; 1990.
[45] Carney, J. M.; Starke-Reed, P. E.; Oliver, C. N.; Landum, R. W.;
Cheng, M. S.; Wu, J. F.; Floyd, R. A. Reversal of age-related
increase in brain protein oxidation, decrease in enzyme activity,
and loss in temporal and spatial memory by chronic administra-
tion of the spin-trapping compound N-tert-butyl-alpha-phenylni-
trone. Proc. Natl. Acad. Sci. USA 88:3633–3636; 1991.
[46] Pogrebniak, H. W.; Merino, M. J.; Hahn, S. M.; Mitchell, J. B.;
Pass, H. I. Spin trap salvage from endotoxemia: the role of
cytokine down-regulation. Surgery 112:130 –139; 1992.
[47] Miyajima, T.; Kotake, Y. Spin trapping agent, phenyl N-tert-butyl
nitrone, inhibits induction of nitric oxide synthase in endotoxin-
induced shock in mice. Biochem. Biophys. Res. Commun. 215:
114 –121; 1995.
[48] Sang, H.; Wallis, G. L.; Stewart, C. A.; Kotake, Y. Expression of
cytokines and activation of transcription factors in lipopolysac-
charide-administered rats and their inhibition by phenyl N-tert-
butylnitrone (PBN). Arch. Biochem. Biophys. 363:341–348;
1999.
[49] Stewart, C. A.; Hyam, K.; Wallis, G.; Sang, H.; Robinson, K. A.;
Floyd, R. A.; Kotake, Y.; Hensley, K. Phenyl-N-tert-butylnitrone
demonstrates broad-spectrum inhibition of apoptosis-associated
gene expression in endotoxin-treated rats. Arch. Biochem. Bio-
phys. 365:71–74; 1999.
[50] Hensley, K.; Williamson, K.; Gabbita, S. P.; Grammas, P.; Floyd,
R. A. Determination of biological oxidative stress using high
performance liquid chromatography with electrochemical detec-
tion (HPLC-ECD). J. High Res. Chromatogr. 22:429 – 437; 1999.
[51] Hensley, K.; Maidt, M. L.; Yu, Z. Q.; Sang, H.; Markesbery,
W. R.; Floyd, R. A. Electrochemical analysis of protein nitroty-
rosine and dityrosine in the Alzheimer brain indicates region-
specific accumulation. J. Neurosci. 18:8126 – 8132; 1998.
[52] Hensley, K.; Maidt, M. L.; Pye, Q. N.; Stewart, C. A.; Wack, M.;
Tabatabaie, T.; Floyd, R. A. Quantitation of protein-bound 3-ni-
trotyrosine and 3,4-dihydroxyphenylalanine by high-performance
liquid chromatography with electrochemical array detection.
Anal. Biochem. 251:187–195; 1997.
[53] Arterbery, V. E.; Pryor, W. A.; Jiang, L.; Sehnert, S. S.; Foster,
W. M.; Abrams, R. A.; Williams, J. R.; Wharam, M. D. Jr.; Risby,
T. H. Breath ethane generation during clinical total body irradi-
ation as a marker of oxygen-free-radical-mediated lipid peroxi-
dation: a case study. Free Radic. Biol. Med. 17:569 –576; 1994.
[54] Montine, T. J.; Beal, M. F.; Cudkowicz, M. E.; O’Donnell, H.;
Margolin, R. A.; McFarland, L.; Bachrach, A. F.; Zackert, W. E.;
Roberts, L. J.; Morrow, J. D. Increased CSF F2-isoprostane
concentration in probable AD. Neurology 52:562–565; 1999.
[55] Roberts, L. J. II; Montine, T. J.; Markesbery, W. R.; Tapper,
A. R.; Hardy, P.; Chemtob, S.; Dettbarn, W. D.; Morrow, J. D.
Formation of isoprostane-like compounds (neuroprostanes) in
vivo from docosahexaenoic acid. J. Biol. Chem. 273:13605–
13612; 1998.
[56] Sano, M.; Ernesto, C.; Thomas, R. G.; Klauber, M. R.; Schafer,
K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C. W.;
Pfeiffer, E.; Schneider, L. S.; Thal, L. J. A controlled trial of
selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s
disease. The Alzheimer’s Disease Cooperative Study. N. Engl.
J. Med. 336:1216 –1222; 1997.
ABBREVIATIONS
RNS—reactive nitrogen species
ROS—reactive oxygen species
PBN—phenyl-tert-butylnitrone
NAC—N-acetyl cysteine
␣-toc—alpha tocopherol
␥-toc—gamma;-tocopherol
IL1
—interleukin-1
p38
MAPK
—p38-mitogen activated protein kinase
PTP—protein tyrosine phosphatase
HPLC-ECD— high performance liquid chromatography
with electrochemical detection
AD—Alzheimer’s disease
1462
K. H
ENSLEY
et al.
Wyszukiwarka
Podobne podstrony:
A protocol for polymerase chain reaction detection of Enterococcus faecalis and Enterococcus faec
15 Multi annual variability of cloudiness and sunshine duration in Cracow between 1826 and 2005
Chapter 15 Diseases of the Urinary Tract and Kidney
93ZJ Secc 8J Turn Signals and Hazard Warning Flashes
Big Profit Patterns Using Candlestick Signals And Gaps Stephen W Bigalow
96ZJ 8J TURN SIGNAL AND HAZARD WARNING SYSTEMS
STM 12 6 Pyrotechnic distress signals and line throwing apparatus
15 The Long Passing – The Low and Lofted Passes
Apoptosis Induction, Cell Cycle Arrest and in Vitro Anticancer Activity
Apoptosis, Cytotoxicity and Cell Proliferation
AN Increased Osteoprotegerin Serum Release Characterizes The Early Onset of Diabetes Mellitus and Ma
Bearden Tech papers Extending The Porthole Concept and the Waddington Valley Cell Lineage Concept
Physiological strains remodel extracellular matrix and cell
Morphogenesis and cell cycle progression in Candida albicans
Simulation for Fuel Cell Inverter using Simplorer and Simulink
więcej podobnych podstron