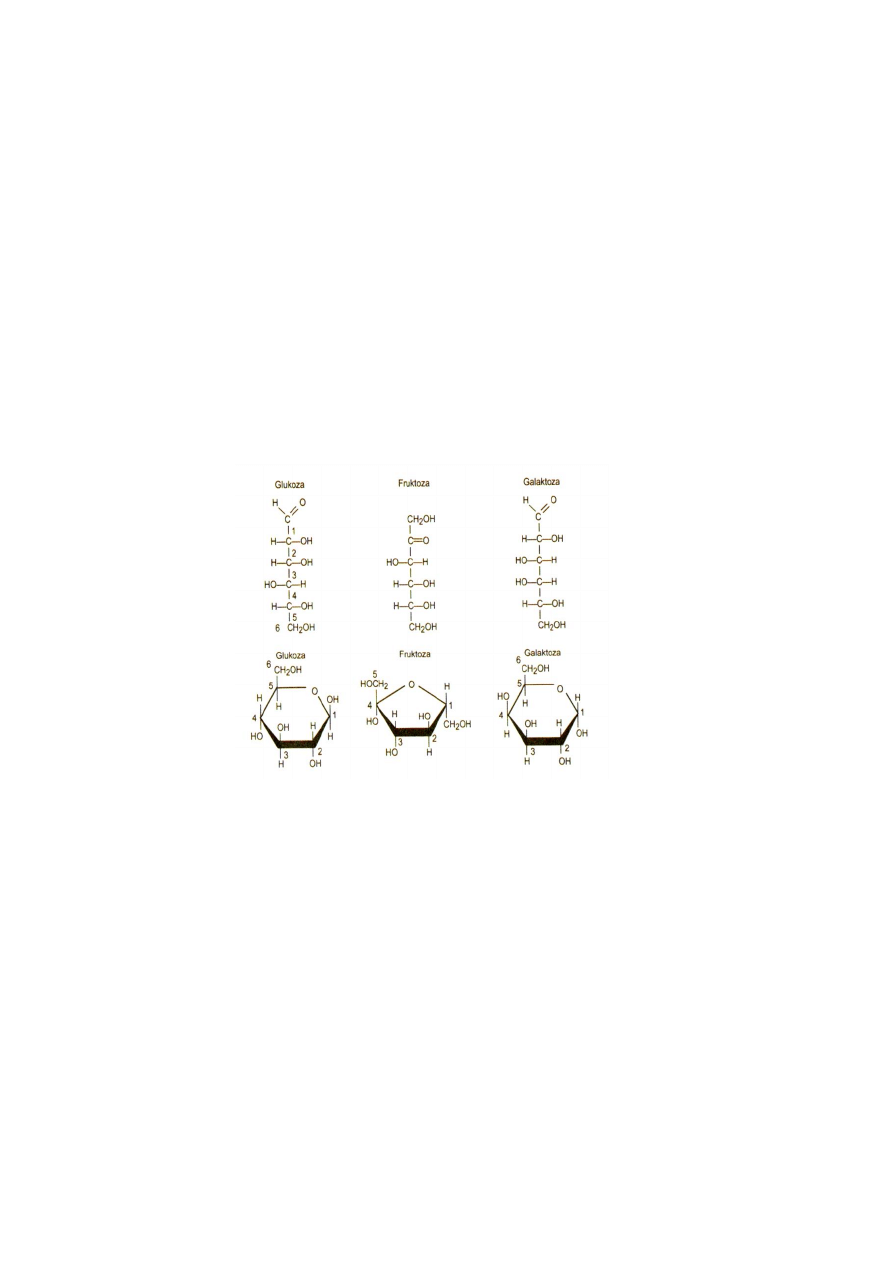
1. PRZEDSTAW KLASYFIKACJĘ SACHARYDÓW
W środkach spożywczych scharakteryzowano ponad 100 sacharydów.
Monosacharydy w swoich cząsteczkach zawierają 3 do 7 atomów węgla. W zależności od
tego określamy je jako triozy, tetrozy, pentozy, heksozy, heptozy.
Wśród cukrów prostych ważną rolę dla człowieka odgrywają jedynie heksozy C
6
H
12
O
6
oraz
pentozy C
5
H
10
O
5
– monosacharydy występujące powszechnie w przyrodzie. Do heksoz należą
glukoza, fruktoza, galaktoza i mannoza, zaś do pentoz: ryboza, ksyloza, ksyluloza, rybuloza i
arabinoza.
Glukoza i fruktoza mają ten sam wzór sumaryczny (C
6
H
12
O
6
), ale różny wzór strukturalny.
Występują one w przyrodzie w dwóch formach, tj. łaocuchowej i pierścieniowej.
Aldopentozy, np. D-Ksyloza,
pochodne aldopentoz: D-Apioza
Aldoheksozy, np. D-Galaktoza, D-Glukoza,
Ketoheksozy, np. D-Fruktoza, L-Sorboza
Pochodne ketoheksoz: D-Glukozamina, chondrozamina
Oligosacharydy
Są zbudowane z 2 – 10 cząsteczek monosacharydów. Wyróżniamy disacharydy (sacharoza –
glukoza+fruktoza; laktoza – glukoza+galaktoza; maltoza – glukoza+glukoza; ), trisacharydy
(melezytoza – glukoza+fruktoza+glukoza; rafinoza - glukoza+galaktoza+fruktoza),
tetrasacharydy (stachioza – glukoza+galaktoza+galaktoza+fruktoza), pentasacharydy
(maltopentaoza, izomaltopentaoza).
Polisacharydy są zbudowane z wielu cząsteczek cukrów prostych. Zalicza się do nich m.in.
skrobię, glikogen, dekstryny, celulozę, agar, hemicelulozy, pektynę.
2. OPISZ BUDOWĘ SKROBI
Skrobia wyizolowana z materiału roślinnego - skrobia natywna – ma budowę ziarnistą.
Kształt ziaren, ich wielkośd, skład, właściwości fizyczne i podatnośd na reakcje chemiczne
zależą od odmiany botanicznej. Najczęściej wykorzystywanymi źródłami skrobi są zboża
(skrobie zbożowe), ziemniaki oraz tapioka – skrobie bulwiaste.
Otoczki ziaren skrobiowych są zbudowane z wysokospolimeryzowanych sacharydów,
prawdopodobnie amylopektyny. Otoczki skrobi mogą mied budowę anizotropową (skrobia
ziemniaczana, tapiokowa, ryżowa) lub izotropową (kukurydziana).
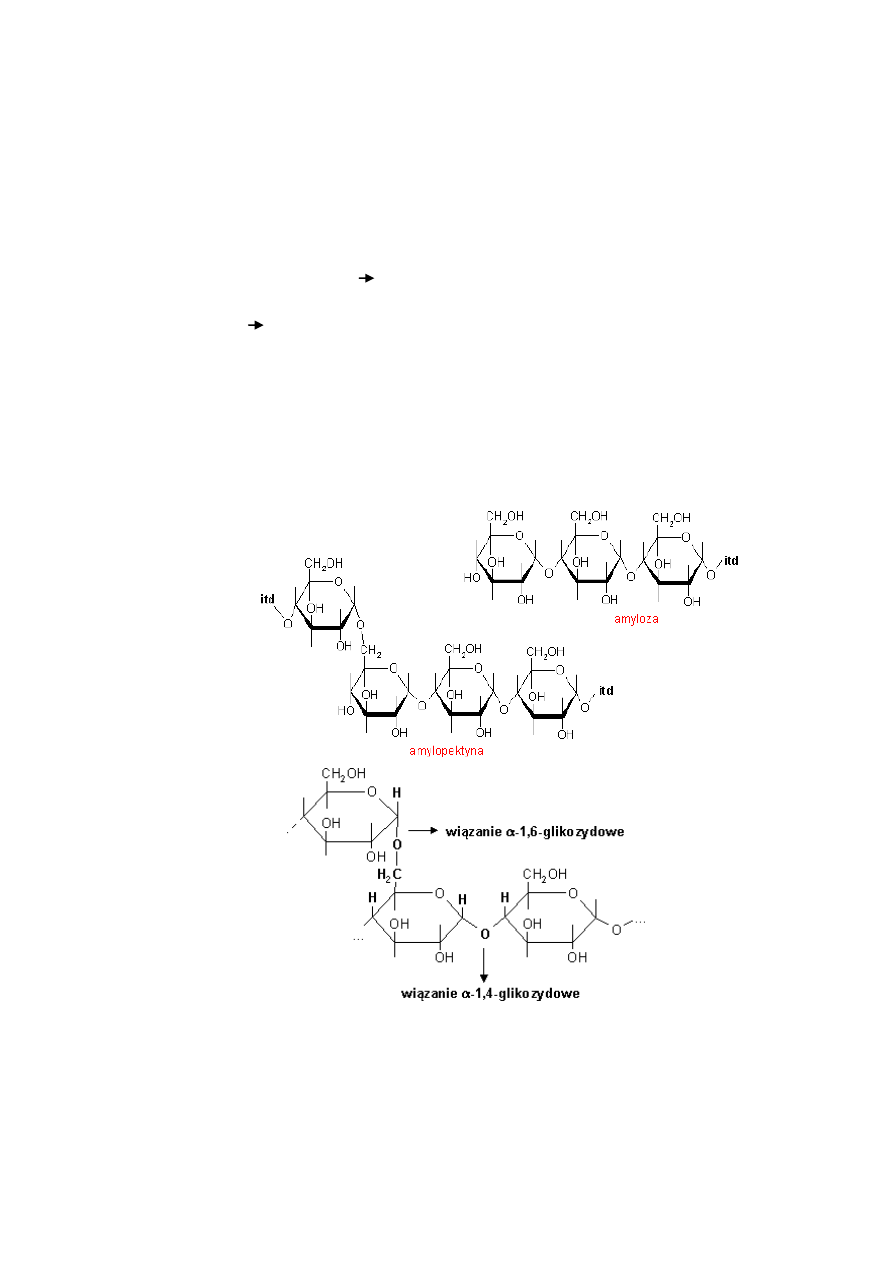
Krystaliczne i bezpostaciowe frakcje ziarna są zbudowane z dwóch polisacharydów: amylozy i
amylopektyny, wzajemnie ze sobą splątanych i poprzetykanych nitkami lipidów i/lub białek,
w zależności od odmiany skrobi.
Amyloza jest w zasadzie liniowym polimerem α-D-glukozy, której pierścienie są połączone
wiązaniami glikozydowymi 1 4. Czasami spotyka się amylozę rozgałęzioną. Jej odgałęzienia
zawierają jeden lub dwa mery glukozowe. Są one połączone z głównym łaocuchem amylozy
wiązaniami 6 1. Z wodą amyloza tworzy roztwory koloidalne. Jej łaocuchy łagodnie i
nieregularnie się zwijają, ale jeśli pojawią się sprzyjające okoliczności pozwalające obniżyd
energię wewnętrzną układu, zwijają się w helisę.
Amylopektyna zbudowana jest z takich samych merów. Od jej trzonu utworzonego przez
liniowo, jak w amylozie, połączone mery α-D-glukozowe odchodzą rozgałęzienia zbudowane
podobnie, lecz połączone z nim wiązaniami 1, 6 glikozydowymi. Takie rozgałęzienia zawierają
od 8 do 12 merów i pojawiają się przeciętnie co 8 jednostek glukozowych. Koocowe gałęzie
mogą się zwijad w helisy o jednym do dwóch zwojów.
3. NA CZYM POLEGA DEHYDRATACJA SACHARYDÓW?
Sacharydy ogrzewane powyżej ich temperatury topnienia zaczynają się lekko pienid z powodu
wewnątrzcząsteczkowego wydzielania się wody. Towarzyszy temu powstawanie
anhydrosacharydów, tj. układów dwupierścieniowych. Kierunek takiego odwodnienia jest
uwarunkowany względami sterycznymi i energetycznymi. Ze względów energetycznych
uprzywilejowane jest wydzielanie się wody z utworzeniem pierścieni pięcio- i

sześcioczłonowych. W przypadku D-glukozy może powstawad 1,6-anhydro-D-glukoza i 3,6-
anhydro-D-glukoza. Pierwszemu etapowi odwodnienia nie towarzyszy zmiana barwy
stopionego sacharydu. Dalsze ogrzewanie zwiększa pienienie się stopu i powoduje zmianę
jego zabarwienia najpierw na żółte, następnie czerwonobrunatne do brunatnego i czarnego.
Towarzysz tym zmianom wydzielane się ostrego zapachu typowego dla palonego cukru, który
coraz bardziej się nasila.
Powstające produkty scharakteryzowano pod względem ich składu ilościowego, a ich
struktura chemiczna nie została dotychczas dokładnie poznana. Związki te nazwano
karamelanem, karamelenem i karamelinem.
6 C
12
H
22
O
11
– 12 H
2
O = 6 C
12
H
18
O
9
6 C
12
H
22
O
11
– 18 H
2
O = 2 C
36
H
48
O
24
6 C
12
H
22
O
11
– 27 H
2
O = 3 C
24
H
26
O
13
Ostry zapach towarzyszący odwadnianiu sacharydów pochodzi od pochodnych furanu.
Odwodnienie pentoz prowadzi do powstania furano-2-aldehydu, a odwodnienie heksoz daje
5-hydroksymetylofurano-2-aldehyd.
Przez odwodnienie powstają też izomaltol i maltol.
Związki te częściowo odpowiadają za tzw. wtórne aromaty żywności, za zapachy pojawiające
się przy termicznej obróbce artykułów spożywczych, szczególnie za aromat świeżego
pieczywa.
Reakcje tworzenia się karamelanu, karamelenu i karamelinu zachodzą w czasie produkcji
karmelu.
4. CO TO SĄ REAKCJE MAILLARDA? W JAKI SPOSÓB POWSTAJĄ AROMATY POCHODZENIA
SACHARYDOWEGO?
Reakcjami Maillarda nazywa się zespół wielu przemian, w których uczestniczą sacharydy i
zasadowe związki azotowe, takie jak aminokwasy, aminy, amoniak. Reakcje te mogą
przebiegad w dwojaki sposób: albo sacharydy reagują ze związkami azotowymi, po czym
ulegają dalszym przemianom do pochodnych pirazyny, pirolu i pirydyny. To te
małocząsteczkowe związki są odpowiedzialne za charakterystyczny zapach produktów. W
reakcjach Maillarda powstają też melanoidyny, polimery o rozległym układzie
chromoforowym, który nadaje produktom reakcji barwę brunatną.
Tradycyjnie tego rodzaju reakcje uważano za reakcje Maillarda, jeśli przebiegały pod
wpływem enzymów. To one odpowiadały za brunatnienie owoców i warzyw. Obecnie do
reakcji Maillarda zalicza się też reakcje zachodzące pod wpływem obróbki termicznej, np.
podczas wypieku chleba i ciasta, pieczenia ziemniaków, palenia ziarna kawowego i
kakaowego, smażenia i pieczenia mięsa, fermentacji tytoniu.
Większośd sacharydów, hemicelulozy i skrobia ogrzewana z aminokwasami i białkami
wytwarza wtórne aromaty żywności. Powstający aromat bardziej zależy od użytego
aminokwasu niż od sacharydu. Aromaty wytwarzane z celulozy, która łatwiej reaguje niż
skrobia, bardziej przypominają aromaty owocowe i kwiatowe. Podobne reakcje zachodzą
między sacharydami i hydroksykwasami; powstają wówczas aromaty zbliżone do roślinnych,
np. zapach świeżo ściętej trawy, świeżo krojonych ogórków, suszonych śliwek, wywaru z
maku.

5. FERMENTACJA JAKO PRZYKŁAD ENZYMATYCZNEJ PRZEMIANY SACHARYDÓW
Oprócz nielicznych wyjątków enzymatyczne przekształcenia sacharydów polegają na ich
degradacji. W praktyce, w procesach takich wykorzystuje się bakterie albo dostępne w
handlu enzymy.
Jeśli prowadzi się fermentację przy dostępie powietrza (proces aerobowy) jest to fermentacja
alkoholowa, w wyniku której powstają etanol, ditlenek węgla i woda. Proces ten prowadzony
bez dostępu powietrza (proces anaerobowy) daje w wyniku kwasy: mlekowy, bursztynowy,
propionowy i mrówkowy, a ponadto butanol, aceton, diacetyl, acetoinę, butandiale, wodór i
metan.
Fermentacja kwasu mlekowego przebiega anaerobowo. Produktem jest mieszanina
racemiczna kwasu D-(-)- i L-(-)-mlekowego i tylko bakterie Streptococcus wytwarzają czysty
enancjometr L-(-).
6. CO ROZUMIESZ POD POJĘCIEM KLEIKOWANIE SKROBI?
Kleikowanie jest jedną z podstawowych fizycznych przemian skrobi. Po spęcznieniu ziarna w
wodzie w podwyższonej temperaturze (zazwyczaj ok. 65
0
C) zaczyna się tworzyd żel.
Jednocześnie rozpoczyna się współzawodnictwo o cząsteczki wody między polisacharydami,
które już opuściły ziarno skrobiowe i budują wraz z wodą żel, a ziarenkami skrobi, które
jeszcze nie spęczniały. Zazwyczaj kleikowanie przeprowadza się w zawiesinach ziarnistej
skrobi w wodzie o stężeniu 5-12%, a i wtedy w żelu pozostają jeszcze liczne ziarna
nieskleikowane. W zasadzie do kleikowania skrobi ziarnistej potrzeba co najmniej 30% wody.
Taka ilośd wody odpowiada naturalnej wodnej pojemności skrobi, tzn. tyle skrobia ta może
przyjąd wody z zachowaniem swej sypkości.
Z kleików skrobiowych wytrąca się (zazwyczaj etanolem) skrobię o strukturze
semikrystalicznej. W skrobi takiej, wskutek kleikowania, została rozerwana większośd
międzycząsteczkowych wiązao wodorowych, dzięki czemu skrobia taka jest rozpuszczalna w
wodzie. Zżelowana skrobia jest o wiele aktywniejsza w trakcie przemian fizykochemicznych, a
zwłaszcza chemicznych
7. OPISZ MECHANIZM ODCZUWANIA SMAKU
Odczucie smaku jest wynikiem oddziaływania między kwasowymi (A) i zasadowymi (B)
ośrodkami w receptorach smakowych na języku a zasadowymi i kwasowymi ośrodkami w
związku wywołującym smak (testancie). Podczas kontaktu produktu spożywczego z językiem
zachodzi więc reakcja zobojętnienia. Na języku, w różnych jego strefach znajdują się
receptory A i B sześciu podstawowych smaków, a odległości między nimi są swoiste dla
danego smaku. Tak więc do oddziaływao między receptorem a testantem może dojśd wtedy,
kiedy odległośd między A i B w receptorze i testancie są identyczne i swoiste dla danego
smaku.
8. OMÓW SKALĘ SŁODKOŚCI
Za wzorzec smaku słodkiego przyjmuje się 10-proc. wodny roztwór sacharozy. Jednostka
słodkości, tzw. względna słodkośd (RS; ang. relative sweetness) takiego roztworu wynosi 1.
Słodkośd maleje ze wzrostem liczby członów monosacharydowych w cząsteczce. Ma to
związek ze zwiększaniem się szans na oddziaływania wewnątrzcząsteczkowe w takiej
cząsteczce kosztem oddziaływao międzycząsteczkowych związek słodki – receptor, a także ze
zwiększoną liczbą konformacji przyjmowanych przez taki związek, co z kolei zmniejsza
możliwośd dopasowania się związku słodkiego do receptora. Poza tym słodkośd sacharydów

można zwiększad przez ich modyfikacje chemiczne, np. chlorowanie. O wiele słodsze od
sacharydów są niektóre białka, np. monelina i taumatyna (białka naturalne), talina (sól
glinowa taumatyny, dotychczas najsłodszy znany związek – RS – 3500), aspartam
(syntetyczny dipeptyd).
9. WYMIEO ZNANE CI NATURALNE SACHARYDOWE ŚRODKI SŁODZĄCE
W Polsce są w użyciu następujące naturalne sacharydowe śr. słodzące:
D-Glukoza, środek słodzący do piwa, napojów orzeźwiających i czekolady. Ze względu na
łatwą przyswajalnośd stanowi substancję odżywczą (źródło energii) dla rekonwalescentów.
D-Fruktoza, świetnie rozpuszczalna w wodzie, higroskopijna, dzięki czemu stosuje się ją jako
dodatek zapobiegający odwadnianiu przechowywanych środków spożywczych. Służy ona też
do słodzenia soków, owoców kandyzowanych, lodów, napojów alkoholowych, jogurtów i
różnych deserów. Jest metabolizowana do glikogenu, może byd zatem spożywana przez
diabetyków.
Laktoza, cukier mlekowy odznacza się małą rozpuszczalnością w wodzie. Źródłem laktozy do
celów przemysłowych. Polepsza ona smak wyrobów mlecznych oraz wygląd produktów
ogrzewanych w polu mikrofalowym.
Sacharoza jest najczęściej stosowanym środkiem słodzącym z racji dostępności i przyjemnego
smaku. Do jej metabolizowania potrzebna jest insulina. Sacharoza jest łatwo metabolizowana
i ma wysoką wartośd odżywczą. Jest często stosowana jako konserwant. Jej 30-proc. roztwór
(w przeliczeniu na suchą masę) nie fermentuje, a roztwór 60-proc. jest odporny na wszystkie
drobnoustroje, oprócz Zygosaccharomyces.
Maltoza ma łagodny słodki smak, jest nieco higroskopijna.
Syropy skrobiowe są produktami scukrzania(kwasowej lub enzymatycznej hydrolizy) skrobi.
Ich słodkośd zależy od stopnia scukrzenia. Najpierw tworzy się syrop maltotetraozowy o
słodkości stanowiącej 1/5 słodkości sacharozy. Dalsze scukrzanie prowadzi do syropów
maltozowych. Ostatecznie otrzymuje się syropy glukozowe. Wykorzystuje się je jako dodatek
tekstu ryzujący, wypełniacz oraz do produkcji cukierków i gum.
Ekstrakty słodowe, będące wodnymi ekstraktami słodu jęczmiennego, zawierają białko, sole
mineralne, 45% sacharozy oraz ślady fruktozy, glukozy i maltozy. Służą one do produkcji
cukierków i jako pożywka do hodowli drożdży piekarskich.
Alkohole cukrowe służą do słodzenia żywności dla diabetyków, są bowiem metabolizowane
bez udziału insuliny. Są one odporniejsze niż sacharydy na niskie pH, higroskopijne, mają
przyjemny, długo utrzymujący się słodki smak dający odczucie chłodu.
Miód, syrop klonowy
10. CO TO SĄ BARWNIKI POCHODZENIA SACHARYDOWEGO? W JAKI SPOSÓB SIĘ JE
OTRZYMUJE?
Barwnikami sacharydowymi są karmele. Mają one charakter micelarny i tylko wtedy spełniają
swoje zadanie. Przy niewłaściwym pH micele ulegają zniszczeniu i karmel się wytrąca. Dlatego
do barwienia różnych produktów spożywczych stosuje się karmele o zróżnicowanym punkcie
izoelektrycznym. Otrzymuje się je w ściśle określony sposób, szczególnie istotny jest dobór
katalizatora. Wprawdzie najsilniej barwiący karmel otrzymuje się wówczas, gdy katalizatorem
jest amoniak (dodatkową zaletą jest niska temperatura prowadzenia procesu), ale ze względu
na punkt izoelektryczny takiego karmelu nie może on byd powszechnie używany.
Klasa I – karmel naturalny – napoje alkoholowe, leki, ciasta, aromaty, przyprawy.

Klasa II – karmel siarczanowy (IV) – alkohole specjalne.
Klasa III – karmel amoniakalny – piwo, chleb, ciasta, zupy, sosy, mięso, konserwy, tytoo,
przyprawy.
Klasa IV – karmel amoniakalno-siarczanowy (IV) – coca-cola, pepsi-cola, wermut, ocet winny.
11. PRZEDSTAW KLASYFIKACJĘ LIPIDÓW
Ze względu na pewne podobieostwa strukturalne lipidy podzielono na trzy zasadnicze grupy:
lipidy proste, lipidy złożone i lipidy pochodne (wtórne). Schemat klasyfikacji lipidów można
przedstawid następująco:
Lipidy proste – estry kwasów tłuszczowych i alkoholi.
Lipidy właściwe – estry kwasów tłuszczowych i glicerolu (acyloglicerole)
Woski – estry wyższych kwasów tłuszczowych i alkoholi innych niż glicerol.
Lipidy złożone – związki zawierające oprócz kwasów tłuszczowych i alkoholi również inne
składniki.
Fosfolipidy – lipidy zawierające kwas fosforowy jako mono- lub diester.
Glicerofosfolipidy – pochodne kwasu glicerofosforowego mające
przynajmniej
jedną O-acylową, O-alkinową lub O-(1-alkenylową) grupę przyłączoną do
reszty
glicerolu.
Sfingofosfolipidy – pochodne 1-fosfoceramidu.
Glikolipidy (glikozylolipidy) - związki zawierające co najmniej jeden cukier połączony
wiązaniem glikozydowym z częścią lipidową.
Glikoglicerolipidy – glikolipidy zawierające jedną lub więcej reszt
glicerolowych.
Glikosfingolipidy – związki mające co najmniej jeden cukier i sfingoid
(sfingoidową
zasadę).
Inne lipidy złożone – np. sulfolipidy.
Lipidy pochodne (wtórne) – pochodne lipidów prostych i złożonych powstałe przede
wszystkim w wyniku ich hydrolizy, zachowujące ogólne właściwości lipidów.
Kwasy tłuszczowe.
Alkohole (inne niż glicerol).
Węglowodory.
12. OMÓW ZASADY NOMENKLATURY KWASÓW TŁUSZCZOWYCH
Nazwa „kwas tłuszczowy” oznacza każdy alifatyczny kwas monokarboksylowy, który może
byd uwolniony w reakcji hydrolizy z naturalnie występujących tłuszczów.
Nazwy systematyczne, powstałe z wykorzystaniem nomenklatury zaproponowanej przez
IUPAC, określają długośd łaocucha kwasu tłuszczowego, pozycję, rodzaj i konfigurację
nienasyconego wiązania oraz pozycję i rodzaj podstawnika.
Nazwy kwasów wyprowadza się od nazw odpowiednich węglowodorów. Kwasy nasycone o
prostym łaocuchu są nazywane n-alkanowymi. W ich przypadku do nazwy rdzenia

macierzystego węglowodoru (nazwa łacioska oznaczająca liczbę atomów węgla w cząsteczce)
dodaje się koocówkę „owy”, a całośd poprzedza słowem kwas (kwas dekanowy pochodzi od
dekanu, a kwas heksadekanowy od heksadekanu).
Nazwy systematyczne kwasów nienasyconych (alkenowych) pochodzą od nazw
macierzystych węglowodorów nienasyconych.
Często w odniesieniu do kwasów tłuszczowych stosuje się nomenklaturę skrótową, używając
liczb i symboli do określenia ich struktury. Symbol 16:1 oznacza kwas szesnastowęglowy,
prostołaocuchowy, niezawierający podwójnych wiązao (kwas palmitynowy), natomiast
18:1 oznacza kwas osiemnastowęglowy, prostołaocuchowy, mający jedno wiązanie
podwójne. Poszerzona symbolika, np. 18:1(9c) albo 18:1(9t) oznacza, że jest to kwas
osiemnastowęglowy, prosto łaocuchowy, zawierający jedno wiązanie podwójne przy C-9, o
kofiguracji cis (c) lub trans (t).
13. KWASY TŁUSZCZOWE NASYCONE – BUDOWA CHEMICZNA, GŁÓWNI PRZEDSTAWICIELE,
WŁAŚCIWOŚCI FIZYCZNE, WYSTĘPOWANIE
Najważniejsze kwasy tłuszczowe nasycone to:
masłowy - butanowy,
kapronowy,
kaprylowy,
kaprynowy
laury nowy - dodekanowy,
mirystynowy
palmitynowy - heksadekanowy
stearynowy - oktadekanowy
arachidowy - ikozanowy
behenowy
lignomerowy
Pierwsza nazwa to nazwa zwyczajowa, druga – systematyczna!
Kwasy laurynowy i mirystynowy występują głównie w olejach kokosowym (odpowiednio 45-
50% i 15-19%) i w oleju z ziarn palmowych (45-50% i 15-18%).
Kwas palmitynowy jest głównym kwasem nasyconym w większości olejów z nasion roślin. W
oleju bawełnianym jego zawartośd wynosi 22-28%, a w oleju palmowym 35-45%. W innych
tłuszczach roślinnych występuje w mniejszych ilościach. W tłuszczach zwierzęcych, w tym
mlecznych, jego zawartośd wynosi 20-30%, a w wielu olejach rybnych 12-20%.
Kwas stearynowy jest obecny przede wszystkim w tłuszczach zapasowych przeżuwaczy (łój
wołowy – 15-30%) oraz w maśle kakaowym (ok. 35%)
Kwasy tłuszczowe o krótkich łaocuchach (C
4
– C
10
)występują w większych ilościach w
tłuszczach mlecznych, głównie przeżuwaczy.
Większośd nasyconych kwasów tłuszczowych występuje w temperaturze pokojowej w stanie
stałym.
14. KWASY TŁUSZCZOWE MONOENOWE - BUDOWA CHEMICZNA, GŁÓWNI PRZEDSTAWICIELE,
WŁAŚCIWOŚCI FIZYCZNE, WYSTĘPOWANIE
Naturalne kwasy monoenowe (alkenowe) najczęściej mają strukturę n-9 oraz konfigurację
cis. Kwasy o konfiguracji trans w naturze występują rzadko (np. tłuszcz mleczny).
Przedstawiciele:

Kwas oleinowy – cis-9-oktadecenowy,
Kwas oleopalmitynowy – cis-9-heksadecenowy,
Kwas wakcenowy,
Kwas elaidynowy,
Kwas erukowy – cis-13-dekozenowy.
Kwas oleinowy jest kwasem najbardziej rozpowszechnionym w naturze. Przyjmuje się, że
stanowi on ok. 40% ilości wszystkich kwasów tłuszczowych. Występuje prawie we wszystkich
tłuszczach. W oliwie jest podstawowym kwasem (ok. 75%). Kwasy heksadecenowe
(oleopalmitynowe) są bardzo rozpowszechnione w tłuszczach zwierzęcych, rzadko jednak
występują w dużych stężeniach. W olejach z tradycyjnych odmian rzepaku kwas erukowy jest
dominującym kwasem (ok. 50%). Kwasy monoenowe o długich łaocuchach (C
20:1
i C
22:1
) są
obecne również w większych ilościach w tłuszczach zwierząt morskich.
15. KWASY TŁUSZCZOWE POLIENOWE - BUDOWA CHEMICZNA, GŁÓWNI PRZEDSTAWICIELE,
WŁAŚCIWOŚCI FIZYCZNE, WYSTĘPOWANIE
Naturalnie występujące kwasy polienowi zawierają 2 – 6 podwójnych wiązao, przeważnie w
konfiguracji cis, ułożonych w ten sposób, że każda para jest przedzielona jedną grupą
metylenową (-CH=CH-CH
2
-CH=CH-), tak jak w kwasie linolowym lub arachidonowym. Ze
względu na strukturę łaocucha między terminalną grupą metylową, a najbliższym względem
niej podwójnym wiązaniem, polienowi kwasy tłuszczowe dzieli się zwykle na rodziny kwasów:
n-9, n-6, n-3. Niektóre z kwasów n-6 i n-3 należą do NNKT.
Główni przedstawiciele:
kwas linolowy – cis, cis-9,12-oktadekadienowy,
kwas linolenowy - all cis-9,12,15-oktadekatrienowy,
kwas arachidonowy – all cis-5,8,11,14-ikozatetraenowy ,
EPA,
DHA.
Kwas linolowy należy do najpowszechniej występujących kwasów. Jest on obecny prawie we
wszystkich tłuszczach, głównie jednak w olejach roślinnych. W niektórych z nich jest głównym
kwasem, np. w oleju krokoszowym (60 – 80%), sojowym (48 – 58%), słonecznikowym (20 –
75%).
Kwas linolenowy zwany jest również kwasem α-linolenowym. W większych ilościach
występuje on Ostatnio ze względów żywieniowych, duże zainteresowanie wywołuje kwas γ-
linolenowy. Występuje on w tkankach zwierzęcych oraz w nasionach niektórych roślin, np.
wiesiołka, ogórecznika, czarnej porzeczki. Bardzo aktywny biologicznie kwas arachidonowy
jest obecny przede wszystkim w fosfolipidach zwierzęcych (np. wątrobowych).
16. NNKT - BUDOWA CHEMICZNA, GŁÓWNI PRZEDSTAWICIELE, WŁAŚCIWOŚCI FIZYCZNE,
WYSTĘPOWANIE
Niektóre spośród kwasów polienowych są niezbędne do prawidłowego rozwoju i normalnego
funkcjonowania organizmu. Dlatego mają szczególne znaczenie w żywieniu człowieka.
Polienowe kwasy tłuszczowe mające właściwości NNKT należą do dwóch rodzin kwasów: n-6 i
n-3.
Przedstawiciele:
Kwasy tłuszczowe n-6: linolowy, γ-linolenowy, arachidonowy

Kwasy tłuszczowe n-3: α-linolenowy, EPA, DHA.
W obrębie każdej grupy kwasy powstają w wyniku biosyntezy i w określonej sekwencji z
pierwszych przedstawicieli na liście.
Kwasy z rodziny n-6 wykazują większą aktywnośd biologiczną niż kwasy z rodziny n-3, stąd
dawniej tych drugich nie zaliczano do NNKT. Obecnie wielu autorów, uwzględniając wyniki
ostatnich badao włącza również kwas linolenowy (n-3) i jego metabolity do kwasów
niezbędnych.
Bardzo ważną rolę odgrywa również w organizmie kwas arachidonowy, m.in. jako prekursor
ikozanoidów (eikozanoidów), związków o bardzo dużej aktywności fizjologicznej i
farmakologicznej, wśród nich prostaglandyn, tromboksanów i leukotrienów. Kwas ten
powstaje w wyniku przemiany kwasu linolowego, polegającej na odwodornieniu (desaturazy)
cząsteczki zachodzącym na przemian z wydłużeniem łaocucha (elongazy). Reakcja obejmuje
fragment cząsteczki między grupą karboksylową a pierwszym (najbliższym) wiązaniem
podwójnym. Fragment łaocucha (grupa metylowa i najbliższe względem niej wiązanie
podwójne) określający przynależnośd kwasu do rodziny, w danym przypadku n-6, nie ulega
zmianie. W ustroju kwas arachidonowy może powstad również w wyniku retro konwersji.
Powstające metabolity n-3 wykazują aktywnośd biologiczną kwasu linolenowego.
Tkanki zwierząt i człowieka, ze względu na brak odpowiednich układów enzymatycznych
zdolnych do tworzenia w łaocuchu kwasów tłuszczowych wiązao podwójnych w położeniu
dalszym niż przy C-9, nie mają możliwości syntezowania kwasów linolowego i linolenowego.
Kwasy te, wytwarzane wyłącznie przez rośliny, muszą byd dostarczone do organizmu
człowieka w pożywieniu.
17. OMÓW TEORIE DOTYCZĄCE ROZMIESZCZENIA KWASÓW TŁUSZCZOWYCH W
TRIACYLOGLICEROLACH
Przez wiele lat, w zależności od stosowanych metod badawczych, wysuwano różne teorie
struktury naturalnych triacylogliceroli. Początkowo sądzono, że tłuszcze są mieszaniną
jednokwasowych acylogliceroli. Później zaproponowano hipotezę równomiernego rozkładu
kwasów tłuszczowych, tj. możliwie najszerszego rozmieszczenia kwasów między wszystkie
cząsteczki triacylogliceroli. Oznacza to np., że jeżeli dany kwas „S” w mieszaninie wszystkich
obecnych kwasów tłuszczowych stanowi mniej niż 1/3, to nie powinien on wystąpid więcej
niż jeden raz w każdej cząsteczce triacyloglicerolu. W miarę postępu wiedzy o tłuszczach
teorię równomiernego rozkładu zastąpiono teorią statystycznego rozkładu. Według niej
kwasy rozkładają się zupełnie przypadkowo zarówno między cząsteczki triacylogliceroli, jak i
wewnątrz nich. Gdy okazało się, że zawartośd całkowicie nasyconych triacylogliceroli jest
zwykle mniejsza niż to wynika z przewidywao, zaproponowano teorię ograniczonej
przypadkowości. Według tej teorii, triacyloglicerole kwasów nasyconych powstają w tłuszczu
zwierzęcym zgodnie z regułą statystycznego rozkładu dopóty, dopóki tłuszcz ten in vivo
pozostaje ciekły.
Odkrycie, że lipaza trzustkowa dzięki swej dużej specyficzności pozycyjnej deacyluje
triacyloglicerole tylko w pozycjach zewnętrznych (sn-1,3) umożliwiło określanie ich struktury
z dużą precyzją. Znajomośd składu kwasów tłuszczowych w pozycjach zewnętrznych (sn-1,3)
oraz wewnętrznych (sn-2) triacylogliceroli umożliwiła wysunięcie hipotezy o 1,3-
przypadkowym-2-przypadkowym rozmieszczeniu kwasów tłuszczowych. Teoria ta zakłada,
że dwie grupy kwasów tłuszczowych o różnym składzie są przypadkowo (statystycznie)
rozmieszczone oddzielnie w pozycjach zewnętrznych (sn-1,3) i wewnętrznych (sn-2)

wszystkich cząsteczek triacylogliceroli, przy założeniu, że pozycje sn-1 i sn-3 są równocenne
(enzym nie rozróżnia ich).
Ostatecznie przyjęto teorię 1-przypadkowego-2-przypadkowego-3-przypadkowego
rozmieszczenia kwasów tłuszczowych w naturalnych triacyloglicerolach. Oznacza to, że trzy
różne grupy kwasów tłuszczowych są oddzielnie, lecz przypadkowo (statystycznie)
rozmieszczone w każdej z trzech pozycji w cząsteczkach triacylogliceroli naturalnych
tłuszczów.
18. MONO- I DIACYLOGLICEROLE – BUDOWA CHEMICZNA, WYSTĘPOWANIE, OTRZYMYWANIE,
ZASTOSOWANIE W PRZEMYŚLE SPOŻYWCZYM
Mono- i diacyloglicerole należą do tzw. niepełnych acylogliceroli. Mają odpowiednio dwie lub
jedną wolną grupę hydroksylową. Mogą to byd sn-1, sn-2 i sn-3 monoacyloglicerole oraz sn-
1,2, sn-2,3 i sn-1,3 diacyloglicerole. W tłuszczach naturalnych występują w nieznacznych
ilościach, z wyjątkiem tych, które uległy częściowej hydrolizie. Zatem te tłuszcze, które uległy
zepsuciu i zawierają dużo wolnych kwasów tłuszczowych mają dużą koncentrację
odpowiednio di- i monoacylogliceroli. Na przykład lipidy w wilgotnych nasionach roślin
oleistych składowanych przez dłuższy czas lub uszkodzonych mogą podlegad głębokim
przemianom hydrolitycznym z powstaniem dużych ilości niepełnych acylogliceroli.
Przemysłowe mono- i diacyloglicerole otrzymywane przez bezpośrednią estryfikację glicerolu
są mieszaniną mono-, di- i triacylogliceroli, zależnie od wzajemnych proporcji użytych
substratów. Zawierają także pewne ilości wolnego glicerolu.
Inną ważną metodą otrzymywania niepełnych acylogliceroli, a zwłaszcza monoacylogliceroli,
jest reakcja glicerozy odpowiednich tłuszczów. W zależności od wzajemnych proporcji
tłuszczu i glicerolu zmienia się zawartośd monoacylogliceroli w produkcie. W handlu są to
produkty o nazwach np. „40% monoglicerydy” czy „50% monoglicerydy”. Destylacja
molekularna umożliwia oddzielenie monoacylogliceroli od di- i triacylogliceroli. W ten sposób
można otrzymad preparaty o koncentracji ponad 90% monoacylogliceroli.
Produkty te reprezentują bardzo ważną klasę emulgatorów spożywczych, stosowanych
między innymi w produkcji margaryn, lodów i wielu innych produktów, do których
wytwarzania niezbędne są emulgatory.
19. WOSKI – BUDOWA CHEMICZNA, WŁAŚCIWOŚCI FIZYCZNE, WYSTĘPOWANIE
Estry innych alkoholi niż glicerol i długołaocuchowych kwasów tłuszczowych klasyfikuje się
jako woski (lipidy proste) i tak ta nazwa powinna byd rozumiana w sensie chemicznym.
Związki te najczęściej występują na powierzchni organizmów zwierzęcych lub roślinnych w
mieszaninie z innymi związkami, m.in. z długołaocuchowymi węglowodorami, wolnymi
alkoholami, kwasmi tłuszczowymi. Dlatego też najczęściej w języku potocznym nazwą wosk
jest określana cała ta mieszanina.
Woski w naturze spełniają wiele ważnych funkcji. Dzięki swej odporności na działanie różnych
czynników fizycznych i chemicznych chronią tkanki przed utratą wody, przed wpływem
wilgoci zewnętrznej, a także przed drobnoustrojami oraz przed szkodliwym działaniem
czynników mechanicznych.
Niekiedy występują jako zapasowe lipidy w organizmach zwierząt i roślin zastępując
triacyloglicerole. W świecie roślinnym woski występują prawie wyłącznie na powierzchni liści,
łodyg, kwiatów, owoców i nasion.

Większośd nasion oleistych zawiera wosk na okrywie nasiennej. Podczas tłoczenia lub
ekstrakcji tłuszczów z nasion woski te zostają zmieszane z triacyloglicerolami.
Jednym ze składników wosku są estry woskowe. Są to estry długołaocuchowych kwasów
tłuszczowych i długołaocuchowych hydroksyzwiązków. Mogą to byd np. mono- lub
dihydroksyalkany albo hydroksykwasy. Typowym woskiem roślinnym jest np. wosk Carnauba,
zawierający ok. 36% wosków estrowych (głównie C
54
– C
60
). Są to przede wszystkim estry
alkoholi C
30
– C
34
i odpowiednich kwasów tłuszczowych. Ważnymi woskami zwierzęcymi są
wosk wełny (lanolina) i wosk pszczeli. W wielu woskach charakterystycznym składnikiem są
wolne i zestryfikowane sterole. W surowcach zwierzęcych jest to zwykle cholesterol,
natomiast w roślinnych – stigmasterol i β-sitosterol.
20. WYMIEO GRUPY ZWIĄZKÓW ZALICZANYCH DO FOSFOLIPIDÓW, KRÓTKO OPISZ ICH
BUDOWĘ I WYSTĘPOWANIE
Do fosfolipidów zalicza się glicerofosfolipidy i sfingofosfolipidy. Są to lipidy zawierające kwas
fosforowy w formie mono- lub diestru . Są one składnikiem każdej komórki roślinnej i
zwierzęcej. Szczególnie dużo znajduje się ich w tkance nerwowej oraz w dojrzałych nasionach
roślin oleistych.
W naturze występują głównie w postaci kompleksów z białkami (lipoproteiny). W tej postaci
są bardzo ważnym składnikiem osocza krwi.
Glicerofosfolipidy
Zgodnie z terminologią IUPAC-IUB, glicerofosfolipidem określa się każdą pochodną kwasu
glicerofosforowego, która zawiera przynajmniej jedną grupę O-acylową, o-alkinową lub O-(1-
alkylenową) przyłączoną do reszty glicerolu.
Charakterystyczną cechą glicerofosfolipidów, odróżniającą je od innych lipidów, jest ich
nierozpuszczalnośd w acetonie.
Glicerofosfolipidy są pochodną kwasu glicerofosforowego,
ściślej sn-glicero-3-fosforanu.
Glicerofosfolipidy ssaków zawierają kwasy C
16
– C
20
, głównie palmitynowy, stearynowy,
oleinowy, linolowy i arachidonowy. Glicerofosfolipidy zwierząt morskich charakteryzują się
dużą zawartością kwasów C
20:5
i C
22:6
. W roślinnych glicerofosfolipidach kwasy są mniej
zróżnicowane. Są to przede wszystkim kwasy: palmitynowy, oleinowy, linolowy.
Oleje zawierające fosfolipidy, np. oleje surowe, mogą ulegad procesowi brunatnienia,
spowodowanego m.in. reakcją aminoaldehydową.
Kwas fosfatydowy jest pochodną sn-glicero-3-fosforanu, w którym dwie grupy hydroksylowe
są zacylowane długołaocuchowymi kwasami tłuszczowymi, najczęściej różnymi. Kwasy
fosfatydowe mogą występowad w tłuszczach w postaci wolnej lub jako sole. Należą do tzw.
niehydratujących się fosfolipidów. W przyrodzie odgrywają ważną rolę jako związki pośrednie
w biosyntezie innych glicerofosfolipidów i triacylogliceroli. Są monoestrami trójzasadowego
kwasu fosforowego, który może tworzyd dalsze wiązania estrowe. W tych związkach reszta
kwasu fosfatydowego zwana jest fosfatydylem. Reagując z odpowiednimi związkami
zawierającymi grupę hydroksylową, tworzą estry fosfatydylowe. Stanowią ważną i bardzo
rozpowszechnioną grupę produktów naturalnych.
Kwas fosfatydowy z choliną (HOCH
2
CH
3
NMe
3
) tworzy fosfatydylocholinę (lecytyna), z
etanoloaminą fosfatydyloetanoloaminę , z seryną fosfatydyloserynę i z inozytolem –
fosfatydyloinozytol.
Stosunkowo niedawno poznano fosfatydyloglicerole i difosfatydyloglicerole.
Fosfatydyloglicerol jest ważnym składnikiem fosfosyntezującej tkanki a difosfatydyloglicerol
jest głównym lipidowym składnikiem mitochondriów.

Ważną grupą glicerofosfolipidów występujących głównie w świecie zwierzęcym są
plazmalogeny.
Sfingolipidy.
Są pochodnymi sfinganiny lub jej homologów oraz nienasyconych pochodnych tych
związków. Są to jednak przede wszystkim pochodne sfingozyny, która występuje głównie w
świecie zwierzęcym. W świecie zwierzęcym jest rozpowszechniona fitosfingozyna oraz jej
pochodna dehydrofitosfingozyna. N-Acylowane formy tych związków są nazywane
ceramidami. Ceramidy wchodzą w połączenia poprzez pierwszorzędową grupę hydroksylową
przy C-1 albo z jednostką cukrową, albo z fosforanowymi estrami, tworząc ważne grupy
związków naturalnych, jak np. glikosfingolipidy i sfingofosfolipidy.
Glikosfingolipidy należą do dużej grupy związków zwanej glikolipidami. Termin
glikosfingolipid obejmuje wszystkie związki zawierające przynajmniej jeden monosacharyd
oraz sfingoid. Określane są one jako glikozylosfingoidy lub glikozyloceramidy. Reszta
cukrowa jest dołączona do grupy hydroksylowej przy C-1 ceramidu. Cukier występujący w
połączeniu z ceramidem może byd prosty (galaktoza, glukoza) albo złożony.
Monoglikozyceramidy zwane są cerebrozydami (występują w tkance mózgowej i innych
tkankach; rozpowszechnione są także w świecie roślinnym), a bardziej złożone związki
gangliozydami.
Ważną grupę związków należących do sfingolipidów stanowią sfingofosfolipidy. Kwas
fosforowy w sfingofosfolipidach estryfikuje pierwszorzędową grupę hydroksylową w
ceramidzie, a następnie łączy się z odpowiednim amino alkoholem lub amino alkoholem.
Sfingofosfolipidy pochodzenia zwierzęcego zawierające fosfocholinę, fosfoetanolaminę są
nazywane sfingomielinami. Wchodzą one w skład osłonki mielinowej włókien nerwowych
białej substancji mózgu. Występują również w lipidach wątroby, śledziony i nerek.
21. LECYTYNA – OTRZYMYWANIE I ZASTOSOWANIE W PRZEMYŚLE SPOŻYWCZYM
Szlamy pohydratacyjne olejów roślinnych, głównie oleju sojowego, stanowią surowiec do
otrzymywania handlowych preparatów fosfolipidów, potocznie zwanych „lecytyną”. W
surowej lecytynie, stanowiącej mieszaninę różnych lipidów, fosfolipidy są dominującym
składnikiem (ponad 50%). Skład fosfolipidów zależy głównie od rodzaju i jakości
hydratowanego surowca tłuszczowego. Dla przykładu w lecytynie sojowej fosfatydylocholina,
fosfatydyloetanoloamina i fosfatydyloinozytol występują prawie w równych ilościach, z
przewagą pierwszego składnika. Zawartośd pozostałych fosfolipidów jest niewielka.
Właściwości emulgujące lecytyny można poprawid np. przez frakcjonowanie jej etanolem.
Frakcja rozpuszczalna wzbogacona w fosfatydylocholinę jest efektywna w tworzeniu i
stabilizacji emulsji typu o/w, z kolei nierozpuszczalna, wzbogacona głównie w
fosfatydyloinozytol, w tworzeniu emulsji typu w/o.
Oczyszczanie i uszlachetnianie surowej lecytyny można przeprowadzid przez m.in.
frakcjonowanie jej rozpuszczalnikami, ekstrakcję gazami w stanie nadkrytycznym oraz
metodami chromatograficznymi i adsorpcyjnymi. Działania te mają głównie na celu
zwiększenie koncentracji fosfatydylocholiny poprzez eliminowanie frakcji kefalinowej.
Modyfikacje lecytyny .
Lecytynę najczęściej modyfikuje się przez acetylowanie, hydroksylowanie, hydrolizę i
uwodornienie.
Spośród związków zawartych w lecytynie acetylowaniu podlegają fosfatydyloetanoloamina i
fosfatydyloseryna. Utworzony produkt jest bardziej hydrofilowy niż surowiec. Podobny efekt

można osiągnąd w wyniku hydroksylowania. Hydroksylowana lecytyna ma jaśniejszą barwę
jest bardziej plastyczna i charakteryzuje się lepszymi właściwościami emulgującymi.
Częściowa deacylacja fosfolipidów metodą hydrolizy chemicznej lub enzymatycznej prowadzi
do powstania lizo fosfolipidów. Tak modyfikowane lecytyny są lepiej rozpuszczalne w wodzie
i mają lepsze właściwości emulgujące w układach o/w. Z kolei katalityczne uwodornienie
nienasyconych wiązao w kwasach tłuszczowych naturalnych lecytyn daje produkt
charakteryzujący się zwiększoną odpornością na utlenianie.
Lecytyna, głównie sojowa, dzięki jej właściwościom emulgującym, zwilżającym, koloidalnym,
przeciwutleniającym i fizjologicznym znalazła zastosowanie m.in. w przemyśle spożywczym i
farmaceutycznym. W połączeniu z innymi emulgatorami, zwykle mono- i diacyloglicerolami,
jest dodawana do osnowy tłuszczowej podczas produkcji margaryn. Przeciwdziała ona m.in.
zjawisku wydzielania się kropel wody z gotowego produktu, jest czynnikiem
przeciwrozpryskowym podczas smażenia, poprawia właściwości reologiczne ciasta, działa
wspomagająco w ochronie wit. A przed utlenieniem. Lecytyna dodana do masy czekoladowej
używanej w produkcji cukierków wydatnie zmniejsza jej lepkośd, umożliwiając tym samym
skrócenie czasu mieszania różnych składników oraz uzyskania równomiernej warstwy
polewy. Polewa czekoladowa z dodatkiem lecytny jest bardziej stabilna, w tym bardziej
odporna na tzw. „wykwity” i cukrowe oraz zjawisko szarzenia.
Lecytyna jest również bardzo wartościowym emulgatorem w różnych wyrobach piekarskich
(chleb, herbatniki, krakersy). Ułatwia wprowadzenie szorteningów do ciasta, poprawia
procesy fermentacji i ułatwia absorpcję wody. Lecytyna jest także stosowana w produkcji
wyrobów typu „instant”, jak np. mleka w proszku i kakao.
22. STEROLE – BUDOWA CHEMICZNA, GŁÓWNI PRZEDSTAWICIELE, WYSTĘPOWANIE
Sterole należą do alkoholi alicyklicznych z grupy steroidów. Są to związki krystaliczne o
wysokich temperaturach topnienia (cholesterol 150,8
0
C). Występują one we wszystkich
organizmach roślinnych i zwierzęcych, są nierozpuszczalne w wodzie, natomiast dobrze
rozpuszczają się w rozpuszczalnikach tłuszczowych. W tłuszczach występują w stanie wolnym
lub w postaci estrów z kwasami tłuszczowymi (wosków). Stanowią główny składnik substancji
niezmydlających się. Podczas rafinacji tłuszczów usuwa się główną masę steroli. Mydła
porafinacyjne oraz kondensaty po procesie odwaniania są stosunkowo bogatymi źródłami
steroli.
Sterole należą do dużej i bardzo ważnej, ze względu na metabolizm, grupy związków ogólnie
nazywanej steroidami. Nazwą „steroid” określa się każdą substancję, która jest pochodną
układu skondensowanych pierścieni cyklopentanoperhydrofrenantrenu.
Wszystkie sterole mają grupę hydroksylową przy C-3 w konfiguracji β (także w pozycji cis do
grupy metylowej przy C-10) oraz łaocuch boczny przy C-17. Poszczególne pierścienie mogą
się znajdowad względem siebie w konfiguracji cis lub trans.
Sterole najczęściej różnią się obecnością lub brakiem takich elementów strukturalnych, jak
podwójne wiązania w pierścieniu B, podwójne wiązanie w łaocuchu bocznym, rozgałęzienie
przy C-24.
W zależności od występowania sterole często dzieli się na trzy zasadnicze grupy: zoosterole,
fitosterole i mykosterole. Pierwsze z nich zawierają przeważnie 27 atomów węgla w
cząsteczce, pozostałe przeważnie 28 i 29.podziału takiego nie należy traktowad zbyt ściśle,

gdyż niektóre ze steroli można zaliczyd do różnych grup. Na przykład cholesterol, typowy
sterol zwierzęcy, może występowad również w mniejszych ilościach w roślinach i grzybach.
Głównym sterolem pochodzenia zwierzęcego jest cholesterol (5-cholesten-3β-ol). Jest on
jednym z najważniejszych oraz najwcześniej i najlepiej poznanych steroli. Występuje we
wszystkich komórkach ssaków; szczególnie dużo jest go w tkance nerwowej i w wątrobie.
Najczęściej występuje w formie wolnej lub zestryfikowanej kwasami tłuszczowymi.
Do najważniejszych steroli roślinnych należą sitosterole, głównie β- sitosterol, stigmasterol,
kampesterol i brassikasterol. W olejach roślinnych, ogólnie, najwięcej jest β-sitosterolu (55-
75% ilości wszystkich steroli). Brassikasterol jest charakterystycznym sterolem oleju
rzepakowego.
Spośród steroli wytwarzanych przez grzyby najbardziej rozpowszechniony jest ergosterol.
Szczególnie bogatym źródłem ergosterolu, surowca do otrzymywania wit. D
2
, są niektóre
drożdże. Występuje on także w surowcach roślinnych i zwierzęcych.
Sterole i estry ulegają stosunkowo łatwo odwodnieniu (dehydratacji) pod wpływem
stężonych kwasów mineralnych, w wyniku czego tworzą się m.in. węglowodory i etery
steroidowe.
23. OPISZ PROCES HYDROLIZY TRIACYLOGLICEROLI. CO ROZUMIESZ POD POJĘCIEM JEŁCZENIE
HYDROLITYCZNE?
Reakcję hydrolizy wiązania estrowego lipidów można przedstawid na przykładzie
triacylogliceroli. Reakcja przebiega przez kolejne stadia (di- i monoacyloglicerole) i jest
odwracalna. Jeżeli lub produkty nie są usuwane z obszaru reakcji, to ustala się stan
równowagi.
W przemysłowym rozszczepianiu tłuszczów wysoki stopieo hydrolizy uzyskuje się przez
stosowanie nadmiaru wody i okresowe usuwanie wodnego roztworu glicerolu. Wysoka
temperatura i ciśnienie przyspieszają hydrolizę. Ze względu na strukturę kwasów
tłuszczowych (np. kwasy Wielonienasycone mogą polimeryzowad), należy optymalizowad
parametry. Normalnie hydroliza jest katalizowana przez kwasy lub zasady. Całkowita
hydroliza acylogliceroli z użyciem kwasów mineralnych prowadzi do powstania kwasów

tłuszczowych i glicerolu. Produktami hydrolizy alkalicznej, zwanej też zmydlaniem, są glicerol
oraz mydła (sole kwasów tłuszczowych). Ostatnio wiele uwagi poświęca się hydrolizie
enzymatycznej – lipolizie, ponieważ może ona przebiegad w warunkach zbliżonych do
normalnych.
Podczas składowania surowców tłuszczowych oraz wydobywania tłuszczów może zachodzid,
zwłaszcza pod wpływem enzymów, częściowa hydroliza triacylogliceroli i innych lipidów
zawierających reszty acylowe (np. fosfolipidów) z uwolnieniem kwasów tłuszczowych. Te
niepożądane przemiany wymagają stosowania procesów rafinacyjnych i prowadzą do strat
substancji tłuszczowej. Niepożądany proces hydrolizy tłuszczów i produktów tłuszczowych
oraz produktów spożywczych zawierających substancje tłuszczowe o charakterze estrowym
jest niekiedy zwany jełczeniem hydrolitycznym. Sensoryczne skutki tego typu jełczenia są
szczególnie uciążliwe w artykułach i przetworach mlecznych. Krótkołaocuchowe kwasy
tłuszczowe (zwłaszcza masłowy) są bowiem przyczyną powstawania nieprzyjemnego zapachu
i smaku. Niekiedy uwalnianie tych kwasów jest sensorycznie pożądane (dojrzewanie serów).
24. WYMIEO ZNANE CI TYPY REAKCJI OTRZYMYWANIA TRIACYLOGLICEROLI
Estry kwasów tłuszczowych można uzyskiwad metodą bądź bezpośredniej estryfikacji
alkoholu kwasem, bądź interestryfikacji, zwanej też trans estryfikacją lub
przeestryfikowaniem.
Estryfikacja w technologii tłuszczów jadalnych jest rzadko wykorzystywana. Często natomiast
jest stosowana w analizie lipidów w celu uzyskania estrów metylowych kwasów
tłuszczowych. Otrzymuje się je z kwasów tłuszczowych i metanolu zawierającego katalizator,
np.H
2
SO
4
, HCL, albo w reakcji z diazometanem. Reakcja estryfikacji stanowi odwrócenie
reakcji hydrolizy. Estryfikując kwasy tłuszczowe bezpośrednio glicerolem, użytym w
odpowiednich proporcjach, otrzymuje się mieszaninę mono-, di- i triacylogliceroli. Produkt
ten ma zastosowanie m.in. jako emulgator.
Alkoholiza bardzo często jest stosowana do przekształcania acylogliceroli w estry metylowe
(metanoliza), bez konieczności wstępnego izolowania wolnych kwasów, oraz do uzyskiwania
niepełnych acylogliceroli w reakcji triacylogliceroli z glicerolem (gliceroliza). W procesie
metanolizy tłuszcz rozpuszcza się w nadmiarze metanolu (przesunięcie równowagi) w
obecności katalizatora.
Katalizator może byd kwaśny (HCL) lub zasadowy (NaOH).
Gliceroliza triacylogliceroli zachodzi podczas ogrzewania ich z glicerolem w obecności
katalizatora (NaOH, CH
3
ONa).
Triacyloglicerol + glicerol monoacyloglicerol + diacyloglicerol
Stopieo glicerozy zależy od ilości dodanego glicerolu (ściślej rozpuszczonego w tłuszczu) i
parametrów reakcji. Stan równowagi jest określony przez skład mieszaniny reakcyjnej. Jest to
ważna metoda preparowania mono- i diacylogliceroli (otrzymywanie emulgatora).
Acydoliza obejmuje wzajemne oddziaływanie estrów z kwasem karboksylowym w obecności
katalizatora (H
2
SO
4
, ZnO, CaO). Może byd wykorzystana np. do zastąpienia kwasów C
16
i C
18
kwasem C
12
. W technologii tłuszczów jadalnych praktycznie nie ma ona zastosowania.

25. NA CZYM POLEGA PROCES INTERESTRYFIKACJI WŁAŚCIWEJ TRIACYLOGLICEROLI?
Reakcja intensyfikacji właściwej (przeestryfikowania właściwego) ma największe znaczenie
technologiczne. Przeprowadzając tę reakcję można dokonad zmiany sposobu rozkładu
kwasów tłuszczowych w triacyloglicrolach, uzyskując tłuszcze zmodyfikowane o pożądanym
zakresie temp. topnienia i właściwościach krystalizacyjnych. W praktyce proces przebiega w
obecności katalizatorów alkalicznych (np. NaOH) w temp. często poniżej 100
0
C. Wówczas
następuje zmiana pozycji grup acylowych zarówno wewnątrz poszczególnych cząsteczek
triacylogliceroli (przeestryfikowanie wewnątrzcząsteczkowe – intraestryfikacja), jak i między
różnymi cząsteczkami ( międzyczcząsteczkowe) aż do osiągnięcia stanu statystycznego
rozmieszczenia kwasów tłuszczowych we wszystkich możliwych pozycjach. W wyniku tak
prowadzonej modyfikacji specyficzne rozmieszczenie kwasów tłuszczowych w naturalnych
acyloglicerolach przekształca się w rozmieszczenie statystyczne (randomizacja). W ten sposób
zmieniają się nie tylko chemiczne, ale przede wszystkim fizyczne właściwości tłuszczów. Na
przykład randomizacja oleju bawełnianego zmienia jego temp. topnienia z 10 do 30
0
C.
Proces
przeestryfikowania może byd zastosowany do mieszaniny różnych tłuszczów – w wyniku
przeestryfikowania bardzo twardych tłuszczów zwierzęcych o małej wartości żywieniowej z
olejami ciekłymi uzyskuje się nowe tłuszcze o pożądanych właściwościach.
Proces przeestryfikowania zmienia strukturę i skład triacylogliceroli, nie zmienia natomiast
naturalnej budowy występujących w nich kwasów tłuszczowych.
Interestryfikacja tłuszczów może do pewnego stopnia zachodzid bez udziału katalizatora, ale
za to w ekstremalnych warunkach, wymagających bardzo wysokiej temp. i długiego czasu.
Proces przeestryfikowania wykorzystuje się w przemyśle do otrzymywania
wysokowartościowych tłuszczów plastycznych, np. do produkcji margaryn.
W praktyce proces przeestryfikowania jest realizowany jako okresowy, półciągły lub ciągły.
26. NA CZYM POLEGA PROCES UWODORNIENIA TŁUSZCZÓW?
Proces uwodornienia tłuszczów jest często stosowany w przemyśle tłuszczowym do zmiany
ich charakteru fizycznego i chemicznego oraz składu występujących w nich kwasów
tłuszczowych. Uwodornienie prowadzi się w dwóch celach – zwiększenia ich stabilności
oksydatywnej oraz przekształcenia olejów w produkty plastyczne, które są bardziej przydatne
do wyrobu margaryn i innych tłuszczów jadalnych.
Uwodornienie jest reakcją katalityczną, którą można przyspieszad, stosując heterogeniczne
lub homogeniczne katalizatory zawierające takie metale, jak Pt, Pd, Ni, Cu i Co. Spośród tych
metali tylko nikiel ma znaczenie przemysłowe.
Uwodornienie przebiega w układzie niejednorodnym, składającym się z trzech faz: stałej
(katalizator), ciekłej (olej) i gazowej (wodór).
Procesy zachodzące podczas katalitycznego uwodorniania można podzielid na przebiegające
w fazie ruchomej (dyfuzja) i na powierzchni katalizatora (adsorpcja, reakcje powierzchniowe,
desorpcja).
Olej do celów spożywczych poddaje się tylko częściowemu uwodornieniu. Dlatego ważnymi
cechami tak prowadzonego procesu są selektywnośd i izomeryzacja. Selektywnośd wiąże się z
różnymi szybkościami reakcji uwodornienia nienasyconych kwasów tłuszczowych o różnym
stopniu nienasycenia. Izomeryzacja wynika z tego, że podwójne wiązania pozostałe w
częściowo uwodornionym tłuszczu mogą mied zmienioną pozycję lub konfigurację (cis –
trans).

Reakcja uwodornienia, w której biorą udział kompleksy aktywne: metal-alken i metal-wodór,
przebiega przez tworzenie się związku półuwodornionego. Wodór z powierzchni metalu
zostaje przeniesiony do jednego z atomów węgla podwójnego wiązania, natomiast drugi
atom węgla wiąże się z powierzchnią metalu. W wyniku przyłączenia się drugiego atomu
wodoru do związku półuwodornionego powstaje związek całkowicie uwodorniony. Reakcja ta
jest praktycznie nieodwracalna.
Ponieważ reakcja powstawania związku półuwodornionego jest odwracalna, związek ten po
odszczepieniu atomu wodoru i przeniesieniu go do metalu może powrócid do stanu
alkenowego. Odszczepienie wodoru może nastąpid od atomu węgla z lewej lub prawej strony
względem atomu węgla związanego z powierzchnią metalu. W ten sposób może byd
odtworzone wiązanie podwójne oryginalne (przed adsorpcją) lub przesunięte (izomeryzacja
pozycyjna).
Wobec swobodnej rotacji wokół wiązao między atomami węgla w związku
półuwodornionym, powstające na skutek odwodornienia wiązania podwójne mogą mied
konfigurację cis lub trans.
Uwodornienie, zmieniając strukturę kwasów tłuszczowych, nie zmienia struktury
triacylogliceroli.
27. OPISZ PROCES AUTOOKSYDACJI LIPIDÓW. SCHEMATYCZNIE ZAPISZ RÓWNANIA REAKCJI
Autooksydacja jest rodnikową reakcją łaocuchową, w której można wyróżnid trzy
podstawowe etapy.
Inicjacja (zapoczątkowanie reakcji) – homolityczne oderwanie wodoru i utworzenie
węglowego rodnika alkilowego w obecności inicjatora
Propagacja (rozwijanie reakcji) – reakcja rodnika z O
2
i utworzenie rodnika nadtlenlowego,
który następnie reaguje z nienasyconym lipidem (alken – RH) i tworzy się wodorotlenek oraz
nowy rodnik lipidowy. Nowo powstały rodnik reaguje z O
2
, tworząc rodnik nadtlenkowy itd.
W ten sposób autooksydacja staje się rodnikowym procesem łaocuchowym.
Terminacja (zakooczenie reakcji). Reakcja łaocuchowa może byd zakooczona (co oznacza
przerwanie łaocucha) na skutek rekombinacji rodników i tworzenia się nierodnikowych
produktów, które nie są ani inicjatorami, ani propagatorami reakcji.
Mimo, że wszystkie nienasycone lipidy ulegają utlenianiu, to z praktycznego punktu widzenia
problem autooksydacji dotyczy nienasyconego łaocucha węglowodorowego kwasów
tłuszczowych, zwłaszcza kwasów polienowych. Szybkośd reakcji rośnie wraz ze stopniem

nienasycenia. Kwas linolowy utlenia się 10-40-krotnie szybciej niż oleinowy, natomiast
linolenowy 2-4-krotnie szybciej niż linolowy.
Koocowymi produktami rodnikowej reakcji łaocuchowej są wodoronadtlenki. SA one
prekursorami zjełczałego zapachu i smaku.
28. NA CZYM POLEGA FOTOSENSYBILIZOWANE UTLENIANIE TŁUSZCZÓW?
Fotoutlenianie obejmuje reakcję alkenu z tlenem w obecności światła i odpowiedniego
sensybilizatora (uczulacza). Sensybilizatory przekształcają tlen
3
O
2
w jego bardziej reaktywny
stan singletowy
1
O
2
. W tłuszczach i produktach spożywczych zawierających tłuszcze
występuje wiele substancji mogących pełnid funkcje fotosensybilizatorów. Są to m.in.
naturalne barwniki, jak chlorofil, feofityna a oraz barwniki hemowe – mioglobina albo
hemoglobina lub ich pochodne. Również erytrozyna, syntetyczny barwnik, jest aktywnym
uczulaczem. Tlen w stanie singletowym reaguje z alkenem bez wytwarzania rodnika (reakcja
„enowa”) przyłączając się do jednego z węgli olefinowych (podwójnego wiązania). Ta reakcja,
połączona z migracją podwójnego wiązania i zmianą jego konfiguracji cis w trans, jest
niezależna od ciśnienia tlenu i nie wykazuje wyraźnie mierzalnego okresu indukcji. Jest
inhibowana przez „wygaszacze” tlenu singletowego (β-karoten, tokoferole, syntetyczne
związki – BHA i BHT). Nie oddziałują na nią przeciwutleniacze (brak rodników). W wyniku
ataku tlenu singletowego na podwójne wiązanie tworzą się wodoronadtlenki alkilowe w
konfiguracji trans.
Fotosensybilizowane utlenianie jest dużo szybsze niż autooksydacja, a różnice w
reaktywności pomiędzy kwasami oleinowym, linolowym i linolenowym są w przybliżeniu
proporcjonalne do liczby występujących w nich wiązao podwójnych.
29. OPISZ REAKCJE ZACHODZĄCE PODCZAS OGRZEWANIA TŁUSZCZÓW W WARUNKACH
BEZTLENOWYCH
Przebieg reakcji zachodzących podczas ogrzewania tłuszczów zależy od składu lipidów oraz
od warunków obróbki termicznej.
W warunkach beztlenowych jest wymagana stosunkowo wysoka temperatura (ponad 200
0
C),
aby nastąpił rozkład nasyconych triaclogliceroli. Produktami takiej reakcji są m.in. normalne
alkany i alkeny akroleina i CO
2
.
Ogrzewanie nienasyconych lipidów w warunkach beztlenowych powoduje przede wszystkim
powstawanie dimerów i związków cyklicznych. Jeden z głównych mechanizmów to
homolityczny rozpad wiązania C-C w położeniu α lub β do podwójnego wiązania i utworzenie
odpowiednich rodnikowych fragmentów.
Bezpośrednie połączenie tych rodników może prowadzid do powstania kwasów krótko- i
długołaocuchowych, kwasów dikarboksylowych oraz węglowodorów. Fragmenty rodnikowe
mogą również odbierad wodór z innej cząsteczki kwasu tłuszczowego (np. oleinowego), w
wyniku czego powstają rodniki allilowe. Powstałe rodniki allilowe o różnej strukturze mogą
reagowad między sobą, co prowadzi do powstania różnych acyklicznych dimerów dienowych
.
W przypadku acylogliceroli może się utworzyd wiązanie C-C pomiędzy dwoma acylami dwóch
różnych cząsteczek lub dwoma acylami tej samej cząsteczki. Może również w obrębie tej
samej cząsteczki acyloglicerolu nastąpid addycja rodnika allilowego do podwójnego wiązania
z utworzeniem cyklicznego monomeru.

Rodnik allilowy może także ulegad dysproporcjonowaniu (dysmutacji) do kwasów mono- i
dienowych .
W przypadku kwasu linolowego zachodzą podobne reakcje, jednakże produkty są złożoną
mieszaniną dimerów: acyklicznych, bicyklicznych i tricyklicznych o różnym stopniu
nienasycenia. Podobne reakcje przebiegają również pomiędzy grupami acylowymi w różnych
acyloglicerolach, dając np. dimery i trimery triacylogliceroli.
Może również zachodzid dimeryzacja z utworzeniem monocyklicznej struktury w następstwie
reakcji Dielsa-Aldera między sprzężonym dienem, np. powstałym z kwasu linolowego, i grupą
alkenową innej cząsteczki. W ten sposób utworzony zostaje czteropodstawiony cykloheksen.
W przypadku acylogliceroli dimeryzacja może zachodzid pomiędzy grupami acylowymi dwóch
różnych cząsteczek lub dwoma grupami acylowymi tej samej cząsteczki.
30. OPISZ REAKCJE ZACHODZĄCE PODCZAS OGRZEWANIA TŁUSZCZÓW PRZY DOSTĘPIE TLENU
Nasycone kwasy tłuszczowe i ich estry wykazują nieporównywalnie większą stabilnośd niż ich
nienasycone analogi. Jednakże ogrzewane w atmosferze powietrza, w temperaturach
wyższych niż 150
0
C, mogą ulegad utlenieniu i złożonym procesom rozkładu. Utlenianie
nasyconych kwasów tłuszczowych następuje na ogół przy węglu α, β lub γ z utworzeniem
odpowiednich rodników alkoksylowych. Termiczny rozpad rodników pomiędzy węglami α, β i
γ daje mieszaninę różnych węglowodorów, aldehydów i ketonów.
Nienasycone kwasy tłuszczowe są bardziej podatne na utlenianie niż nasycone analogi. Ich
oksydatywne przemiany w podwyższonej temperaturze następują z dużą szybkością.
Większośd związków powstałych w wysokiej temp. jest jakościowo podobna do tych, które
powstają w temp. pokojowej. Jednakże rozpad wodoronadtlenków oraz wtórne utlenianie
zachodzi z ekstremalną szybkością w podwyższonej temp. Ogrzewanie w wyższej temp.
nienasyconych kwasów tłuszczowych w atmosferze powietrza prowadzi do tworzenia
oksydimerów lub polimerów z grupami wodoronadtlenkowymi, hydroksylowymi,
epoksydowymi i karbonylowymi.
Oksydatywnotermiczny rozpad nienasyconych kwasów tłuszczowych najczęściej prowadzi do
utworzenia dimerów, trimerów i tetramerów z grupami polarnymi.
31. JAKIM PRZEMIANOM ULEGAJĄ TŁUSZCZE W PROCESIE GŁĘBOKIEGO SMAŻENIA?
Większośd procesów smażenia odbywa się w temp. 170 – 190
0
C. w tych warunkach w
tłuszczu smażalniczym zachodzą wielokierunkowe przemiany fizykochemiczne, wśród których
dominują:
hydroliza,
utlenianie,
przemiany termiczne, w tym polimeryzacja i cyklizacja.
Efektem tego jest powstanie w medium tłuszczowym wielu różnych związków, często o
bardzo złożonej i nie zawsze ustalonej strukturze. Ogólnie związki te można podzielid na lotne
(m.in. aldehydy, ketony, węglowodory, laktony, alkohole, kwasy i estry) i nielotne (m.in.
niepolimerowe związki polarne, cykliczne monomery, dimery i polimery kwasów i
acylogliceroli).
Powstałe podczas smażenia produkty rozpadu i przemiany lipidów powodują m.in. wzrost
poziomu WKT w oleju, obniżenie jego temp. dymienia, zwiększenie zdolności pienienia się,
wzrost lepkości i pociemnienie barwy tłuszczu.

32. WYMIEO I KRÓTKO OPISZ ZNANE CI GRUPY TŁUSZCZÓW MODYFIKOWANYCH
Tłuszcze uwodornione
Katalityczne uwodornienie przekształca oleje w półstałe plastyczne tłuszcze odpowiednie do
wytwarzania m.in. szorteningów, tłuszczów smażalniczych i cukierniczych oraz margaryn.
Tłuszcze przeestryfikowane
Interestryfikacja jest procesem technologicznym stosowanym głównie w celu zmiany
właściwości fizycznych tłuszczów. W wyniku interestryfikacji otrzymuje się tłuszcze
modyfikowane o pożądanym zakresie temp. topnienia i właściwościach krystalizacyjnych,
wykorzystywanych np. w produkcji margaryn, szorteningów i tłuszczów cukierniczych, oraz
tłuszcze specjalne (strukturyzowane) o dużej wartości żywieniowej.
Tłuszcze fizycznie modyfikowane
Tłuszcze frakcjonowane. Nazwa ta, użyta bez dodatkowego określenia, odnosi się do
tłuszczów otrzymywanych metodą krystalizacji frakcjonowanej. Frakcjonowanie tłuszczów
przeprowadza się z kilku powodów, m.in. w celu usunięcia niektórych substancji
towarzyszących, ograniczających wykorzystanie tłuszczu (oddzielanie substancji woskowych –
uszlachetnianie oleju słonecznikowego), wzbogacenia tłuszczów w niektóre triacyloglicerole,
rozdzielenia tłuszczu na dwie lub więcej frakcji o bardziej pożądanych właściwościach
użytkowych niż substrat (np. frakcjonowanie łoju, oleju z ziaren palmowych, otrzymywanie
stearyny i oleiny palmowej).
Proces krystalizacji obejmuje dwa etapy: krystalizację w celu wytworzenia stałych kryształów
i ciekłej matrycy oraz oddzielanie kryształów od frakcji ciekłej. Krystalizacji poddaje się bądź
bezpośrednio stopiony tłuszcz (frakcjonowanie suche), bądź tłuszcz rozpuszczony w
odpowiednim rozpuszczalniku (frakcjonowanie mokre). Metoda krystalizacji frakcjonowanej
jest szczególnie przydatna do pozyskiwania składników tłuszczowych o specyficznych
właściwościach, odpowiednich w produkcji tłuszczów alternatywnych dla masła kakaowego.
Tłuszcze mieszane. Są to mieszanki tłuszczowe nazywane również blendami. Mieszając różne
tłuszcze w odpowiednich proporcjach, można otrzymad nowy tłuszcz (mieszankę tłuszczową)
o pożądanych właściwościach, np. chemicznych i fizycznych. Na przykład mieszając ciekłe
oleje z tłuszczami twardymi, można otrzymad mieszanki o zróżnicowanej konsystencji w
zależności od proporcji składników. Tą metodą można także uzyskad tłuszcze smażalni cze o
dobrej stabilności, a przy tym stosunkowo tanie.
33. OMÓW STRUKTURĘ PIERWSZORZĘDOWĄ BIAŁEK
Struktura pierwszorzędowa białka mówi o sekwencji aminokwasów w cząsteczce. Tę
strukturę bada się metodami chemicznymi.
struktura
pierwszorzędowa
-
określa, w jaki sposób atomy w cząsteczkach białka są z sobą
połączone wiązaniami kowalencyjnymi, czyli jak tworzą się łańcuchy. Inaczej struktura
pierwszorzędowa określa kolejność aminokwasów w łańcuchu białkowym.
Struktura pierwszorzędowa
Jak wiemy białka są produktami kondensacji wielu aminokwasów. Z dotychczasowych
doświadczeń wynika, że aminokwasy nie są połączone między sobą w sposób przypadkowy,
lecz kolejność ich jest specyficzna i charakterystyczna dla określonego białka. Ta
uprządkowana kolejność nazywana jest sekwencją aminokwasów w białku. Sekwencja
aminokwasów może na przykład wyglądać następująco:
H
2
N Tyr-Tre-Wal-Asp-Leu-Gli-Gli-Cys-His COOH
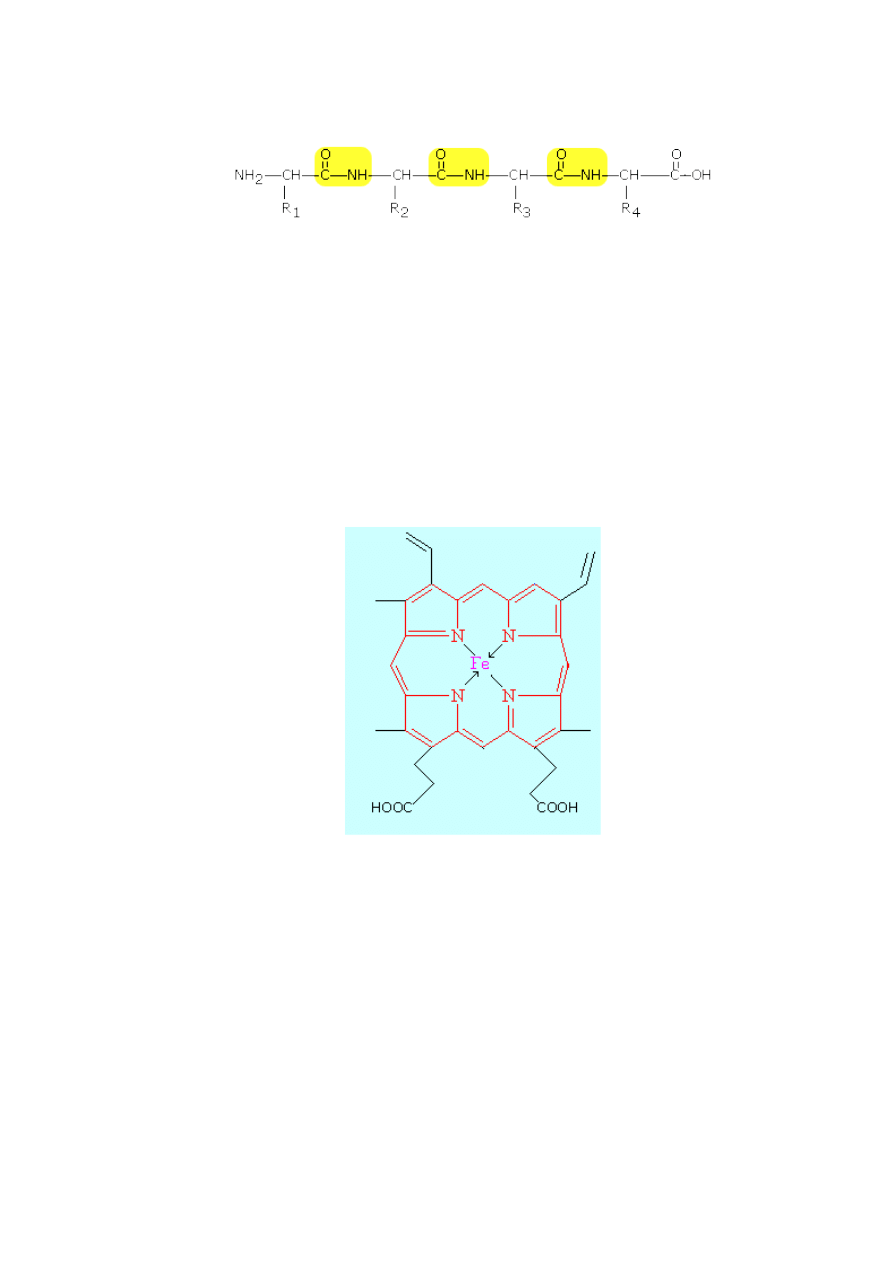
Białka zbudowane są z łańcuchów peptydowych w którym do co trzeciego atomu jest
przyłączony łańcuch boczny (R
1
, R
2
, R
3
, R
4
,...).
Struktura łańcucha bocznego zależy od reszty określonego amionokwasu, np. w przypadku
glicyny jest to atom - H, alaniny - grupa (-CH
3
), waliny - grupa (-CH(CH
3
)
2
), itd
Niektóre z tych bocznych łańcuchów zawierają grupy zasadowe, np. grupę -NH
2
i grupy
kwasowe -COOH (patrz tablica 13.1).
Ze względu na obecność tych kwasowych i zasadowych łańcuchów bocznych wzdłuż
łańcucha peptydowego rozmieszczone są grupy naładowane dodatnio lub ujemnie.
I właśnie ta charakterystyczna dla określonego białka sekwencja łańcuchów bocznych, nadaje
mu charakterystyczne właściwości
Łańcuchy boczne wywierają wpływ na właściwości białek nie tylko dzięki swej kwasowości lub
zasadowości, ale również poprzez inne właściwości chemiczne, a nawet poprzez wielkość i
kształt. Na przykład obecność grupy wodorotlenowej (-OH) i grupy tiolowej (-SH) przyczynia
się do reakcji tworzenia estrów.
Niektóre cząsteczki białek zawierają fragmenty niepeptydowe (nazywane
grupą prostetyczną
).
Grupa prostetyczna je
st ściśle powiązana ze specyficzną biologiczną funkcją białka.
Na przykład grupą prostetyczną hemoglobiny jest
hem
Jak wynika ze wzoru, hem zawiera atom żelaza związany z układem pirolowym, znanym jako
porfina
. To właśnie utworzenie odwracalnego kompleksu tlen-hem umożliwia hemoglobinie
przenoszenie tlenu z płuc do tkanek.
Tlenek węgla tworzy podobny, ale bardziej trwały kompleks, dzięki czemu wiąże on
hemoglobinę uniemożliwiając transport tlenu, co powoduje śmierć.
Hem jest połączony z peptydowym fragmentem białka (
globiną
) w wyniku chelatowania atomu
żelaza przez histydynowe atomy azotu białka, a także za pomocą wiązań wodorowych oraz sił
van der Waalsa działających pomiędzy hydrofobowymi fragmentami dwóch cząsteczek.
34. OMÓW STRUKTURĘ DRUGORZĘDOWĄ BIAŁEK
Struktura drugorzędowa białka określa kształt łaocucha polipeptydowego, czyli konformację
(łaocuch rozciągnięty lub zwinięty w spiralę). W tej strukturze istotną rolę odgrywają wiązania
wodorowe.
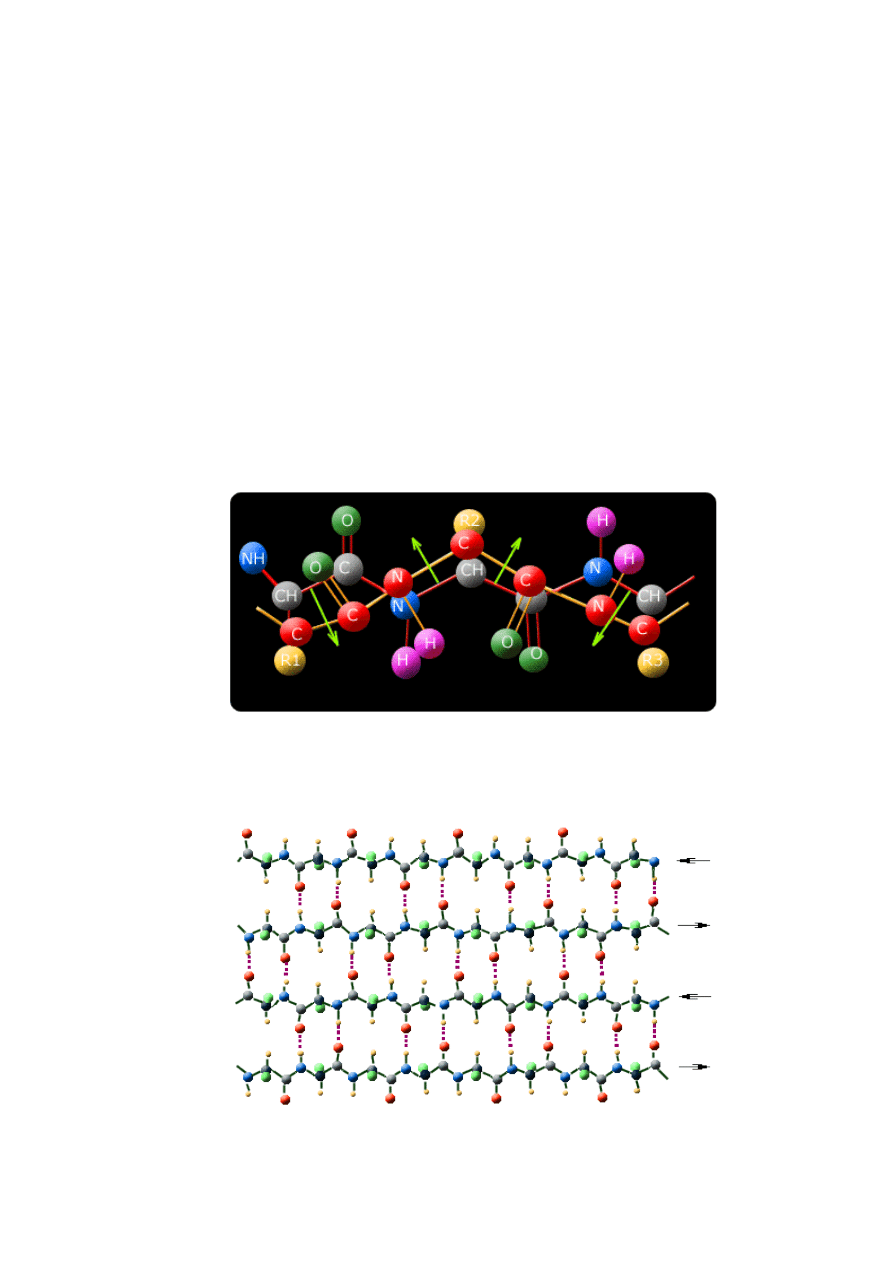
struktura
drugorzędowa
-
określa, w jaki sposób utworzone łańcuchy są ułożone w
przestrzeni, czyli jakie formy przestrzenne (spirale, arkusze albo kule) tworzą one za pomoca
wiązań wodorowych, łączących różne łańcuchy lub różne części tego samego łańcucha
Struktura drugorzędowa
Termin "struktura drugorzędowa" określa wzajemne, przestrzenne ułożenie aminokwasów w
łańcuchu białkowym o określonej sekwencji. Badania prowadzone metodami rentgenowskimi
udowodniły, że nie wszystkie możliwe struktury łańcucha białkowego są jednakowo cenne pod
wzgledem trwałości.
Najtrwalsze muszą zawierać maksymalną liczbę wiązań wodorowych między grupami
karbonylowymi -C=O i grupami -N-
H występującymi w wiązaniu peptydowym. Wiązania
wodorowe będą silnie stabilizować strukturę, jednakże aby mogły powstać, odpowiednie grupy
muszą znaleźć się w odległości oddziaływań wodorowych.
W przypadku białek z grupy skleroproteidów trwała struktura osiągana jest dzięki
oddziaływaniom wodorowym między dwoma łańcuchami białkowymi biegnącymi równolegle
do siebie. Tworzą one wtedy tzw. strukturę "pofałdowanej kartki (harmonijki)". Taka struktura
nazywana jest również
strukturą beta
.
To pofałdowanie powstaje w wyniku ściągnięcia łańcuchów peptydowych, przez co zmienia
się geometria wiązania peptydowego aminokwasu z płaskiej na pofałdowaną (rysunek 13.4).
Uzykujemy wtedy bardziej korzystną strukture do rozmieszczenia małych lub średnich
łańcuchów bocznych.
Na rysunku 13.4 przedstawiono płaskie wiązanie peptydowe w kolorze czarnym, natomiast
kolorem różowym efekt ściągnięcia łańcuchów peptydowych w strukturze beta.
Zmiana geometrii wiązania peptydowego podczas tworzenia strubtury beta
Na rysunku 13.5 przedstawiono pełny obraz struktury harmonijkowej (beta). W tej strukturze
każdy łańcuch jest połączony z innymi łańcuchami wiązaniem wodorowym (=O -- H-). Na
rysunku wiązanie wodorowe zaznaczone jest kolorem różowym.
Struktura harmonijkowa (struktura beta). Ściągnięte łańcuchy stwarzają miejsce dla małych
łańcuchów bocznych; sąsiednie łańcuchy są rozwinięte w przeciwnych kierunkach

Struktura pofałdowana jest korzystna dla białek w których łańcuchy boczne są małe.
Gdy łańcuchy boczne są bardzo duże, wówczas najlepsze rozmieszczenie zapewnia struktura
innego rodzaju, w której każdy łańcuch jest zwinięty i tworzy
heliks (struktura alfa)
(rysunek
13.6)
Struktura alfa
Łańcuch peptydowy jest tu spiralnie owinięty wokół hipotetycznego walca z taką gęstością
zwojów, aby grupy -C=O i -N-H zwojów sąsiadujących ze sobą znalazły się w odległości
odpowiedniej do utworzenia wiązań wodorowych. Z tego wynika, że różne fragmenty tego
samego łańcucha są połączone wiązaniami wodorowymi, które pomagają utrzymać strukturę
heliksu. Na rysunku 13.7 przedstawiono pełną strukturę heliksu.
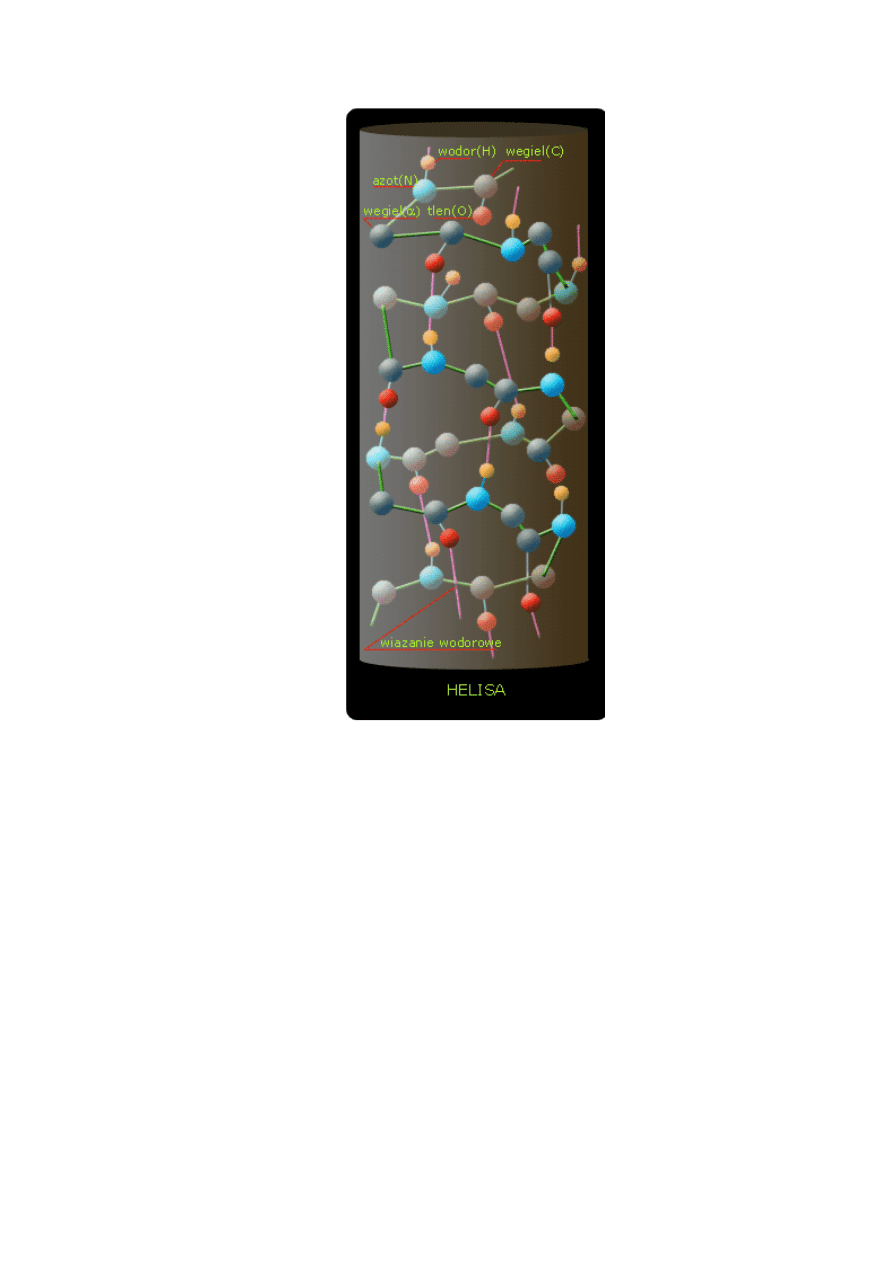
Struktura heliksu (struktura alfa). W prawoskretnym heliksie na każdy skręt przypada 3,6
reszt; wiązania wodorowe znajdują się wewnątrz łańcucha
Posługując się modelami można wykazać, że właśnie taki heliks stwarza wystarczającą ilość
miejsca dla łańcuchów bocznych i pozwala utworzyć się wszystkim możliwym wiązaniom
wodorowym. Wyjaśnia to powtarzającą sie odległość 0,15 nm, która jest odległością pomiędzy
resztami aminokwasowymi, zmierzoną wzdłuż osi heliksu.
Każdy heliks może być sam skręcony w
superheliks
. Dowiedziono, że struktura heliksu
odgrywa kluczową rolę w konstrucji białek spotykanych w przyrodzie. Ale tym zajmuje się
struktura trzeciorzędowa
35. OMÓW STRUKTURĘ TRZECIORZĘDOWĄ BIAŁEK
Struktura trzeciorzędowa białka mówi o kształcie cząsteczki, czyli o zwinięciu łaocucha
polipeptydowego. W tej strukturze istotną rolę odgrywają wiązania dwusiarczkowe.
struktura
trzeciorzędowa
-
określa najbardziej korzystne uporządkowanie przestrzenne
poszczególnych części cząsteczki białka z punktu widzenia energetycznego; zależy od
oddziaływań między łańcuchami bocznymi jednej lub większej liczby makrocząsteczek.
Struktura trz
eciorzędowa
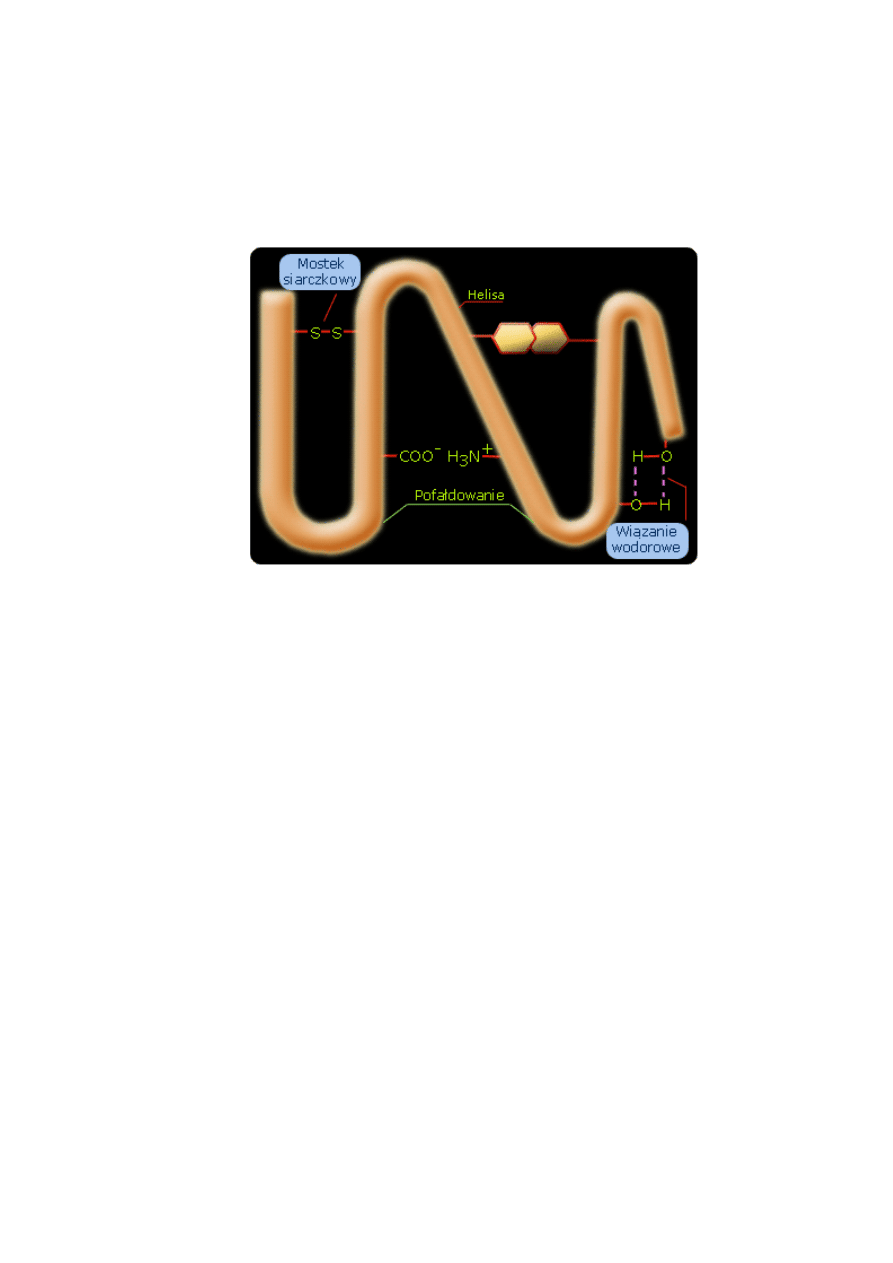
Struktura trzeciorzędowa określa sposób w jaki układają się i fałdują w przestrzeni łańcuchy
białkowe o określonej strukturze drugorzędowej. Zwoje i fałdy jakie tutaj się obserwuje są
utrzymywane różnego typu wiązaniami. Tymi wiazaniami są:
wiązania wodorowe
-
które mogą
powstawać między resztami aminokwasów zawierających grupy funkcyjne, nie związane
wiązaniami peptydowymi (seryna, arginina, treonina, kwas glutaminowy),
mostki siarczkowe
-
powstające między resztkami cysteiny, które łączą różne punkty spirali, zaginając ją w
odpowiedni sposób,
prolina (aminokwas z grupą aminową umieszczoną w pierścieniu)
-
która
może w różny sposób odziaływać na strukturę drugorzędową.
Model struktury trzeciorzędowej
36. OMÓW STRUKTURĘ CZWARTORZĘDOWĄ BIAŁEK
struktura czwartorzędowa
-
określa sposób przestrzennego powiązania kilku cząsteczek w
jedną złożoną strukturę białka.
Struktura czwartorzędowa białka – wzajemne położenie łaocuchów polipeptydowych oraz
ewentualnie struktur niebiałkowych (grupa prostetyczna):
a. cukrów w glikoproteidach
b. lipidów w lipoproteidach
c. kwasów nukleinowych w nukleoproteidach
d. barwników w chromoproteidach
e. resztę kwasu fosforowego w fosfoproteidach
St
ruktura czwartorzędowa
Struktura czwartorzędowa określa występowanie niektórych białek w postaci agregatów kilku
podobnych lub nawet identycznych podjednostek o charakterze białkowym. Przykładem jest
hemoglobina
(rysunek 13.9), gdzie cztery pofałdowane łańcuchy hemogloginy są do siebie
dopasowane i tworzą w przybliżeniu kulistą cząsteczkę (o wymiarach 6,4x5,5x5,0).
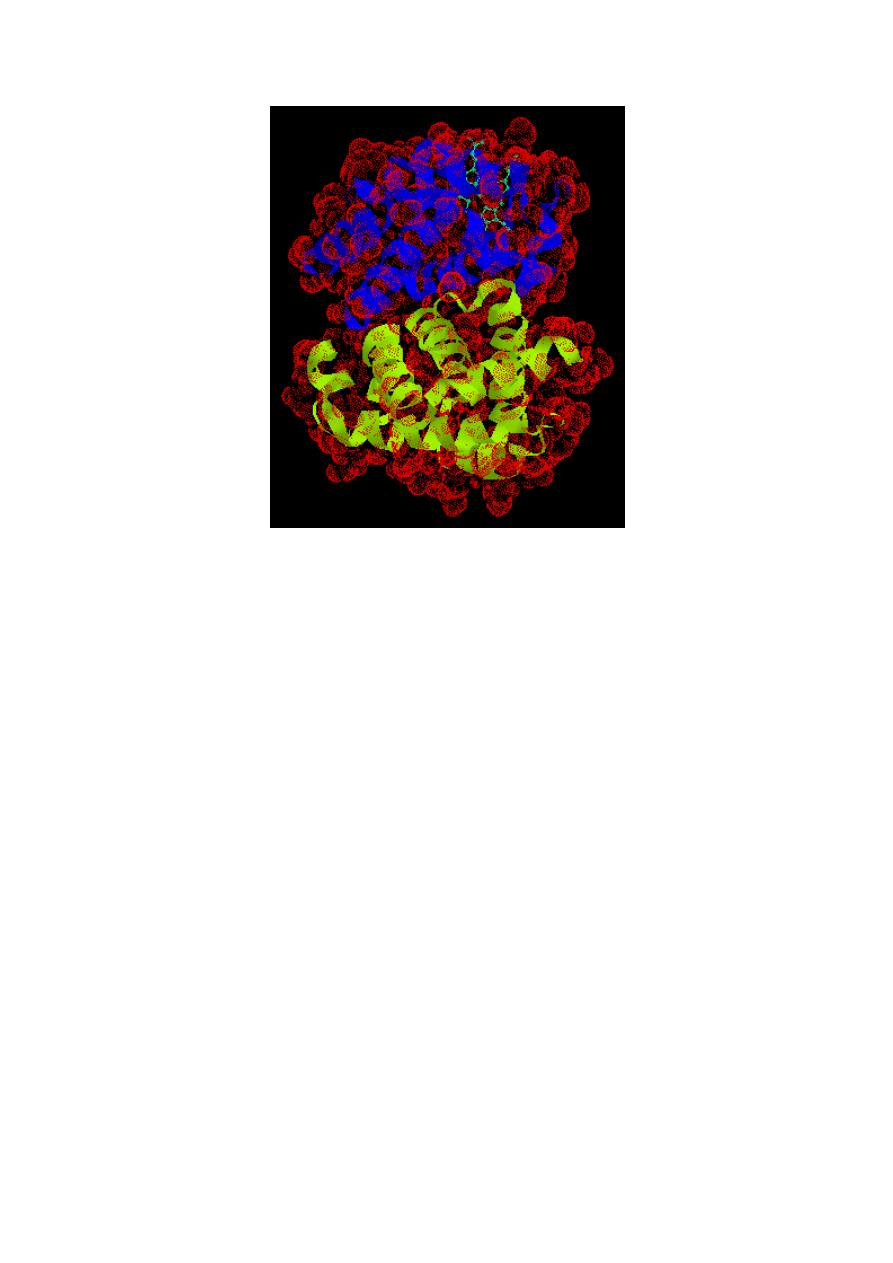
Cząsteczka hemoglobiny
Cztery płaskie grupy hemowe, każda zawierająca atom żelaza, który może wiązać cząsteczkę tlenu,
mieszczą się w oddzielnych "kieszeniach" tej kuli.
Budowę podjednostkową ma również wiele białek enzymatycznych.
37. OPISZ PROCES DENATURACJI BIAŁEK
Denaturacją nazywa się rozpad wiązao stabilizujących drugo- i trzeciorzędową strukturę
białka. Niewielkie siły wywołują w białku nieznaczne zmiany stanu reszt aminokwasów i
rozluźnienie struktury. Ze wzrostem energii działania destabilizacyjnego, np. wskutek
ogrzewania do temp. ok. 60
0
C, dużej zmiany odczynu środowiska lub w obecności
rozpuszczalnika organicznego albo mocznika następuje dysocjacja oligomerów na
podjednostki, rozwinięcie łaocuchów polipeptydowych oraz zniszczenie struktur fałdowych i
heliksowych. Substancje redukujące niszczą w białkach wiązania disulfidowe, wewnątrz- i
międzyłaocuchowe. Po przekroczeniu granicznych naprężeo denaturacja ma charakter
procesów sprzężonych. Denaturacja udostępnia do reakcji ze środowiskiem wiele grup
funkcyjnych ukrytych w białku w stanie rodzimym we wnętrzu cząsteczki. Niektóre białka
mają zdolnośd renaturacji w korzystnych warunkach, zwłaszcza gdy w zdenaturowanej
postaci cząsteczka zachowuje stabilne fragmenty rodzimej konformacji. Renaturacja jest
bardziej prawdopodobna w czystych roztworach białek niż w środowisku produktu
żywnościowego, w którym jest wiele reaktywnych związków.
Bardzo często wykorzystuje się w przetwórstwie żywności denaturujące działanie obróbki w
celu unieczynnienia enzymów katalizujących niepożądane reakcje, np. enzymatyczne
ciemnienie owoców i warzyw przy udziale oksydazy o-difenolowej , mięknienie kwaszonych
ogórków pod wpływem enzymów pektynolitycznych.
38. OD CZEGO ZALEŻY WODOCHŁONNOŚD BIAŁEK?

Dzięki hydrofilowym właściwościom białek liczne produkty żywnościowe przejawiają dużą
wodochłonnośd, tj. zdolnośd utrzymywania w swej strukturze wody własnej lub dodanej,
wbrew działaniu sił zewnętrznych, np. grawitacji, siły odśrodkowej, ciśnienia. Ilośd wody
utrzymywana w strukturze różnych produktów dochodzi nawet do ok. 10 g/g białka.
Wodochłonnośd białek zależy od: ich ogólnej hydrofobowości i konformacji, uwikłania w
struktury komórkowe i tkankowe, interakcji z innymi, wielkocząsteczkowymi związkami,
odczynu oraz oddziaływania jonów. Wszystkie czynniki zacieśniające strukturę białek
zmniejszają wodochłonnośd.
Wodochłonnośd ma bardzo duże znaczenie w przetwórstwie mięsa, ryb i produktów
roślinnych, gdyż decyduje o ich soczystości i właściwościach reologicznych, a także o ubytku
masy wskutek ogrzewania. Dlatego służy ona do oceny wpływu różnych parametrów obróbki
na właściwości białek i polisacharydów.
39. NA CZYM POLEGA ŻELOWANIE PRZY UDZIALE BIAŁEK?
Żelowanie polega na tworzeniu uporządkowanej, przestrzennej struktury cząsteczek
polimerów, zdolnej do zatrzymania rozpuszczalnika i substancji rozpuszczonych albo
wypełniaczy. W produktach żywnościowych taką strukturę mogą utworzyd polisacharydy albo
białka lub obydwa polimery wspólnie.
Żelowanie przebiega na ogół dwustopniowo. Najpierw dysocjują struktury czwartorzędowe i
rozfałdowują się łaocuchy polipeptydowe wskutek denaturacji lub częściowej hydrolizy.
Zdenaturowane cząsteczki, oddziałując między sobą, tworzą trójwymiarową strukturę;
oziębienie układu zazwyczaj ją stabilizuje. Jeśli szybkośd tworzenia struktury jest mniejsza niż
szybkośd denaturacji, to powstaje uporządkowana sied przezroczystego, odwracalnego żelu.
Jeżeli zaś drugi etap procesu przebiega bardzo szybko, przypadkowe interakcje cząsteczek
zdenaturowanego białka prowadzą do wydzielenia się koagulatu, który na ogół jest mętny i
nie rozpuszcza się po ponownym ogrzaniu.
Żelowanie powoduje, że lepka ciecz staje się lepko – elastycznym ciałem stałym. Dzięki
żelowaniu tworzy się pożądana struktura drobnorozdrobnionych wędlin parzonych oraz
galaretek mięsnych i rybnych, wiążą się kawałki mięsa w różnych przetworach oraz powstają
charakterystyczne, reologiczne cechy wielu innych artykułów żywnościowych. Soczystośd i
reologiczne właściwości żeli zależą w dużym stopniu od wodochłonności białek.
Przy określonym stężeniu białka zdolnośd żelowania zależy przede wszystkim od wielkości i
kształtu łaocuchów polipeptydowych w strukturze siatki przestrzennej. Na ogół białka
globularne o masie cząsteczkowej mniejszej niż 23 kDa nie tworzą żelu. Interakcje różnych
białek mogą zmniejszad twardośd żelu, mogą nie mied żadnego znaczenia lub mogą działad
synergicznie, zależnie od właściwości tych białek i różnych czynników.
Żele mogą powstawad w roztworach lub dyspersjach białek oraz w mieszaninach
rozdrobnionego mięsa, wody i soli, jak np. w wędlinach.
Sied przestrzenna żelu utrzymuje się dzięki różnym wiązaniom. Ogrzewanie sprzyja tworzeniu
oddziaływao hydrofobowych, zapoczątkowujących sied, a niska temperatura zwiększa
stabilnośd żelu dzięki wiązaniom wodorowym. Wiązania kowalencyjne nadają żelom
stabilnośd cieplną i sprężystośd. Zatem żele utrzymywane głównie wiązaniami wodorowymi
są stabilne w niskiej temp., topnieją po ogrzaniu i mogą byd odtworzone po oziębieniu, a żele
usieciowane wiązaniami kowalencyjnymi są odporne na ogrzewanie.
40. OPISZ PROCES EMULGOWANIA LIPIDÓW PRZEZ BIAŁKA

Białka ułatwiają tworzenie emulsji niemieszających się cieczy i stabilizują powstający układ.
Zmniejszenie średnicy kropel cieczy, np. lipidu, rozproszonych w fazie ciągłej, np. w wodzie,
powoduje wykładniczy wzrost powierzchni międzyfazowej. Pracę niezbędną do zwiększenia
powierzchni kropli lipidu można zmniejszyd, obniżając napięcie powierzchniowe przez
przyłączenie cząsteczek białka do powierzchni kropli. Błona białkowa tworząca się wokół
kropel lipidu, mająca ładunek elektryczny, zapobiega fluktuacji, tzn. tworzeniu agregatów
kropel i podstojowi, a także koalescencji, tzn. tworzeniu ciągłej fazy lipidowej. Ponadto białko
w roztworze, zwiększając lepkośd fazy rozpraszającej, zmniejsza szybkośd koalescencji i
podstoju – w mleku jest to szybkośd samorzutnego zbierania się śmietanki w jego górnej
warstwie.
Wydajnośd białka jako emulgatora zależy od jego hydrofobowości powierzchniowej i
powierzchniowego ładunku elektrostatycznego od sztywności cząsteczki oraz lepkości
środowiska. Białka stabilizowane hydrofilową warstwą powierzchniową mają dużą zdolnośd
emulgowania tylko po rozfałdowaniu. Przydatnośd białka jako emulgatora nie wzrasta
jednakże proporcjonalnie do hydrofobowości, lecz zależy od równowagi hydrofilowo-
lipofilowej w cząsteczce. Rozpuszczalnośd wpływa na zdolnośd białka do migracji w kierunku
kuleczek lipidów. Ilośd białka niezbędna do stabilizowania emulsji wzrasta ze zwiększeniem
udziału fazy rozproszonej i ze zmniejszeniem średnicy kuleczek lipidowych. Stężenie białka w
jednocząsteczkowej warstwie na granicy faz wynosi ok. 0,1 mg/m
2
, a stężenie skutecznie
stabilizujące emulsję 0,5 – 20 mg/m
2
. Aby zapewnid dużą szybkośd tworzenia ochronnej
warstwy wokół rozproszonych kropel lipidu, trzeba wprowadzid do emulsji 0,5 – 5% białka.
41. OMÓW PROCES TWORZENIA PIANY PRZY UDZIALE BIAŁEK
W produktach żywnościowych piana powstaje wskutek zdyspergowania pęcherzyków
powietrza w fazie ciągłej ciekłej lub półstałej masie zawierającej białko. Zdolnośd tworzenia
piany ma znaczenie w powstawaniu pożądanych właściwości sensorycznych, np. tekstury
chleba, pieczywa cukierniczego, bitej śmietany i lodów. W pianach występujących w żywności
dwa pęcherzyki powietrza SA oddzielone od siebie dwoma błonami białkowymi
zaadsorbowanymi na granicy faz, rozgraniczonymi cienką warstwą cieczy. Objętośd
pęcherzyków powietrza może stanowid nawet 99% ogólnej objętości piany. Zawartośd białka
w różnych spienionych produktach żywnościowych wynosi od 0,1 do 10% i ok. 1 mg/m
2
, przy
czym białko nie musi byd rozpuszczone – wystarczy żeby było silnie zdyspergowane.
Pianotwórcze właściwości białka można zwiększyd przez krótkotrwałe ogrzanie. Optymalne
warunki ogrzewania zależą od rodzaju i stężenia białka. stabilizacja piany polega na
zmniejszeniu napięcia powierzchniowego na granicy faz gaz-ciecz i utworzenia elastycznej
błony otaczającej pęcherzyki, odpornej na zniszczenie. Stabilizacji układu sprzyja duża lepkośd
fazy ciągłej. Piana opada wskutek ubytku cieczy z przestrzeni między błonami pod wpływem
grawitacji wirowania, ciśnienia albo odparowania oraz przez dyfundowanie gazu z małych do
większych pęcherzyków i przez koalescencję pęcherzyków po zniszczeniu błon białkowych.
42. OPISZ ZMIANY ENZYMATYCZNE BIAŁEK WYSTĘPUJĄCE WSKUTEK OGRZEWANIA ŻYWNOŚCI
Jednym z celów ogrzewania w przetwarzaniu żywności jest zapewnienie optymalnej
temperatury dla wybranych procesów enzymatycznych, przede wszystkim hydrolizy
polisacharydów, białek i lipidów, a także estryfikacji, utleniania i glikozylacji. Temperaturę i
czas ogrzewania dobiera się do charakterystyki stosowanych enzymów i pożądanych
produktów przemian. Natomiast w przemyśle mięsnym, rybnym, w mleczarstwie i w
przetwórstwie nasion roślin oleistych i strączkowych ogrzewanie ma bardzo często na celu

unieczynnienie endogennych enzymów, które mogłyby wywoływad niekorzystne przemiany.
Przykładem może byd inaktywacja lipoksygenazy, której działanie zapoczątkowuje reakcje
prowadzące do powstawania niepożądanego zapachu fasoli w mleku sojowym. Na ogół
pasteryzacja inaktywuje enzymy, dzięki czemu testy enzymatyczne można stosowad jako
miernik skuteczności pasteryzacji.
Warunki obróbki cieplnej stosowane w danym procesie technologicznym mogą niekiedy
przyspieszad niepożądane przemiany biochemiczne, jak np. niszczenie struktury żelu rybnego
wskutek działania proteinaz mięśniowych aktywnych w temp. 60-65
0
C.
43. OPISZ ZMIANY WŁAŚCIWOŚCI REOLOGICZNYCH I UWODNIENIA ZWIĄZANE Z
OGRZEWANIEM ŻYWNOŚCI
Reologiczne właściwości produktów żywnościowych poddanych obróbce cieplnej zależą
przede wszystkim od histologicznej struktury danego materiału oraz od zdolności białek do
utrzymywania wody i tworzenia żelu. Bardzo silnie zmienia się wskutek ogrzewania tekstura
produktów mięsnych i rybnych. Już w temp. 30-40
0
C następuje rozfałdowanie miozyny mięśni
ryb. Denaturacja cieplna wywołuje skurcz włókien mięśniowych, co przejawia się
twardnieniem mięsa. Natomiast kolagen przy ogrzewaniu w wilgotnym środowisku traci
strukturę fibrylarną i przemienia się w rozpuszczalną żelatynę, żelującą po oziębieniu, co
zmniejsza twardośd.
Wiele produktów żywnościowych traci dużo wody wskutek cieplnej denaturacji białek i
powstania nowych wiązao sieciujących. Przy ogrzewaniu mięsa ryby największy ubytek wody
następuje w zakresie temp. 30-60
0
C. Ilośd wycieku zależy od rodzaju mięsa. Wieconym mięsie
straty wskutek parowania i wycieku wynoszą do 40%pierwotnej masy surowca. Ze wzrostem
temperatury zmniejsza się wpływ odczynu w mięsie na wyciek. Parowanie stosowane przy
produkcji konserw rybnych powoduje ok. 20% wyciek, zawierający 1-3% białka, a kryl ogrzany
do temp. 70
0
C traci nieco ponad 30% wody.
44. OPISZ PROCES POWSTAWANIA WIĄZAO SIECIUJĄCYCH WSKUTEK OGRZEWANIA ŻYWNOŚCI
Ogrzewanie produktów bogatych w białko może prowadzid do sieciowania polimerów w
reakcji Maillarda, do tworzenia wiązao ε-N-(γ-glutamylo)lizyloamidowych oraz reakcji
transamidacji. Reakcje wymiany grup tiolowych i disulfidowych oraz utleniania grup
tiolowych, zachodzące w podwyższonej temperaturze, uczestniczą w tworzeniu sieciujących
wiązao międzycząsteczkowych. Można je wykorzystad do wytwarzania jadalnych błon
białkowych o określonej przepuszczalności tlenu i pary wodnej. Kilkugodzinne ogrzewanie w
temp. 80
0
C przy wilgotności względnej powietrza ok. 80%, korzystnie wpływa na
wytrzymałośd i właściwości barierowe błon z izolatów białek serwatki. W białkach i
produktach bogatych w białko, ogrzewanych w zasadowym środowisku, powstają
usieciowania przez lizynoalaninę .
W drastycznie ogrzewanych białkach istnieje także możliwośd tworzenia wiązao imidowych,
estrowych i tioestrowych.
45. JAKIE ZMIANY BIAŁEK W ŻYWNOŚCI POWODUJE PROMIENIOWANIE JONIZUJĄCE?
Promieniowanie jonizujące powoduje dekarboksylację lub utlenienie aminokwasów w stopniu
zależnym od ich stężenia w roztworze, od obecności substancji ochronnych i sensybilizatorów
oraz od wielkości pochłoniętej dawki. Rodniki aminokwasowe, powstające w reakcji z
rodnikami
.
OH tworzącymi się w wyniku radiolizy wody, uzyskują stabilizację przez

dysproporcjonację lub dimeryzację. Aminokwasy aromatyczne ulegają hydroksylacji, a
pierścienie aromatyczne rozerwaniu. Szczególnie wrażliwe na promieniowanie są aminokwasy
siarkowe. Grupa tiolowa cysteiny utlenia się w środowisku tlenowym, a w warunkach
beztlenowych powstaje siarkowodór lub wolna siarka.
Metanotiol, produkt radiolizy metioniny, uczestniczy w tworzeniu typowego zapachu
napromieniowanej żywności. Reszty aminokwasów w białkach są odporniejsze na
promieniowanie niż wolne aminokwasy.
46. OPISZ ZASTOSOWANIE HYDROLIZY BIAŁEK W TECHNOLOGII ŻYWNOŚCI
W przemyśle żywnościowym wykorzystuje się enzymatyczną hydrolizę białek, np. przy
wytwarzaniu serów, tradycyjnych przetworów z nasion soi, solonych i marynowanych ryb oraz
izolatów białek kryla. Proteolityczne przemiany uczestniczą też w dojrzewaniu mięsa,
wytwarzaniu piwa i pieczywa oraz w modyfikowaniu funkcjonalnych właściwości izolatów
białkowych. W tych wszystkich przypadkach współdziałają w hydrolizie na ogół liczne,
endogenne proteazy, a stopieo przemiany białek jest niewielki.
W wyniku częściowej proteolizy zmieniają się reologiczne właściwości oraz powstają
charakterystyczne cechy smakowe i zapachowe produktów, zmienia się rozpuszczalnośd
izolatów białkowych oraz ich zdolnośd żelowania, emulgowania i wytwarzania piany, ulegają
inaktywacji składniki niekorzystne biologicznie. Daleko idącą hydrolizę białka, prowadzącą do
utrwalenia krótkich peptydów i aminokwasów stosuje się w celu uzyskania hydrolizatów do
celów żywnościowych, paszowych lub technicznych albo peptydów aktywnych biologicznie.
Hydrolizaty przyprawowe otrzymuje się głównie z kazeiny, albuminy mleka, odtłuszczonych
skwarek, glutenu pszenicy, drożdży lub mąki z nasion roślin strączkowych, zazwyczaj w
roztworze kwasu solnego albo siarkowego w temp. ok. 110
0
C.
Po neutralizacji poddaje się hydrolizaty dalszej obróbce i dojrzewaniu celem otrzymania
przypraw albo wykorzystuje się je do wytwarzania, w reakcjach z sacharydami, preparatów o
charakterystycznym aromacie gotowanego lub pieczonego mięsa.
Enzymatyczna hydroliza nie powoduje rozkładu aminokwasów, a właściwości produktu można
modyfikowad, dobierając do danego surowca odpowiednie enzymy i warunki procesu.
Hydrolizaty wytwarzane „na miarę”, o pożądanym stopniu hydrolizy stosuje się jako
funkcjonalne dodatki o dużej zdolności emulgowania lipidów i tworzenia piany. Produkty o
określonym składzie peptydowym i aminokwasowym wykorzystuje się w diecie osób po
zabiegach operacyjnych przewodu pokarmowego, cierpiących na niedostateczne wydzielanie
enzymów trawiennych, na fenyloketonurię lub alergie pokarmowe. Hydrolizaty z białek mleka,
mięsa, soi, fibrynogenu i kolagenu zawierają biologicznie aktywne peptydy hamujące wzrost
bakterii i wirusów, zmniejszające ciśnienie krwi, działające przeciwzakrzepowo, neuroaktywne
lub regulujące metabolizm.
47. CO TO SĄ PLASTEINY, JAK SIĘ JE OTRZYMUJE I DO CZEGO SIĘ JE WYKORZYSTUJE?
Katalizę enzymatyczną wykorzystuje się do przekształcania białek w reakcji polegającej na
amidowym wiązaniu pożądanych reszt aminokwasów do peptydów hydrolizatu białkowego.
Inkubując hydrolizat białka o dużym stężeniu 30-50%, wzbogacony w estry etylowe tych
aminokwasów z odpowiednią endopeptydazą w stosunku ok. 1:50 przy pH 4-7 i temp. ok.
37
0
C, otrzymuje się po kilku dobach produkty zwane plasteinami, o masie cząsteczkowej 2-3
kDa, wzbogacone o pożądane aminokwasy. Za pomocą reakcji plasteinowej można z białka o
składzie ubogim w reszty aminokwasów niezbędnych wytworzyd produkt o większej wartości
biologicznej. Można również dla chorych na fenyloketonurię otrzymad plasteiny wolne od

reszt fenyloalaniny. Szybkośd wbudowywania aminokwasów w łaocuch peptydowy wzrasta z
hydrofobowością reszty. Można zatem dzięki reakcji plasteinowej zmniejszyd gorzkośd
hydrolizatu przez „ukrycie” hydrofobowych aminokwasów w długich peptydach plastein.
48. OMÓW WPŁYW FOSFORANÓW NA WŁAŚCIWOŚCI FUNKCJONALNE BIAŁEK
Fosforany w różnych warunkach środowiska mogą reagowad z białkami albo wpływad na
właściwości białek przez oddziaływania na inne składniki żywności. Dzięki temu mają one duże
zastosowanie jako dodatki do żywności. Zależnie od składu i warunków środowiska mogą one
służyd jako dodatki buforujące, podwyższające lub obniżające pH, wiążące jony metali,
zwiększające zdolnośd białek do emulgowania lipidów, stabilizujące białka w roztworach lub
koagulujące je, a także zwiększające wodochłonnośd produktów mięsnych, drobiowych i
rybnych. Do spełniania różnych funkcji w żywności dobiera się fosforany zależnie od ich
kwasowości w roztworze wodnym i możliwości tworzenia wiązao wodorowych z cząsteczkami
wody.
Fosforany, a zwłaszcza wydajne polifosforany, wiążą się elektrostatycznie z dodatnio
naładowanymi resztami aminokwasów. Wskutek tego ich wolne grupy hydroksylowe, dzięki
wiązaniom wodorowym z cząsteczkami wody, tworzą warstwę hydratacyjną zapobiegającą
koagulacji białek. Wykorzystuje się to do ekstrahowania białek z różnych surowców i do
modyfikowania funkcjonalnych właściwości izolatów i preparatów białkowych.
Dzięki sieciowaniu polifosforanami można przyspieszyd koagulację białek przy przetwarzaniu
serwatki. Kompleksowanie polifosforanami w kwaśnym środowisku może byd
skuteczniejszym sposobem strącania białek z odcieków w przemyśle żywnościowym niż
działanie mieszaniną wodorotlenków wapnia i sodu lub pochodnymi kwasu lignosulfonowego.
Fosforany wykorzystuje się również w celu ulepszania lub zmiany sensorycznych właściwości
wielu produktów żywnościowych. Tworząc wiązania jonowe z resztami aminokwasów,
zwiększają one zdolnośd białek do emulgowania lipidów, , a wiążąc się z wapniem zmieniają
strukturę miceli kazeinowych i ich zdolnośd uczestniczenia w oddziaływaniach hydrofobowych
z lipidami. Dlatego fosforany zapobiegają rozwarstwianiu się fazy tłuszczowej i wodnej w
zagęszczonym mleku, stabilizują strukturę topionych serów, ulepszają teksturę i zwiększają
soczystośd przetworów mięsnych.
Zwiększenie wodochłonności, polepszenie tekstury i zmniejszenie wycieku w przetworach
mięsnych przez dodatek polifosforanów wynika ze zwiększenia rozpuszczalności i zdolności
emulgowania białek mięsa przez podwyższenie pH, związania w kompleksy jonów wapnia i
magnezu oraz zniszczenia wiązao sieciujących między miozyną i aktyną. W przetwórstwie
mięsnym mieszaniny fosforanów stosuje się w solach i solankach peklujących celem
przyspieszenia peklowania oraz związania kawałków mięsa w wyrobach.
Fosforany spełniają szczególnie istotną rolę w dietetycznych przetworach ubogich w sól.
49. WOLNE AMINOKWASY I PEPTYDY – WYSTĘPOWANIE W ŻYWNOŚCI
Wolne aminokwasy w różnych surowcach i produktach żywnościowych są produktami
metabolizmu komórek, powstają wskutek procesów mikrobiologicznych oraz ulegają
przemianom w wyniku stosowanych w przemyśle procesów technologicznych i w czasie
obróbki kulinarnej. Ich ogólna zawartośd i skład w niektórych przypadkach są
charakterystyczne dla danego rodzaju surowca.
W mięsie zwierząt rzeźnych i ryb zawartośd wolnych aminokwasów zmienia się bardzo istotnie
wskutek dojrzewania po uboju lub śnięciu. W mięsie dorsza wynosi ona ok. 2,5% w stosunku
do aminokwasów zawartych w białkach.
W bulwach ziemniaka azot niebiałkowy stanowi ok. 50% ogólnej ilości azotu, a na azot
wolnych aminokwasów przypada 28-48% N x 6,25, zależnie od odmiany i warunków uprawy.
Ok. 50% ogólnej ilości wolnych aminokwasów w bulwach to kwas asparaginowy, kwas
glutaminowy i walina. Utlenianie wolnej tyrozyny pod wpływem oksydazy polifenolowej
powoduje ciemne przebarwienia bulw ziemniaków.
Miód w 100 g suchej substancji zawiera ok. 0,12 g wolnych aminokwasów, w tym 50-85%
stanowi prolina. W zielonej herbacie najwyższej jakości jest ok. 4,8%, a w czarnej ok. 1,6%
wolnych aminokwasów w suchej substancji. W wielu artykułach roślinnych i zwierzęcych
występują trimetylowe pochodne aminokwasów znane pod wspólną nazwa betain.
50. GLUTAMINIAN SODU – BUDOWA, WŁAŚCIWOŚCI I ZASTOSOWANIE W TECHNOLOGII
ŻYWNOŚCI
(2S)-2-amino-5-hydroksy-5-oxo-pentanonian sodu NaC
5
NO
4
H
8
Od początku ubiegłego stulecia jest znane korzystne, sensoryczne działanie glutaminianu sodu,
występującego w stanie wolnym w wielu artykułach żywnościowych. Należy on do potencjatorów
smakowych – już w minimalnym stężeniu wzmaga smakowitośd różnych produktów, a sam
wywołuje wrażenie smakowe określane jako umami. Kwas glutaminowy został wyizolowany w
1866 r. z glutenu pszennego przez Ritthausena. Glutaminian sodu obecnie wytwarza się
metodami biotechnologicznymi w ilości kilkuset tysięcy ton rocznie, przy użyciu Brevibacterium.
Wolny glutaminian sodu występuje w artykułach żywnościowych na ogół w ilościach mniejszych
niż 0.1 g/100g, natomiast szczególnie dużo glutaminianu zawierają sery i pomidory, odpowiednio
ok. 0,6 i 0.25 g/100 g. Celem wzbogacenia smaku dodaje się do potraw glutaminan sodu zwykle
w ilościach 0,2-0,8 g/100 g.
Jest szeroko stosowany jako dodatek do żywności, jest np. składnikiem zup instant, sosów,
przypraw, konserw rybnych, jako wzmacniacz smaku i zapachu.
51. PRZEMIANY AMINOKWASÓW I PEPTYDÓW W TRAKCIE PRZECHOWYWANIA I
PRZETWARZANIA ŻYWNOŚCI
Pod wpływem endogennych enzymów, obróbki w procesach technologicznych oraz
działalności mikroflory zachodzą w żywności pożądane i niekorzystne przemiany wolnych
aminokwasów i peptydów. Znane są reakcje syntezy i deaminacji aminokwasów, eliminacji
amoniaku, jak np. przy udziale wykrytej w mięśniach ryb makrelowatych amoniakoliazy,
uwalniania amoniaku z amidów i deaminacji przez odwodornienie lub przy udziale tlenu.
Wskutek bakteryjnego rozkładu tryptofanu w nieświeżych produktach rybnych nagromadzają
się indol i skatol o odrażającej woni kału i powstaje m.in. kwas indolilopirogronowy, kwas
indolilooctowy i amoniak. Lotne związki o nieprzyjemnej woni tworzą się również wskutek
mikrobiologicznego rozkładu aminokwasów siarkowych. Duży wpływ na sensoryczne i

zdrowotne cechy żywności wywierają endogenne i bakteryjne dekarboksylazy aminokwasów.
Są one przyczyną m.in. bombaży przeterminowanych marynat rybnych w hermetycznie
zamkniętych opakowaniach.
52. AMINY – WYSTĘPOWANIE W ŻYWNOŚCI
Wolne aminy są naturalnymi, powszechnie występującymi składnikami żywności. Znaczne
ilości amin znajdują się m.in. w produktach piekarskich, piwie, winach, herbacie, czekoladzie,
kawie, owocach, warzywach, przetworach zbożowych, serach i przetworach mlecznych, w
mięsie, rybach i grzybach.
Prekursorami wolnych amin w żywności mogą byd aminokwasy, TMAO i fosfolipidy. Z
aminokwasów aminy mogą powstawad wskutek dekarboksylacji pod wpływem endogennych
enzymów, ale głównie przy udziale dekarboksylaz drobnoustrojów, a także w wyniku rozkładu
cieplnego i w złożonych przemianach nieenzymatycznego brązowienia. Aminy powstają w
żywności także jako produkty aminacji i transaminacji aldehydów.
Aminy mają bardzo istotny wpływ na jakośd żywności. Aminy lotne uczestniczą w tworzeniu
typowego, dojrzałego lub nieświeżego zapachu wielu artykułów żywnościowych. Zawartośd
niektórych lotnych związków azotowych wykorzystuje się jako wskaźnik świeżości, zwłaszcza
ryb i produktów rybnych.
53. CO TO SĄ N-NITROZOAMINY, W JAKI SPOSÓB POWSTAJĄ I JAKIE MAJĄ WŁAŚCIWOŚCI?
W latach 60-tych ubiegłego wieku zauważono, że w żywności mogą powstawad w reakcji amin
z azotanami (III) toksyczne związki N-nitrozowe. Wśród zbadanych ok. 300 takich związków ok.
90% przejawia działanie rakotwórcze w stosunku do różnych zwierząt doświadczalnych.
Rakotwórcza aktywnośd N-nitrozoamin (NNA), zalezy od ich budowy. Najsilniejsze działanie
rakotwórcze wywiera N-nitrozodimetyloamina (NDMA) – ok. 10 μg tej substancji w 1 kg paszy
wywołuje nowotwór u myszy, podczas gdy N-nitrozopirolidyna ma 100-krotnie mniejszą
aktywnośd.
Nitrozowaniu w reakcji z azotanami (III) ulegają drugorzędowe i trzeciorzędowe aminy oraz
amidy.
Wydajnośd nitrozowania amin trzeciorzędowych jest ok. 10-krotnie mniejsza niż amin
drugorzędowych. Udział w reakcji biorą tylko wolne, nieproponowane aminy. W zakresie pH
5-9 szybkośd nitrozowania dimetyloaminy zwiększa się 10-krotnie przy obniżeniu pH o 1
jednostkę. Szybkośd nitrozowania jest największa przy pH 2-4, gdyż ze wzrostem pH
wprawdzie zwiększa się udział niezdysocjowanych amin, lecz jednocześnie maleje stężenie
czynników nitrozujących. W kwaśnym środowisku powstaje bowiem z azotanu (III) słaby,
nietrwały HNO
2
, a z niego bezwodnik N
2
O
3
. Czynnikami nitrozującymi są bezwodnik N
2
O
3
,
bezwodnik protonowany i kation nitrozoniowy oraz halogenki nitrozylu O=N-X i tiocyjanian
nitrozoniowy O=N-NCS.
Wpływ temperatury ogrzewania żywności na zawartośd nitrozo amin jest złożony. W wielu
produktach warunkiem nitrozowania jest obróbka cieplna – w surowym bekonie są jedynie
ślady, natomiast w smażonym bekonie znacznie większe ilości NNA. Ilośd powstających NNA
zwiększa się z czasem i temperaturą smażenia. Prawdopodobnie powstający w peklowanym
mięsie N
2
O
3
tworzy addukty z nienasyconymi grupami lipidów. Te rozkładają się w
podwyższonej temperaturze wydzielając tlenki azotu, które nitrozują obecne w środowisku
wolne aminy. W wielu artykułach żywnościowych NNA tworzą się nawet w warunkach
zamrażalniczych. W temperaturze stosowanej przy gotowaniu i pieczeniu niektóre NNA
ulatniają się lub rozkładają.

Liczne NNA wystepują również w środowiskach glebowych ekosystemów polowych i
trawiastych oraz w wodach powierzchniowych. W ich powstawaniu uczestniczą bakterie
obecne w glebie. Tworzeniu się NNA sprzyja intensywne nawożenie azotowe związkami
mineralnymi. Konsekwencją nagromadzania się tych związków w glebie może byd ich
obecnośd w paszach i w mięsie zwierząt rzeźnych. Pastwiskowe żywienie zwierząt zwiększa
zawartośd NNA w mięsie.
54. OPISZ W JAKI SPOSÓB POWSTAJĄ I JAKIE MAJĄ WŁAŚCIWOŚCI HETEROCYKLICZNE AMINY
AROMATYCZNE
Heterocykliczne aminy aromatyczne (HAA) powstają w ogrzewanych, głównie pieczonych i
smażonych produktach żywnościowych bogatych w białka. dotychczas zidentyfikowano ok. 20
takich amin. Związki te mają działanie mutagenne i rakotwórcze. Są to produkty pirolizy
aminokwasów i białek oraz pochodne wytwarzane w reakcji Maillarda z kreatyniny,
aminokwasów i sacharydów.
Ilośd HAA powstających w żywności zależy od obecności prekursorów, aktywatorów i
inhibitorów oraz temperatury, czasu reakcji, aktywności wody i odczynu środowiska.
Największy wpływ na wydajnośd tworzenia się tych związków w czasie przemysłowej lub
kulinarnej obróbki cieplnej ma temperatura. W ogrzewanych układach modelowych i w
pieczonym mięsie, w temp. 150-230
0
C, zawartośd HAA zwiększa się ze wzrostem temperatury,
a w danej temperaturze stabilizuje się po upływie kilku minut. Dłuższe ogrzewanie prowadzi
do ubytków niektórych związków. Tak wysoka temperatura występuje przy pieczeniu tylko w
zewnętrznych warstwach żywności. Dlatego na ogół zawartośd HAA jest największa w
spieczonej, zewnętrznej warstwie produktu. Niektóre HAA wykryto jednakże również w
produktach żywnościowych ogrzewanych w temp. niższej niż 100
0
C, np. w mięsie ryb
wędzonych na gorąco.
W obecności kwasów tłuszczowych w środowisku reakcji zwiększa się wydajnośd tworzenia się
niektórych HAA. Niektóre przyprawy, m.in. czosnek, rozmaryn i tymianek, a także solanka
peklująca zawierająca azotan (III), zmniejszają wydajnośd tworzenia niektórych HAA o ok. 50%.
Inhibujący efekt wywierają również przeciwutleniacze zawarte w niektórych tłuszczach do
smażenia.
Zawartośd HAA w różnych, pieczonych produktach mięsnych, drobiowych i rybnych jest od
poniżej jednego do kilku μg/100 g.
55. JAKIE MUTAGENY MOGĄ POWSTAWAD W ŻYWNOŚCI PODDANEJ OBRÓBCE TERMICZNEJ?
Mutagenami nazywamy czynniki i substancje, które mogą powodowad zmiany w zapisie
genetycznym. W żywności poddanej obróbce termicznej mogą powstawad:
Wielopierścieniowe węglowodory aromatyczne (WWA), zawierające układ
skondensowanych pierścieni aromatycznych; tworzą się w wyniku niepełnego spalania materii
organicznej. Silnie rakotwórczym przedstawicielem tej grupy jest najlepiej zbadany
kancerogen benzo[a]piren. W produktach żywnościowych WWA powstają przede wszystkim
podczas smażenia oraz pieczenia, zwłaszcza nad otwartym ogniem, np. na ruszcie. Tworzą się
one nie tyle wskutek samego ogrzewania, co pirolizy tłuszczu. W wędzonym mięsie i
wędzonych rybach źródłem WWA jest dym użyty do wędzenia.
Heterocykliczne aminy aromatyczne (HAA) powstają w wyniku obróbki cieplnej wielu
rodzajów żywności, przede wszystkim o dużej zawartości białka. Związki te są niezwykle

silnymi mutagenami. Badania na zwierzętach wykazały, iż są one także rakotwórcze, chod nie
należą do silnych kancerogenów.
Temperatura obróbki cieplnej ma zasadniczy wpływ na rodzaj HAA powstających w
przetwarzanej żywności. Produkty pirolizy aminokwasów i białek powstają w temp. > 300
0
C.
dlatego też powstają one przede wszystkim na powierzchni mięsa i ryb pieczonych nad
otwartym ogniem. W niższych temperaturach (150-200
0
C) powstają pochodne chinoliny,
chinoksaliny i pirydyny w wyniku reakcji kreatyniny, aminokwasów i cukrów obecnych w
mięsie. Związki te należą do najbardziej mutagennych. Ponieważ temperatury potrzebne do
ich utworzenia leżą w zakresie temperatur, które mogą działad na powierzchnię produktów
ogrzewanych i smażonych, są obecne w smażonym i pieczonym mięsie, smażonych rybach, a
także wywarach mięsnych
W konserwach powstawanie mutagenów wiąże się również z obróbką cieplną, chod
stosowane przy produkcji konserw temperatury są stosunkowo niskie, 110-120
0
C. mutageny
powstające w tych procesach dotychczas nie zostały jeszcze scharakteryzowane chemicznie.
W przypadku innych wysokobiałkowych produktów spożywczych, takich jak mleko, sery, jaja
oraz warzywa strączkowe, obecnośd mutagenów stwierdza się wyłącznie po obróbce cieplnej
prowadzącej do zmiany koloru wskutek np. przypalenia.
56. CO TO SĄ MIKOTOKSYNY?
Mikotoksyny są silnie toksycznymi związkami wytwarzanymi przez pleśnie, przede wszystkim
Aspergillus, Penicillium i Fusarium. Stanowią one najgroźniejsze zanieczyszczenie pojawiające
się przede wszystkim w czasie przechowywania licznych produktów żywnościowych, wśród
których najczęściej wymienia się kukurydzę i orzechy ziemne. Szczególnie dużo mikotoksyn
powstaje w klimacie podzwrotnikowym i zwrotnikowym, przy nieodpowiednich metodach
zbioru i przechowywania plonów.
Spośród wielu klas związków należących do grupy mikotoksyn tylko dla trzech udowodniono,
że mają właściwości rakotwórcze. Są to alfatoksyny i sterigomatocystyna indukujące
nowotwory wątroby oraz ochratoksyna A wywołująca nowotwory nerek u zwierząt
doświadczalnych. Najbardziej rakotwórcza jest alfatoksyna B
1
, która jako jedyny związek
została uznana za kancerogen (hepatokancerogen) u ludzi.
57. WYMIEO ZNANE CI MUTAGENY WYSTĘPUJĄCE W UŻYWKACH
Kawa palona, rozpuszczalna oraz bezkofeinowa wykazują silne działanie mutagenne. Oprócz
mutagenów naturalnych, np. kwasu chlorogenowego , zawierają powstający wskutek pirolizy
metyloglioksal oraz mniej aktywne glioksal i biacetyl .
Substancje te są obecne także w herbacie, sosie sojowym oraz napojach alkoholowych typu
whisky i brandy. Ponadto w wyniku metabolizmu alkoholu etylowego powstaje mutagenny
aldehyd octowy oraz inne szkodliwe aldehydy, natomiast kawa i herbata zawierają kofeinę,
która jest inhibitorem syntezy naprawczej DNA i może się przyczyniad do zwiększenia ryzyka
chorób nowotworowych (co zresztą okazało się nieprawdą).
58. OPISZ CZYNNIKI O DZIAŁANIU PRZECIWRAKOTWÓRCZYM WYSTĘPUJĄCE W ŻYWNOŚCI
POCHODZENIA ROŚLINNEGO
Związki przeciwrakotwórcze, zarówno pochodzenia naturalnego, jak i przemysłowego, mają
zdolnośd zapobiegania powstawaniu lub rozrostowi nowotworów. Dzieli się je na trzy grupy:
czynniki blokujące działające na najwcześniejszych etapach kancerogenezy – chronią
one komórkę przed substancjami, które mogłyby zainicjowad w niej zmiany

prowadzące do zezłośliwienia. Istnieją trzy podstawowe mechanizmy ich działania.
Niektóre z nich uniemożliwiają aktywację związków rakotwórczych lub
promotorowych. Przykładem może byd wit. C, która hamuje tworzenie się
rakotwórczych nitrozo amin z amin i azotanów(III) obecnych w żywności. Drugi
mechanizm polega na obniżeniu aktywności enzymów odpowiedzialnych za aktywację
kancerogenów (enzymy fazy I) oraz indukcję enzymów zaangażowanych w
detoksykację (enzymy fazy II). Tego typu aktywnością charakteryzują się m.in.
ditiolotiony oraz izotiocyjaniany obecne w warzywach krzyżowych, a także katechiny
będące składnikami herbaty. Trzeci mechanizm działania czynników blokujących
polega na wychwytywaniu rakotwórczych metabolitów. W tym przypadku największą
efektywnośd mają związki zawierające siarkę, w szczególności glutation, obecne
przede wszystkim w warzywach z rodziny Allium (czosnek, cebula).
czynniki supresorowe odgrywające rolę na etapie promocji i niekontrolowanego
wzrostu zainicjowanych komórek - wpływają na proces przemiany komórki
przedrakowej w komórkę w pełni zezłośliwioną. Wiele nieodżywczych składników
żywności pochodzenia roślinnego wykazuje właściwości spowalniające lub hamujące
rozrost nowotworowy. Mechanizmy działania ochronnego obejmują stymulację
różnicowania komórek (retinol), hamowanie aktywacji onkogenów (izotiocyjaniany w
brokułach), selektywną inhibicję proliferacji komórek nowotworowych oraz
hamowanie procesu tworzenia naczyo krwionośnych guza niezbędnych do jego
wzrostu (genistein obecny w nasionach soi).
czynniki „uodparniające”, które czynią komórkę mniej podatną na transformację
nowotworową – są jeszcze słabo poznane. Proponowane mechanizmy to stymulacja
dojrzewania tkanek. Uważa się, że taki mechanizm przyczynia się do ograniczenia
wzrostu raka piersi na skutek działania izoflawonów obecnych w nasionach soi oraz
hamowanie podziałów komórkowych w tkance docelowej. Do tej grupy czynników
przeciwrakotwórczych (oprócz wit. D, wapnia i fosforu) można zaliczyd pewne
składniki czosnku wykazujące działanie bakteriobójcze w stosunku do Helicobacter
pylori. Bakterie te wywołują chorobę wrzodową u ludzi co, jak wykazano, przyczynia
się do rozwoju raka żołądka. Czosnek hamując ich wzrost, przeciwdziała
uszkodzeniom nabłonka i zwiększa jego odpornośd na szkodliwe działanie substancji
rakotwórczych.
59. OMÓW BUDOWĘ ORAZ PODSTAWOWE WŁAŚCIWOŚCI FIZYKOCHEMICZNE WODY
Cząsteczka wody H
2
O, składa się z jednego atomu tlenu i dwóch atomów wodoru połączonych
polarnym wiązaniem kowalencyjnym. Właściwości fizyczne i chemiczne wody są bardzo
odmienne od związków o podobnej, prostej strukturze, jak np. HF lub H
2
S. Aby zrozumied
przyczyny charakterystycznych właściwości wody, trzeba szczegółowo zapoznad się ze
strukturą jej cząsteczki. Jej budowę najlepiej oddaje nieregularny czworościan z atomem tlenu
w środku. Dwa wiązania atomu tlenu z atomami wodoru są skierowane w stronę dwóch
naroży czworościanu, a niepodzielne pary elektronów na hybrydyzowanych orbitalach sp
3
w
stronę dwóch pozostałych.
Cząsteczka wody jest elektrycznie obojętna (ma taką samą liczbę elektronów i protonów) i
polarna, ponieważ podział elektronów tworzących wiązanie kowalencyjne, między atomem O
i H, jest asymetryczny. Jądro atomu tlenu przyciąga elektrony silniej niż jądro atomu wodoru.
Nadmierne zagęszczenie elektronów na atomie tlenu wytwarza słabo ujemny obszar w

obrębie dwóch kątów czworościanu. Zatem w cząsteczce wody powstają dwa dipole
elektryczne wzdłuż każdego z wiązao H-O. atom tlenu uzyskuje częściowy ładunek ujemny, a
każdy z atomów wodoru częściowy ładunek dodatni. Dzięki temu cząsteczki wody mogą
oddziaływad między sobą. W wyniku przyciągania elektrostatycznego między atomem tlenu
jednej cząsteczki i atomem wodoru drugiej powstaje wiązanie wodorowe. Tetraedryczne
ukierunkowanie orbitali wokół atomu tlenu umożliwia każdej cząsteczce wody utworzenie
wiązao wodorowych z czterema sąsiednimi cząsteczkami.
W porównaniu z innymi cieczami następujące parametry wody wyróżniają się dużymi
wartościami:
Temperatura topnienia – 0
0
C,
Temperatura wrzenia - 100
0
C,
Ciepło właściwe – 4kJ/mol
Ciepło parowania – 2260 kJ/mol
Ciepło topnienia – 333 kJ/mol
Stała dielektryczna – 80.
Wymienione właściwości wynikają ze spójności ciekłej wody, zwanej kohezją, spowodowanej
silnym oddziaływaniem między cząsteczkami H
2
O przez sied wiązao wodorowych. Cząsteczki
wody przylegają również do powierzchni tych substancji, na których występują grupy polarne
lub zjonizowane. Działające w tym przypadku siły adhezji tłumaczą zdolności zwilżające wody.
Siły adhezji i kohezji wpływają na zjawiska kapilarne, polegające na podnoszeniu się wody w
rurkach o małej średnicy. Zjawisko to ma ogromne znaczenie w biologii, jest bowiem
wykorzystywane przez rośliny jako sposób transportu substancji odżywczych od korzeni do
liści w procesie transpiracji.
Istnienie międzycząsteczkowych wiązao wodorowych jest przyczyną dużego ciepła właściwego
wody, co oznacza, że do podniesienia temperatury nawet o jeden stopieo Celsjusza potrzebna
jest znaczna ilośd energii.
Ciepło parowania jest bezpośrednią miarą ilości energii potrzebnej do pokonania w cieczy sił
przyciągania między sąsiednimi cząsteczkami, tak aby pojedyncze cząsteczki mogły się uwolnid
i przejśd w stan gazowy. Nawet w temp. 100
0
C woda ma znaczną liczbę wiązao wodorowych,
dlatego ciepło parowania wody jest znacznie większe niż innych znanych cieczy.
Zagęszczenie cząsteczek wody na granicy faz z powietrzem na skutek ich silniejszego
oddziaływania między sobą niż z cząsteczkami powietrza wyjaśnia wysokie napięcie
powierzchniowe wody.
Inną cechą charakterystyczną ciekłej wody jest jej mała lepkośd. Decyduje o tym labilnośd
wiązao wodorowych.
Rozpatrując właściwości wody należy uwzględnid jej skłonnośd do nieznacznej jonizacji. Woda
może działad jako bardzo słaby kwas, uwalniając proton i tworząc jon hydroksylowy, lub jako
bardzo słaba zasada, akceptując proton i tworząc jon oksoniowy. Stopieo jonizacji wody w
stanie równowagi jest mały i w temp. 25
0
C tylko 1 na 10
7
cząsteczek jest zjonizowana.
60. OPISZ WŁAŚCIWOŚCI WODY JAKO ROZPUSZCZALNIKA
O właściwościach wody jako rozpuszczalnika decyduje polarna natura jej cząsteczek oraz
zdolnośd do tworzenia wiązao wodorowych. Woda jest dobrym rozpuszczalnikiem dla
związków polarnych i zjonizowanych, a złym dla węglowodorów. Związki, które chętnie
rozpuszczają się w wodzie, noszą nazwę hydrofilowych, a te, które środowiska wodnego nie
lubią – hydrofobowych.

Dodanie jakiejkolwiek substancji do wody powoduje zmianę właściwości tej substancji, jak
również właściwości środowiska wodnego. Substancja rozpuszczona wymusza
uporządkowanie cząsteczek wody, dlatego struktura wody w bezpośrednim sąsiedztwie
cząsteczek substancji rozpuszczonej jest mniej labilna niż struktura pozostałej wody. Na
skutek tego właściwości roztworów są inne niż właściwości czystej wody i zależą od natury
substancji rozpuszczonej oraz ich stężenia..
Oddziaływanie cząsteczek wody z różnymi substancjami jest znane jako hydratacja. Rozmiary
i trwałośd powłok hydratacyjnych zależą od struktury substancji rozpuszczonej, pH i
temperatury, a także od obecności innych związków w środowisku.
Większośd związków ulegających dysocjacji łatwo rozpuszcza się w wodzie, gdyż procesowi
temu towarzyszy uwalnianie dużej ilości energii.
Woda jest dobrym rozpuszczalnikiem dla większości cząsteczek wchodzących w skład
organizmów żywych, gdyż zazwyczaj są one polarne lub mają zjonizowane grupy funkcyjne.
Cząsteczki amfipatyczne są to takie cząsteczki, które mają grupy polarne lub zjonizowane i
jednocześnie fragmenty hydrofobowe. W wodzie ulegają dyspersji, jeśli siły przyciągania
między ich grupami polarnymi i cząsteczkami wody przeważają nad oddziaływaniami
hydrofobowymi ich fragmentów apolarnych. Wiele związków biologicznie czynnych ma
właściwości amfipatyczne: fosfolipidy, sterole, aminokwasy, niektóre witaminy i barwniki.
Gdy związek amfipatyczny znajdzie się w kontakcie z wodą, jego dwa fragmenty wykazują
tendencje przeciwstawne: fragment hydrofilowy lub grupa zjonizowana chętnie oddziałuje z
wodą i stara się w niej rozpuścid, a jednocześnie fragment hydrofobowy stara się od niej
uciec. Powoduje to agregację cząsteczek w taki sposób, aby powierzchnia hydrofobowa
eksponowana do środowiska wodnego była jak najmniejsza.
Wiele związków amfipatycznych w środowisku wodnym tworzy trwałe struktury składające
się z tysięcy cząsteczek. Przykładem są micele oraz wszelkiego rodzaju sztuczne błony
lipidowe.
61. PODAJ DEFINICJĘ WODY ZWIĄZANEJ W ŻYWNOŚCI
Istnieje wiele różnych definicji wody związanej. Jedne biorą pod uwagę temperaturę
zamarzania, inne dostępnośd wody jako rozpuszczalnika. Fennema przedstawił najwłaściwszą
definicję wody związanej: woda związana to ta, która jest zlokalizowana w bezpośrednim
sąsiedztwie substancji rozpuszczonych lub zawieszonych, ma zmniejszoną aktywnośd,
odmienne właściwości od pozostałej masy wody zawartej w danym materiale i nie zamarza
do temp. -40
0
C.
62. JAKIE ZNASZ RODZAJE WODY W ŻYWNOŚCI?
Najczęściej wyróżnia się następujące rodzaje wody w żywności:
woda strukturalna (krystaliczna, związana chemicznie) (<0,03%) – jest integralną
częścią składników niewodnych, ulokowana w wolnych przestrzeniach
makrocząsteczek lub związana w postaci wodzianów,
woda związana w postaci monowarstwy (0,1 – 0,9%) – silnie oddziałuje z grupami
polarnymi i zjonizowanymi składników niewodnych,
woda związana w dalszych kilku warstwach (1 – 5%) – o strukturze uporządkowanej
wokół hydrofilowych grup składników niewodnych, stabilizowana wiązaniami
wodorowymi, utworzonymi między tymi grupami a wodą oraz między cząsteczkami
samej wody,

woda nie związana (wolna) (5 - 96%) – o właściwościach zbliżonych do właściwości
wody w rozcieoczonych roztworach soli, powiązana siecią wzajemnych wiązao
wodorowych, ruchliwa,
woda uwięziona (5 – 96%) – o właściwościach wody wolnej, ale uwięziona w
niewypełnionych przestrzeniach składników strukturalnych lub w żelach, przez co jej
przepływ jest utrudniony.
63. WYJAŚNIJ POJĘCIE AKTYWNOŚD WODY
Woda zawarta w materiale biologicznym jest roztworem wielu związków chemicznych.
Prężnośd pary roztworów jest mniejsza niż prężnośd pary czystego rozpuszczalnika. Stopieo
zmniejszenia prężności pary wyraża prawo Raoulta
p/p
o
= n
2
/n
1
+ n
2
= a
w
gdzie p i p
o
to odpowiednio prężnośd par roztworu i czystego rozpuszczalnika w danej
temperaturze, n
1
i n
2
– stężenie molowe substancji rozpuszczonej i rozpuszczalnika, a
w
–
aktywnośd wody.
Aktywnośd wody może przyjmowad wartości od 1 dla czystej wody do ok. 0 dla układów o
niewielkiej zawartości wody.
Aktywnośd wody jest miarą zawartości wody wolnej w danym materiale, umożliwia więc
określenie intensywności z jaką woda asocjuje z różnymi niewodnymi składnikami.
Aktywnośd wody lub inaczej względną prężnośd pary oznacza się przez umieszczenie małej
próbki substancji badanej w zamkniętej komorze na czas wystarczający do osiągnięcia
równowagi, a następnie mierzy się ciśnienie lub wilgotnośd względną w komorze.
64. JAKI MA WPŁYW AKTYWNOŚD WODY NA TRWAŁOŚD ŻYWNOŚCI?
Aktywnośd wody ma wpływ na wiele czynników decydujących o trwałości żywności.
Bezpośredni związek z aktywnością wody ma rozwój mikroflory. Drobnoustroje nie mogą się
rozmnażad, gdy a
w
< 0,6. Zawartośd wody ma wpływ na konformację białek enzymatycznych,
które decydują o aktywności i zdolności katalitycznej enzymów. Brak fazy wodnej
uniemożliwia transport substratów i produktów reakcji.
Woda może sprzyjad procesom utleniania przez zwiększenie ruchliwości i rozpuszczalności
jonów metali uczestniczących w tych reakcjach, jak też przez pęcznienie białek, które
ułatwiają działanie rodników powstających w procesach utleniania lipidów.
Przemiany fizyczne związane z aktywnością wody i mające wpływ na jakośd i trwałośd
produktów żywnościowych to krystalizacja lub rozpuszczanie niektórych składników
żywności. Krystalizacji ulegają przede wszystkim sacharydy. Woda uwalniana w procesie
krystalizacji powoduje jakościowe zmiany produktu, polegające na utracie kruchości
wyrobów piekarniczych, zbrylaniu produktów suszonych oraz przyspieszaniu reakcji
chemicznych.
Zakres zmian fizycznych, biochemicznych, chemicznych i mikrobiologicznych zachodzących w
czasie przechowywania żywności w znacznym stopniu można ograniczyd przez zmniejszenie
zawartości wody lub przeprowadzenie wody wolnej w wodę związaną.
Najczęściej stosowanymi metodami są: suszenie, wędzenie, zamrażanie i dodawanie
substancji zmniejszających a
w
(metody osmoaktywne – częściowe odwodnienie –
zagęszczenie, lub dodanie substancji mających zdolnośd trwałego wiązania wody –
sacharydy, NaCl, białko mleka lub soi, glicerol, sorbitol).

65. WYMIEO PODSTAWOWE WSKAŹNIKI JAKOŚCI WODY PITNEJ
Do wskaźników jakości wody pitnej należą: barwa, mętnośd, chlorki, siarczany, sód, glin,
azotany (V), azotany (III), amoniak, kadm, ołów, występowanie bakterii Escherichia coli, i
Enterobacter.
Nie wiem czy to pytanie tak ma byd opracowane!!!
66. JAK DEFINIUJE SIĘ SUBSTANCJE DODATKOWE W ŻYWNOŚCI, KIEDY MOGĄ BYD STOSOWANE
W ŻYWNOŚCI?
Substancja dodatkowa to substancja, która nie jest zwyczajowo traktowana jako żywnośd, nie
będąca typowym składnikiem żywności, niezależnie od tego czy posiada wartośd odżywczą,
czy nie, której celowe użycie technologiczne w procesie produkcji, przetwarzania,
przygotowywania, pakowania, przewozu i przechowywania żywności spowoduje lub może
spowodowad, że substancja ta stanie się bezpośrednio lub pośrednio składnikiem środka
spożywczego albo półproduktów będących jego komponentami.
Substancje dodatkowe mogą byd stosowane w żywności, jeżeli:
przy dozwolonym poziomie nie stanowią zagrożenia dla zdrowia lub życia,
ich stosowanie jest uzasadnione technologicznie, a cel ich stosowania nie może byd
osiągnięty w inny sposób, praktycznie możliwy z punktu widzenia technologicznego i
ekonomicznego,
ich użycie nie wprowadza konsumenta w błąd.
67. PRZEDSTAW KLASYFIKACJĘ SUBSTANCJI DODATKOWYCH DODAWANYCH DO ŻYWNOŚCI
Substancje dodatkowe można podzielid na:
- substancje słodzące,
- barwniki
- substancje inne: substancje konserwujące, kwasy, przeciwutleniacze, regulatory
kwasowości, stabilizatory, emulgatory, sole emulgujące, zagęstniki, substancje żelujące,
substancje wzmacniające smak i zapach, skrobie modyfikowane, substancje wypełniające,
substancje wiążące, subst. utrzymujące wilgotnośd, subst. spulchniające, subst. glazurujące,
subst. przeciwzbrylające, nośniki, gazy do pakowania, gazy nośne, subst. pianotwórcze, subst.
przeciwpianotwórcze, sekwestranty, , emulgatory, środki do przetwarzania mąki.
68. JAKIE GRUPY ZWIĄZKÓW ZALICZAMY DO DODATKÓW ZWIĘKSZAJĄCYCH TRWAŁOŚD
ŻYWNOŚCI?
Do dodatków zwiększających trwałośd żywności zaliczamy:
konserwanty – inaktywacja enzymów lub drobnoustrojów,
przeciwutleniacze – zapobieganie tworzeniu się nadtlenków,
kwasy – obniżenie pH, hamuje rozwój drobnoustrojów i aktywnośd enzymów,
synergenty – wspomagają i przedłużają działanie przeciwutleniaczy. Polega to na
aktywowaniu funkci przeciwutleniacza i kompleksowaniu śladów metali ciężkich,
które katalizują procesy utlenienia. Synergenty tworzą z metalami trwałe kompleksy,
tzw. chylaty. Do ważniejszych zalicza się: EDTA – wersenian wapniowo-sodowy,
kwasy: cytrynowy, winowy, jabłkowy oraz di fosforany, aminokwasy, peptydy.
69. JAKIE GRUPY ZWIĄZKÓW ZALICZAMY DO DODATKÓW KSZTAŁTUJĄCYCH CECHY
SENSORYCZNE?
Do tej grupy związków należą:

barwniki – do barwienia żywności stosuje się: barwiące części roślin jadalnych,
barwniki organiczne naturalne, barwniki organiczne syntetyczne identyczne z
naturalnymi, barwniki organiczne syntetyczne, barwniki nieorganiczne (pigmenty),
substancje słodzące ,
substancje wzmacniające smak i zapach– przyprawy naturalne, aromaty naturalne,
esencje spożywcze, aromaty identyczne z naturalnymi, aromaty syntetyczne,
substancje wzmacniające smak, ,
70. JAKIE GRUPY ZWIĄZKÓW ZALICZAMY DO DODATKÓW TEKSTUROTWÓRCZYCH?
Zaliczamy tu substancje, które kształtują i utrzymują pożądaną strukturę produktu lub
współdziałają w tworzeniu albo wzmocnieniu naturalnego żelu:
emulgatory,
sole emulgujące,
zagęstniki,
substancje żelujące,
skrobie modyfikowane,
substancje wypełniające,
substancje wiążące,
substancje utrzymujące wilgotnośd,
substancje spulchniające,
substancje przeciwzbrylające.
71. W JAKI SPOSÓB PRZEPROWADZA SIĘ OCENĘ TOKSYKOLOGICZNĄ DODATKÓW DO
ŻYWNOŚCI?
Proste badania toksykologiczne (toksycznośd ostra), są prowadzone na specjalnie do tego celu
selekcjonowanych zwierzętach doświadczalnych, głównie szczurach. W kolejnych etapach
badao eksperyment jest prowadzony porównawczo na kilku gatunkach zwierząt. Już badając
toksycznośd podprzewlekłą w teście 90-dniowym stosuje się szczury i psy. Szczególnie
długotrwałe są badania toksyczności przewlekłej, toksyczności chronicznej i rakotwórczości,
które wymagają prowadzenia eksperymentu ponad 18 miesięcy na myszach, 30 miesięcy na
szczurach i 12(?) miesięcy na psach. Toksycznośd reprodukcyjną bada się na płodach królików
i szczurów oraz prowadząc testy na wielu generacjach szczurów.
Na podstawie tych badao ustala się, czy badana substancja może byd szkodliwa dla zdrowia
oraz określa wartośd ADI dla dodatków, która wyraża ile mg danej substancji, na kg wagi ciała,
dziennie bez szkody dla organizmu może spożyd człowiek przez całe życie. Przy hipotetycznym
ustalaniu wartości ADI jako wartośd wyjściową, przyjmuje się maksymalną dawkę, która nie
spowodowała jakichkolwiek zmian w czasie badao chronicznej toksyczności (szczury). Dzieli się
ją przez 10 jako współczynnik bezpieczeostwa przeniesienia wyników ze zwierzęcia na
człowieka i ponownie przez 10, aby uwzględnid wrażliwośd poszczególnych osobników
(chorzy, kobiety ciężarne, dzieci). Określone w ten sposób wartości ADI mają ogromny
margines bezpieczeostwa i przekraczają wielokrotnie normalne ilości spożywanych dodatków.
Wyszukiwarka
Podobne podstrony:
chemia żywności egzamin 1 moje
CHEMIA-ŻYWNOŚCI-sem.-IV, STUDIA PŁ, TECHNOLOGIA ŻYWNOŚCI I ŻYWIENIA CZŁOWIEKA, ROK II, SEM 4, Chemia
chemia zywnosci egzamin
Chemia żywności egzamin 15
moja sciaga, technologia żywności, chemia żywności, chemia żywności egzamin
Chemia żywności egzamin 15
EGZAMIN CHEMIA ŻYWNOŚCI, chemia żywności
EGZAMIN Z CHEMII ŻYWNOŚCI (wersja C), CHEMIA, Żywności
egzamin - chemia żywności 2002 (A), CHEMIA, Żywności
chemia, ywno ci pytania na egzamin www.przeklej.pl, Chemia Żywności – pytania na egzamin
chemia zywnosci pytanka egzamin
chit egzaminy 2013, TŻ 2, Chemia Żywności - CHiT
więcej podobnych podstron