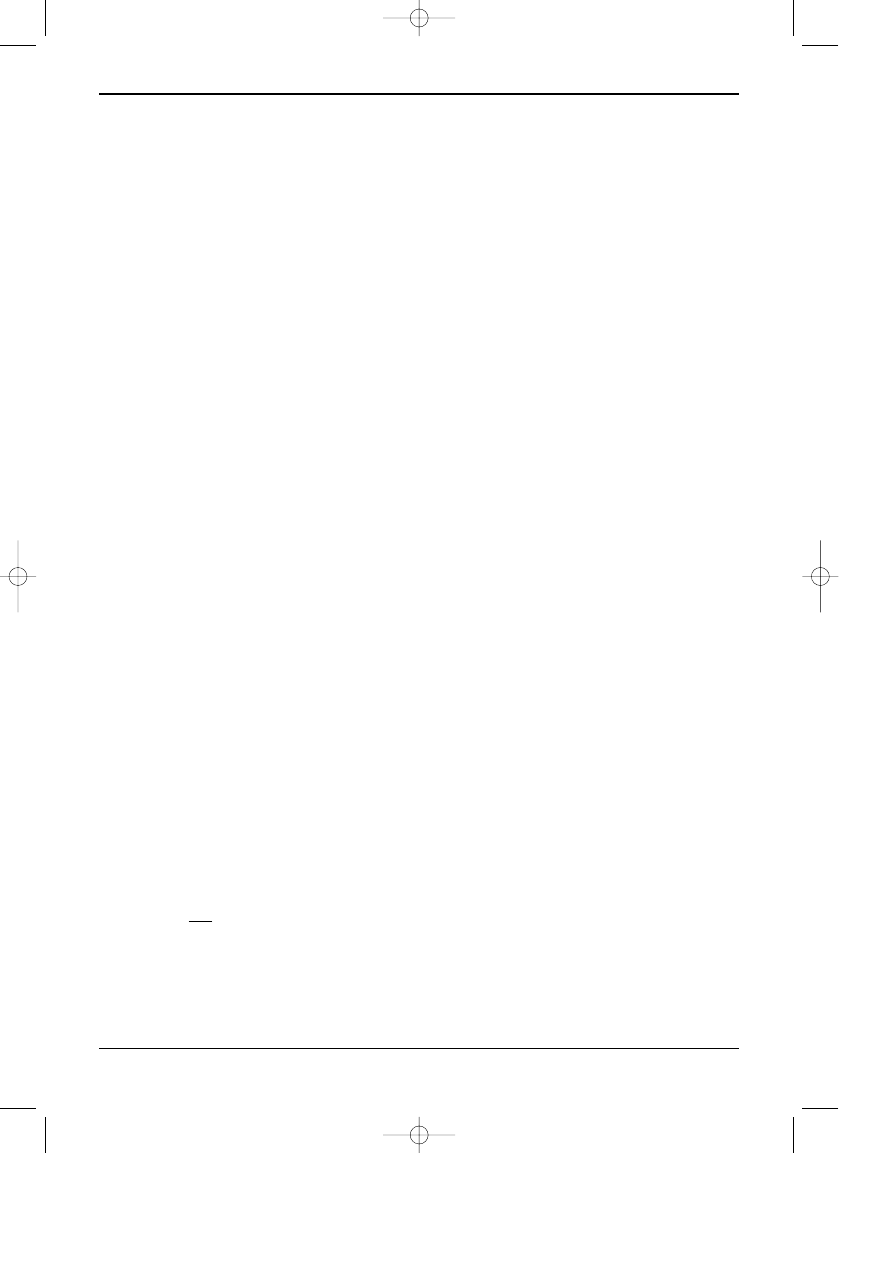
Celem rozdzielania mieszaniny substancji na poszczególne sk³adniki, b¹dŸ oddzielenia
tylko wybranych substancji od innych, jest najczêœciej ich iloœciowe oznaczenie, tj. ustalenie
zwartoœci lub masy analitu w bardzo du¿ej próbce.
Wysokosprawna chromatografia cieczowa (HPLC) jest wykorzystywana do oznaczenia
zawartoœci sk³adników g³ównych, t.j. wystêpuj¹cych w próbce na poziomie stê¿eñ w zakresie od
1 do 100 %, sk³adników poœrednich - od 0.001 do 1% oraz substancji obecnych w analizowanej
próbce poni¿ej 0.001%, tj. sk³adników œladowych.
Oznaczenie iloœciowe w HPLC polega na ustaleniu zale¿noœci pomiêdzy sygna³em detek-
tora, czyli powierzchni¹ pod pikiem lub ewentualnie wysokoœci¹ piku, a stê¿eniem lub mas¹
sk³adnika, którego zawartoœæ zamierzamy oznaczyæ. W praktyce mamy do dyspozycji cztery
metody oznaczania: metodê wzorca zewnêtrznego (metodê krzywej kalibracyjnej), metodê wzor-
ca wewnêtrznego, metodê dodatku wzorca (metodê fortyfikacji) oraz metodê prostej normaliza-
cji lub normalizacji ze wspó³czynnikami korekcyjnymi.
11.1.
METODA WZORCA ZEWNÊTRZNEGO
(METODA KRZYWEJ KALIBRACYJNEJ, EXTERNAL STANDARD)
Istot¹ metody wzorca zewnêtrznego jest wyznaczenie zale¿noœci pomiêdzy powierzchni¹
(wysokoœci¹) piku dla ka¿dej z oznaczanych substancji i ich stê¿eniem lub mas¹.
W tym celu przygotowuje siê roztwory kalibracyjne, tzn. roztwory substancji oznaczanej
albo mieszaniny analizowanych substancji w kilku ró¿nych stê¿eniach (np. 1.0, 2,0, 3.0, 4.0
mg/mL). Ka¿dy z przygotowanych roztworów dozowany jest do kolumny chromatograficznej.
Na podstawie powierzchni lub wysokoœci pików, na uzyskanych chromatogramach wyznacza siê
przebieg krzywej kalibracyjnej b¹dŸ oblicza siê wartoœci wspó³czynników w równaniu kalibra-
cyjnym dla ka¿dej z oznaczanych substancji. Nastêpnie dozowane do kolumny i rozdzielane s¹
próbki, w których oznaczona ma byæ zawartoœæ substancji. Uzyskane powierzchnie pików sub-
stancji oznaczanych w nieznanej próbce umo¿liwi¹ oznaczenie ich zawartoœci, tj. stê¿enia
(masy). W praktyce wykonuje siê to metod¹ graficznej interpolacji na wykresie krzywej kalibra-
cyjnej, w oparciu o równanie krzywej kalibracyjnej lub z wykorzystaniem równania (1).
(1)
gdzie: C
x
- oznaczona zawartoϾ, A
x
- powierzchnia piku, R
f
- wspó³czynnik odpowiedzi, tj. ilo-
raz powierzchni piku i stê¿enia substancji o znanym stê¿eniu w roztworze wzorcowym. Gdy
równanie kalibracyjne wyra¿one jest funkcj¹ typy f(x) = a
1
x, to wspó³czynnik odpowiedzi jest
wspó³czynnikiem kierunkowym a
1
tej prostej, dla których wykonano kalibracjê.
171
11. OZNACZANIE ILOŒCIOWE W HPLC
Rafa³ Kartanowicz
f
x
x
R
A
C
=
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:44 Page 171
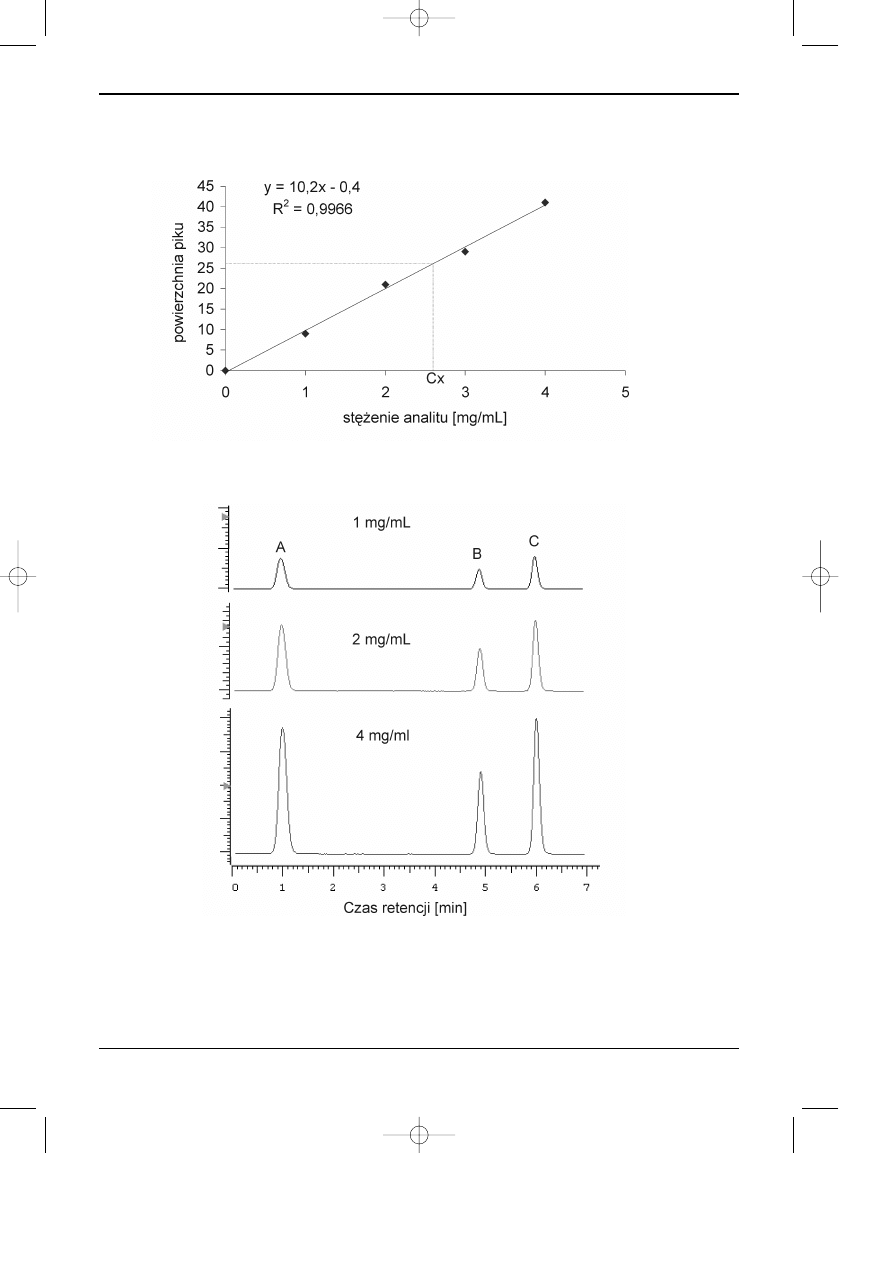
Na rysunku 1 i 2 przedstawiono odpowiednio: typow¹ krzyw¹ kalibracyjn¹ uzyskan¹ metod¹
wzorca zewnêtrznego oraz chromatogramy roztworów wzorcowych.
172
Rys. 11.1. Wykres krzywej kalibracyjne otrzymanej metod¹ wzorca zewnêtrznego.
Rys. 11.2. Chromatogramy roztworów wzorcowych substancji A, B, C o stê¿eniach 1.0, 2.0 i 4.0
mg/mL.
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:44 Page 172
Stosowanie tej metody kalibracji wymaga, aby zarówno roztwory wzorcowe jak i anali-
zowane próbki by³y rozdzielane ("chromatografowane") w tych samych warunkach temperatury,
objêtoœciowego natê¿enie przep³ywu eluentu, typu i wymiarów kolumny chromatograficznej, z
regu³y tej samej kolumny, sk³adu eluentu oraz takiego samego programu elucji. Ponadto trzeba
braæ te¿ pod uwagê nastêpuj¹ce czynniki:
z
Równanie kalibracyjne jest najczêœciej zale¿noœci¹ prostoliniow¹ typu y = a
1
x + a
2
, ale
niekiedy do opisu przebiegu krzywej kalibracyjnej wykorzystuje siê inne funkcje:
gdzie: y - powierzchnia piku(wysokoœæ), x - stê¿enie (masa) substancji,
a
1,2,3,4
- wspó³czynniki.
z
Dozowanie do kolumny chromatograficznej powinno byæ powtarzalne.
z
Oznaczanie zawartoœci substancji w próbkach nieznanych mo¿e byæ wykonywane tylko
w zakresie stê¿eñ, w jakim zosta³a wykona kalibracja, dotyczy to tak¿e innych metod tj.
metody wzorca wewnêtrznego czy fortyfikacji. Ekstrapolacja przebiegu krzywej kali-
bracyjnej poza zakres wykonanej kalibracji, mo¿e byæ Ÿród³em niedok³adnych wyników
oznaczeñ ze wzglêdu na nieliniowy przebieg odpowiedzi detektora.
11.2.
METODA WZORCA WEWNÊTRZNEGO
Jest to metoda polegaj¹ca na dodaniu do próbki znanej iloœci sk³adnika, tzw. wzorca
wewnêtrznego (IST), który jest jednak inny od substancji oznaczanych i nie mo¿e byæ obecny w
analizowanych próbkach przed jego dodaniem.
W celu uzyskania krzywej kalibracyjnej do kilku roztworów wzorcowych o ró¿nych stê¿e-
niach substancji oznaczanej dodaje siê najczêœciej sta³¹ i znan¹ iloœæ wzorca wewnêtrznego. Na
podstawie uzyskanych chromatogramów roztworów wzorcowych wykreœla siê zale¿noœæ stê¿e-
173
2
1
4
3
2
2
3
1
3
2
2
1
1
,
a
x
a
y
a
xa
x
a
x
a
y
a
xa
x
a
y
x
a
y
=
+
+
+
=
+
+
=
=
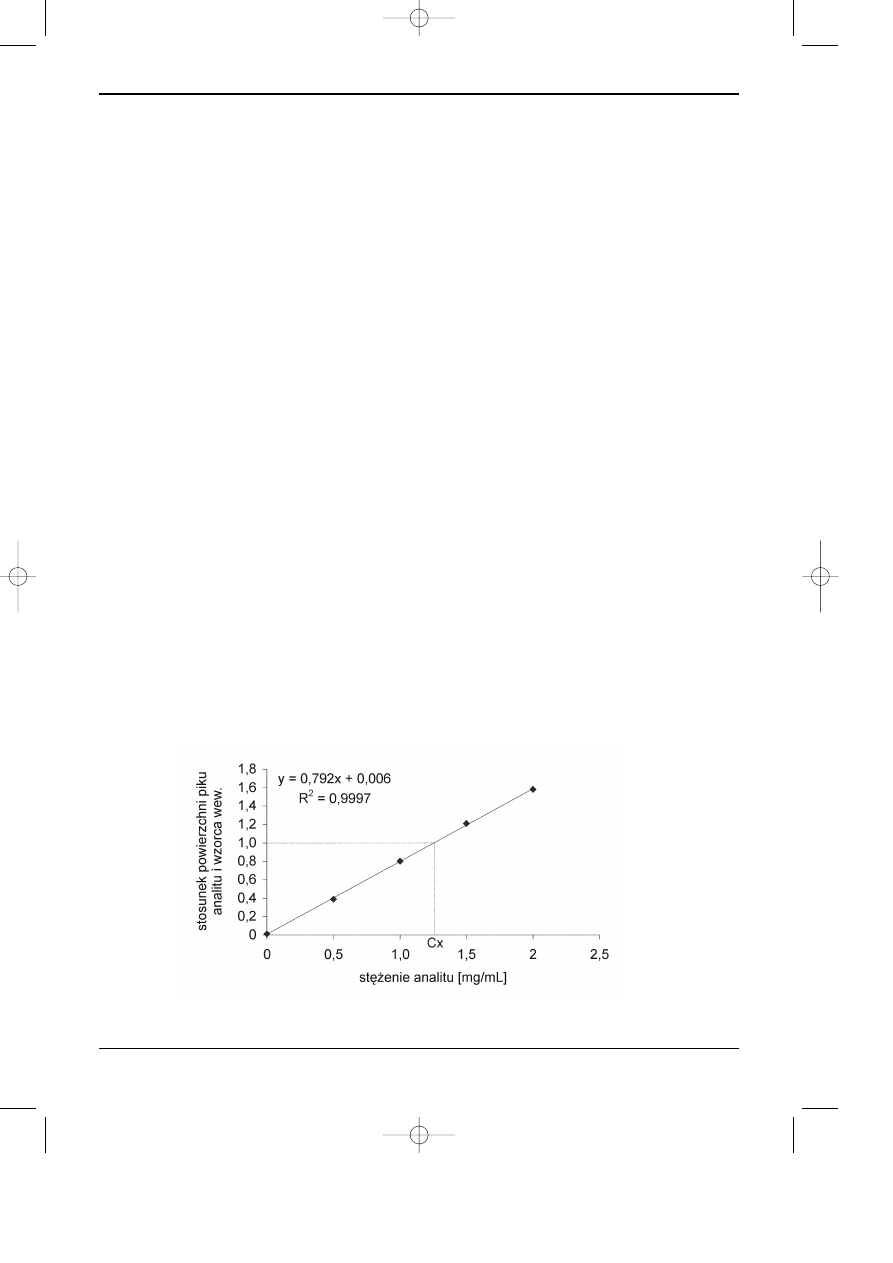
Rys. 11.3. Wykres krzywej kalibracyjnej otrzymanej przy zastosowaniu techniki wzorca
wewnêtrznego.
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 173
nia analitu w funkcji stosunku powierzchni piku analitu i wzorca wewnêtrznego. Na rysunku 11.3
przedstawiono krzyw¹ kalibracyjn¹ dla substancji oznaczanej metod¹ wzorca wewnêtrznego.
W celu oznaczenia zawartoœci substancji metod¹ wzorca wewnêtrznego w próbce badanej
(o znanej masie lub objêtoœci), dodaje siê do niej znan¹ iloœæ wzorca wewnêtrznego. Nastêpnie
z zarejestrowanego chromatogramu oblicza siê stosunek powierzchni piku analitu i wzorca
wewnêtrznego. Zawartoœæ substancji wyznacza siê metod¹ graficznej interpolacji, oblicza z rów-
nania krzywej kalibracyjnej lub z wykorzystaniem zale¿noœci (2) i (3) w sytuacji, kiedy krzywa
kalibracyjna jest zale¿noœci¹ liniow¹ typu f(x) = a
1
x:
(2)
(3)
gdzie: C
x
- oznaczona zawartoϾ, A
x
- powierzchnia piku analitu, A
ist
- powierzchnia piku wzor-
ca wewnêtrznego, R
f
- wspó³czynnik odpowiedzi, A
i
, A
ist(i)
- powierzchnia piku analitu i wzorca
wewnêtrznego w roztworze kalibracyjnym, c
i
- stê¿enie analitu w roztworze kalibracyjnym.
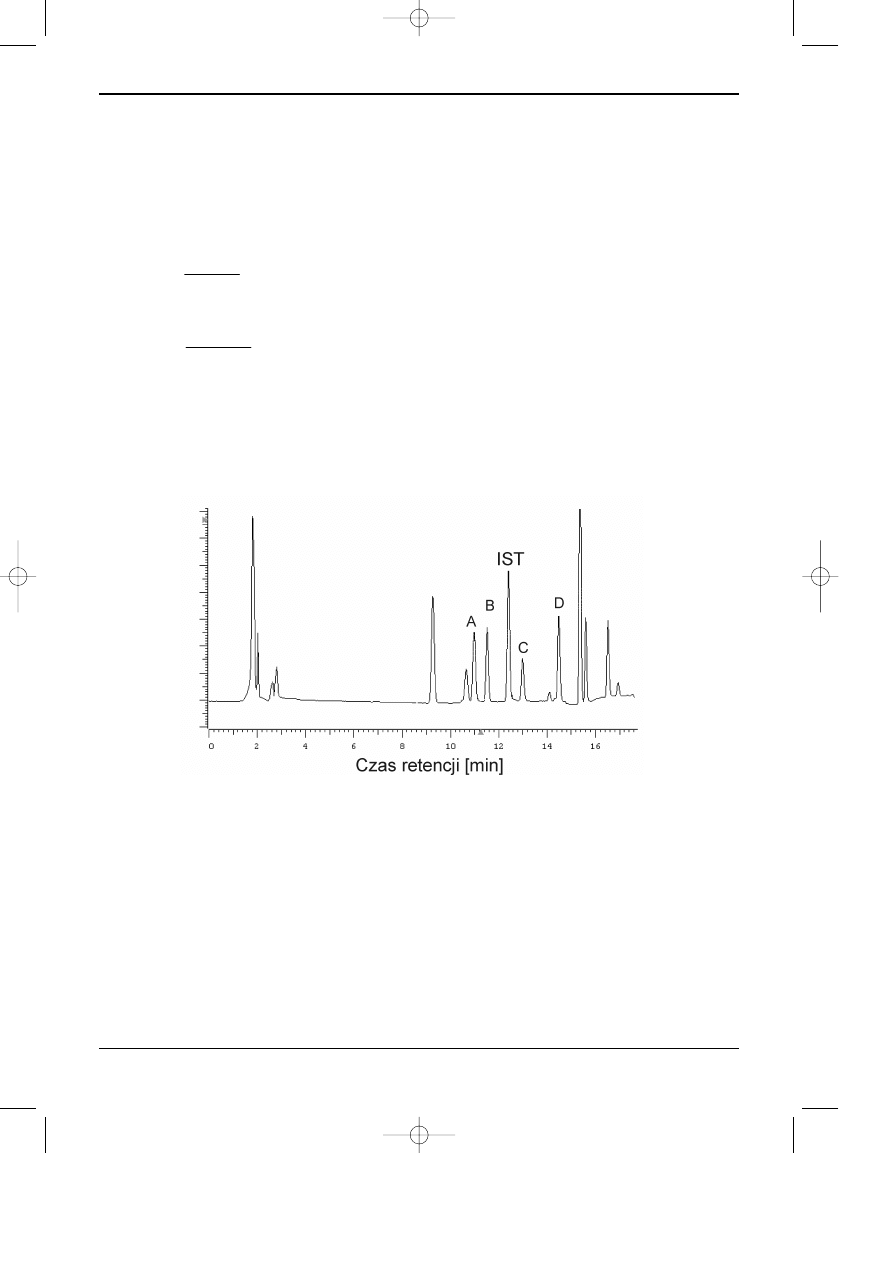
Na rysunku 11.4 przedstawiono chromatogram mieszaniny wzorcowej substancji
oznaczanych oraz wzorca wewnetrznego.
Wzorzec wewnêtrzny powinien spe³niaæ nastêpuj¹ce wymagania:
z
musi byæ rozdzielony od innych sk³adników wystêpuj¹cych w próbce
z
czas retencji powinien byæ zbli¿ony do czasu retencji analitu(ów)
z
nie wystêpuje w próbkach pierwotnych
z
w³aœciwoœci fizyko-chemiczne s¹ podobne do analitu. Jest to szczególnie istotne na
etapie przygotowania próbek (oczyszczania, wzbogacania czy derywatyzacji)
z
powinien byæ mo¿liwie wysokiej czystoœci
z
stabilny chemicznie
z
odpowiedŸ detektora dla wzorca wewnêtrznego powinna byæ zbli¿ona do odpowiedzi
substancji oznaczanych
i
i
ist
i
f
c
A
A
R
)
(
/
=
f
ist
x
x
R
A
A
C
/
=
174
Rys. 4. Chromatogram mieszaniny substancji oznaczanych A, B, C, D oraz wzorca
wewnêtrznego (IST).
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 174
Metoda ta ma zastosowanie szczególnie w przypadku metod analitycznych wymagaj¹cych
z³o¿onej, wieloetapowej procedury przygotowania próbki (izolacja, wzbogacanie, derywatyzac-
ja itp.), co mo¿e powodowaæ straty analitów. Dodanie wzorca wewnêtrznego do badanej próbki
jeszcze przed przyst¹pieniem do przegotowania próbki do analizy chromatograficznej pozwala
na skorygowanie tych strat.
Stosowanie metody wzorca wewnêtrznego pozwala równie¿ na uniezale¿nienie otrzymy-
wanych wyników od wahañ iloœci dozowanej próbki. Ograniczaniem w stosowaniu tej metody
mo¿e byæ bardzo bogata matryca, w której znajduj¹ siê anality. Trudne mo¿e byæ dobranie
odpowiedniej substancji jako wzorca wewnêtrznego. W praktyce metoda ta jest te¿ bardziej pra-
coch³onna ni¿ metoda krzywej kalibracyjnej.
11.3.
METODA DODATKU WZORCA
Metoda ta polega na dodaniu do próbki znanych iloœci substancji oznaczanych (analitów)
np. 0.1, 0.2, 0.3, 0.4 i 0.5 mg/mL, zwykle jest to od 50 do 150% oczekiwanej zawartoœci
oznaczanej substancji. Po zanalizowaniu tych roztworów, na podstawie uzyskanych chro-
matogramów, wykreœla siê krzyw¹ kalibracyjn¹, tj. zale¿noœæ powierzchni piku analitu w funkcji
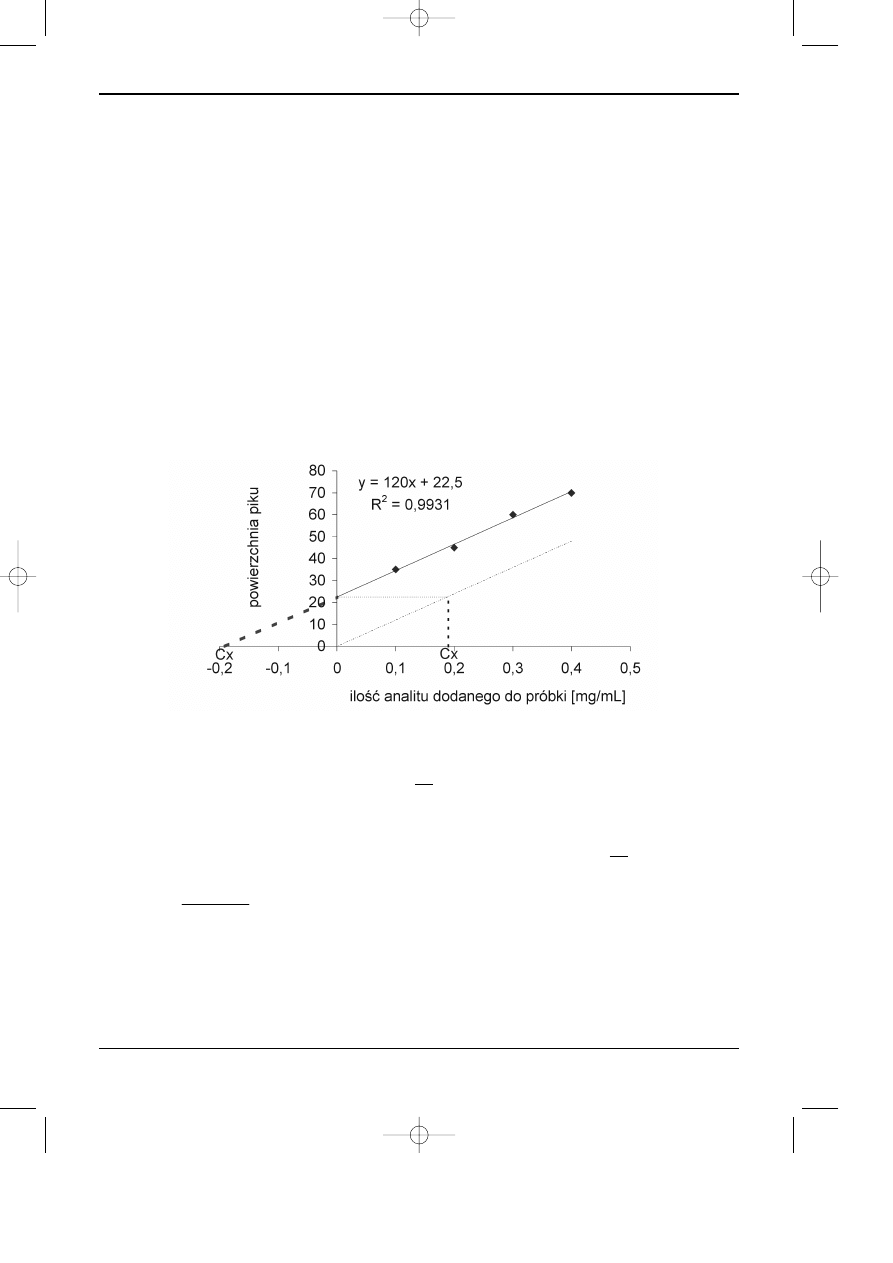
iloœci analitu dodanej do próbki (Rysunek 11.5).
Nastêpnie przeprowadza siê graficzn¹ ekstrapolacjê krzywej kalibracyjnej do punkt prze-
ciêcia z osi¹ odciêtych i odczytuje siê wartoœæ Cx. Innym sposobem okreœlenia zawartoœci sub-
stancji oznaczanej w próbce bez dodatku wzorca jest graficzne wyznaczenie przebiegu nowej
linii kalibracyjnej typu f(x) = a
1
x, o tym samym wspó³czynniku nachylenia prostej, co krzywa
kalibracyjna. Przeprowadza siê wówczas graficzn¹ interpolacjê, b¹dŸ oblicza siê stê¿enie anali-
tu w próbce bez dodatku substancji oznaczanej z równania (4), na przyk³ad: Cx=22.5/120=0.19.
(4)
gdzie: a
1
- wspó³czynnik kierunkowy prostej, a
2
- wartoœæ w punkcie przeciêcia siê prostej z osi¹
rzêdnych.
175
Rys. 11.5. Wykres krzywej kalibracyjnej otrzymanej przy zastosowaniu metody dodatku wzorca.
1
2
)
0
(
a
x
a
c
x
=
=
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 175
Zalet¹ metody dodatku wzorca jest wykonanie kalibracji w takich warunkach, ¿e anality
znajduj¹ siê w rzeczywistej matrycy. Szczególnie, kiedy jest bardzo trudne b¹dŸ niemo¿liwe
otrzymanie matrycy (placebo), w której nie znajdowa³aby siê substancja oznaczana, np. w przy-
padku próbek klinicznych i œrodowiskowych. Szczególn¹ cecha tej metody jest jej wysoka
"odpornoœæ" na sytuacjê niepe³nego rozdzielenia analitu oraz gdy pik substancji oznaczanej nie
jest rozdzielony do linii podstawowej od innych substancji, których piki s¹ jednak znacznie
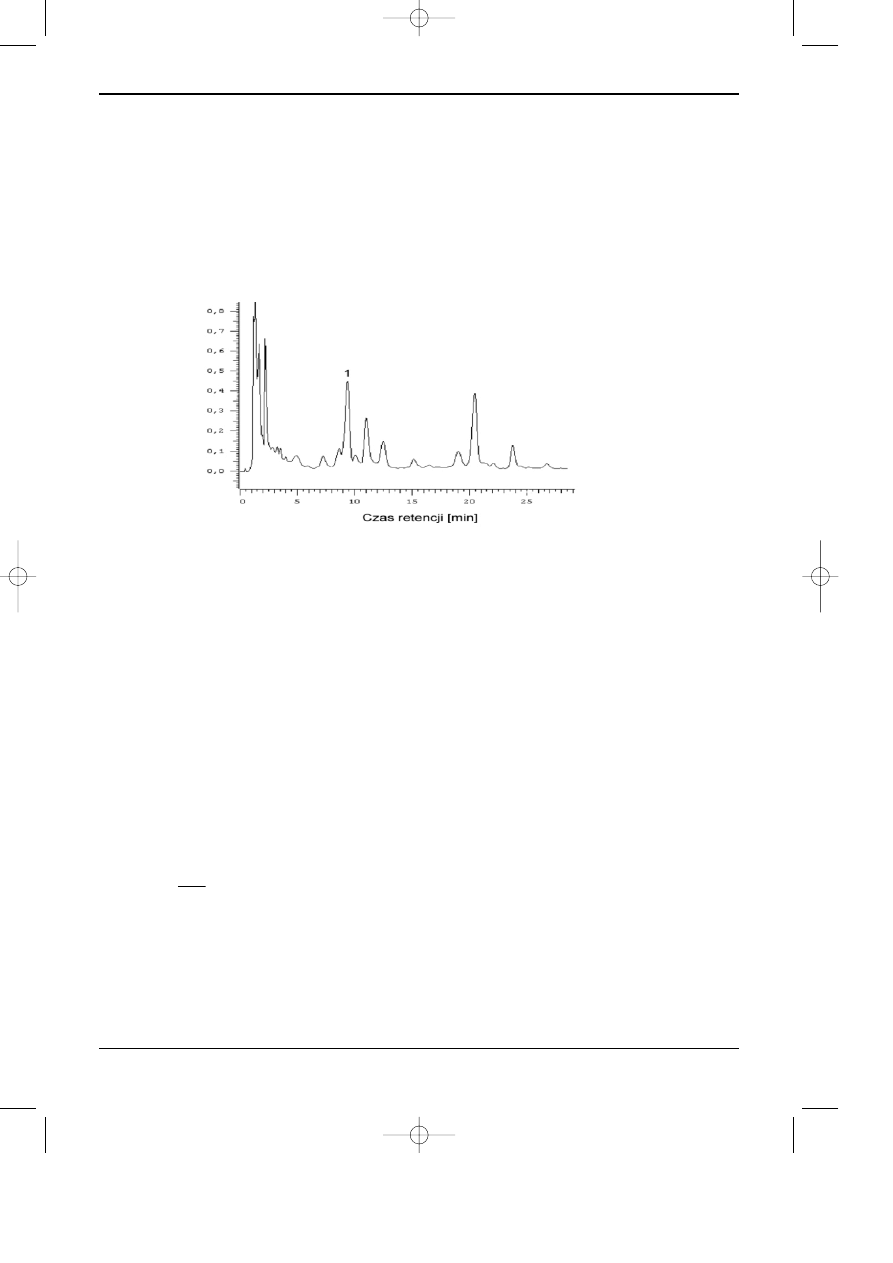
mniejsze w stosunku do substancji oznaczanej. Na rysunku 11.6 przedstawiono przyk³ad takiego
chromatogramu, na którym pik substancji oznaczanej zaznaczono symbolem "1". W wy¿ej
wymienionych sytuacjach tylko metoda dodatku wzorca pozawala na oznaczenie zawartoœci sub-
stancji w próbce. Wykonanie oznaczenia zawartoœci substancji metod¹ dodatku wzorca jest jed-
nak pracoch³onne.
11.4.
METODA PROSTEJ NORMALIZACJI
Metoda prostej normalizacji nie jest metod¹ kalibracji. Nie stosuje siê w niej odniesienia
do znanej iloœci wzorca. Jednak¿e metoda ta umo¿liwia oszacowanie wzglêdnych iloœci sub-
stancji np. zawartoœci zanieczyszczeñ w badanej próbce.
Na podstawie chromatogramu oblicza siê sumaryczn¹ powierzchniê pików, któr¹ traktuje
siê jak 100%. Nastêpnie oblicza siê udzia³ powierzchni okreœlonego piku wzglêdem sumy
powierzchni wszystkich lub tylko wybranych pików na chromatogramie. Otrzymanej wartoœci
dla danej substancji przypisuje siê procentow¹ zawartoœci wzglêdem innych substancji obecnych
w mieszaninie. Obliczenia wykonuje siê zgodnie z poni¿szym równaniem (5).
(5)
gdzie: c
i
- udzia³ % substancji i wzglêdem innych pików na chromatogramie, A
i
- powierzchnia
piku,
ΣA
i
- suma powierzchni wszystkich lub wybranych pików na chromatogramie
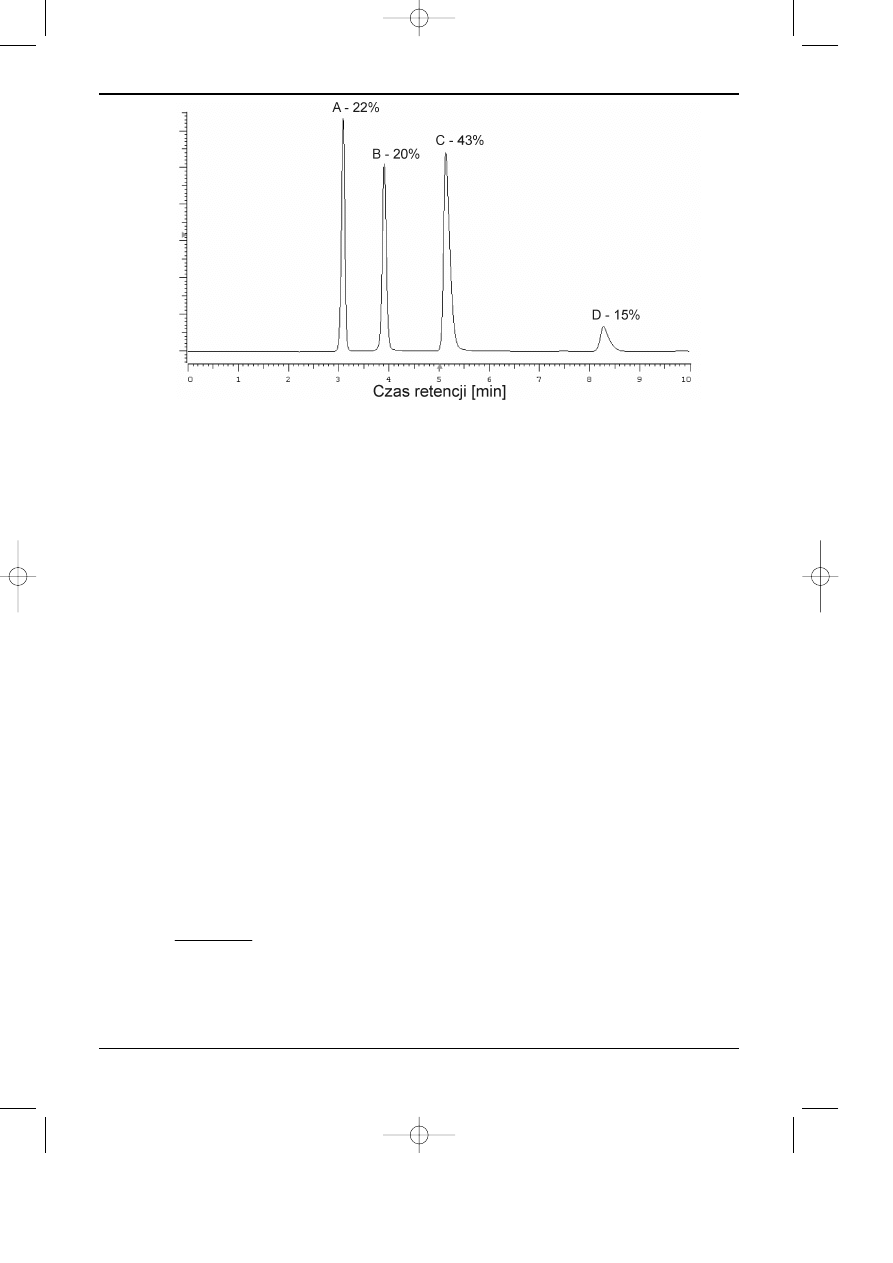
Na rysunku 11.7 chromatogram mieszaniny substancji oznaczanych oraz ich udzia³y pro-
centowe w próbce uzyskanê metod¹ prostej normalizacji.
100
×
Σ
=
i
i
i
A
A
c
176
Rys. 11.6. Chromatogram mieszaniny substancji, w której analit (1) nie jest w rozdzielony do linii
bazowej od innych substancji.
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 176
Metoda prostej normalizacji opiera siê na za³o¿eniu, ¿e wspó³czynniki odpowiedzi dla
ka¿dego sk³adnika próbki s¹ takie same, tzn. takie same stê¿enia ró¿nych substancji odpowiada-
j¹ takim samym powierzchniom ich pików chromatograficznych. Tak¹ charakterystyk¹ cechuje
siê detektor FID (p³omieniowo-jonizacyjny) stosowany w chromatografii gazowej do oznacza-
nia wêglowodorów. W chromatografii cieczowej metoda uproszczonej normalizacji jest
stosowana bardzo rzadko. Wynika to st¹d, ¿e detektory stosowane w HPLC charakteryzuj¹ siê
zró¿nicowan¹ czu³oœci¹ wzglêdem substancji. Stosowanie detektora typu UV-VIS i metody
prostej normalizacji jest nie celowe, poniewa¿ nawet substancje nale¿¹ce do tego samego szeregu
homologicznego charakteryzuj¹ siê zró¿nicowanymi molowymi wspó³czynnikami absorbancji
(
ε). Zatem odpowiedŸ detektora UV-VIS zale¿y od stê¿enia substancji jak i molowego
wspó³czynnika absorbancji
Korzystne mo¿e byæ stosowanie metody prostej normalizacji, gdy wykorzystywany jest
detektor refraktometryczny lub detektor œwiat³a rozproszonego. Detektory te wykazuj¹ zbli¿on¹
odpowiedŸ dla ró¿nych substancji o zbli¿onej strukturze i masie cz¹steczkowej.
11.5.
METODA NORMALIZACJI ZE WSPÓ£CZYNNIKAMI
KOREKCYJNYMI
Metoda ta, podobnie jak metoda prostej normalizacji, polega na okreœleniu wzglêdnej
powierzchni piku substancji badanej wzglêdem wybranych b¹dŸ wszystkich pików na chro-
matogramie z uwzglêdnieniem wspó³czynników korekcyjnych, uwzglêdniaj¹cych zró¿nicowan¹
odpowiedŸ detektora dla ró¿nych sk³adników próbki.
Wspó³czynniki korekcyjne wykorzystuje siê w celu oznaczenia rzeczywistego udzia³u
procentowego poszczególnych substancji w próbce analizowanej zgodnie z równaniem (6).
(6)
gdzie: c
i
- udzia³ % substancji i, A
i
- powierzchnia piku,
ΣA
i
- suma powierzchni wszystkich lub
wybranych pików na chromatogramie, R
f,i
- wspó³czynnik odpowiedzi (wspó³czynnik korek-
cyjny) dla substancji i
177
Rys. 11.7. Chromatogram mieszaniny substancji, na którym podano udzia³y procentowe sk³adników
otrzymane metod¹ prostej normalizacji.
100
)
(
×
×
Σ
×
=
fi
i
fi
i
i
R
A
R
A
c
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 177
W celu wyznaczenia wspó³czynników korekcyjnych postêpuje siê w sposób nastêpuj¹cy.
Na chromatogramie mieszaniny o znanych zawartoœciach substancji badanych oblicza siê pola
powierzchni pików, a nastêpnie oblicza siê wzglêdny udzia³ procentowy poszczególnych sub-
stancji metod¹ prostej normalizacji. W dalszej kolejnoœci oblicza siê wartoœæ wspó³czynnika
korekcyjnego dla danej substancji korzystaj¹c z równania (7).
(7)
gdzie: c
i
(znane) - zawartoœæ sk³adnika i w roztworze wzorcowym, c
i
(obliczone) - udzia³ sk³ad-
nika i w rozworze wzorcowym obliczony metod¹ prostej normalizacji.
W praktyce, czêsto zak³ada siê, ¿e dla wybranej substancji w mieszaninie wspó³czynnik
korekcyjny przyjmuje wartoϾ 1.
Na przyk³ad, dla substancji A, B, C, D przedstawionych na chromatogramie na rysunku 7
wyznaczone wspó³czynniki korekcyjne wynosz¹: R
fA
= 1, R
fB
= 1.1, R
fC
= 1.1, R
fD
= 1.2, st¹d
uzyskane zawartoœci po uwzglêdnieniu wspó³czynników korekcyjnych poszczególnych sk³ad-
ników wynosz¹: A - 20.1, B - 20.0, C - 43.5, D - 16.4 [%].
178
)
(
)
(
obliczone
c
znane
c
R
i
i
fi
=
Oznaczanie ilo ciowe w HPLC.qxp 2004-06-04 00:45 Page 178
Wyszukiwarka
Podobne podstrony:
MP3, fenol katechol-krzywa-4, KRZYWA KALIBRACYJNA DLA OZNAZCZANIA STĘŻENIA FENOLU
MP3, fenol katechol-krzywa-4, KRZYWA KALIBRACYJNA DLA OZNAZCZANIA STĘŻENIA FENOLU
krzywa kalibracyjna lab 5
krzywa kalibracyjna lab 5
krzywa kalibracyjna (3)
krzywa kalibracyjna 1
Krzywa opytu w modelu chamberlena
3 1 Krzywa podazy AS ppt
Krzywa doświadczeń
KRZYWA MOZLIWOŚCI PRODUKCYJNYCH
krzywa Phillipsa
KRZYWA PHILLIPSA, ● STUDIA EKONOMICZNO-MENEDŻERSKIE (SGH i UW), ekonomia matematyczna
kalibracja
krzywa wydatku
Krzywa wieża, katecheza, scenariusze
Kalibracja ekranu
Karabinek granatnik wz 1960 kalibru 7,62 mm
więcej podobnych podstron