
Instrukcja ćwiczeń laboratoryjnych – analityka zanieczyszczeń środowiska
Oznaczanie herbicydów z grupy triazyn z zastosowaniem techniki HPLC
WSTĘP
Herbicydy - środki chwastobójcze, stosowane do selektywnego lub całkowitego hamowania
rozwoju albo niszczenia roślin.
Herbicydy można klasyfikować ze względu na:
1)
budowę chemiczną
2)
działanie na rośliny
3)
sposób stosowania.
Jako herbicydy stosuje się głównie związki organiczne należące do różnych klas, m.in. związki
mocznikowe, związki triazynowe, karbaminiany, pochodne kwasu fenoksyoctowego. Działanie
chwastobójcze (fitotoksyczne) herbicydów polega głównie na zaburzaniu procesów fotosyntezy
oraz przemian enzymatycznych u roślin, a także hamowaniu podziału komórek (kiełkowania,
rozrostu pędów, kłączy), degradacji chlorofilu bądź wzroście transpiracji.
Zawartość herbicydów w wodzie można oznaczyć z wykorzystaniem metody HPLC w układzie faz
odwróconych.
CEL ĆWICZENIA
Celem ćwiczenia jest zapoznanie się z głównymi technikami przygotowania próbek do analizy
chromatograficznej oraz z zakresem zastosowań chromatografii cieczowej. Podczas zajęć
laboratoryjnych zostanie wykonany eksperyment polegający na izolacji i ilościowym oznaczeniu
symazyny, atrazyny i propazyny z próbki wody.
WARUNKI ANALIZY CHROMATOGRAFICZNEJ
Kolumna: Lichrospher 100 RP-18e 5 um (125x4 mm i.d.)
Faza ruchoma: MeOH:H2O 60:40, 1.0 mL/min,
Detekcja: UV długość fali: 220 – 450 nm
WYKONANIE ĆWICZENIA
1.Kalibracja
•
Przygotować roztwory kalibracyjne SYMAZYNY, ATRAZYNY, PROPAZYNY o stężeniu
10, 5, 2.5, 1, 0.5 ug/mL w fazie ruchomej poprzez rozcieńczenie roztworów podstawowych
o stężeniu 1000 ug/mL (stosować kolbki miarowe o pojemności max 10 mL i
mikrostrzykawki o pojemności 100 uL, 50 ul i 10 uL).
•
Ustawić stałą prędkość przepływu eluentu (patrz: warunki rozdzielania)
•
Ustabilizować warunki rozdzielania (stabilna linia podstawowa)
•
Po ustaleniu warunków wstrzyknąć do kolumny kolejne roztwory kalibracyjne symazyny,
atrazyny i propazyny począwszy od najniższego stężenia
•
Zarejestrować chromatogramy i wyznaczyć powierzchnie pików poszczególnych
składników roztworów kalibracyjnych
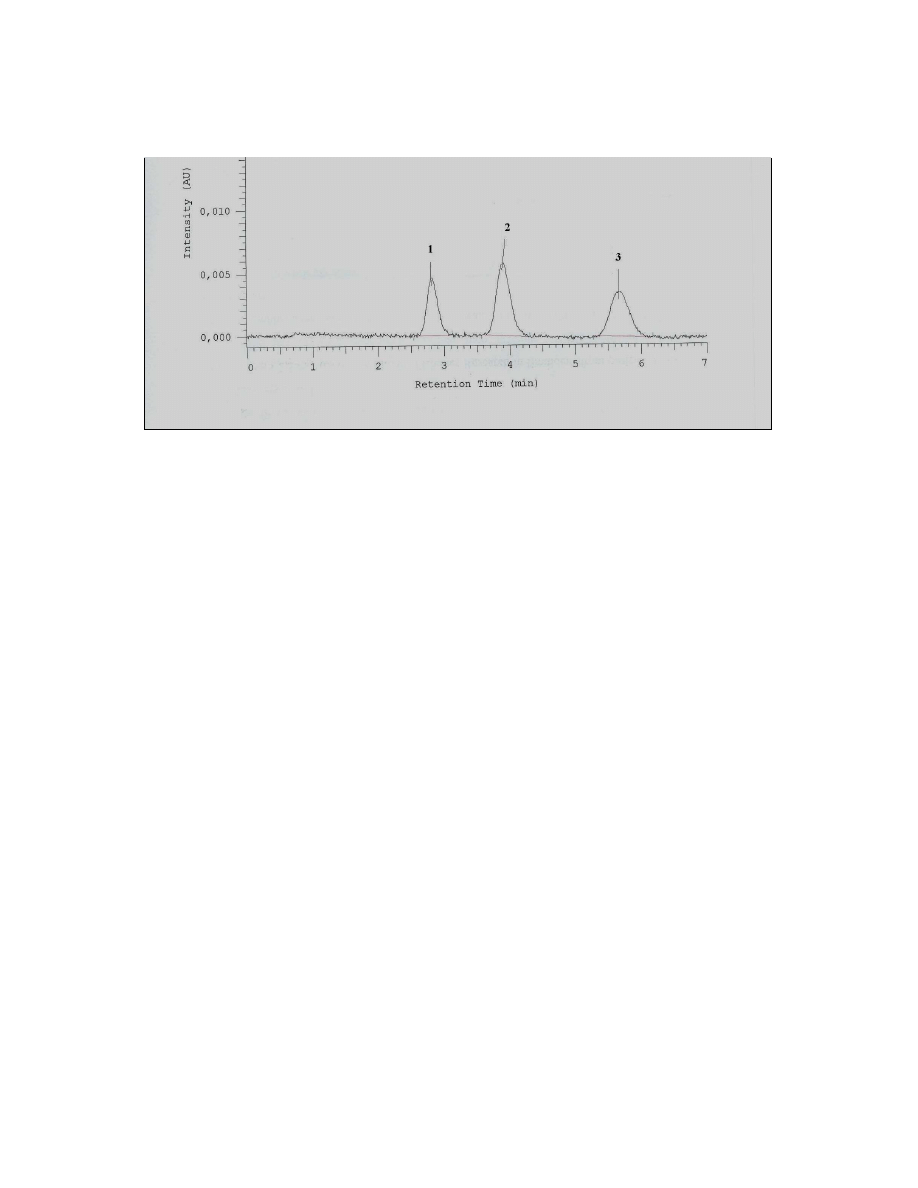
Rys. 1 Przykład chromatogramu przedstawiający rozdzielenie herbicydów z grupy triazyn
znajdowanych w glebie
1 – symazyna Kolumna: Lichrospher 100 RP-18e 5 um (125x4 mm i.d.)
2 - atrazyna Faza ruchoma: MeOH:H2O 60:40, 1.0 mL/min,
3 - propazyna Detekcja: UV długość fali: 270 nm
2. Przygotowanie próbek gleby do analizy chromatograficznej - ekstrakcja.
6 g próbki gleby ekstrahować 2-krotnie 10 ml rozpuszczalnika (n-heksan, n-heptan, chloroform,
eter metylowo-tertbutylowy) – grupa laboratoryjna zostanie podzielona na 4 podgrupy; każda z
podgrup ekstrahuje przy użyciu jednego z wymienionych rozpuszczalników. Uzyskuje się około 20
ml ekstraktu, który należy przesączyć przez sączek karbowany. Należy też wykonać ślepe próby z
zastosowaniem poszczególnych ekstrahentów.
Uzyskany ekstrakt odparować prawie do sucha w próżniowej wyparce obrotowej, a następnie, po
ilościowym przeniesieniu do sucha w strumieniu azotu. Pozostałość rozpuścić w 1 ml fazy
ruchomej.
3. Wykonanie oznaczenia
•
Nanieść na kolumnę chromatograficzną określoną objętość przygotowanej próbki
•
Zarejestrować chromatogramy, zidentyfikować piki symazyny, atrazyny i propazyny
•
Odczytać powierzchnie pików symazyny, atrazyny i propazyny
OPRACOWANIE WYNIKÓW
1.
Wykonać oznaczenie zawartości SYMAZYNY, ATRAZYNY, PROPAZYNY w próbce
gleby dostarczonej przez prowadzącego
OBOWIĄZUJĄCY ZAKRES MATERIAŁU
1.
Pojęcia: układ chromatograficzny - faza stacjonarna, faza ruchoma, substancje rozdzielane
2.
Układ faz normalnych i odwróconych podstawowe pojęcia

3.
Zjawiska decydujące o rozdzielaniu substancji metodą chromatografii cieczowej.
4.
Mechanizmy rozdzielania substancji w chromatografii
5.
Podstawowe parametry układu chromatograficznego – czas retencji, czas martwy kolumny,
objętość retencji, objętość martwa, współczynnik rozdzielenia, wysokość półki teoretycznej,
liczba półek teoretycznych
6.
Pojęcie selektywności, sprawności i rozdzielczości układu chromatograficznego
7.
Metody przygotowania próbek do analizy chromatograficznej: ekstrakcja ciecz-ciecz,
ekstrakcja ciecz-ciało stałe, ekstrakcja ciecz-gaz, ekstrakcja nadkrytyczna
LITERATURA
1.
M. Kamiński (ed.), „Chromatografia cieczowa”, CEEAM, Gdańsk, 2004.,
2.
Z. Witkiewicz, „Podstawy chromatografii”, Wydawnictwo Naukowo – Techniczne,
Warszawa, 2004

OZNACZANIE WIELOPIERŚCIENIOWYCH WĘGLOWODORÓW
AROMATYCZNYCH (WWA) W PRÓBKACH WODY METODĄ HPLC Z
DETEKCJĄ FLUORESCENCYJNĄ PO EKSTRAKCJI
CIECZ – CIECZ
WSTĘP
Wielopierścieniowe węglowodory aromatyczne (WWA) występują prawie we wszystkich rodzajach
wód . Związki te są zaadsorbowane na cząstkach stałych (osady, zawiesiny) a także są
rozpuszczone w fazie wodnej. Wielopierścieniowe węglowodory aromatyczne mają właściwości
rakotwórcze lub są o nie podejrzewane.
CEL ĆWICZENIA
Celem ćwiczenia jest poznanie w praktyce zasad postępowania analitycznego w celu oznaczenia
ś
ladowych zawartości nisko polarnych i wysoce hydrofobowych substancji z zastosowaniem
ekstrakcji ciecz – ciecz, jako techniki przygotowania próbki oraz techniki wysokosprawnej
chromatografii cieczowej w układach faz odwróconych (RP-HPLC) z elucją gradientową i detekcją
UV-DAD oraz fluorescencyjną dla rozdzielenia, identyfikacji i ilościowego oznaczenia substancji
stanowiących
zanieczyszczenia
ś
rodowiska
na
przykładzie
oznaczania
zawartości
wielopierścieniowych węglowodorów aromatycznych w próbkach wody.
ZASADA METODY
Zasada postępowania analitycznego w czasie ćwiczenia oparta jest na metodyce opisanej w normie
PN-EN ISO 17993 „Oznaczanie 15 wielopierścieniowych węglowodorów aromatycznych (WWA)
w wodzie metodą HPLC z detekcją fluorescencyjną po ekstrakcji ciecz-ciecz”.
Oznaczane składniki analitu (WWA) są ekstrahowane z wody za pomocą n-heksanu. Ekstrakt jest
zatężany przez odparowanie n-heksanu a pozostałość rozpuszczana w rozpuszczalniku
odpowiednim do wykonania analizy chromatograficznej w warunkach chromatografii w układach
faz odwróconych. WWA są rozdzielane z zastosowaniem techniki elucji gradientowej.
Identyfikacja i oznaczenie ilościowe są wykonywane za pomocą detekcji UV-VIS/DAD, w
przypadku oznaczania szczególnie wysokich zawartości WWA, albo szczególnie wysokiego stopnia
wzbogacenia i fluorescencyjnej, w przypadku oznaczania zawartości śladowych.
PRZYGOTOWANIE PRÓBKI
•
Do 1000 ml zhomogenizowanej próbki wody dodać 25 ml heksanu i dokładnie wstrząsnąć,
mieszać badaną próbkę przez ok. 60 min.
•
Przenieść badaną próbkę do lejka rozdzielczego i odczekać co najmniej 5 min. Na
rozdzielenie faz.
•
Warstwę heksanową przenieść do kolby stożkowej i ekstrakt suszyć przez co najmniej 30
min za pomocą bezwodnego siarczanu sodu.
•
Osuszony ekstrakt zdekantować, kolbę przepłukać dwukrotnie porcjami po 5 ml heksanu i
dodać je do ekstraktu.
•
Osuszony ekstrakt heksanowy odparować za pomocą wyparki próżniowej na łaźni wodnej o
temp. 30
°
C do momentu gdy ekstrakt pozostanie tylko w zwężonej końcówce naczynia
redukcyjnego
•
W celu oczyszczenia ekstraktu dodać 250 ul N, N – dimetyloformamidu i zhomogenizować
mieszaninę za pomocą 500 ul acetonu

•
Za pomocą strumienia azotu usunąć całkowicie heksan i aceton, tak aby objętość ekstraktu
zmniejszyła się i wynosiła między 200 a 250 ul
•
Rozcieńczyć ekstrakt do znanej objętości (np. 500 ul) za pomocą acetonitrylu.
•
Do kolejnego oczyszczania ekstraktu użyć kolumienek zawierających co najmniej
0,5 g żelu krzemionkowego
•
Ż
el krzemionkowy w kolumience przemyć mieszaniną dichlorometan/heksan (1:1) o
objętości pięciokrotnie wyższej niż objętość złoża, następnie kondycjonować kolumienkę
taką samą objętością heksanu.
•
Za pomocą pipety Pasteura przenieść ekstrakt heksanowy na szczyt kolumienki i pozwolić
mu prawie całkowicie wniknąć w złoże żelu.
•
Frakcję zawierającą WWA eluować z kolumny za pomocą mieszaniny dichlorometan :
heksan (1:1) (co najmniej 3 ml)
•
Do eluatu dodać 250 ul N, N – dimetyloformamidu, zhomogenizować przez wytrząsanie i
zatężyć do objętości między 200 a 250ul (do ok. 2 ml za pomocą wyparki próżniowej a
następnie w strumieniu azotu)
•
Rozcieńczyć ekstrakt do ok. 2 ml za pomocą acetonitrylu
•
Tak przygotowaną próbkę poddać analizie chromatograficznej
WYKONANIE OZNACZENIE
1.
Warunki analizy chromatograficznej
Natężenie przepływu fazy ruchomej – ok. 0,5 ml / min
Temperatura pokojowa
Kolumna: wypełnienie: LiChrospher 100 C18 – 5 um, wymiary: 250 x 4 mm
Eluent: A: acetonitryl, B: woda; Program elucji gradientowej:
•
Czas [min.] 0
35
45
Program elucji: 60:40(A:B v/v) do 100 B (liniowo)
100 B (izokratycznie)
Detektor UV-VIS typu DAD (z tablicą elementów fotoczułych) i szeregowo przyłączony
detektor fluorescencyjny: długość fali wzbudzenia: 270 nm; długość fali emisji: 400 nm
2.
Wzorcowanie
•
Sporządzić roztwory standardów wielopierścieniowych węglowodorów
aromatycznych w acetonitrylu (stężenie roztworu powinno wynosić ok. 10 ug/ml
każdego składnika)
•
Tak przygotowany roztwór rozcieńczyć 10 -, 30 -, 50 – krotnie.
•
Po ustaleniu warunków oznaczania dozować do kolumny kolejno roztwory
wzorcowe WWA o wzrastającym stężeniu wielopierścieniowych węglowodorów
aromatycznych;
•
Zarejestrować chromatogramy z zastosowaniem obu detektorów i odczytać wartości
czasu retencji oraz powierzchnię piku każdego wzorca.

Rys. 1 Przykład chromatogramu przedstawiający rozdzielenie WWA znajdowanych w
wodzie do spożycia, detektor fluorescencyjny dług. fali wzb. 270 nm, długość
fali emisji 400nm.
3. Wykonanie oznaczenia
•
Po ustabilizowaniu warunków oznaczania, identycznych jak te, które stosowano
podczas wzorcowania, dozować do kolumny roztwór przygotowanej wcześniej
próbki
OPRACOWANIE WYNIKÓW
•
Zidentyfikować poszczególne substancje i odpowiadające im piki chromatogramu na
podstawie widm w świetle UV w zakresie 200 do 400 nm
•
Sporządzić wykres zależności stężenia substancji w próbce dozowanej do kolumny
od powierzchni piku dla każdego wzorca wielopierścieniowych węglowodoru
aromatycznych
•
Na postawie uzyskanych krzywych kalibracyjnych wyznaczyć zawartość
poszczególnych WWA w próbkach wody wyrażoną w ug/ml
OBOWIĄZUJACY ZAKRES MATERIAŁU TEORETYCZNEGO
8.
Pojęcia układu chromatograficznego: faza stacjonarna, faza ruchoma, substancje
rozdzielane, retencja, selektywność, sprawność, rozdzielczość pików;
9.
Konkurencja
oddziaływań
sorpcyjnych
/
zróżnicowanie
potencjałów
termodynamicznych / zróżnicowanie szybkości dyfuzji jako mechanizmy rozdzielania
chromatograficznego;

10.
Układ faz normalnych / układ faz odwróconych - zjawiska i mechanizmy
fiykochemiczne decydujące o retencji i selektywności:
- Zjawiska decydujące o rozdzielaniu substancji w chromatografii cieczowej w układach faz
odwróconych (RP),
- Zjawiska decydujące o rozdzielaniu substancji w chromatografii cieczowej w układach faz
normalnych (NP);
4. Pojęcie selektywności, sprawności i rozdzielczości układu chromatograficznego;
5. Podstawowe parametry układu chromatograficznego – czas retencji, czas martwy,
objętość retencji, objętość martwa, współczynnik retencji, współczynnik rozdzielenia,
wysokość półki teoretycznej, liczba półek teoretycznych, prędkość przepływu eluentu,
„zredukowane” parametry sprawności i prędkości przepływu;
6. Elucja gradientowa – Co oznacza to pojęcie ? Jaki jest cel stosowania elucji gradientowej
? Podstawowe parametry programu elucji gradientowej; Wpływ parametrów programu
elucji na wyniki rozdzielania; Na czym polega postępowanie w celu doboru optymalnego
programu elucji;
LITERATURA
3.
M. Kamiński (ed.), „Chromatografia cieczowa”, CEEAM, Gdańsk, 2004.,
4.
Z. Witkiewicz, „Podstawy chromatografii”, Wydawnictwo Naukowo – Techniczne,
Warszawa 2004.,
5.
PN-EN ISO 17993 – Oznaczanie 15 wielopierścieniowych węglowodorów aromatycznych
(WWA) w wodzie metodą HPLC z detekcją fluorescencyjną po ekstrakcji ciecz-ciecz

Instrukcja ćwiczenia laboratoryjnego – Analityka zanieczyszczeń środowiska
OZNACZANIE PRODUKTÓW FARMACEUTYCZNYCH W PRÓBKACH STAŁYCH Z
WYKORZYSTANIEM
PRZYSPIESZONEJ
EKSTRAKCJI
ZA
POMOCĄ
ROZPUSZCZALNIKA (ASE) I OZNACZENIEM KOŃCOWYM TECHNIKĄ HPLC – DAD
WSTĘP
Postęp cywilizacyjny jest nieodłącznie związany ze wzrostem ilości chorób, które dręczą
ludzkość. Stanowi to siłę napędową działań w zakresie ciągłego unowocześniania leków już
dostępnych na rynku oraz projektowania i wdrażania do produkcji nowych preparatów. Jednak
przemysł farmaceutyczny zajmuje się wytwarzaniem, nie tylko leków przeznaczonych dla ludzi, ale
także dla zwierząt. Jedną z grupy takich leków są leki kokcydiostatyczne, stosowane jako dodatki
paszowe. Kokcydiostatyki są to substancje stosowane w profilaktyce kokcydiozy, pasożytniczej
choroby układu pokarmowego, występującej wśród drobiu, królików, szynszyli i innych zwierząt
hodowlanych. Za zastosowaniem tej grupy związków przemawia fakt, że należą one do substancji
charakteryzujących się m.in.: brakiem toksycznego wpływu na zwierzęta, szybką eliminacją z
organizmu (krótki czas retencji) oraz stabilnością podczas produkcji mieszanek paszowych ich
magazynowania. Stosowanie kokcydiostatyków jak i innych środków leczniczych nie pozostaje
jednak bez wpływu na skład chemiczny żywności, a tym samym zdrowie człowieka, stąd też
Dyrektyw Unii Europejskiej nakładają na producentów konieczność kontroli zawartości wszystkich
dodatków stosowanych przy ich produkcji. Wynikiem tych działań jest opracowywanie wielu
metodyk oznaczania produktów farmaceutycznych w próbkach środowiskowych. Jedną z nich jest
metodyka oznaczania Robenidyny w próbkach pasz zwierzęcych, która wykonywana będzie na
ć
wiczeniu.
Cl
N
N
Cl
NH
H
H
N
N
Rys. 1 Wzór strukturalny Robenidyny
CEL ĆWICZENIA
Celem ćwiczenia jest poznanie zasad postępowania analitycznego w celu przygotowania próbki i
wykorzystania techniki HPLC do oznaczania zawartości dodatków do pasz i żywności na
przykładzie Robenidyny.
ZASADA METODY
Robenidyna (ROB) obecna w próbce paszy ekstrahowana jest z niej za pomocą metanolu
zakwaszonego CH
3
COOH. Następnie ekstrakt jest oczyszczany z zastosowaniem techniki SPE
(kolumienek Pasteura wypełnionych Al
2
O
3
). Robeniyna wymywana jest z kolumienek za pomocą
metanolu cz.d.a. Identyfikację i oznaczanie ilościowe jest wykonane z zastosowaniem
Wysokosprawnej Chromatografii Cieczowej z detektorem UV typu DAD.

WYKONANIE
Ć
WICZENIA
Przygotowanie krzywej kalibracyjnej
•
Dozować kolejno roztwory wzorcowe o określonych stężeniach (0,21µg/ml, 0,51 µg/ml,
0,62 µg/ml).
•
Wyznaczyć zależność: pole powierzchni w funkcji stężenia robenidyny w próbce.
Warunki rozdzielania i detekcji robenidyny
Kolumna: Purospher ODS RP-18e (125 x 3 mm i.d., 5
µ
m)
Natężenie przepływ eluentu: 0,7 ml/min
Faza ruchoma: warunki izokratyczne (70% metanol, 30% woda z dodatkiem 0,1 % v/v kwasu
triflorooctowego)
Detektor: DAD, 317 nm
Przygotowanie próbek do ekstrakcji
Paszę w postaci granulowanej zmielić na drobną frakcję (średnica cząstek
≤
1, 0 mm). Około
8g zmielonej paszy zmieszać z ok. 8 g piasku kwarcowego, umieścić w naczyniu do przyspieszonej
ekstrakcji za pomocą rozpuszczalnika (ASE) o pojemności 22 ml i przeprowadzić ekstrakcję w
podwyższonej temperaturze i w podwyższonym ciśnieniu, w wybranym medium ekstrakcyjnym.
Parametry ekstrakcji ASE przedstawiono w tabeli 1
Tabela 1 Warunki ekstrakcji ASE robenidyny z paszy
Parametry ekstrakcji ASE
Wartość zadana
Czas ekstrakcji statycznej [min]
3
Temperatura [
o
C]
100
Ciśnienie [psi]
1500
Ilość cykli ekstrakcyjnych
3
Objętość rozpuszczalnika do płukania [%]
100
Czas płukania azotem [s]
60
Rozpuszczalnik
MeOH + 1%CH
3
COOH
1 psi = 6,8948 kPa
Uzyskany ekstrakt zebrać w naczyniu, w którym było sito molekularne typu 5A, stosowane jako
ś
rodek osuszający (5g). Po około 5 minutach wytrząsania pobrać z naczynia 2 ml ekstraktu i poddać
go oczyszczeniu na kolumience wypełnionej 1g Al
2
O
3
. Robenidynę wyeluować przy użyciu 10 ml
metanolu, eluat zebrać do kolby miarowej. Tak uzyskany ekstrakt poddać analizie
chromatograficznej (RP – HPLC –DAD).
OBLICZYĆ
•
Efektywność ekstrakcji dla próbek bez ROB z dodatkiem wzorca z paszy.
•
Odzysk Robenidyny z próbek pasz zwierzęcych, (w których zawartość Robenidyny jest
podana przez producenta).
OBOWIĄZUJĄCY ZAKRES MATERIAŁU TEORETYCZNEGO
•
Znajomość technik ekstrakcji próbek stałych, ich wad i zalet, możliwość stosowania.
•
Chromatografia cieczowa w układzie faz odwróconych (schemat blokowy chromatografu,
pojecie selektywności, sprawności i rozdzielczości układu chromatograficznego, detekcja
oraz oznaczanie ilościowe w HPLC)

LITERATURA
M. Kamiński, „Chromatografia Cieczowa”, CEEAM, Gdańsk 2004.
J. Namieśnik, Z. Jamrógiewicz, M. Pilarczyk, L. Torres, „Przygotowanie próbek środowiskowych
do analizy”, WNT, Warszawa 2002
Wyszukiwarka
Podobne podstrony:
Chrom gaz I
chrom
Blender 3D Materiały Shadery Chrom
zadania chrom, Analityka semestr IV, Analiza Instumentalna
Acetonitryl do chrom (HPLC)
chrom i nie tylko
Chrom
alfa Pikolina?,9 wzorzec do chrom gaz
Metylocykloheksan?,9 wzorzec do chrom gaz
chrom, nauka
Benzen do chrom (HPLC)
CHROM (Cr)
instrukcja chrom, rekultywacja i ochrona gruntów - RiOG
Etylu octan do chrom (HPLC)
n Heksan do chrom (HPLC)
więcej podobnych podstron