
424
MOLECULAR RECOGNITION IN DENDRIMERS
Vol. 10
MOLECULAR
SELF-ASSEMBLY
Introduction
Molecular self-assembly is an extremely versatile tool for the thermodynamically
controlled generation of higher order constructs (1). Nature uses self-assembly
extensively in biological systems with “lock and key” specificity to give the di-
verse range of highly ordered systems observed in living organisms. Applying the
self-assembly strategy to the reversible synthesis and controlled aggregation of
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.

Vol. 10
MOLECULAR SELF-ASSEMBLY
425
macromolecules has garnered considerable interest in recent years. The utility of
directional and highly specific noncovalent interactions has been demonstrated
to be a particularly attractive method of controlling polymer aggregation, using
the abundance of recognition elements developed by supramolecular chemists (1).
The high degree of modularity makes this approach extremely versatile in that
the assembling units can be precisely tuned with directed synthesis to customize
the selectivity, directionality, and association strength of the noncovalent inter-
action. Also, the strength of the noncovalent interactions that embody molecular
self-assembly is thermally dependent, allowing for reversible control over the as-
sembly process which contributes to the design of unique material properties with
defect correction and self-healing capabilities.
Recently, a number of contributions have been published that detail a grow-
ing field of synthetic polymer chemistry: supramolecular polymers/polymerization
(2,3). Supramolecular polymers are macromolecules containing monomers held to-
gether by noncovalent interactions. As listed in Table 1, these interactions can be
divided into six commonly used categories which include hydrogen bonding (4)
(often multiple hydrogen bonds acting in tandem),
π stacking of aromatic systems
(5), metal–ligand interactions (6), London dispersion forces (7), electrostatics, and
hydrophobics. As formed, these novel supramolecular polymers should exhibit
similar material properties to their covalently bound counterparts with the added
functionality of reversible binding. In addition to the supramolecular polymeriza-
tion of main chain polymers, it is also conceivable to modify covalently bound linear
polymers with pendant sidechains capable of noncovalent binding. In theory, this
combination provides the most advantageous properties from both components:
intrinsic materials properties dictated by the choice of polymer backbone with the
reversible lock and key nature of supramolecular interactions.
Main-Chain Versus Side-Chain Supramolecular Polymers
The present concept of supramolecular polymers containing multiple hydrogen
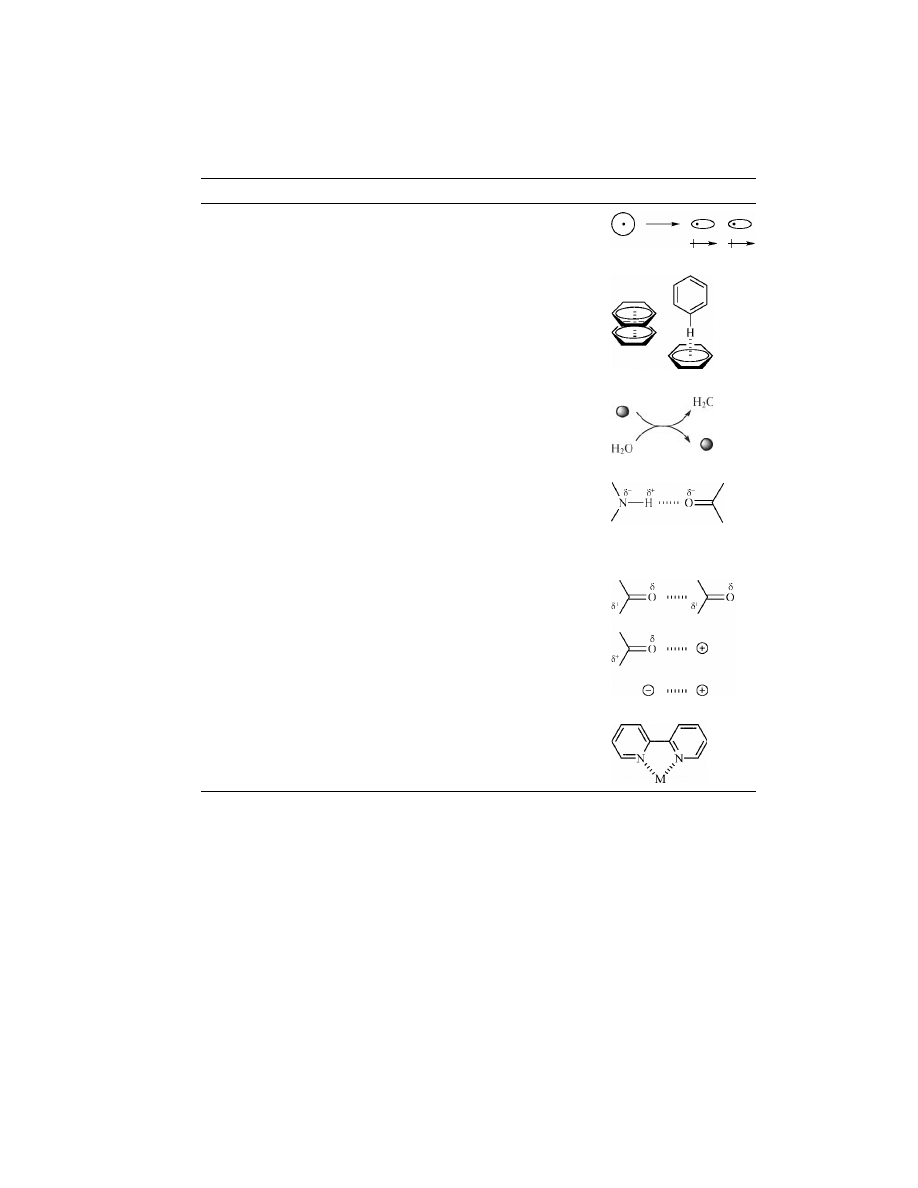
bonding units was first introduced over a decade ago by Jean-Marie Lehn (8).
Three-point hydrogen bonds between bisfunctional diaminopyridine and thymine
derivatives form supramolecular polymer chains exhibiting liquid crystalinity
(Fig. 1a). Lehn and co-workers expanded the scope of this methodology to in-
clude bisfunctional molecules joined by chiral tartaric acid spacers (9) and rigid
anthracene-based linkers (10). The utility of this approach has been demonstrated
many times over in the years since, as can be ascertained by numerous contribu-
tions from many researchers (11,12).
Building upon Lehn’s three-point hydrogen bound supramolecular poly-
mers is the work done by E. W. Meijer using self-complementary ureidopyrimidi-
nones, recognition elements capable of quadruple hydrogen bonding (Fig 1b). The
quadruple hydrogen bonding system has two major differences with complemen-
tary three-point hydrogen bonding between thymine and diamidopyridines: an
extremely high degree of association (K
dim
>10
6
M
− 1
) and self-complementarity
that eliminates stoichiometric concerns. The high dimerization constant of urei-
dopyrimidinone makes this an excellent system for supramolecular polymeriza-
tions, as the resulting polymers will possess a high degree of polymerization in
426
MOLECULAR SELF-ASSEMBLY
Vol. 10
Table 1. Six Categories of Noncovalent Intermolecular Interactions
a
Interaction
Description and bond strengths
Selected example
(London) Dispersion
forces
> 4 kJ/mol. Temporarily induced
dipole–dipole interactions
π–π stacking
8–12 kJ/mol (face–face), 12–21
kJ/mol (edge–face). Attractive
forces between electron-rich
interior with electron-poor exterior
Hydrophobic
4–42 kJ/mol. Association of nonpolar
complements in aqueous or polar
media
Hydrogen bonding
> 4kJ/mol (weak), 4–17 kJ/mol
(moderate), 21–42 kJ/mol (strong).
Donor–acceptor interaction
involving hydrogen atom as proton
donor and base (electron pair) as
proton acceptor
Electrostatic
4–42 kJ/mol (dipole–dipole), 42–125
kJ/mol (ion–dipole),
>188 kJ/mol
(ion–ion). Coulombic attraction
between opposite charges
Dative bonding
21–377 kJ/mol. Metal–ligand
coordination. Ligand donates
electron pair(s) to center
a
Association strengths are for systems in chloroform.
solution. The resulting macromolecules generate excellent materials that have
many advantages, such as strongly temperature-dependent viscoelastic proper-
ties. However, substantially more energy is required to induce complete dissocia-
tion to overcome the high degree of association.
In contrast to the above supramolecular polymers, recognition groups co-
valently attached as pendant polymer side chains (13) have been investigated
for their potential elastomeric properties, first by Stadler and co-workers (14)
in 1986 to increase miscibility between incompatible polybutadiene and poly-
isoprene blends by employing the relatively weak dimerization of urazole moi-
eties (K
dim
< 10
2
M
− 1
), and later in 2001 where G. W. Coates and co-workers
Vol. 10
MOLECULAR SELF-ASSEMBLY
427
Fig. 1.
Supramolecular polymers developed by (a) Lehn employing three-point hydro-
gen bonds between diamidopyridine and thymine residues and (b) Meijer employing
self-complementary, quadruple hydrogen bonds. (c) A simplified schematic depicting the
extended chain of repeating bisfunctional monomers fashioning the backbone of a
supramolecular polymer.
(15) demonstrated the synthesis of elastomeric polyolefins equipped with ther-
moreversible cross-links between ureidopyrimidinone pendant groups. Kato and
co-workers (16,17) have made large contributions to this field by exploring side-
chain polymers as liquid crystalline materials. Furthermore, these motifs have
been investigated by M. Weck and co-workers (18) for their dual functionality in
catalysis and self-assembly, as well as by Sleiman and co-workers for their “DNA-
mimetic” supramolecular aggregation properties yielding potentially functional
morphologies (19).
As will be detailed in the following section, polymer assemblies can be formed
very efficiently with moderate strength association constants
∼500–600 M
− 1
(
∼14.6 kJ/mol = 3.5 kcal/mol), especially when many of these interactions are
working cooperatively. These moderate strength interactions contribute to over-
all constructs that are quite dynamic and require minimal amounts of energy for
disassembly. These characteristics provide an elegant and potent means for con-
trolling solution-state polymer structure, and applications are envisioned ranging
in diversity including, but not limited to transport, encapsulation, delivery sys-
tems, sensors, devices, and other environmentally responsive materials.
428
MOLECULAR SELF-ASSEMBLY
Vol. 10
“Plug and Play” Polymers
As mentioned earlier, the recognition elements inducing main-chain supramolecu-
lar polymerizations appear in many varieties. Analogously, these assembling units
can also be effectively utilized as assembling units on polymer side chains. There
exist two methods of generating pendant side chains containing supramolecular
elements: (1) polymerizing or copolymerizing the monomer with a presynthesized
recognition moiety, and (2) polymerizing a monomer with an active site that does
not participate in the polymerization but is capable of post-polymerization modi-
fication. The latter scenario will be discussed throughout the rest of this article,
as it avoids issues of blockiness (nonrandom blocks of functionality due to cooper-
ativity during polymerization) and solubility associated with aggregation during
copolymerization processes.
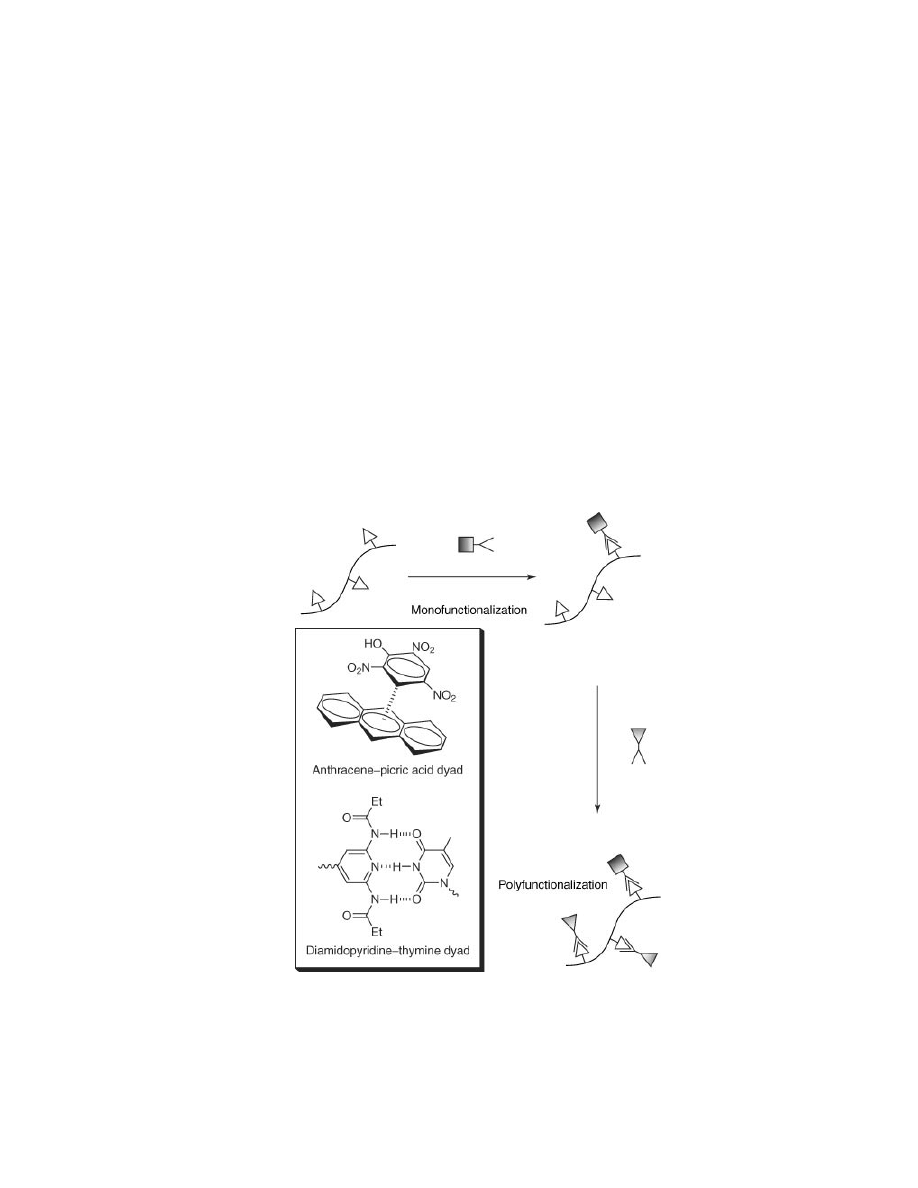
Supramolecular assembly is dynamic by nature. Further enhancing this
characteristic is the modular nature of noncovalent interactions that permit a
divergent approach to polymer structure, allowing multiple systems to be assem-
bled using a single polymer backbone (Fig. 2). These polymers provide flexible
backbones, are synthesized readily using free-radical–initiated polymerizations
Fig. 2.
Schematic representation of versatility of reversible, supramolecular side-chain
modification, and selected examples of interactions that can be employed (inset).
Vol. 10
MOLECULAR SELF-ASSEMBLY
429
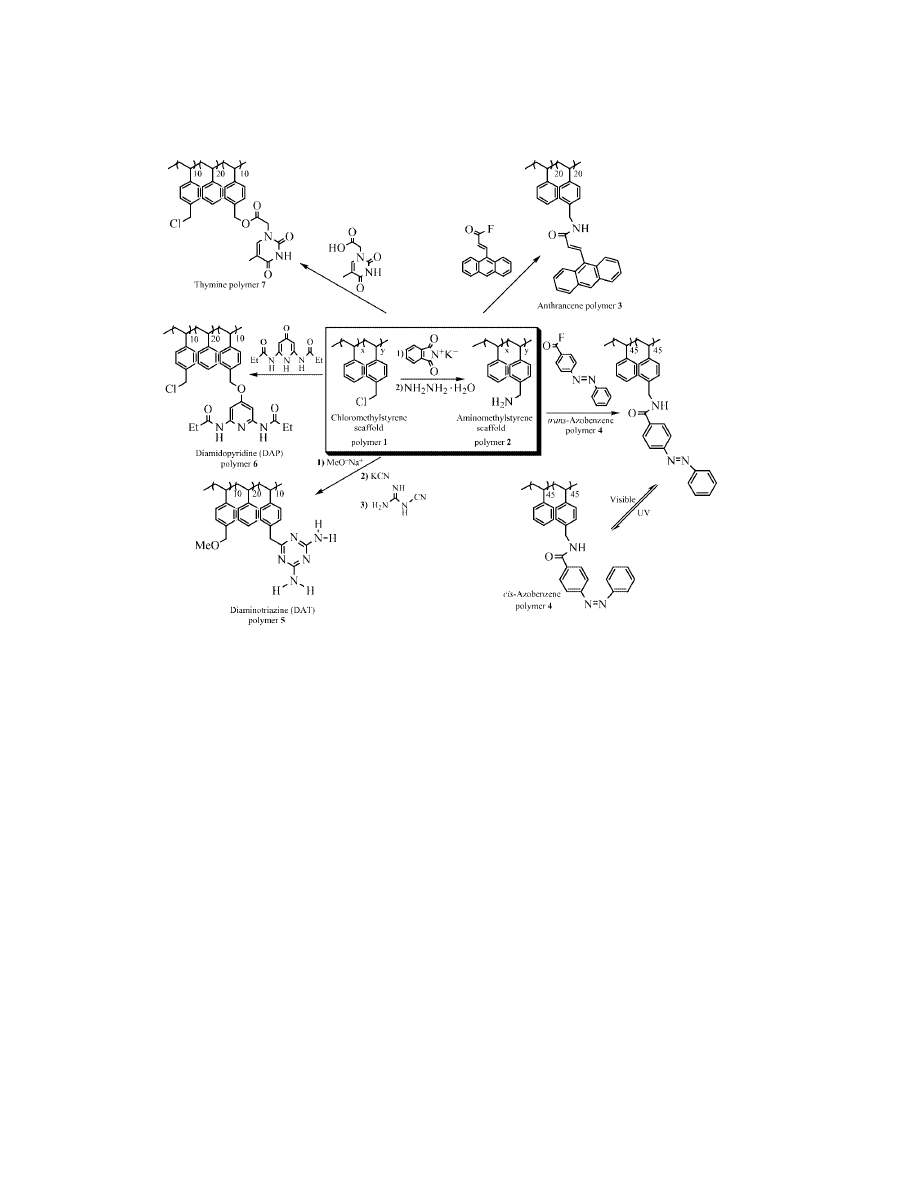
Fig. 3.
Schematic illustration of versatile polymer scaffolds providing a divergent ap-
proach to polymer structure.
in multigram scales, and contain numerous monomers with sites for facile substi-
tution. [Styrene and p-(chloromethyl)styrene polymerize with similar reactivity
rates; therefore the desired 1:1 monomer ratio was achieved by initiating the poly-
merization in the presence of a 1:1 molar ratio of monomers]. The versatility of this
polymer is demonstrated in Figure 3. For example, the chloromethyl sites can be
directly functionalized via nucleophilic substitution, or easily converted to amines
which react quantitatively with active esters, acid halides, or other electrophiles.
Our first investigations into polymer structural control were achieved utiliz-
ing aromatic stacking interactions. Employing polymers 3 and 4, obtained from
amine-functionalized polymer 2, anthracene and azobenzene moieties were in-
troduced quantitatively onto the styrene backbone. Anthracene polymer 3 adopts
a highly folded, compact globular structure in solution because of numerous fa-
vorable anthracene–anthracene and anthracene–benzene interactions. (20) Cap-
italizing on the electron-rich nature of the pendant anthracene side chains,
electron-deficient picric acid (Fig. 4a) was introduced into the system to induce a
swelling of the polymer structure. The increased electrostatic interaction between
the electron-rich anthracenes and electron-poor picric acid causes picric acid to
430
MOLECULAR SELF-ASSEMBLY
Vol. 10
intercalate between anthracenes, affecting a swelling of the polymer globule. The
hydrodynamic radius of the polymeric globule is dependent upon the concen-
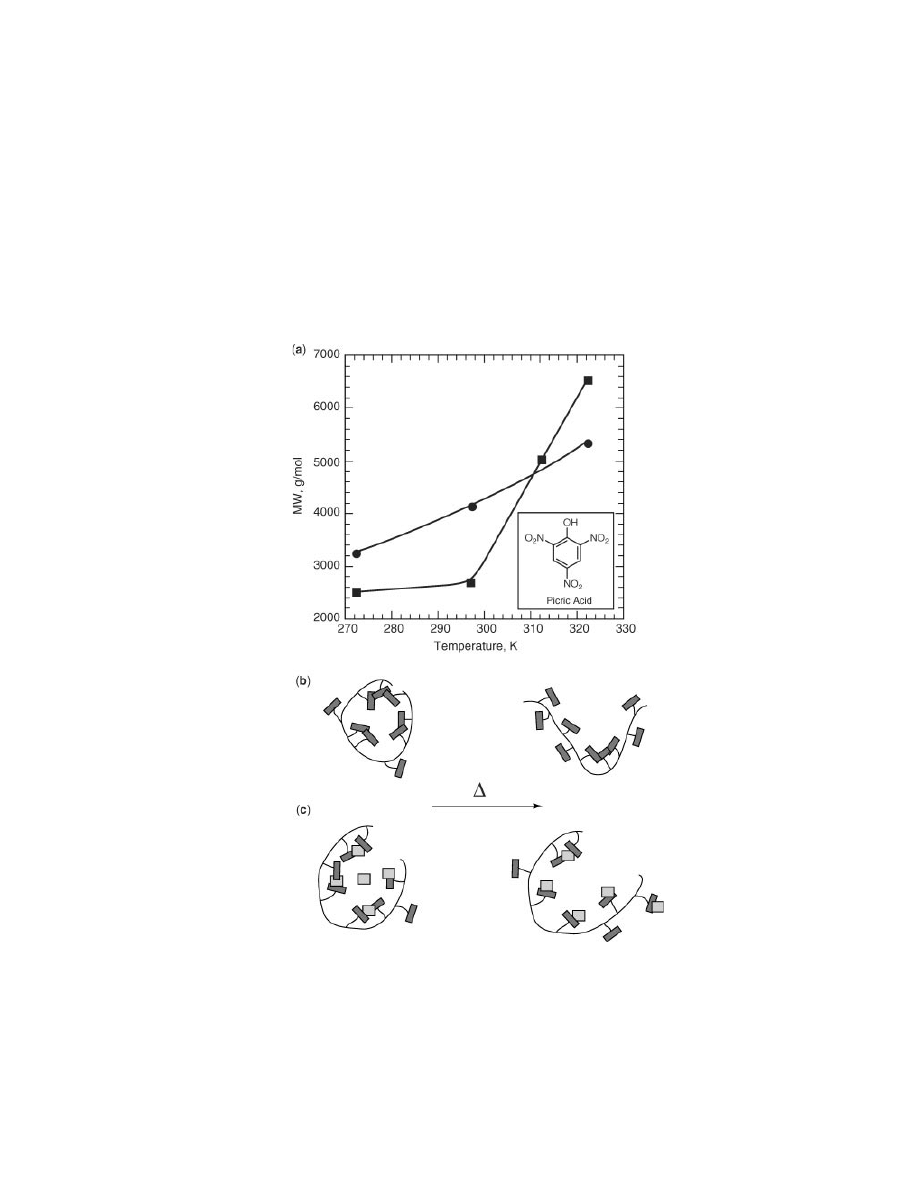
tration of picric acid, allowing for facile control over polymer size. Also, addi-
tion of picric acid causes this system to exhibit an increased thermal stability
due to increased electrostatic interactions between anthracene–picric acid over
anthracene–anthracene (Fig. 4).
Later, it was also demonstrated that pendant azobenzene units provide for
photochemical control over polymer structure (21). Azobenzene is an extremely
versatile photochrome; its structure can be controlled by the absorption of light,
Fig. 4.
Graphical and schematic illustrations of thermal unfolding process. (a) Variations
in relative sizes of polymer 3 with and without picric acid. Apparent molecular weight
obtained relative to polystyrene standards, (b) polymer 3, and (c) polymer 3 with picric
acid. Picric acid was introduced as component of elution solvent, 1.0
× 10
− 4
M.

Vol. 10
MOLECULAR SELF-ASSEMBLY
431
Fig. 5.
(a) Azobenzene polymer 4 and reversible photochemical behavior. (b) Molecular
dynamics simulation (300 K, CHCl
3
) of trans and cis forms of polymer 4.
often used for the creation of light-driven devices. (22) Photochemical isomeriza-
tion of the azobenzene chromophore (UV irradiation induces trans to cis isomeriza-
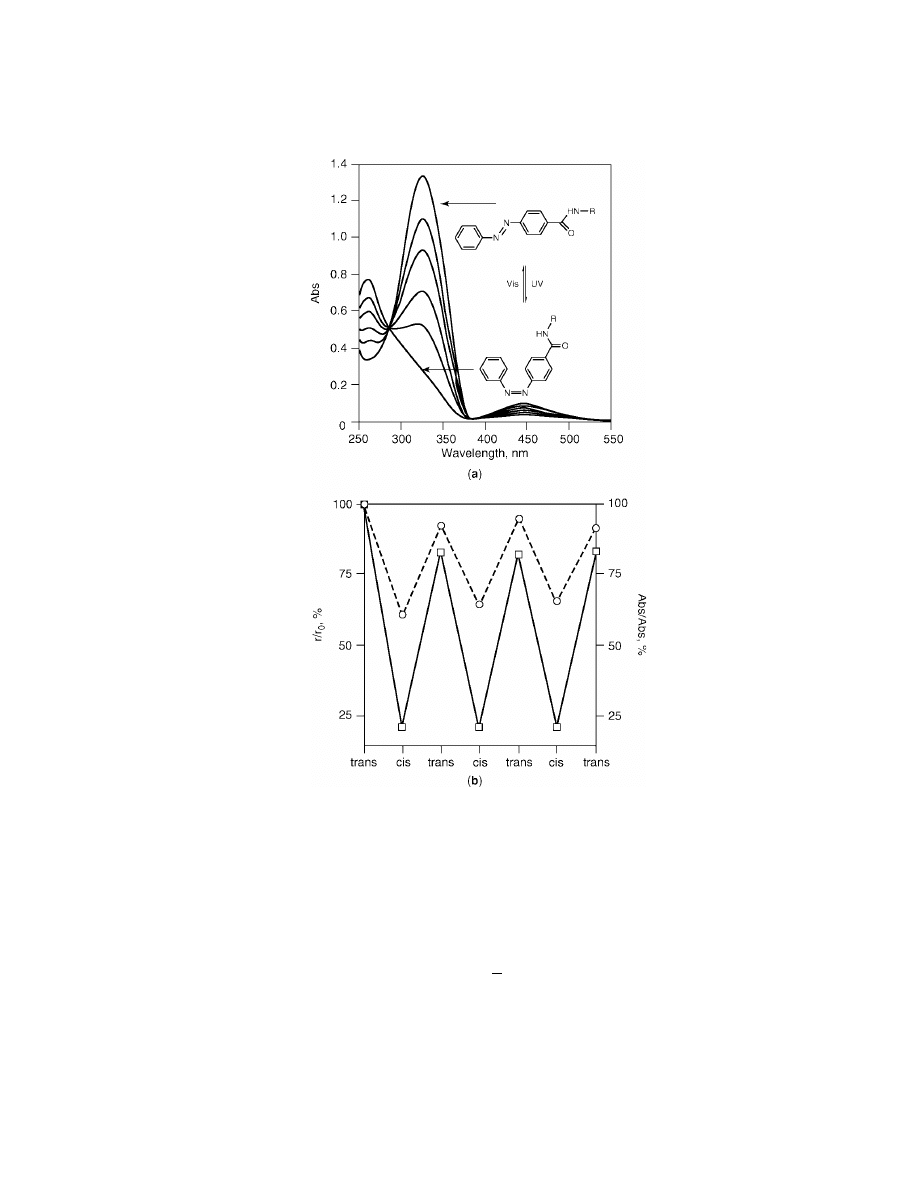
tion) causes the hydrodynamic radius to decrease as a result of the more compact
cis isomer allowing for a greater degree of side-chain–side-chain aromatic stack-
ing (Figs. 5 and 6). This isomerization process is reversible and directly linked to
the UV–vis behavior of azobenzene.
To extend the methodology and demonstrate the modularity of reversible
side-chain modification using specific noncovalent interactions, recognition el-
ements capable of three-point hydrogen bonding were attached to the polymer
scaffold. Diaminotriazine is a suitable recognition element for noncovalent side-
chain modification through its donor–acceptor–donor hydrogen bonding units.
However, attachment of triazine to the chloromethylstyrene scaffold (polymer
1), fashioning polymer 5, generates a polymer with a highly folded, micelle-like
structure in noncompetitive solvents such as CHCl
3
as a result of the multiple
modes of intrapolymeric hydrogen bonding between diaminotriazine moieties (23).
This compact macromolecular conformation creates a system that inefficiently
432
MOLECULAR SELF-ASSEMBLY
Vol. 10
Fig. 6.
(a) Absorption spectra variations of polymer 4 upon irradiation with UV and
visible lights, and (b) variations in the relative radius of gyration (solid line) and absorption
(dashed line) of 4 during irradiation.
complexes external guests because of competition between polymer–polymer and
polymer–guest interactions. However, this conformation is very attractive for en-
capsulation since site isolation is a major advantage in catalysis and data storage.
Specifically, ferroceneuracil 8 is able to penetrate the highly compact structure of
5 and bind multiple triazine units in the polar core (24). This guest is equipped
with an additional hydrogen bond donor, N H(1) in Figure 7, not in a linear array
with the three-point DAD motif, creating a “wraparound” effect that maximizes
favorable contacts between host and guest.

Vol. 10
MOLECULAR SELF-ASSEMBLY
433
Fig. 7.
Molecular dynamics runs (300 K, CHCl
3
) of polymers (a) 5 and (b) 6 (40 repeat
units each, schematically represented in Figure 3). Electroactive guest 8 and fluorescent
guest 11 employed to probe binding efficiencies of 5 and 6.
Efforts to provide more efficiency in guest complexation resulted in the
replacement of diaminotriazine with diacyldiamidopyridine. Diacyldiamidopyri-
dine is a highly analogous molecule containing the very same donor–acceptor–
donor motif as diaminotriazine; however, diacyldiamidopyridine self-associates to
a lesser extent resulting in a less-folded polymer capable of more efficient guest
complexation.
To probe the binding efficiencies with the polymers above, a guest,
isobutylflavin 11, was employed. Isobutylflavin 11 contains the complementary
acceptor–donor–acceptor recognition moiety required for specific complexation.
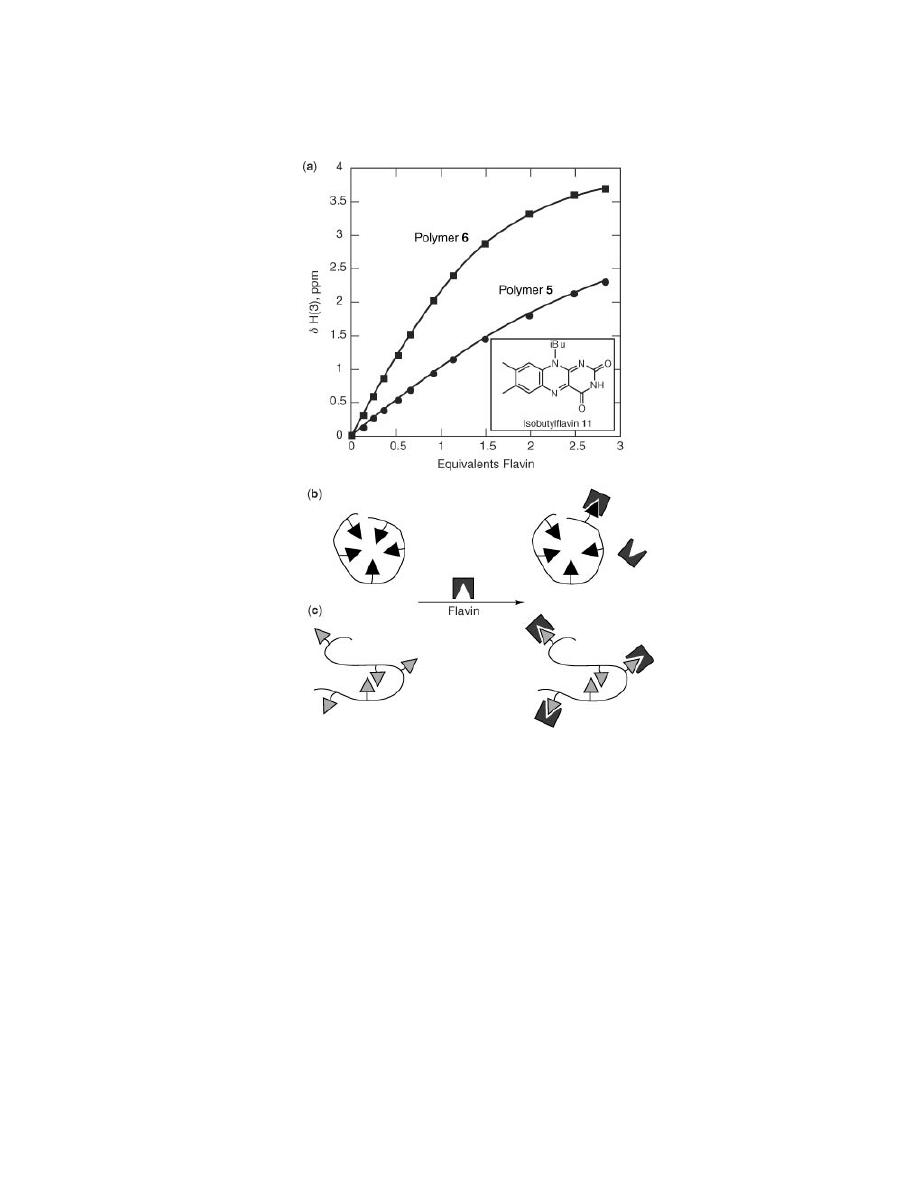
Figure 8a shows that there is a significant reduction in binding efficiency of the tri-
azine derivative compared to the diamidopyridine derivative. As previously men-
tioned, implicit in the bimolecular recognition of intrapolymerically associated
triazine polymers is a decrease in the efficiency of interpolymeric binding. This
inefficiency is explained by the requirement for polymer 5 to first uncoil prior to
the recognition event (Fig. 8b).
The previous paragraphs have outlined a few methodologies by which we
have demonstrated control over solution-state polymer structure by reversibly
434
MOLECULAR SELF-ASSEMBLY
Vol. 10
Fig. 8.
(a) Graphical representation of the downfield shift of the imide proton of polymer
11 upon introduction to polymers 5 and 6. Schematic representations of the recognition
behavior of (b) polymers 5 and (c) 6 with 11.
binding small guests to the pendant side chains of a flexible polymer scaffold or
through reversible interactions of the pendant side chains with themselves. As
such, this “plug and play” methodology has proven to be a very versatile approach
to supramolecular polymer engineering.
Formation of Highly Structured, Functional Microscale Systems
Reversible side-chain modification has been demonstrated to be quite useful and
very versatile at controlling the structure of single polymer strands in solution.
This approach can also be very effectively used for solution-state assembly of
Vol. 10
MOLECULAR SELF-ASSEMBLY
435
many polymer strands into higher order structures such as vesicles (25), micelles
(26), and gels (27). As specific molecular recognition processes provide a tool for
obtaining molecular-level control over higher order assembly processes (28), so too
will integration of the “lock and key” selectivity with the phase-separated, three-
dimensional nature of vesicular and micellar templates present a new opportunity
for the creation of systems featuring novel structural and dynamic properties.
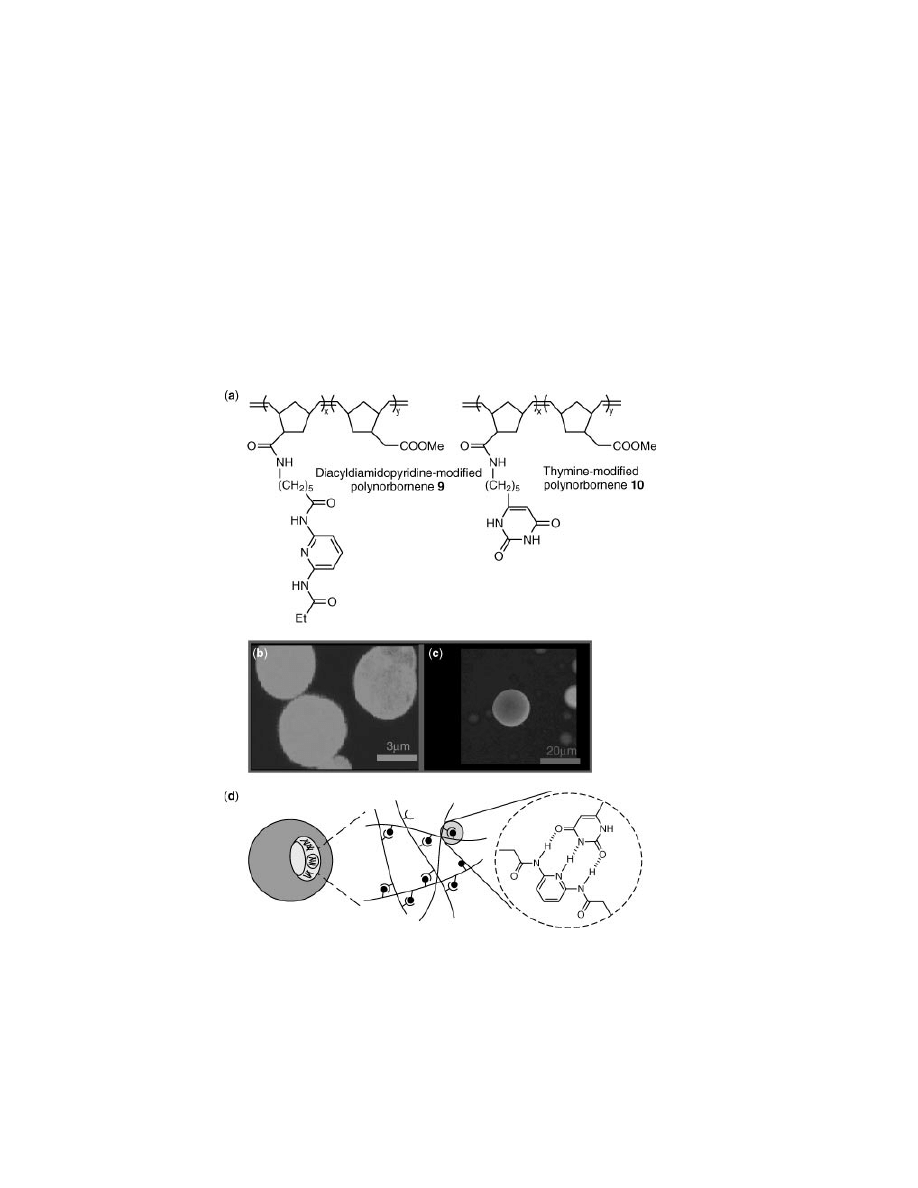
Combination of complementary diacyldiamidopyridine- and thymine-
functionalized polymers 6 and 7, respectively, in nonpolar media results
spontaneously in the formation of giant vesicular aggregates, or recognition-
induced polymersomes (RIPs). (Fig. 9) (29). In contrast to reports of di-
block copolymer-based polymersomes that rely upon solvophobicity and phase
Fig. 9.
(a) Random polynorbornene-based copolymers 9 and 10 that assemble into RIPs
analogous to copolymers 6 and 7. Laser confocal scanning micrographs of polymersomes
formed through combination of complementary polymers (b) 6 and 7, and (c) 9 and 10.
(d) Schematic illustration of specific interstrand hydrogen bonding driving polymersome
formation.
436
MOLECULAR SELF-ASSEMBLY
Vol. 10
segregation for self-assembly (18), these RIPs are formed through highly specific
interactions between the complementary pendant side chains (Fig. 9d) as the poly-
mers do not aggregate unless functionalized post-polymerization with recognition
elements. Interestingly, these highly unusual, ordered microstructures from dis-
ordered, random copolymers are not unique to flexible polystyrene backbones; the
more rigid polynorbornene scaffold, when derivatized with diacyldiamidopyridine
(polymer 9) and thymine (polymer 10), also self-assembles in nonpolar media into
RIPs (30) (Fig. 9b).
The formation of vesicular architectures from random copolymers lacking
well-defined headgroups was unprecedented, proving a new method for the cre-
ation of supramolecular assemblies. This mode of assembly provides a new tool
for the control of vesicle structure; complementary monovalent and multivalent
guests would be expected to distort or disrupt vesicle structure through competi-
tive binding to the polymer recognition sites (31). (see P
OLYMER
V
ESICLES
).
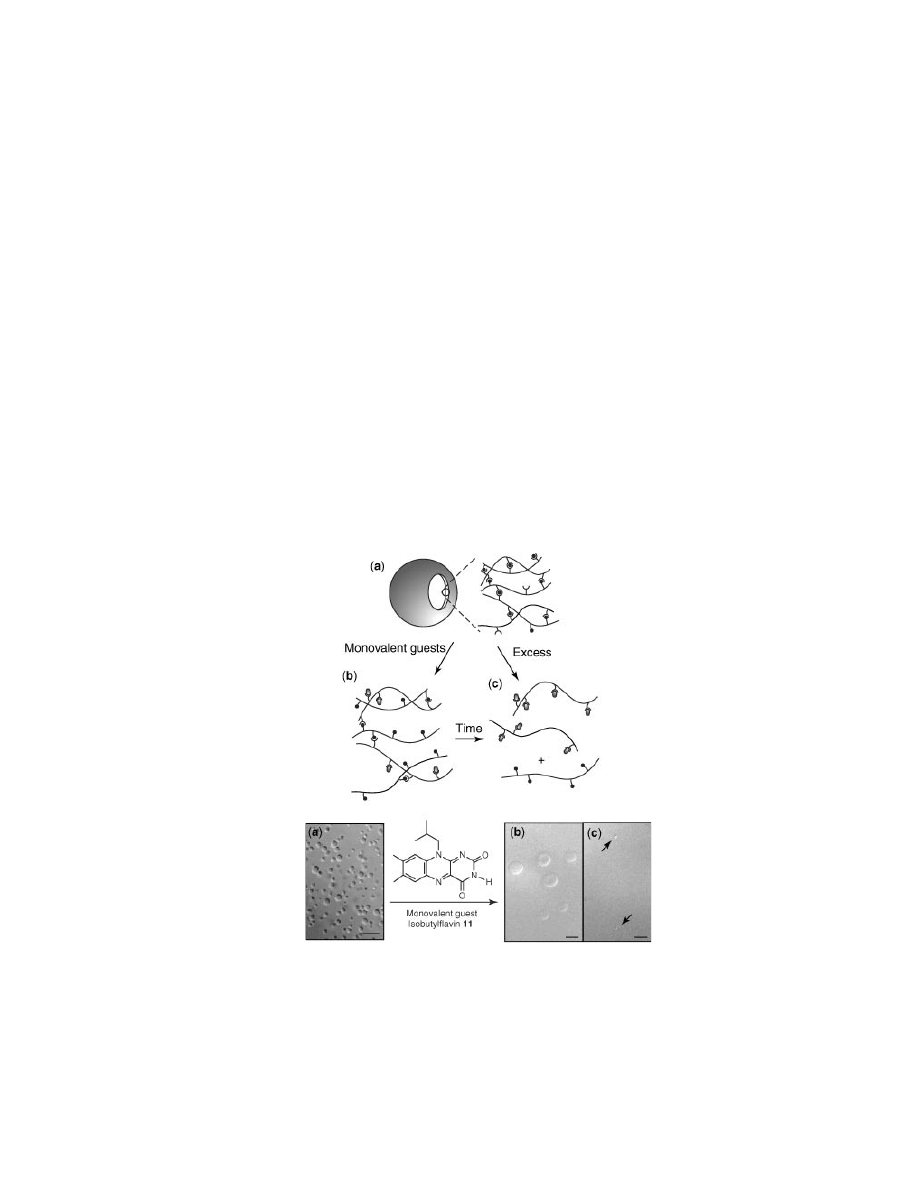
Initially, flavin was introduced into a solution of preformed RIPs to specifi-
cally and reversibly incorporate a fluorescent tag into the vesicular structure as
an extension of our plug and play approach to polymer side-chain modification. As
expected, the monodentate flavin caused a disruption in RIP structure appearing
as an immediate swelling upon contact (Fig. 10b). However, this result was briefly
lived as RIP diameter slowly returned to that of original, preformed RIPs (Fig. 11).
Fig. 10.
Differential interference contrast micrographs of RIP–guest interactions with
corresponding schematics: (a) native, undoped RIPs; (b) swollen RIPs 30 s after monovalent
guest 11 addition; and (c) equilibrated RIPs 50 min after addition of 11. Scale bars represent
10
µm.
Vol. 10
MOLECULAR SELF-ASSEMBLY
437
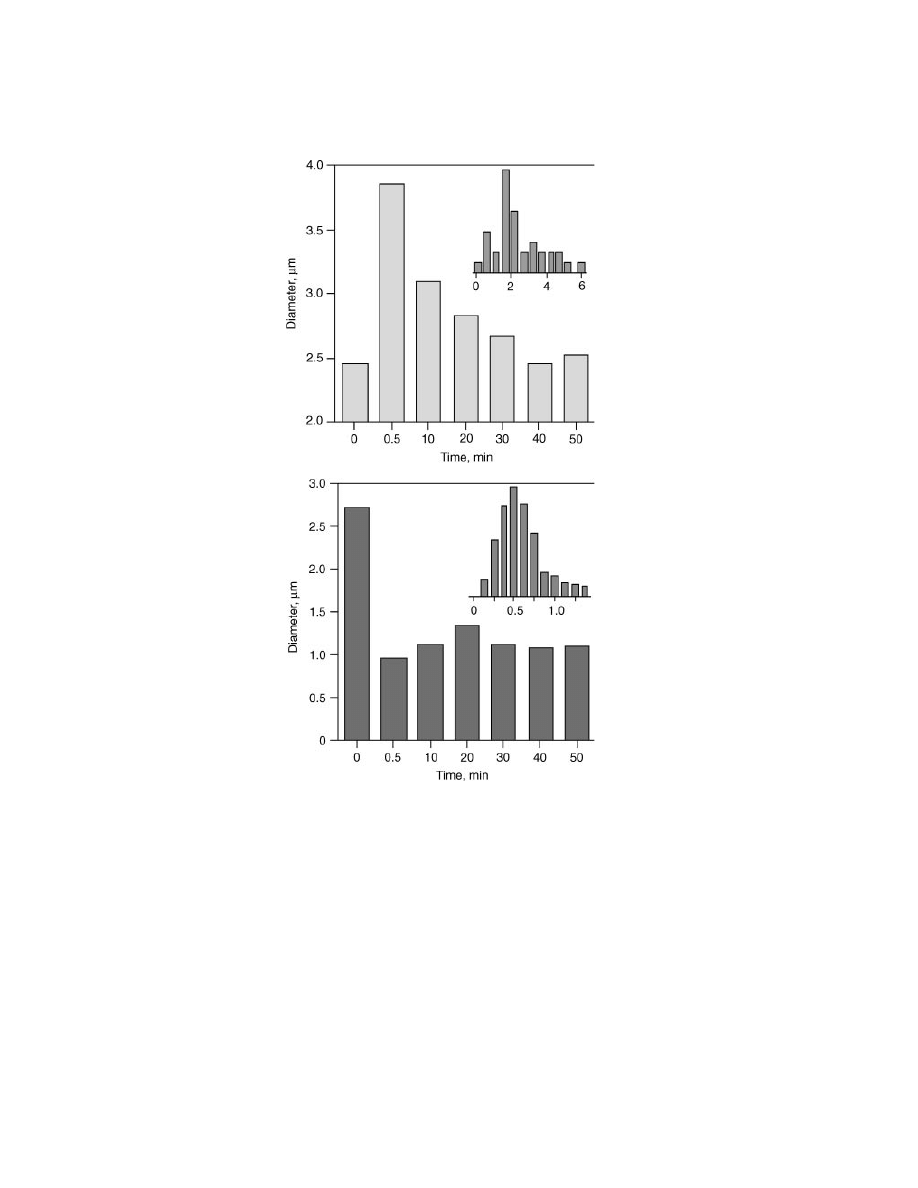
Fig. 11.
Graphical representations of size changes occurring upon introduction of mono-
and multivalent guests. Flavin (top) affects an immediate but temporary RIP swelling. Thy–
Au (bottom) constricts polymer strands within RIPs resulting in submicrometer constructs.
The insets on each graph are histograms of the equilibrated system 50 min after guest
addition.
Presumably, this is due to the dynamic nature of these specific recognition pro-
cesses. Flavin competitively binds with the DAP moiety of polymer 6; over time,
strands of polymer 6 become saturated with 11, causing these polymers to leave
the aggregate, in turn resulting in a return to original RIP size. Since RIP size
distribution (Fig. 11a) and average diameter return to that of original, undoped
RIPs firmly suggest that flavin is not incorporated into the final, equilibrated RIP
system.
438
MOLECULAR SELF-ASSEMBLY
Vol. 10
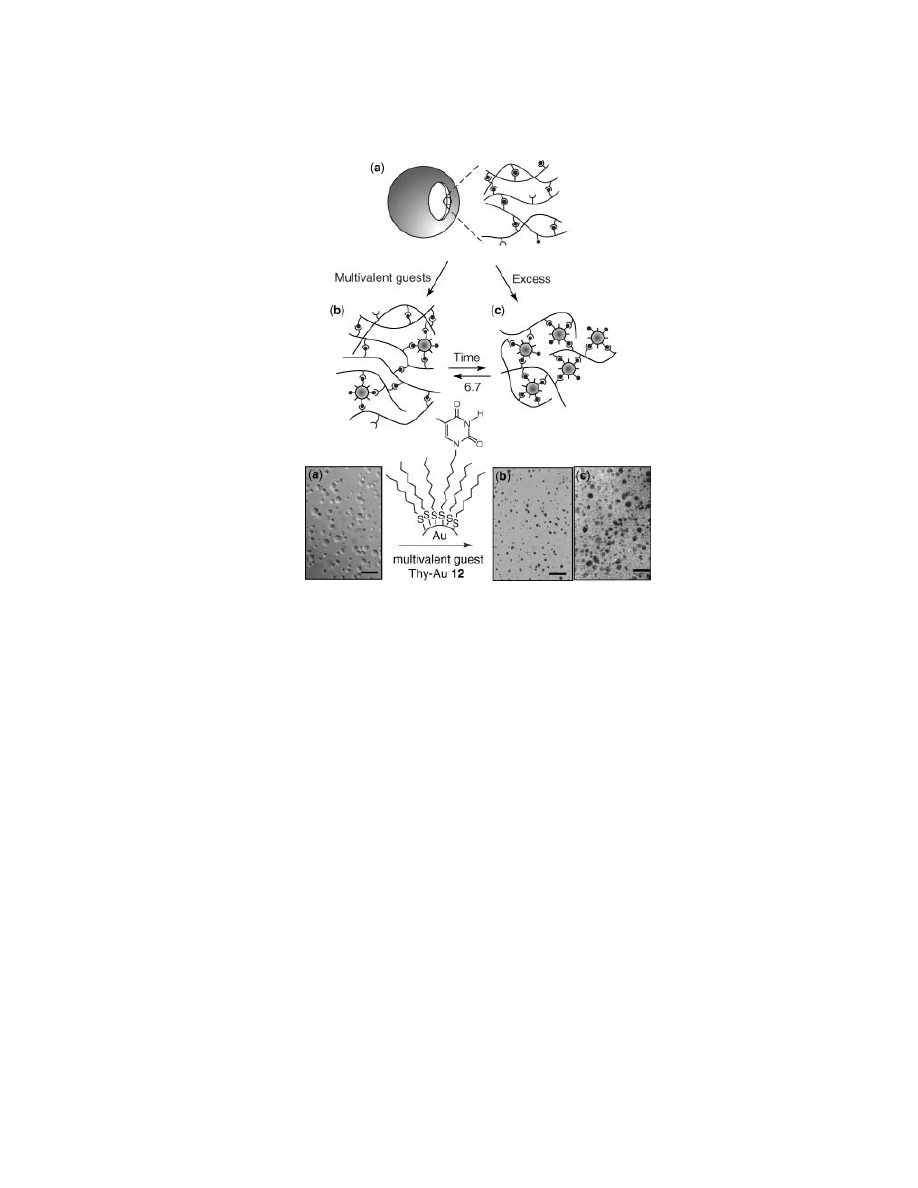
Fig. 12.
Differential interference contrast micrographs of RIP–guest interactions with
corresponding schematics: (a) native, undoped RIPs; (b) smaller RIPs 30 s after multivalent
guest 12 addition (scale bars represent 10
µm); (c) submicrometer polymer–nanoparticle
assemblies 50 min after Thy–Au addition. Scale bar represents 1
µm.
As already mentioned, multivalent guests were expected to act very differ-
ently with RIPs because of the greater number of contacts that, potentially, can
be formed between the multifunctional Thy–Au and RIP (Fig. 12). In previous
studies, we have demonstrated that analogous polymers interact specifically with
Thy–Au to form solid spherical assemblies (32). The gold nanoparticles are excel-
lent guests for these investigations because of their discrete structure, solubility in
nonaqueous media, facile place exchange of ligands, and desired multifunctional-
ity. From this, we predicted and confirmed that Thy–Au specifically embeds within
the RIP and then causes a major reorganization of the component polymers. This
process can followed qualitatively by phase contrast microscopy as RIPs are ini-
tially observed to be translucent in solution. The solution darkened upon addition
of an aliquot of Thy–Au, but the solution cleared very rapidly (
∼30 s), indicating
Thy–Au is rapidly incorporated into the RIP wall. However, this rapid incorpora-
tion was followed by an overall decrease in average diameter (Fig. 12b). Addition
of Thy–Au constricts the component polymers, resulting in an immediate decrease
in RIP diameter. However, the structures do not equilibrate back to original size,
as seen in the flavin system, but rather new self-assembled spheres on the sub-
micrometer scale forms.
Vol. 10
MOLECULAR SELF-ASSEMBLY
439
To demonstrate the specific nature of these observed effects, highly anal-
ogous N-methylated versions of guests were synthesized and employed in the
same capacity. The methyl groups cause only minimal changes in guest struc-
ture; however, these groups drastically change guest activity in solution as the
methyl group effectively disrupts three-point hydrogen bonding, making these
excellent molecules for control experiments. In both cases, no size changes or mor-
phological effects were observed, clearly indicating the specific nature of these
interactions.
Extension of this self-assembly approach to supramolecular engineering has
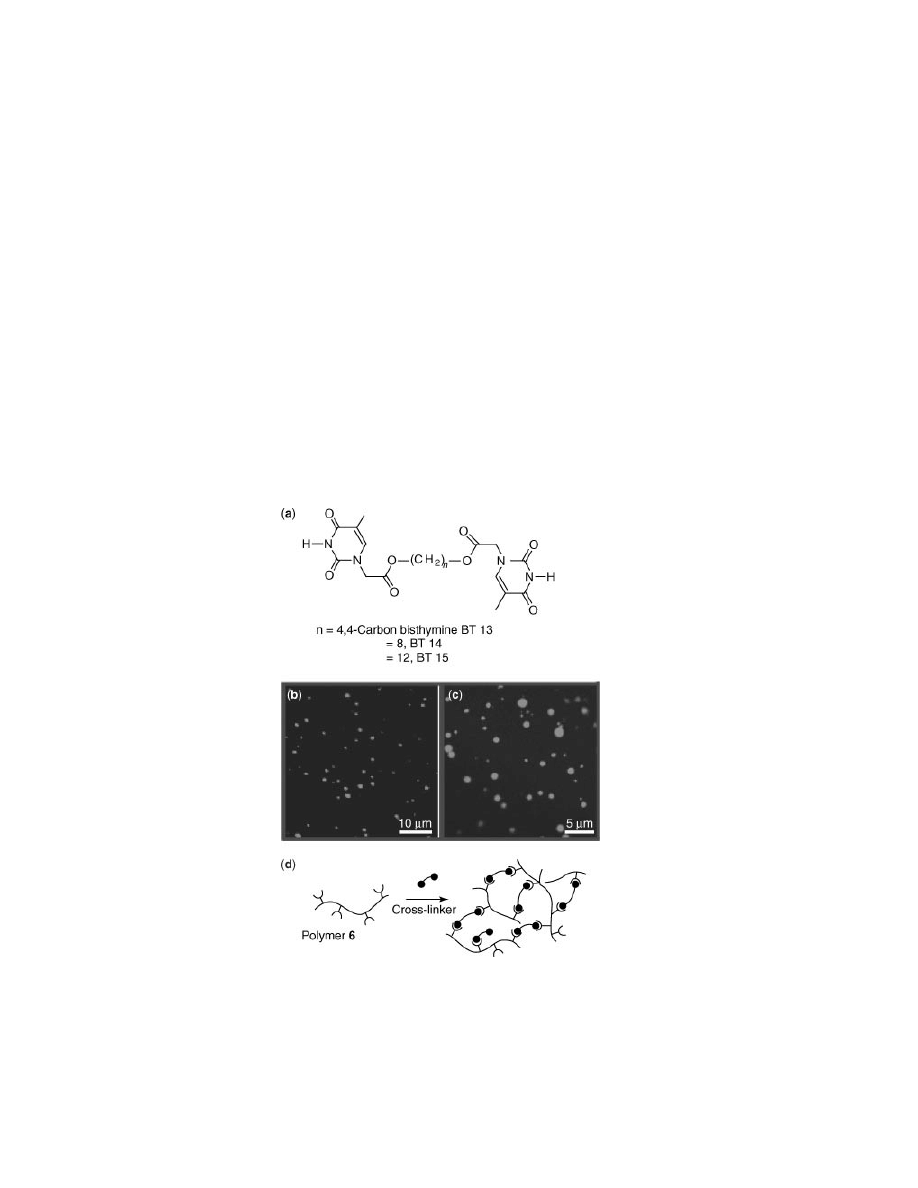
led to an alternate motif for noncovalent cross-linking, a series of bisthymines that
are complementary to the diamidopyridine side chains of polymer 6 (33). Upon
combination in non-polar media thermally reversible, micrometer scale spherical
aggregates were formed (Figs. 13b and 13c).
From the LCSM micrographs in Figure 13, the solid fluorescence profile of
these spherical assemblies is indicative of a filled sphere rather than the vesicu-
lar morphology. Moreover, it is apparent that the fluorescence emission tapers off
with microsphere radius; fluorescence is more intense in the microsphere center
Fig. 13.
(a) Bisthymine cross-linkers 13–15. (b,c) LCSM micrographs of microspheres
generated from combination of polymer 6 and bisthymine (BT) 16. (d) Schematic depiction
of noncovalent polymer cross-linking.
440
MOLECULAR SELF-ASSEMBLY
Vol. 10
indicating the presence of a microgel morphology. Also, the highly localized fluo-
rescence observed in the micrographs indicates that very little flavin-tagged poly-
mer exists free in solution, demonstrating the effectiveness of the cross-linking
process.
Fig. 14.
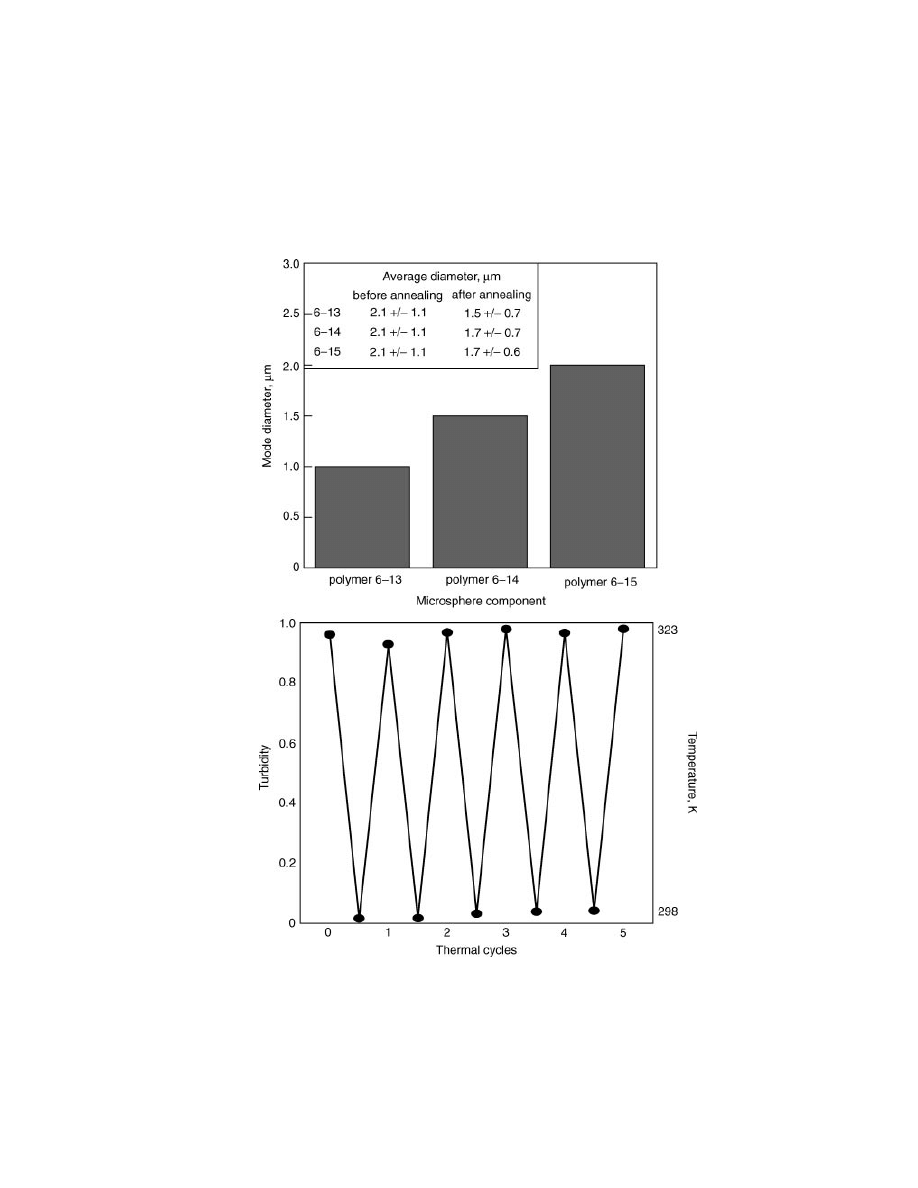
Graphical representations of increasing microsphere diameter with increasing
cross-linker length and average diameters (top) before and after thermal annealing cycles
(inset), and solution turbidity during annealing cycles (bottom).
Vol. 10
MOLECULAR SELF-ASSEMBLY
441
Interestingly, the overall size distribution of the resulting microspheres was
found to be dependent on cross-linker preorganization; varying the alkyl spacer
between thymines on the cross-linker translates into a stepwise growth in overall
diameter on the microscale (Fig. 14). In accord with self-assembled systems, these
microspheres could be thermally cycled, with complete disassociation of system
components at elevated temperatures. Although small variations in turbidity are
detected, microspheres after thermal annealing appear to retain less polydisperse,
smaller overall diameters. Presumably, this change in distribution is the result of
thermal cycling of the initial kinetic structures to thermodynamically more stable
aggregates.
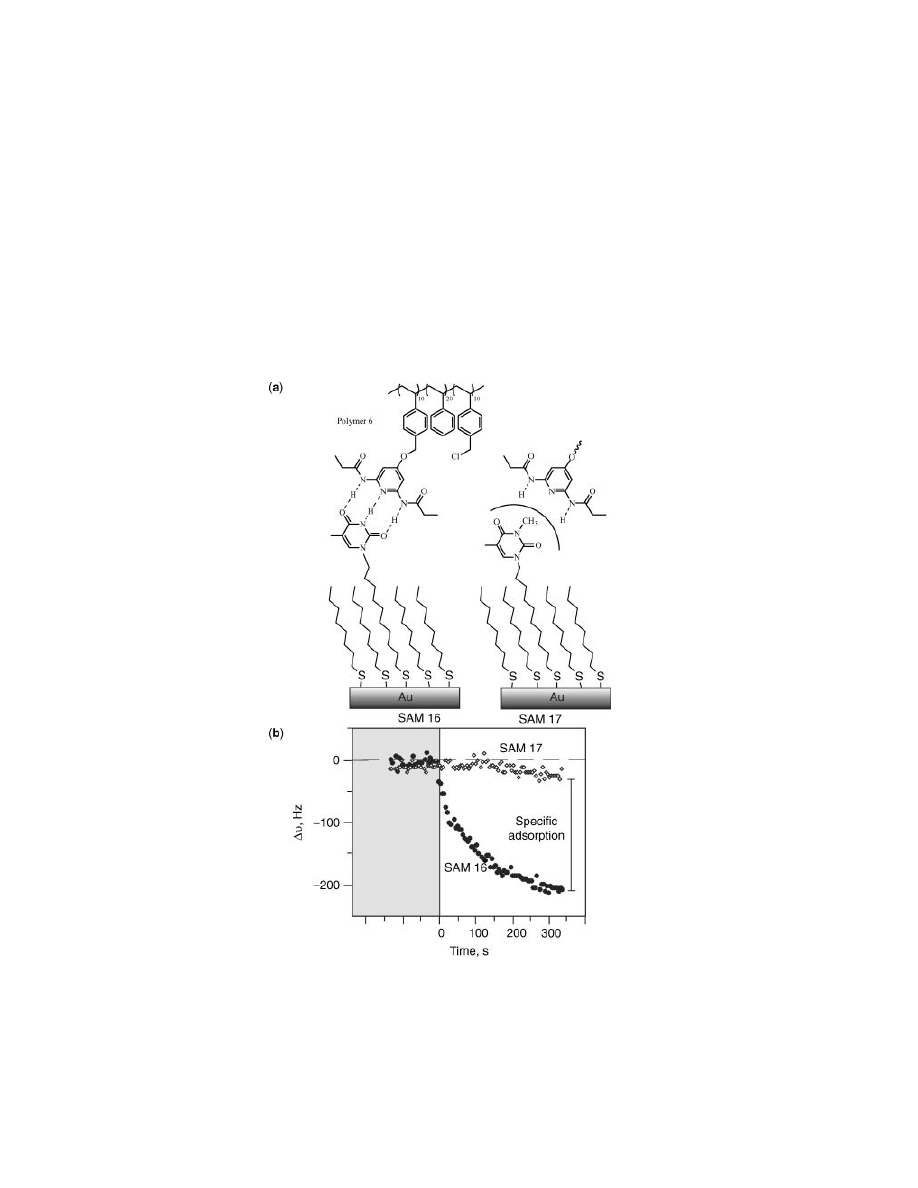
Fig. 15.
(a) Self-assembled monolayers (SAMs) featuring imide functionality 16 and N-
methylated control 17. (b) Graphical representation clearly illustrating the essential role
specific hydrogen bonding plays in polymer deposition.

442
MOLECULAR SELF-ASSEMBLY
Vol. 10
Surface Modification
The concepts of supramolecular chemistry are also well suited to interactions
at the solid–liquid interface. This provides an effective and efficient means of
reversibly controlling the physical and chemical properties of a surface. There
currently exist many well-established techniques for surface modification, such
as electrostatics, ionic coordination, chemisorption, and Langmuir–Blodgett de-
position; however, introducing specific hydrogen bonding interactions allows one
to enhance the interfacial specificity between the surface and adsorbate while
maintaining surface-charge neutrality, a prerequisite when designing materials
with biocompatibility. To this end, self-assembled monolayers (SAMs) allow the
incorporation of specific amounts of diverse functionality to investigate polymer
deposition using noncovalent interactions (Fig. 15) (34).
Quartz crystal microbalance (QCM) is a well-established technique for moni-
toring surface deposition as the natural frequency of quartz decreases when mass
is deposited onto the chip. This frequency change can be directly related to film
formation and can be monitored in real time. The QCM experiment in Figure
15b depicts this phenomenon and indicates that polymer is rapidly adsorbed onto
SAMs with the selectively expected from the DAP–Thy recognition dyad. These
data also provide a method for determining the varying contributions of adsorp-
tion from nonspecific or specific interactions. Any frequency change in the case
of N-methylated SAM 17 is due to nonspecific adsorption and can be subtracted
from the response observed with SAM 16 to isolate the contribution of specific
adsorption processes.
BIBLIOGRAPHY
1. L. J. Prins, D. N. Reinhoudt, and P. Timmerman, Angew. Chem., Int. Ed. 40, 2383–2426
(2001).
2. J. M. Lehn, Supramolecular Chemistry–Concepts and Perspectives, VCH, Weinheim,
1995.
3. R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, Jhkk Hirschberg, R. F. M.
Lange, J. K. L. Lowe, and E. W. Meijer, Science 278, 1601–1604 (1997); L. Brunsveld,
B. J. B. Folmer, E. W. Meijer, and R. P. Sijbesma, Chem. Rev. 101, 4071–4097 (2001);
A. Ciferri, Macromol. Rapid Commun. 23, 511–529 (2002);
J. S. Moore, Curr. Opin.
Colloid Interface Sci. 4, 108–116 (1999);
J. Huff, J. A. Preece, and J. F. Stoddart,
Macromol. Symp. 102, 1–8 (1996);
V. Percec, J. Heck, G. Johansson, D. Tomazos,
M. Kawasumi, and G. Ungar, J. Macromol. Sci., Pure Appl. Chem. A31, 1031–1070
(1994);
H. A. Klok, J. J. Hwang, J. D. Hartgerink, and S. I. Stupp, Macromolecules
35, 6101–6111 (2002);
R. K. Castellano, D. M. Rudkevich, and J. Rebek Jr., Proc.
Natl. Acad. Sci. U.S.A. 94, 7132–7137 (1997);
R. K. Castellano, K. Clark, S. L.
Craig, C. Nuckolls, and J. Rebek Jr., Proc. Natl. Acad. Sci. U.S.A. 97, 12418–12421
(2000).
4. R. P. Sijbesma and E. W. Meijer, Chem Commun. 5–16 (2003); D. C. Sherrington and
K. A. Taskinen, Chem. Soc. Rev. 30, 83–93 (2001).
5. D. H. Zhao and J. S. Moore, Macromolecules 36, 2712–2720 (2003);
S. Tsuzuki,
K. Honda, and R. Azumi, J. Am. Chem. Soc. 124, 12200–12209 (2002);
D. J. Hill
and J. S. Moore, Proc. Natl. Acad. Sci. U.S.A. 99, 5053–5057 (2002); J. D. Hartgerink,

Vol. 10
MOLECULAR SELF-ASSEMBLY
443
E. R. Zubarev, and S. I. Stupp, Curr. Opin., Solid State Mater. Sci. 5, 355–361 (2001);
V. Berl, I. Huc, R. G. Khoury, M. J. Krische, and J. M. Lehn, Nature 407, 720–723
(2000).
6. B. G. G. Lohmeijer and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem. 41, 1413–
1427 (2003);
K. J. Calzia and G. N. Tew, Macromolecules 35, 6090–6093 (2002);
Y. Chujo, K. Sada, and T. Saegusa, Macromolecules 26, 6320–6323 (1993); Y. Chujo,
K. Sada, and T. Saegusa, Macromolecules 26, 6315–6319 (1993).
7. A. D. Stroock, R. S. Kane, M. Weck, S. J. Metallo, and G. M. Whitesides, Langmuir 19,
2466–2472 (2003); J. Slager and A. J. Domb, Adv. Drug Deliv. Rev. 55, 549–583 (2003);
A. A. Louis, Philos. Trans. R. Soc. London, Ser. A 359, 939–960 (2001); E. Pefferkorn,
J. Coll. Interface Sci. 216, 197–220 (1999);
J. Li, L. Wang, Y. H. Ying, and Y. Yang,
J. Acta Chim. Sin. 60, 1700–1706 (2002).
8. J. M. Lehn, Science 260, 1762–1763 (1993);
J. M. Lehn, Makromol. Chem. Macro-
mol. Symp. 69, 1–17 (1993);
J. M. Lehn, Polym. Int. 51, 825–839 (2002);
V. Berl,
M. Schmutz, M. J. Krische, R. G. Khoury, and J. M. Lehn, Chem. Eur. J. 8, 1227–1244
(2002).
9. T. Gulikkrzywicki, C. Fouquey, and J. M. Lehn, Proc. Natl. Acad. Sci. U.S.A. 90, 163–167
(1993).
10. M. Kotera, J. M. Lehn, and J. P. Vigneron, Tetrahedron 51, 1953–1972 (1995);
M. Kotera, J. M. Lehn, and J. P. Vigneron, J. Chem. Soc. Chem. Commun. 2, 197–199
(1994).
11. H. W. Gibson, L. Hamilton, and N. Yamaguchi, Polym. Adv. Technol. 11, 791–797
(2000);
K. Yamauchi, J. R. Lizotte, D. M. Hercules, M. J. Vergne, and T. E. Long,
J. Am. Chem. Soc. 124, 8599–8604 (2002); K. Yamauchi, J. R. Lizotte, and T. E. Long,
Macromolecules 35, 8745–8750 (2002);
E. A. Archer, N. T. Goldberg, V. Lynch, and
M. J. Krische, J. Am. Chem. Soc. 122, 5006–5007 (2000);
K. M. Chen, X. P. Jiang,
L. C. Kimerling, and P. T. Hammond, Langmuir 16, 7825–7834 (2000);
T. H. Galow,
A. K. Boal, and V. M. Rotello, Adv. Mater. 12, 576–579 (2000).
12. S. C. Zimmerman and L. J. Lawless, Supramolecular Chemistry of Dendrimers,
Vol. 217, Springer-Verlag, Berlin, 2001, pp. 95–120.
13. R. F. M. Lange and E. W. Meijer, Macromolecules 28, 782–783 (1995);
M. Born and
H. Ritter, Makromol. Chem. Rapid Commun. 12, 471–476 (1991); H. Ritter, Macromol.
Symp. 77, 73–38 (1994); C. B. St. Pourcain and A. C. Griffin, Macromolecules 28, 4116–
4121 (1995).
14. R. Stadler and J. Burgert, Makromol. Chem. Macromol. Chem. Phys. 187, 1681–1690
(1986); C. Hilger and R. Stadler, Polymer 32, 3244–3249 (1991); C. Hilger, M. Drager,
and R. Stadler, Macromolecules 25, 2498–2501 (1992);
C. Hilger and R. Stadler,
Macromolecules 25, 6670–6680 (1992).
15. L. R. Rieth, R. F. Eaton, and G. W. Coates, Angew. Chem., Int. Ed. Engl. 40, 2153–2156
(2001).
16. T.
Kato,
Molecular
Self-Assembly,
Vol.
96,
Springer-Verlag,
Berlin,
2000,
pp. 95–146.
17. T. Kato and J. M. J. Frechet, J. Am. Chem. Soc. 111, 8533–8534 (1989);
T. Kato,
H. Kihara, U. Kumar, T. Uryu, and J. M. J. Frechet, Angew. Chem., Int. Ed. Engl. 33,
1644–1645 (1994);
T. Kato, H. Kihara, T. Uryu, A. Fujishima, and J. M. J. Frechet,
Macromolecules 25, 6836–6841 (1992);
T. Kato, Kobunshi Ronbunshu 54, 855–862
(1997).
18. J. M. Pollino and M. Weck, Synthesis (Stuttgart) 1277–1285 (2002); J. M. Pollino and
M. Weck, Org. Lett. 4, 753–756 (2002).
19. H. S. Bazzi and H. F. Sleiman, Macromolecules 35, 9617–9620 (2002);
J. Dalphond,
H. S. Bazzi, K. Kahrim, and H. F. Sleiman, Macromol. Chem. Phys. 203, 1988–1994
(2002).

444
MOLECULAR SELF-ASSEMBLY
Vol. 10
20. F. Ilhan, M. Gray, K. Blanchette, and V. M. Rotello, Macromolecules 32, 6159–6162
(1999).
21. G.
Clavier,
F.
Ilhan,
and
V.
M.
Rotello,
Macromolecules
33,
9173–9175
(2000).
22. M. S. Vollmer, T. D. Clark, C. Steinem, and M. R. Ghadiri, Angew. Chem., Int. Ed. Engl.
38, 1598–1601 (1999); I. Willner, D. Rubin, R. Shatzmiller, and T. Zor, J. Am. Chem.
Soc. 115, 8690–8694 (1993);
M. Ueda, N. Fukushima, K. Kudo, and K. Ichimura,
J. Mater. Chem. 7, 641–645 (1997).
23. F. Ilhan, M. Gray, and V. M. Rotello, Macromolecules 34, 2597–2601 (2001).
24. T. H. Galow, F. Ilhan, G. Cooke, and V. M. Rotello, J. Am. Chem. Soc. 122, 3595–3598
(2000).
25. B. M. Discher, Y. Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher, and
D. A. Hammer, Science 284, 1143–1146 (1999);
B. M. Discher, D. A. Hammer,
F. S. Bates, and D. E. Discher, Curr. Opin. Coll. Interface Sci. 5, 125–131 (2000);
F. Checot, S. Lecommandoux, H. A. Klok, and Y. Gnanou, Eur. Phys. J. 10, 25–35 (2003);
H. Kukula, H. Schlaad, M. Antonietti, and S. Forster, J. Am. Chem. Soc. 124, 1658–1663
(2002); F. H. Meng, C. Hiemstra, G. H. M. Engbers, and J. Feijen, Macromolecules 36,
3004–3006 (2003).
26. S. E. Webber, J. Phys. Chem. B 102, 2618–2626 (1998); K. L. Wooley, J. Poly. Sci., Part
A: Polym. Chem. 38, 1397–1407 (2000);
K. B. Thurmond, H. Y. Huang, C. G. Clark,
T. Kowalewski, and K. L. Wooley, Coll. Surf. B: Biointerfaces 16, 45–54 (1999); H. Y.
Huang, E. E. Remsen, and K. L. Wooley, Chem. Commun. 1415–1416 (1998); Y. Zheng,
Y. Y. Won, F. S. Bates, H. T. Davis, L. E. Scriven, and Y. Talmon, J. Phys. Chem. B 103,
10331–10334 (1999).
27. M. Annaka and T. Tanaka, Nature 355, 430–432 (1992);
A. P. Nowak, V. Breedveld,
L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan, and T. J. Deming, Nature 417, 424–428
(2002); Y. Osada and J. P. Gong, Adv. Mater. 10, 827–837 (1998); S. Varghese, A. K.
Lele, D. Srinivas, M. Sastry, and R. A. Mashelkar, Adv. Mater. 13, 1544–1548 (2001).
28. T. A. Taton, C. A. Mirkin, and R. L. Letsinger, Science 289, 1757–1760 (2000);
T. H.
Galow, A. K. Boal, and V. M. Rotello, Adv. Mater. 12, 576–579 (2000).
29. F. Ilhan, T. H. Galow, M. Gray, G. Clavier, and V. M. Rotello, J. Am. Chem. Soc. 122,
5895–5896 (2000).
30. U. Drechsler, R. J. Thibault, and V. M. Rotello, Macromolecules 35, 9621–9623 (2002).
31. R. J. Thibault, T. H. Galow, E. T. Turnberg, M. Gray, P. H. Hotchkiss, and V. M. Rotello,
J. Am. Chem. Soc. 124, 15249–15254 (2002).
32. A. K. Boal, F. Ilhan, J. E. DeRouchey, T. Thurn-Albrecht, T. P. Russell, and V. M. Rotello,
Nature 404, 746–748 (2000).
33. R. J. Thibault, P. Hotchkiss, M. Gray, and V. M. Rotello, J. Am. Chem. Soc. 125,
11249–11252 (2003).
34. T. Norsten, E. Jeoung, R. J. Thibault, and V. M. Rotello, Langmuir 19, 7089–7093
(2003).
R
AYMOND
J. T
HIBAULT
V
INCENT
M. R
OTELLO
Department of Chemistry, University of Massachusetts
MOLECULARLY IMPRINTED POLYMERS.
See Volume 7.
MORPHOLOGY.
See Volume 7.
Wyszukiwarka
Podobne podstrony:
assembler
Molecular Toxicology 8
Assembler ENG
Molecular evolution of FOXP2, Nature
Assembly Language for Kids Commodore 64 Addendum
P000718 A Eng Vertical shaft assembly
M001882 B Eng Lower assembly
mb star c3 self test manual
arm assembly
bushwarbler assembly
Ch18 Assemble Complex Models
P000724 A Eng Lower assembly
assembler model, Programowanie
ARTICLE BRAKES PEDAL ASSEMBLY SERVICE
Atmel AVR Assembler id 71678 Nieznany (2)
SELF ASSESSMENT SHEET
Assembler Intel Code Table
więcej podobnych podstron