
1
Ćwiczenie 4
PREPARATYKA DNA Z JĄDER KOMÓRKOWYCH IZOLOWANYCH
Z ETIOLOWANYCH SIEWEK PSZENICY
Część doświadczalna obejmuje:
−
izolowanie jąder komórkowych z etiolowanych siewek pszenicy
−
preparatykę DNA z wyizolowanych jąder
−
ocenę czystości preparatu DNA na podstawie analizy spektrofotometrycznej
WPROWADZENIE
Izolacja jąder komórkowych
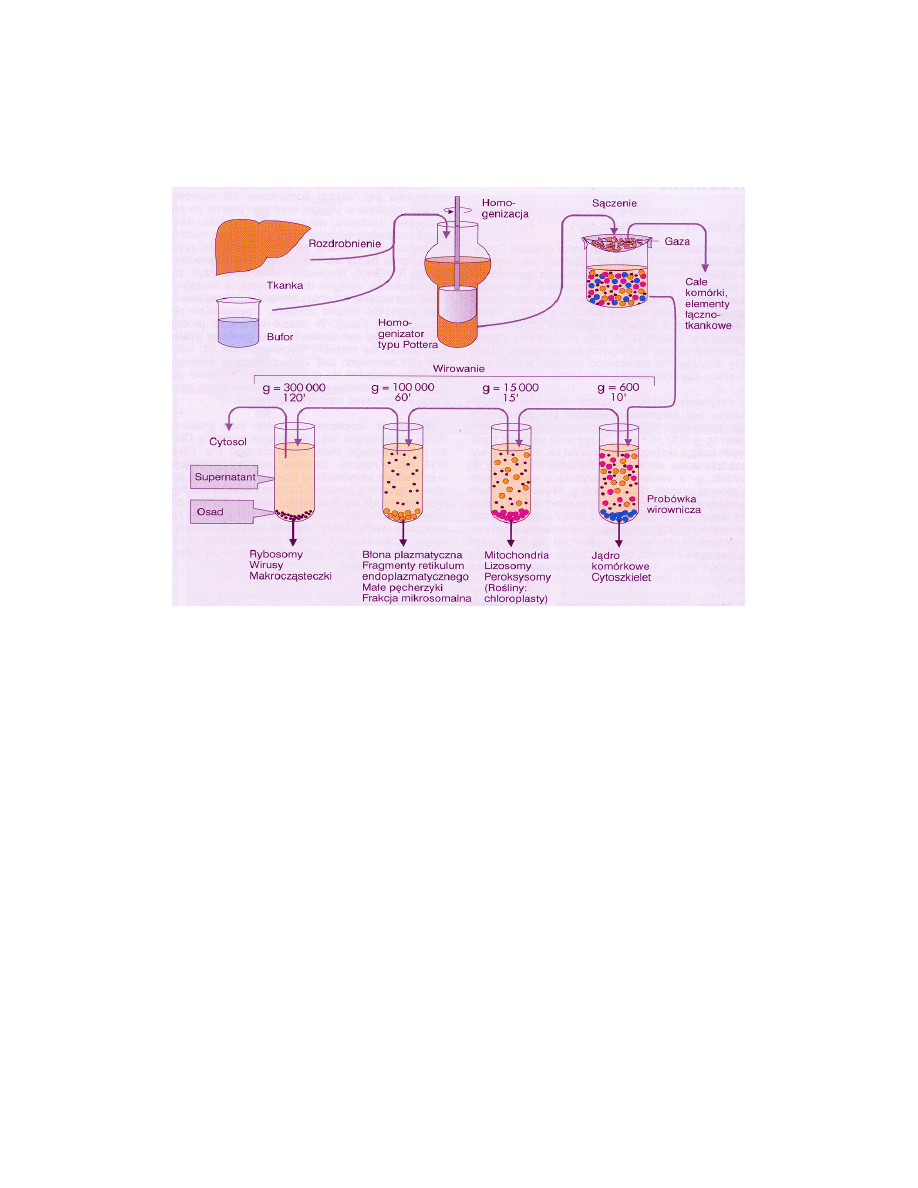
Wyodrębnienie poszczególnych struktur subkomórkowych rozpoczyna się od roz-
drobnienia badanego materiału i homogenizacji (roztarcia komórek) w odpowiednim środo-
wisku (roztworze homogenizacyjnym). W pozyskiwaniu nieuszkodzonych organelli używa
się roztworu izotonicznego tzn. takiego, którego ciśnienie osmotyczne jest równe ciśnieniu
osmotycznemu wewnątrz komórki. W roztworach hipotonicznych organelle wchłaniają wodę
i pękają, natomiast w roztworach hipertonicznych kurczą się.
Homogenizacja tkanki umożliwia zgrubną filtrację homogenatu przez gazę lub nylon,
pozwalającą oddzielić nieroztarte, całe komórki czy fragmenty tkanek. Frakcjonowanie
składników komórkowych można przeprowadzić w drodze tzw. wirowania różnicowego
polegającego na kolejnych wirowaniach przy odpowiednio wzrastających przyspieszeniach
podawanych jako wielokrotność przyspieszenia ziemskiego wywołanego przyciąganiem
ziemskim (g = 9,81 m x s
-2
). Ze względu na zróżnicowaną wielkość, kształt i gęstość po-
szczególnych organelli można je w ten sposób wyodrębnić z zawiesiny (Ryc. 1).
Jądra komórkowe osadzają się już przy małych przyspieszeniach (600 x g), które
można osiągnąć w prostych wirówkach stołowych. Przez ostrożne usunięcie roztworu znajdu-
jącego się nad osadem (supernatantu) i zawieszenie osadu jąder w izotonicznym roztworze
można uzyskać frakcję wzbogaconą w dużą liczbę jąder komórkowych. Taka „surowa” frak-
cja jądrowa zawiera jeszcze inne komponenty komórkowe jako „zanieczyszczenia”. Oczysz-
czoną frakcję jądrową można otrzymać na drodze wirowania w skokowym lub ciągłym gra-
diencie gęstości np. roztworu sacharozy lub glicerolu. W tym wypadku, rozdzielające się w
2
czasie wirowania frakcje są stabilizowane przez gradienty gęstości środowiska, które hamują
przepływy konwekcyjne utrudniające rozdzielanie cząstek.
Ryc. 1. Schemat frakcjonowania struktur subkomórkowych metodą wirowania różnicowego
(Koolman, Röhm 2005)
Przy frakcjonowaniu struktur subkomórkowych duże znaczenie ma analiza stopnia
czystości otrzymanych frakcji. Obecność określonych organelli w wyizolowanej frakcji moż-
na potwierdzić oznaczając w niej aktywność odpowiednich enzymów markerowych lub in-
nych cząsteczek znacznikowych. Np. DNA jest znacznikiem jąder komórkowych, rRNA –
rybosomów, a mitochondria można identyfikować oznaczając dehydrogenazę bursztynianową
lub oksydazę cytochromową. Stopień jednorodności izolowanej frakcji można określić ozna-
czając w niej aktywność enzymów lub zawartość cząsteczek znacznikowych charakterystycz-
nych dla innych frakcji subkomórkowych.
Poszczególne etapy izolacji organelli komórkowych wykonuje się w niskich tempera-
turach (przeważnie przy 0-5
0
C) spowalniając w ten sposób reakcje trawienia przez uwolnione
enzymy proteolityczne oraz ograniczając inne niekorzystne procesy np. utlenianie grup hy-
drosulfidowych w białkach.
3
Preparatyka DNA z izolowanych jąder komórkowych
Organizacja genomów roślinnych
Genom roślin składa się z trzech części: genomu jądrowego, genomu mitochondrial-
nego i genomu chloroplastowego. Genom jądrowy zawarty jest w chromosomach, których
liczba u różnych organizmów waha się od kilku do kilkudziesięciu. Genomy organelli są w
większości koliste, chociaż niektóre doniesienia sugerują, że obok genomów kolistych koeg-
zystują wersje liniowe, a w przypadku chloroplastów mniejsze fragmenty koliste, które zawie-
rają część całego genomu. Genomy organelli mogą występować pojedynczo lub, jak to
stwierdzono w mitochondriach ludzkich, nawet w 10, a w przypadku innych organizmów
nawet w 100 kopiach w jednym mitochondrium.
Genomy jądrowe
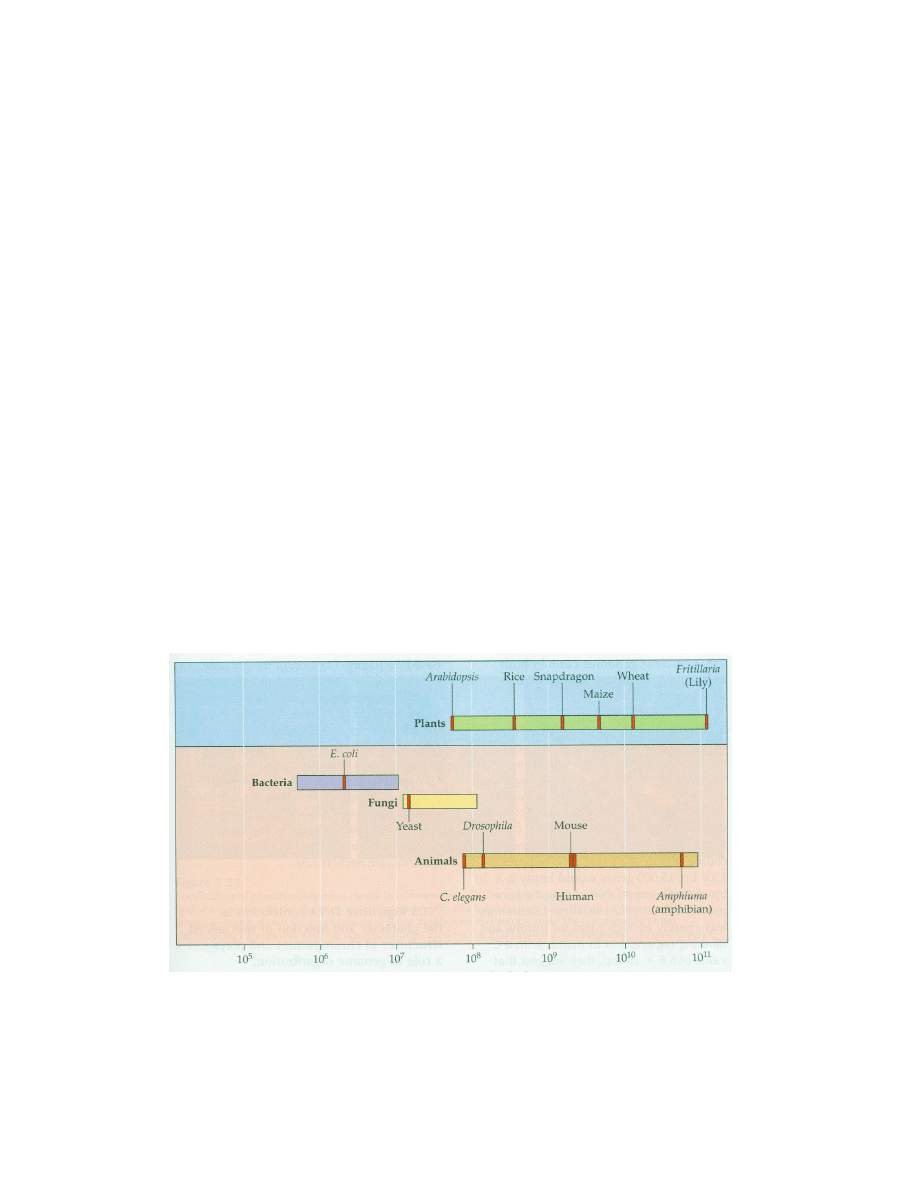
Na Ryc. 2 pokazano zakres wielkości haploidalnych genomów jądrowych roślin
oraz, dla porównania, rozpiętość wielkości genomów bakterii, grzybów i zwierząt. Wielkość
genomów jądrowych roślin wyższych jest niezwykle zróżnicowana, gdyż najmniejszy genom
rzodkiewnika (Arabidopsis thaliana) zawarty w pięciu chromosomach liczy 7x 10
7
par zasad
(70 Mpz), a największy genom szachownicy (Fritillaria assyriaca) ma 1,2x 10
11
par zasad
(120 000 Mpz).
pz
Ryc. 2. Porównanie wielkości haploidalnych genomów bakterii, grzybów, roślin i zwierząt
(Buchanan, Gruissem, Jones 2000).

4
Na Ryc. 3 pokazany jest pokrój rzodkiewnika (A. thaliana), rośliny o najmniejszym
genomie i szachownicy (F. assyriaca), rośliny z rodziny liliowych, której genom jest około
1700x większy od genomu rzodkiewnika. Porównując wielkość genomów różnych organi-
zmów zwraca uwagę brak bezpośredniego związku między wielkością genomu, a pozio-
mem komplikacji organizmu.
Ryc. 3. Porównanie wyglądu: A, rzodkiewnika (Arabidopsis thaliana) (genom jądrowy – 70
Mpz) i B, szachownicy (Fritillaria assyriaca) (genom – 20 000 Mpz) (Buchanan, Gruissem,
Jones, 2000)
Brak dokładnej korelacji pomiędzy złożonością organizmu i wielkością jego genomu określa-
ny jest mianem paradoksu wartości C. Obecnie już wiemy, że w genomach mniej złożo-
nych organizmów (drożdże, C. elegans), przestrzeń jest wykorzystywana oszczędniej, gdyż
ich geny leżą bliżej siebie. Inaczej jest w przypadku genomu kukurydzy (Zea mays) (6 600
Mpz) zawartego w 10 chromosomach i genomu pszenicy (Triticum aestivum) (17 000 Mpz),
które są odpowiednio ponad dwu- i prawie sześciokrotnie większe od genomu ludzkiego. Ge-
nomy jądrowe tych roślin zdominowane są przez elementy powtarzające się, które obejmują
cztery główne rodzaje rozproszonych sekwencji powtarzających się: długie rozproszone se-
kwencje jądrowe LINE (ang. long interspersed nuclear elements); krótkie rozproszone se-
kwencje jądrowe SINE (ang. short interspersed nuclear elements); długie powtórzenia koń-
cowe LTR (ang. long terminal repeats) i transpozony DNA zawierające sekwencje każdego
z pozostałych typów.
Genom A. thaliana, małego chwastu z rodziny krzyżowych, zawiera około 70 000 kilo
par zasad (70 Mpz) w przeliczeniu na haploidalny garnitur chromosomów. Genom ten jest
tylko około sześciokrotnie większy od genomu drożdży i piętnastokrotnie większy od genomu
E. coli
. Szacuje się, że rzodkiewnik ma około 26 500 genów. Liczba genów u pozostałych

5
roślin, wg aktualnych poglądów, jest podobna, a podstawowa różnica między rzodkiewnikiem
a innymi roślinami polega na tym, że u rzodkiewnika około 80% całkowitego jądrowego
DNA stanowią sekwencje występujące w jednej lub w kilku kopiach i są to w większości se-
kwencje kodujące białka. Niemal całkowity brak w genomie rzodkiewnika rozproszonych
sekwencji powtarzających się ułatwia znajdowanie genów, co m. in. zdecydowało o tym, że
ten mały chwast stał się modelową rośliną w biologii molekularnej roślin.
Genomy organellarne roślin
Wielkość genomów mitochondrialnych jest zróżnicowana i niepowiązana ze stop-
niem złożoności organizmu. Genom mitochondrialny człowieka, liczący 16 569 par zasad
(16,5 kpz), znacząco odbiega wielkością od genomu mitochondrialnego roślin, który u rzod-
kiewnika liczy 367 kpz, 570 kpz u kukurydzy, a u melona aż 2 500 kpz. Wielkość genomu
chloroplastowego różnych gatunków roślin jest zbliżona i mieści się w przedziale od 120 do
160 kpz (około 200 genów). Przyjmując, że komórka roślinna ma przeciętnie od 200 do nawet
3000 mitochondriów i od kilkudziesięciu do kilkuset chloroplastów, to całkowita ilość DNA
w tych organellach jest porównywalna z ilością DNA jądrowego. Np. zakładając, że komórka
rzodkiewnika zawiera 500 mitochondriów i 50 chloroplastów, a genomy w tych organellach
występują tylko w pojedynczych kopiach, to całkowita ilość organellarnego DNA w 1 komór-
ce będzie wynosić około 189 500 kpz (189,5 Mpz) (183 500 kpz w mitochondriach i 6000 kpz
w chloroplastach), co znaczy, że jest niemal 1,5x większa od ilości DNA zawartego w jądrze
każdej komórki diploidalnej (2 x 70 Mpz).
Preparatyka DNA
W preparatyce DNA wykorzystuje się zdolność białek i cukrowców do tworzenia nie-
rozpuszczalnych kompleksów z dodecylosiarczanem potasu, co umożliwia rozdzielenie tych
związków od kwasów nukleinowych. Ponadto, użycie w ostatnim etapie oczyszczania 0,3 M
octanu sodowego i 40% izopropanolu pozwala na oddzielenie DNA o wysokiej masie czą-
steczkowej od pozostałych w roztworze cukrowców oraz pozwala na uzyskanie DNA w po-
staci długich włókien, które łatwo można przemyć i wysuszyć w alkoholu etylowym. Otrzy-
many w tej procedurze wielkocząsteczkowy DNA jest dobrym substratem dla większości en-
zymów restrykcyjnych używanych w biologii molekularnej i może być wykorzystany do ana-
lizy struktury genomu roślinnego. Metoda ta pozwala na preparatykę jądrowego DNA bez
ultrawirowania preparatu w gradiencie CsCl. Szybkość oczyszczania oraz niewielka ilość
materiału potrzebna do izolacji DNA (mniej niż 1 g tkanki) daje możliwość analizy tego

6
związku w bardzo wczesnym stadium rozwoju rośliny. Nieskomplikowana procedura pozwa-
la na jednoczesną analizę dużej ilości prób. Metodę można stosować do izolacji DNA z ko-
mórek większości roślin.
MATERIAŁ I METODY
Odczynniki:
A.
Homogenizacja tkanki
1.
Bufor do homogenizacji (BH): 50 mM Tris-HCl pH 7,5 zawierający 0,3 M sacharozę,
5mM MgCl
2
i 3 mM CaCl
2
2.
Bufor do ekstrakcji (BE): 100 mM Tris-HCl pH 8.0 zawierający 50 mM EDTA i 500
mM NaCl
B.
Denaturacja białek
1.
20% SDS (siarczan dodecylu sodu)
2.
5 M octan potasu
C.
Wytrącanie DNA
Bufor (B1): 50 mM Tris-HCl, pH 8.0 zawierający 10 mM EDTA
3 M octan sodu
Izopropanol
D.
Oczyszczanie DNA
1.
Bufor (B2): 10 mM Tris-HCl, pH 8.0 zawierający 1 mM EDTA
Materiał:
Siewki pszenicy rosnące 5 dni na podłożu z trocin w temperaturze 25-27
0
C w całkowitej
ciemności.
WYKONANIE
A. Homogenizacja tkanki i izolacja jąder komórkowych
5g świeżo ściętych etiolowanych siewek pszenicy homogenizować ręcznie w wychło-
dzonym moździerzu wyłożonym nylonem i gęstą nylonową siatką z 30 ml roztworu homoge-
nizacyjnego o następującym składzie: 50 mM bufor Tris-HCl pH 7,5, 0,3 M sacharoza, 3 mM
CaCl
2
i 5 mM MgCl
2
(6 ml roztworu na 1 g tkanki). Po przesączeniu homogenatu przez
cztery warstwy gazy, całą objętość rozlać do dwóch probówek wirówkowych, które następnie

7
należy dokładnie zrównoważyć. Zrównoważone probówki umieścić w wirówce MPW-350 i
wirować 10 minut przy 700 x g (2200 obr/min). W tych warunkach w osadzie otrzymamy
„surową” frakcję jądrową wzbogaconą w jądra komórkowe. Uzyskaną frakcje jąder ko-
mórkowych możemy dalej oczyszczać na drodze wirowania przez skokowy gradient gęsto-
ści zgodnie z procedurą opisaną poniżej.
UWAGA! Wykonywane ćwiczenie nie obejmuje oczyszczania jąder
Oczyszczanie jąder z„surowej” frakcji jądrowej
Osad uzyskany po wirowaniu przy 700xg zawiesza się w niewielkiej objętości (1 ml) buforu
homogenizacyjnego, a następnie wolno nawarstwia na umieszczony w probówkach wirówko-
wych roztwór 0,3 M sacharozy i 40% glicerolu przygotowany w 10 mM buforze Tris-HCl, pH
7,5 zawierający dodatkowo 10 mM KCl, 10 mM MgCl
2
i 10 mM 2-merkaptoetanol. Wirowa-
nie „surowej” frakcji jądrowej przez skokowy gradient gęstości umożliwia usunięcie większo-
ś
ci „zanieczyszczeń”, które pozostają w nadsączu i osadzenie frakcji jąder komórkowych o
stosunkowo wysokiej „czystości”. Po ostrożnym zebraniu nadsączu, osad jąder zawiesza się
w odpowiednim środowisku, którego skład zależy od tego do jakich celów będą wykorzystane
wyizolowane jądra. Ryc. 4 przedstawia zdjęcie z mikroskopu świetlnego i mikroskopu fluore-
scencyjnego frakcji jąder uzyskanych z etiolowanych koleoptyli owsa.
Ryc. 4. Jądra komórkowe izolowane z etiolowanych siewek owsa. A – obraz z mikroskopu
ś
wietlnego, B – obraz z mikroskopu fluorescencyjnego (Hetmann – Praca doktorska 2005)

8
B. Preparatyka DNA z izolowanych jąder komórkowych
Wytrącanie białek z ekstraktu jądrowego
1.
Osad „surowej” frakcji jąder uzyskany po wirowaniu przy 700 x g zawiesić w 5 ml buforu
ekstrakcyjnego (BE) o następującym składzie: 100 mM Tris-HCl, pH 8,0, 50 mM EDTA
i 500 mM NaCl. Po dokładnym rozprowadzeniu osadu, uzyskaną zawiesinę jąder podzielić
na 5 porcji o objętości 1 ml (każda para ćwiczeniowa pobiera po 1ml zawiesiny do pro-
bówki Eppendorfa).
2.
Do 1 ml zawiesiny dodać 70 µµµµl 20% roztworu SDS, dobrze wytrząsnąć i umieścić na 10
min w łaźni wodnej o temp. 65
0
C.
3.
Do próby dodać 300 µµµµl 5 M roztworu octanu potasu, dokładnie wytrząsnąć, a następnie
probówkę umieścić na 10 min w lodzie.
4.
Zdenaturowane białka oddzielić wirując próbę 10 min przy 17 500 x g (15 000 obr/min)
Wytrącanie DNA
1.
Supernatant uzyskany po odwirowaniu zdenaturowanych białek przenieść do suchej pro-
bówki Eppendorfa, a następnie dodać 600 µ
µ
µ
µl wychłodzonego do –20
0
C izopropanolu.
2.
Probówkę dokładnie wytrząsnąć, a następnie umieścić na 10 min w zamrażarce w tempera-
turze –20
0
C. Strącający się w tych warunkach DNA osadzić wirując próbę 10 min przy
17 500 x g (15 000 obr/min) .
3.
Pipetą ściągnąć ostrożnie supernatant znajdujący się nad osadem DNA (UWAGA! Na-
leży to robić bardzo uważnie, tak żeby nie poruszyć osadu DNA). W tym celu najpierw
dokładnie obejrzeć dno probówki, znaleźć osad DNA, a następnie koniec pipety włożyć
na dno probówki obok osadu DNA.
4.
Resztki supernatantu usunąć pozostawiając na jakiś czas probówkę z osadem DNA na
bibule filtracyjnej odwróconą do góry dnem. (UWAGA! Nie wytrząsać resztek roztwo-
ru, gdyż można odkleić osad DNA od ścianki probówki).

9
Oczyszczanie DNA
1.
Osad DNA rozpuścić w 0,7 ml roztworu B1 zawierającego: 50 mM bufor Tris-HCl, pH
8,0 i 10 mM EDTA.
2.
Nierozpuszczony materiał odwirować w mikrowirówce (wirować 3 min).
3.
Ściągnąć ostrożnie pipetą supernatant i przenieść go do suchej probówki Eppendorfa, a
następnie dodać 75 µ
µ
µ
µl octanu sodu i 500 µ
µ
µ
µl izopropanolu schłodzonego do –20
0
C. Pro-
bówkę starannie wytrząsnąć, a następnie strącający się DNA odwirować w mikrowirów-
ce (wirować 5 min).
4.
Uzyskany osad DNA rozpuścić w 100 µµµµl roztworu B2 (10 mM bufor Tris-HCl, pH 8,0
zawierający 1 mM EDTA).
Analiza spektrofotometryczna uzyskanego preparatu DNA – ocena czystości preparatu
1.
Do suchej probówki Eppendorfa pobrać 50 µl otrzymanego roztworu DNA i dodać 950
µl roztworu B2 (10 mM bufor Tris-HCl, pH 8,0 zawierający 1 mM EDTA) (roztwór DNA
zostanie rozcieńczony 20-krotnie).
2.
W spektrofotometrze Schimadzu UV-160A wyznaczyć widmo absorpcyjne w zakresie
240 – 350 nm, a następnie mierząc absorbancję przy 260 nm i 280 nm wyznaczyć współ-
czynnik K
A
(A260 nm /A280 nm ). Jako próby odniesienia używać roztworu B2 (10
mM bufor Tris-HCl, pH 8.0 zawierający 1 mM EDTA). Wartość współczynnika K
A
określa czystość preparatu DNA. DNA o wysokiej czystości charakteryzuje się wartością
K
A
zbliżoną do 2.
Zagadnienia do przygotowania:
−
budowa chemiczna i struktura DNA i RNA
−
genomy jądrowe i organellarne
−
budowa chromatyny organizmów eukariotycznych
Literatura:
Biochemia – JM Berg, JL Tymoczko, L Stryer PWN, Warszawa, 2005

10
Genomy – TA Brown PWN, Warszawa 2001
Biochemistry & Molecular Biology of Plants – BB Buchanan, W Gruissem, RL Jones, Ame-
rican Society of Plant Physiologist, Rockville, Maryland, 2000
Wyszukiwarka
Podobne podstrony:
Ocena ilo ciowa i jako ciowa preparat w DNA i RNA
(), Biochemia L, sprawozdanie Preparacja DNA z grasicy cielęcej (ćw J)
Elektroforeza DNA komórkowego BioAut1, BioAut2 i Ch1
Sprawozdanie z izolacji genomowego DNA z komórki roślinnej
IG.4 - Uszkodzenia i naprawa DNA w komórkach nowotworowych, Genetyka, Inżynieria genetyczna
DNA, Medycyna, Biologia medyczna, 1) Genetyka 1 i komórka
Replikacja DNA w komórkach prokariotycznych
preparatyka plazmidowego DNA
Elektroforeza DNA komórkowego BioAut1, BioAut2 i Ch1
Niszczący DNA czip będzie wbudowywany w telefony komórkowe
Niespodzianki w szczepionkach Świński wirus DNA i komórki nerek małpy
Dr Berrenda Vox przedstawia materiał dowodowy o DNA i zmianach na poziomie komórkowym w wywiadzie ud
Komórkowe usługi EDGE
Replikacja DNA i choroby związane
Cw 1 ! komorki
więcej podobnych podstron