
1
Ćwiczenie 3
Metody oznaczania stężenia białka
Wyciąg z kart charakterystyki substancji niebezpiecznych
•
Kwas ortofosforowy – C
•
Wodorotlenek sodu – C
•
Miedzi (II) siarczan kryst. 5 hydrat – Xn, N
•
Etanol 96% - F
SPEKTROFOTOMETRIA ABSORPCYJNA
W metodach spektrofotometrycznych wykorzystuje się zjawisko pochłaniania (selektywnej absorpcji)
promieniowania przez badane substancje w celu ich wykrycia, identyfikacji lub ilościowego oznaczenia, gdyż
cechą charakterystyczną każdej substancji chemicznej jest zdolność do absorpcji ściśle określonych kwantów
promieniowania z zakresu widma elektromagnetycznego. Ze względu na wykorzystywany zakres widma (Tab. 1)
dokonuje się podziału na spektrofotometrię absorpcyjną w świetle widzialnym, zwaną kolorymetrią
i absorpcjometrię w nadfiolecie i podczerwieni. W badaniach analitycznych zastosowanie znalazła
absorpcjometria w zakresie światła widzialnego (VIS) oraz bliskiego ultrafioletu (UV). Absorpcja promieniowania
z tych przedziałów długości fal zależy głównie od ilości i rozmieszczenia elektronów w badanej cząsteczce, oraz
powoduje wzbudzenie cząsteczki danego związku polegające na przeniesieniu elektronów na wyższy poziom
energetyczny.
Rodzaj promieniowania
Długość fali
promienie
γ
poniżej 10 pm
promienie X
10 pm-10 nm
ultrafiolet daleki
10-200 nm
ultrafiolet bliski
200-390 nm
światło widzialne
390-800 nm
podczerwień
800 nm-1 mm
Mikrofale
1 mm-30 cm
Tabela 1 Widmo promieniowania elektromagnetycznego.
Ilościową miarą charakteryzującą zdolność pochłaniania promieniowania przez dany związek jest absorbancja
oznaczana literą A (Uwaga: absorbancja zwana jest też ekstynkcją i oznaczana literą E). Pomiar absorbancji
oparty jest na prawie Lamberta-Beera, które określa zależność pomiędzy natężeniem promieniowania
padającego na roztwór danego związku (I
o
) a natężeniem promieniowania przechodzącego (I) przez warstwę
badanej próbki zgodnie z poniższym wzorem:
A = log (I
o
/ I) = kcl
2
A – absorbancja (ekstynkcja)
I
o
– natężenie światła monochromatycznego o określonej długości fali padającego na roztwór
I – natężenie światła po przejściu przez roztwór
k – współczynnik absorbancji właściwej dla danej długości fali, wielkość charakterystyczna dla danej
substancji (niezależna od jej stężenia) i rozpuszczalnika dla danej długości fali, liczbowo równa
absorbancji roztworu o stężeniu 1 mol/l i grubości warstwy l = 1 cm
c – stężenie danego związku (mol/l)
l – grubość warstwy absorbującej, standardowo l = 1 cm, gdyż w badaniach biochemicznych grubość
stosowanych kuwet wynosi zwykle 1 cm
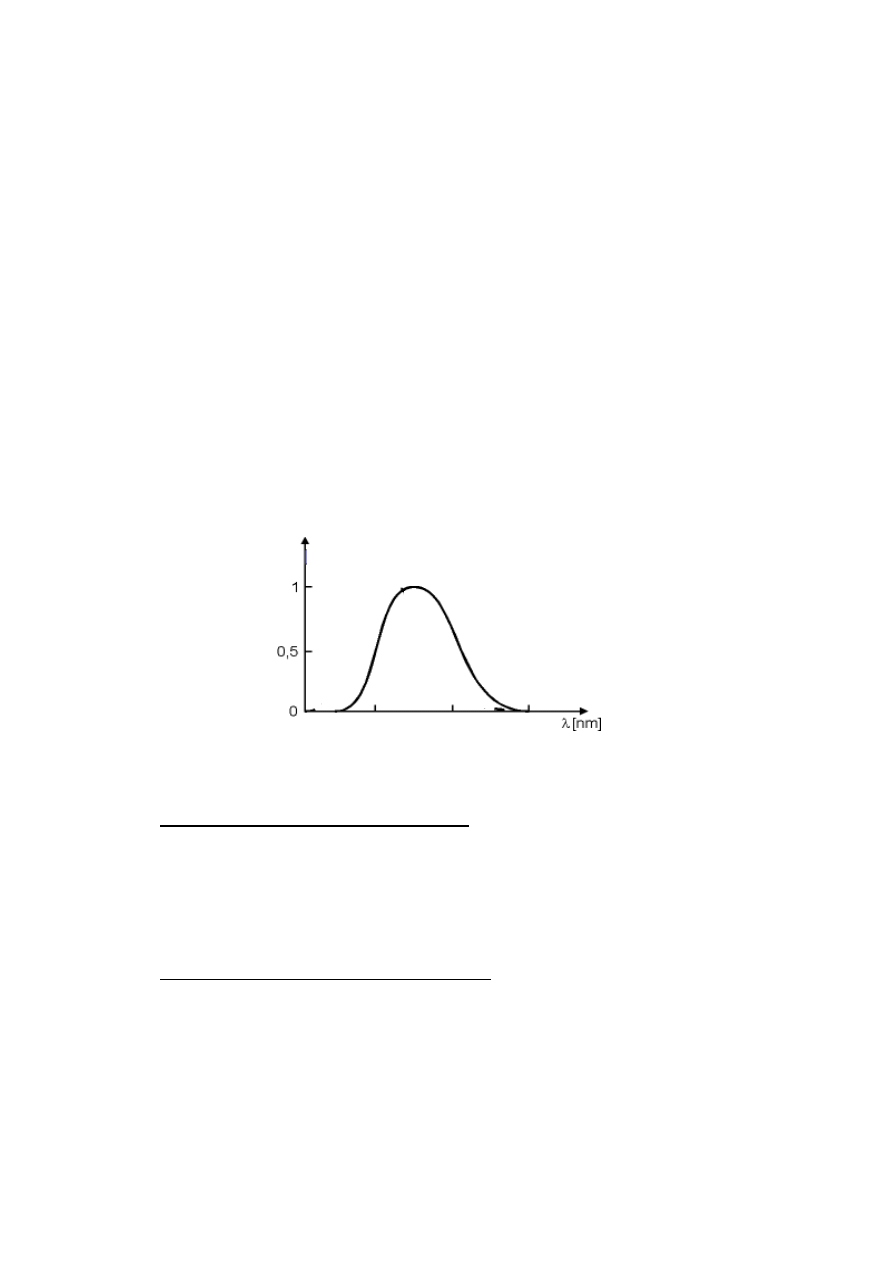
Badanie selektywnego pochłaniania promieniowania przez dany związek w zależności od długości fali
λ
pozwala
na wyznaczenie jego widma absorpcyjnego. Rozkład maksimów i minimów w widmie zależy od właściwości
barwnych związku (maksimum absorbancji określa barwę). Gdy zakres długości fal promieniowania
absorbowanego przez dany związek jest dostatecznie wąski, to związek ten ma barwę dopełniającą do barwy
absorbowanego promieniowania. Jeśli dany związek absorbuje promieniowanie o kilku barwach, to jego
ostateczna barwa jest kombinacją barw dopełniających do tych absorbowanych. Związki bezbarwne nie
pochłaniają fal w widzialnym obszarze widma, a z kolei związki czarne pochłaniają wszystkie długości fal. Dzięki
znajomości widma absorpcyjnego badanego związku możliwe jest oznaczenie jego stężenia (ilości) w roztworze,
gdyż można wybrać światło monochromatyczne o takiej długości fali, która jest maksymalnie absorbowana przez
dany związek. Do oznaczania stężenia bezbarwnych związków stosuje się specjalne odczynniki, z którymi tworzą
one barwne pochodne.
Rys. 1 Widmo absorpcyjne
Stężenie badanej substancji w roztworze można obliczyć:
1. korzystając bezpośrednio z prawa Lamberta-Beera dokonując pomiaru absorbancji próby badanej (A
b
)
i absorbancji próby wzorcowej o znanym stężeniu (A
w
):
A
b
/A
w
= kc
b
l/kc
w
l = c
b
/c
w
⇒
⇒
⇒
⇒
c
b
= (A
b
/A
w
)c
w
Uwaga: Powyższą zależność można stosować tylko wtedy, gdy obserwuje się prostoliniową zależność
między stężeniem danego związku a mierzoną absorbancją.
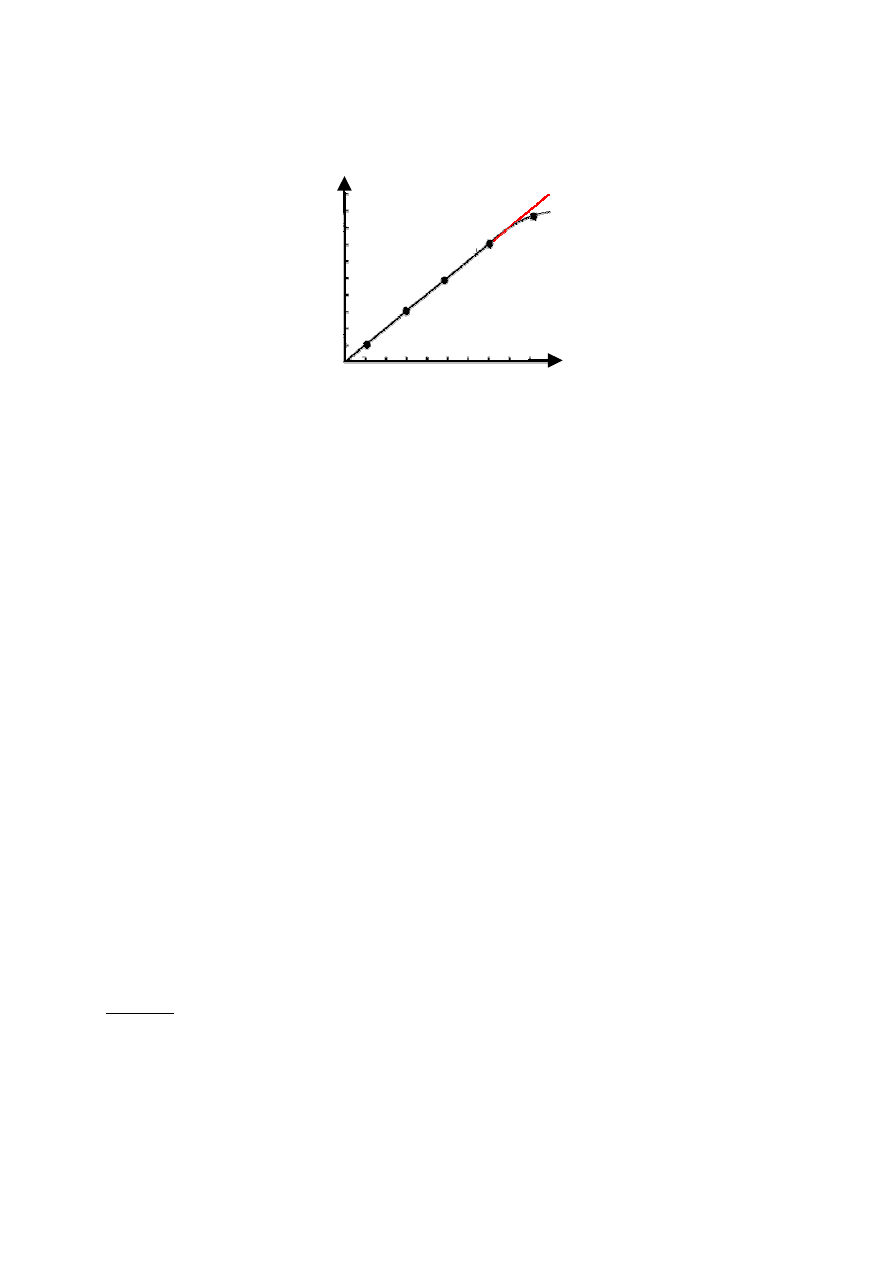
2. stosując krzywą kalibracyjną (wzorcową, standardową)
Dokonuje się pomiaru absorbancji próby badanej (A
b
) i absorbancji kilku prób wzorcowych o znanym
stężeniu (A
w1
...A
wn
), a po wykreśleniu zależności absorbancji próbek wzorcowych (A
w
) od ich stężenia
(c
w
) otrzymuje się prostą przechodząca przez początek układu współrzędnych. Znając absorbancję badanej
próby z krzywej wzorcowej odczytuje się stężenie zawartego w niej związku, ale jedynie w jej prostoliniowym
zakresie.
Uwaga 1: Na osi odciętych odkłada się wartości stężeń, a na osi rzędnych – uzyskane wartości absorbancji.
Uwaga 2: Przyjmuje się, że absorbancja czystego rozpuszczalnika (w którym rozpuszczony się dany
związek) i względem, którego wykonuje się pomiary spektrofotometryczne wynosi zero. Ślepa odczynnikowa
A
3
(ślepa, próbka kontrolna) składa się ze wszystkich odczynników dodawanych w takiej samej ilości oraz takiej
samej kolejności jak miało to miejsce w przypadku próbki właściwej.
Rys. 2 Krzywe wzorcowe dla: a – roztworu stosującego się do prawa Lamberta-Beera, b – roztworu
wykazującego przy dużych stężeniach odchylania od praw absorpcji.
1. Oznaczanie zawartości białka metodą pomiaru w nadfiolecie
Aminokwasy nie absorbują światła widzialnego (tj. są one bezbarwne) i z wyjątkiem aminokwasów
aromatycznych (Trp, Tyr, Phe, His) nie absorbują również światła nadfioletowego o długości powyżej 240 nm.
Białka natomiast absorbują światło nadfioletowe w paśmie 260-290 nm, ze względu na obecność w ich
łańcuchach aminokwasów aromatycznych (głównie reszt Trp i Tyr odpowiedzialnych za pochłanianie większości
światła ultrafioletowego). Zwykle odczytu absorbancji białek dokonuje się przy długości fali 280 nm, gdyż Trp i Tyr
wykazują maksimum absorbancji właśnie przy tej długości fali. Przyjmuje się, że ilość zaabsorbowanego światła
jest proporcjonalna do ilości białka, ale różne ilości Trp i Tyr w poszczególnych białkach powodują utrudnienie
w ilościowej ocenie stężenia białka w roztworze tą metodą. Dodatkowo w tym samym zakresie światła UV
absorbują również kwasy nukleinowe i nukleotydy, których maksimum pochłaniania jest przy długości fali 260 nm.
W zmodyfikowanej metodzie spektrofotometrycznej stężenie białka w próbce określa się na podstawie
różnicy absorbancji mierzonych przy długościach fal 260 i 280 nm stosując kuwetę kwarcową o grubości 1 cm,
a stężenie białka wyrażone w mg/ml oblicza się wg wzoru podanego przez Layne’a:
C
białka
= 1,55
*
A280 – 0,76
*
A260
Zaletą zmodyfikowanej metody spektrofotometrycznej jest możliwość szybkiego określenia stężenia białka w
dużej ilości próbek bez konieczności korzystania z odnośnika białkowego i krzywej kalibracyjnej, również przy
dokonywaniu pomiarów w małej ilości materiału w obecności dużych stężeń NaCl czy (NH
4
)
2
SO
4
. Jest to metoda
wykorzystywana na przykład przy oznaczaniu stężenia białka we frakcjach uzyskiwanych podczas rozdziału
chromatograficznego na kolumnach. Ponadto metoda ta eliminuje wkład kwasów nukleinowych
w wartość mierzonej absorbancji, o ile ich zawartość w mieszaninie nie jest większa niż 20%. Niemniej jednak
metoda ta jest obarczona dużym błędem i daje tylko przybliżone wyniki, gdyż zarówno białka, jak i kwasy
nukleinowe, nie dają identycznych wartości absorbancji maksymalnych przy jednakowych stężeniach. Ponadto,
również inne związki małocząsteczkowe (puryny, pirymidyny, fenole) cechują się duża absorbancją przy 260 nm
i 280 nm.
Wykonanie
Zmierzyć wartość absorbancji próbki badanej przy długościach fal 260 i 280 nm wobec ślepej odczynnikowej tj.
roztworu/rozpuszczalnika, w którym jest rozpuszczone białko; w kuwecie kwarcowej o grubości 1 cm. Obliczyć
orientacyjną zawartość białka korzystając z podanego powyżej wzoru. Do kuwety odpipetować po 0,5 ml
roztworów próbek badanych P1, P2, P3 i P4.
2. Oznaczanie zawartości białka metodą mikrobiuretową
Zasada oznaczania – patrz metoda biuretowa Piotrowskiego (Ćwiczenie 2).
Stężenie [mg/ml]
Absorbancja
a
b

4
Dzięki zastosowaniu spektrofotometrycznego pomiaru absorbancji fiołkowego kompleksu powstającego na
skutek reakcji jonów miedzi w środowisku zasadowym z wiązaniami peptydowymi, przy długości fali 310 nm
uzyskuje się 50-krotny wzrost czułości oryginalnej metody biuretowej opracowanej przez Itzhaki i Gill (Itzhaki RF,
Gill DM (1964) Anal Biochem 9: 401-10). Dodatkowo, opcjonalne traktowanie oznaczanej próby 0,5 M roztworem
NaOH zamiast wody, umożliwia oznaczenie zawartości białka w płynach biologicznych, homogenatach
komórkowych czy frakcjach subkomórkowych. Rodzaj oznaczanego białka nie ma wpływu na wartość mierzonej
absorbancji.
Wykonanie
Przygotować próbki serii A i B wg Tabeli 1. Do każdej probówki serii A dodać po 0,25 ml 0,21% roztworu
CuSO
4*
5H
2
O w 30% roztworze NaOH, a do probówek serii B – po 0,25 ml 30% roztworu NaOH. Zawartość
eppendorfów wymieszać i po upływie 5 minut zmierzyć w spektrofotometrze UV-VIS absorbancję wszystkich
próbek przy długości fali 310 nm. Absorbancję próbek serii A odczytać wobec ślepej odczynnikowej A
o
(parametr
A dla każdej próbki serii A), natomiast absorbancję próbek serii B wobec ślepej odczynnikowej B
o
(parametr B).
Absorbancję właściwą wszystkich oznaczanych prób obliczyć z różnicy A-B. Na podstawie otrzymanych wartości
absorbancji właściwej dla wzorcowych roztworów BSA uzupełnić Tabelę 1 i wykreślić krzywą kalibracyjną
zależności zmierzonych absorbancji od stężenia białka. Z krzywej kalibracyjnej odczytać zawartość białka
w badanych próbkach P1, P2, P3 i P4.
Uwaga: wszystkie oznaczenia wykonywać w trzech równoległych powtórzeniach.
3. Oznaczanie zawartości białka metodą Bradforda
Bradford MM (1976) Anal Biochem 72: 248-54.
W metodzie Bradforda wykorzystywana jest zdolność wiązania się barwnika zwanego błekitem
kumazyny (ang. Coomassie Brilliant Blue G-250) z białkiem za pomocą wiązań jonowych i hydrofobowych.
Z barwnikiem reagują głównie reszty argininy, a w minimalnym stopniu reszty histydyny, lizyny, proliny, tryptofanu
i tyrozyny. W środowisku kwaśnym (w roztworze kwasu H
3
PO
4
) błękit kumazyny przyjmuje zabarwienie brunatne.
Dodanie błękitu kumazyny do roztworu białka powoduje powstawanie kompleksu o intensywnie niebieskim
zabarwieniu i przesunięcie maksimum absorbancji barwnika z 465 nm do 595 nm. Efekt ten jest konsekwencją
występowania barwnika w dwóch różnych formach – brunatnej i niebieskiej. Forma brunatna ulega konwersji do
niebieskiej po związaniu się barwnika z białkiem. Wiązanie to jest procesem bardzo szybkim (poniżej 2 minut), a
kompleks barwnik-białko pozostaje trwały i nie agreguje w roztworze przez stosunkowo długi czas (do 60 minut).
Natężenie barwy niebieskiej jest proporcjonalne do zawartości białka w roztworze. Optymalny czas pomiaru
wynosi od 5 do 20 minut od dodania odczynnika Bradforda do próbki. Zaletami metody są prostota
i szybkość wykonania, oraz jej czułość (białko może być wykryte przy jego stężeniu w granicach 2-8
µ
g/ml).
Oznaczenie można wykonywać w obecności powszechnie stosowanych buforów, wersenianu sodu (EDTA),
jonów magnezu, sacharozy, glikolu i związków tiolowych. Wadą metody jest różne wiązanie barwnika przez różne
białka, a ponadto fakt, że aceton, siarczan dodecylu sodu (SDS) i Triton X-100 dają w połączeniu
z barwnikiem dodatkowy interferujący kolor, co zwiększa natężenie barwy niebieskiej i przeszkadza w uzyskaniu
wiarygodnych wyników.
Wykonanie
Przygotować ślepą odczynnikową, wzorce oraz próbki badane wg Tabeli 2. Do każdej próbki dodać po 250
µ
l 5-
krotnie rozcieńczonego odczynnika Bradforda. Po upływie minimum 5 minut (a przed upływem 20 minut) od
momentu dodania odczynnika Bradforda, zmierzyć w czytniku do płytek titracyjnych wartości OD wszystkich
próbek przy długości fali 595 nm. Na podstawie otrzymanych wartości OD
595
uzupełnić Tabelę 2. Od wartości
OD
595
dla wzorców i próbek badanych odjąć wartość OD
595
ślepej odczynnikowej. Wykreślić krzywą kalibracyjną
zależności zmierzonych OD
595
od stężenia białka. Z krzywej kalibracyjnej odczytać zawartość białka w badanych
próbkach P1, P2, P3 i P4.
Uwaga: wszystkie oznaczenia wykonywać w trzech równoległych powtórzeniach.
Odczynniki
wzorcowe roztwory BSA o stężeniu 1mg/ml i 0,5 mg/ml; roztwory badanych próbek P1, P2, P3 i P4;
30% roztwór NaOH ; 0,21% roztwór CuSO
4*
5H
2
O w 30% NaOH; odczynnik Bradforda: 0,01% (w/v) błękit
kumazyny, 4,7% (w/v) etanol, 8,5% (w/v) kwas ortofosforowy
Wyszukiwarka
Podobne podstrony:
cw.3-oznaczanie stezenia bialka
Kolorymetryczne oznaczanie stężenia białka przez reakcję z?rwnikiem Coomassie Brillant Blue(1)
Ilościowe oznaczanie stężenia białka metodą Lowry
2.oznaczenie stezenia roztworu hcl, Chemia-labolatoria
KWASY I ZASADY OZNACZENIE STĘŻENIA
Ćw 1 Oznaczenie wilgotności gruntu
mikrobiologia ćw 4, Oznaczanie liczebności Escherichia coli w hodowli nocnej
57. Pomiar widm absorpcji i oznaczanie stężenia ryboflawiny w roztworach wodnych za pomocą spektrofo
57. Pomiar widm absorpcji i oznaczanie stężenia ryboflawiny w roztworach wodnych za pomocą spektrofo
9.Oznaczanie stężenia badanego roztworu metodą miareczkową., Państwowa Wyższa Szkoła Zawodowa
OZNACZANIE STĘŻENIA?NOLU METODĄ MIARECZKOWANIA KONDUKTOMETRYCZNEGO
OZNACZANIE ZAWARTOŚCI BIAŁKA W MLEKU METODĄ KJELDAHLA, Chemia analityczna
sprawko oznaczanie zawartości białka, TŻ UR, II rok, Analiza i ocena jakości żywności
Ćw 5 Oznaczenie wilgotności optymalnej
Metody oznaczania stężenia D dimerów przydatne w diagnostyce żylnej choroby zakrzepowo zatorowej
Ćw 4 Oznaczenie stopnia zagęszczenia
Cw 2 Oznaczenie gestosci grunt Nieznany
więcej podobnych podstron