
1
Chemia Analityczna
Chromatografia
Tłumaczyła: inż. Karolina Hierasimczyk
Korekta:
dr hab. inż. Waldemar Wardencki, prof. nadzw. PG
prof. dr hab. inż. Jacek Namieśnik
Część II
Terminy i definicje.
Katedra Chemii Analitycznej
Wydział Chemiczny
Politechnika Gdańska
2002

2
SPIS TREŚCI
Wprowadzenie
1. Co to jest chromatografia?
1.1. Proces chromatograficzny
1.2. Podział metod chromatograficznych
1.3. Co to jest chromatografia gazowa?
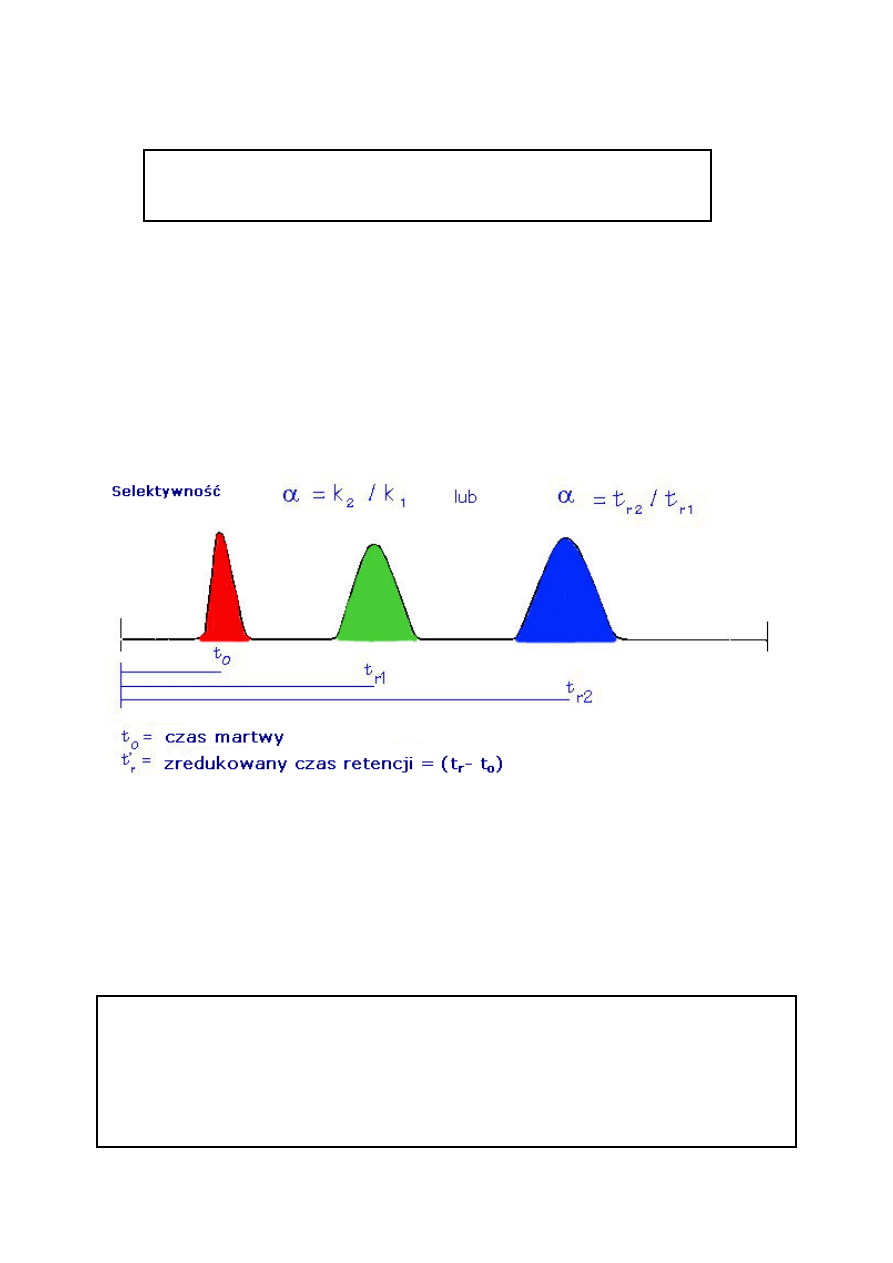
2. Terminy i definicje .........................................................................................................II/2
2.1. Czas retencji (t
R
) .........................................................................................................II/3
2.2. Współczynnik retencji (k) ............................................................................................II/3
2.3. Indeks retencji (I) ......................................................................................................II/12
2.4. Współczynnik rozdzielenia .......................................................................................II/14
2.5. Teoretyczna liczba półek (N) lub sprawność kolumny .............................................II/14
2.6. Rozdzielczość (R
S
) ....................................................................................................II/17
2.7. Stosunek faz (β) .........................................................................................................II/21
3. Kolumny kapilarne do chromatografii gazowej
3.1. Fazy stacjonarne
3.1.1. Polisiloksany
3.1.2. Glikole polietylenowe
4. Gazy nośne
5. Dozowniki
5.1. Dozowniki wykorzystujące odparowanie
5.2. Dyskryminacja związków dozowanych
5.3. Opłukiwanie membrany
5.4. Dozowanie na kolumnę typu „Megabore”
5.5. Dozowniki z dzieleniem strumienia gazu (split)
5.6. Dozownik bez podziału strumienia gazu
6. Detektory w GC
6.1. Detektor cieplno-przewodnościowy (TCD)
6.2. Detektor płomieniowo – jonizacyjny (FID)
6.3. Detektor wychwytu elektronów (ECD)
6.4. Detektor azotowo fosforowy (NPD)
6.5. Detektor płomieniowo – fotometryczny (FPD)
6.6. Detektor fotojonizacyjny (PID)
6.7. Spektrometr mas (MS)
7. Analiza ilościowa
3
2. Terminy i definicje
2.1. Czas retencji (t
R
)
Czas retencji (t
R
) jest to czas, w którym substancja przechodzi przez kolumnę. Czas retencji
przypisany jest pikowi odpowiadającemu danej substancji. Czas zatrzymania składnika jest
miarą ilości czasu jaki substancja rozpuszczona spędza w kolumnie. Jest sumą czasu
spędzonego w fazie stacjonarnej i w fazie ruchomej.
Czas retencji nie zatrzymywanego związku
Czas retencji substancji nie zatrzymywanej lub czas martwy (t
M
lub t
O
) to czas w jakim nie
zatrzymywany związek przechodzi przez kolumnę. Nie zatrzymane cząsteczki substancji
rozpuszczonej nie oddziałują z fazą stacjonarną, lecz przemieszczają się w dół kolumny z tą
samą szybkością z jaką przemieszcza się strumień gazu nośnego. Jest to równoważne z
czasem jaki związek spędza w fazie ruchomej. Czas martwy piku otrzymany jest poprzez
dozowanie substancji nie zatrzymywanych i określeniu ich czasu retencji.
2.2. Współczynnik retencji (k)
Współczynnik retencji (k) jest inną miarą retencji. Jest to stosunek
ilości czasu jaki substancja rozpuszczona spędza w fazie stacjonarnej i
ruchomej (gaz nośny).

4
Obliczony jest za pomocą równania 1. Współczynnik retencji wcześniej nazywany był
współczynnikiem podziału lub współczynnikiem pojemnościowym. Ponieważ wszystkie
substancje rozpuszczone spędzają tę samą ilość czasu w fazie ruchomej, współczynnik
retencji jest miarą retencji przez fazę stacjonarną. Jest pomiarem względnym i ma charakter
liniowy. Na przykład, substancja o k = 6 jest podwójnie tak zatrzymana przez fazę stacjonarną
(nie przez kolumnę) jak substancja o k = 3. Współczynnik retencji nie dostarcza wszystkich
informacji o zatrzymaniu składnika; dostarcza informacje o względnej retencji. Dla nie
zatrzymywanej substancji k = 0.
Współczynnik retencji (k)
k = (t
R
– t
M
) / t
M
= t’
R
/ t
M
(1)
t
R
= czas retencji
t’
R
= redukowany czas retencji
t
M
= czas retencji związku nie zatrzymywanego
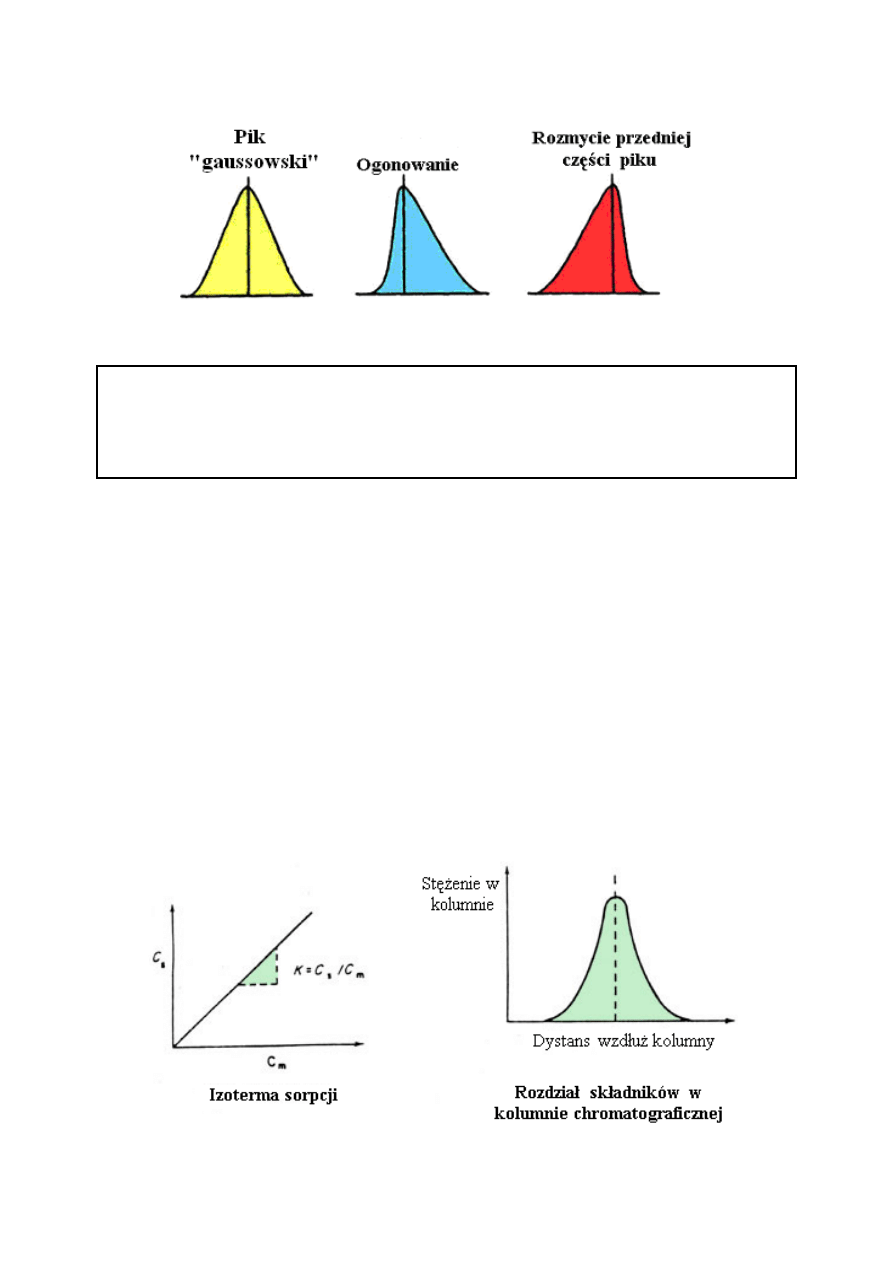
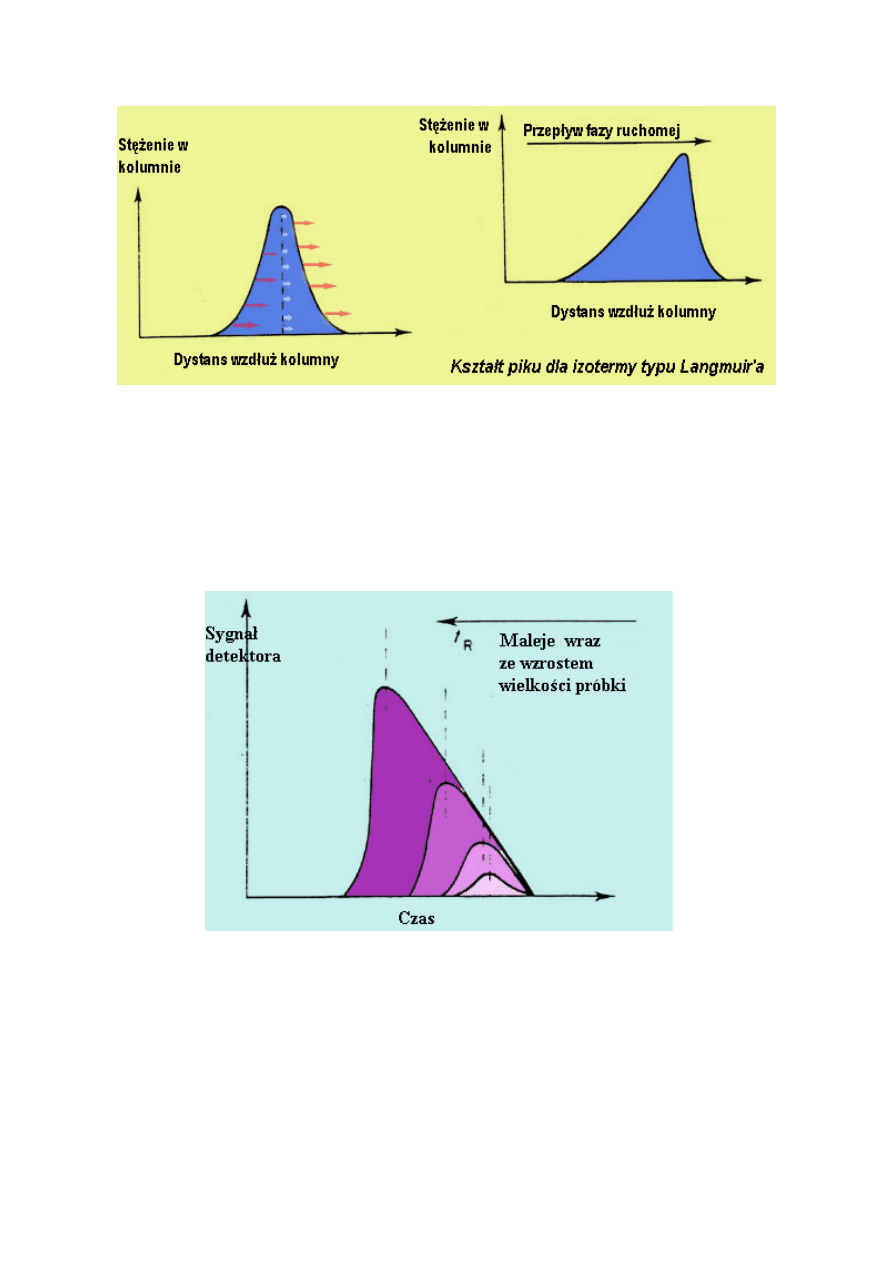
KSZTAŁT PIKU
Dyfuzja jest powszechnie obserwowanym zjawiskiem. Jakakolwiek część niezwiązanych
cząsteczek będzie dążyć do rozproszenia w zajmowanej objętości. Jeżeli tak się dzieje to
następuje spadek ich stężenia i wzrasta ich entropia. Dlatego spełnione jest jedynie drugie
prawo termodynamiki. Mówiąc o dyfuzji, często myśli się o cząsteczkach poruszających się z
obszaru o wysokim stężeniu do obszaru o niskim stężeniu. Takie założenie, z góry przyjmuje,
iż cząsteczki są ‘świadome’ obszarów o wysokim i niskim stężeniu. W rzeczywistości dyfuzja
odbywa się we wszystkich kierunkach ponieważ ruchy cząsteczek są przypadkowe.
Ostatecznie jednak, obszar o niższym stężeniu cząsteczek zostanie w nie wzbogacony a
obszar o wyższym stężeniu ulegnie zubożeniu. Więcej cząsteczek popłynie z obszaru o
wyższym stężeniu do obszaru o niższym stężeniu, i nastąpi ogólne wyrównanie poziomu
stężenia.
Na początku profile substancji rozpuszczonej mają kształt symetrycznych pików, profile
stężenia jednak ulegają w czasie zmianom i przybierają odpowiednie kształty:
5
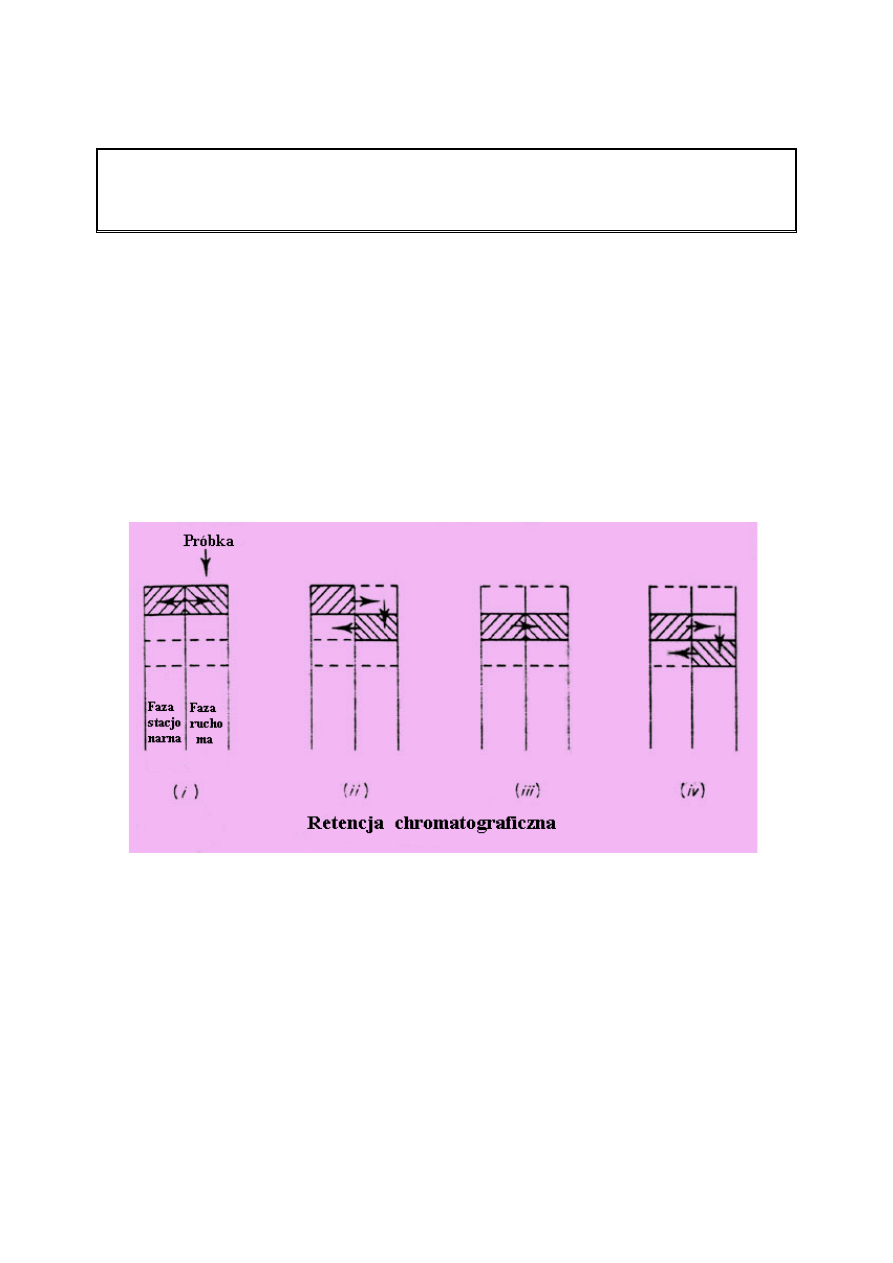
IZOTERMA SORPCJI
Ilość poszczególnej substancji zasorbowanej przez fazę stacjonarną zależy od stężenia w fazie
ruchomej. Stosunek pomiędzy ilością zasorbowaną a stężeniem w fazie ruchomej, przy stałej
temperaturze, nazywany jest izotermą sorpcji.
Sorpcja jest ogólnym pojęciem obejmującym kilka mechanizmów, takich jak, proces
adsorpcji (np. oddziaływanie pomiędzy cząsteczkami substancji rozpuszczonej a
powierzchnią ciała stałego), podział (rozcieńczenie cząsteczek substancji rozpuszczonej w
cieczy), wymianę jonową (wymiana jonów substancji rozpuszczonej z jonami w fazie
stacjonarnej) i wykluczanie (ograniczenie dyfuzji cząsteczek substancji rozpuszczonej
poprzez porowatą fazę stacjonarną).
Izoterma sorpcji i kształt piku
Poniższe rysunki przedstawiają przebieg izotermy gdy stosunek pomiędzy stężeniem badanej
substancji w fazie stacjonarnej (Cs) i stężeniem substancji rozpuszczonej w fazie ruchomej
(Cm) jest liniowy. Stosunek Cs / Cm określa nachylenie krzywej.
6
Jest on określony jako współczynnik podziału K.
System chromatograficzny, w którym relacja ta jest liniowa opisany jest jako chromatografia
liniowa, której efektem jest rozdzielenie substancji i uzyskanie pików typu gaussowskiego.
Powyższe rysunki ukazują izotermę liniową i charakterystykę pasma chromatograficznego z
którym jest związana.
Retencja w chromatografii
Poniższy rysunek przedstawia sytuację, w której próbka wprowadzona jest do kolumny w
fazie ruchomej. W praktyce, faza stacjonarna będzie rozproszona całkowicie w kolumnie ale
dla wygody możemy wyobrazić sobie, że zajmuje ona połowę kolumny i że faza ruchoma
zajmuje drugą połowę kolumny.
Gdy próbka (niesiona przez fazę ruchomą) zetknie się z fazą stacjonarną, rozdzieli się miedzy
te dwie fazy zgodnie z wartościami ich współczynników podziału (K
D
lub K). Jest to
równoważne z separacją w pierwszym „lejku” separacyjnym. Jednakże w przeciwieństwie do
lejka separacyjnego, nie trzeba czekać aby otworzyć zawór lejka i pozwolić próbce
przepłynąć do drugiego lejka. Nie musimy także czekać aż próbka osiągnie równowagę
pomiędzy dwoma fazami zanim przejdzie do drugiego kroku separacji. Oznacza to, że jeżeli
równowaga ma być osiągnięta, musi to nastąpić natychmiastowo. Gdy już jest rozdzielona
pomiędzy dwie fazy, próbka pozostająca w fazie ruchomej będzie poruszać się dalej w dół
kolumny napotykając „nową” fazę stacjonarną. Próbka ponownie poruszać się będzie z fazy
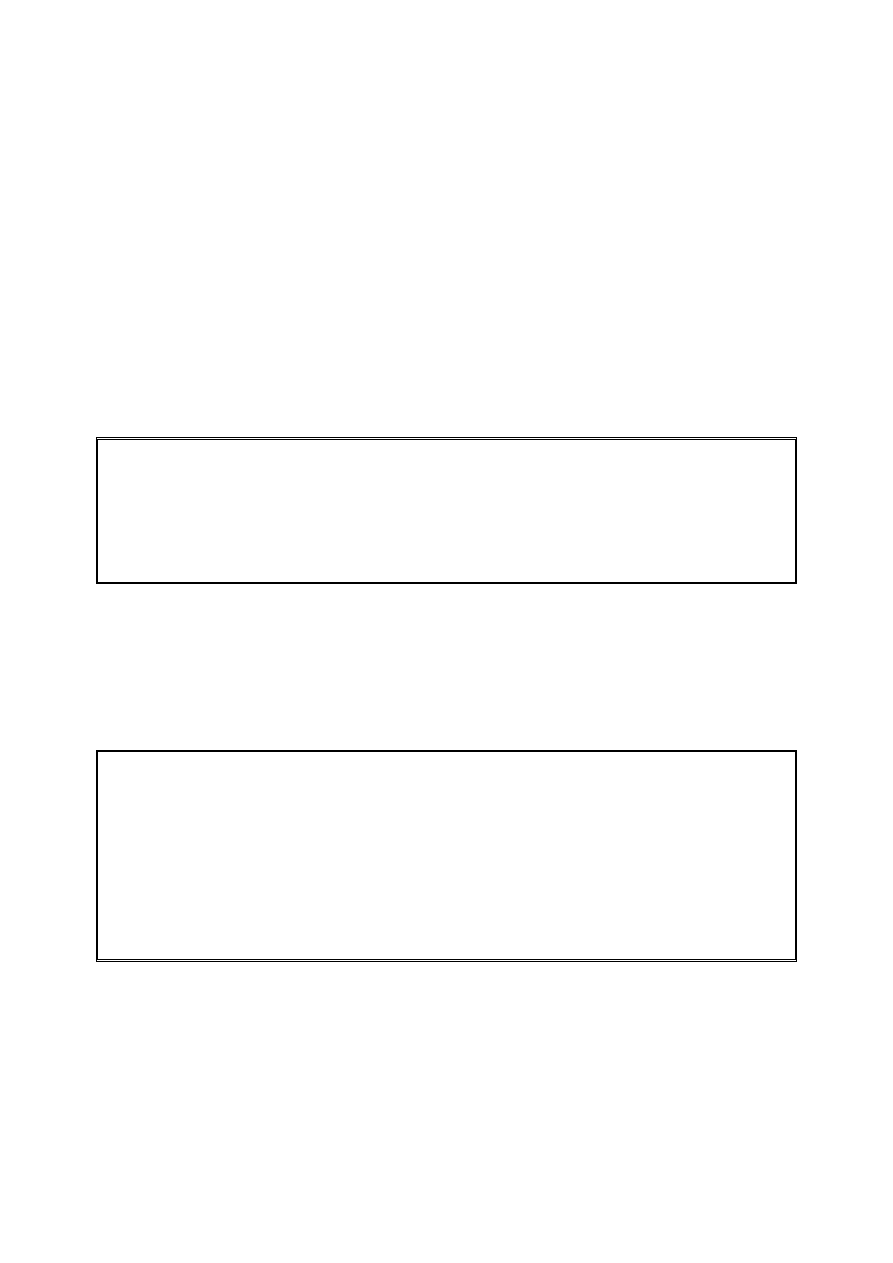
7
ruchomej do fazy stacjonarnej (proces ten zwany jest sorpcją) aż ponownie zostanie
osiągnięta równowaga. Ponieważ stężenie próbki w fazie ruchomej w pierwszej sekcji zostało
zmniejszone, próbka przemieszczać się będzie z fazy stacjonarnej z powrotem do fazy
ruchomej (proces ten nazywany jest desorpcją) w celu utrzymania stałej wartości
współczynnika podziału. Proces ten zobrazowany jest na Rysunku (ii). W bardzo krótkim
czasie cała próbka zostanie zdesorbowana z fazy stacjonarnej w pierwszej sekcji kolumny i
porwana przez fazę ruchomą do drugiej sekcji kolumny. Ostatecznie, równowaga znowu
zostanie ustalona a próbka w całości znajdzie się w drugiej sekcji kolumny (Rys. (iii)). Proces
sorpcji i desorpcji będzie trwał (Rys. (iv)) aż próbka dojdzie do końca kolumny i zostanie
wymyta z kolumny.
Chromatografiści nie stosują terminu – lejki separacyjne lub sekcje kolumn ale zapożyczają
terminologię z techniki destylacji, z którą chromatografia ma pewne cechy wspólne. Przez
analogię z destylacją, każda sekcja kolumny chromatograficznej w której przyjmuje się, iż
równowaga została osiągnięta znana jest jako półka teoretyczna.
Przyjrzeliśmy się procesowi chromatograficznemu, który przebiega jako seria dyskretnych
kroków tak jak w przypadku lejka separacyjnego. Ale chromatografia jest dynamicznym
procesem a nie serią statycznych kroków. Jednakże, czyniąc nasze kroki lub ‘teoretyczne
półki’ bardzo małymi, procesy statyczne i dynamiczne stają się równoważne.
Długość kolumny odpowiadająca teoretycznym półkom nazwana jest Wysokością
Równoważną Półce Teoretycznej (Height Equivalent to a Theoretical Plate - HETP lub
H). Im mniejsza wartość H tym więcej kroków równoważnych zawiera kolumna (jest to
równoważne z przeprowadzaniem dużych ilości kolejnych separacji w lejku) i przyjmuje się,
że kolumna posiada dużą sprawność. W praktyce, oznacza to że kolumna sprawdziłaby się
lepiej przy separacji składników mieszanin.
Odnosząc się ponownie do rysunków przedstawianych powyżej, jeżeli strzałki wskazują
kierunek, w którym poruszają się cząsteczki substancji, obserwujemy iż w fazie stacjonarnej
nie odbywa się ruch bezpośrednio wzdłuż kolumny. Cząsteczki poruszają się tylko przez
granicę pomiędzy fazą stacjonarną i fazą ruchomą, a wzdłuż kolumny tylko w czasie kiedy
znajdują się w fazie ruchomej. W fazie ruchomej, wszystkie cząsteczki poruszają się z taką

8
samą prędkością (µ), gdzie µ = prędkość liniowa fazy ruchomej, a więc czas spędzony w fazie
ruchomej jest taki sam dla wszystkich cząsteczek. Czas ten nazwany jest czasem retencji
substancji nie zatrzymywanej lub martwym czasem kolumny (t
M
).
Zróżnicowanie migracji wynika z czasu (t
S
) jaki różne substancje spędzają w fazie
stacjonarnej. Składnik (A) z niskim współczynnikiem podziału, K, w jakimkolwiek czasie
będzie posiadał mniej cząsteczek w fazie stacjonarnej w porównaniu do innego składnika (B)
z wyższym współczynnikiem podziału, dlatego będzie mniej opóźniony i szybciej przemieści
się przez kolumnę. W związku z tym, stosunek przemieszczania się składnika, (Rm), jest
odwrotnie proporcjonalny do wartości jego współczynnika podziału; stosunek
przemieszczania się
Rm proporcjonalne do 1 / K
Składniki mieszaniny będą odseparowane tylko wtedy gdy ich współczynniki podziału
pomiędzy fazą stacjonarną a fazą ruchomą będą się różniły.
Całkowity czas retencji składnika ( t
R
) jest sumą czasów spędzonych w fazie ruchomej i
fazie stacjonarnej:
t
R
= t
m
+ t
S
t
S
– zazwyczaj zwane jest zredukowanym czasem retencji, t’
R
W chromatografii gazowej przyjmuje się, że faza ruchoma (zazwyczaj azot, hel lub wodór)
nie wykazuje oddziaływania z próbką, np. nie wpływa na rozdzielenie cząsteczek próbki
pomiędzy fazami, które zależy tylko od oddziaływań międzycząsteczkowych między próbką a
fazą stacjonarną. Jedyną funkcją fazy ruchomej jest transport cząsteczek substancji
rozpuszczonej wzdłuż kolumny. W chromatografii cieczowej faza ruchoma opisana jest jako
interaktywna i odgrywa ważną rolę w rozdzielaniu składników próbki. Poprzez zmianę składu
chemicznego fazy ruchomej, rozkłady składników pomiędzy fazy stacjonarną i ruchomą
przyczyniają się do tego, iż stosunek ich migracji może być znacznie zmieniony. Dlatego w
chromatografii cieczowej, analityk może dodatkowo kontrolować selektywność.
Stosunek przemieszczania się pasma badanej substancji przez kolumnę jest odwrotnie
proporcjonalny do wartości współczynnika podziału. W wypadku izotermy liniowej, K nie
zależy od stężenia (tj. nachylenie krzywej jest stałe).

9
Dlatego też, chociaż powyższy rysunek przedstawia gradient stężenia w poprzek piku i
maksymalne stężenie znajdujące się w centrum piku, wartość K pozostanie stała w poprzek
piku. Oznacza to, że cały pik przemieszczać się będzie w tym samym stosunku a podstawowy
kształt pozostanie bez zmian tj. będzie miał charakter gaussowski. Jak zostało to już
przedstawione, w zasadzie pik będzie się rozmywał przechodząc wzdłuż kolumny z powodu
zjawiska dyfuzji, ale w obecnej dyskusji możemy pominąć ten fakt. W takim przypadku mówi
się o idealnej liniowej chromatografii, przy czym termin ‘idealna’ odnosi się do faktu, iż
dyfuzja została zignorowana.
W chromatografii adsorpcyjnej, izoterma ma kształt „wypukły” w stosunku do osi Cm
(izoterma typu Langmuir). Takie zachowanie ma miejsce w wypadku silnych oddziaływań
między analizowaną substancją a fazą stacjonarną, ale oddziaływania substancja /substancja
są stosunkowo słabe. Początkowo, ilość substancji zaadsorbowanej przez fazę stacjonarną
wzrasta szybko wraz ze wzrostem stężenia w fazie ruchomej, tak długo aż jednocząsteczkowa
warstwa substancji uformuje się na adsorbencie. Ponieważ oddziaływania pomiędzy
cząsteczkami substancji są słabe, przypadki adsorpcji, w których formowane są warstwy
jednocząsteczkowe i pewna ilość substancji zostaje zaadsorbowana, pozostają stałe nawet gdy
stężenie w fazie ruchomej później wzrasta. Dlatego współczynnik podziału jest wysoki przy
niskich wartościach Cm, ale maleje wraz ze wzrostem Cm.
Poniższy rysunek przedstawia kształt piku w odniesieniu do izotermy tego typu.
10
Oznacza to, że stosunek przemieszczania się nie jest teraz taki sam. Jeżeli oznaczy się stopień
przemieszczania się za pomocą strzałek, brzegi pasma, gdzie stężenie jest najniższe, będą
miały najwyższą wartość K i dlatego poruszanie się tej strefy następuje stosunkowo wolno w
porównaniu do centrum pasma gdzie panuje najwyższe stężenie a wartość K jest najniższa.
Dlatego centrum pasma porusza się szybciej docierając do rozmytej przedniej części piku i
pozostawiając za sobą ogon.
W układach podziałowych, izoterma zazwyczaj jest wklęsła do osi Cm (izoterma typu anty-
Langmuira). Ten rodzaj izotermy powstaje w wyniku silnych oddziaływań między
cząsteczkami substancji, a oddziaływania pomiędzy substancją a fazą stacjonarną są słabe.
Dlatego, przy niskich stężeniach, rozpuszczalność cząsteczek badanej substancji jest mała i
parametr K ma niską wartość, ale gdy kilka cząsteczek substancji rozpuści się w fazie
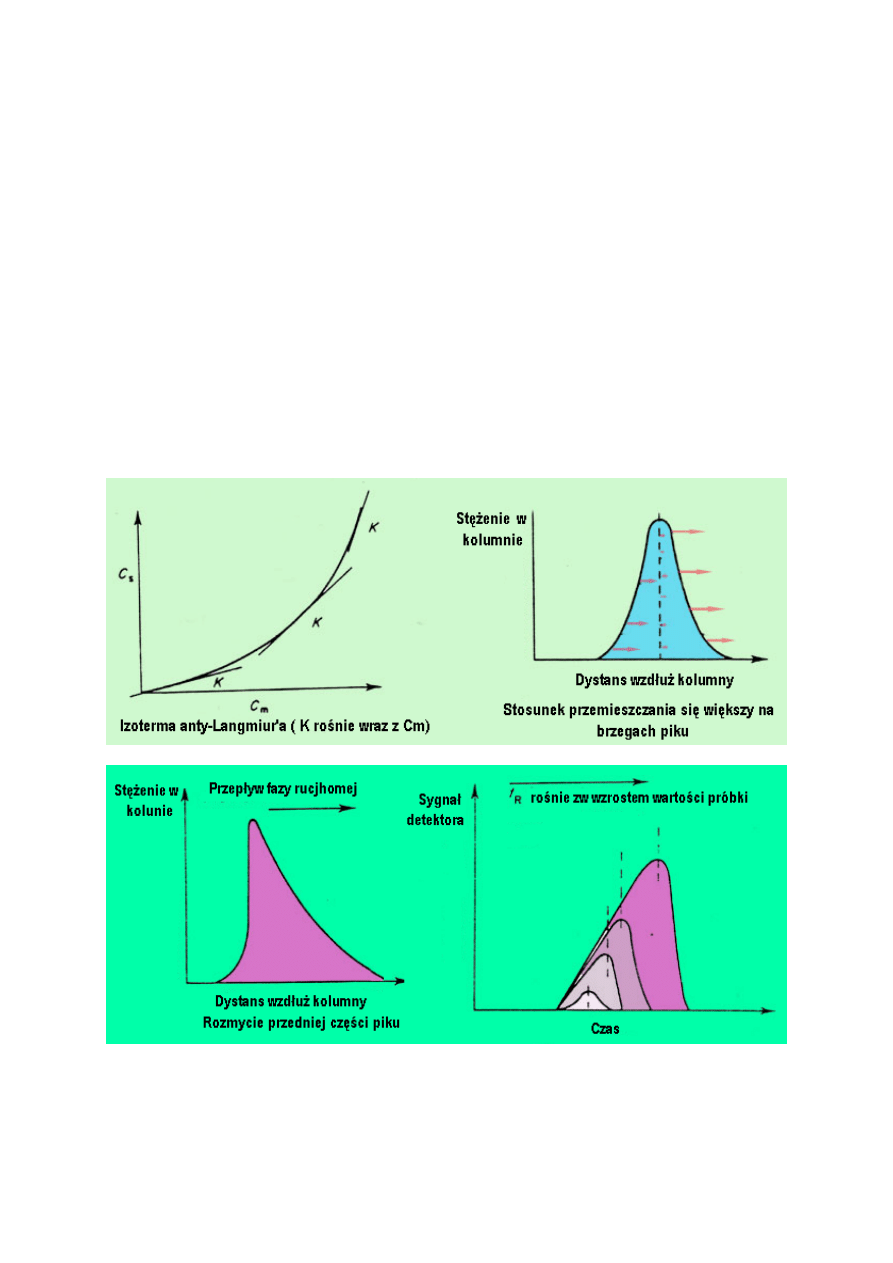
11
stacjonarnej, silne oddziaływania substancja /substancja wciągają kolejne cząsteczki do fazy
stacjonarnej i K wzrasta gwałtownie.
Jakiego kształtu piku należy się spodziewać w układzie podziałowym? Pik z rozproszonym
przodem i ostrym ogonem nazywany jest pikiem z rozmytą częścią przednią.
Należałoby rozważyć, że w przypadku izotermy typu anty-Langmuira K wzrośnie wraz ze
wzrostem stężenia w poprzek pasma, więc najniższe będzie na brzegach a najwyższe w
centrum piku. Dlatego stosunek przemieszczania się będzie najniższy w centrum a najwyższy
na brzegach. Ogon będzie dążył do środka a przód będzie odbiegał od środka przyczyniając
się do powstania piku z ostrym ogonem i rozmytym przodem (tj. rozmycie przedniej części
piku). Proces ten przedstawiono na rysunku.
1. Do asymetrycznego wzrostu piku przyczynić się może także kilka innych czynników,
a w szczególności powolne dozowanie do kolumny, ale mogą one zostać ograniczone
w wyniku dobrania odpowiednich warunków pracy.
12
Generalnie, ogonowanie i rozmycie przedniej części piku są szersze niż odpowiadający
kształt piku typu gaussowskiego. Poniżej widać, iż poszerzony pik nie sprzyja pełnemu
rozdzieleniu dwóch związków, więc takie zjawisko powinno być unikane.
W jaki sposób można temu przeciwdziałać?
Obie formy izoterm mogą być postrzegane jako liniowe jeżeli stężenie substancji jest
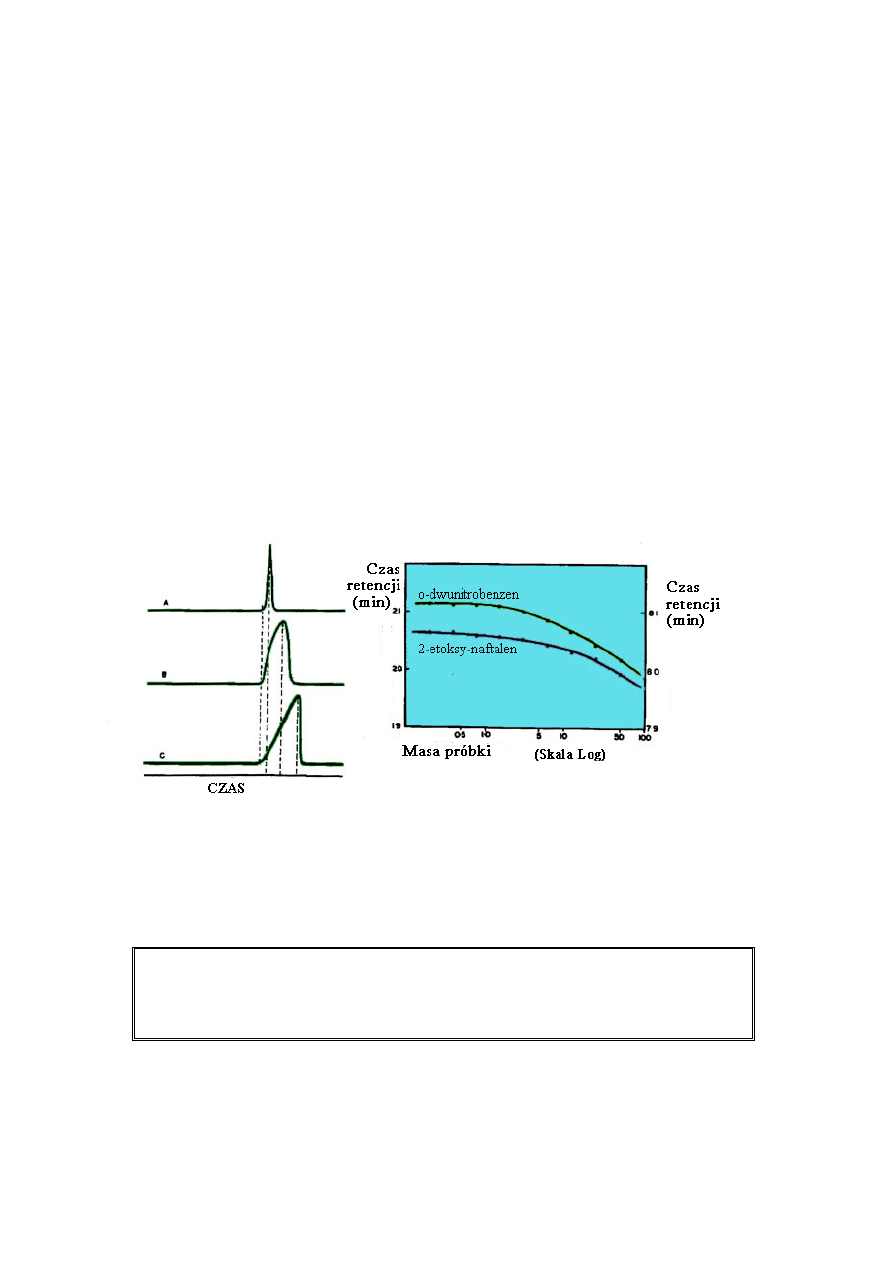
stosunkowo niskie. Dlatego, jeżeli dozowana ilość próbki do kolumny jest utrzymana na
niskim poziomie, piki typu gaussowskiego zazwyczaj będą otrzymane w przypadku izotermy
nieliniowej. W chromatografii gazowej i cieczowej dla typowej kolumny z wypełnieniem (4
mm śr.) wielkość próbki danego składnika w mieszaninie powinna być utrzymana poniżej 1
mg (0,1 µL jeżeli M = 100). W chromatografii gazowej z wypełnieniem stałym, wielkość
próbki musi być znacznie mniejsza jeżeli dąży się do otrzymania piku typu gaussowskiego.
Jeżeli wielkość próbki jest zbyt duża piki stają się zniekształcone a kolumnę uważa się za
przeładowaną.
Wpływ wielkości próbki na czas retencji analitów: A - kolumna nie przeładowana;
B – kolumna lekko przeładowana; C – kolumna mocno przeładowana;
2.3. Indeks retencji (I)
Indeks retencji (I) jest miarą retencji badanej substancji względem retencji
normalnych alkanów (łańcuch prosty) w danej temperaturze i dla danej kolumny.
Do obliczania indeksów retencji w warunkach temperatury izotermicznej stosuje się równanie
2a. W warunkach z programowaniem temperatury, można użyć równania 2b.
Masa próbki (Skala logarytmiczna)
13
Indeks retencji dla n-alkanów to liczba węgli pomnożona przez 100. Na przykład, n-dodekan
(n-C
12
H
26
) ma I = 1200. Jeżeli badana substancja posiada I = 1478, eluowana jest po n-C
14
a
przed n-C
15
, i bliższa jest n-C
15
. Indeksy retencji normalizują zmienne parametrów
przyrządów tak, że dane retencji mogą być porównywane dla różnych systemów GC. Indeksy
retencji dobrze służą również przy porównywaniu charakterystyk różnych kolumn.
Indeks retencji (I) w warunkach izotermicznych (2a)
I = 100y + [100 (z-y) (log t’
R(x)
– log t’
R(y)
)] / [ log t’
R(z)
– log t’
R(y)
]
Indeks retencji (IT) dla warunków z programowaniem temperatury (2b)
IT = 100 [ (t
R(x)
– t
R(y)
) / (t
R(z)
– t
R(y)
] + 100y
t
R
= czas retencji
x = badana substancja (analit)
y = n-alkany z liczbą y atomów węgla eluowane przed substancją rozpuszczoną x
z = n-alkany z liczbą z atomów węgla eluowane przed substancją rozpuszczoną x
z – y = różnica w liczbie węgli pomiędzy dwoma n-alkanami
14
2.4. Współczynnik rozdzielenia
α
Współczynnik rozdzielania jest miarą czasu lub odległości między
maksymalnymi wartościami dwóch pików.
Oblicza się go za pomocą równania 3. Jeżeli
α = 1, piki mają tę samą retencję i koeluują.
Współczynnik rozdzielania (
α)
α= k
2
/ k
1
(3)
k
1
= współczynnik retencji pierwszego piku
k
2
= współczynnik retencji drugiego piku
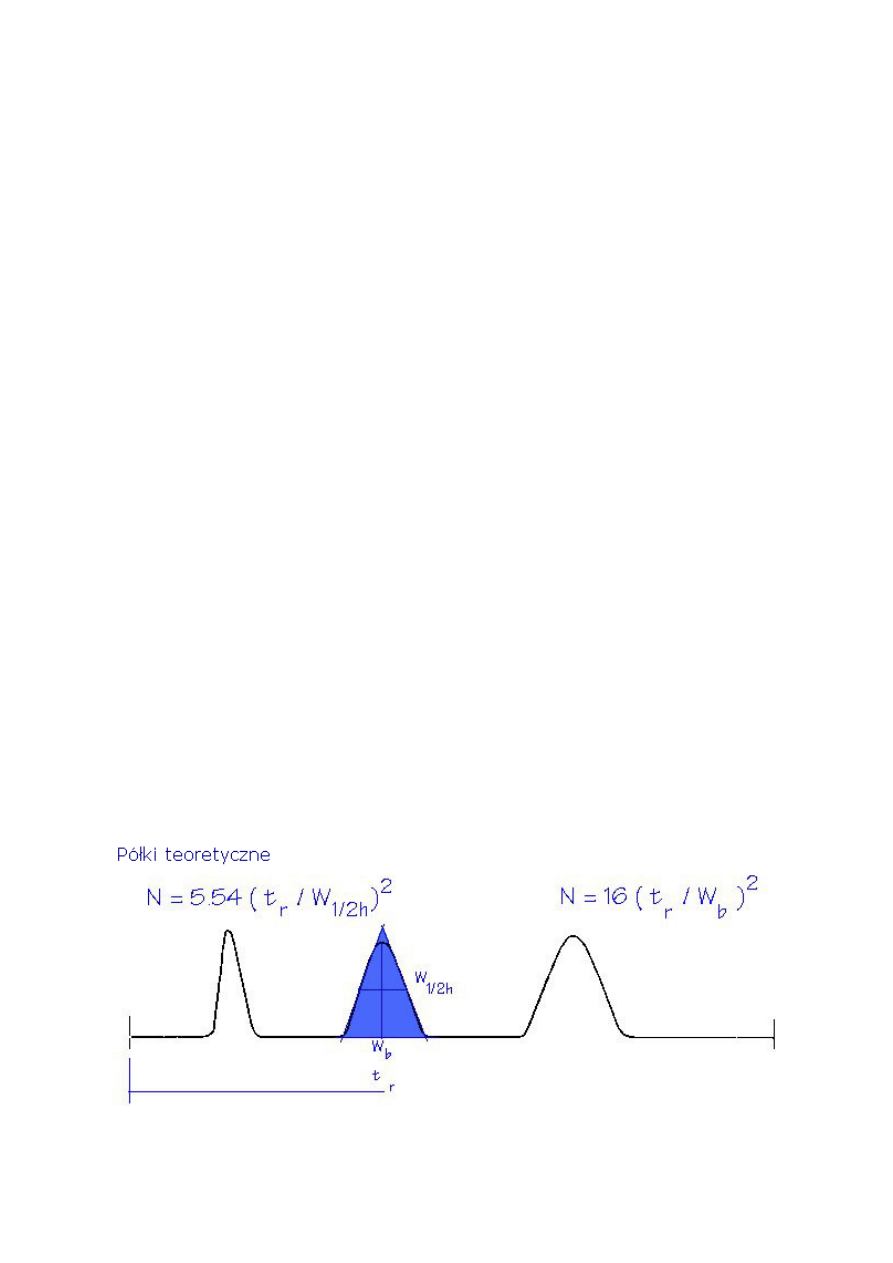
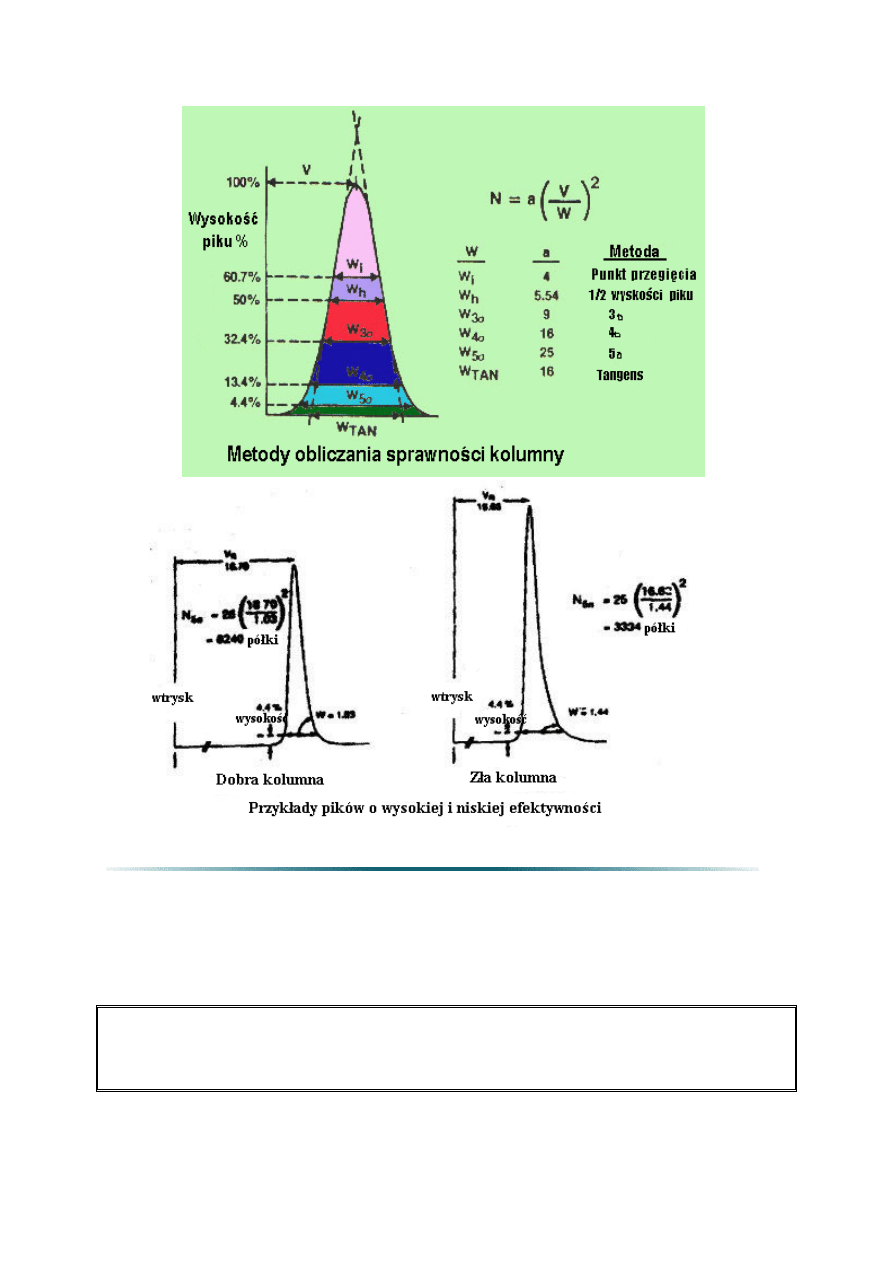
2.5. Teoretyczna liczba półek (N) lub sprawność kolumny
Sprawność kolumny wyrażona jest liczbą półek teoretycznych (N). Liczba półek
teoretycznych może być obliczona stosując równanie 4. Teoretyczne półki są pojęciem
umownym, ponieważ kolumna nie zawiera niczego co mogłoby przypominać fizycznie półki
destylacyjne albo inne podobne elementy.
Liczba teoretycznych półek jest pośrednią miarą szerokości piku dla piku o specyficznym
czasie retencji. Przyjmuje się, że kolumny z wyższą liczbą półek są bardziej sprawne niż
kolumny z mniejszą liczbą półek. Kolumna z dużą liczbą półek „wytwarzać” będzie węższe
piki w danym czasie retencji w porównaniu z kolumną o mniejszej liczbie półek.
15
Półki teoretyczne lub sprawność (N)
N = 5.545 [(t
R
) / w
h
]
2
(4)
N = 16[t
R
/ w
b
]
2
N = liczba półek teoretycznych
t
R
= czas retencji
w
h
= szerokość piku w połowie wysokości (w jednostkach czasu)
w
b
= szerokość piku w podstawie (w jednostkach czasu)
Wysoka sprawność kolumny ma szczególne znaczenie gdy należy rozdzielić nieduże piki.
Gdy stężenia substancji są większe, można użyć mniej efektywnych kolumn. Sprawność
kolumny jest funkcją wymiarów kolumny (średnicy, długości, i grubości filmu), rodzaju
gazu nośnego i jego natężenia przepływu lub średniej liniowej prędkości, oraz związku i jego
retencji. W celu porównania kolumn, często stosowana jest liczba teoretycznych półek na
metr (N/m).
Liczba teoretycznych półek ma znaczenie tylko w przypadku określenia konkretnych
warunków. Zwykle niezbędne są warunki temperatury izotermicznej a programowanie
temperatury przyczynia się do powstania nadmiernych półek. Także, współczynnik retencji
(k) testowanej substancji w celu obliczenia liczby półek powinien być większy od 5. Bowiem
mniej „zatrzymywane” piki przyczyniają się do nadmiernej liczby półek. Porównując liczby
półek teoretycznych dla różnych kolumn, wymaga się tych samych warunków
temperaturowych i retencji piku (k) aby porównanie było poprawne.
16
Wysokość równoważna półce teoretycznej (H)
Inną miarą sprawności kolumny jest wysokość
równoważna półce teoretycznej (H).
Jest ona obliczona za pomocą równania 5 i zazwyczaj podawana jest w mm. Im krótsza
każda z teoretycznych półek, tym więcej półek zawiera się w danym odcinku kolumny.
Przekłada się to na więcej półek teoretycznych na metr i wyższą sprawność. Małe wartości
wysokości równoważnych półce wskazują na wyższą sprawność.
Wysokość równoważna półce teoretycznej (H) (5)
H = L / N
L = długość kolumny (mm)
N = liczba półek teoretycznych
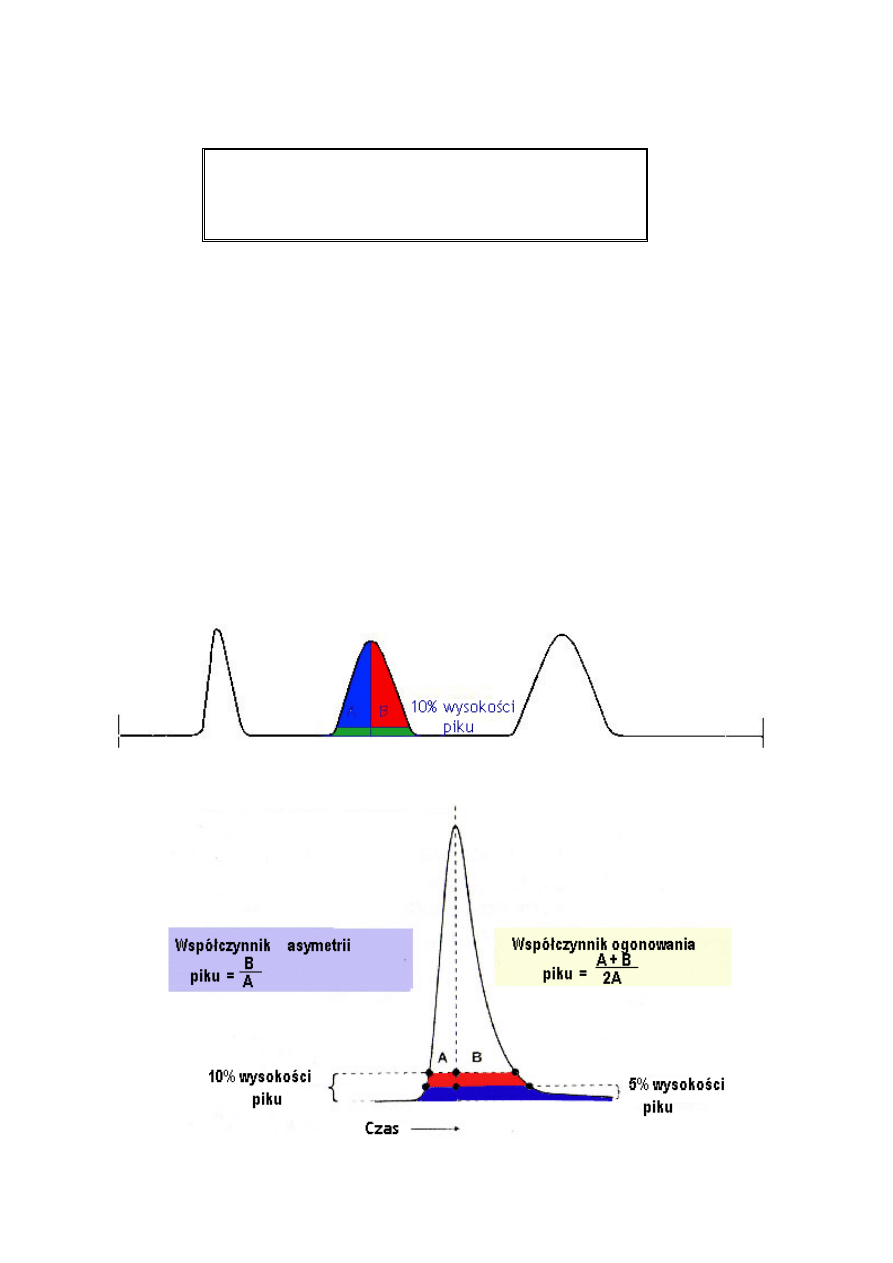
Asymetria
17
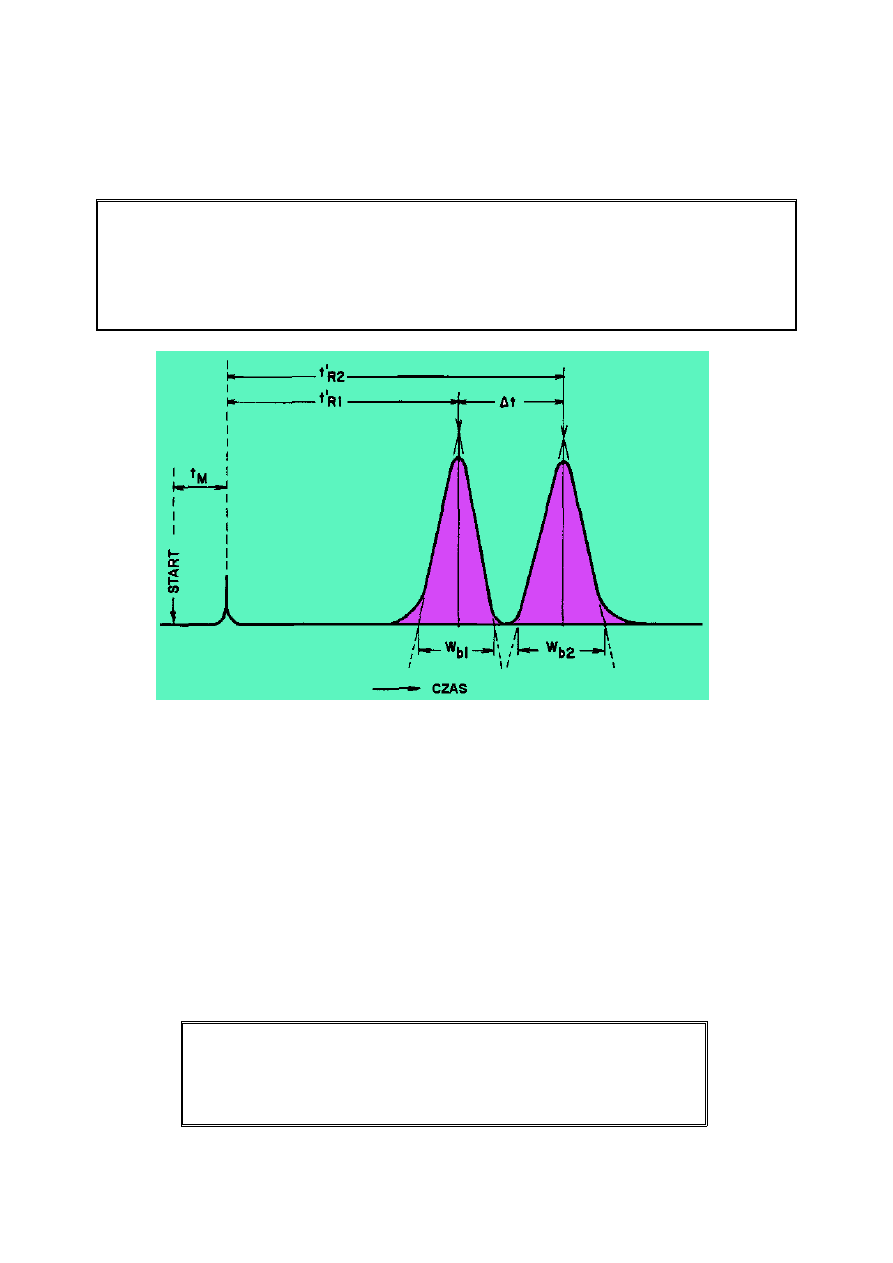
2.6. Rozdzielczość (R
S
)
Im wyższa rozdzielczość tym dwa piki nakładają się na siebie mniej.
Rozdzielenie to tylko odległość lub czas pomiędzy maksymalnymi wartościami (a) dwóch
pików. Rozdzielczość dotyczy zarówno odległości (a) i szerokości pików.
Może być ona obliczana z dwóch równań 6 (a i b).
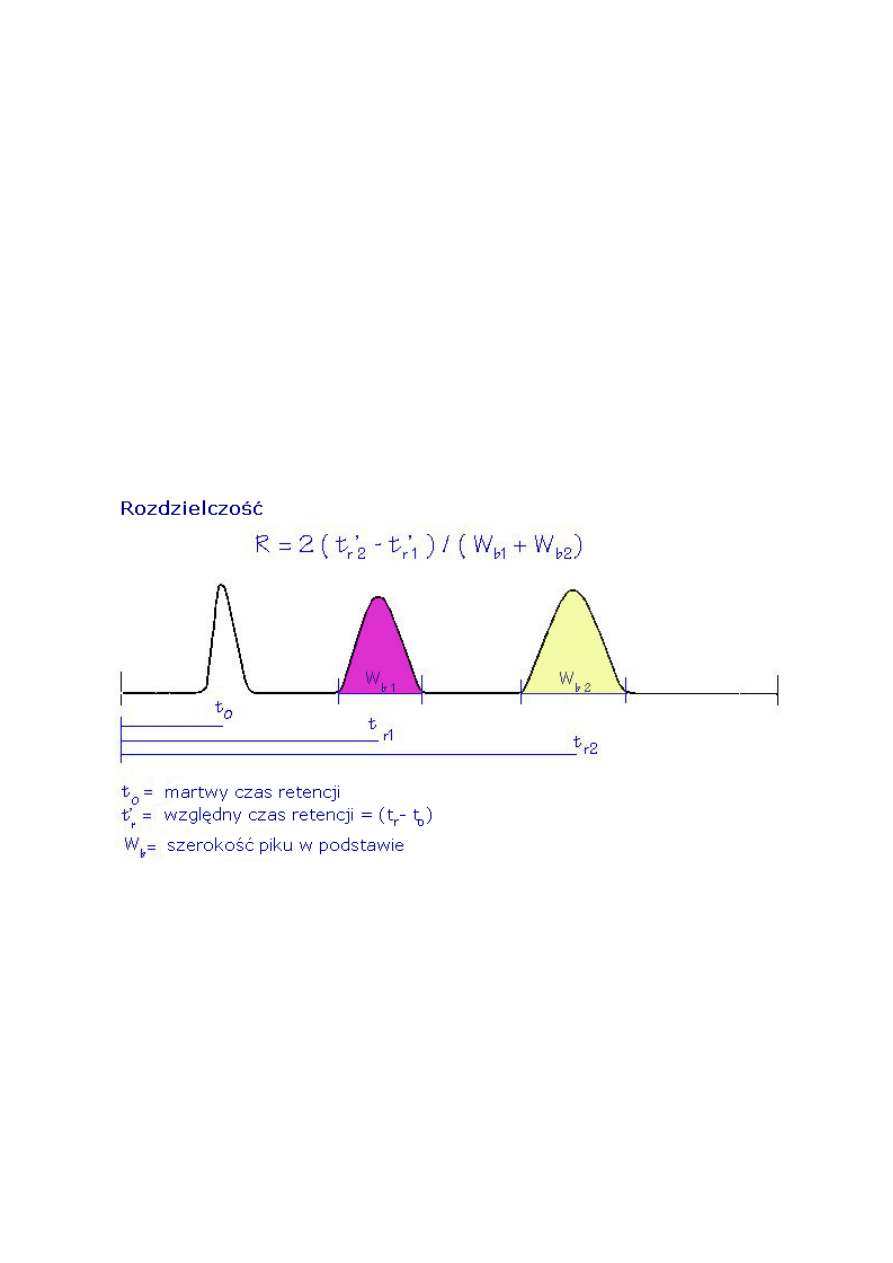
18
Rozdzielczość (R
S
)
R = 1.18 [ (t
R2
-t
R1
) / (w
h1
+ w
h2
)] (6 a)
R = 2 (t
r2
-t
R1
) / (w
b1
+ w
b2
) (6 b)
t
R1
= czas retencji pierwszego piku
t
R2
= czas retencji drugiego piku
w
h1
= szerokość piku w połowie wysokości (w jednostkach czasu) – pik pierwszy
w
h2
= szerokość piku w połowie wysokości (w jednostkach czasu) - pik drugi
w
b1
= szerokość piku przy podstawie (w jednostkach czasu) – pik pierwszy
w
b2
= szerokość piku przy podstawie (w jednostkach czasu) – pik drugi
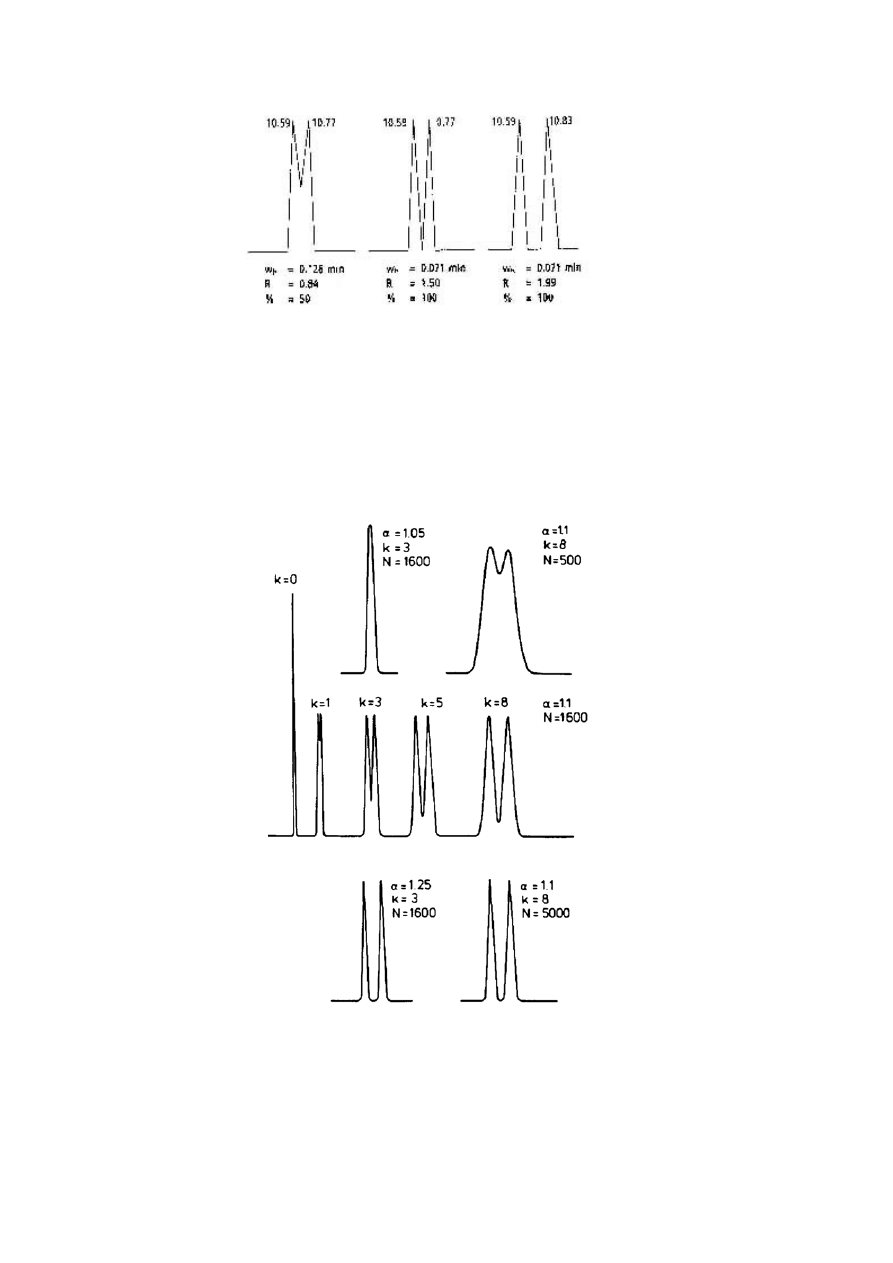
Rozdzielczość „do linii podstawowej” zazwyczaj ma miejsce przy wartości R 1.50, jednakże,
nie ma wtedy linii podstawy pomiędzy pikami. Wartości większe od 1.50 wskazują na linię
podstawy między pikami. Wartości mniejsze niż 1.50 wskazują na koelucję piku. Przykłady
można znaleźć na poniższym rysunku.
Przykłady rozdzielczości
19
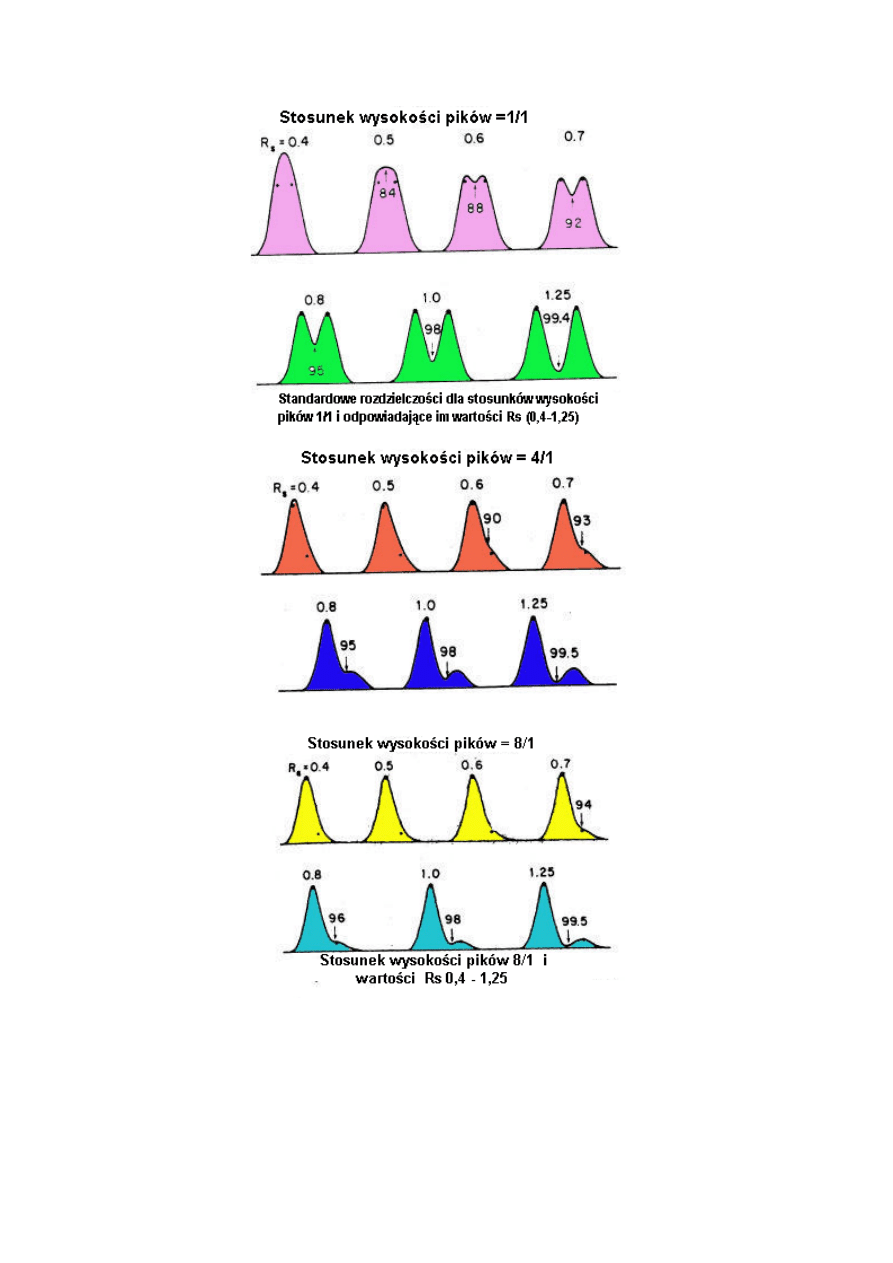
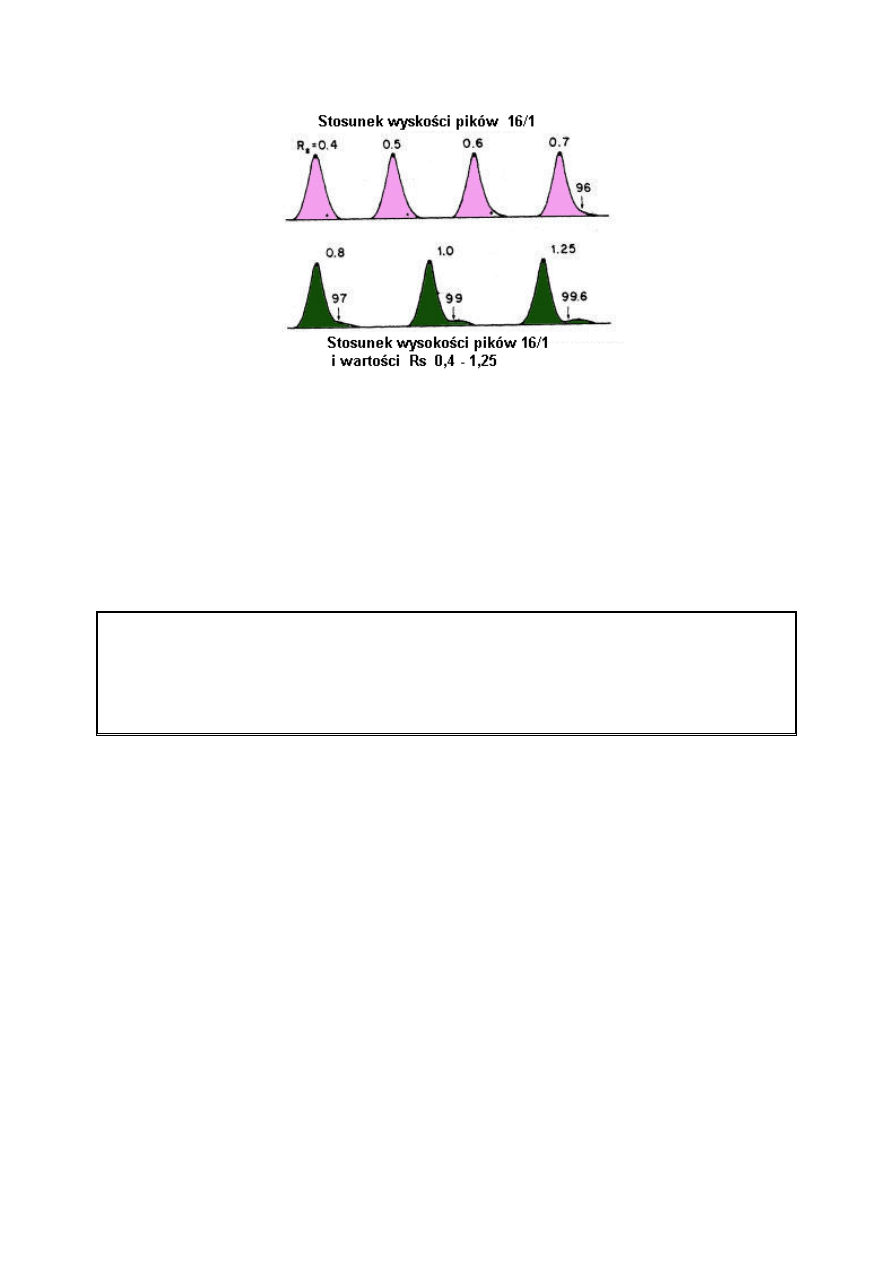
Czasami wartości rozdzielczości wyraża się w procentach. Są one obliczone ze stosunku
wysokości wgłębień pomiędzy pikami do całkowitej wysokości piku. Wartość tę łatwiej
można sobie wyobrazić niż liczbę rozdzielczości; jednakże, niemożliwe jest odróżnienie
poziomu rozdzielenia do linii podstawy.
Zmiany rozdzielczości jakie obserwuje się na chromatogramie przedstawiającym dwa piki dla
różnych wartości współczynnika separacji lub liczby teoretycznych półek. Średnia wartość
współczynnika separacji wskazana jest przez k.
20
21
2.7. Stosunek faz (β)
Stosunek faz kolumny (β) obliczony jest z równania 7. Dla tej samej fazy stacjonarnej i
temperatury kolumny (programowana lub izotermiczna), zmiana stosunku faz może być
wykorzystana w celu obliczenia zmiany retencji substancji. Zależność ta wyrażona jest za
pomocą równania 8.
Stała podziału (Kc) wyraża stosunek stężenia badanej substancji w fazie stacjonarnej i
ruchomej (C
S
/ C
M
). Stałe podziału są niezmienne dla tej samej fazy stacjonarnej, temperatury
kolumny i substancji rozpuszczonej.
Stosunek faz (β)
β = r / (2 d
f
) (7)
r = promień kolumny ( µm)
d
f
= grubość warstwy (µm)
Stała podziału (Kc)
Kc = C
s
/ C
M
(8)
Kc = k β = k [ r / (2 d
f
)]
C
s
= stężenie substancji rozpuszczonej w fazie stacjonarnej
C
M
= stężenie substancji rozpuszczonej w fazie ruchomej
22
Dlatego, dla danej fazy stacjonarnej i temperatury kolumny, można określić ilość i kierunek
jakiejkolwiek zmiany retencji w zależności do zmian średnicy kolumny lub grubości filmu
fazy stacjonarnej. Równanie 8 wykazuje, iż wzrost stosunku faz daje odpowiedni spadek
retencji (k) gdy Kc jest wartością stałą. Odwrotnie, spadek stosunku faz przyczynia się do
odpowiedniego wzrostu retencji (k).
Równanie 8 wskazuje na to, że stosunek faz maleje wraz ze zmniejszeniem się średnicy lub
ze wzrostem grubości warstwy. Każda z tych zmian daje wzrost retencji badanej substancji.
Stosunek faz wzrasta ze wzrostem średnicy lub spadkiem grubości warstwy. Każda z tych
zmian daje spadek retencji substancji. Czasami pożądana jest zmiana średnicy kolumny lub
grubości warstwy po to aby otrzymać specyficzny efekt (wzrost sprawności), bez zmiany
retencji. Można tego dokonać poprzez proporcjonalną zmianę średnicy kolumny i grubości
warstwy.
Na przykład, jeżeli średnica kolumny zostanie zmniejszona z 0.25 do 0.18 mm, odpowiednia
zmiana grubości warstwy ( np. 0.25 µm – 0.18µm) utrzymuje ten sam stosunek faz. Starania
ogólne mają przyczynić się do utrzymania tej samej retencji osiągając zarazem wyższą
sprawność dzięki zmniejszeniu średnicy kolumny. W tabela 1 przedstawiono stosunki faz dla
kolumn o najczęściej spotykanych średnicach.
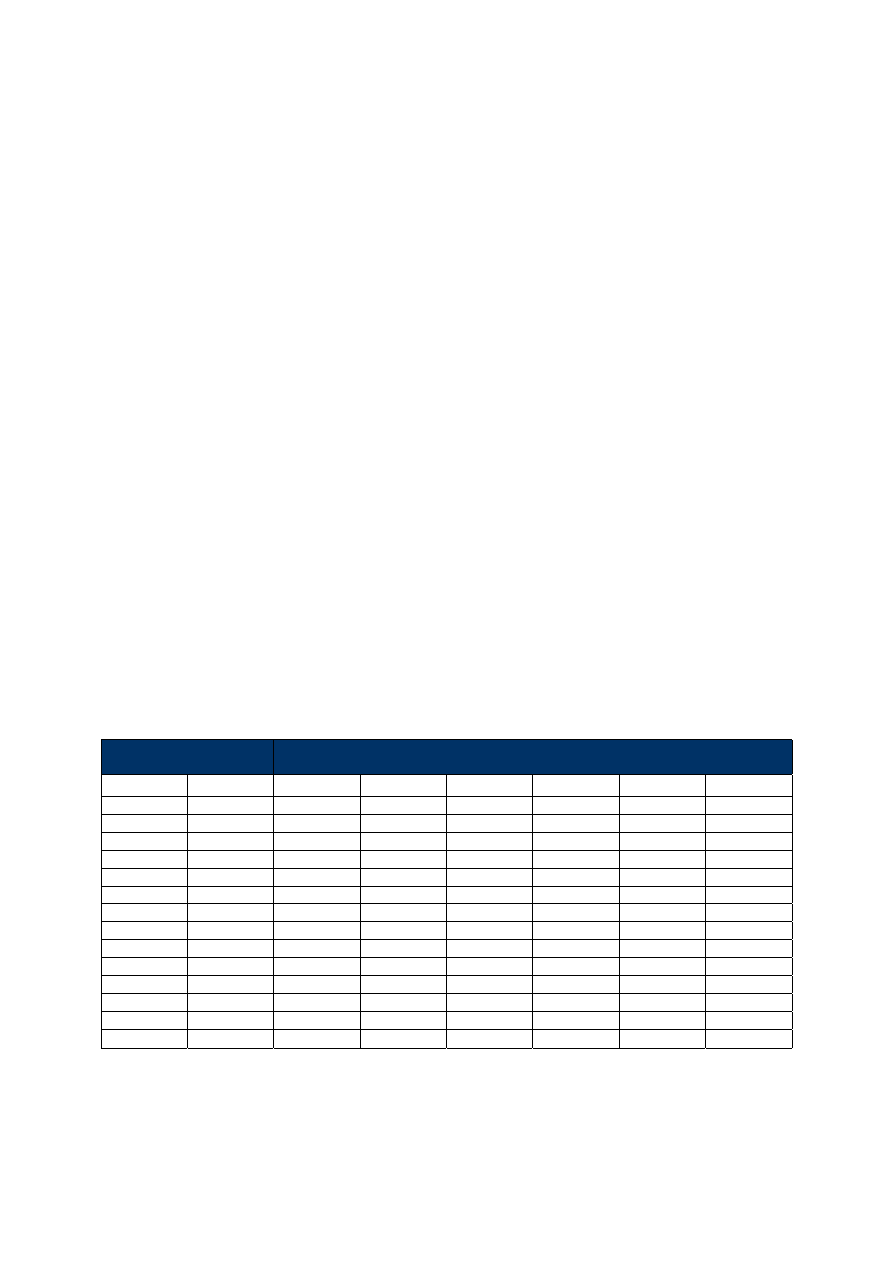
Tabela 1. Stosunek faz dla kolumn o najczęściej spotykanych średnicach.
Grubość warstwy
d
f
(µm)
Średnica kolumny
(mm)
0.10
0.18
0.20
0.25
0.32
0.45
0.53
0.10
250
450
500
625
800
1125
1325
0.18
139
250
278
347
444
625
736
0.25
100
180
200
250
320
450
530
0.40
63
113
125
156
200
281
331
0.42
107
119
149
190
268
315
0.50
90
100
125
160
225
265
0.83
60
75
96
136
160
0.85
59
74
94
133
156
1.00
50
63
80
113
133
1.27
49
63
88
104
1.50
42
53
75
88
2.55
25
31
44
52
3.00
21
27
38
44
5.00
13
16
23
27
23
Wyszukiwarka
Podobne podstrony:
Cz II Terminy i definicje
I,II,III termin cz Ö Ť ç II 2006,07
socjologia cz II
BADANIA DODATKOWE CZ II
Wykład 5 An wsk cz II
AUTOPREZENTACJA cz II Jak w
Podstawy Pedagogiki Specjalnej cz II oligo B
J Poreda Ewangelia zdrowia, cz II
mmgg, Studia PŁ, Ochrona Środowiska, Chemia, fizyczna, laborki, wszy, chemia fizyczna cz II sprawka
!Spis, ☆☆♠ Nauka dla Wszystkich Prawdziwych ∑ ξ ζ ω ∏ √¼½¾haslo nauka, hacking, Hack war, cz II
UE szczepienia i racjonalne stosowanie antybiotyków, Zdrowie publiczne, W. Leśnikowska - Ścigalska -
Dziady cz. II jako dramat, j.polski - gimnazjum
MIKROEKONOMIA cz.II
wskaźniki - zadania1, FIR UE Katowice, SEMESTR V, Analiza finansowa, Analiza finansowa1, Analiza fin
patomorfa, pytania 11 II termin
więcej podobnych podstron