
CHEMIA GLEBY
Ćwiczenie laboratoryjne.
Dr hab. inż. Agata Kot-Wasik
Wstęp
Gleba - biologicznie czynna powierzchniowa warstwa litosfery, powstała
ze skały macierzystej pod wpływem czynników glebotwórczych (głównie
organizmów żywych, klimatu i wody) i podlegająca stałym przemianom.
Powstała ona w wyniku długotrwałych procesów, które przebiegały na
powierzchni Ziemi. O długości tego procesu świadczy fakt, iż warstwa ziemi o
grubości 2-3cm kształtuje się od 200 do 1000 lat. Proces ten polega na
oddziaływaniu czynników klimatycznych, które powodują wietrzenie skał, jak i
na oddziaływaniu organizmów. Rozdrobniona skała zatrzymuje cząstki wody i
powietrze. Z czasem pojawiają się rośliny utrwalające glebę. Bardzo ważną
funkcję pełnią drobnoustroje, których zadaniem jest rozkładanie szczątków
roślinnych i zwierzęcych, wzbogacając w ten sposób glebę w próchnicę i
minerały.
Gleba składa się z trzech faz:
•
stałej – obejmującej cząstki mineralne, organiczne i organiczno-mineralne
o różnym stopniu rozdrobnienia; materia organiczna stanowi 5%, zaś
minerały 45%;
•
ciekłej – wody, w której są rozpuszczone związki mineralne i organiczne
tworzące roztwór glebowy; woda stanowi ok. 25%;
•
gazowej – mieszaniny gazów i pary wodnej; powietrze stanowi ok. 25%.
Składniki chemiczne
Wyróżniamy następujące pierwiastki: węgiel C, glin Al., wapń Ca, żelazo
Fe, chlor Cl, wodór H, potas K, magnez Mg, azot N, sód Na, tlen O, fosfor P,
siarka S, krzem Si + mangan Mn, molibden Mo, miedź Cu, kobalt Co, ołów Pb,
cynk Zn, bor B, tytan Ti.
Część mineralna to przede wszystkim: krzem Si, glin Al, żelazo Fe, wapń Ca.
Krzem Si występuje w 60 do 90 % pod postacią krzemionki. Glin Al występuje w
granicach 5 do 12%. Żelazo Fe występuje w postaci Fe
2+
oraz Fe
3+
. Natomiast
wapń Ca w postaci węglanu wapnia CaCO
3
. Zawartość CaCO
3
waha się
pomiędzy ilościami śladowymi, a kilkudziesięcioma procentami. Pozostałe
pierwiastki mierzymy w ppm; mogą sięgać najwyżej 1% zawartości.
1

Fizyczne właściwości gleby
Priorytetem przy ustalaniu właściwości fizycznych jest układ trójfazowy
tej gleby. Na fazę stałą przypadają cząstki mineralno-organiczne, mineralne i
organiczne. Fazę ciekłą stanowi roztwór glebowy, natomiast gazową powietrze.
Powietrze na przemian z roztworem glebowym wypełnia pory. Zasadniczymi
właściwościami fizycznymi są:
•
skład granulo-metryczny
Podstawowymi składnikami gleby są cząstki mineralne, które powstają w
wyniku erozji wietrznej działającej na skałę macierzystą. Do mineralnych
składników zaliczamy minerały: ilaste (np. illit), krzemianowe (np. kwarc i
skalenie) i bezkrzemowe (kalcyt i gips). Do części spławianych zaliczamy ił
pyłowy koloidalny, drobny i gruby. Składem granulometrycznym nazywamy
udział danej frakcji w definicyjnej jednostce masy gleby. Uznaje się to za
zasadniczą cechę gleby, mającą związek z materiałem macierzystym i jego
jakością.
•
gęstość gleby
Gęstością gleby nazywamy masę jednego metra sześciennego suchej gleby,
nienaruszonej strukturalnie. Jest zależna od uziarnienia oraz struktury gleby.
•
porowatość
Suma wolnych przestrzeni gleby. Wyróżniamy porowatość kapilarną oraz
niekapilarną.
•
zwięzłość
Jest to siła, będąca miarą spojenia cząsteczek. Mierzy się ją poprzez określenie
siły potrzebnej do ich rozdzielenia.
•
plastyczność
Jest cechą umożliwiającą przybieranie glebie różnych kształtów, gdy jest
wilgotna. Jest zależna od wielkości cząsteczek.
•
lepkość
Jest wyrażana zdolnością przylegania gleby. Zależy od składu mechanicznego
oraz wilgotności gleby.
•
pęcznienie, kurczenie
Zachodzi w glebach zawierających dużo cząstek koloidalnych. Zwiększenie
objętości przez gęstość, przy nawilgotnieniu to właśnie pęcznienie, a kurczenie
przebiega w drugim kierunku.
•
wodne właściwości
Woda może przyjmować różne postacie: wolną (kiedy przepływa z góry w dół
gleby, determinowana własną masą), kapilarną (wnikającą do najcieńszych
kanalików glebowych; jest rezerwuarem wilgoci w glebie i porusza się w
każdym kierunku), błonkową (trudnodostępna dla roślin; powleka cząsteczki),
higroskopową (silnie związaną i dostającą się do gleby z atmosfery; jest obecna
w ciężkich oraz próchniczych typach gleb), molekularną (zatrzymuje się na
cząsteczkach gleby, w wyniku działania sił adhezji), pary wodnej (ta znajduje
się w porach i jest częścią składową powietrza glebowego).
2

•
cieplne właściwości
Mają związek z przewodnictwem i pojemnością cieplną. Intensywność
nagrzewania oraz szybkość utraty ciepła gleby mają związek z barwą oraz
wilgotnością tej gleby. Ciepło może dostarczać słońce, procesy biologiczne i
powietrze.
Właściwości chemiczne oraz fizyko-chemiczne gleby
Skład chemiczny, formy, związki i przemiany pierwiastków określamy
mianem właściwości chemicznych gleby. Zwykle badania są prowadzone aby
oznaczyć:
•
zawartość materii organicznej gleby. Jeśli gleba jest prawidłowo
użytkowana powinna występować równowaga pomiędzy substancjami
organicznymi i tworzącymi się związkami próchnicowymi. W przypadku
przyspieszonej mineralizacji możemy wnioskować, iż doszło do
zakwaszenia lub akumulacji toksycznych związków.
•
zawartość próchnicy, a także węgla organicznego utlenialnego. Węgiel i
próchnica pozwalają oszacować zawartość substancji organicznej w
glebie, a także stopień jej humifikacji. Zawartość węgla w glebie świadczy
o zawartości próchnicy. Sposób oznaczenia opiera się na utlenianiu węgla
C do dwutlenku węgla CO
2
(oznaczenie przebiega w środowisku
kwaśnym).
•
zawartość azotu. Jest zależna od jakości oraz ilości substancji organicznej,
a także od stopnia rozkładu (C\N). Zawartość azotu w glebie to zawartość
azotu organicznego + zawartość związków mineralnych azotu.
•
ilość ołowiu, kobaltu, kadmu, niklu, magnezu i manganu.
Odczyn pH, sorpcyjność i właściwości ohydo-redukcyjne określają właściwości
fizyko-chemiczne.
Odczyn gleby – jest zależny od stężenia jonów wodorowych H
+
i
zasadowych OH
-
. Odczyn pH ma związek z aktywnością biologiczną. Jeśli
stosunek jonów kwasowych do zasadowych jest równy 1 to pH jest neutralne.
W środowisku kwaśnym występuje przewaga jonów H
+
, a w środowisku
zasadowym jonów OH
-
. Do oznaczenia odczynu gleby używa się różnych metod
(
jakich? ) .
Zdolność sorpcyjna – to zdolność absorbenta do absorpcji par, gazów,
cząsteczek niezdysocjonowanych oraz jonów pochodzących z roztworu
glebowego. Polega to na pochłanianiu wymienionych substancji, które zachodzi
na powierzchni tego absorbenta. W przypadku gleby sorpcja zależy od
koloidalnej fazy stałej (są to cząsteczki 2*10
-3
mm). Wyróżniamy: koloidy
glebowe, Fe(OH)
2
, Fe(OH)
3
, Al.(OH)
3
, minerały ilaste, kompleksy ilasto-
próchnicze i próchnicę. Wyróżniamy trzy typy sorpcji: biologiczną, chemiczną i
wymienną. W glebie funkcjonuje ta ostatnia. Jej istotą jest wymiana wcześniej
zaabsorbowanych jonów na te znajdujące się w roztworze glebowym.
Maksymalną ilość kationu H
+
, którą jest w stanie zaabsorbować 100 g
materiału glebowego nazywa się pojemnością sorpcyjną gleby. W czasie
3

zachodzących reakcji redoks dochodzi do przyłączania lub oddawania
elektronów. W czasie przemian materii organicznej w glebie dominują procesy
utleniania (są nieodwracalne).
4

Cel ćwiczenia
Celem ćwiczenia jest porównanie różnych właściwości fizyko-
chemicznych gleb. Materiałem badawczym będą próbki gleb przyniesione przez
studentów. Porównaniu poddane gleby piaszczyste (pobrane z plaży),
ogrodnicze, gliniaste i pobrane z okolic tras komunikacyjnych.
OZNACZANIE WILGOTNOŚCI WAGOWEJ GLEBY
Zakres tego pomiaru obejmuje wyznaczenie wilgotności wagowej materiałów
glebowych. Wilgotność wagową oznacza się metodą straty masy podczas suszenia.
Wykonanie pomiaru
1.
Suche naczynka wagowe (np. szkiełka zegarkowe) po odpowiednim
oznaczeniu zważyć
(m
n
) przy pomocy wagi analitycznej z dokładnością do
0,001g. Wynik zapisać.
2.
Próbki gleb umieścić w naczynkach wagowych i zważyć przy pomocy wagi
analitycznej
(do oznaczenia pobrać ok. 5 g gleby). Wynik zapisać (m
gwn
).
3.
Naczynka z glebą umieścić w suszarce i suszyć do stałej masy (ok. 1
godzinę).
4.
Po wysuszeniu próbki gleb z naczynkami ostudzić.
5.
Po ostudzeniu ponownie zważyć naczynka z glebą a wyniki zapisać (m
sn
).
6.
Obliczyć masę gleby wilgotnej m
gw
(m
gwn
-m
n
) i masę gleby suchej m
s
(m
sn
m
n
).
7.
Obliczyć masę wody zawartej w próbce m
w
(m
gw
-m
s
).
8.
Obliczyć wilgotność wagową gleby W :
W= (m
w
/ m
s
) * 100 [%] = ((m
gw
-m
s
)/m
sn
-m
n
) * 100 [%]
5
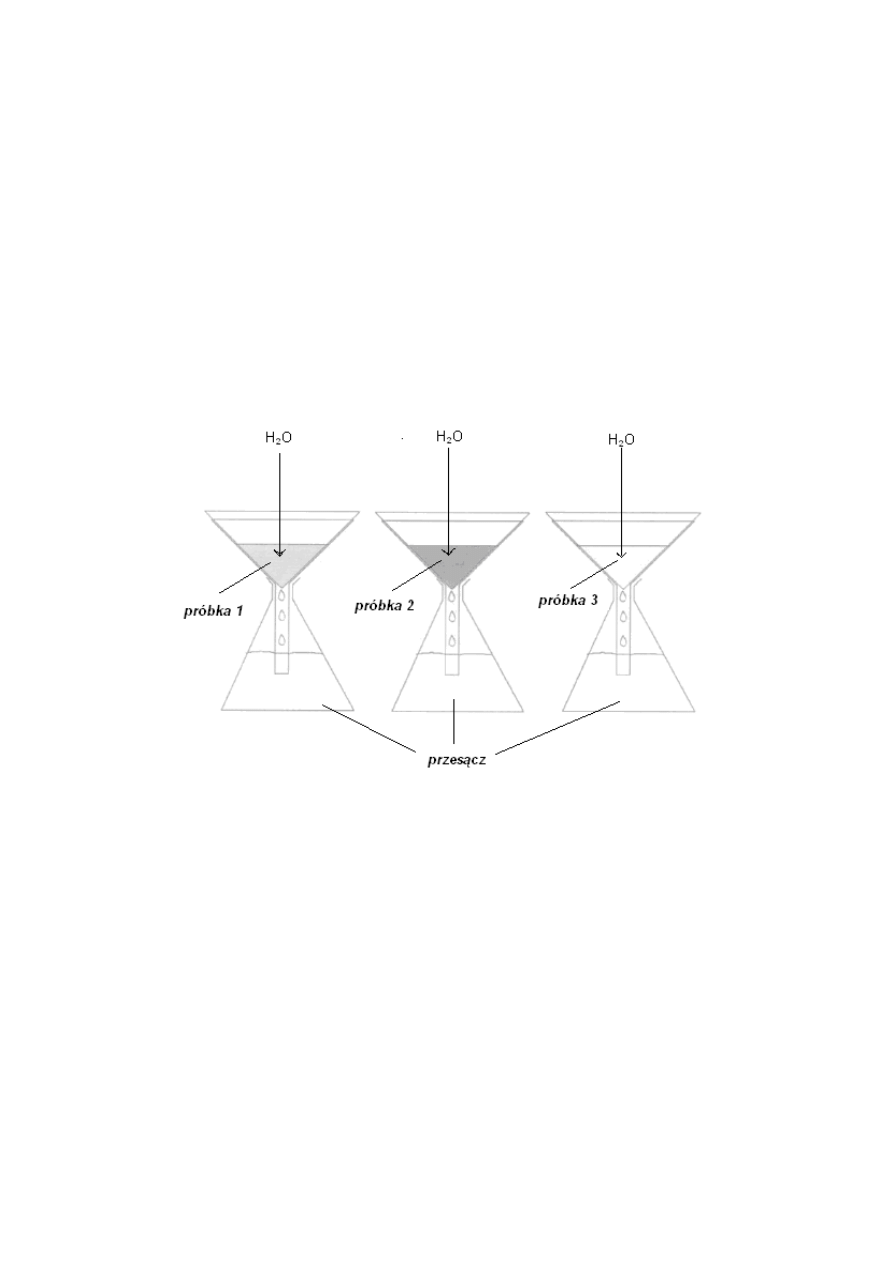
WYZNACZANIE POJEMNOŚCI WODNEJ GLEBY
Celem tego pomiaru jest określenie zdolności gleby do zatrzymania wody na
podstawie wyznaczenia jej maksymalnej pojemności wodnej.
Wykonanie pomiaru
1.
Na dno lejka włożyć po kawałeczku waty (lub umieścić saczek) oraz
wsypać 10 g
(m
s
) suchej gleby (najlepiej w dwóch powtórzeniach).
2.
Lejki wstawić do cylindra miarowego i przesyć przez glebę równą ilość
wody (30g (m
1
)) UWAGA! Wlewamy wodę tak, aby zwilżyć równomiernie całą
powierzchnię
gleby, kilkakrotnie przelewając przesącz przez próbkę.
3.
Po ustaniu odcieku odczytać ilość wody, która wypłynęła z badanej gleby
do cylindra miarowego (m
2
)
4.
Na podstawie uzyskanych wyników obliczyć pojemność wodną PW
PW = (m
1
-m
2
)/m
s
*100 [%]
gdzie:
PW - pojemność wodna gleby [% wag],
m
1
- masa wody, jaka zalano glebę (np. 300 g),
m
2
– masa wody grawitacyjnej, która przeszła przez glebe,
m
s
- masa suchej gleby.
MIERZENIE WŁAŚCIWOŚCI KAPILARNYCH GLEB
6
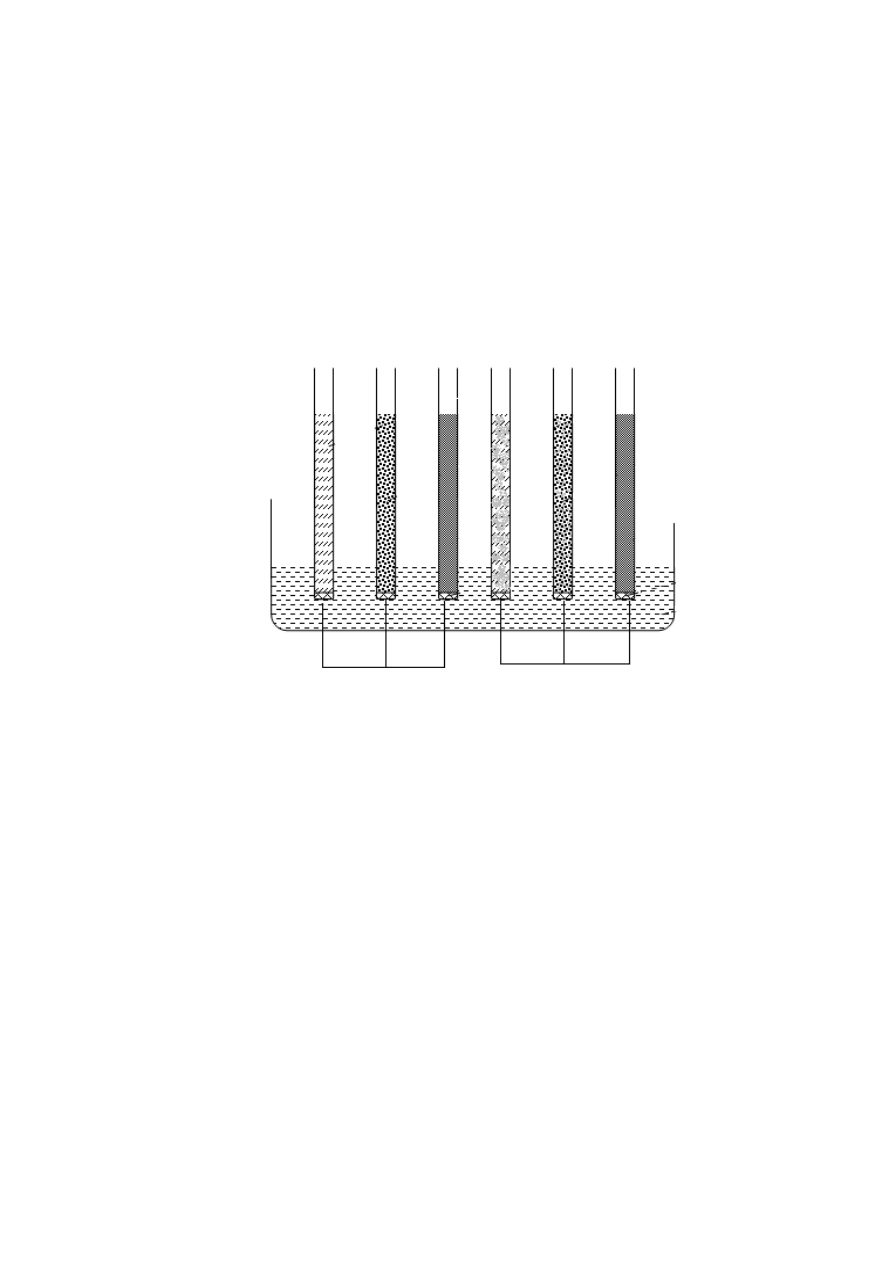
Celem tej części ćwiczenia jest wyznaczenie wysokości
maksymalnej i szybkości
uzyskania podsiąku maksymalnego gleb.
Wykonanie pomiaru
Do rurek szklanych o jednakowej średnicy (ok. 1 cm) i przekroju, z
jednakowym końcem obwiązanym gazą, wsypać na jednakową wysokość różne
rodzaje wysuszonych gleb. Rurki z glebą umocować pionowo w statywie tak,
aby dolny koniec każdej rurki zanurzony był w wodzie (na wysokość 3-5 cm).
Po upływie 10, 20, 30, 40, 50, 60 i 90 minut oznaczać poziom, do jakiego
podnosi się woda. Wyniki zapisać (w tabeli), porównać i skomentować.
7
tu opis próbek gleby

OZNACZANIE KWASOWOŚCI GLEB
Odczyn gleby mówi nam o tak zwanym zakwaszeniu gleby.
Jony wodorowe zakwaszające glebę pochodzą z
różnego rodzaju kwasów
organicznych i nieorganicznych oraz fizjologicznie kwaśnych soli. Większość
kwasów glebowych powstaje w wyniku procesów życiowych mikroorganizmów.
Wszystkie elektrolity (kwasy, zasady i sole) dysocjują w wodzie na jony:
HCl = H
+
+ Cl
–
NaOH = Na
+
+ OH
–
Mocne kwasy (np. kwas solny, azotowy i siarkowy) dysocjują prawie
całkowicie i o tych kwasach mówimy, że są silnie kwaśne.
Przeciwieństwem tych kwasów są kwasy słabo zdysocjowane. Należą do nich
wszystkie kwasy organiczne (np. kwas octowy, mrówkowy, cytrynowy) i inne, w
których roztworach obecne są małe ilości jonów wodorowych H
+
.
Odpowiednikami mocnych i słabych kwasów są mocne i słabe zasady.
O ile jony wodorowe decydują o kwasowości danego roztworu, o tyle jony
wodorotlenowe są odpowiedzialne za jego zasadowość.
Jeżeli w roztworze znajduje się tyle samo jonów wodorowych i
wodorotlenowych mówimy o środowisku obojętnym. Dla scharakteryzowania
kwasowości lub zasadowości danego roztworu można podawać stężenie tego
samego rodzaju jonów, mianowicie jonów wodorowych. Stężenie to jest małą
liczbą i dlatego dla wygody podaje się ujemny logarytm dziesiętny ze stężenia
jonów wodorowych, oznaczony skrótem pH. I tak stężenie jonów wodorowych w
1 litrze destylowanej wody w temp. 25
o
C wynosi 7. Czyli:
-
środowisko obojętne ma pH = 7
-
środowisko kwaśne, poniżej 7
-
środowisko zasadowe, powyżej 7
Wartość pH jest wielkością logarytmiczną. Stąd zmianie pH o jednostkę
odpowiada 10-ciokrotny wzrost stężenia jonów wodorowych.
W glebie można wyróżnić dwa rodzaje kwasowości: aktywną i
potencjalną. Przez pojęcie kwasowości aktywnej rozumie się ilość
zdysocjowanych wolnych jonów wodorowych występujących w roztworze
glebowym; ich miarą jest pH oznaczone
w zawiesinie wodnej. Z
kolei Kwasowość potencjalna obejmuje niezdysocjowane jony wodorowe
związane z kompleksem sorpcyjnym gleby czyli bardzo drobnymi cząsteczkami
i fazy stałej gleby. Jony te mogą być ujęte przy pomiarze pH po uprzednim ich
przeprowadzeniu z
kompleksu sorpcyjnego do roztworu glebowego przy użyciu
roztworu
chlorku potasowego. Dla charakterystyki gleb z punktu widzenia
gleboznawczego mierzy się pH w zawiesinie wodnej. Natomiast dla celów
rolniczych oznacza się pH w roztworze chlorku potasowego (0,1 - 1,0 mol/dm
3
).
Można także mierzyć pH gleby w roztworze CaCl
2
o stężeniu 0,01 - 0,1
mol/dm
3
.
8

Do pomiaru pH roztworu glebowego próbki gleby należy pobierać z
warstwy 0-20 cm. Pomiar pH gleby mierzy się w zawiesinie glebowej o stężeniu
2,5 części roztworu na 1 część gleby.
Najczęściej do oznaczania potencjometrycznego pH stosuje się elektrodę
kombinowaną.
Wykonanie oznaczenia pH gleby
W zawiesinie wodnej: odważyć 10 g gleby (roztartej i przesianej przez
sito o średnicy oczek 1 lub 2 mm) w zlewce o pojemności 50 ml. Dodać 25
ml
wody destylowanej i wymieszać. Następnie zmierzyć temperaturę zawiesiny
glebowej. UWAGA: ten roztwór posłuży do pomiaru zawartości substancji
odżywczych – nie wylewać po zrobieniu pomiaru pH !
W roztworze CaCl
2
lub KCl: przygotować roztwór 0.1 M CaCl
2
i/lub 1M
KCl. Następnie należy sporządzić zawiesinę w stosunku 10g gleby + 25mL
roztworu CaCl
2
(10g gleby oraz 25mL roztwory KCl). Glebę z roztworem CaCl
2
(ew. Kcl) należy mocno wstrząsnąć kilka razy i pozostawić do odstania.
Najwcześniej po 10 minutach w zawiesistej cieczy może się ustabilizować
równowaga. Następnie w zawiesinie gleby zanurzamy filtr, tak aby czysty
roztwór zebrał się we wnętrzu filtra. W tym czystym roztworze zmierzymy
wartość pH.
Pomiar papierkiem wskaźnikowym:
Pomiar potencjometryczny: podłączyć do aparatu elektrodę,
przepłukując ją wodą destylowaną i zanurzyć w roztworze buforowym o
znanym pH do kalibracji aparatu. Wybrać bufor o odczynie bliskim
spodziewanej wartości pH badanej zawiesiny (pH~7). Po kalibracji aparatu
odstawić bufor, przemyć elektrodę wodą destylowaną, wymieszać zawiesinę
glebową i zmierzyć pH zanurzając elektrodę w zawiesinie glebowej. Po
zakończeniu oznaczeń elektrodę należy przepłukać wodą destylowaną. W celu
przechowania elektrody do następnych pomiarów elektrodę umieścić w
9

nasyconym roztworze chlorku potasowego.
10

Wyniki pH gleby wskazują na :
pH < 3,5 →
gleba ekstremalnie kwaśna
pH ~ 4,5 →
gleba silnie kwaśna
pH ~ 5,5 →
gleba kwaśna
pH ~ 6,5 →
gleba lekko kwaśna
pH ~ 7,2 →
gleba obojętna
pH ~ 8,5 →
gleba zasadowa
pH > 8,5 →
gleba silnie zasadowa
Ocena: obojętna po lekko kwaśną wartość pH gleby (pH 6 do 7)
- pozytywnie wpływa na aktywność biologiczną gleby a więc także
tworzenie próchnicy
- dobrze wpływa na rozpuszczalność substancji odżywczych
- zapobiega uwalnianiu trujących jonów glinu
- oraz wspiera wzrost roślin.
11
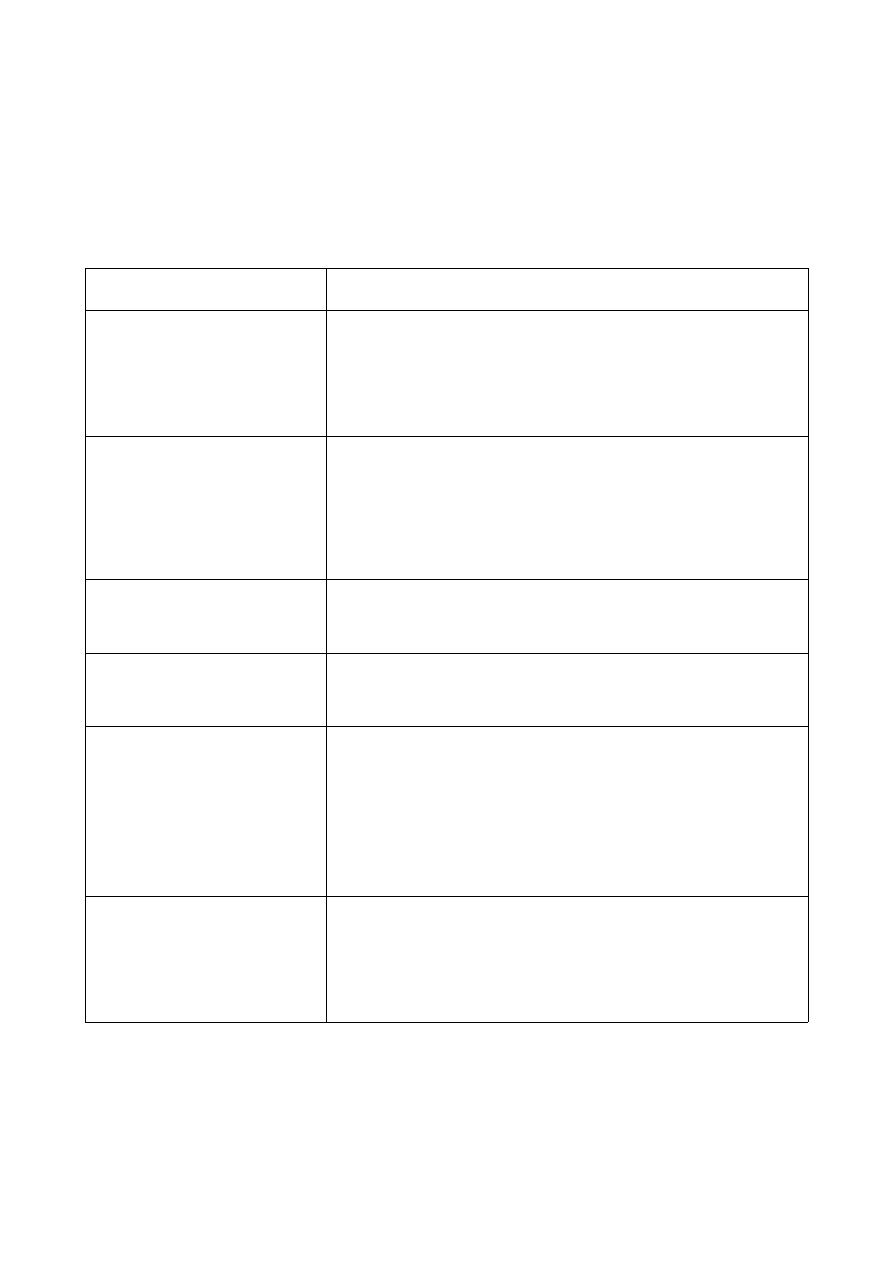
POMIAR ZAWARTOŚCI SUBSTANCJI ODŻYWCZYCH
Pomiaru zawartości substancji odżywczych (pokarmowych, nutrientów)
dokonuje się w roztworze wodnym uzyskanym podczas pomiaru pH gleby.
Substancje odżywcze
Procedura
NH
4
+
4 – 80 mg/L
0.5 – 16 mg/L
0.2 – 8 mg/L
Ekstrakt o objętości 0.1 - 1mL (pH = 4 - 13) wlać do naczynia
zawierającego odczynnik i wymieszać. Następnie wsypać 1
łyżeczkę odczynnika (umieszczoną w zakrętce odczynnika
NH4-1K). Zawartość naczynia wymieszać do całkowitego
rozpuszczenia. Próbkę pozostawić na 15 minut, po czym
należy dokonać pomiaru.
NO
3
-
2 – 80 mg/L
Wsypać 1 łyżeczkę (umieszczoną w zakrętce odczynnika NO3-
1K) do naczynia z roztworem. Zawartość naczynia wymieszać
do całkowitego rozpuszczenia. Ekstrakt o objętości 1.5mL (pH
= 1 - 3) wlewać powoli do naczynia zawierającego mieszaninę,
zamknąć i wymieszać. UWAGA: naczynie będzie gorące.
Próbkę pozostawić na 10 minut, po czym należy dokonać
pomiaru.
NO
2
-
0.05 – 2 mg/L
Ekstrakt o objętości 5mL (pH = 3 - 11) ostrożnie wlać do
naczynia zawierającego odczynnik i wymieszać. Próbkę
pozostawić na 10 minut . Dokonać pomiaru.
P
0.5 – 25 mg/L
Ekstrakt o objętości 5mL (pH = 5 - 8) wlać do naczynia
zawierającego odczynnik i dokładnie wymieszać. Dokonać
pomiaru.
SO
4
2-
100 – 1000 mg/L
Przygotowanie próbki odniesienia: ekstrakt o objętości 5mL
wlać do naczynia z białą nakrętką.
Przygotowanie próbki pomiarowej: ekstrakt o objętości 1mL
wlać do naczynia zawierającego odczynnik i wymieszać.
Następnie wsypać 1 łyżeczkę odczynnika (zieloną,
umieszczoną w zakrętce odczynnika SO4-1K). Zawartość
naczynia wymieszać do całkowitego rozpuszczenia. Próbkę
pozostawić na 10 minut, po czym należy dokonać pomiaru.
SO
3
2-
1-25 mg/L
Wsypać 1 łyżeczkę (umieszczoną w zakrętce odczynnika SO3-
1K) do naczynia z roztworem. Zawartość naczynia wymieszać
do całkowitego rozpuszczenia. Ekstrakt o objętości 3mL (pH =
7 - 9) wlać do naczynia zawierającego mieszaninę, zamknąć i
wymieszać. Próbkę pozostawić na 2 minuty, po czym należy
dokonać pomiaru.
12

OZNACZANIE METALI BIOPRZYSWAJALNYCH
W przypadku stałych próbek środowiskowych przeprowadzenie do
roztworu konieczne jest nie tylko w celu oznaczenia całkowitej zawartości
pierwiastków, ale również określenie udziału poszczególnych form chemicznych
i fizycznych analitu poprzez poznanie stężeń poszczególnych związków
chemicznych i form fizycznych, w których dany pierwiastek występuje w
badanej próbce. Zastosowanie technik ekstrakcyjnych daje możliwość
oddzielania analitu od matrycy, eliminacji lub redukcji interferencji
pochodzących od innych składników, a także wzbogacenie próbki (analitu) do
poziomu umożliwiającego oznaczenie. Procedury ekstrakcji sekwencyjnej
(wieloetapowej) pozwalają na wyodrębnienie kilku frakcji metali. Dzięki
wykorzystaniu procedur symulujących przebieg naturalnych zjawisk
przyrodniczych (np. kwaśne deszcze) ułatwiają określenie biodostępności i
mobilności metali. Tego typu procedury mają zastosowanie w badaniu:
materiałów geologicznych, osadów rzecznych, osadów morskich, osadów z
lagun, osadów kanalizacyjnych oraz popiołów.
Przy tym sposobie postępowania różnicowanych jest kilka frakcji:
1.
frakcja wymienna - metale zaadsorbowane;
2.
frakcja węglanowa - metale związane z węglowodorami;
3.
frakcja podatna na redukcję - metale związane z tlenkami Fe i Mn;
4.
frakcja utlenialna (utleniająca się) - metale związane ze składnikami
organicznymi i
siarczkowymi;
5.
frakcja szczątkowa (pozbawiona struktury) - metale związane ze
szkieletem mineralnym.
Wpływ na wydajność i powtarzalność techniki ekstrakcji wieloetapowej
mają:
1. właściwości chemiczne, selektywność i wydajność wybranych ekstrahentów,
2. kolejność poszczególnych kroków,
3. warunki pracy, takie jak: pH ekstrahentów, ich stężenia, czas ługowania,
stosunek mas ciała stałego do roztworu, temperatury ekstrakcji, rodzaj
atmosfery nad roztworem (powietrze, azot), sposób rozdziału faz tj. wpływ
prędkości wirowania, czasu, wirowania, rodzaju użytych sączków, itp.,
4. „efekty matrycowe” związane z zawartością faz i pierwiastków oraz
readsorpcją.
Przykład ekstrakcji sekwencyjnej :
1. frakcja wymienna: 1 g próbki (gleby, osadu) ekstrahować z 8 ml 1 M MgCl
2
(pH = 7,0) wytrząsając 1 godzinę w temperaturze pokojowej;
13

2. frakcja węglanowa: osad z punktu 1. ługować w temperaturze pokojowej z 8
ml 1 M CH
3
COONa (pH = 5,0 - regulowane CH
3
COOH) przez 5 godzin ciągle
mieszając;
3. frakcja podatna na redukcję: do pozostałości z punktu 2. dodać 20 ml 0,04M
chlorowodorku hydroksyaminy w 25 % CH
3
COOH, ekstrakcję prowadzić w
temperaturze 96 °C przez 6 godzin;
4. frakcja utlenialna: do osadu z etapu 3 dodać 3 ml 0.02 M HNO
3
i 5 ml 30%
wody utlenionej, mieszaninę ogrzewać w temperaturze 85 °C przez 2
godziny, po tym czasie dodać 3 ml 30% H
2
O
2
i próbkę ponownie ogrzewać
przez 3 godziny, po ostudzeniu do próbki dodać 5 ml 3,2 M CH
3
COOHNH
4
w
20 % HNO
3
, próbkę rozcieńczono do objętości 20ml; i wytrząsano 30 min;
5. pozostałość: 1 gram próbki roztworzyć w mieszaninie stężonych kwasów:
nadchlorowego (2 ml) i fluorowodorowego (10 ml) w tyglu platynowym, po
odparowaniu prawie do sucha dodać 1 ml HClO
4
i ogrzewać do pojawienia
się białych dymów, pozostałość rozpuścić w 12 M HCl i uzupełnić do
objętości 25 ml.
Pomiędzy poszczególnymi etapami próbki należy odwirowywać 30 min z
prędkością 10000 obrotów na minutę, roztwór znad osadu oddzielać przy
pomocy pipety i poddawać analizie, natomiast osad przemywać za pomocą 8
ml wody dejonizowanej, którą po odwirowaniu (30 min) należy odrzucić.
W poszczególnych fakcjach oznaczyć można zawartość: Cd, Co, Cu, Fe,
Mn, Ni, Pb i Zn z zastosowaniem atomowej spektrometrii absorpcyjnej.
Zamiast konwencjonalnego wytrząsania próbek można wykorzystać
energię mikrofalową i ultradźwięki, co znaczne skraca czas ekstrakcji.
Do zidentyfikowania frakcji metali bioprzyswajalnych wystarczającym
sposobem postępowania jest zastosowanie ekstrakcji za pomocą kwasu
octowego.
Wykonanie oznaczenia metali z próbek gleby
Odczynniki:
Roztwór kwasu octowego, 0,11 mol/dm
3
: do kolby o pojemności 100 cm
3
zawierającej ok. 50 cm
3
wody destylowanej wlać 2,5 cm
3
roztworu kwasu
octowego. Uzupełnić do kreski. 25,0 cm
3
tego roztworu (kwas octowy o
stężeniu 0,43 mol/dm
3
) rozcieńczyć do objętości 100 cm
3
uzyskując tym samym
roztwór
o
stężeniu
0,11 mol/dm
3
.
Procedura ekstrakcji :
Ługowanie frakcji metali bioprzyswajalnych - do naczynia zawierającego
ok. 1 g (dokładnie odważonego) analizowanej próbki gleby dodać 20 cm
3
0,11
M kwasu octowego. Naczynie zamknąć (parafilmem) i umieścić w myjce (łaźni)
ultradźwiękowej na okres 5 minut, po czym odwirować (5 minut, 6000
14

obr./min.); roztwór z nad osadu zlać do plastikowego pojemnika i poddać
analizie na zawartość Fe, Cd, Zn.
15

Oznaczanie metali w ekstraktach z gleby
Metal
Procedura
Zn
0.025 – 1.00 mg/L
Ekstrakt o objętości 10 mL (pH=1 - 7) wlać do naczynia
zawierającego KCN, wsypać 1 łyżeczkę (szarą; umieszczoną w
zakrętce odczynnika Zn-1K) odczynnika. Zawartość naczynia
wymieszać do całkowitego rozpuszczenia. Następnie do
pustego naczynia wlać 0.5 mL odczynnika Zn-2K, naczynie
zamknąć szczelnie i wytrząsać. Do niego wlać potem 2 mL
mieszaniny z odczynnikiem Zn-1K i ponownie wymieszać. Po
czym dodać 5 kropli odczynnika ZN-3K i znowu wymieszać.
Próbkę pozostawić na 15 minut, po czym należy dokonać
pomiaru.
Cd
0.025 – 1.00 mg/L
Ekstrakt o objętości 5 mL (pH=5 - 9) wlać do naczynia, dodać
3 krople odczynnika Cd-1K i wymieszać. Dodać 1 łyżeczkę
(szarą; umieszczoną w zakrętce odczynnika Cd-2K)
odczynnika. Zawartość naczynia wymieszać do całkowitego
rozpuszczenia. Próbkę pozostawić na 5 minut, po czym należy
dokonać pomiaru.
Fe
0.1 – 4.0 mg/L
Ekstrakt o objętości 5 mL (pH=1 - 7) wlać do naczynia, dodać
1 łyżeczkę (niebieską; umieszczoną w zakrętce odczynnika Fe-
1K) odczynnika. Zawartość naczynia wymieszać do
całkowitego rozpuszczenia. Próbkę pozostawić na 3 minuty, po
czym należy dokonać pomiaru.
Literatura
1. Tessier, A., Campbell, P. G. C. i Bisson, M.,
Analytical Chemistry, 1979, 51
(7), 844-851
2. Namieśnik J., Jamrógiewicz Z., Pilarczyk M., Torres L.,
Przygotowanie
próbek środowiskowych do analizy, Wydawnictwa Naukowo-Techniczne,
Warszawa 2000
3. Kot A., Namieśnik J., Trends in Anal. Chem., 2000, 19, 69-79
16
Po wykonaniu oznaczenia uporządkować stanowisko
laboratoryjne !
Wyszukiwarka
Podobne podstrony:
cw 7 wstęp, chemia środowiska
cw 6 wstep, chemia środowiska
cw 1 wstep, chemia środowiska
PYTANIA Z CHEMII ROLNEJ CW, Studia, Semestr III, Chemia srodowiska
Ćw 3, AGH, SEMESTR 4, CHEMIA ŚRODOWISKA, 3. Oznaczanie zawartości siarki w węglu metodą Eschki
cw 7 wstęp, chemia środowiska
chemia kliniczna cw 1 2011 id Nieznany
Chemia i środowisko(2)
Akwakompleksy metali, OŚ, sem II 2 SOWiG, Chemia Środowiska, Seminarium ChŚ
Chemia środowiska
Chemia fizyczna - Ćw. 13 i 14 - Dysocjacja, Dysocjacja, hydroliza, pH,
Chemia środowiska?ŁOŚĆ
Chemia środowiska 4
pytania kontrolne CHEMIA SR, Studia, UR OŚ INŻ, semestr IV, chemia środowiska
moje typy, Studia, UR OŚ INŻ, semestr IV, chemia środowiska, ćwiczenia
chsr, Studia, UR OŚ INŻ, semestr IV, chemia środowiska, ćwiczenia
Chemia Srodowiska warunki zaliczenia
więcej podobnych podstron