
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
1
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
Rola wody w środowisku geologicznym bierze się z ABSOLUTNIE UNIKALNYCH WŁASNOŚCI
fizycznych i chemicznych wody w porównaniu z innymi substancjami, z ktorych zbudowana jest i który-
mi otoczona jest kula ziemska. Dość zauważyć, że woda występuje na Ziemi w trzech stanach skupienia:
w postaci gazu (para wodna), cieczy (wody rzek, jezior, mórz, oceanów, wody podziemne) i w postaci
stałej (śniegi i lody obszarów podbiegunowych i wysokogórskich). I te trzy formy skupienia wsółwystę-
pują ze sobą, dynamicznie przechodząc jedna w drugą w każdej chwili na Ziemi. Czy jest w tym coś wy-
jątkowego? Wiele minerałów na Ziemi występuje w postaci cieczy (stopy magmowe) i ciał stałych (skały
magmowe), ale ich pary można zaniedbać. Rtęć występuje w postaci ciekłej i gazowej (pary rtęci), ale w
warunkach ziemskich nie występuje jako ciało stałe. Poza wodą, żaden inny minerał czy substancja mine-
ralna budujące nasz świat na Ziemi nie jest spotykany w trzech stanach skupienia. Nic więc dziwnego, że
woda pełni wyjątkową rolę w naszym ziemskim świecie. To woda jest podstawą życia na Ziemi. To woda
kształtuje wygląd powierzchni Ziemi będąc głównym medium erozji, wietrzenia, transportu i sedymenta-
cji. To woda jest głównym czynnikiem wpływającym na klimat na Ziemi łagodząc ekstremalne upały czy
srogie zimy. Jesteśmy tak przyzwyczajeni do jej obecności, że nie dostrzegamy jej roli a jeszcze rzadziej
zastanawiamy się, z czego ta wyjątkowość wynika. Geochemia wody i roztworów wodnych jest niezwy-
kle bogatą i ciekawą dziedziną geochemii bardzo blisko związaną z naszym życiem i bardzo szeroko po-
wiązaną z innymi dziedzinami nauki.
6.1. Powstanie i ewolucja oceanów
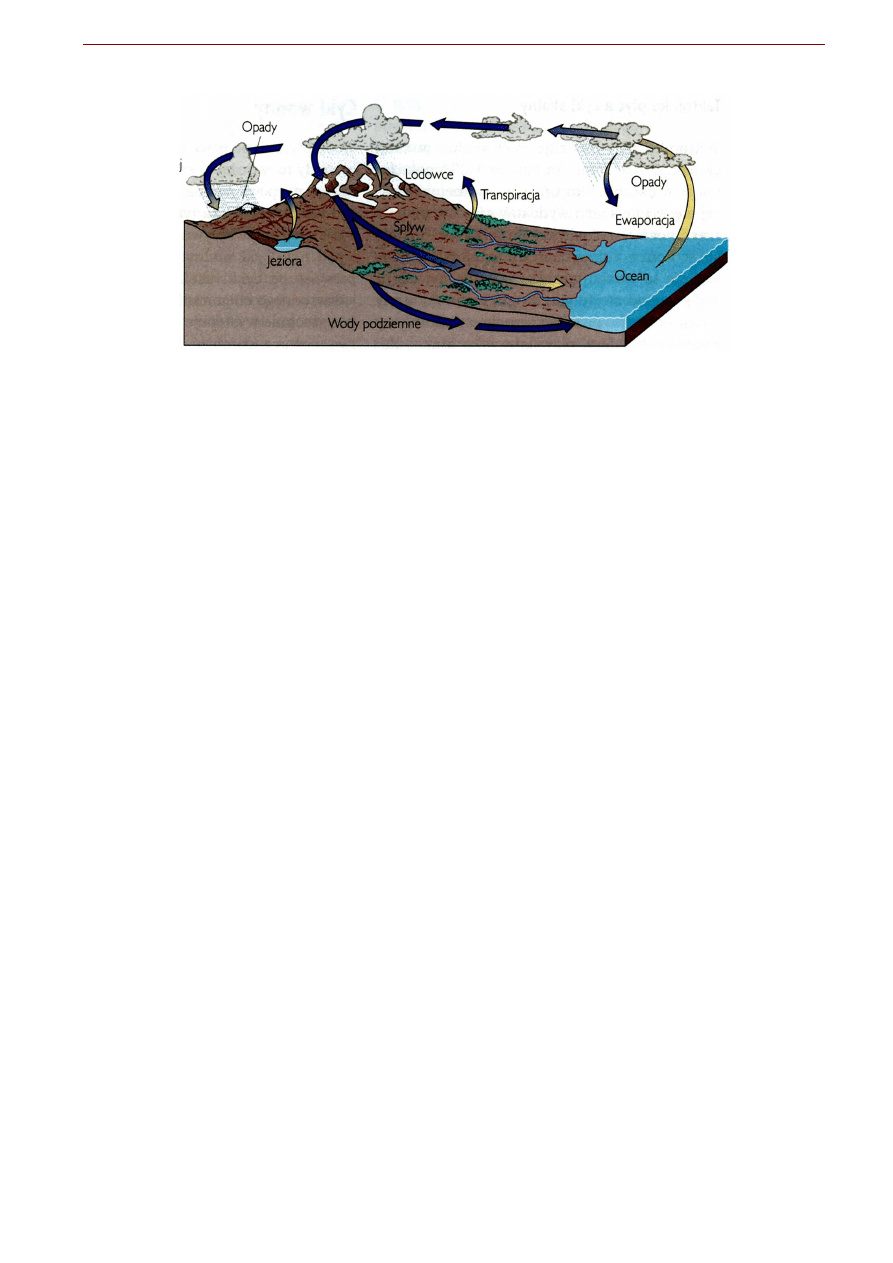
Zdecydowana większość zasobów wody na Ziemi zawarta jest w oceanach (Tab. 6.1.), które zajmują
71% powierzchni Ziemi a ich średnia głębokość wynosi 3730 m. W szybkim obiegu (w atmosferze, rze-
kach, jeziorach, glebach, lodowcach, wodach podziemnych, organizmach żywych i in.) znajduje się tylko
około 2% wody. Skąd się wzięła woda na Ziemi? Od jak dawna istnieją oceany? Czy zasolenie oceanów
wzrasta czy jest stałe? Dlaczego?
Tabela 6.1. Dystrybucja wody na Ziemi
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
2
Fig 6.1. Obieg wody na Ziemi (cykl hydrologiczny). Zdecydowana większość wody zawarta jest w oceanach.
Najstarsze fragmenty skał i minerałów noszące ślady działalności wody datuje się na ok. 3,9 miliarda
lat. Nie ma bezpośrednich dowodów na istnienie wody w okresie od powstania Ziemi do tego czasu. Stąd
nasza wiedza na temat powstania i ewolucji wczesnych oceanów na Ziemi jest bardzo spekulatywna i
powiązana raczej z teoriami o powstaniu Ziemi niż z faktami. Meteoryty z grupy chondrytów, które są
uważane za fragmenty protoplanety podobnej do tej, z której powstała Ziemia, zawierają kilka do kilku-
nastu procent wody. Wydaje się więc, że gdy Ziemia przechodziła przez okres roztopienia, dyferencjacji
chemicznej towarzyszyło odgazowanie lotnych składników, w tym gazów szlachetnych i pary wodnej.
Zdają się to potwierdzać współczesne pomiary zawartości argonu w atmosferze oraz oszacowania jego
ilości powstałej na przestrzeni dziejów z rozpadu promieniotwórczego izotopu potasu
40
K. Wydaje się, że
większość wody będącej obecnie w obiegu została wtedy uwolniona do gorącej atmosfery. Od tego czasu
tylko znikome ilości wody są uwalniane w procesach wulkanicznych (tzw. wody juwenilne) albo bez-
powrotnie tracone w wyniku fotodysocjacji na atomowy tlen i wodór w najwyższych warstwach atmosfe-
ry. Nieco wody dostarczyły też niewątpliwie komety licznie uderzające w Ziemię we wczesnym okresie
jej istnienia. Jednakże bezpośrednie badania składu chemicznego i izotopowego komet bezpośrednio
przeprowadzone przez sondę kosmiczną wykazały, że woda zawarta w lodzie budującym komety ma inną
proporcję izotopów od wody ziemskiej. A więc komety nie były głównym źródłem wody na Ziemi.
Trudno jest odtworzyć początki dziejów oceanów na Ziemi. Przedstawiona w dalszym ciągu historia
jest jedynie jednym z możliwych scenariuszy. W trakcie odgazowania, atmosfera nad rozgrzaną do co
najmniej 600
o
C powierzchnią Ziemi zawierała parę wodną H
2
O, dwutlenek węgla CO
2
, chlorowodór
HCl, azot, dwutlenek siarki SO
2
i trochę innych gazów w tym gazy szlachetne. Nie było w niej wolnego
tlenu. W miarę stygnięcia woda uległa skropleniu a gazy zawarte w atmosferze rozpuściły się tworząc
ocean gorący i kwaśny, który szybko reagował z minerałami skał magmowych powodując wzrost zasole-
nia. Zaczęły powstawać pierwsze skały osadowe:
skały magmowe + kwasy nieorganiczne (H
2
CO
3
, H
2
SO
4
, HCl) + H
2
O <=> skały osadowe + słona woda oceaniczna + atmosfera

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
3
W wyniku powstawania węglanu wapnia CaCO
3
oraz soli chlorkowych i siarczanowych (NaCl, KCl, Ca-
SO
4
i in.) wyrastało pH i zasolenie wody oceanicznej. Przypuszcza się, że oceany osiągnęły już wtedy
zasolenie i skład zbliżony do dzisiejszego. Przykładowe reakcje pierwotnych minerałów skał magmo-
wych ze składnikami pierwotnych oceanów to:
CaSiO
3
+ CO
2
=> CaCO
3
+ SiO
2
piroksen
kalcyt
kwarc
CaAl
2
Si
2
O
8
+ CO
2
+ H
2
O => CaCO
3
+ Al
2
Si
2
O
5
(OH)
plagioklaz
kalcyt kaolinit
2NaAlSi
3
O
8
+ 2HCl => 2NaCl + Al
2
Si
4
O
10
(OH)
2
+ SiO
2
plagioklaz zasolenie montmorillonit kwarc
CaSiO
3
+ H
2
SO
4
+ H
2
O => CaSO
4
.
2H
2
O + SiO
2
piroksen
gips
kwarc
Podstawową różnicą warunków w stosunku do dzisiejszych był brak tlenu w atmosferze. A więc woda
oceaniczna mogła zawierać znacznie więcej żelaza, które nie ulegało utlenieniu a w formie zredukowanej
Fe
2+
tworzy sole rozpuszczalne w wodzie. Zamiast tlenu, atmosfera zawierała dużo dwutlenku węgla.
Dlatego woda oceaniczna miała prawdopodobnie niższe pH niż dzisiejsza.
Upraszczając, ewolucja wczesnego oceanu na powierzchni wczesnej Ziemi sprowadza się do reakcji
skały bazaltowej z roztworem kwasu solnego przy obecności nadmiaru CO
2
i braku dostępu tlenu. A więc
teoretycznie, jeśli zakwasimy kwasem solnym silnie gazowaną wodę mineralną, wrzucimy do niej trochę
pokruszonego bazaltu, zamkniemy szczelnie żeby nie było dostępu powietrza i podgrzejemy, to mamy
model analogowy praoceanu. Minerały wchodzące w skład bazaltu wejdą w reakcje podobne do wypisa-
nych powyżej, uwalniając jony Na, K, Ca, Mg, Al i Fe. Spowoduje to stopniową neutralizację kwaśnego
odczynu. Krzemionka SiO
2
w dużym stopniu wytrąci się jako galaretowaty osad zamieniający się później
w krzemienie, a częściowo wejdzie w skład powstających autogenicznych minerałów jak krzemiany czy
glinokrzemiany Fe i Mg np. chloryty. Obecność dwutlenku węgla spowoduje wytrącanie się CaCO
3
i
CaMg(CO
3
)
2
obniżając zawartość Ca
2+
a głównymi jonami wody oceanicznej pozostaną Cl
-
i Na
+
, tak jak
to ma miejsce w dzisiejszych oceanach.
Patrząc na skład chemiczny wody oceanicznej widać, że nie jest ona skoncentrowanym przez parowanie
roztworem wody rzecznej. Mimo to, kiedyś przypuszczano, że zasolenie wody oceanicznej jest wynikiem
systematycznie wnoszonych składników jonowych przez rzeki. Sądzono też w związku z tym, że zasole-
nie powoli ale stale wzrasta. W 1899 roku niejaki Jon Joly z Dublina oszacował roczny transport Na
przez rzeki świata do oceanów i obliczył „wiek Ziemi”: ok. 90 milionów lat. Wydawało się to wtedy nie-
prawdopodobnie dużo. Był to przyczynek do trwającej wtedy debaty na temat darwinistycznej ewolucji,
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
4
wieku ziemi i rozbieżności rzeczywistości z biblijnymi opisami początków świata. Nieco wcześniej Lord
Kelvin obliczał kilkakrotnie wiek Ziemi na podstawie bilansu cieplnego i prędkości stygnięcia kuli ziem-
skiej. W zależności od przyjętych założeń czasem wychodziło mu 20 milionów a czasem 200 milionów
lat. Ale ponieważ Kelvin był bardzo religijny, więc do opisów biblijnych bardziej pasowało mu 20 milio-
nów.
Z czasem zrozumiano, że skład wody oceanicznej jest od co najmniej 2 milionów lat stały (fluktuujący
nieco wokół średniego) a kontrolowany jest przez skomplikowane bilansujące się mechanizmy dopływu i
odpływu składników utrzymujące stan równowagi dynamicznej (ang. steady state). To tak jak nalewana
woda do wanny: gdy wanna się wypełni przez górny odpływ wylewa się tyle samo wody co wpuszczane
jest z kranu. Pomimo stałego przepływu przez zbiornik poziom wody w zbiorniku się nie zmienia. Po-
dobnie poziom stężeń składników wody oceanicznej, pomimo stałego dopływu nie ulega zmianie.
Czas
pobytu składnika w wodzie
od momentu dostarczenia przez rzeki do momentu usunięcia przez jeden z
procesów geologicznych (coś jakby „staż” składnika w oceanie) określa się angielskim terminem „
resi-
dence time
”. Obliczany jest przez podzielenie całkowitej zawartości w oceanach świata przez ilość do-
starczaną rocznie rzekami do mórz. Obie liczby, a w szczególności ta druga, są z natury rzeczy bardzo
szacunkowe, wiec otrzymane liczby należy traktować orientacyjnie.
Tabela 6.2. Porównanie średniego składu wody rzecznej i oceanicznej (w ppm)
Składnik
Woda oceaniczna
Woda rzeczna
Na
+
10 800
5,15
K
+
407
1,3
Ca
2+
413
13,4
Mg
2+
1 296
3,35
Cl
-
19 010
5,75
SO
4
2-
2 717
8,25
HCO
3
-
137
52,0
SiO
2
~5
~10
Są dwie kategorie składników rozpuszczonych w wodzie morskiej: s
kładniki konserwatywne
(zacho-
wawcze) i składniki niekonserwatywne. Do pierwszych zalicza się
te składniki, których proporcje są
stałe we wszystkich wodach oceanicznych
. Należą do nich Na, K, Mg, Ca, Cl, SO
4
i BO
4
. Są one sto-
sunkowo niereaktywne (mają też bardzo długi czas przebywania residence time)a ich stężenia w wodzie
morskiej są setki razy wyższe niż stężenia w wodach słodkich. Natomiast stosunki stężeń pierwiastków
niekonserwatywnych zmieniają się w szerokim zakresie, szczególnie z głębokością. Należą do nich REE i
inne pierwiastki śladowe oraz fosforany PO
4
3-
, azotany NO
3
-
, wodorowęglany HCO
3
-
oraz rozpuszczona
krzemionka SiO
2
i rozpuszczony tlen. Ich stężenia nie są wiele wyższe od stężeń w rzekach co wskazuje
na szybkie usuwanie z wody morskiej. Czynnikiem wpływającym i kontrolującym stężenia tych składni-

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
5
ków nie jest rozpuszczalność ich soli czy minerałów. Są one usuwane z wody morskiej głównie poprzez
procesy sorpcji i wymiany jonowej na materii organicznej, podstawienia izomorficzne w krystalizujących
minerałach węglanowych, fosforanowych czy krzemianowych (powstających często z udziałem organi-
zmów), przez procesy hydrotermalne wzdłuż stref spreadingu na grzbietach oceanicznych i przez podsta-
wienia i krystalizację podczas powstawania ewaporatów.
6.2. Woda – substancja magiczna: dlaczego ręce myjemy w ciepłej wodzie?
Woda występuje na Ziemi w trzech stanach skupienia: stałym, ciekłym i gazowym. Trudno jest wska-
zać jakąś inną substancję, która by również spotykana była w trzech stanach skupienia. Jedynie siarka
naturalna może bywać w stanie stopionym, gazowym lub krystalicznym w pobliżu wyziewów gazów
wulkanicznych. Woda jest podstawą życia na Ziemi. Kształtuje wygląd powierzchniowych stref i jest
głównym czynnikiem wpływającym na klimat. To nie przypadek. Bierze się to z faktu, że woda posiada
wiele wyjątkowych właściwości chemicznych i fizycznych.
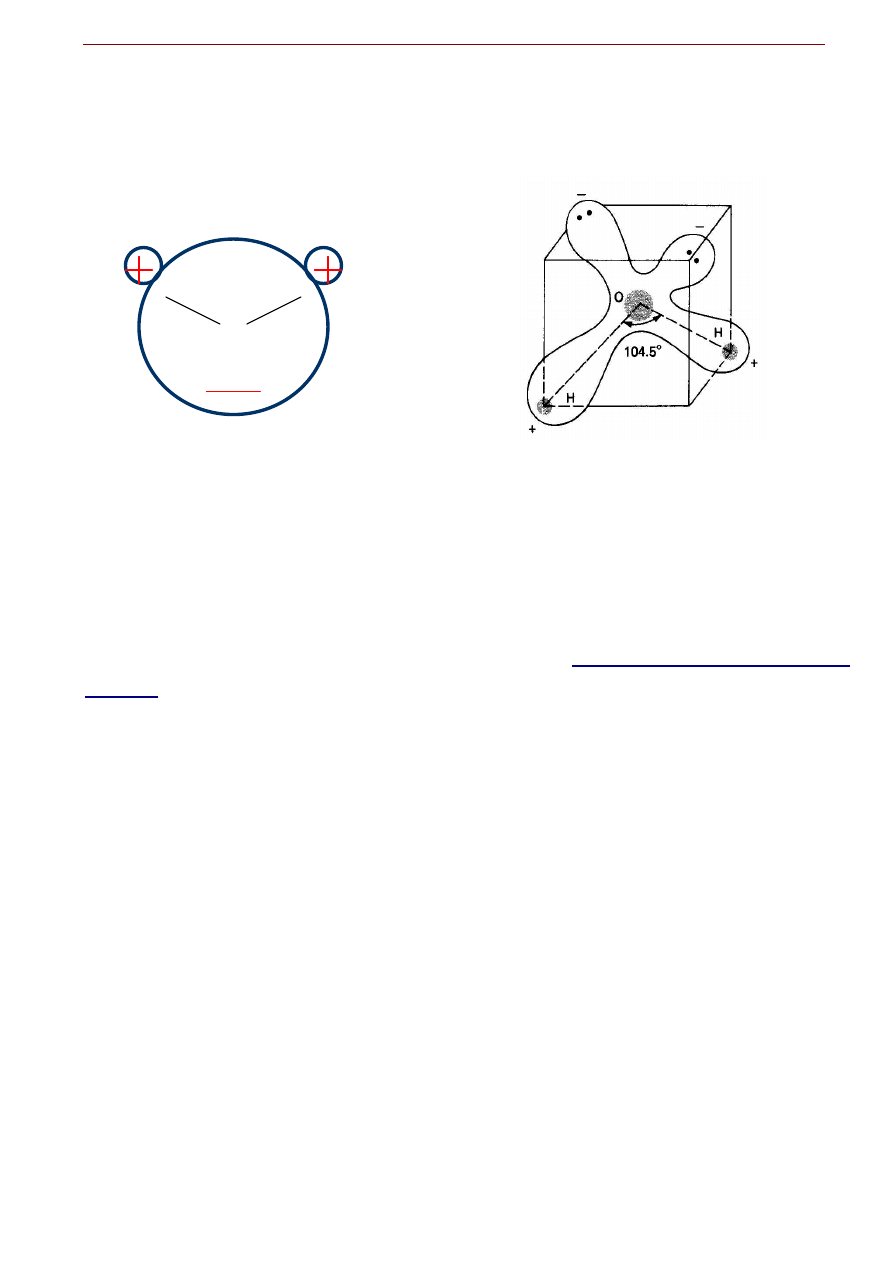
Fig. 6.2. Trzy stany skupienia wody na Ziemi.
Dipolowa budowa cząsteczki wody H
2
O
Większość szczególnych właściwości wody bierze się z charakterystycznej struktury cząsteczkowej:
dwa atomy wodoru połączone są z atomem tlenu wiązaniami kowalencyjnymi nie w linii prostej lecz pod
kątem. Atom tlenu, będąc w szóstej grupie układu okresowego, ma sześć elektronów walencyjnych. Każ-
dy z atomów wodoru wnosi do wiązania atomowego po jednym elektronie. Spośród tych wymienionych
ośmiu elektronów, dwie pary elektronowe użytkowane są przez atom tlenu wspólnie z atomami wodoru:
tworzą one wiązania atomowe cząsteczki wody. Pozostają jeszcze dwie wolne pary elektronowe w pobli-
Woda
lód
ciecz
para wodna
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
6
ż
u atomu tlenu. Ale ponieważ wiązania atomowe tworzą kąt 104,5o a „po przeciwnej stronie” występują
dwie wolne pary elektronowe, powstaje niesymetryczne rozmieszczenie ładunku elektrycznego w obrębie
obojętnej skądinąd cząsteczki (Fig. 6.3).
Fig. 6.3. Asymetryczna przestrzenna budowa cząsteczki wody: wielki atom tlenu połączony z dwoma malutkimi atomami
wodoru wiązaniami atomowymi.
Zarówno kąt wiązań atomowych z atomami wodoru różny od 180
o
jak i asymetryczne
rozmieszczenie dwóch wolnych par elektronowych atomu tlenu powodują, że cząsteczka wody jest dipolem elektrycz-
nym zdolnym do oddziaływań elektrostatycznych w tym m.in. do tworzenia wiązań wodorowych
.
W trzech wymiarach można to przedstawić umieszczając cząsteczkę wody wewnątrz zniekształconego
nieco sześcianu, z tlenem w środku, atomami wodoru na przekątnych narożach jednej ściany a niesparo-
wanymi elektronami na narożach przeciwległych. Daje to pojęcie o nierównomiernym rozmieszczeniu
ładunków elektrycznych, co w rezultacie prowadzi do faktu, że
cząsteczka wody jest dipolem elek-
trycznym
z przewagą ujemnego ładunku na tlenie a dodatniego ładunku od strony atomów wodoru. Aby
się przekonać o istnieniu ładunków elektrycznych w wodzie można wykonać proste doświadczenie: do
cieniutkiego strumienia wody wypływającej z kranu pionowo w dół przysuń nadmuchany balon, potarty
uprzednio o sukno dla naelektryzowania. Pod wpływem oddziaływania ładunków elektrycznych nagro-
madzonych na powierzchni balonu na dipole cząsteczek wody strumień odchyli się.
Wiązanie wodorowe
Dipolowy charakter cząsteczki jest przyczyną powstawania
WIĄZAŃ WODOROWYCH
pomiędzy
sąsiadującymi cząsteczkami wody:
ładunek dodatni jednego dipola przyciąga się z ładunkiem ujem-
nym drugiego
. Wiązanie wodorowe nie jest więc wiązaniem per se, jest raczej oddziaływaniem elektro-
statycznym. I choć jest 10 do 50 razy słabsze niż wiązanie atomowe, jest wystarczająco silne by skupiać
cząsteczki wody z dodatkową siłą, nieobecną u większości innych substancji. To właśnie wiązania wodo-
rowe, czyli elektrostatyczne oddziaływania pomiędzy dipolami elektrycznymi, są odpowiedzialne za
większość niezwykłych właściwości wody jako substancji.

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
7
Temperatura wrzenia i topnienia
Przyczyną wysokiej temperatury wrzenia i topnienia wody są wiązania wodorowe. Jak zachowywałaby
się woda, gdyby nie było wiązań wodorowych? Cząsteczki każdej cieczy oddziaływają ze sobą siłami van
der Waalsa, które utrzymują ciecz w naczyniu zapobiegając natychmiastowemu wyparowaniu. W miarę
podgrzewania, dostarczmy energii, która w postaci drgań termicznych przeciwstawia się przyciąganiu
cząsteczek do siebie. W temperaturze wrzenia siły Wan der Waalsa nie są już w stanie utrzymać cząstek
cieczy przy sobie i zaczyna sie parowanie w całej objętości: powstają bąble par. Siły van der Waalsa są
proporcjonalne do masy cząsteczkowej cząsteczkowej: czym lżejsza cząsteczka tym słabsze oddziaływa-
nie i niższa temperatura wrzenia. Dlatego można z grubsza przewidzieć temperaturę wrzenia (i topnienia)
wody badając inne, podobne chemicznie związki. Woda jest związkiem wodoru i pierwiastka z VI grupy
układu okresowego – tlenu. Możemy więc porównać własności związków wodoru z innymi pierwiastka-
mi VI grupy: siarką S, selenem Se i tellurem Te (Fig. 6.4). Na wykresie widać, że w miarę spadku masy
cząsteczkowej spada również proporcjonalnie temperatura wrzenia i topnienia tych związków. Gdyby w
wodzie występowały wyłącznie oddziaływania van der Waalsa przewidywana temperatura wrzenia wy-
nosiłaby około -70
o
C a temperatura zamarzania -90
o
C. Nie tylko byłyby znacznie niższe, ale i różnica
między nimi byłaby pięciokrotnie mniejsza.
Obecność dodatkowych, względnie silnych wiązań powo-
duje, że aby je zrównoważyć i umożliwić swobodne parowanie cieczy w całej objętości (wrzenie)
musimy dostarczyć znacznie więcej energii i podnieść energię termiczną drgających cząstek (tem-
peraturę) znacznie wyżej
. Dzięki temu, że woda ma wysoką temperaturę topnienia i dużą różnicę po-
między punktem wrzenia i zamarzania, Ziemia jest wyjątkową planetą w Układzie Słonecznym posiada-
jącą obfitość ciekłej wody. Trudno sobie wyobrazić oparte na wodzie życie na Ziemi czy innej planecie,
gdyby potrzebna do jego istnienia ciecz była płynna tylko w małym zakresie 20 stopni w okolicach minus
stu stopni Celsjusza. Ale wtedy pewnie i skala Celsjusza wyglądałaby inaczej.
Fig. 6.4. Zależność temperatury wrzenia i topnienia od masy atomowej w podobnych chemicznie związkach wodoru z pier-
wiastkami z grupy tlenowców. Woda H
2
O odbiega od trendu zmienności ze względu na obecność wiązań wodorowych
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
8
Tabela 6.3. Opis wybranych fizycznych i chemicznych właściwości wody
Właściwość
Opis
Przyczyna i znaczenie
Własności rozpusz-
czalnikowe
Najlepszy rozpuszczalnik pod
względem ilości i różnorodno-
ś
ci, doskonały rozpuszczalnik
soli, związków o budowie
jonowej czy polarnej
Przyczyną jest dipolowa budowa cząsteczek powodująca po-
wstawanie oddziaływanie elektrostatycznego z jonami kryszta-
łów osłabiając wiązania jonowe. Woda jest rozpuszczalnikiem
płynów fizjologicznych organizmów, i znaczącym czynnikiem
skałotwórczym przez rozpuszczanie, transport i krystalizację np.
ewaporatów, skał węglanowych, cementów skał klastycznych
czy nagromadzeń złożowych pierwiastków.
Temperatura wrze-
nia i temperatura
topnienia
Nienormalnie wysokie.
Wyjątkowo duża różnica wy-
nosząca 100
o
C
Przyczyną są wiązania wodorowe. Wielka różnica temperatur
pozwala na istnienie wody w trzech stanach skupienia na naszej
planecie oraz istnienie ciekłej wody (a wiec i życia) w szerokim
zakresie warunków klimatycznych.
Gęstość i zmienność
gęstości z tempera-
turą
Maksimum gęstości o 4 stopnie
powyżej temperatury zamarza-
nia (a nie w punkcie zamarza-
nia)
Głównymi przyczynami są wiązania wodorowe, struktura lodu i
struktura ciekłej wody. W efekcie woda zwiększa objętość przy
zamarzaniu (powodując m.in. wietrzenie przez zamróz) a lód
pływa po wodzie (jeziora nie zamarzają od dna w górę).
Napięcie po-
wierzchniowe
7,2
.
10
9
N/m
Najwyższe z cieczy występują-
cych naturalnie na Ziemi
Przyczyną są wiązania wodorowe zwiększające kohezję cieczy.
Umożliwia tworzenie się kropli (chmury, deszcz), podsiąkanie
kapilarne w glebach i roślinach i wiele procesów komórkowych.
Ciepło właściwe
Najwyższe z substancji wystę-
pujących naturalnie na Ziemi
Przyczyną są wiązania wodorowe zwiększające siłę oddziaływań
międzycząsteczkowych i utrudniające wzbudzenie drgań ter-
micznych. Dzięki temu woda jest wielkim regulatorem i maga-
zynem ciepła łagodzącym klimat
Ciepło parowania
540 cal/g
Najwyższe z cieczy występują-
cych naturalnie na Ziemi
Przyczyną są wiązania wodorowe zwiększające siłę oddziaływań
międzycząsteczkowych i utrudniające uwolnienie cząsteczek
pary. Powoduje regulację globalnej wymiany ciepła między
oceanami, atmosferą i strefami klimatycznymi. Parowanie chło-
dzi też nasze spocone czoło.
Selektywne pochła-
nianie promienio-
wania
Silne w zakresie podczerwo-
nym i ultrafioletowym, słabsze
w zakresie widzialnym
Woda jest bezbarwna. Umożliwia przenikanie światła w głąb
wód pozwalając na rozwój fotosyntetyzującego planktonu w
oceanach.
Ciepło zamarzania
Niewielkie, zaledwie 1/7 ciepła
parowania
Przyczyną są wiązania wodorowe ułatwiające uporządkowanie
cząsteczek w strukturę lodu. Umożliwia łatwe zmiany stanu
skupienia wody.
Lepkość
10
-3
N s/m
2
Niewielka
Woda jest cieczą łatwo przepływającą dla wyrównania nawet
niewielkich różnic ciśnienia
Gęstość i zmiany gęstości z temperaturą
We wszystkich cieczach wzrost temperatury powoduje wzrost ruchów termicznych cząstek a więc i
zwiększenie się średnich odległości pomiędzy cząsteczkami. W efekcie, ze wzrostem temperatury maleje
gęstość substancji. Dzięki temu unoszą się w górę balony na gorące powietrze a prądy konwekcyjne po-
ruszają masy w płaszczu Ziemi. Ten efekt, choć obecny, jest niezauważalny w wodzie w zakresie tempe-
ratur od 0
o
C do 4
o
C. W temperaturze 0
°
C woda zamarza w lód, który ma gęstość mniejszą niż faza ciekła,
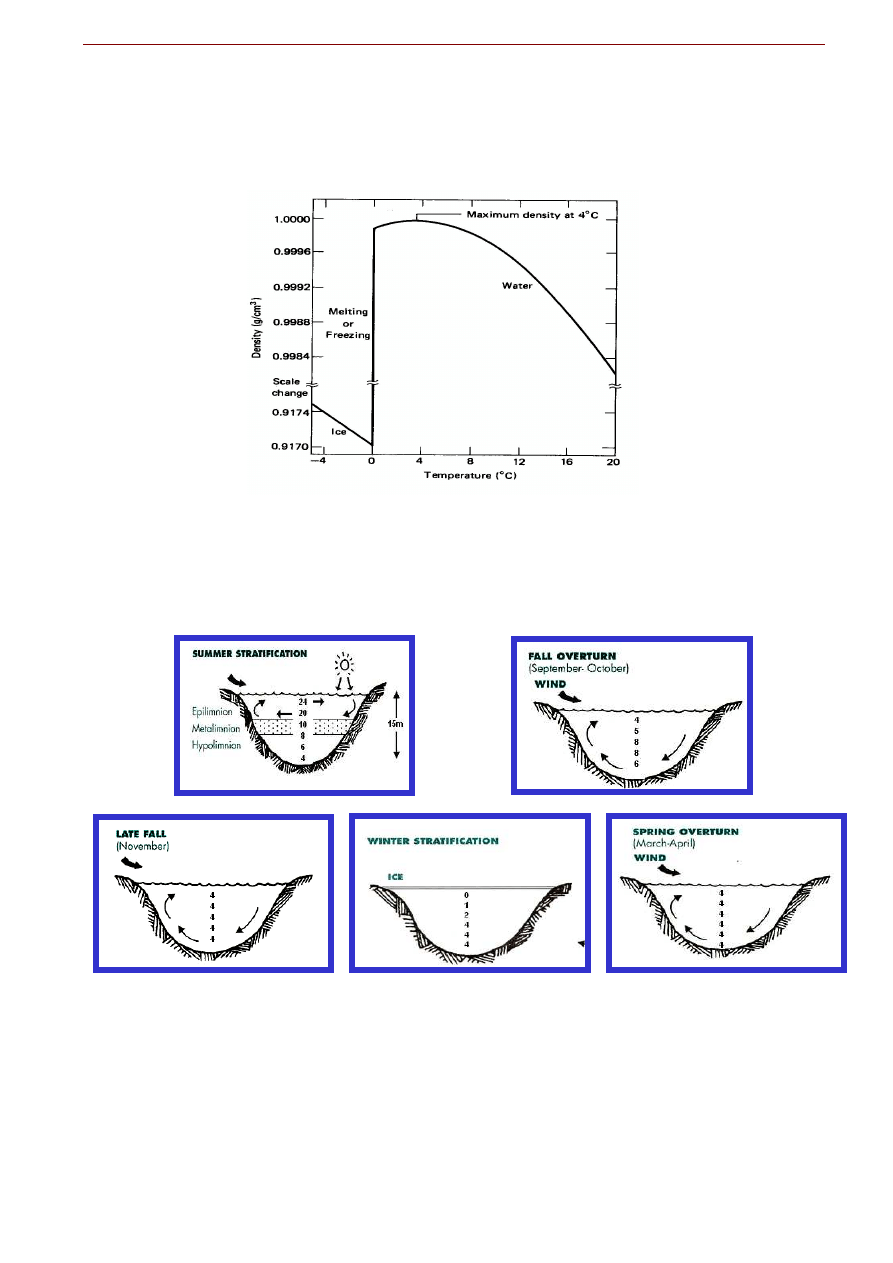
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
9
z której powstaje. Między 0
°
C a 4
°
C gęstość wody wzrasta ze wzrostem temperatury (cieplejsza woda
jest cięższa), osiągając maksimum w 3.94
°
C (Fig. 6.5). Dopiero powyżej 4
°
C gęstość maleje z temperatu-
rą tak jak u wszystkich innych substancji.
Fig. 6.5. Zależność gęstości lodu i wody od temperatury. W temperaturze 0
°
C woda zamarza w lód, który ma gęstość mniejszą
niż faza ciekła, z której powstaje. Między 0
°
C a 4
°
C gęstość wody wzrasta ze wzrostem temperatury, osiągając maksimum w
3.94
°
C. Dopiero powyżej 4
°
C gęstość maleje z temperaturą tak jak u wszystkich innych substancji.
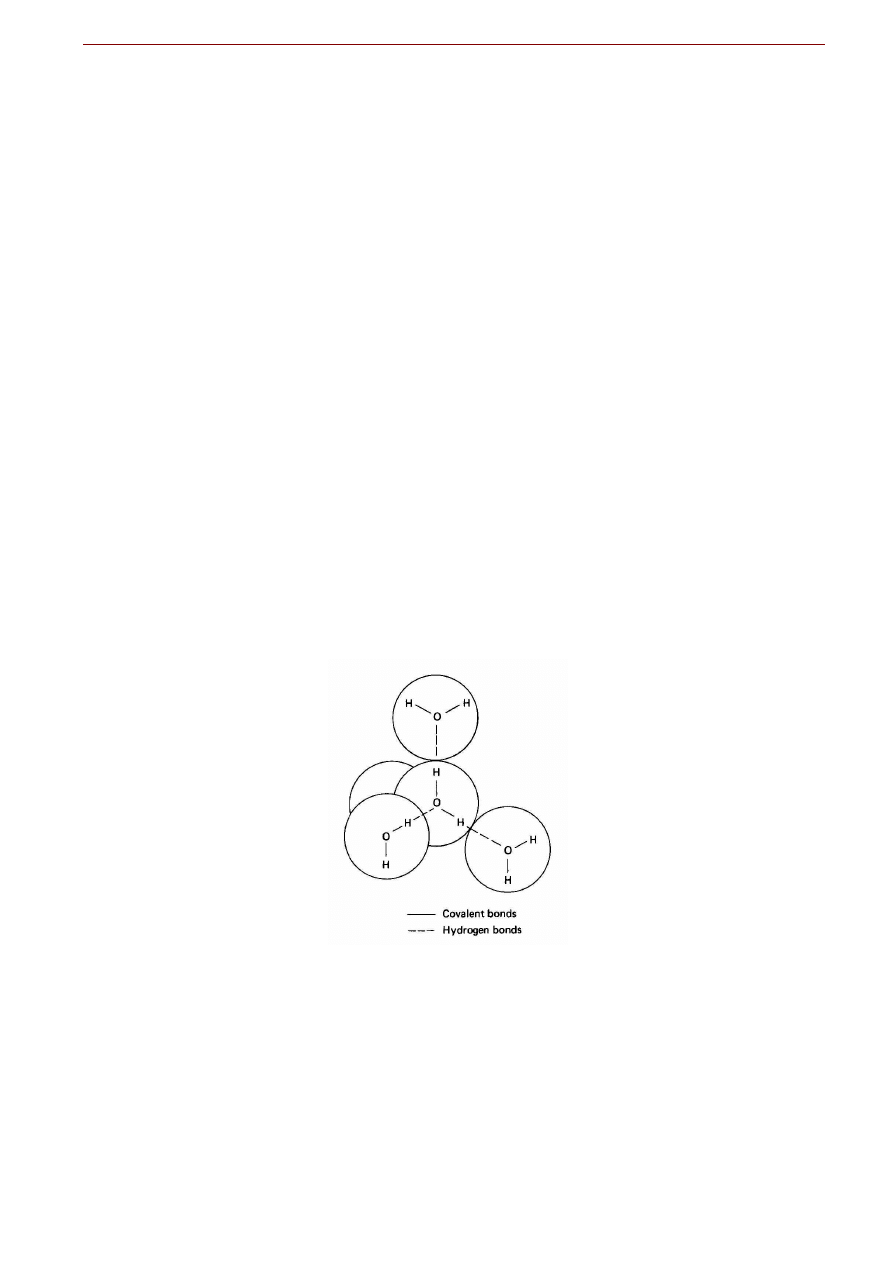
Fig. 6.6. Sezonowa zmienność stratyfikacji w zbiornikach wód powierzchniowych. 1. W lecie górne warstwy zbiorników
wodnych (epilimnion) nagrzewają się znacznie szybciej niż dolne (hipolimnion). Ciepła, górna warstwa ma mniejszą gęstość i
nie miesza się z cięższą warstwą dolną. Ustala się stagnacja letnia. W dolnych warstwach brakuje tlenu. 2. W okresie jesien-
nym spada insolacja, a rośnie siła wiatrów. Warstwa powierzchniowa się schładza i przemieszcza w głąb zapoczątkowując
mieszanie warstw. 3. Temperatura epilimnionu i hipolimnionu wyrównują się. Woda cyrkuluje w całym jeziorze (cyrkulacja
jesienna, w dużym stopniu wymuszona wiatrem i falowaniem), co sprzyja natlenieniu głębszych warstw. 4. Gdy wskutek po-
stępujących chłodów temperatura wierzchnich warstw spadnie poniżej 4
°
C, ustaje cyrkulacja. Woda o temperaturze poniżej
4
o
C ma mniejszą gęstość i utrzymuje się przy powierzchni a cieplejsza woda pozostaje w głębi. Przy dalszym spadku tempera-
tury jezioro zamarza na powierzchni. Ustala się stratyfikacja zimowa. 5. Wiosną lód topnieje, a woda się nagrzewa. Sprzyja to
ponownemu wyrównaniu temperatur w całej objętości zbiornika i uruchomieniu cyrkulacji wiosennej.
1
.
2.
3.
4.
5.
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
10
Podczas topienia się lodu jego struktura krystaliczna nie ulega natychmiast doszczętnemu zburzeniu.
W
temperaturach pomiędzy 0
o
C a 4
o
C w ciekłej wodzie występują jeszcze ugrupowania cząsteczek
połączonych ze sobą wiązaniami wodorowymi w struktury przestrzenne podobne do tych, jakie
występują w krystalicznym lodzie, przez co odległość między cząsteczkami wody jest większa (czyli
mniejsza gęstość)
. W miarę podgrzewania jest ich coraz mniej i gęstość wody rośnie. Dopiero powyżej
4
o
C te struktury znikają, efekt termiczny bierze górę i woda zaczyna zachowywać się tak jak przystało: jej
gęstość maleje ze wzrostem temperatury. Należy przy tym pamiętać, że opisana zależność gęstości od
temperatury jest cechą czystej wody (o niewielkiej ilości rozpuszczonych substancji) i nie występuje w
wodzie morskiej. Środowiskową konsekwencją tak specyficznej zmienności gęstości wody z temperaturą
jest m.in. jest sezonowa zmienność stratyfikacji wód powierzchniowych umożliwiająca mieszanie a co za
tym idzie natlenienie i wymianę składników odżywczych (Fig. 6.6).
Struktura lodu
Cząsteczki wody w strukturze lodu nie są ułożone jak ciasno upakowane kule, lecz pod wpływem
wiązań wodorowych układają się w przestrzenne struktury tworząc tetraedry: każdy atom tlenu
sąsiaduje z czterema innymi atomami tlenu poprzez atom wodoru (Fig. 6.7)
. Tetraedry łączą się ze
sobą tworząc heksagonalne przestrzenne pierścienie. Odległość pomiędzy atomami tlenu wynosi 2,76A a
kąt 109
o
.
Taka struktura, utrzymywana wiązaniami wodorowymi, jest dość obszerna wpływając na
względnie niską gęstość lodu
.
Fig. 6.7. Struktura cząsteczki lodu.
Podczas topienia lodu przestrzenna struktura krystaliczna lodu ulega zburzeniu. Cząsteczki wody przy-
ciągane wiązaniami wodorowymi już bez ograniczeń geometrycznych mogą się zbliżyć do siebie. Obję-
tość zmniejsza się o około 9%. W rezultacie, gęstość wody kapiącej z topniejącego lodu jest większa.
Fakt, że lód pływa po wodzie ma kolosalne konsekwencje środowiskowe. Dość wyobrazić sobie, że w
przeciwnym razie zbiorniki wodne zamarzałyby od dna! Nie dość, że spowodowałoby to całkowite za-

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
11
marznięcie małych zbiorników wodnych a wszelkie stworzenia można by koszami zbierać zimą po po-
wierzchni lodu, to jeszcze większość raz utworzonego lodu na dnie nigdy by nie rozmarzała izolowana
wodą z góry. Implikacje są zresztą znacznie szersze i trudno wyobrazić sobie, żeby żywe istoty przeżyły
w takich warunkach.
Pojemność cieplna
Pojemność cieplna to ilość ciepła potrzebna do ogrzania jednostkowej masy substancji o 1 sto-
pień
. Wzrost temperatury ciał polega na zwiększeniu energii termicznych drgań cząsteczek.
W przypad-
ku wody trudniej jest zwiększyć energię drgań termicznych gdyż trzeba przezwyciężyć dodatkowo
oddziaływanie wiązań wodorowych
. Woda ma najwyższą pojemność cieplną ze wszystkich naturalnych
substancji znanych na Ziemi. Oznacza to, że potrzeba dużo ciepła, aby wodę ogrzać, ale za to po ogrzaniu
woda „trzyma” ciepło i wolno stygnie. To dlatego przy przeziębieniu mama wkłada dziecku pod kołdrę
termofor z gorącą wodą. Dla porównania, żelazko do prasowania ma małe ciepło właściwe: chcemy, żeby
zaraz po włączeniu szybko się zagrzało i umożliwiło prasowanie. Natomiast piec kaflowy ma duże ciepło
właściwe: jak napali się w piecu wieczorem, to nawet po wygaśnięciu ognia piec zostaje ciepły do rana.
Ponieważ woda ma dużą pojemność cieplną (większą od kafli piecowych i wszystkich skał na Ziemi)
obecność dużych zbiorników wodnych (jezior, oceanów) wpływa łagodząco na klimat Ziemi, minimali-
zując szybkie ekstremalne zmiany temperatur.
Przykład. Zapytajcie żeglarza, kiedy wychodzi w morze: odpowie, że o świcie, kiedy wieje poranna
bryza od lądu. A kiedy zawija z powrotem do portu? O zachodzie słońca, kiedy wieje wiatr od morza. I
tak jest codziennie.
Fig. 6.8. Powstawanie bryzy morskiej m.in. w wyniku różnic w pojemności cieplnej zbiornika wodnego i lądu (do samodziel-
nego opracowania na podstawie wykładu).
Przeanalizujmy to zjawisko. Wieczorem, po upalnym słonecznym dniu, nagrzane są od słońca zarówno
skały wybrzeża jak i wody oceanu. Przez noc ich temperatura systematycznie spada. Ale ponieważ woda
ma pięciokrotnie wyższe ciepło właściwe niż skały, temperatura wody spada wolniej. W efekcie gdzieś
nad ranem temperatura wody jest wyraźnie wyższa niż temperatura skał wybrzeża. Powietrze nad cie-
płym morzem ogrzewa się i unosi powodując powstawanie wiatru od lądu. Następnie wschodzi słońce i
przychodzi następny letni wakacyjny dzień. Promienie słoneczne znacznie szybciej nagrzewają skały lądu
niż wodę morską, bo woda ma duże ciepło właściwe. Dlatego poranna bryza szybko ustaje, a po południu
zostaje zastąpiona wiatrem przeciwnym. Powstaje on przez to, że powietrze ogrzewa się od nagrzanej
powierzchni lądu i unosi do góry a z nad chłodniejszego morza wieje wiatr, z którym wracają do portu
ż
eglarze.

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
12
Ciepło parowania
Ciepło parowania
wody w 100oC wynosi 540 cal/cm
3
, jedno z najwyższych ze wszystkich znanych
cieczy.
Jest to energia niezbędna cząsteczkom, aby przeciwstawić się przyciąganiu innych cząste-
czek i wyrwać się „na wolność” w stan gazowy
. W przypadku wody, łączy się to z koniecznością ze-
rwania (poza normalnymi oddziaływaniami międzycząsteczkowymi) dodatkowych oddziaływań, jakimi
są wiązania wodorowe. Podczas skraplania woda oddaje tę samą ilość energii. W ten sposób zużywane
jest około 23% energii słonecznej padającej na Ziemię. Wysokie ciepło parowania wpływa łagodząco na
klimat Ziemi oraz wpływa na globalną dystrybucję ciepła w atmosferze i oceanach. Ciepło pochłaniane
podczas parowania wody (powstawanie chmur) jest w całości oddawane do otoczenia podczas skraplania
(powstawania deszczu i śniegu) dzięki czemu część ciepła z nad tropików dociera w podbiegunowe ob-
szary Ziemi (Fig. 6.9).
Fig. 6.9. Jeden z mechanizmów globalnej dystrybucji ciepła dzięki wysokiemu ciepłu parowania wody.
Napięcie powierzchniowe
Napięcie powierzchniowe jest miarą siły przyciągania się cząsteczek na granicy faz, np. na granicy
ciecz-gaz. Obecność wiązań wodorowych powoduje wyjątkowo silną tendencję wody do zbierania się w
krople. Odgrywa to istotną rolę przy powstawaniu chmur, rosy i deszczu. Ma to też ogromną rolę w kapi-
larnym podsiąkaniu wody w glebach i w roślinach. Ale wysoka kohezja spowodowana wiązaniami wo-
dorowymi maleje ze wzrostem temperatury. Woda wtedy trudniej tworzy krople a więc lepiej zwilża sub-
stancje. Dlatego mycie i pranie jest efektywniejsze w ciepłej wodzie.
Selektywne pochłanianie promieniowania
Ciekła woda dość dobrze przepuszcza widzialne promieniowanie, niezbędne do fotosyntezy. Dzięki
temu światło słoneczne, niezbędne m.in. do fotosyntezy, może w zbiornikach wodnych przenikać na
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
13
znaczną głębokość (do 100, czasem nawet 300 m) w głąb jezior i mórz. Natomiast woda silnie absorbuje
promieniowanie podczerwone i ultrafioletowe. Para wodna w atmosferze, absorbując promieniowanie
podczerwone, jest jednym z najistotniejszych „gazów cieplarnianych”.
Fig. 6.10. Dzięki wiązaniom wodorowym woda ma duże napięcie powierzchniowe i tendencję do tworzenia kropel. I choć jest
to bardzo pożądany efekt w środowisku, utrudnia on pranie i mycie. Na szczęście napięcie powierzchniowe maleje ze wzro-
stem temperatury. Dlatego mysie rąk i pranie są efektywniejsze w ciepłej wodzie.
Woda jako rozpuszczalnik
Cząsteczki wody mają budowę dipolową. Należy więc oczekiwać, że obecność zewnętrznego pola elek-
trycznego będzie powodować, że te dipole będą się ustawiały w sposób zorientowany. I tak rzeczywiście
jest. Jednakże elektrostatyczne oddziaływanie wiązań wodorowych przeciwstawia się temu uporządko-
waniu: wielkość tego wpływu podaje stała dielektryczna. Woda ma wysoką stałą dielektryczną. Powoduje
to, że obecność cząsteczek wody silnie osłabia (do 80-ciu razy!) przyciąganie elektrostatyczne pomiędzy
przeciwnie naładowanymi jonami substancji ulegającej rozpuszczeniu, ułatwiając ich rozdzielenie. W
efekcie
woda jest doskonałym rozpuszczalnikiem dla stałych i ciekłych substancji o budowie jono-
wej (takich jak sól NaCl) lub polarnej (takich jak alkohol)
. Rozpadają się one na naładowane frag-
menty (jony), które mogą tworzyć oddziaływania elektrostatyczne z dipolami wody. Natomiast praktycz-
nie nie rozpuszczają się w wodzie substancje o wiązaniach atomowych. Dlatego rozpuszczające się mine-
rały węglanowe dysocjują na jon wapnia Ca
2+
i jon węglanowy CO
3
2-
, natomiast kowalencyjne połącze-
nie węgla z tlenem wewnątrz anionu węglanowego jest „wodoodporne”:
CaCO
3
= Ca
2+
+ CO
3
2-
wiązanie jonowe wiązanie kowalencyjne
Fig. 6.11. Uproszczony schemat mechanizmu rozpuszczania kryształu jonowego przez elektrostatyczne oddziaływania dipo-
lowych cząsteczek wody.
napięcie
powierzchniowe
roztwór
kryształ

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
14
Dipolowa budowa cząsteczek wody ma dalsze konsekwencje dla roztworów elektrolitów.
Jony w roz-
tworze wodnym są w otoczce hydratacyjnej
. Powstaje ona przez zorientowanie się cząsteczek wody
znajdujących się najbliżej jonu pod wpływem sił elektrostatycznych. Tak więc kationy otoczone są chmu-
rą dipoli H
2
O zorientowanych swym ujemnym biegunem a aniony przeciwnie. Powstawanie otoczki hy-
dratacyjnej zwiększa rozpuszczalność substancji jonowych. Ponadto, w efekcie elektrostatycznego przy-
ciągania i pewnego uporządkowania się cząsteczek wody wokół jonów,
objętość roztworu wodnego
zmniejsza się podczas rozpuszczania w nim substancji (tzw. kontrakcja)
.
Przykład. Zmierzona objętość jednego kilograma roztworu NaCl o stężeniu 0,5 m (m oznacza stężenie
molalne, czyli ilość moli NaCl na kilogram roztworu) wynosi 983 cm
3
. Jest nieco mniej niż powinna z
wyliczeń teoretycznych. Teoretyczną objętość tego roztworu można wyliczyć korzystając z następujących
danych:
masa molowa NaCl = 58,44 g/mol, gęstość NaCl = 2,165 g/cm
3
masa molowa H
2
O = 17,01 g/mol, gęstość wody = 0,997 g/cm
3
Do sporządzenia 1 kilograma roztworu o zadanym stężeniu 0,5 m musimy wziąć pół mola czyli 29,22 g
NaCl i dopełnić do kilograma dolewając 970,78g wody. Dzieląc te masy przez odpowiednie gęstości mo-
ż
emy obliczyć objętość użytych składników: 29,22 g dzielone przez 2,165 g/cm
3
= 13,50 cm
3
dla NaCl
oraz 970,78g/0,997 g/cm
3
= 973,70 cm
3
dla wody. Po zmieszaniu, objętości te dodają się dając 13,50 +
973,70 = 987,2 cm
3
, o 4,2 cm
3
więcej, niż objętość wyznaczona pomiarem eksperymentalnym. A więc
roztwór „skurczył się” o około 0,5 %.
Woda jest doskonałym rozpuszczalnikiem dla wszystkich substancji o budowie polarnej: cukrów, alko-
holi, aminokwasów, amoniaku itp., co jest gwarancją procesów życiowych. Rozpuszczalnikowe własno-
ś
ci wody determinują cykl geologiczny i przebieg ewolucji na Ziemi. Woda nie rozpuszcza natomiast
zupełnie substancji niejonowych, takich jak węglowodory (metan CH
4
, etan C
2
H
6
etc.). Wzajemne od-
działywanie wody i cząsteczek substancji rozpuszczanej jest tak słabe, że nie wydziela się dostatecznie
dużo energii do zniszczenia struktury rozpuszczanej substancji.
Podsumowując, dzięki temu, że woda ma wyjątkowe właściwości jako substancja Ziemia jest wyjątko-
wą planetą w Układzie Słonecznym posiadającą obfitość ciekłej wody – podstawy życia.
6.3. Elementy hydrogeochemii
Skład i parametry fizykochemiczne wód naturalnych
Aby ocenić przydatność wody do określonego celu trzeba zbadać jej skład chemiczny oraz parametry
fizykochemiczne i pewne inne właściwości. Do najważniejszych i najczęściej oznaczanych parametrów
wód naturalnych należą:
a)
fizykochemiczne: temperatura, gęstość, przewodnictwo elektryczne, barwa, przezroczystość (męt-
ność), zapach, smak
b)
chemiczne: odczyn pH, potencjał utleniająco-redukcyjny Eh, mineralizacja TDS, alkaliczność, twar-
dość, stężenie głównych kationów i anionów: Ca
2+
, Mg
2+
, K
+
, Na
+
,HCO
3
-
, CO
3
2-
, SO
4
2-
, Cl
-

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
15
c)
dodatkowe parametry w razie potrzeby: stężenie SiO
4
, NO
3
-
, F
-
, wybrane metale, zawartość rozpusz-
czonego tlenu, zawartość węgla organicznego, bakterie, i wiele innych
Przewodność elektrolityczna właściwa
Zdolność do przewodzenia prądu elektrycznego przez roztwory wodne jest proporcjonalna do
stężenia rozpuszczonych w nich substancji wnoszących jony – nośniki ładunku elektrycznego.
Za-
tem pomiar przewodności od razu informuje nas o tym czy woda jest słabo czy silnie zmineralizowana.
Jednostką przewodnictwa jest odwrotność ohma – siemens (1 S = 1/
Ω
). Do pomiaru wykorzystuje się
konduktometr zbudowany z elektrody i z miernika, który po zanurzeniu elektrody w roztworze mierzy
przepływ prądu pomiędzy elektrodami oddalonymi o 1cm. Przewodności wód są zazwyczaj niewielkie,
więc jednostkami używanymi w praktyce są milisiemensy mS i mikrosiemiensy µS. Czysta woda desty-
lowana ma ok. 18 µS/cm.
Wyższa przewodność wskazuje na wyższą mineralizacje (czy zasolenie)
wody.
Szacunkowo można przyjąć, że przewodność wyrażona w µS odpowiada w przybliżeniu minera-
lizacji wyrażonej w mg/L (zwanej po angielsku TDS czyli total dissolved solids). Przewodność jest ła-
twym do zmierzenia parametrem wrażliwym na zmiany dynamicznie zachodzące w roztworach, dlatego
ś
wietnie się nadaje do monitoringu wód lub prospekcji geochemicznej, szczególnie w ochronie środowi-
ska.
Zawiesiny, mętność, zapach
Obecność w wodzie zawiesin związana jest z unoszonymi przez nią cząstkami mułu, iłu oraz różnych
organicznych i nieorganicznych substancji koloidalnych. Najdrobniejsze zawieszone w wodzie cząstki
wpływają na jej mętność.
Mętność oznacza się porównując badaną wodę z odpowiednio przygotowa-
nymi roztworami zawierającymi zawiesinę koloidalnej krzemionki i stąd jego wartość podaje się w
mg SiO
2
/dm
3
. Barwa naturalnej wody może być lekko żółtawa, co wiąże się z obecnością rozpuszczo-
nych kwasów organicznych.
Pomiaru intensywności barwy dokonuje się przez porównanie próbki z
wzorcami otrzymanymi przez rozpuszczenie związków platyny i kobaltu
(wynik podawany jest w mg
Pt/dm
3
). Oznaczenie wykonuje się kolorymetrycznie przez pomiar wielkości absorpcji światła przy uży-
ciu spektrofotometru. Z kolei
zapach określa się organoleptycznie
(czytaj – węsząc własnym nosem),
oceniając jego charakter (np. roślinny, gnilny, specyficzny) i moc (np. słabo wyczuwalny, bardzo wyraź-
ny etc.) lub krotność. Krotność oznacza stopień rozcieńczenia próbki, przy którym zapach przestaje być
wyczuwalny. Niektóre rodzaje zapachów – np. gnilne, specyficzne – wykluczają możliwość wykorzysta-
nia wody do celów pitnych.
Odczyn kwasowo-zasadowy pH
Woda jest słabym elektrolitem: niewielka część cząsteczek wody jest stale zdysocjowana na
jony hy-
droniowe H
+
i hydroksylowe OH
-
. Równanie dysocjacji cząsteczki wody można zapisać tak:

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
16
H
2
O < = > H
+
+ OH
-
lub poprawniej 2H
2
O < = > H
3
O
+
+ OH
-
Stała równowagi tej reakcji (tzw. iloczyn jonowy wody) dla rozcieńczonych roztworów w temperaturze
25
o
C wynosi K
w
= a
H+
.
a
OH-
= 10
-14
. W roztworach obojętnych tyle samo jest jonów wodorowych H
+
(czy poprawniej – hydroniowych H
3
O
+
) co jonów hydroksylowych OH
-
. W roztworach kwaśnych prze-
ważają jony H
+
a w roztworach zasadowych jony OH
-
, ale ich stężenia zmieniają się proporcjonalnie do
siebie tak, aby iloczyn ich aktywności a
H+
.
a
OH-
wynosił stale 10
-14
. Przyjęło się mierzyć aktywność jo-
nów wodorowych i wyrażać odczyn w jednostkach pH.
Z definicji, pH jest to ujemny logarytm dzie-
siętny z aktywności (czyli skorygowanego stężenia) jonów hydroniowych: pH = -log[H
+
].
Skala pH
obejmuje wartości od 0 do 14. Roztwory kwaśne mają pH<7 a roztwory alkaliczne pH>7. Pomiaru pH
dokonuje się za pomocą pH-metru, czyli elektrody selektywnie wrażliwej na stężenie jonów hydronio-
wych podłączonej do miernika, który wyświetla wartość mierzonego pH w roztworze. Odczyn pH zależy
silnie od temperatury i dlatego wskazane jest, aby pomiar pH próbek naturalnych wód powierzchniowych
czy podziemnych był wykonywany w terenie. Dla wód naturalnych pomiaru należy dokonać w terenie w
momencie pobrania próbki, ponieważ przechowywanie i transport próbki zazwyczaj zaburza wynik. Do-
kładność pomiaru w terenie zazwyczaj nie przekracza 0,05 jednostek pH.
Fig. 6.12. Pomiar pH przy użyciu pH-metru
Zazwyczaj w środowisku spotykamy roztwory o pH w granicach 4 do 9. Każda woda na powierzchni
Ziemi jest w kontakcie z powietrzem i zawiera nieco rozpuszczonego dwutlenku węgla, który tworząc
kwas węglowy zakwasza ją lekko. Dlatego woda deszczowa ma zazwyczaj pH pomiędzy 5 a 6. Natomiast
woda morska ma pH ponad 8 ze względu na alkalizujące działanie rozpuszczonego w niej węglanu wap-
nia CaCO
3
. Od wody pitnej oczekuje się pH w zakresie 6,5 – 8,5.
Większość minerałów lepiej rozpusz-
cza się w kwaśnych roztworach.
Gdy pH wzrasta, wiele metali ma tendencję do wytrącania się z roz-
tworów w postaci wodorotlenków. Wyjątkiem jest kwarc i substancje zawierające SiO
2
:
rozpuszczalność
SiO
2
rośnie ze wzrostem pH a spada w roztworach kwaśnych
. Dlatego nie można przechowywać roz-
tworów silnych zasad (jak NaOH) w naczyniach szklanych.
Potencjał utleniająco-redukcyjny Eh
Podwyższenie stopnia utlenienia pierwiastka (oddanie elektronów) to proces utleniania
, na przy-
kład Cu
o
-> Cu
2+
czy Fe
2+
-> Fe
3+
, co w przypadku metali wiąże się najczęściej z utworzeniem kationu i

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
17
przejściem do roztworu. Ale również utlenianiem jest spalanie węgla gdy zmienia on stopień utlenienia z
zerowego na plus czwarty C
o
-> C
4+
co wcale nie oznacza, że powstają takie kationy w roztworze. Wręcz
przeciwnie, węgiel takich kationów nie tworzy, ale podczas spalania utlenia się do plus czwartego stopnia
utleniania tworząc np. CO
2
. Podobnie siarka S
o
-> S
2+
spalając się ulega utlenieniu tworząc np. SO
2
.
Ob-
niżenie stopnia utlenienia (pobranie elektronów) to proces redukcji
. Na przykład Fe
3+
-> Fe
2+
albo
S
6+
-> S
4+
. W reakcjach utleniania-redukcji (reakcjach redoks) zawsze obecne są obie formy pierwiast-
ków: utleniona i zredukowana.
Potencjał oksydacyjno-redukcyjny Eh jest miarą potencjału elektrycznego (mierzonego w vol-
tach V) powstającego podczas przebiegu reakcji utleniania lub redukcji, mierzonego względem
elektrody, na której zachodzi reakcja redukcji wodoru H
+
-> H
o
.
Potencjał redox jest ilościową miarą
tendencji środowiska wodnego do utleniania lub redukowania jego składników. Ponieważ procesy utle-
niania-redukcji polegają na wymianie elektronów w większości wypadków zaangażowane w proces są
również protony H
+
co wpływa na odczyn pH. Utlenianie często zmniejsza pH (np. utlenianie pirytu i
zakwaszanie wód kopalnianych) a redukcja podwyższa pH. Wszystkie reakcje redox opisują jednocześnie
utlenianie jednego składnika i redukcję innego, mogą więc być rozbite na dwie reakcje połówkowe, które
muszą się zbilansować liczbą wymienianych elektronów
Reakcja połówkowa redukcji:
Fe
3+
+ e
-
= Fe
2+
Reakcja połówkowa utleniania:
4H
+
+ O
2
o
(gaz)
+ 4e
-
= 2H
2
O
Dla zbilansowania ilości elektronów użytych w obydwu reakcjach, tę ostatnią modyfikujemy:
H
+
+ ¼ O
2
o
(gaz)
+ e
-
= ½ H
2
O
Zbilansowane reakcje połówkowe można teraz połączyć w jedną zapisując tak, aby części z elektronami
znalazły się po przeciwnej stronie równania, przez co elektron znika z zapisu:
Fe
3+
+ ½ H
2
O < = > Fe
2+
+
H
+
+ ¼ O
2
o
(gaz)
Eh jest teoretycznym potencjałem w voltach odpowiadającym konkretnej reakcji połówkowej
cC + ne
-
= dD
Eh(V) = E
o
+ RT/nF ln(a
c
c
/a
D
d
)
Gdzie E
o
= -∆G
o
/nF to potencjał standardowy czyli wyznaczony dla warunków normalnych 25
o
C i 1 atm,
R – stała gazowa 0,001987 kcal/mol
.
K, F – stała Faradaya 23,061 kcal/V, a – aktywność (skorygowane
stężenie molowe) formy utlenionej C i zredukowanej D w roztworze. W większości współczesnej litera-
tury Eh zastępowane jest (przez analogie do odczynu pH) jednostką pE:
pE = -log
10
(e
-
).
Przykładowe wartości potencjału redoks (E
0
oznacza, że potencjał wyznaczono dla warunków normal-
nych 25
o
C i 1 atm) podaje tabela 6.4.

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
18
Tabela 6.4. Przykładowe potencjały redoks różnych reakcji
reakcja
E
0
[V]
Au
+
+ e
-
⇒ Au
+ 1,68
Pt
2+
+ 2e
-
⇒ Pt
+ 1,20
Ag
+
+ e
-
⇒ Ag
+ 0,80
Cu
+
+ e
-
⇒ Cu
+ 0,52
2H
+
+ 2e
-
⇒ H
2
0,00
Fe
2+
+ 2e
-
⇒ Fe
- 0,44
Mg
2+
+ 2e
-
⇒ Mg
- 2,37
Mn
3+
+ e
-
⇔
Mn
2+
+ 1,51
MnO
2
+ 4H
+
+ 2e
-
⇔
Mn
2+
+ 2H
2
O
+ 1,28
O
2
+ 4H
+
+ 4e
-
⇔
2H
2
O
+ 1,23
Fe
3+
+ e
-
⇔
Fe
2+
+ 0,77
Cu
2+
+ e
-
⇔
Cu
+
+ 0,15
Mn(OH)
3
+ e
-
⇔
Mn(OH)
2
+ OH
-
+ 0,10
Fe(OH)
2
+ 2H
+
+ 2e
-
⇔
Fe + 2H
2
O
- 0,05
Patrząc na powyższe równanie i tabelkę warto zapamiętać kilka ogólnych zależności:
-
gdy [red] = [utl], wówczas E
h
= E
0
(bo ln 1 = 0)
-
wysokie wartości E
h
sprzyjają utlenianiu, niskie sprzyjają redukcji;
-
wyższe wartości E
0
danej reakcji oznaczają wyższe wartości E
h
potrzebne do utlenienia (czyli środo-
wisko musi być bardziej utleniające), tak więc w przyrodzie łatwiej przebiegają reakcje charakteryzu-
jące się niższymi E
0
. Dobrym przykładem ilustrującym tą prawidłowość jest oddzielenie żelaza od
manganu w strefie wietrzenia. Gdy migrujące w wodzie Fe
2+
i Mn
2+
dostają się w strefę oddziaływa-
nia warunków utleniających (np. przy wypływie zmineralizowanej wody źródlanej na powierzchnię),
jako pierwsze będą się wytrącać związki Fe
3+
, gdyż potencjał redoks utleniania Fe
2+
do Fe
3+
jest mniej
więcej dwukrotnie niższy niż dla reakcji utleniania Mn
2+
do Mn
3+
. Konsekwencją tego może być dal-
sza „samotna” migracja manganu do strefy gdzie będą panować silniej utleniające warunki;
-
w przypadku metali, im wyższy jest E
0
jego jonizacji, tym metal jest bardziej „szlachetny” (Au > Pt >
Ag > Cu > Fe), czyli tym trudniej go utlenić. W skałach znajdujących się na powierzchni Ziemi znaj-
dujemy rodzime złoto, platynę i srebro, znacznie rzadziej miedź, natomiast występowanie np. magne-
zu w formie metalicznej jest niemożliwe.
-
pierwiastki o niższych E
0
wypierają pierwiastki o wyższych E
0
z roztworów ich soli, np. CuSO
4
+ Fe
⇒ FeSO
4
+ Cu
Pomiar Eh wykonujemy w terenie w czasie pobierania próbki, ponieważ parametr ten bardzo szybko się
zmienia. Do pomiaru używa się sprzętu bardzo podobnego do pH-metru: jest to elektroda Eh podłączona
do miernika. Wysokie wartości potencjału redox świadczą o środowisku utleniającym. Występują one w

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
19
wodach opadowych i powierzchniowych bogatych w tlen. Niskie wartości potencjału redox świadczą o
ś
rodowisku redukującym i występują często w bagnach bogatych w rozkładającą się substancję orga-
niczną lub w strefach utlenienia minerałów siarczkowych. Jednak ze względu na trudne do przewidzenia
nakładające się wpływy różnych składników wód naturalnych, zawiesin, rozpuszczonego powietrza, tem-
peratury i pH, pomiar potencjału redoks nie jest tak dokładny jak pomiar pH i powinien być traktowany
bardziej orientacyjnie niż do dokładnych ilościowych obliczeń. W roztworach wód naturalnych zazwy-
czaj przebiega jednocześnie kilka lub kilkanaście reakcji redoks i zmierzone Eh jest orientacyjnym chwi-
lowym potencjałem wypadkowym tych reakcji. Dlatego w przeciwieństwie do pH, które odczytujemy z
dokładnością do dwóch miejsc po przecinku, odczytana wartość Eh jest mniej precyzyjna, odczyt mniej
powtarzalny i przydatny głównie do porównań. Podaje nam orientacyjnie czy środowisko jest bardziej
redukcyjne czy mniej względem próbki z innego miejsca lub względem pomiaru wykonanego w prze-
szłości. Wyższe wartości zmierzonego Eh wskazują na środowisko o większej tendencji utleniającej. Po-
miar jest szczególnie użyteczny do kontroli jakości wód i ścieków, gdzie wykonywany jest systematycz-
nie na jednakowych próbkach w jednakowy sposób i stanowi parametr wrażliwy na wszelkie zaburzenia
przebiegu procesu uzdatniania czy oczyszczania.
Mineralizacja
Mineralizacja ogólna
(wspomniane powyżej TDS czyli total dissolved solids)
to suma rozpuszczo-
nych składników stałych wyrażona w mg/dm
3
. Oznaczana jest bezpośrednio przez pomiar masy sub-
stancji pozostałej po odparowaniu porcji wody (sucha pozostałość), przez wykonanie pełnej analizy che-
micznej wody i zsumowanie składników, lub przez oszacowanie z pomiaru przewodnictwa elektryczne-
go, które jest proporcjonalne do zawartości rozpuszczonych jonów. Woda pitna nie powinna mieć mine-
ralizacji przekraczającej 600 mg/dm
3
. Woda deszczowa ma mineralizację poniżej 100 mg/dm
3
a woda
morska ma mineralizację ok. 35 000 mg/dm
3
. Wody rzek i jezior mają zazwyczaj niska mineralizację. Z
podwyższoną mineralizacją możemy mieć do czynienia w przypadku wód podziemnych. Na podstawie
mineralizacji ogólnej wody podziemne dzielimy na wody zwykłe (słodkie, mineralizacja < 500 mg/dm
3
),
akratopegi (wody o podwyższonej mineralizacji, mineralizacja 500-1000 mg/dm
3
) i wody mineralne (sło-
ne, mineralizacja > 1000 mg/dm
3
). W wodach podziemnych rozpuszczone są też nieraz duże ilości dwu-
tlenku węgla CO
2
. Wody o znacznej zawartości tego gazu są nazywane wodami kwasowęglowymi (500-
1000 mg/dm
3
), natomiast jeszcze silniej nasycone CO
2
noszą nazwę szczaw (powyżej 1000 mg/dm
3
).
Alkaliczność
Alkaliczność (zasadowość) wody określa ilościowo jej zdolność do przyjęcia protonów (zobojęt-
nienia kwasowości)
dzięki obecności rozpuszczonych wodorotlenków, węglanów, fosforanów, krzemia-
nów, soli kwasów organicznych i in. Oznaczana jest przez miareczkowanie kwasem solnym HCl. Podaje

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
20
się ją zazwyczaj przeliczoną na mg/L CaCO
3
. W większości wód słodkich na alkaliczność A składają się
głównie jony węglanowe i wodorowęglanowe:
A = 2[CO
3
2-
] + [HCO
3
-
].
Skład chemiczny (związki nieorganiczne)
Przy badaniu składu chemicznego wody ważne są dwie grupy składników. Główne kationy i aniony
występują w największych ilościach, decydując o mineralizacji wody oraz o jej typie hydrogeochemicz-
nym. Z kolei składniki występujące w podrzędnych lub nawet śladowych ilościach często warunkują
możliwość wykorzystania wody do określonego celu – np. jako wody pitnej.
Głównymi kationami wy-
stępującymi w wodach śródlądowych są Ca
2+
, Mg
2+
, Na
+
i K
+
, natomiast podstawowe aniony to
HCO
3
-
, SO
4
2-
i Cl
-
(zwykle, choć nie zawsze w takiej kolejności).
W podrzędnych ilościach pojawiają się w wodach związki azotu i fosforu. Mają one bardzo duży (ko-
rzystny lub niekorzystny) wpływ na jakość wody. Podstawowymi formami, w jakich te pierwiastki wy-
stępują w wodach są następujące jony: NO
3
-
,
NO
2
-
, NH
4
+
i PO
4
3-
. Ich nadmiar związany np. ze spłukiwa-
niem do wód nadmiernych ilości nawozów, może prowadzić do eutrofizacji zbiorników. W przypadku
azotu, proporcje różnych jego form pozwalają niekiedy wnioskować o źródłach zanieczyszczenia wód. W
większości wód naturalnych obecny jest również krzem pod postacią różnych rozpuszczonych związków
chemicznych. Jego zawartość podaje się jako
mg SiO
2
/L
.
Pozostałe nieorganiczne jony naturalnych wód to
mikroskładniki
– pojawiają się w stężeniach rzędu
ułamków mg/dm
3
do kilku mg/dm
3
. Należą do nich m.in. Sr
2+
, Ba
2+
, Fe
2+
, Mn
2+
, B
3+
, Al
3+
, Zn
2+
, Cu
2+
,
Li
+
, F
-
, Br
-
, I
-
. W jeszcze mniejszych ilościach występują
pierwiastki toksyczne
, np. As
3+
, As
5+
, Cr
6+
,
Cd
2+
, Hg
+
.
Związki organiczne
Badając jakość wód powierzchniowych, czy podziemnych nie można zapomnieć o
substancji orga-
nicznej
, gdyż bardzo często to właśnie ona jest głównym czynnikiem zanieczyszczającym.
Pod tym po-
jęciem rozumiemy zarówno mikroorganizmy obecne w wodach, jak również produkty ich metabo-
lizmu oraz rozkładu, a także różnorakie substancje pochodzenia antropogenicznego
. Do tych ostat-
nich należą np.
fenole, węglowodory aromatyczne (BTX – benzen, toluen, ksylen), wielopierścienio-
we węglowodory aromatyczne (WWA), pestycydy i inne
. Ze względu na trudności analityczne oraz tak
szeroki wachlarz możliwych substancji, bardzo często na etapie badań wstępnych szacuje się stopień za-
nieczyszczenia wód substancjami organicznymi w sposób uproszczony, wprowadzając pewne umowne
wskaźniki. Należą do nich ogólny węgiel organiczny, oraz chemiczne i biochemiczne zapotrzebowanie na
tlen.
Ogólny węgiel organiczny (OWO, albo TOC – ang. total organic carbon) to stężenie węgla orga-
nicznego zawartego we wszystkich związkach organicznych w wodzie
(a nie związanego w jonach

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
21
węglanowych CO
3
2-
czy HCO
3
-
). Podczas jego oznaczania, w celu wyeliminowania węgla nieorganiczne-
go (TIC – ang. total inorganic carbon) próbkę wody zakwasza się, ponieważ powoduje się tym samym
usunięcie węglanów, w głównej mierze odpowiadających za węgiel nieorganiczny. Wartości OWO waha-
ją się w bardzo szerokich granicach, dochodząc w szczególnych przypadkach do kilkuset mg/dm
3
.
Chemiczne zapotrzebowanie na tlen (ChZT, albo COD – ang. chemical oxygen demand) oznacza
ilość mocnego utleniacza (dwuchromianu potasu K
2
Cr
2
O
7
lub manganianu(VII) potasu KMnO
4
)
niezbędną do rozkładu substancji organicznych występujących w wodzie
. Parametr ten wyznacza się
miareczkując próbkę wody dwuchromianem lub nadmanganianem w obecności kwasu siarkowego i kata-
lizatora, a wynik podaje się w przeliczeniu na tlen cząsteczkowy, czyli w mg O
2
/dm
3
.
Aby oszacować
ilość mikroorganizmów w wodzie określa się jeszcze jeden wskaźnik – biochemiczne zapotrzebowa-
nie na tlen (BZT albo BOD – ang. biochemical oxygen demand)
. Idea określania tego wskaźnika opie-
ra się na spostrzeżeniu, że organizmy wodne zużywają rozpuszczony w wodzie tlen. Zatem jego ilość w
zamkniętej porcji wody powinna z czasem maleć. Oznaczenie jest dwuetapowe. Mierzy się stężenie tlenu
rozpuszczonego w wodzie przed (tuż po pobraniu próbki) i po pięciodniowej inkubacji w warunkach la-
boratoryjnych w ciemności. W takich warunkach ubytek tlenu jest wynikiem zużywania go przez mikro-
organizmy wodne. Np. jeśli początkowe stężenie O
2
w wodzie wynosiło 8,3 mg/dm
3
, a po pięciu dniach
spadło do 5,0 mg/dm
3
, to BZT
5
jest równe 3,3 mg/dm
3
.
Bardzo istotne z punktu widzenia użytkowania wody, szczególnie do celów pitnych, są ponadto wskaź-
niki biologiczne, a szczególnie stan bakteriologiczny. Biologiczna ocena jakości wody opiera się na fak-
cie, że skład i właściwości wody wpływają na rodzaj i liczebność zamieszkujących ją organizmów. Zwy-
kle wykorzystuje się do tego celu tzw. gatunki wskaźnikowe, które dzieli się na trzy główne grupy:
-
saprokseniczne – występują tylko w wodach czystych, a unikają zanieczyszczonych
-
saprofilne – występują w wodach czystych, ale w zanieczyszczonych masowo
-
saprobiontyczne – występują tylko w wodach zanieczyszczonych
Zatem badając populacje biologiczne w wodach poprzez szacowanie liczebność i proporcji gatunko-
wych, można wnioskować o stanie czystości wód.
Spośród wskaźników mikrobiologicznych, wskazu-
jących na stan bakteriologiczny wody, najczęściej stosuje się ilość pałeczek okrężnicy (Escherichia
coli) w określonej objętości próbki
. Można ją też podawać jako miano coli, czyli najmniejszą objętość
wody (w cm
3
), na jaką przypada jedna bakteria okrężnicy.
Twardość wody
Twardość określa ilościowo obecność jonów, które w reakcji z mydłem sodowym wytworzą nie-
rozpuszczalny osad
. Twardość jest parametrem praktycznym mającym znaczenie wyłącznie dla wód
wykorzystywanych przez człowieka i określa łatwość spłukiwania mydła czy tworzenia kamienia kotło-
wego (osadu w czajniku).
Twardość wiąże się z obecnością w wodzie rozpuszczonych jonów dwuwar-
tościowych, głównie Ca
2+
, Mg
2+
, Sr
2+
i Ba
2+
(a także Fe
2+
, Zn
2+
) i trójwartościowych (Al
3+
, Fe
3+
). Jeśli

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
22
metale te występują w postaci wodorowęglanów, węglanów lub wodorotlenków, wówczas mówimy o
tzw. twardości węglanowej (przemijającej). Zwykle zanika ona w trakcie gotowania wody przez wytrące-
nie minerałów węglanowych (głównego składnika „kamienia kotłowego”):
Ca(HCO
3
)
2
⇒ H
2
O + CO
2
+ CaCO
3
⇓
Gdy w wodzie obecne są siarczany i chlorki tych metali, wówczas mamy do czynienia z twardością nie-
węglanową (trwałą). Taka twardość nie zanika w trakcie gotowania, lecz wymaga stosowania specjalnych
metod chemicznych. Suma twardości węglanowej i niewęglanowej daje twardość ogólną.
Jednostkami twardości wody są mval/dm
3
(milirównoważnik), lub stopnie twardości
wg skali nie-
mieckiej (
°
N, 1 stopień odpowiada 10 mg CaO w dm
3
), francuskiej (
°
Fr, 1 stopień odpowiada 1 g CaCO
3
w 100 dm
3
), angielskiej (
°
Ang, 1 stopień odpowiada 1 g CaCO
3
w 70 dm
3
) oraz amerykańskiej (
°
USA) i
WHO (1
°
= 500 mg CaCO
3
/dm
3
). W Polsce stosowane są najczęściej mval/dm
3
, stopnie skali niemieckiej
lub jednostki WHO. W zależności od twardości ogólnej wyróżniamy (1 mval/dm
3
= 2,8
°
N):
-
wodę bardzo miękką;
< 0,5
°
N
-
wodę miękką;
5-10
°
N
-
wodę średnio twardą;
10-20
°
N
-
wodę twardą;
20-30
°
N
-
wodę bardzo twardą
> 30
°
N
Twardość wody ma wpływ na możliwości jest wykorzystania. Z wód twardych łatwo wytrąca się ka-
mień, więc nie powinno się ich wykorzystywać np. w sieciach centralnego ogrzewania. Nie powinno się
jej również stosować w gospodarstwie domowym, gdyż zwiększa użycie mydła i środków piorących, a
niekiedy nawet zmienia smak mięsa, kawy i herbaty. Może też powodować podrażnienia skóry. Z kolei
wody zbyt miękkie sprzyjają np. zachodzeniu procesów korozyjnych w rurach wodociągowych oraz
zwiększają ryzyko chorób serca.
Twardość wody można wyznaczyć eksperymentalnie lub obliczyć z wyników analizy chemicznej
wody
. W takim przypadku mnoży się stężenia kationów powodujących twardość (podane w mg/dm
3
)
przez odpowiednie współczynniki przeliczeniowe (Tabela 2) otrzymując wyniki w mval/dm
3
.
Tabela 6.5. Współczynniki przeliczeniowe do obliczania twardości wody
Kation
Mnożnik
Kation
Mnożnik
Ca
2+
0,04990
Fe
3+
0,05372
Mg
2+
0,08224
Al
3+
0,1112
Sr
2+
0,02282
Zn
2+
0,03059
Fe
2+
0,03581
Mn
2+
0,07281

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
23
Pobór próbek wód
Przy pobieraniu próbek wód do analiz chemicznych i fizykochemicznych musimy zdawać sobie sprawę
z faktu, że właściwości wody ulegają bardzo szybkim zmianom. Zmiany te mogą być związane m.in. z
takimi zjawiskami jak:
-
zużywanie niektórych składników pokarmowych przez bakterie, glony itp., wydzielanie produk-
tów metabolizmu, reakcje fotosyntezy;
-
utlenianie organiki, Fe(II), siarczków;
-
wytrącanie niektórych składników (CaCO
3
, Fe(OH)
3
, Mg
3
(PO
4
)
2
etc.);
-
ulatnianie się niektórych składników (O
2
, H
2
S, CN
-
, Hg, związków organicznych);
-
adsorpcja CO
2
z powietrza - zmiana pH, przewodnictwa, zawartości CO
2
etc.;
-
adsorpcja na ściankach naczyń lub na obecnych w wodzie zawiesinach.
Z tego względu niektóre oznaczenia powinno wykonywać się od razu, w terenie. Dotyczy to m.in.
temperatury, pH, Eh, przewodności, zawartości rozpuszczonego tlenu. W innych przypadkach można
wodę analizować po przetransportowaniu do laboratorium, ale wymaga to odpowiedniego sposobu pobo-
ru, przechowywania a w wielu przypadkach również tzw. utrwalania próbek. Zapobieganie zmianom
składu i właściwości próbek obejmuje:
-
odpowiedni dobór i przygotowanie przyrządów i naczyń (np. nie można przechowywać w naczy-
niach szklanych próbek do oznaczania śladowych zawartości Si, Al, Na, B, a w plastikowych pró-
bek, w których oznaczane będą niektóre składniki organiczne – np. WWA);
-
sączenie w przypadku próbek przeznaczonych do badania składników rozpuszczonych;
-
schładzanie/zamrażanie próbek (wstrzymuje to proces rozmnażania bakterii oraz sprzyja zatrzy-
maniu substancji lotnych w próbce – pamiętamy, że rozpuszczalność gazów rośnie ze spadkiem
temperatury)
-
całkowite napełnianie naczyń – czyli bez powietrza (ogranicza to wstrząsy w trakcie transportu,
zapobiega wymianie składników z powietrzem, spowalnia reakcje redoksowe)
-
dodawanie odpowiednich środków utrwalających
Rodzaj dodawanych środków utrwalających zależy od tego, jakie oznaczenia chcemy wykonać.
Bardzo często może się więc okazać, że do laboratorium przywozimy kilka różnych naczyń zawierają-
cych próbkę tej samej wody, ale utrwaloną w różny sposób. Do najczęściej stosowanych „utrwalaczy”
zaliczamy:
-
stężone kwasy (gł. HNO
3
, H
2
SO
4
– oznaczanie metali, N-NH
4
, N
org
, ChZT, fenoli); HNO
3
utrzy-
muje metale w stanie rozpuszczonym, H
2
SO
4
działa jako bakteriocyd i tworzy siarczany z lotnymi
zasadami (aminy, amoniak);
-
chloroform (oznaczanie N-NO
3
, N-NO
2
, detergentów anionowych, cukrów, WWA, zawiesin);
-
NaOH (oznaczanie CN
-
, fenoli lotnych); tworzy sole sodowe lotnych kwasów;

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
24
-
HgCl
2
(bakteriocyd – do próbek zawierających biodegradowalne związki organiczne oraz różne
formy azotu i fosforu).
Należy pamiętać, że utrwalenia próbki dokonuje się PO pomiarach wstępnych (pH, przewodnictwo
itd.)!!. Warto również podkreślić, że utrwalenie próbki wody pozwala jedynie na niewielką zwłokę w
wykonaniu analizy. Większość składników powinno się oznaczyć przed upływem 24 lub 48 h. Stosunko-
wo najodporniejsze na zmiany są (oczywiście przy odpowiednim utrwaleniu i przechowywaniu próbki)
tzw. metale ciężkie – w niektórych przypadkach dopuszcza się nawet kilkumiesięczny okres przechowy-
wania próbek.
Sposoby przedstawiania wyników analiz chemicznych wód
Analizy chemiczne wód przedstawia się w różnych jednostkach. Najczęściej stosuje się mg/dm
3
(mg/L)
i
µ
g/dm
3
(
µ
g/L), rzadziej w mg/kg czy
µ
g/kg. Dla wód mineralnych, zawierających duże ilości rozpusz-
czonych substancji, często ich ilość wyraża się w g/dm
3
. W przypadku substancji o szczególnie niskich
stężeniach (np. WWA) wykorzystuje się bardzo małe jednostki, jak ng/dm
3
.
Większość składników rozpuszczonych w wodach występuje w formie jonowej. Sumaryczny dodatni
ładunek kationów musi być równoważony przez sumaryczny ładunek ujemny anionów. Z tego względu,
do celów prezentowania składu wód oraz ich klasyfikacji, zawartość (stężenie) każdego kationu i anionu
przedstawia się w formie równoważnikowej (np. mval/dm
3
, meq/L), uwzględniającej jego masę molową i
wartościowość.
Stężenie równoważnikowe dla danego jonu otrzymujemy mnożąc jego stężenie mo-
lowe przez ładunek
.
Przykład. Poniższe wyniki analizy chemicznej wody zestawione w mg/dm
3
przedstaw w mmol/dm
3
i
mval/dm
3
.
Na
+
13,70
HCO
3
-
79,9
K
+
1,18
Cl
-
31,2
Ca
2+
42,50
SO
4
2-
39,0
Mg
2+
3,21
NO
3
-
1,3
1 mol Na ma masę 22,99 g, czyli 1 mmol Na to 22,99 mg. Zatem jeśli stężenie sodu w wodzie wynosi
13,70 mg/dm
3
, to z proporcji otrzymujemy:
1 mmol Na – 22,99 mg
x mmol Na – 13,70 mg
x = 0,60 mmol
Stężenie sodu wynosi 0,60 mmol/dm
3
, a ponieważ sód występuje w postaci kationu jednowartościowego
więc 1 mmol = 1 mval, czyli 0,60 mmol/dm
3
= 0,60 mval/dm
3
.
W przypadku wapnia masa jednego milimola to 40,08 mg, więc stężenie molowe Ca w tej wodzie to
42,50mg/40,08=1,06 mmol/dm
3
. Ale wapń występuje w postaci kationu dwuwartościowego, więc rów-
noważnik jest dwukrotnie większy, czyli 1,06 mmol/dm
3
×
2 = 2,12 mval/dm
3
. W ten sposób przeliczamy
wszystkie kationy i aniony na „stężenia ładunków” wyrażone w mval/dm
3
otrzymując:
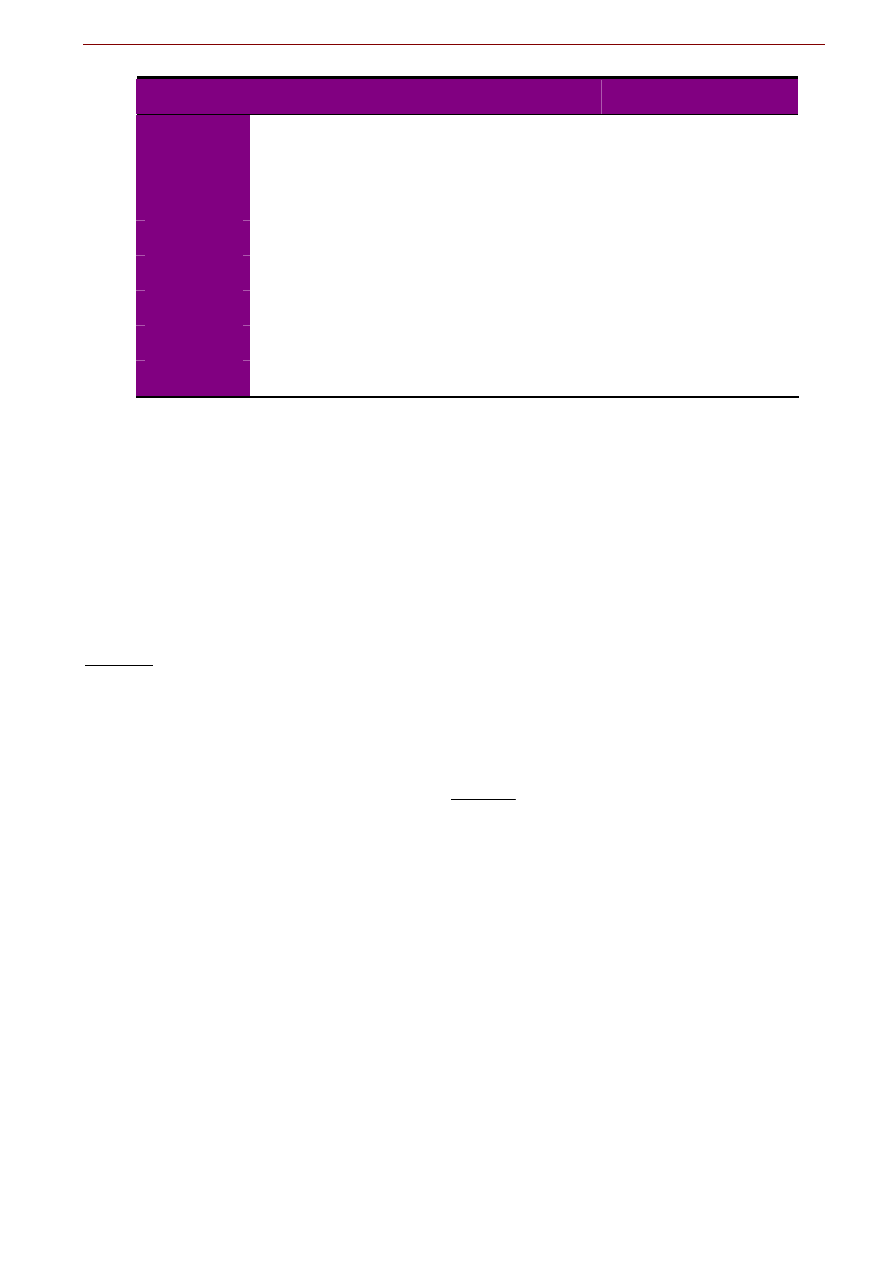
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
25
Jon
mg/dm
3
Masa molowa
mmol/dm
3
Ładunek
mval/dm
3
Na
+
13,70
22,99
0,60
1
0,60
K
+
1,18
39,1
0,03
1
0,03
Ca
2+
42,50
40,08
1,06
2
2,12
Mg
2+
3,21
24,31
0,13
2
0,26
HCO
3
-
79,9
61,02
1,31
1
1,31
SO
4
2-
39,0
96,06
0,41
2
0,82
Cl
-
31,2
35,45
0,88
1
0,88
NO
3
-
1,3
62,00
0,02
1
0,02
Przedstawiając wyniki analizy chemicznej można od razu w prosty sposób sprawdzić jej popraw-
ność.
Ponieważ sumaryczny dodatni ładunek kationów musi być równoważony przez sumaryczny
ładunek ujemny anionów, więc suma (mili)równoważników kationów powinna być (mniej więcej)
równa sumie (mili)równoważników anionów
. Wyraźna różnica pomiędzy tymi wartościami może wy-
nikać albo z błędu analitycznego (błędne oznaczenie któregoś ze składników), albo z faktu, że badana
woda ma nietypowy skład chemiczny – zawiera jakieś składniki, których rutynowo się nie oznacza. Ten
drugi przypadek ma często miejsce przy badaniach wód silnie zanieczyszczonych i ścieków.
Przykład. Na podstawie bilansu jonowego sprawdź poprawność analizy chemicznej wody z poprzedniego
przykładu.
Sumujemy stężenia równoważnikowe kationów i anionów:
Σ
K = 0,60 + 0,03 + 0,26 + 2,12 = 3,01
Σ
A = 0,88 + 1,31 + 0,82 + 0,02 = 3,03
Widać, że
Σ
K ≈
Σ
A, ale lepiej to wyrazić obliczając błąd analizy (nie powinien on przekraczać 5%):
%
100
⋅
Σ
+
Σ
Σ
−
Σ
=
A
K
A
K
x
w naszym przypadku błąd analizy jest bardzo mały i wynosi:
x = (3,01+3,03)/3,01-3,03
⋅
100% = - 0,3%
Graficzne sposoby prezentacji składu chemicznego wód
W celu szybkiego zorientowania się w typie hydrogeochemicznym danej grupy wód, ich składy che-
miczne często przedstawia się graficznie – na diagramach kołowych, słupkowych, trójkątnych i innych.
Do najpopularniejszych i najczęściej stosowanych sposobów takiej prezentacji należą diagramy Pipera,
Stiffa i Udlufta.
Diagram Pipera składa się z trzech części – dwóch trójkątów i rombu (rysunek). Lewy trójkąt podaje
względne proporcje głównych kationów (Ca
2+
, Mg
2+
oraz sumy Na
+
i K
+
) przeliczone z wyników analiz
wyrażonych w mval/dm
3
, a prawy przedstawia w ten sam sposób główne aniony (HCO
3
-
+ CO
3
2-
, SO
4
2-
i
Cl
-
). Punkty odpowiadające proporcjom kationów i anionów rzutuje się następnie prostymi na diagram
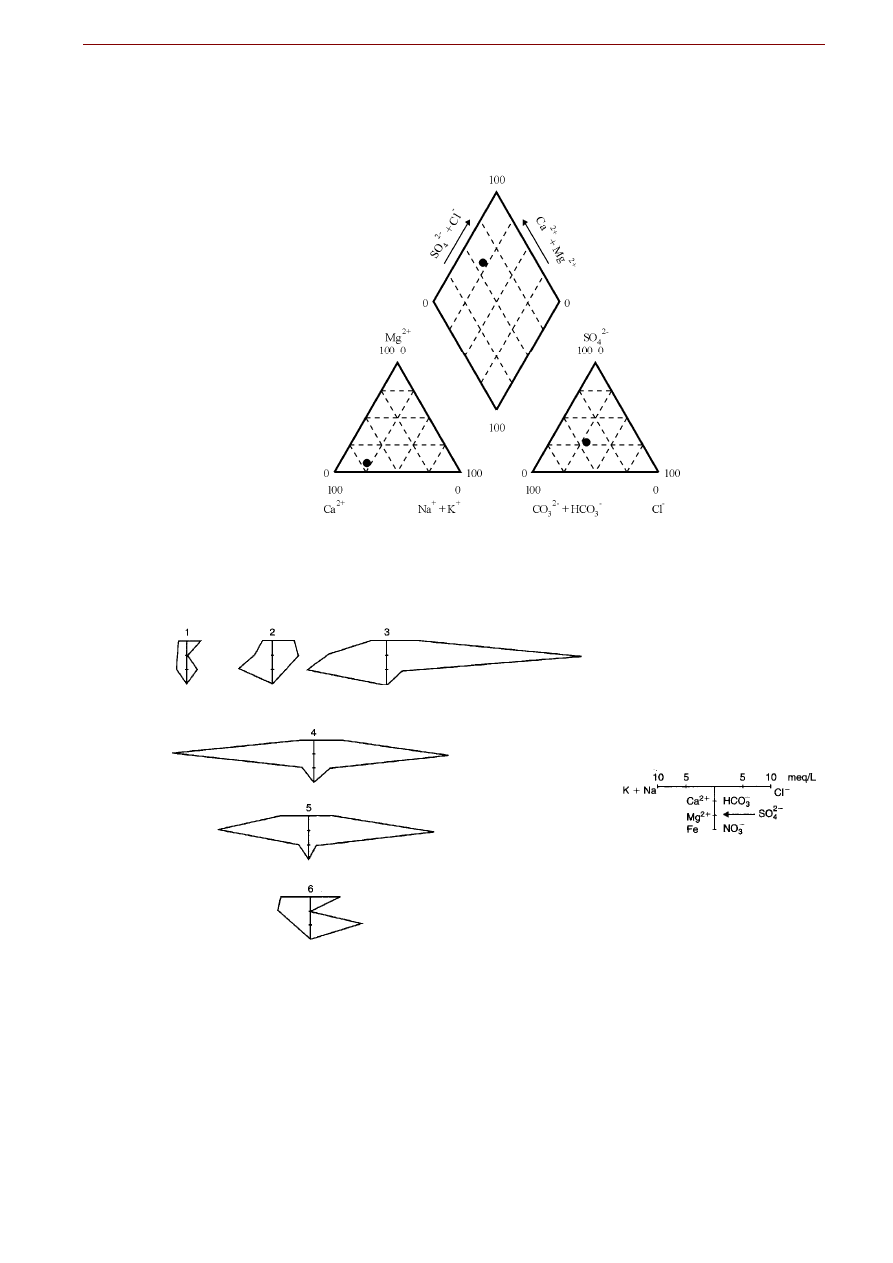
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
26
rombowy, uzyskując w ich przecięciu punkt projekcyjny reprezentujący badaną wodę. Dla pełniejszego
zobrazowania jej składu zwykle różnicuje się średnicę kółka w zależności od wielkości mineralizacji wo-
dy.
Rys. . Diagram Pipera.
Rys. . Przykładowe diagramy Stiffa.
Diagram Stiffa często stosuje się do przedstawiania składu chemicznego wód podziemnych w profilach
pionowych. Na tym wykresie pionową oś pomocniczą (przy profilu wyskalowaną jako głębokość otworu)
przecinają cztery (najczęściej) równoległe osie, dające w efekcie osiem półosi, na których nanosi się stę-
ż
enia kationów (po lewej stronie osi pionowej) i anionów (po jej prawej stronie). Zawsze tym samym
osiom przyporządkowuje się określone jony, a stężenia, podobnie jak w diagramie Pipera nanosi się w
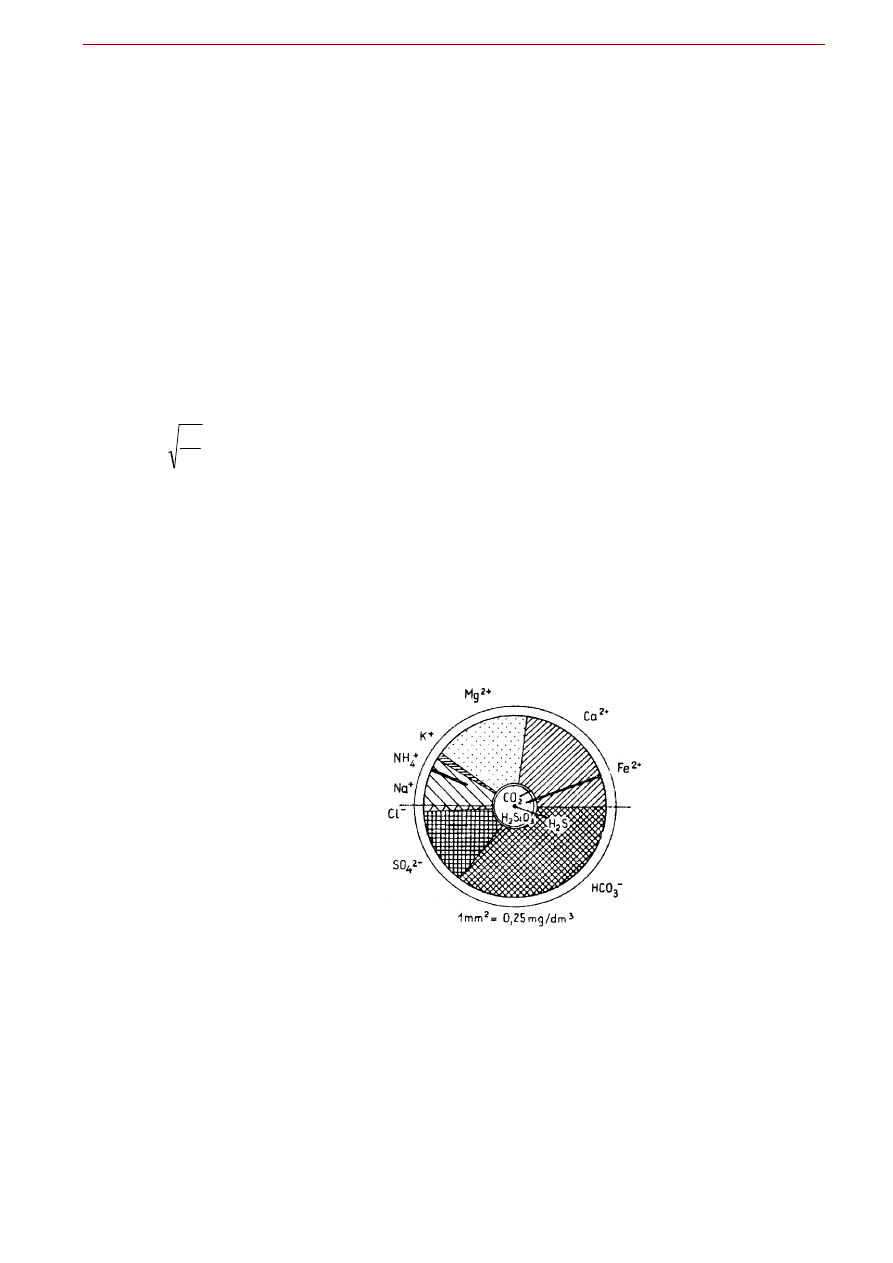
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
27
mval/dm
3
, przypisując pionowej osi pomocniczej stężenie zerowe. W oryginalnej wersji diagramu lewym
półosiom przypisuje się (idąc od góry): Na
+
+ K
+
, Ca
2+
, Mg
2+
i Fe
2+
, zaś prawym półosiom, odpowiednio,
Cl
-
, HCO
3
-
, SO
4
2-
i CO
3
-
. Naniesione na półosiach punkty stężeń łączy się ze sobą otrzymując nieregular-
ny wielobok.
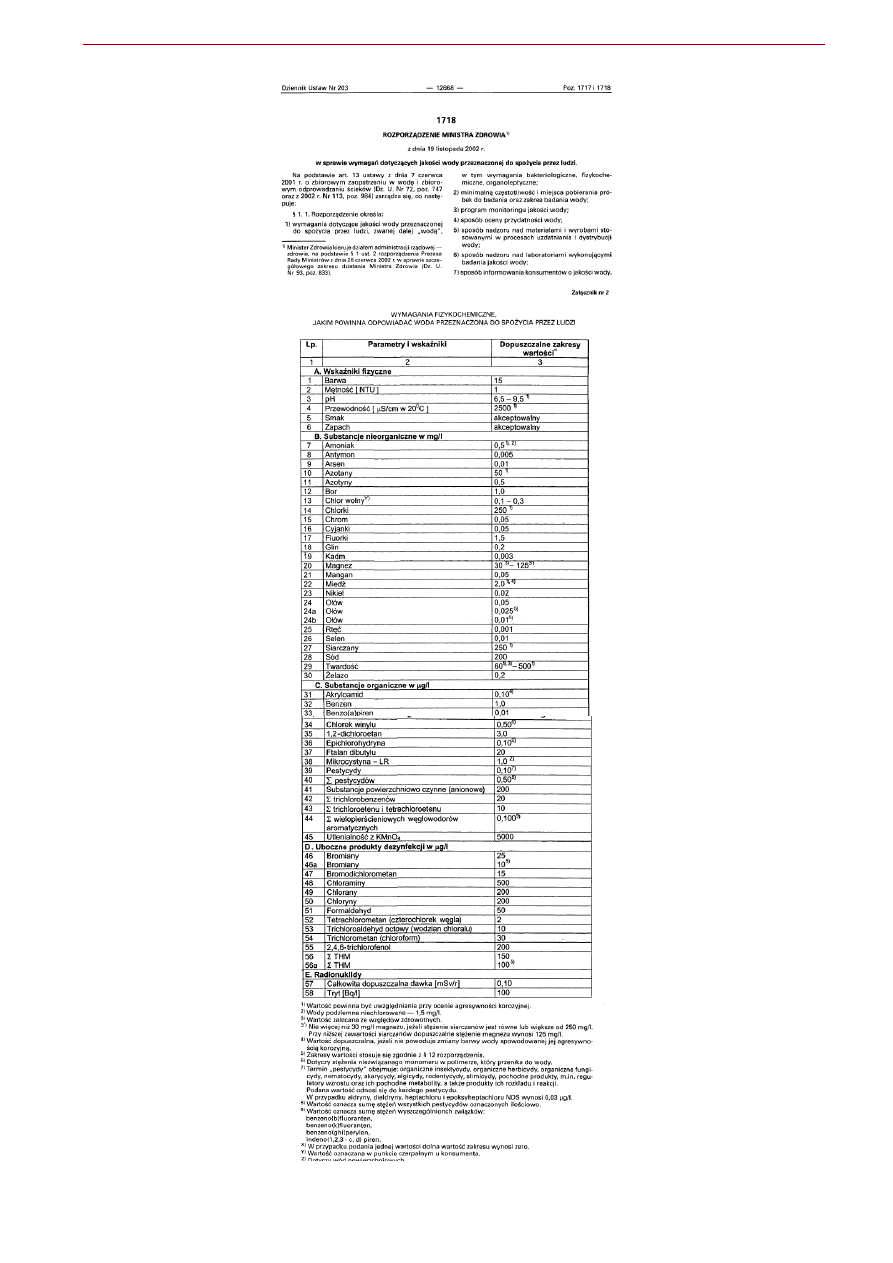
Diagram Udlufta jest wykresem kołowym, uwzględniającym nie tylko główne kationy i aniony, ale
również substancje gazowe rozpuszczone w wodzie. Przy jego sporządzeniu przyjmujemy, że pole koła
wyraża sumaryczną ilość składników stałych i gazowych zawartych w wodzie (M). W zależności od
przyjętej skali wykresu, 1 mm
2
powierzchni koła może odpowiadać np. 1, 4, 9, 16 itd. mg/dm
3
sumy tych
składników. Przyjmując jednostkową zależność 1 mg/dm
3
= 1 mm
2
otrzymujemy:
M =
Π
r
2
stąd:
Π
=
M
r
gdzie M to łączna masa składników stałych i gazowych rozpuszczonych w 1 litrze wody a r to promień
koła [mm] obrazującego skład chemiczny wody.
Składniki stałe i nie zdysocjowane zaznacza się w postaci współśrodkowych kół, których promień obli-
cza się zgodnie z powyższym wzorem, przyjmując zamiast sumarycznego M masę składników gazowych
i niezdysocjowanych. W górnej połowie koła lub pierścienia przedstawia się wycinkami procenty mili-
gramorównoważników podstawowych kationów, w dolnej połowie podstawowych anionów.
Rys. . Przykładowy diagram Udlufta
Woda pitna
Skład i metodyka kontroli jakości wody pitnej jest regulowana odpowiednimi ustawami. Na figurze
poniżej przedstawiony jest przykład fragmentu takiego rozporządzenia, w którym podany jest jego zakres
oraz jeden z załączników podający normy regulujące maksymalne dopuszczalne zawartości niektórych
składników wód, które są istotne z punktu widzenia konsumpcji.
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
28
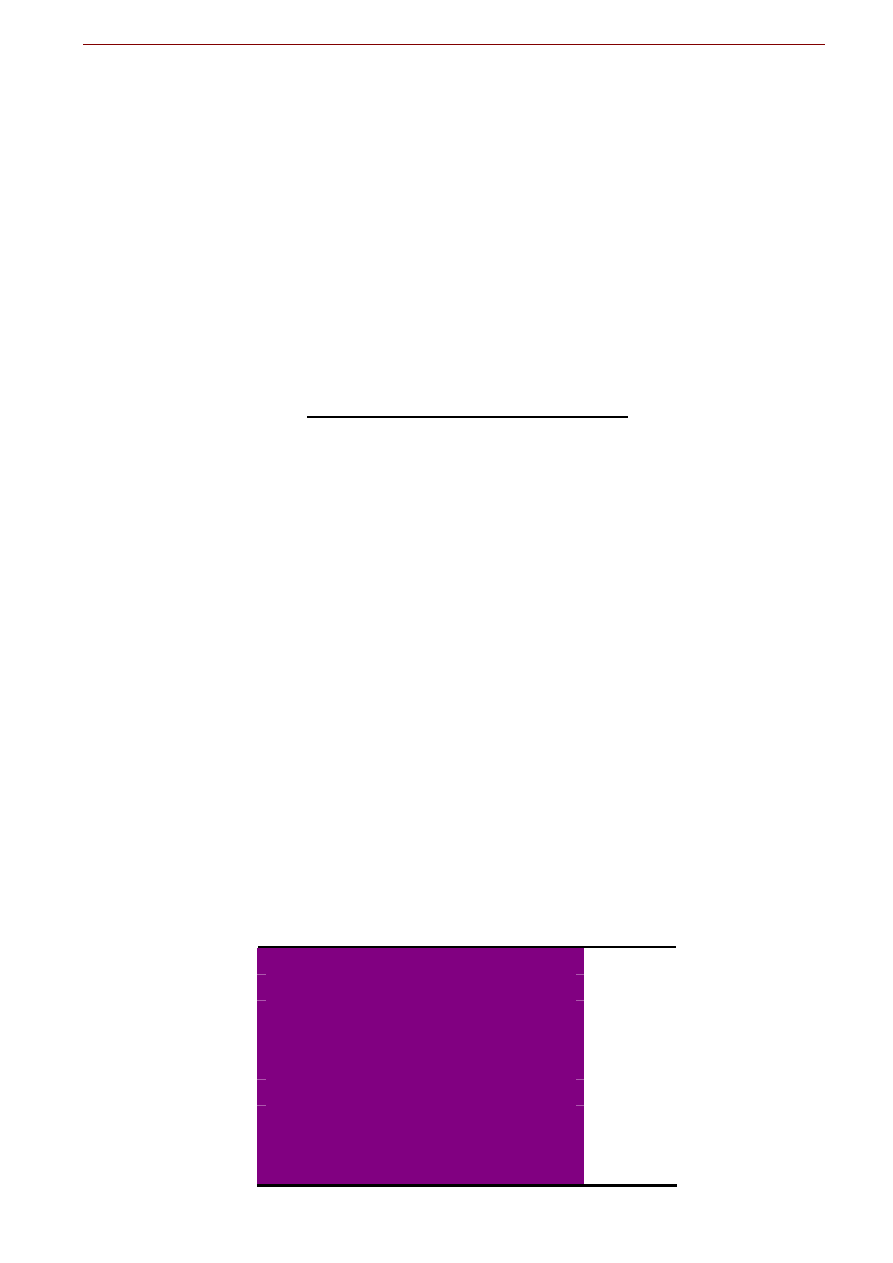
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
29
Źródłem wody pitnej są głównie studnie wiercone i kopane, wody rzek i jezior. Wody podziemne czę-
sto nie wymagają kosztownych procesów uzdatniania, może istnieć potrzeba zmiękczania, które może
być przeprowadzone w niewielkich domowych instalacjach montowanych w piwnicy. Wody rzeczne i
jeziorne do celów komunalnych są uzdatniane głównie przez usunięcie zawiesin oraz kontrolę i zabezpie-
czenie bakteryjne. W większości krajów określenie „woda pitna” oznacza, że można nalać sobie z kranu
wodę do szklanki i wypić bez konsekwencji w pieluchach. Nie radzę jednak próbowania takiego ekspe-
rymentu w Krakowie gdyż grozi to dość rzadką (!) chorobą na siedząco. Najwięcej zabiegów jednak wy-
maga oczyszczenie ścieków przed odprowadzeniem ich z powrotem do środowiska. Dotyczy to zarówno
zakładów przemysłowych, instalacji komunalnych jak i pojedynczych gospodarstw domowych.
6.4. Elementy geochemii wód naturalnych
Aby spowolnić postępującą degradację zasobów wodnych w wielu krajach rozwiniętych stosowane jest
odpowiednie prawodawstwo. Na przykład w niektórych krajach istnieje przepis, że wodę z rzeki można
pobierać tylko PONIŻEJ ujścia własnych ścieków. Są też przepisy stanowiące, że gospodarstwa domowe
czy gminy nie mogą uruchomić wodociągu dopóki nie maja wykonanej instalacji kanalizacyjnej obejmu-
jącej utylizację ścieków. Powszechnie uważa się jednak, że najskuteczniejszym sposobem ochrony i
optymalnego wykorzystania zasobów wodnych na Ziemi jest edukacja społeczeństw. Znacznie taniej jest
oszczędzać wodę i unikać zanieczyszczeń ściekami niż likwidować skutki takich zanieczyszczeń. Czy
używasz wody godnie? Przyglądnij się poniższej tabelce z oszacowaną objętością zużywanej przez nas
każdego dnia wody w cywilizowanym świecie. Pamiętaj o tym, że z każdej kropli wody użytej z kranu
powstanie kropla ścieków do oczyszczenia w oczyszczalni. Ta oczywista prawda dla wielu nie jest tak
oczywista. Na przykład przez wiele lat różne organizacje międzynarodowe niosące pomoc krajom Trze-
ciego Świata finansowały powstawanie ujęć wody i studni w krajach tropikalnych, gdzie dostęp do wody
jest utrudniony. Nie zawsze jednak starczało zdrowego rozsądku lub pieniędzy, aby zbudować kanaliza-
cję, przez co zdarza się, że ścieki komunalne płyną ulicami.
Tabela. Czy używasz wody godnie? W tabeli podana jest szacunkowa objętość zużywanej przez nas wody przy codziennych
czynnościach. Zużywając takie ilości wody produkujemy tą samą objętość ścieków wymagających utylizacji w oczyszczalni.
Prysznic, 5 min.
100-150 L
Wanna
120 L
Mycie zębów przy otwartym kurku
8 L
Golenie przy otwartym kurku
20 L
Mycie naczyń pod strumieniem wody
120 L
Zmywarka do naczyń
5 – 15 L
Mycie rąk pod strumieniem wody
8 L
Spłukanie ubikacji
4 – 10 L
Podlewanie kwiatów w ogródku 5 min.
150–200L

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
30
Fig. Nie brak już wody lecz wciąż brak kanalizacji…
Jednym z doniosłych problemów zanieczyszczeń wód powierzchniowych m.in. ze względu na olbrzy-
mie objętości trudne do opanowania działaniami neutralizującymi są bardzo kwaśne ścieki wód kopalnia-
nych
AMD
(ang. Amid Mine Drainage). Głównym powodem ich powstawania jest obecność minerałów
siarczkowych, towarzyszących w większości złóż węgli i metali.
Przy eksploatacji następuje ekspozy-
cja tych minerałów powodując utlenianie pod wpływem tlenu z powietrza. Wskutek ich utlenienia
powstaje kwas siarkowy
, na przykład w myśl reakcji podanych poniżej (po ich prawej stronie zapisany
jest bilans elektronowy niezbędny do uzgodnienia reakcji).
2FeS
2
+ 2H
2
O + 7O
2
⇒ 2FeSO
4
+ 2H
2
SO
4
4S
1-
- 28e
-
⇒ 4S
6+
7O
2
+ 28e
-
⇒ 14O
2-
4FeSO
4
+ 6H
2
O + O
2
⇒ 4FeOOH + 4H
2
SO
4
4Fe
2+
- 4e
-
⇒ 4Fe
3+
goethyt
O
2
+ 4e
-
⇒ 2O
2-
2Fe
2+
+ 3H
2
O + ½O
2
→
2FeOOH + 4H
+
2Fe
2+
- 2e
-
⇒ 2Fe
3+
goethyt
½O
2
+ 2e
-
⇒ O
2-
Odczyn pH kwaśnych wód AMD jest znacznie niższy od środowisk nieskażonych i może wynosić mię-
dzy 1 a 3, a czasem nawet mniej. Jest to bardzo kwaśny odczyn rzadko spotykany w innych ekstremal-
nych środowiskach. Dla porównania, jest to pH zbliżone lub niższe od kwasu solnego HCl używanego
powszechnie przez geologów terenowych do identyfikacji kalcytu. Każdy student, któremu na praktyce
wypadły dziury w spodniach po kapnięciu takiego kwasu wie, co mam na myśli. Produktem tych reakcji
są m.in. nierozpuszczalne minerały żelaza na trzecim stopniu utlenienia, na przykład goethyt FeOOH,
które powodują powstawanie charakterystycznych żółtych i brunatnych osadów zwanych po angielsku
yellow boy. W warunkach naturalnych reakcje utleniania siarczków zachodzą niemal zawsze przy udziale
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
31
bakterii, które przyspieszają ich przebieg dziesiątki i setki razy (!). Najbardziej znaną bakterią tego typu
jest kwasolubna Acidothiobacillus ferrooxidans.
Fig. AMD w zbiornikach utworzonych w zalanych wyrobiskach i w odpływach ścieków kwaśnych wód kopalnianych. Bakte-
rie kwasolubne Acidothiobacillus ferrooxidans biorące udział w procesach utleniania siarczków prowadzących do zakwaszenia
wód ściekowych.
Diagramy pH-Eh
Diagramy pH-Eh, konstruowane dla różnych układów, pozwalają na przewidywanie zachowania się
pierwiastków w trakcie zmian warunków środowiska. Do ich skonstruowania konieczne jest przyjęcie
różnych założeń, dotyczących stężeń, ciśnień parcjalnych gazów, temperatury etc.
Fig. Przykład diagramu pH-Eh z naniesionymi zakresami warunków dla wybranych środowisk na powierzchni Ziemi: 1 - wo-
dy deszczowe; 2 - wody rzeczne; 3 - wody morskie; 4 - granica warunków silnie utleniających
Jednym z powodów łącznego rozpatrywania odczynu pH i warunków utleniająco-redukcyjnych Eh
przy analizowaniu wód naturalnych jest fakt, że stopień utlenienia pierwiastków w środowisku jest
silnie zależny zarówno od pH jak i potencjału redoks Eh
. Wystarczy zwrócić uwagę na ukośny prze-
bieg linii – granic pól trwałości na diagramie pH-Eh. Na przykład żeby utlenienie Fe
2+
do Fe
3+
(przekro-
czenie linii będącej granica pól trwałości tych jonów na diagramie pH-Eh) może być spowodowane bądź
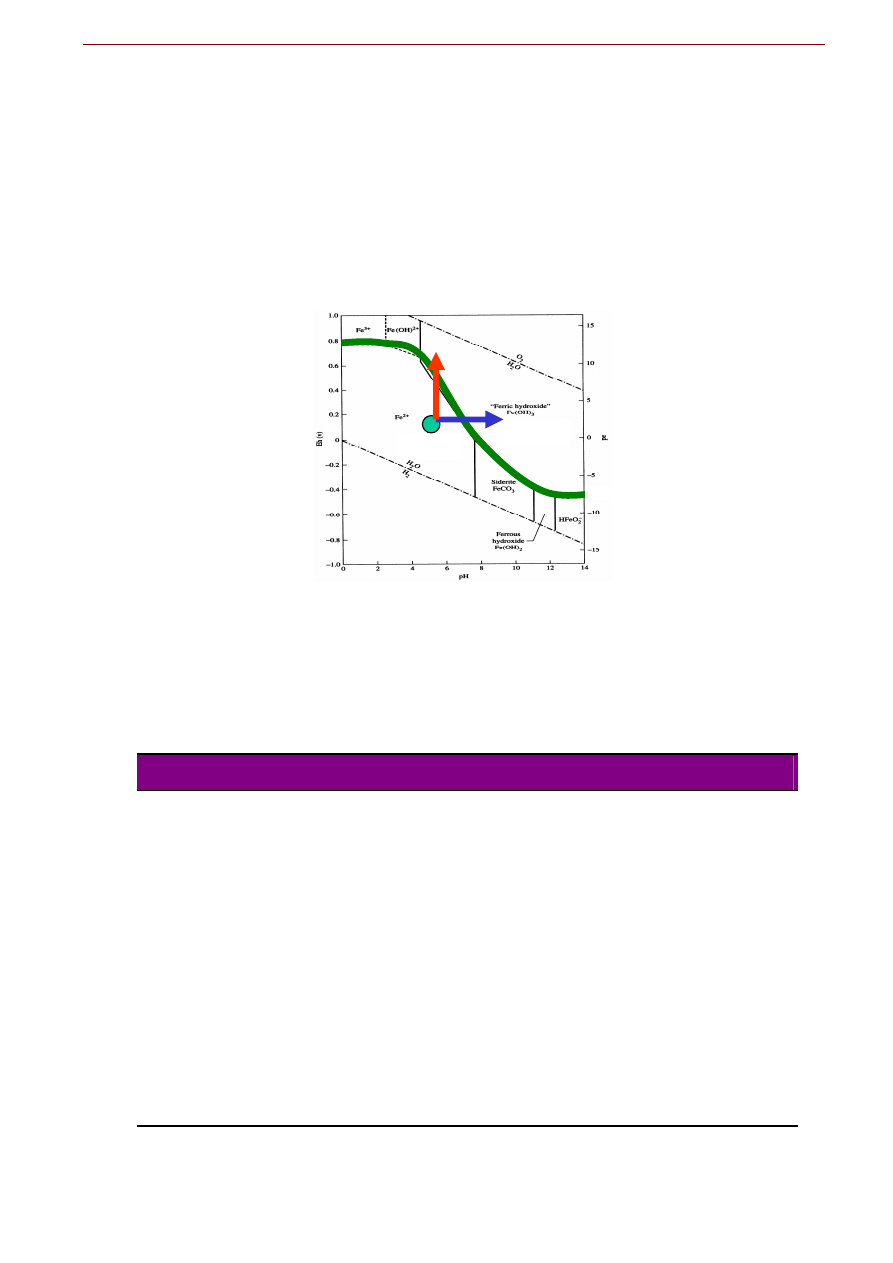
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
32
to podniesieniem utleniającego charakteru środowiska (na diagramie: zwiększeniem Eh) bądź też zalkali-
zowaniem środowiska (na diagramie: zwiększeniem pH). Obrazują to odpowiednie strzałki na Fig. poni-
ż
ej. Żelazo migruje w wodzie głównie w formie Fe
2+
. Jego utlenienie prowadzi do natychmiastowego
wytrącenia FeOOH. Może do tego dojść wskutek zmiany warunków z redukcyjnych na utleniające (np.
pod wpływem tlenu z powietrza czy działalności bakterii, jak to ma miejsce w przypadku AMD) lub
wskutek wzrostu pH (np. wytrącanie tlenków Fe przy ujściach rzek do morza). Przykłady reakcji wpły-
wających na pH środowisk naturalnych podane są w tabeli poniżej.
Fig. Stopień utlenienia pierwiastków w środowisku jest silnie zależny zarówno od pH jak i potencjału redoks Eh (zwróć uwagę
na ukośny przebieg linii na diagramie pH-Eh). Np. żeby utlenić Fe
2+
do Fe
3+
można zwiększyć Eh lub podwyższyć pH (zalkali-
zować środowisko).
Tabela 6.. Przykłady reakcji kontrolujących pH w wodach naturalnych
Przykłady reakcji kontrolujących pH w wodach naturalnych
Rozpuszczanie CO
2
w wodzie ( -> obniża pH)
CO
2(g)
+ H
2
O < = > HCO
3
-
+ H
+
Rozpuszczanie minerałów węglanowych ( -> podnosi pH)
CaCO
3
+ H
+
< = > Ca
2+
+ HCO
3
-
Wytrącanie węglanów na skutek reakcji z osadem np. z gipsem ( -> obniża pH)
CaSO
4
.
2H
2
O + HCO
3
-
< = > CaCO
3
+ SO
4
2-
+ H
2
O + H
+
Wietrzenie chemiczne glinokrzemianów ( -> podnosi pH)
2KAlSi
3
O
8
+ 2H
2
CO
3
+ 9H
2
O < = > Al
2
Si
2
O
5
(OH)
4
+ 2K
+
+ 4H
4
SiO
4
+ 2HCO
3
-
Redukcja siarczanów z udziałem mikroorganizmów ( -> podnosi pH)
SO
4
2-
+ 8H
+
+ 6e
-
< = > S
o
+ 4H
2
O
SO
4
2-
+ 9H
+
+ 8e
-
< = > HS
-
+ 4H
2
O
Fe
3+
Fe
2+

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
33
6.5. Elementy termodynamiki roztworów wodnych
Aktywność jonów w roztworze i obliczanie współczynnika aktywności
W chemii i geochemii najpospolitszym i jednocześnie najbardziej użytecznym sposobem wyrażania
stężenia substancji w roztworze jest stężenie molowe, biorące pod uwagę nie tylko bezwzględną ilość
rozpuszczonej substancji w określonej objętości roztworu, ale również rodzaj tej substancji.
Stężenie mo-
lowe oznacza ilość moli danego składnika zawartą w 1 litrze (lub jednym kilogramie) roztworu
.
Jednakże do obliczeń geochemicznych znajomość stężeń składników roztworu okazuje się często niewy-
starczająca czy wręcz niezbyt reprezentatywna dla jego właściwości. Jony w roztworach elektrolitów od-
działywają między sobą oraz z cząsteczkami wody m. in. siłami elektrostatycznymi. Dlatego aktywność
jonów w roztworze jest zwykle mniejsza niż jego stężenie:
[Me+2] = a =
γ
. m
γ
< 1
Aktywność jest to skorygowane stężenie
. Niższa aktywność niż stężenie oznacza, że część z jonów
obecnych w roztworze, na skutek oddziaływań elektrostatycznych z innymi jonami, nie będzie brała
udziału w reakcjach chemicznych i procesach geochemicznych (krystalizacja, sorpcja itd.). Takie oddzia-
ływanie wpływające na aktywność jonów wzrasta ze wzrostem stężeń. Przez analogię, pojęcie wzajemnej
relacji stężenia do aktywności można przedstawić na przykładzie sali wykładowej. Jeśli ilość studentów
w sali jest niewielka (niskie „stężenie” studentów) wykładowca ma bezpośredni kontakt wzrokowy ze
wszystkimi słuchaczami, którzy aktywnie uczestniczą w zajęciach. A więc
przy niskim stężeniu aktyw-
ność jonów jest równa stężeniu
. Natomiast, gdy sala wykładowa jest przepełniona, studenci siłą rzeczy
przeszkadzają sobie albo zwyczajnie grają w ostatnich rzędach w karty i liczba osób aktywnie uczestni-
czących w zajęciach jest wyraźnie mniejsza niż liczba obecnych na sali (aktywność niższa niż stężenie).
A więc
przy wyższych stężeniach aktywność jonów jest niższa niż stężenie i musi zostać obliczona
przez znalezienie współczynnika aktywności γ
. Dla cząsteczek nieposiadających ładunku oddziaływa-
nia elektrostatyczne praktycznie nie występują w ogóle a więc dla nich nie istnieje różnica pomiędzy stę-
ż
eniem a aktywnością.
Zazwyczaj nie da się zmierzyć wprost aktywności jonów w roztworze. Możemy tylko wyznaczyć stę-
ż
enie, a współczynnik aktywności trzeba obliczyć z założeń teoretycznych. Współczynnik aktywności γ
jest poprawką, którą się wprowadza aby skorygować zmierzone stężenie i wyliczyć rzeczywistą aktyw-
ność chemiczną składnika. Dla rozcieńczonych roztworów elektrolitów (wody deszczowej, czystych wód
podziemnych, rzecznych czy jeziornych) można w większości przypadków przyjąć, że aktywność równa
jest stężeniu (współczynnik aktywności γ wynosi 1). W przypadku wód o wyższej mineralizacji (wielu
wód źródlanych, roztworów glebowych, zanieczyszczonych wód i ścieków itp.) do obliczenia współ-
aktywność
stężenie molowe
współczynnik
aktywności
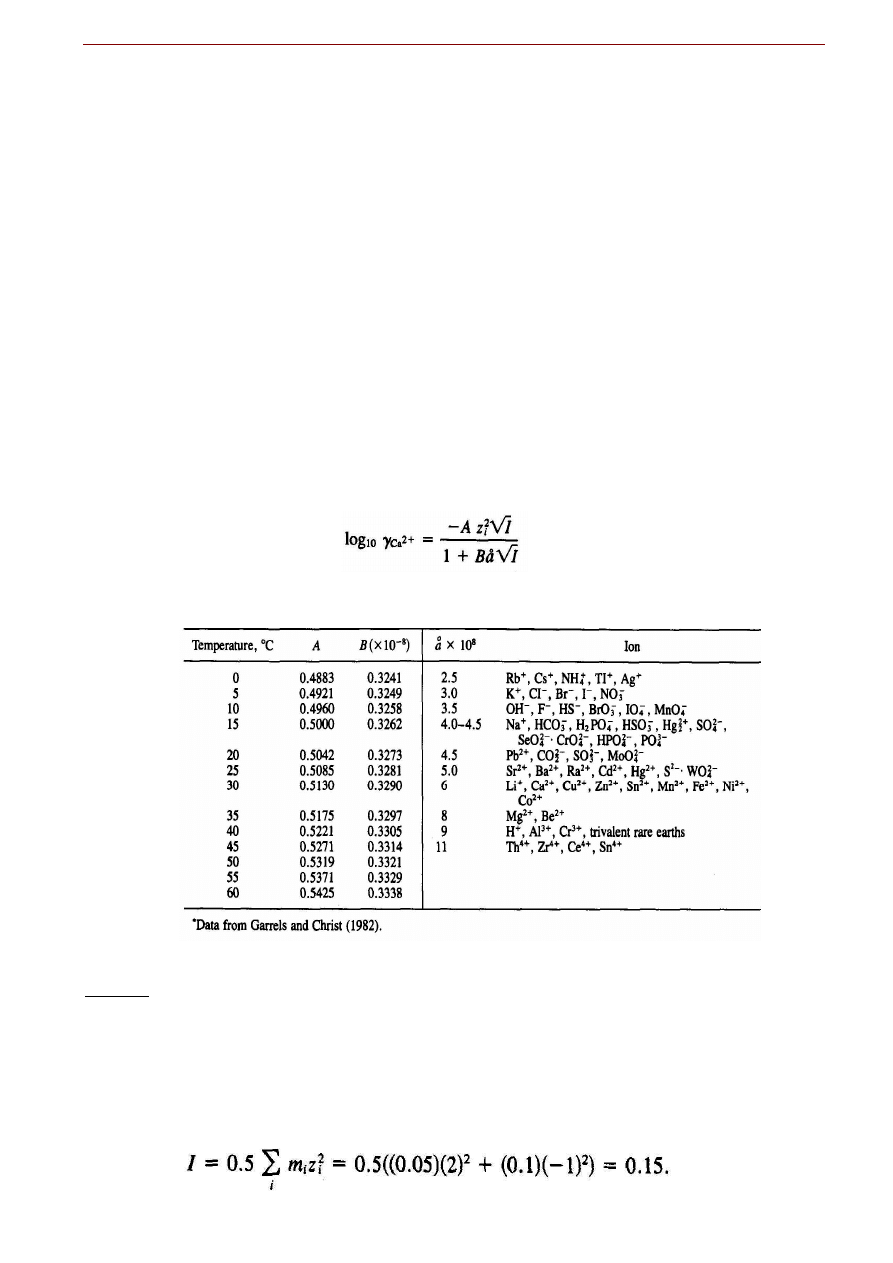
6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
34
czynników aktywności stosuje się na przykład model Debye-Hückel’a. Procedura obliczeń jest dość
ż
mudna, ponieważ trzeba dla każdego interesującego nas jonu przeprowadzić obliczenia osobno, a same
obliczenia wykonuje się metodą kolejnych przybliżeń (metoda iteracyjną). Na szczęście wykonują to za
nas programy komputerowe stworzone do hydrochemicznego modelowania specjacji roztworów wod-
nych (
specjacja to jest ilościowe rozpisanie każdego składnika roztworu na jego formy występowa-
nia w roztworze, a więc jony, związki kompleksowe, formy zaadsorbowane i in.
).
Aby obliczyć wartość współczynnika aktywności jonu w roztworze musimy wykonać analizę che-
miczną i poznać zawartość wszystkich głównych składników roztworu. Pozwala to obliczyć
siłę jonową
roztworu I
(wstawiając do wzoru oznaczone stężenia wszystkich składników jonowych i ich ładunki):
I = 0.5
.
Σ
m
i
z
i
2
gdzie: m – stężenie, z – ładunek
Następnie oblicza się współczynnik aktywności osobno dla każdego jonu w roztworze stosując wzór wy-
nikający z teoretycznie przewidzianych oddziaływań pomiędzy jonami. Wzór ten zawiera siłę jonową I,
ładunek jonu z oraz kilka parametrów empirycznych (A, B, å), które są stabelaryzowane w podręczni-
kach (patrz tabela poniżej). Wzór (model oddziaływań) Debyle-Hückel’a jest następujący:
Tabela ... Stałe potrzebne do obliczenia współczynnika aktywności jonu metodą Debye-Huckel’a
Przykład. Wyznaczyć współczynnik aktywności dla Ca
+2
w 0,05 M roztworze CaCl
2
w 25
o
C.
Najpierw zapisujemy równanie reakcji rozpuszczania CaCl
2
:
CaCl
2
<=> Ca
+2
+ 2 Cl
-
Dzięki temu możemy określić jakie są stężenia jonów w roztworze. Jeśli podczas rozpuszczania z każde-
go mola CaCl
2
powstaje jeden mol jonów Ca
2+
i dwa razy tyle moli jonów Cl
-
, to z 0,05 moli CaCl
2
, które
mamy w zadaniu powstanie 0,05 mola jonów Ca
2+
i 2
.
0,05=0,1 mola jonów Cl
-
. Teraz możemy już wyli-
czyć siłę jonową roztworu:

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
35
Następnie z wzoru Debye-Hückel’a, korzystając z danych tabelarycznych, wyliczamy współczynnik ak-
tywności γ:
Rozpuszczalność i stała rozpuszczalności
Substancje o wiązaniach jonowych rozpuszczają się w wodzie. Proces rozpuszczania w zamkniętym
zbiorniku zachodzi do momentu osiągnięcia stanu nasycenia roztworu. Jeśli pozostało jeszcze w roztwo-
rze nieco nierozpuszczonego minerału, to powstaje stan równowagi dynamicznej.
W stanie równowagi
dynamicznej, tyle samo substancji jednocześnie rozpuszcza się, co krystalizuje z roztworu
. Dla
ogólnej odwracalnej reakcji
aA + bB
⇔
cC + dD
w stanie równowagi prędkość zachodzenia reakcji w prawo jest równa prędkości zachodzenia reak-
cji w lewo. Oznacza to, że w praktyce stężenia się nie zmieniają,.
Z pozoru nic się nie dzieje.
Stosunek
ilości produktów do substratów w stanie równowagi pozostaje stały. Wyraża go stała równowagi
reakcji K
. Dla reakcji zapisanej powyżej, stała równowagi wynosi:
gdzie nawiasy kwadratowe oznaczają aktywności (czyli skorygowane stężenia) poszczególnych jonów
podniesione do potęgi równej współczynnikom w reakcji.
Przyglądnijmy się równowadze reakcji rozpuszczania i krystalizacji na przykładzie fluorytu CaF
2
. Eks-
peryment zobrazowany jest na Fig. poniżej: garść kryształków fluorytu wrzucono do wody w wystarcza-
jącym nadmiarze, że po pewnym czasie część minerału uległa rozpuszczeniu, roztwór się nasycił i układ
osiągnął stan równowagi. Obecnie w roztworze na dnie zlewki mamy trochę nierozpuszczonych kryształ-
ków, roztwór zawiera jony Ca
2+
i F
-
i ustawicznie zachodzi jednoczesne rozpuszczanie fluorytu i jego
krystalizacja tak, że ani ilość kryształków ani stężenia w roztworze nie ulegają zmianie. Mamy stan rów-
nowagi dynamicznej. Reakcja rozpuszczania jest prosta:
CaF
2
<=> Ca
2+
+ 2F
-
Korzystając z ogólnego wzoru na stałą równowagi reakcji podanego powyżej, możemy zapisać stałą re-
akcji rozpuszczania fluorytu:
b
a
d
c
B
A
D
C
K
]
[
]
[
]
[
]
[
⋅
⋅
=
2
2
2
2
2
]
[
]
[
]
[
]
[
]
[
−
+
−
+
⋅
=
⋅
=
F
Ca
CaF
F
Ca
K
so

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
36
W liczniku są aktywności jonów a w mianowniku jest „aktywność kryształków fluorytu”. Czyste fazy
biorące udział w reakcjach rozpuszczania mają zawsze aktywność równą 1, a więc mianownik nam znika
ze wzoru i pozostaje jedynie iloczyn aktywności jonów.
Iloczyn stężeń (aktywności) jonów pozostają-
cych w równowadze z osadem nazywa się iloczynem rozpuszczalności
. Jest to jedna z najczęściej wy-
korzystywanych stałych równowagi w obliczeniach geochemicznych równowag w roztworach wodnych.
Pozwala m.in. wyrazić ilościowo rozpuszczalność substancji w wodzie. W temperaturze 25
°
C rozpusz-
czalność CaF
2
wynosi 0,017 g/kg. Wykorzystując masę molową fluorytu (40 g + 2
×
19 g = 78 g/mol) mo-
ż
emy ją przeliczyć na rozpuszczalność w molach/kg. Wynosi ona 0,00022 mol/kg (0,22 mmol/kg). Pa-
trząc na napisane powyżej równanie reakcji rozpuszczania fluorytu możemy stwierdzić, że na każde
0,00022 mola CaF
2
, które uległo rozpuszczeniu w litrze wody (a więcej już nie mogło) powstało 0,00022
mola jonów Ca
2+
i dwa razy tyle, czyli 0,00044 mola jonów F
-
:
CaF
2
<=> Ca
2+
+ 2F
-
0,00022 0,00022 0,00044 mola
Wstawiając te stężenia do wzoru na stałą równowagi reakcji, możemy obliczyć iloczyn rozpuszczalności
fluorytu, który wynosi:
K
so
= [Ca
2+
][F
-]
2 = 0,00022 · (0,00044)
2
= 4,26
×
10
-11
= 10
-10,4
Iloczyn rozpuszczalności to parametr szczególnie użyteczny dla minerałów słabo rozpuszczalnych. Dla
minerałów dobrze rozpuszczalnych wygodniejsze jest stosowanie rozpuszczalności.
Rozpuszczalność
jest to stężenie procentowe roztworu nasyconego, czyli maksymalna ilość substancji, jaka może się
rozpuścić w jednym litrze roztworu
(lub w jednym kilogramie wody).
Fig. Eksperyment rozpuszczania fluorytu CaF
2
w wodzie
Współczynnik nasycenia roztworu
Współczynnik nasycenia roztworu podaje zależność między zmierzonymi stężeniami jonów w roz-
tworze
(w postaci iloczynu jonowego IAP)
a tablicową stałą rozpuszczalności K
sp
:
IAP
SI > 0 roztwór przesycony
SI = log --------
SI = 0 roztwór nasycony
Ksp
SI < 0 roztwór niedosycony
Ca
2+
i F
-
w roztworze

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
37
gdzie IAP to iloczyn aktywności jonów zmierzony w terenie czy laboratorium a Ksp to tabelaryczny ilo-
czyn rozpuszczalności.
Na przykład dla kalcytu w stawie: CaCO
3
<=> Ca+2 + CO3-2
Iloczyn rozpuszczalności kalcytu wzięty z tabel wynosi Ksp = 10
-8.48
Analiza wody stawowej pozwoliła oznaczyć stężenia molowe (i wyliczyć aktywności) jonów dla wyli-
czenia IAP
IAP = [Ca
2+
] . [CO
3
2-
] (lub poprawniej a
Ca
. a
CO3
)
SI = log (IAP/K
sp
)
• Jeśli roztwór jest nasycony to kalcyt jest prawdopodobnie obecny w roztworze (w równowadze z roz-
tworem) a stężenia Ca
2+
i CO
3
2-
są takie, że ich iloczyn jest równy K
sp
. Uwaga! W roztworze nasyconym
stężenia jonu Ca
2+
nie muszą być równe stężeniu jonów CO
3
2-
, w przyrodzie trafiają się dowolne kombi-
nacje stężeń w roztworach nasyconych, ale ich iloczyn zawsze wynosi tyle ile K
sp
.
• Jeśli roztwór jest nienasycony to kalcytu nie ma w roztworze, cały uległ rozpuszczeniu zanim roztwór
osiągnął stan równowagi a stężenia jonów nie wzrosły wystarczająco aby ich iloczyn osiągnął K
sp
. współ-
czynnik nasycenia SI < 0 a roztwór nie jest w stanie równowagi z kalcytem, bo kalcytu nie ma.
• Gdy roztwór jest przesycony kalcyt jest obecny, właśnie krystalizuje, a iloczyn stężeń Ca
2+
i CO
3
2-
jest
większy niż stała rozpuszczalności K
sp
=> SI > 0. Złapaliśmy roztwór na gorącym uczynku w trakcie
ustalania się równowagi: na przykład w upalne latu nieco wody ze stawu wyparowało co wytrąciło układ
z równowagi bo roztwór uzyskał stan przesycenia.
Przykłady prostych obliczeń rozpuszczalności
I. Obliczenie stężenia jonów w roztworze. Oblicz stężenie Pb+2 w roztworze w równowadze z PbSO
4
.
Rozpuszczalność dla PbSO
4
wyrażona jako pK (pK = -logK) wynosi pK=7.7 (to jest równe Ksp = 10
-7.7
bo –log 10
-7.7
= 7.7).
PbSO
4
< = > Pb+2 + SO
4
-2
W stanie równowagi roztwór jest nasycony:
SI = log (IAP/Ksp) = 0 and
Ksp = [Pb][SO
4
] = 10-7.7 mol/L.
Jeśli nie ma innych jonów w roztworze, z bilansu ładunków (roztwór musi być elektrycznie obojętny)
wynika:
[Pb+2] = [SO
4
-2]
K = 10-3.85 mol/L.
Wynik można przeliczyć na stężenie wyrażone w ppm (mg/kg) wiedząc, że masa molowa Pb wynosi
207.2 g/mol:

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
38
1.4 . 10-4 mola Pb 207.2 g Pb 1 litr H2O 1000 mg
litr H2O mol Pb kg H2O g
= 29.3 mg Pb/kg H2O (ppm)
Proste obliczenia rozpuszczalności: efekt wspólnego jonu
II. Efekt wspólnego jonu. Oblicz stężenie Pb+2 w roztworze będącym w równowadze z PbSO
4
i CaSO
4
.
Rozpuszczalność dla PbSO
4
wynosi pK=7.7, a dla CaSO
4
wynosi pK= 4.2.
PbSO
4
< = > Pb+2 + SO
4
-2
K
1
= [Pb][SO
4
] = 10-7.7CaSO
4
< = > Ca+2 + SO
4
-2
K
2
= [Ca][SO
4
] = 10-4.2
Z bilansu ładunków: [Ca+2] + [Pb+2] = [SO4 -2]
Ponieważ jednak CaSO4 jest znacznie bardziej rozpuszczalne niż PbSO4 suma stężeń jonów [Ca+2] +
[Pb+2] wynosi praktycznie tyle co samo [Ca+2] (wkład od [Pb+2] jest pomijalnie mały),
a więc [Ca+2] = [SO4-2] = x
Ponieważ
K2 = [Ca][SO4] = 10-4.2
więc [SO4] = 10-2.1 M (pierwiastek z 10-4.2).
Teraz wstawiamy stężenie [SO4] do zależności:
K1 = [Pb][SO4] = 10-7.7 => [Pb] . 10-2.1 = 10-7.7 = 10-4.6 M
Obliczone stężenie ołowiu [Pb] = 10-4.6 M jest znacznie niższe niż w przykładzie I-szym.Wniosek: roz-
puszczalność PbSO4 została obniżona przez obecność innej substancji, która wniosła wspólny jon do
roztworu (w tym wypadku jon siarczanowy). Efekt wspólnego jonu jest przejawem reguły przekory: re-
akcja rozpuszczania cofa się w lewo pod wpływem podwyższenia stężenia jednego z jonów po prawej
stronie równania reakcji.
III. Efekt pH. Ile wynosi rozpuszczalność Pb(OH)2 przy różnych wartościach pH: 2, 7, i 9?
Pb(OH)2 < = > Pb+2 + 2 OH-
Stała równowagi (rozpuszczalność wzięta z tablic)
K = [Pb][OH]2 = 10-15.6
(uwaga: stężenie OH- jest wzięte do kwadratu ponieważ w równaniu reakcji występują 2 mole OH-).

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
39
a) Przy pH = 2 stężenie jonów wodorowych [H+] = 10-2
a ponieważ stała dysocjacji wody Kw = [H][OH]=10-14
więc [OH] = Kw/(10-2) = 10-14/(10-2) = 10-12 mol/L
Wstawiając do K: [Pb] = 10-15.6 / (10-12 )2 = 108.38 M
Pb(OH)2 jest bardzo rozpuszczalne w pH=2.
b) Przy pH = 7 stężenie jonów wodorowych [H+] = 10-7
[OH] = Kw/(10-7) = 10-14/10-7 = 10-7
więc
[Pb] = 10-15.6 / (10-7 )2 = 10-1.6 M
Pb(OH)2 jest znacznie mniej rozpuszczalne w pH = 7. Rozpuszczalność (po przeliczeniu stężenia molo-
wego na mg/L) wynosi około 5200 ppm.
c) Przy pH = 9 stężenie jonów wodorowych [H+] = 10-9
[OH] = Kw/(10-7) = 10-14/10-9 = 10-5
więc
[Pb] = 10-15.6 / (10-5 )2 = 10-5.6 M
Pb(OH)2 jest jeszcze mniej rozpuszczalne w pH = 9. Rozpuszczalność (po przeliczeniu stężenia molowe-
go na mg/L) wynosi około 5 ppm.
Zastosowanie ∆G do obliczania stałej rozpuszczalności minerałów
W dowolnym momencie przebiegu ogólnie zapisanej reakcji:
aA + bB <=> Cc + dD
wyrażenie na działanie mas przedstawia proporcję produktów do substratów:
b
a
d
c
B
A
D
C
Q
]
[
]
[
]
[
]
[
=
gdzie nawiasy [ ] oznaczają aktywności (skorygowane stężenia) reagentów. Wyrażenie na działanie mas
powiązane jest z energią swobodną Gibbsa ∆G
reakcji
równaniem:
∆
G
reakcji
= ∆G
o
reakcji
+ RT ln Q
gdzie ∆G
reakcji
jest zmianą energii swobodnej towarzyszącą reakcji w dowolnej temperaturze T (wyrażo-
nej w Kelwinach, K =
o
C + 273,15), natomiast ∆G
o
reakcji
jest zmianą energii swobodnej towarzyszącą
reakcji w warunkach normalnych (25
o
C i 1 atm). R jest stałą gazową równą 8,314 J/mol
.
K (lub 1,987
cal/mol
.
K).

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
40
Kierunek samorzutnego przebiegu powyższej reakcji jest wskazany przez znak ∆G
reakcji
:
∆
G
reakcji
> 0 reakcja biegnie w lewo zmierzając w kierunku równowagi
∆
G
reakcji
= 0 reakcja jest w stanie równowagi
∆
G
reakcji
< 0 reakcja biegnie w prawo zmierzając w kierunku równowagi
Tak więc, w stanie równowagi ∆G
reakcji
= 0 , czyli:
0 = ∆G
o
reakcji
+ RT ln Q
∆
G
o
reakcji
= - RT ln Q
Stan równowagi oznacza, że aktywności (a więc i stężenia) produktów i substratów osiągnęły w wyniku
przebiegu reakcji takie wartości, że energia swobodna, która jest motorem przebiegu reakcji, osiągnęła
poziom zerowy i dalej nie następują żadne zmiany stężeń w czasie. Na przykład minerał rozpuścił się na
tyle w roztworze, że osiągnął stan nasycenia i więcej już rozpuścić się nie może.

6. GEOCHEMIA WÓD POWIERZCHNIOWYCH I PODZIEMNYCH
41
Zastosowanie ∆G do obliczania stałej rozpuszczalności minerałów.
Wyliczone dla stanu równowagi wyrażenie na działanie mas Q nazywa się stałą równowagi reakcji K.
Stała równowagi K jest charakterystyczna dla każdej reakcji. Zależy ona od temperatury reakcji. W po-
wyższym równaniu znaczek
o
przy symbolu energii swobodnej Gibbsa wskazuje na to, że równanie doty-
czy wyłącznie warunków normalnych (25
o
C i 1 atm). A więc stała równowagi dowolnej reakcji (a dla
reakcji rozpuszczania to jest równoznaczne ze stałą rozpuszczalności) może być wyliczona przy użyciu
tablicowych danych termodynamicznych (∆G
reakcji
=∆G
produktów
- ∆G
substratów
) z równania:
∆
G
o
reakcji
= - RT ln K
K = e
-∆G/RT
Jest to fundamentalna i bardzo użyteczna zależność, stanowiąca pomost pomiędzy termodynamiką teore-
tyczną (reprezentowaną przez ∆G
o
reakcji
wyliczaną z danych tablicowych) a eksperymentem i obserwa-
cjami empirycznymi (reprezentowanymi przez rzeczywiste i zmierzone stężenia jonów służące do wy-
znaczenia wyrażenia na działanie mas Q lub stałej równowagi reakcji K). Połączenie wyliczonych zależ-
ności w jedną:
∆
G
reakcji
= -RT lnK + RT ln Q
pozwala ilościowo ocenić (np. przez pomiar chwilowych stężeń w roztworze i wyliczenie Q) na ile aktu-
alny stan reakcji odbiega od równowagi do której zmierza.
Posługując się równaniem K = e
-∆G/RT
można na przykład przewidzieć z termodynamicznych danych ta-
blicowych wielkość rozpuszczalności minerału w temperaturze 25
o
C.
Wyszukiwarka
Podobne podstrony:
,pytania na obronę inż,Układy technologiczne uzdatniania wód powierzchniowych i podziemnych
Charakteryzowanie wód powierzchniowych i podziemnych
2009 Dz U Nr 81 poz 685 monitoring wod powierzch i podziemn
Samooczyszczanie się wód powierzchniowych jest kompleksowym procesem
03 Sklad wod powierzchniowych
Samooczyszczanie wód powierzchniowych, Inżynieria Środowiska PŚk, Semestr 1, Biologia
Środki przeciwdziałania powierzchniowym (2) Podziemny drenaż
termodynamika 2, Ciśnienie - to wielkość skalarna określona jako wartość siły działającej prostopadl
028 Sprawozdanie 2 Zanieczyszczenie wód powierzchniowych Górnego Śląska
06 geochemia wody i roztworów wodnych
monitoring wod powierzchniowych, monitoring środowiska
ochrona wód powierzchniowych (8 str), Pomoce naukowe, studia, ochrona srodowiska
Wody powierzchniowe i podziemne Polski kl IIID
Eutrofizacja wód powierzchniowych
Monitoring wód powierzchniowych i gruntowych wokół składowisk odpadu, Ochrona Środowiska studia, 3 r
06, KATECHEZA DLA DZIECI, PODZIĘKOWANIA MATCE BOŻEJ
SAMOOCZYSZCZANIE WÓD POWIERZCHNIOWYCH
19 Ocena jakości wód powierzchniowych na obszarach
Formy krasu powierzchniowego i podziemnego
więcej podobnych podstron