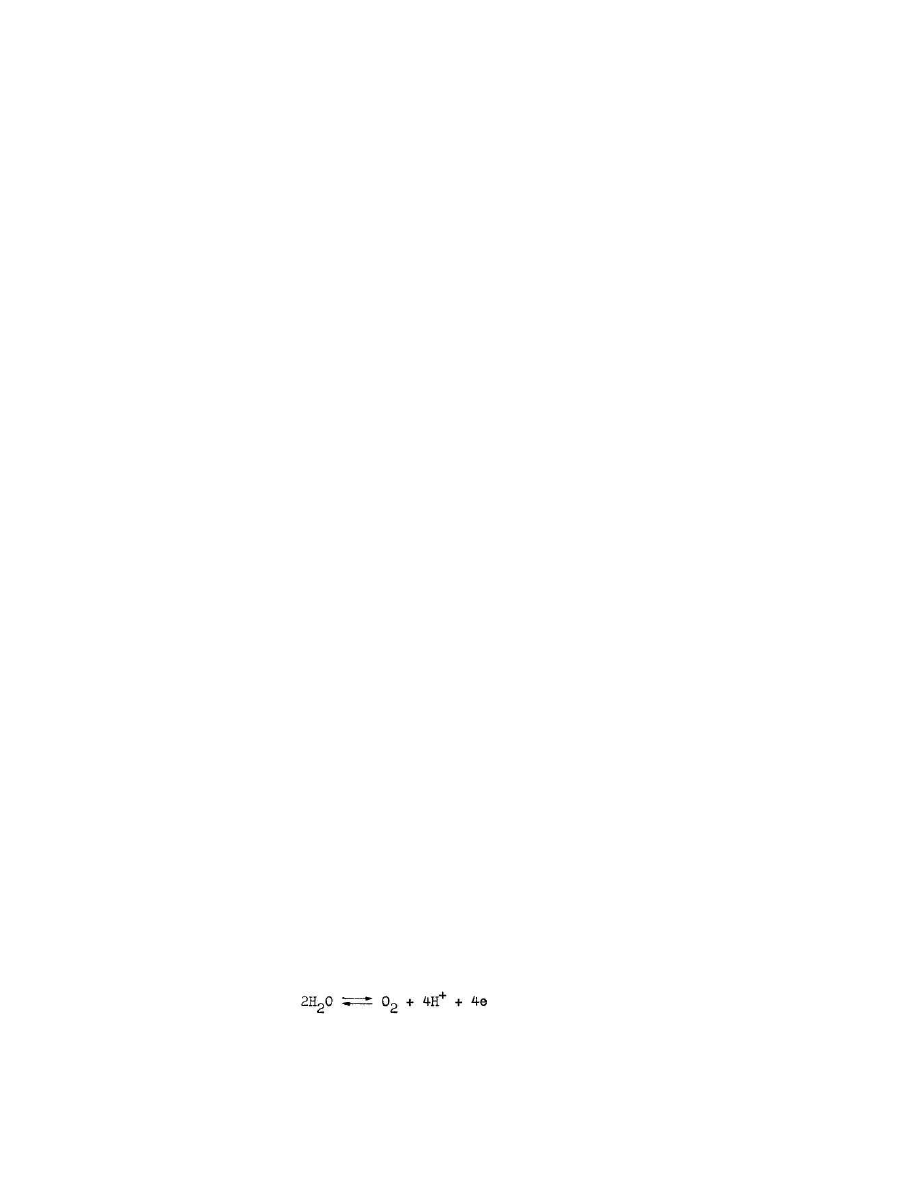
12. KOROZJA I OCHRONA METALI
12.1. CEL WICZENIA
Celem wiczenia jest zapoznanie si z podstawowymi wiadomo ciami z zakresu
korozji metali, przyczynami i skutkami zachodzenia zjawisk korozyjnych oraz
sposobami zabezpieczenia metali przed korozj .
12.2. CZ
TEORETYCZNA
12.2.1. TERMODYNAMICZNY ASPEKT KOROZJI
Podatno
korozyjna wi kszo ci metali zwi zana jest z ich skłonno ci do
reagowania z otaczaj cym rodowiskiem, którym najcz
ciej jest rodowisko wodne
lub powietrze. W tych rodowiskach metale tworz układ termodynamicznie
nietrwały, d
cy do oddawania energii w procesie przechodzenia metalu w jego
zwi zek chemiczny, np. tlenek, tlenek uwodniony lub siarczek. Wi kszo
metali
wydobywa si z ich zwi zków wyst puj cych w przyrodzie - rud - w przemianach
zwi zanych z dostarczeniem energii. Proces korozji wyra a wi c d
no
do powrotu
do stanu naturalnego, jakim jest jego posta utleniona. Dlatego te dla rozwa a
nad korozj istotne jest przedstawienie jej w kategoriach termodynamicznych, przez
co mo na stwierdzi , jakie warunki w wypadku danego metalu powinny sprzyja
korozji a jakie j wyklucza .
W badaniach nad korozj elektrochemiczn w rodowiskach elektrolitów du
pomoc
s wykresy przedstawiaj ce zale no
potencjału od pH. Wykresy te od nazwiska ich
twórcy zwane s wykresami Pourbaix'go (wym.Purbe). Na wykresie rz dna przedstawia
potencjał redoksy układu koroduj cego, a odci ta - skal pH. Wykorzystuj c dane
termodynamiczne oraz równanie Nern-sta do obliczenia potencjałów elektrodowych
mo na wyznaczy granice obszarów, wewn trz których dany metal lub jego pochodne
s trwałe. Na rysunkach 20 i 21 pokazano w postaci uprosz czonej tego typu
wykresy dla miedzi i dla elaza. Wykresy przedstawiaj dane dla temp. 25°C przy
zało eniu, e korozja metalu nie zachodzi (metal jest odporny), gdy jego
st
enie
równowagowe w roztworze jest mniejsze ni 1*10
-6
mol/dm
3
.
Opadaj ce przerywane linie proste na wykresach pokazuj potencjały redoksy
roztworów b d cych odpowiednio w równowadze z tlenem i wodorem. Jak wida z
wykresu, potencjał redoksy roztworu b d cego w równowadze z tlenem jest o 1,23 V
wy szy (bardziej szlachetny) ni w równowadze z wodorem. Wzi to te pod uwag ,
e zarówno potencjał redoksy elektrody tlenowej jak i wodorowej spada o 0,059 V
ze wzrostem warto ci pH o jednostk . Wynika to z charakteru reakcji elektrodowych
zachodz cych na obu elektrodach.
Równanie reakcji elektrodowej elektrody tlenowej ma posta
Rys.20. Potencjały równowagowe dla układu mied -woda w temp. 25°C w funkcji
pH (wykres Pourbaix'go)
Rys.21. Potencjały równowagowe dla
układu elazo-woda w temp. 25°C
w funkcji pH
Zgodnie z równaniem Nernsta potencjał tej elektrody opisany jest równaniem
gdzie:
0
o
2
- potencjał normalny elektrody tlenowej = +1,23 V,
R
- stała gazowa,
T
- temp. w kelwinach,
F
- stała Faradaya,

a
- aktywno
,
a
p
o
2
- aktywno
ci nieniowa tlenu gazowego.
Przy zało eniu, e
i temp. 25°C, mamy
Podobnie dla elektrody wodorowej, na której zachodzi reakcja
potencjał jej zmienia si ze zmian pH w sposób nast puj cy:
gdzie
0
H
2
- potencjał normalny elektrody wodorowej przyj ty
łi
2
jako równy 0,0000 V dla ka dej temperatury.
Ogólnie bior c wszystkie linie pochyłe na wykresach odpowiadaj równowagom
zale nym od pH. Ponadto proste linie poziome odzwierciedlaj równowagi
niezale ne od pH (np.: Cu = = Cu
+2
+ 2e, Fe = Fe
+2
+ 2e, Fe
+2
= Fe
+3
+ e).
Proste pionowe odpowiadaj stanom równowagi bez zachodz cych zmian
warto ciowo ci i dlatego niezale nym od potencjału (np. równowagi rozpuszczalno ci
mi dzy zwi zkiem amfoterycz-nym Cu(0H)
2
a kwasem lub zasad ).
Je li istniej takie warunki, e metal znajduje si w fazie trwałej (stabilnej),
mówi si o stanie odporno ci i korozja nie mo e zachodzi . We wszystkich innych
przypadkach, gdy metal nie jest faz termodynamicznie trwał , korozja mo e wy-
st powa . W pewnych jednak okoliczno ciach słabo rozpuszczalny zwi zek metalu mo e
tworzy warstw ochronn na jego powierzchni, obni aj c szybko
korozji do
znikomych warto ci, metal znajduje si wtedy w stanie pasywnym. Do osi gni cia tego
stanu konieczne jest, by słabo rozpuszczalny zwi zek metalu, np. jego tlenek,
tworzył spoist i przylegaj c warstw na jego powierzchni. W jakim stopniu
warstewka produktu korozji b dzie mie własno ci pasywuj ce, nie mo na przewidzie
jedynie na podstawie rozwa a termodynamicznych. Wynik ostateczny b dzie
empiryczny i zwi zany w wi kszym stopniu z kinetyk reakcji. Informacje o znacznie
wi kszej przydatno ci uzyskuje si wówczas, gdy wnioski wyci gni te z wykresów
Pour-baix`go wzbogaci si o dane empiryczne dotycz ce pasywno ci.
Jak wynika z rys.20, potencjał elektrody wodorowej znajduje si dla wszystkich
warto ci pH w obszarze odporno ci miedzi. Oznacza to, e odpowiednie roztwory nie
utleniaj cych i nie tworz cych kompleksów z miedzi kwasów, zasad lub soli nie
atakuj miedzi metalicznej. Jednak w roztworach napowietrzonych lub w roztworach
utleniaj cych mied jest atakowana: w roztworze kwa nym tworz si jony Cu
+2
,
natomiast w silnie alkalicznym - jony CuO
2
-2
, zgodnie z amfoterycznym charakterem
wodorotlenku miedziowego.

W przedziale pH 6,7-12,7 powierzchni miedzi pokrywa nierozpuszczalny CuO.
Je li ma on tendencje do tworzenia szczelnej i przylegaj cej warstwy, to
reaktywno
miedzi szybko znika. Mied osi ga stan pasywny. Przy warto ciach pH
bliskich granicom obszarów pasywno ci i korozji prawdopodobne jest niecałkowite
pokrycie powierzchni tlenkiem miedziowym CuO, a w zwi zku z tym mo liwe jest
wyst powanie korozji lokalnej.
Wykres na rys.20 nie ma zastosowania, gdy w roztworze s zwi zki kompleksuj ce
jony miedzi, np. amoniak i cyjanki. Zachodz wtedy całkowicie odmienne reakcje
elektrodowe i ustalaj si wła ciwe dla tych reakcji potencjały. W rodowiskach
takich niebezpiecze stwo korozji ro nie, poniewa bardziej ograniczone s obszary
odporno ci, natomiast lepsza jest rozpuszczalno
tlenków miedzi w obecno ci NH, i
CN~.
W przypadku elaza (rys.21) potencjał elektrody wodorowej w całym zakresie pH
le y powy ej obszaru odporno ci, co oznacza, e elazo mo e rozpu ci si z
wydzieleniem wodoru w roztworze wodnym w całym zakresie pH. Pomimo to w
przedziale pH 9,4-12,5 tworzy si warstewka pasywna Fe(0H)
2
. Przy mniejszych
warto ciach pH elazo koroduje z wytworzeniem jonów Fe
+2
, przy wi kszych -
wodorotlenek elazawy rozpuszcza si tworz c jony HFeO
-
2
. W tym ostatnim
przedziale pH wyst puje tzw. krucho
alkaliczna stali, polegaj ca na p kaniu
korozyjnym wyrobów. Przy wy szych potencjałach rodowiska korozyjnego (wy-
stepowanie silnych utleniaczy) warstewka pasywuj ca składa si z Fe
3
O
4
lub Fe
2
O
3
•
n H
2
O.
W bardzo silnie utleniaj cym rodowisku alkalicznym korozja mo e zachodzi z
wytworzeniem jonów elazianowych FeO
4
-2
.
12.2.2. KLASYFIKACJA ZJAWISK KOROZYJNYCH
Zjawiska zwi zane z przebiegiem procesów korozyjnych s bardzo zró nicowane,
ró ne s równie objawy zniszcze korozyjnych, st d te istnieje wiele mo liwo ci
podziału i klasyfikacji nauki o korozji. Najcz ciej stosowanymi kryteriami
podziału s :
1)
mechanizm korozji,
2)
typ zniszcze korozyjnych,
3)
charakter rodowiska korozyjnego.
W zale no ci od mechanizmu procesów korozyjnych rozró niamy korozj
chemiczn i elektrochemiczn .
Korozja wg mechanizmu c h e m i c z n e g o ma miejsce w wypadku, gdy
wymiana elektronów mi dzy metalem a utleniaczem zachodzi w rodowisku nie
wykazuj cym przewodnictwa jonowego
mMe + n Y = Me
m
Y
n
(77)
gdzie Y - utleniacz.
Charakterystyczn cech tych reakcji jest to, e zapocz tkowanie procesu
utleniania i tworzenia nowego zwi zku zachodzi w tym samym miejscu na granicy
zetkni cia metalu z utleniaczem. Je eli produkt reakcji jest lotny lub tworzy
warstw nieci gł , wtedy mo e zachodzi dalsza reakcja na granicy faz. W rezultacie
szybko korozji pozostaje stała. W przypadku gdy powstaje ci gła, szczelna
warstwa produktów korozji, która oddziela reagenty od siebie, reakcja ulega
zwolnieniu lub zahamowaniu.
Korozja metali w wodnych roztworach elektrolitów zachodzi wg mechanizmu
e l e k t r o c h e m i c z n e g o .
Zasadnicza ró nica pomi dzy elektrochemicznym mechanizmem korozji a
mechanizmem czysto chemicznym polega na tym, e ogólna reakcja chemiczna
zachodz ca pomi dzy metalem a utleniaczem w przypadku korozji elektrochemicznej
jest wynikiem dwóch procesów przebiegaj cych niezale nie od siebie:
1)
procesu anodowego - przechodzenie metalu do roztworu
w postaci uwodnionych jonów, przy czym w nierozpuszczalnej
cz
ci metalu pozostaje równowa na ilo
elektronów nadmiaro
wych;
2)
procesu katodowego - asymilacja pojawiaj cych si w me
talu nadmiarowych elektronów przez jakiekolwiek utleniacze
(atomy, cz steczki lub jony roztworu mog ce ulega redukcji
na katodzie).
Istnienie przewodnictwa elektronowego w metalu i przewodnictwa jonowego w
roztworze sprawia ponadto, e procesy anodowe i katodowe mog by rozdzielone pod
wzgl dem przestrzennym, czyli mog przebiega w ró nych miejscach powierzchni.
Warunek ten nie jest niezb dny dla procesu elektrochemicznego, gdy niekiedy
korozyjne procesy katodowe i anodowe mog odbywa si na tej samej powierzchni,
nast puj c kolejno po sobie. Jednak z punktu widzenia energetycznego przestrzenny
rozdział reakcji anodowych i katodowych jest bardziej uprzywilejowany, gdy
reakcje katodowe i anodowe mog si lokalizowa w tych miejscach, w których
przebieg ich jest ułatwiony. Z tego te wzgl du w praktyce przebieg reakcji
elektrochemicznej w wi kszo ci przypadków charakteryzuje si lokalizacj procesów
katodowych i anodowych w poszczególnych miejscach powierzchni -jest to druga
istotna cecha charakteryzuj ca elektrochemiczny mechanizm procesu korozyjnego w
odró nieniu od mechanizmu czysto chemicznego. Ponadto szybko
wszystkich procesów
elektrochemicznych zale y od potencjału istniej cego na granicy faz: metal
elektrody-roztwór elektrolitu (patrz rozdz. 11.2).
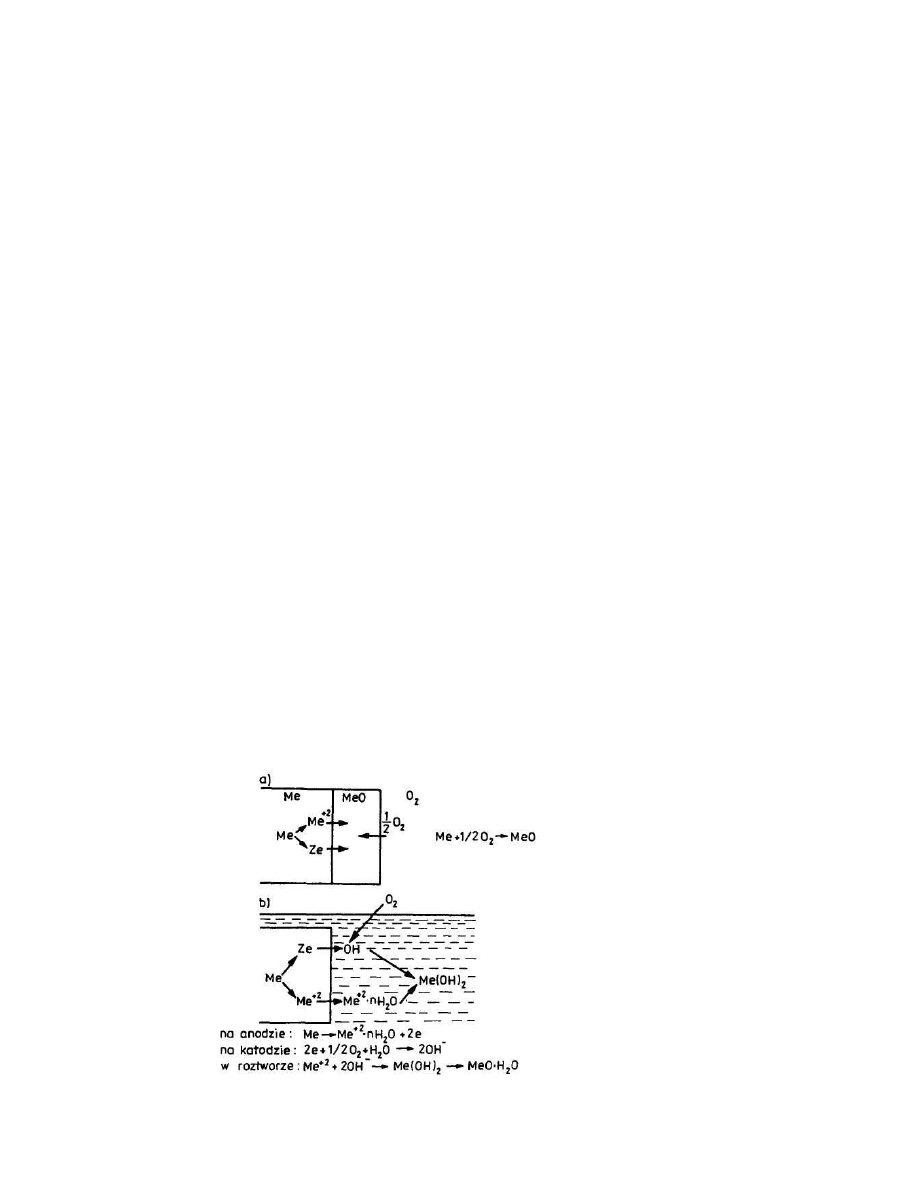
Na rys.22 przedstawiono schematycznie ten sam proces korozyjny - wzajemne
oddziaływanie na siebie metalu i tlenu prowadz ce do powstania tlenku metalu
według mechanizmu chemicznego (a) i elektrochemicznego (b).
Rys.22. Mechanizm reakcji metalu z tlenem wg mechanizmu:
a) chemicznego, b) elektrochemicznego

Wa n cech korozji elektrochemicznej jest to, e sumaryczny ładunek
wymieniony w obu procesach (katodowym i anodowym) musi by taki sam. Wielko
ta
okre la równie szybko
procesu korozyjnego, tzn.
i
k
=i
A
= i
kor
(78)
gdzie: i, - nat
enie pr du katodowego - równe ilo ci ładunków wymienionych w
procesie katodowym na jednostk czasu, i. - nat
enie pr du anodowego,
i, - nat
enie pr du korozyjnego (szybko
procesu korozji).
0 szybko ci ka dej reakcji, a wi c i reakcji elektrochemicznej decyduje
najwolniejszy etap. W procesach korozyjnych o szybko ci całego procesu decyduje
prawie zawsze szybko
redukcji czynnika korozyjnego (proces katodowy). W rodowi-
skach wodnych mamy najcz
ciej do czynienia z redukcj tlenu (w rodowiskach
oboj tnych i zasadowych)
W zale no ci od t y p u z n i s z c z e
k o r o z y j n y c h mo na
wyodr bni nast puj ce rodzaje korozji, które schematycznie przedstawiono na
rys.23.
Korozja o g ó l n a zachodzi równomiernie na całej powierzchni stykania
si metalu ze rodowiskiem korozyjnym.
Rys.23. Typy zniszcze
Korozyjnych: a)
KO
-
rozja równomierna, b)
korozja selektywna, c) korozja w erowa, d) korozja punktowa,
e) korozja mi dzykrystaliczna, f) korozja napr
eniowa
Przy wyst powaniu tego typu korozji powierzchnie anodowe i katodowe s tak małe, e
nie mo na ich do wiadczalnie odró ni . Korozja ogólna atakuje metale głównie w
rodowisku kwa nym.
Korozja w
e r o w a i p u n k t o w a wyst puje przede wszystkim w
wodzie morskiej, w wodach gruntowych i we wszystkich roztworach zawieraj cych jony
chlorowców. Warunkiem koniecznym powstawania tego typu zniszcze jest to, aby
powierzchnie katodowe były nieporównywalnie wi ksze od powierzchni anodowych, co
przy warunku i, = i. prowadzi do bardzo szybkiego rozpuszczania metalu w
miejscach o charakterze anodowym. Korozji w erowej ulegaj metale i stopy, które
swoj du
odporno
korozyjn zawdzi czaj obecno ci na
ich powierzchni warstw pasywnych, a wi c: glin i jego
stopy, tytan i jego stopy, chrom, stale chromowe i
chromowo-nikłowe.
i wodoru (w rodowiskach kwa nych)

Obecno
w roztworze jonów chlorkowych, a w mniejszym
stopniu tak e jonów innych chlorowców, powoduje zanik
własno ci pasywnych lub uniemo liwia tworzenie si warstw
pasywnych na wymienionych metalach i stopach. Jony Cl
-
mog wnika w warstw tlenku przez pory lub defekty
sieciowe i niszczy j łatwiej ni inne jony.
Zniszczenie warstwy pasywnej pod wpływem jonów Cl
-
ma cha-
rakter lokalny i nie obejmuje całej powierzchni metalu,
co prawdopodobnie wynika z drobnych ró nic w grubo ci i
budowie warstwy pasywnej.
Na powierzchni metalu powstaj małe obszary mikroanodowe
(aktywnego metalu) otoczone du ymi obszarami katodowymi spa-
sywowanego metalu. Du a g sto
pr du w obszarach anodowych
powoduje szybkie, lokalne rozpuszczanie metalu i tworzenie
si gł bokich w erów.
Korozja m i
d z y k r y s t a l i c z n a charakte-
ryzuje si selektywnym niszczeniem metalu wzdłu granic
ziaren. Wynikiem takiego procesu jest powa ne pogorszenie
własno ci mechanicznych metalu. W wypadkach kra cowych mo e
nast pi nawet rozsypanie si metalu w proszek. Warunkiem
koniecznym zachodzenia korozji mi dzykrystalicznej jest
nierównocenno
elektrochemiczna granicy mi dzyziarnowej
i obszaru ziarna. Je li obszar mi dzy ziarnami ma
charakter anodowy wzgl dem reszty ziarna, to ze wzgl du
na niekorzystny stosunek powierzchni F
a
<< F
k
nast puje silne rozpuszczenie obszaru anodo-
wego. W przypadku stali chromowych i chromowo-niklowych przyjmuje si , e korozja
mi dzykrystaliczna jest ci le zwi zana ze zjawiskiem wydzielania si w glików
chromu na granicach ziaren i ze zubo eniem przygranicznych stref ziaren w chrom
(proces dechromizacji). Strefa mi dzyziarnowa, o ni szej zawarto ci chromu ni
osnowa, staje si anod i ulega szybkiemu rozpuszczaniu si . Aby poprawi
odporno
stali nierdzewnych na korozj mi dzykrystaliczna, nale y "usun
" z
nich w giel odpowiedzialny za proces dechromizacji. Najcz
ciej dokonuje
si tego przez dodanie do stali niewielkiej ilo ci tytanu lub niobu. S to
pierwiastki łatwo tworz ce bardzo trwałe w gliki, tym samym ich obecno
zapobiega
szkodliwemu zjawisku dechromizacji.
Korozja n a p r
e n i o w a jest specjalnym rodzajem korozji
miejscowej wywołanej równoczesnym działaniem rodowiska korozyjnego i napr
e
(zewn trznych lub wewn trznych), przy czym oddzielne działanie tych czynników nie
powoduje p kania.
Charakterystyczn cech korozji napr
eniowej jest powstawanie szczelin,
tworz cych si w płaszczyznach prostopadłych do kierunku maksymalnych napr
e . Z
tego powodu szczelina korozyjna mo e rozprzestrzenia si nie tylko wzdłu granic
ziaren, czyli mi dzykrystalicznie, ale je3t tak e zdolna przecina poszczególne
krystality, czyli rozprzestrzenia si ródkrystalicznie.
Efektem działania korozji napr
eniowej jest p kanie elementów pracuj cych pod
napr
eniem w rodowiskach, w których odporno
na korozj ogóln pracuj cego
elementu mo e by bardzo du a.
12.2.3. METODY ZABEZPIECZANIA METALI PRZED KOROZJ
Istniej ce metody zabezpieczania metali przed korozj mo na podzieli na

cztery wyra nie ró ni ce si grupy:
1)
metody polegaj ce na modyfikacji rodowiska korozyjne
go poprzez dodatek opó niaczy procesów korozyjnych (inhibito
rów korozji),
2)
metody polegaj ce na modyfikacji samego metalu poprzez
celowy dodatek odpowiednich składników stopowych,
3)
metody oparte na mechanicznym oddzieleniu metalu od
agresywnego rodowiska przez nało enie na metal powłok ochron
nych.
4)
metody elektrochemiczne (ochrona katodowa i anodowa -
rozdz. 13).
I n h i b i t o r y
Inhibitorami korozji nazywamy substancje, które dodane
do rodowiska korozyjnego powoduj wyra ne zmniejszenie
szybko ci korozji w wyniku hamowania procesu anodowego lub
katodowego, albo obu tych procesów ł cznie. W zale no ci od
rodzaju hamowanego procesu rozró niamy odpowiednio inhibitory
anodowe, katodowe lub mieszane.
Inhibitory a n o d o w e w wi kszo ci s anionami.
W roztworach elektrolitów w druj one do powierzchni anody
tworz c z jonami r ozpu szczaj ceg o si m eta lu trud no
ro zpuszcz alne zwi zki, które pasywuj powierzchnie metalu.
Dlatego te inhibitory anodowe cz sto nazywa si
p a s y w a t o r a -
m i . W ród inhibitorów anodowych znajduje si wiele
inhibi-
torów nieorganicznych,jak ortofosforany PO
4
-3
, krzemiany
SiO
3
-2
azotyny N0
2
-
chromiany CrO
4
-2
, oraz organicznych, np.
benzoesany.
Chocia inhibitory anodowe s bardzo efektywne i
znajduj szerokie zastosowanie praktyczne, mog wywoła
równie niepo
dane efekty. Gdy st
enie inhibitora
staje si zbyt małe, aby mógł on pokry cał
powierzchni anodow , powstaj warunki, w których
powierzchnia katody (F
k
) jest du o wi ksza od powierzchni
anody ( F
A
) . W rezultacie mo e to prowadzi do
powstawania korozji w erowej. W takich wypadkach
zastosowanie inhibitora mo e wyrz dzi wi cej szkody ni
po ytku.
Inhibitory k a t o d o w e hamuj szybko
procesu katodowego. Wynik ten mo na osi gn
albo przez
zmniejszenie aktywnej powierzchni katod, albo przez
zmniejszenie st
enia czynnika okre laj cego szybko
procesu
katodowego (np. zmniejszenie st
enia tlenu czy jonów
wodorowych). Pierwszy efekt wywieraj substancje (np.
Ca(HC0
3
)
2
i ZnSO
3
) tworz ce trudno rozpuszczalne w wodzie
zwi zki z jonami 0H~ wydzielaj cymi si na
powierzchni katod. Natomiast do usuwania tlenu z roztworu
korozyjnego mo na stosowa substancje łatwo wi
ce tlen -
tzw. odtleniacze, np. siarczyny, hydrazyn

Inhibitory m i e s z a n e charakteryzuj si
równoczesnym hamowaniem procesu katodowego i anodowego. Takie
dzia-

Wyszukiwarka
Podobne podstrony:
materiały izolacyjne, Technologia maszyn, 15. korozja i ochrona
prezentacja korozja, Technologia maszyn, 15. korozja i ochrona
15 - Korozja metali i ich stopów, Korozja metali i ich stopów
Czasowa ochrona metali przed korozją
Części maszyn 13 - 15 BHP i ochrona środowiska, czesci maszyn
korozja ochrona wirto id 248171 Nieznany
korozja elektrochemiczna metali tom
15 plan (ochrona środowiska)
rodzaje korozji i ochrona, BHP, Techniki wytwarzania
beton i stal Sciaga(korozja ochrona itp), Materiały budowlane
07 Korozja i ochrona przed korozja 26 02 2015
elektrochemia-skrypt, beton, KOROZJA I OCHRONA ZBROJENIA W BETONIE
Korozja i ochrona przed korozją
korozja elektrochemiczna metali
Części maszyn 13 - 15 BHP i ochrona środowiska, czesci maszyn
więcej podobnych podstron