
2. Błony biologiczne
Błony są strukturami charakterystycznymi dla wszystkich komórek, prokariotów
i eukariotów, zwierzęcych oraz roślinnych. Błony nie tylko ograniczają kompart-
menty komórki i organelli komórkowych, ale także kontrolują całą komunikację
pomiędzy wnętrzem i środowiskiem zewnętrznym komórki. Ta komunikacja
przyjmuje formy transportu jonów i cząsteczek do i na zewnątrz komórki, jak
również formy przepływu informacji dokonującego się poprzez indukowane
zmiany konformacyjne składników poszczególnych błon. Błony stanowią miejsce
lokalizacji wielu enzymów kontrolujących reakcje „transbłonowe", tzn. produkt
i substrat znajdują się po przeciwnych stronach błony (takim procesem jest rów-
nież swoisty transport przez błony), są także miejscem koncentracji wielu enzy-
mów katalizujących następujące po sobie etapy konkretnego procesu ułatwiając
w ten sposób ich interakcję. Inne enzymy zaangażowane są w biosyntezę błon i są
przykładami enzymów, których substraty znajdują się w błonie. Większość pod-
stawowych procesów biochemicznych w jakimś punkcie przebiega z udziałem
błon. Przykładami mogą być tak różne procesy, jak replikacja prokariotycznego
DNA, biosynteza białka, wydzielanie białek, reakcje bioenergetyczne oraz
odpowiedź hormonalna.
Pomimo wielkiego zróżnicowania struktur otoczonych błonami podstawy bu-
dowy błon biologicznych są w zasadzie te same. Dotyczy to również błon komó-
rek roślinnych i bakterii.
Zaproponowany w 1972 roku przez Singera i Nicolsona model zakłada, że
błonę stanowi płynna mozaika lipidów w postaci dwumolekularnej warstwy,
w której zanurzone są białka integralne ją penetrujące. Dwumolekularna warstwa
lipidową stanowi barierę przepuszczalności (głównie dla substancji hydrofilnych)
i „rozpuszczalnik" dla białek. Tylko mała część lipidów oddziałuje swoiście
z białkami, co powoduje, że białka mogą swobodnie dyfundować w płaszczyźnie
bocznej (lateralnej) błony. Dyfuzja boczna ograniczana jest poprzez tzw. szkielet
błonowy, strukturę utworzoną przez białka peryferyjne oddziałujące z białkami
integralnymi. Białkami integralnymi określa się te białka, które związane są
mocno z dwumolekularna warstwą lipidową, tzn. penetrują ją przynajmniej jed-
Rys.14. Model mozaiki płynnych lipidów i białek
globularnych Singera i Nicolsona

2. Błony biologiczne 70
nym odcinkiem (ok. 20 reszt aminokwasowych) swojej struktury. Takim białkiem
jest np. glikoforyna A błony erytrocytu. Inne białka mogą penetrować dwuwar-
stwę wielokrotnie, np. dziesięciokrotnie białko przenoszące aniony. Białka te
można wyekstrahować z błony tylko po zastosowaniu odczynników chaotropo-
wych, niektórych rozpuszczalników organicznych lub, a właściwie przede wszy-
stkim, detergentów. Szczególnie użyteczne są tu detergenty niejonowe, które nie
denaturują białek. Białka peryferyjne to te, które związane są luźno z powierzch-
nią (głównie cytoplazmatyczną) błony. Można je wyekstrahować z błony przy
użyciu roztworów wodnych o niskiej lub wysokiej sile jonowej oraz wysokim pH.
Przykłady oczyszczania integralnych i peryferyjnych białek błony erytrocytu po-
dano w skrypcie „Molekularna organizacja komórki", red. J. Szopa, Wrocław,
1992, s. 40-45. Podstawy zasad izolacji poszczególnych organelli komórkowych
i co za tym idzie ich błon przedstawiono tamże („Molekularna...", s. 1-6).
2.1. Białka i lipidy błon erytrocytów
Od początku badań nad strukturą i funkcją błon, erytrocyty ssaków stały się
najpopularniejszym dla nich modelem doświadczalnym z bardzo oczywistego
powodu, a mianowicie posiadają tylko jeden rodzaj błony: błonę plazmatyczną.
Komórka ta uległa bowiem takiej specjalizacji w trakcie ewolucji, że stanowi
rodzaj woreczka wypełnionego hemoglobiną w wysokim stężeniu, co czyni ją
efektywnym nośnikiem tlenu, który musi być dostarczony z płuc do pozostałych
tkanek organizmu. Ponieważ błona erytrocytu jest łatwo przepuszczalna dla wo-
dy, a nieprzepuszczalna dla prawie wszystkich substancji hydrofilnych, erytrocy-
ty pęcznieją i ulegają lizie, kiedy umieści się je w roztworze hypotonicznym.
Dzięki temu można uzyskać duże ilości czystych (jednego rodzaju) błon plazma-
tycznych.
Przybliżony chemiczny skład błon erytrocytu przedstawia się następująco:
50% białka, 40% lipidów i 10% cukrów. Błona komórkowa (plazmolemma)
zwykle zawiera względnie wysokie stężenia cholesterolu. Stosunek molowy fo-
sfolipidów do cholesterolu w błonie erytrocytów wynosi od 0.6 do 0.9. Wolne
węglowodany nie są obecne w błonach. Są kowalencyjnie związane zarówno
z białkami (glikoproteidy), jak i z lipidami (glikolipidy).
Podwójna warstwa lipidową, w której cząsteczki posiadają swobodę szybkiej
dyfuzji lateralnej (w przeciwieństwie do ruchliwości w płaszczyźnie poprzecznej
błony, która jest bardzo ograniczona), stanowi środowisko dla wielu białek inte-
gralnych.
Cztery rodzaje fosfolipidów stanowią 90% tej klasy związków w błonie ery-
trocytu. Trzy z nich są glicerofosfolipidami: fosfatydylocholina (PC, lecytyna),
fosfatydyloetanoloamina (PE) oraz fosfatydyloseryna (PS), a czwarty, sfingomie-
lina (SM), jest fosfolipidem nieglicerolowym. Należy wspomnieć, że PC i SM
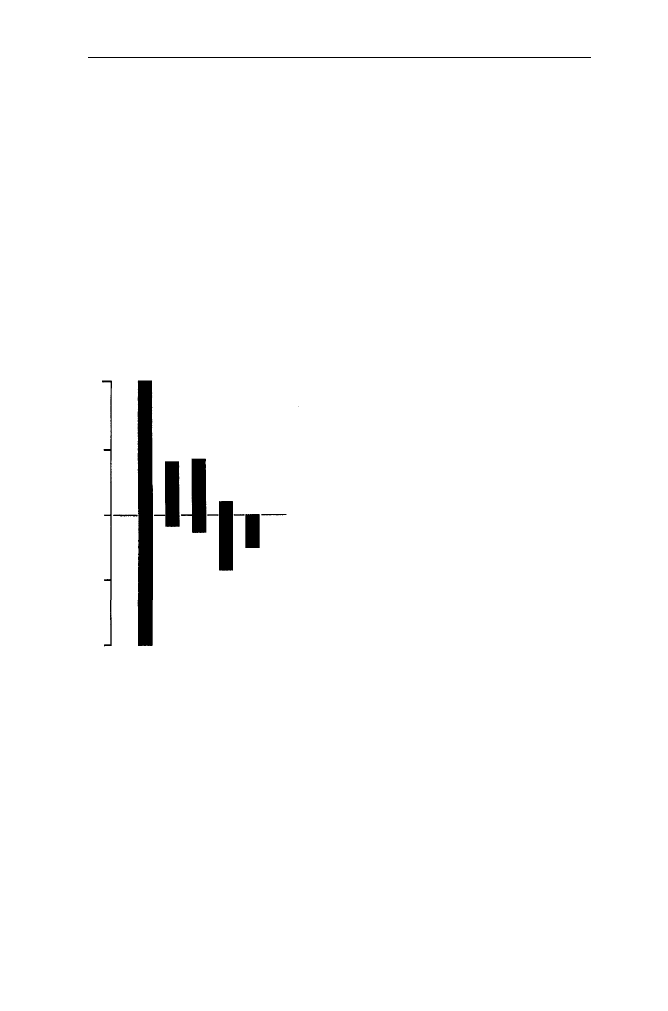
2.1. Białka i lipidy błon erytrocytów 7l
zawierają identyczne główki polarne (cholina), a PE i PS zawierają wolną grupę
aminową, dzięki czemu nazywane są aminofosfolipidami.
Skład fosfolipidowy erytrocytów krowy i innych przeżuwaczy charakteryzuje
się prawie kompletnym brakiem fosfatydylocholiny, która zastąpiona jest sfingo-
mielina.
Od wczesnych lat siedemdziesiątych wiadomo, że błony wykazują asymetrię
składu lipidowego, tzn. obie monowarstwy tworzące dwumolekularna warstwę
lipidową różnią się od siebie składem: absolutną asymetrię wykazują glikolipidy
- występują tylko w zewnętrznej monowarstwie. Fosfolipidy charakteryzuje tak-
że asymetria, aczkolwiek nie absolutna: zewnętrzna monowarstwa zawiera
w większości fosfolipidy cholinowe (PC i SM), a wewnętrzna, cytoplazmatyczną,
głównie aminofosfolipidy - fosfatydyloetanoloaminę i fosfatydyloserynę. Ten
ostatni fosfolipid występuje wyłącznie w cytoplazmatycznej monowarstwie błony
(Rys.
15).
Należy pamiętać, że występowanie fosfolipidów nie jest ograniczone do błon
komórek (krwi). Są one także składnikami osocza stanowiąc element lipoprotei-
dów. Fosfatydylocholina należy do najpowszechniejszych fosfolipidów osocza,
stanowiąc źródło ciągłego uzupełniania tego fosfolipidu w błonie komórek krwi.
Szybkość wymiany PC pomiędzy lipoproteidami i błoną erytrocytu wynosi około
1% całkowitej ilości PC w błonie erytrocytu na godzinę. Innym ważnym nośni-
kiem substancji trudno rozpuszczalnych w wodzie jest albumina, która przenosi
kwasy tłuszczowe i lizofosfolipidy.
Białka błony erytrocytu należą do najlepiej poznanych białek błonowych. Do
białek integralnych należą białka pełniące funkcje przenośników: ATP-aza wa-
pniowa usuwająca jony wapnia z komórki, przenośnik glukozy, a przede wszy-
stkim białko przenoszące aniony (białko 3 pasma elektroforetycznego), które
stanowi nie mniej niż 25-30% białek całkowitych błony erytrocytu. Wiadomo, że
Rys. 15. Asymetria lipidów błony
erytrocytów
monowarstwa wewnętrzna
monowarstwa zewnętrzna
SM PC PE PS
fosfolipidy całkowite
procen
t lipidó
w
całkowityc
h
50
25
25
50
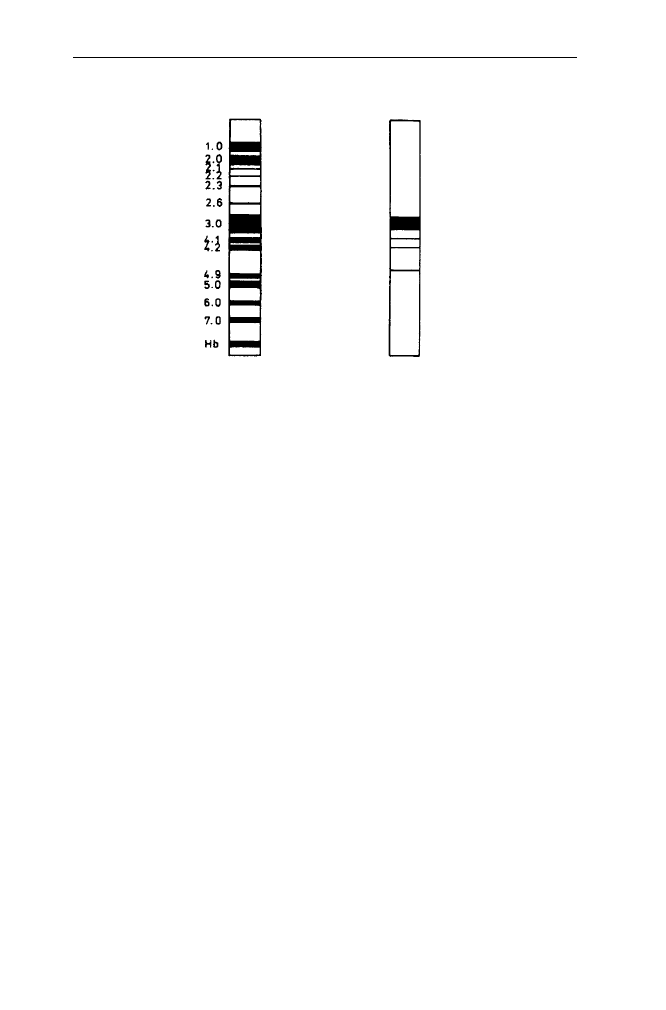
2. Błony biologiczne 72
BIAŁKA PERYFERYJNE BIAŁKA INTEGRALNE
A B
PAS 1 glikoforyna
PAS 2 glikoforyna
PAS 2 glikoforyna
PAS 3 glikoforyna
BIAŁKO
PRZENOSZĄCE
ANIONY
Spektryna {
ankiryna (
dematyna
aktyna
dehydrogenaza aldehydu
3-fosfoglicerynowego
Rys. 16. Rozdział elektroforetyczny błony białek cieni erytrocytów w żelu poliakryloamidowym
w obecności SDS
A. żel barwiony na białka, B. żel barwiony na cukry
białko to odpowiada za obojętną elektrycznie wymianę anionów HCO3
-
/C1
-
mają-
cą kluczowe znaczenie w przenoszeniu CO
2
przez erytrocyty. Na Rys. 16 przed-
stawiono typowy schemat elektroforetogramu białek błon (cieni) erytrocytu w 5%
żelu poliakryloamidowym w obecności SDS. Białka peryferyjne błony erytrocytu
występują na wewnętrznej, cytoplazmatycznej powierzchni błony erytrocytu,
gdzie główne z nich tworzą tzw. szkielet błonowy. Ta dwuwymiarowa sieć utwo-
rzona jest głównie przez tetramery spektryny, z których każdy składa się z dwóch
heterodimerów zbudowanych z α i β podjednostek spektryny. Spektryna jest
białkiem stanowiącym 25% wszystkich białek błony i występuje także powszech-
nie w innych nieerytroidalnych komórkach bardzo wielu organizmów. Tetramery
spektrynowe łączą się ze sobą w tzw. kompleksach węzłowych, których oś stano-
wi krótki filament aktynowy (37 nm, 13 podjednostek aktynowych). W skład
kompleksu wchodzą białka aktywujące oddziaływania spektryny i aktyny: białko
4.1, adducyna oraz białko zwane dematyna (białko 4.9) o nie poznanej dotąd
funkcji. Centrum tetrameru spektrynowego połączone jest przez inne ważne biał-
ko błony erytrocytu ankirynę (występujące także w wielu komórkach nieerytroi-
dalnych) z białkiem przenoszącym aniony. To połączenie, tzn. β-spektryna-anki-
ryna-białko, przenoszące aniony stanowi główne miejsce zakotwiczenia szkieletu
błonowego do zrębu hydrofobowego błony (dwumolekularna warstwa lipidową
z zanurzonymi w niej białkami integralnymi). Szkielet błony odpowiada, jak się
uważa, za dyskoidalny kształt erytrocytu i zapewnia wielką elastyczność tej ko-
mórki, która wielokrotnie przechodzi przez naczynia kapilarne o średnicy znacz-
nie mniejszej od średnicy komórki.
Białka błony charakteryzują się absolutną asymetrią, co wyraża się stałą, jeśli
chodzi o powierzchnię błony, lokalizacją białek peryferyjnych oraz bardzo precy-

2. l. Białka i lipidy błon erytrocytów 73
zyjną orientacją białek integralnych. Podobnie do glikolipidów cukrowe reszty
glikoproteidów błonowych znajdują się zawsze na ekstracytoplazmatycznej po-
wierzchni błony.
2.1.1. Porównanie składu lipidowego błon erytrocytów
przeżuwaczy i nieprzeżuwaczy
Doświadczenie obejmuje: izolację erytrocytów z krwi zwierzęcej, ekstrakcję lipi-
dów z osocza i erytrocytów oraz porównanie składu jakościowego i ilościowego
fosfolipidów erytrocytów przeżuwaczy i nieprzeżuwaczy.
Doświadczenie składa się z następujących etapów:
a) Izolacja erytrocytów
(wg Dodge J. T., Mitchell C., Hanahan D., Arch. Biochem. Biophys. 100 (1963) 119-130) -
zob. skrypt „Molekularna...", s. 13)
b) Ekstrakcja lipidów osocza
(wg Folch J., Lees M., Sloane-Stanley G. H., J. Biol. Chem. 226 (1957) 497-509)
Materiały, odczynniki i roztwory:
świeża krew, chroniona antykoagulantem (3.8% cytrynian sodowy) dodanym do
świeżo pobranej krwi w stosunku objętościowym l cz. na 9 cz. krwi;
chloroform/metanol 2:1 (v/v);
chloroform/metanol/woda 3:48:47 (ν/ν/ν).
Postępowanie:
• W 50 ml kolbce z doszlifowanym korkiem umieścić l ml osocza i zadać 19 ml
mieszaniny chloroform-metanol. Zamknąć kolbkę korkiem i zawartość mie-
szać przez kilka minut.
• Zdenaturowane białka odsączyć przez sączek z bibuły filtracyjnej w szklanym
lejku i 7.5 ml klarownego przesączu przenieść do czystej kolbki na 25 ml.
Dodać 1.5 ml wody dest. i dobrze wymieszać poprzez wytrząsanie.
• Przenieść mieszaninę do szklanej probówki wirówkowej (K-23) i odwirować
przy 2000 obr. /min przez 5 min.
• Ostrożnie usunąć fazę wodną (górną), przenieść dolną fazę ponownie do
czystej kolbki na 25 ml. Dodać 10 ml mieszaniny chloroform/metanol/woda
(3:48:47, ν/ν/ν) i dobrze wstrząsnąć. Rozdzielić fazy przez wirowanie (5 min,
2000 obr.) i usunąć fazę górną.
• Fazę dolną odparować w wyparce próżniowej. Uwaga: temperatura łaźni
wodnej wyparki nie powinna przekraczać 40
0
C. Rozpuścić wyekstrahowa-
ne lipidy w 200 μl mieszaniny chloroform-metanol.

2. Błony biologiczne 74
c) Ekstrakcja lipidów erytrocytów
(wg Rose H. G., Oklander M., J. Lipid Res. 6 (1965) 428-431)
Materiały, odczynniki i roztwory:
zawiesina przemytych i upakowanych erytrocytów;
chloroform/metanol 2:1 (v/v);
l mM EDTA;
izopropanol;
dichlorometan.
Postępowanie:
• 250 μl erytrocytów umieścić w 10 ml szklanej probówce wirówkowej (K-23)
i zmieszać z 250 μl EDTA.
•
Dodać 2 ml izopropanolu podczas wytrząsania na wstrząsarce. Mieszać przez
90 sek., po czym dodać l .25 ml dichlorometanu i znowu zmieszać przez 90
sek. Odwirować przez 5 min przy 2000 obr. /min.
• Supernatant przenieść do czystej probówki i odparować w strumieniu azotu
w łaźni wodnej o temperaturze 37°C. Uwaga: odparowywanie wykonać pod
wyciągiem.
• Rozpuścić wyekstrahowane lipidy w 200 μl mieszaniny chloroform/metanol.
Zamiast
powyższych metod ekstrakcji lipidów można zastosować postępowa-
nie wg N. S. Radin (zob. s. 19).
d) Analiza jakościowa i ilościowa lipidów erytrocytów i osocza w chro-
matografii cienkowarstwowej
Wykonanie jak w doświadczeniach ze s. 25 i s. 26. Nanieść na jednej płytce
następujące preparaty lipidów: standardową mieszaninę, lipidy osocza świni,
lipidy erytrocytów świni, lipidy osocza i lipidy erytrocytów wołu.
2.1.2. Izolacja i analiza ilościowa i jakościowa białek
cieni erytrocytów
Doświadczenie obejmuje izolację cieni erytrocytów, analizę składu białkowego
w elektroforezie w żelu poliakryloamidowym w obecności SDS barwionym na
obecność białek i węglowodanów. Ta sama analiza dotyczyć będzie białek całko-
witych, białek peryferyjnych oraz integralnych, których rozdziału dokonuje się na
drodze selektywnej ekstrakcji cieni erytrocytów roztworem 0.1 M NaOH. Błony
w wyniku takiej ekstrakcji pozbawione są całkowicie białek peryferyjnych, które
znajdują się w supernatancie.
Doświadczenie składa się z następujących etapów:
a) Izolacja cieni erytrocytów krowy lub świni

2.1. Białka i lipidy błon erytrocytów 75
(wg Dodge J. T., Mitchell C., Hanahan D., Arch. Biochem. Biophys. 100 (1963) 119-130);
zob. skrypt „Molekularna...", s. 13.
b) Selektywna ekstrakcja roztworem alkalicznym
zob. skrypt „Molekularna...", s. 15.
W uzyskanych preparatach należy oznaczyć ilościowo białko metodą Low-
ry'ego oraz cukry całkowite metodą fenolową.
c) Elektroforeza w 7% żelu poliakryloamidowym w obecności SDS
zob. skrypt „Molekularna...", s. 50.
d) Barwienie na obecność białek i glikoproteidów
zob. skrypt „Molekularna...", s. 51 i 52.
2.1.3. Oznaczanie białka metodą Lowry'ego
(wg Lowry O. H., Rosebrough A. L., Farr A. L., Randall R. J., J. Biol. Chem. 193 (l951)
265-275).
Roztwory:
2% Na
2
CO
3
w 0.1 M NaOH;
2% winian sodowo-potasowy;
l % CuSO
4
x 5H
2
O.
odczynnik Folina (przygotowanie jak w „Kurs praktyczny z biochemii", W. Mej-
baum-Katzenellenbogen i I. Mochnacka, PWN Warszawa, 1966, s. 175, lub
„Ćwiczenia z biochemii dla biologów", pod red. W. Mejbaum-Katzenellenbogen,
Wrocław, 1992, s. 97);
roztwór albuminy surowicy krwi wołowej 0.5 mg/ml.
Postępowanie:
• Do 200 μl roztworu białka (0-50 μg/próbkę; 0-250 μg/ml) należy dodać 2 ml
odczynnika A świeżo przygotowanego przez zmieszanie: 50 ml 2% Na
2
CO
3
w 0.1 M NaOH, 0.5 ml 2% winianu sodowo-potasowego i 0.5 ml 1%
CuSO
4
x 5H
2
O.
• Po 10 minutach w temperaturze pokojowej dodać 200 μl odczynnika Folina
i bardzo dobrze
wymieszać.
• Po 30 min odczytać absorbancję przy 750 nm wobec ślepej próby odczynniko-
wej, do której dodano 200 μl wody zamiast roztworu białka.
Krzywą standardową sporządzić stosując roztwór albuminy surowicy krwi
wołowej 0.5 mg/ml.

2. Błony biologiczne 76
2.1.4. Oznaczanie cukrów całkowitych
(wg Dubois M., Gilles K. A., Hamilton J. K., Rebers P. A., Smith F., Anal. Chem. 28 (1956)
350-356)
Odczynniki i roztwory:
stężony kwas siarkowy;
5% roztwór wodny fenolu (należy użyć świeżo przedestylowanego fenolu);
standardowy roztwór (1%) glukozy w nasyconym roztworze kwasu benzoesowe-
go (2.5 g kwasu benzoesowego rozpuścić w litrze wrzącej wody i ochłodzić).
Postępowanie:
• Do 0.5 ml roztworu zawierającego 5-40 μg cukru dodać 0.5 ml 5% fenolu
a następnie 2.5 ml stężonego kwasu siarkowego (Uwaga: pipetować ostroż-
nie stosując gumową gruszkę lub dozownik!).
• Próby pozostawić na 10 min w temperaturze pokojowej, po czym wstawić do
łaźni wodnej o 20°C na 20 min.
• Odczytać absorbancję prób w spektrofotokolorymetrze przy 490 nm.
2.2. Właściwości barierowe
błon biologicznych
Błona plazmatyczną jest tą błoną, przez którą muszą przechodzić do komórki
wszystkie związki pokarmowe i przez którą muszą zostać przeniesione cząsteczki
wydalane i wydzielane przez komórkę. Błona ta nie stanowi więc ścisłej bariery
dla przepływu solutów do i z wnętrza komórek.
Poznano trzy typy transportu małych solutów poprzez błony biologiczne:
1. transport bierny - zachodzący na drodze prostej dyfuzji poprzez błonę
biologiczną zachowującą się jak błona półprzepuszczalna,
2. transport ułatwiony - wykorzystujący nośniki lub przenośniki białkowe
obecne w samej błonie,
3. transport aktywny, w którym dla procesu przenoszenia solutów przez błonę
wymagane jest dostarczenie energii, zwykle pochodzącej z hydrolizy ATP.
2.2.1. Transport bierny małych nieelektrolitów
(wg de Gier J., Mandersloot J. G., van Deenen L. L. M., Biochim. Biophys. Acta 150 (1968)
666-675)
Małe niezjonizowane cząsteczki są transportowane przez błonę biologiczną podo-
bnie jak przez półprzepuszczalna błonę. Zdolność przejścia tych związków zależy
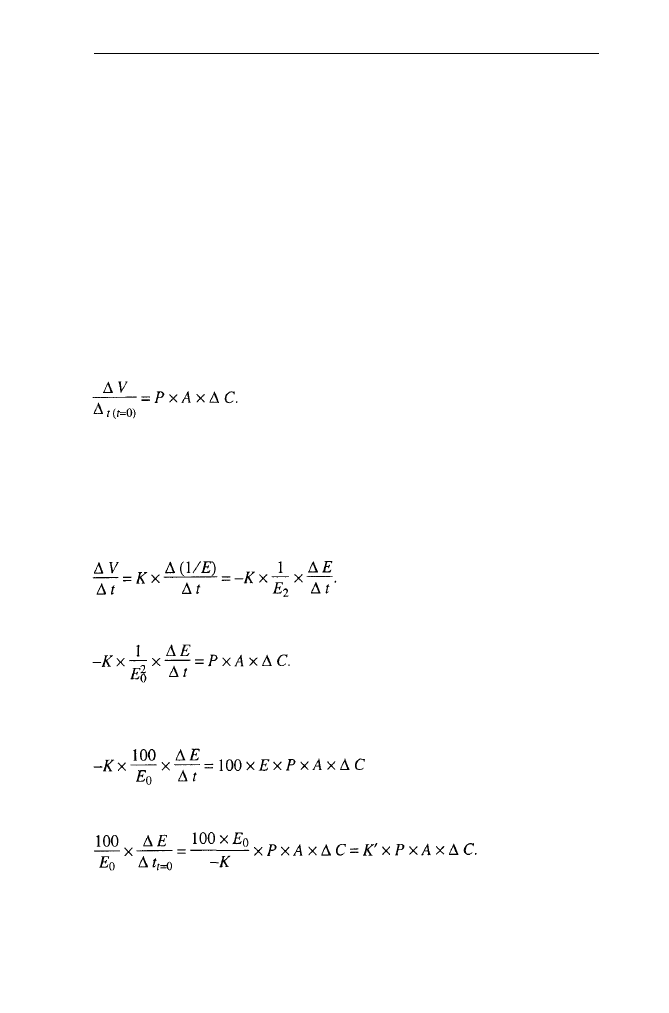
2.2. Właściwości barierowe błon biologicznych 77
od wielkości ich cząsteczek i stopnia ich hydratacji. Do badań procesów transpo-
rtu biernego i roli składu dwuwarstwy fosfolipidowej w tym transporcie wygod-
nym modelem są liposomy. Wykazano, że kiedy liposomy zawierające nie prze-
chodzący przez dwuwarstwę elektrolit umieści się w izoosmotycznym roztworze
nieelektrolitu, który może przechodzić przez barierę dwuwarstwy
,
to obserwuje
się stopniowy przyrost ich objętości (liposomy pęcznieją). Spowodowane to jest
wymuszonym przez gradient stężenia wnikaniem przechodzących przez błonę
cząsteczek do wnętrza liposomu. Podobne zjawisko obserwuje się również
w przypadku komórek. Wnikanie solutu do wnętrza, zaburzając równowagę os-
motyczną, powoduje z kolei zwiększenie ilości cząsteczek wody przechodzących
w tym samym kierunku, co wzmacnia proces osmotycznego zwiększania objęto-
ści. Ponieważ przepuszczalność błon dla wody jest o kilka rzędów wielkości
większa niż przepuszczalność dla solutów, dlatego szybkość zwiększania objęto-
ści będzie proporcjonalna do przepuszczalności P przechodzącego nieelektrolitu.
Szybkość początkową pęcznienia określa równanie:
(1)
gdzie: A - powierzchnia liposomów lub komórek, ΔC - gradient stężenia nieele-
ktrolitu (ΔC = C
zewn.
- C
wewn.
), P -
współczynnik przepuszczalności błony dla
nieelektrolitu.
Wzrost objętości (pęcznienie) może być śledzony metodą optyczną. Wykaza-
no, że pomiędzy objętością a odwrotnością absorbancji zawiesiny liposomów czy
też komórek zachodzi liniowa zależność:
(2)
(3)
Dla badań porównawczych lepiej jest wyrażać ΔΕ jako procentową zmianę
wartości początkowej:
lub
Pomiary E i EIt w czasie t=0 dają wartości przepuszczalności, które mogą być
porównywane tak długo, jak długo A jest wartością stałą.
W czasie t=0 równanie (2) podstawione do (1) daje:
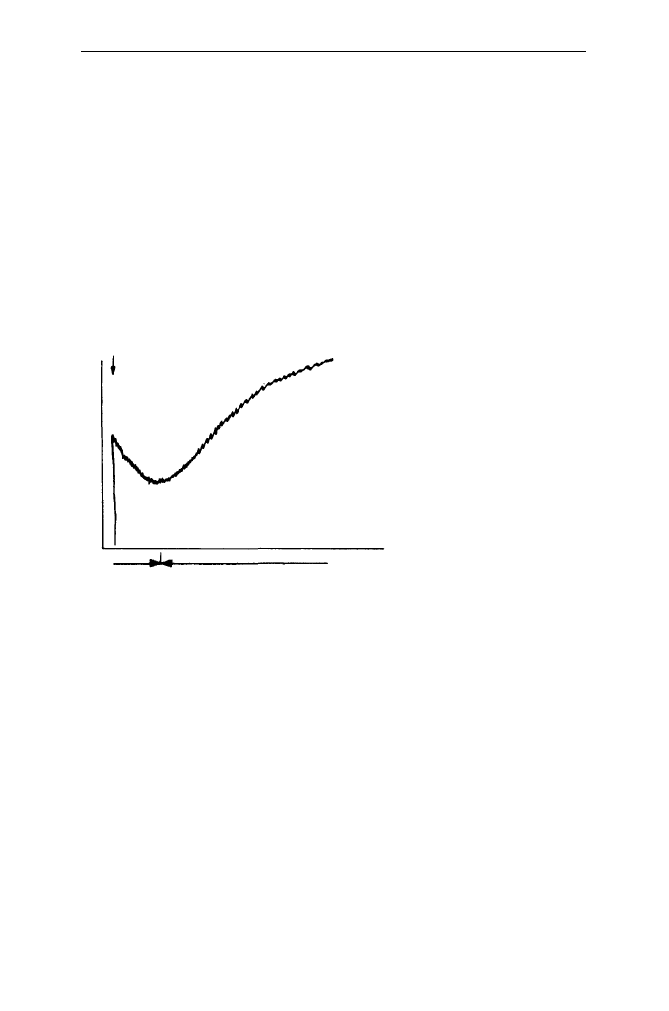
A 4 5 0
wstrzyknięcie
czas
kurczenie się pęcznienie
(wyciek ) (wpływanie do wnętrza )
2. Błony biologiczne 78
Metoda ta może być używana do badań np.:
- wpływu rozmiarów cząsteczek nieelektrolitów na ich przepuszczalność,
- wpływu temperatury na szybkość tego procesu,
- wpływu zmian składu lipidowego błony na jej przepuszczalność.
Jeżeli w liposomach jest zamknięty solut przechodzący przez błonę o prze-
puszczalności większej od przepuszczalności solutu znajdującego się na zew-
nątrz, to obserwuje się efekt kurczenia się tych struktur. Z kolei, po wstrzyknięciu
liposomów lub komórek do hyperosmotycznego roztworu przechodzącego przez
błonę nieelektrolitu obserwuje się najpierw kurczenie się, a następnie pęcznienie
badanych struktur. Kurczenie się i czas jego trwania odzwierciedla wypływanie
wody, pęcznienie natomiast - wpływanie solutu wraz z wodą do wnętrza. Czas
trwania etapu kurczenia się odzwierciedla również zdolność solutu znajdującego
się w środowisku do przekroczenia bariery błony (Rys. 17).
Rys. 17. Zmiany gęstości optycznej zawiesiny liposomów zawierających nieprzechodzący przez
błonę solut po ich wstrzyknięciu do roztworu przechodzącego przez błonę solutu
Ponieważ, jak wspomniano, komórki są podobnie jak liposomy strukturami
zamkniętymi, zawierającymi wysokie stężenia elektrolitów, opisany system eks-
perymentalny może być zastosowany do badań przepuszczalności błon natural-
nych. W przypadku komórek proces silnego pęcznienia prowadzi w końcu do ich
lizy, tj. uwolnienia zawartości do środowiska. Badania tego typu są szczególnie
przydatne dla określania wpływu różnorodnych modyfikatorów błony na jej wła-
ściwości barierowe.
Materiały i roztwory:
krew zebrana do środka zapobiegającego krzepnięciu;
0.14 M NaCl w 10 mM buforze Tris-HCl, pH 7.4;
300 mM roztwór erytrozy w 10 mM buforze Tris-HCl, pH 7.4;
300 mM roztwór glicerolu w 10 mM buforze Tris-HCl, pH 7.4;
300 mM roztwór sacharozy w 10 mM buforze Tris-HCl, pH 7.4;
roztwory modyfikatorów (np. detergentów).

2.2. Właściwości barierowe błon biologicznych 79
Sprzęt
Fotokolorymetr Spekol lub spektrofotometr z przystawką termostatującą, mie-
szadłem oraz rejestratorem.
Przygotowanie zawiesiny krwinek:
• Krew odwirować przy 1500 obr. /min. przez 15 min
• Zlać ostrożnie supernatant, a krwinki przemyć trzykrotnie zbuforowanym roz-
tworem NaCl, stosując każdorazowo zawieszenie krwinek w roztworze oraz
wirowanie przy 1500 obr. /min przez 10 min.
• Po ostatnim przemyciu upakowane krwinki rozcieńczyć dwukrotnie roztwo-
rem NaCl i używać do doświadczeń.
Postępowanie:
• Pomiary prowadzić przy 450 lub 600 nm w temperaturze 37
0
C.
• Wystandaryzować długość odcinka na papierze rejestracyjnym dla wartości
T=5%.
• W kuwecie aparatu umieścić 4 ml odpowiedniego roztworu nieelektrolitu (dla
badania procesu indukowanej hemolizy - 0.14 M NaCl w 10 mM buforze
Tris-HCl, pH 7.4) i Preinkubować go w żądanej temperaturze przez 2 min.
• Doprowadzić wskazania absorbancji w aparacie oraz pisak rejestratora na
„zero", uruchomić mieszadło i rejestrator.
• Za pomocą strzykawki Hamiltona wstrzyknąć szybko do roztworu odpowied-
nią objętość zawiesiny krwinek (zwykle 30-50 μl), zanotować absorbancję
początkową (E
0
).
• Rejestrować zmiany gęstości optycznej zawiesiny przez 5-10 min zależnie od
szybkości zachodzących zmian.
• Przy badaniu wpływu modyfikatorów do kuwety wstrzykiwać ich roztwory
przed wstrzyknięciem zawiesiny krwinek.
Obliczenia:
• Dla każdej krzywej zmian gęstości optycznej zarejestrowanej w czasie do-
świadczenia wykreślić styczną do jej części początkowej (zob. Rys. 18) i obli-
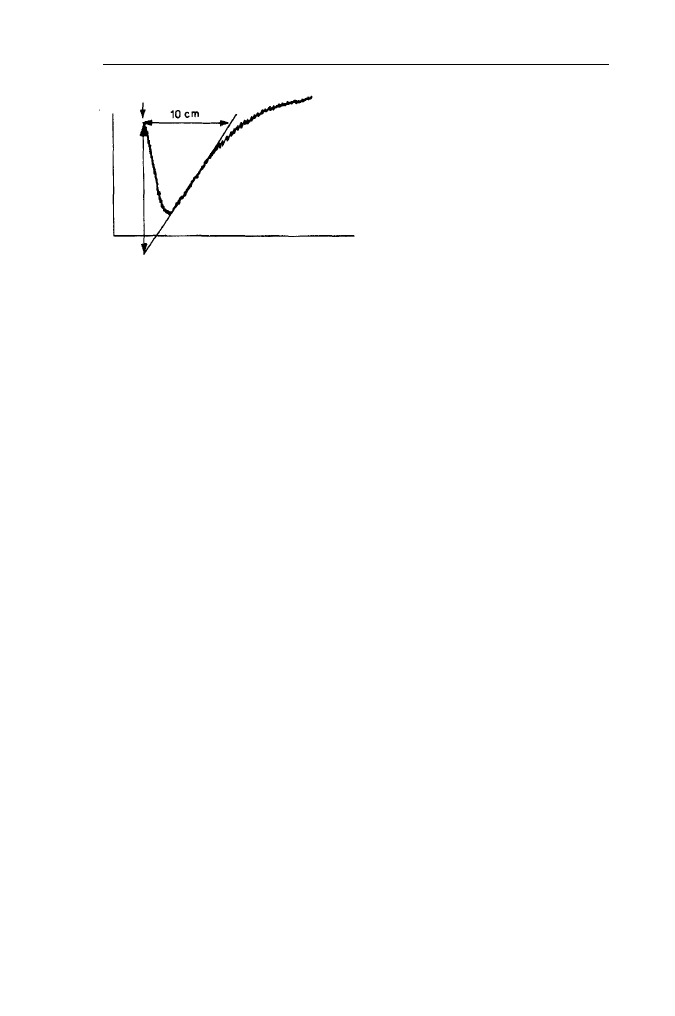
czyć początkowe nachylenie dT/dt wg wzoru:
• Obliczyć szybkość początkową procesu:
• Sporządzić wykresy zależności szybkości początkowych od rodzaju nieele-
ktrolitu i (lub) stężenia detergentu.
wstrzyknięcie
czas
T ( E )
a
Rys. 18. Sposób wyznaczania
nachylenia początkowego krzywej
zmian gęstości optycznej zawiesiny
liposomów do obliczenia dT/dt
2. Błony biologiczne 80
2.2.2. Wybiórczość transportu ułatwionego jonów
poprzez błonę biologiczną
(wg Adams P. Α., Black D. J., Chem. Educ. 67 (1990) 526-527)
Transport
ułatwiony, w którym soluty przenoszone są przez błonę przy udziale
białkowych cząsteczek nośnikowych lub kanałowych, jest transportem swoistym,
wybiórczym. Przykładem ilustrującym tę Wybiórczość jest integralne białko bło-
ny erytrocytów - białko przenoszące aniony wchodzące w skład białek elektro-
foretycznego pasma 3 (zob. Rys. 16). Białko to jest jednym z najlepiej poznanych
białek uczestniczących w procesach transportu ułatwionego. Białko o masie
90-100 kDa transportujące aniony jest najbardziej rozpowszechnionym białkiem
błon erytrocytarnych. Poniższe doświadczenie wykaże, że białko transportujące
aniony pośredniczy w transporcie przez błonę jedno- i dwuwartościowych anio-
nów, natomiast nie przenosi anionów trójwartościowych.
Materiał i roztwory:
świeża krew zebrana do roztworu zapobiegającego krzepnięciu;
145 mM NaCl w 5 mM buforze Hepes, pH 6.4;
0.05 M roztwór nadjodanu sodowego (NaIO
4
) w 5 mM buforze Hepes, pH 6.4;
0.05 M roztwór dwusiarczynu sodowego (Na
2
S
2
O
5
) w 5 mM buforze Hepes pH
6.4;
0.05 M roztwór żelazicyjanku potasowego (K
3
Fe(CN)
6
) w 5 mM buforze Hepes,
pH 6.4;
1% roztwór wodny Tritonu X-100.
Postępowanie:
• l ml krwi odwirować przy 1500 obr. /min przez 10 min.
• Uzyskane krwinki przemyć dwukrotnie 10 obj. roztworu NaCl w buforze,
a następnie zawiesić w 3 ml tego roztworu.
• W trzech probówkach umieścić po 0.5 ml zawiesiny krwinek i po 0.5 ml
roztworów trzech anionów, wymieszać.
2.2. Właściwości barierowe błon biologicznych 8l
• Opisać zmiany w barwie zawiesin po 5 min.
• Odwirować próby przy 1500 obr/min przez l0 min, opisać wynik.
• Próbę zawierającą żelazicyjanek ponownie zawiesić, dodać kilka kropel deter-
gentu, wymieszać i jeszcze raz odwirować. Opisać wynik.
Dlaczego z trzech utleniających oksyhemoglobinę związków jedynie dwa po-
wodowały zmianę zabarwienia zawiesiny erytrocytów? Jak wyjaśnić fakt, że
zmiana w trzeciej próbie następowała dopiero po dodaniu do niej detergentu?
2.2.3. Indukowana przepuszczalność dwuwarstwy
fosfolipidowej dla jonów. Jonofory
(wg Blok M. C., de Gier J., van Deenen L. L. M., Biochim. Biophys. Acta 367 (1974)
202-209 i 210-224)
Czyste dwuwarstwy lipidowe są bardzo słabo przepuszczalne dla jonów. Przepu-
szczalność tę można zwiększyć w swoisty sposób przez dodanie „jonoforów",
związków selektywnie zwiększających przepuszczalność błony dla pewnych jo-
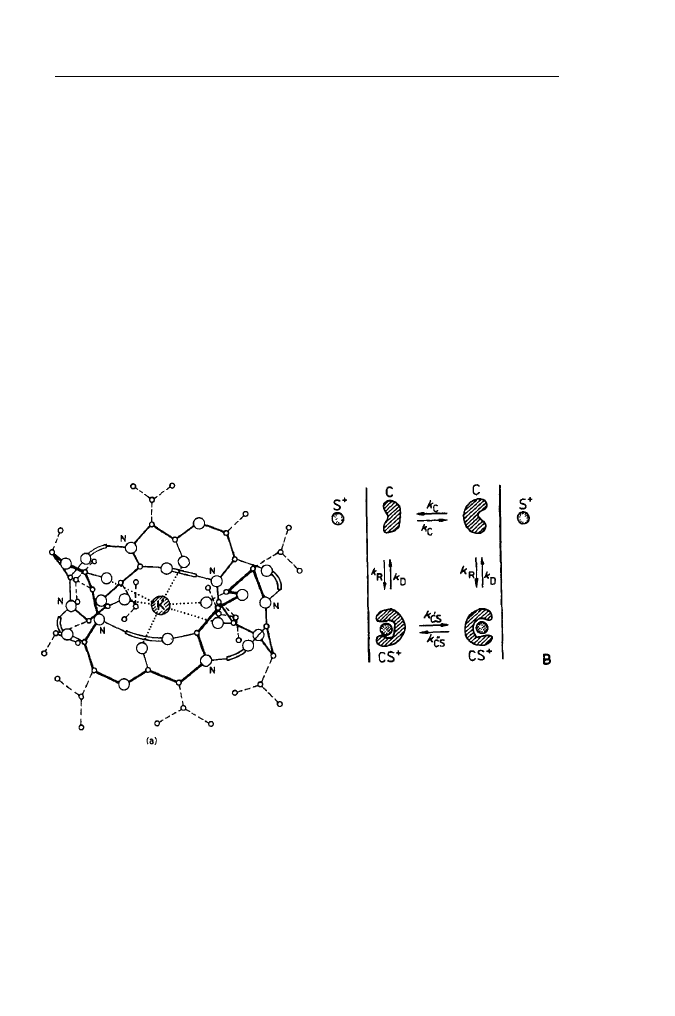
nów. Bardzo dobrym przykładem jonoforu jest występujący naturalnie depsypep-
tyd walinomycyna (Rys. 19).
Jest to mały pierścieniowy peptyd będący efektywnym nośnikiem jonów K
+
i Rb
+
. Tworzy on kompleksy z tymi jonami, które szybko (w przeciwieństwie do
uwodnionych jonów) dyfundują poprzez hydrofobowy rdzeń błon sztucznych
i naturalnych. Ze względu na prostą budowę i łatwą dostępność jonofory peptydo-
we stały się modelami do badań mechanizmów transportu ułatwionego.
Przepuszczalność błony dla protonów można selektywnie zwiększyć przez
dodanie tzw. rozsprzęgaczy, którymi są słabe kwasy o aromatycznym pierścieniu,
np. 2,4-dwunitrofenol (DNP) lub fenylohydrazon trójfluorometoksykarbonylo-
cyjanku (FCCP). Dzięki przemieszczeniu ładunku forma anionowa kwasu może
Rys. 19. Budowa kompleksu
walinomycyny z jonem potasowym
(K) i kinetyczny schemat
mechanizmu translokacji jonu przy
A udziale cząsteczki nośnikowej

2. Błony biologiczne 82
również przechodzić przez błonę. Transport nie zdysocjowanej cząsteczki roz-
sprzęgacza w jednym kierunku, a formy anionowej w kierunku przeciwnym, jest
równoważny z transportem protonów. Wykazano, że wpływ walinomycyny na
wyciek jonów K
+
silnie zależy od natury anionów zamkniętych we wnętrzu
liposomów. Jeżeli anion nie może przekroczyć bariery błony, wtedy mała ilość
jonów K
+
przeniesiona przez błonę z udziałem walinomycyny generuje wysoki
potencjał błonowy hamujący dalszy ich wyciek. Ponowny wyciek tych jonów
można uzyskać dopiero po dodaniu jonoforu protonowego, np. FCCP.
Materiały i roztwory:
lecytyna jajeczna, roztwór chloroformowy 30 μmoli/ml;
cholesterol, roztwór chloroformowy 10 mg/ml;
walinomycyna, roztwór wodny 2.5 μg/ml;
dwunitrofenol,
roztwór wodny l mg/ml;
50 mM
KCl;
150 mM KCl;
150 mM KCNS;
150 mM MgSO
4
w 10 mM Tris-H
2
SO
4
, pH 7.5,
10% Triton X-100.
Sprzęt:
pH-metr z elektrodą selektywną dla jonów K
+
połączony z rejestratorem.
Postępowanie:
• Przygotować w sposób opisany w poprzednim rozdziale liposomy o następu-
jącym składzie:
a) lecytyna (60 μmoli) zawieszona w KCl,
b) lecytyna-cholesterol 70:30, molowo (60 μmoli) zawieszone w KCNS,
c) lecytyna (60 μmoli) zawieszona w KCNS.
Uzyskane liposomy
umieścić w dobrze oznakowanych woreczkach dializacyj-
nych i dializować do 150 mM MgSO
4
w buforze, początkowo zmieniając
roztwór co 30 min (3 x), a następnie pozostawić w świeżym roztworze na noc
w +4°C.
• W naczyńku pomiarowym (zapewniającym termostatowanie i stałe miesza-
nie) umieścić 7.5 ml buforowanego roztworu MgSO
4
, dodać 2 μl 50 mM KCl
i
odczekać aż roztwór osiągnie temperaturę 25°C i uzyska się stabilną linię
podstawową na rejestratorze.
• Dodać 100 μl wydializowanych liposomów i równoważyć przez 2 min. Reje-
strowane zmiany wskazania elektrody odzwierciedlają tzw. bierny wyciek.
• Dodać 10 μl roztworu walinomycyny i rejestrować wyciek jonów potasowych
przez 2 min.
• Następnie dodać 10 μl roztworu DNP i śledzić wyciek przez dalsze 2-3 min.
• Na zakończenie dodać 50 μl roztworu Tritonu X-100 i rejestrować zmiany do
chwili ustabilizowania się odczytu.

2.3. Płynność błony biologicznej 83
• Powtórzyć doświadczenie używając preparatów b i c. Badając preparat b wy-
konać dwa pomiary - z dodatkiem DNP i bez DNP. Po zakończeniu pomiarów
na liposomach wykalibrować układ rejestrując zmiany wskazania elektrody
przy kolejnym dodawaniu l μl porcji 50 mM KCl do 7.5 ml roztworu MgSO
4
.
Obliczyć ilość K
+
zamykanego w badanych typach liposomów.
Podać, czy typ anionu zamkniętego wraz z jonem potasowym jest istotny w procesie
indukowanego przez walinomycynę wycieku. Jak wpływa obecność cholesterolu
w błonie na szybkość wycieku? Czy obecność rozsprzęgacza jest bezwzględnie
konieczna do indukowanego przez walinomycynę wycieku jonów potasowych?
2.3. Płynność błony biologicznej
Błony biologiczne wykazują własności ciekłych kryształów. Stan ciekłego kry-
ształu jest stanem materii pośrednim pomiędzy stanem stałym charakteryzującym
się uporządkowaniem molekularnym a stanem ciekłym charakteryzującym się
ruchliwością cząstek. Cząsteczki, które mogą osiągać ten stan charakteryzują się
wydłużonym kształtem (stosunek osi 4-8:1). Fosfolipidy są takimi cząsteczkami.
Stan ciekłego kryształu może być związany z układem jednoskładnikowym (sy-
stem termotropowy) lub wieloskładnikowym (system liotropowy). Układ lipidy-
woda jest układem liotropowym. Błony biologiczne są liotropowymi, blaszkowa-
tymi (dwuwymiarowymi) układami ciekłokrystalicznymi.
Proste związki amfifilowe, w tym większość lipidów błon, podlegają przejściu
fazowemu: stała faza żelu krystalicznego-ciekły kryształ lub faza uporządkowa-
na-faza nieuporządkowana w określonej temperaturze zwanej temperaturą przej-
ścia fazowego (T
c
), która jest charakterystyczna dla poszczególnego lipidu (zob.
s. 51 oraz Tab. 4).
Do opisu stanu fizycznego płynnej błony, szczególnie naturalnej, stosuje się
pojęcie płynności, które jest odwrotnością lepkości. O ile podczas przejścia fazo-
wego płynność spada o dwa rzędy wielkości, o tyle w przypadku błon naturalnych
interesujące są niewielkie zmiany płynności (mikrolepkości) w obrębie stanu
ciekłego. W znacznej mierze dotyczy ona ruchliwości łańcuchów węglowodoro-
wych wokół osi prostopadłej do płaszczyzny błony, tzw. dyrektora (Rys. 20).
Jedną z miar fluktuacji osi cząsteczek jest parametr uporządkowania S zdefi-
niowany równaniem:
S =1/2 [3(cos
2
Θ)-1],
gdzie Θ jest
kątem pomiędzy drgającą osią cząsteczki a osią odnośnikową (dyre-
ktora). Wyrażenie (cos
2
Θ) jest
uśrednioną czasowo orientacją cząsteczki (lub
dipolu przejściowego, lub wiązania w cząsteczce) w odniesieniu do normalnej do
płaszczyzny błony. Parametr uporządkowania przyjmuje wartości od 0 (układ
nieuporządkowany) do l (układ uporządkowany).

Rys. 20. Modele ilustrujące ruchliwości rotacyjne w błonie
Miarą ruchliwości cząsteczki w błonie jest także czas korelacji rotacyjnej tzw.
τ
c
,
który jest miarą skali czasowej ruchu. Sztywna kula o promieniu a w roztwo-
rze o lepkości η i temperaturze T charakteryzuje się czasem korelacji τ
c
:
Najczęściej stosowanymi technikami oceny płynności błon naturalnych i sztu-
cznych fosfolipidowych są: deuterowy magnetyczny rezonans jądrowy
2
H-NMR,
w której to technice stosuje się znakowane deuterem fosfolipidy (w określonym
miejscu łańcucha węglowodorowego). Widmo
2
H-NMR czułe jest na orientację
wiązania C-D w stosunku do zewnętrznie przyłożonego pola magnetycznego.
Podobnie, widma elektronowego rezonansu paramagnetycznego (EPR lub ESR)
czułe są na orientację wiązania =N O (stabilny rodnik nitroksylowy obecny
w wielu powszechnie stosowanych „sondach", takich jak znakowane spinowo
kwasy tłuszczowe lub lipidy) wobec zewnętrznego pola magnetycznego. Techni-
ka depolaryzacji fluorescencji polega na tym, że polaryzacja wyemitowanego
światła zależy od orientacji momentu molekularnego dipola przejściowego w od-
niesieniu do kierunku zdefiniowanego przez polaryzator użyty do pomiaru. W tej
technice wzbudza się subpopulację sondy fluorescencyjnej (fluoryzujące związki
organiczne lokalizujące się wewnątrz dwumolekularnej warstwy lipidowej; do
najczęściej stosowanych należy difenyloheksatrien, DPH) światłem spolaryzowa-
nym. Jeśli cząsteczki sondy nie zmieniają położenia w przedziale czasowym
pomiędzy absorpcją i emisją (10
-8
sek.), polaryzacja światła wyemitowanego bę-
dzie określana przez światło użyte do wzbudzenia. Jeśli cząsteczki sondy porusza-
ją się swobodnie (izotropowo) i szybko, to nie obserwuje się preferowanej orien-
tacji wektora elektrycznego światła wyemitowanego i polaryzacja równa jest
zero. Wartości polaryzacji pomiędzy tymi ekstremami mogą odpowiadać zarów-
no ograniczeniu szybkości ruchu, tzn. 10
-8
sek. nie wystarcza, aby uśrednić pola-
ryzację światła wyemitowanego, jak i ograniczeniu ruchów sondy w niektórych
kierunkach.
Doświadczalnie w pomiarach płynności błon techniką depolaryzacji fluore-
scencji stosuje się parametr r
2. Błony biologiczne 84

2.4. Liposomy jako modele w badaniach procesu fuzji błon biologicznych 85
gdzie - natężenie fluorescencji przy polaryzatorach światła wzbudzającego
i emitowanego ustawionych równolegle, przy polaryzatorach ustawionych
prostopadle.
2.3.1. Ocena płynności błon pęcherzyków lipidowych
sporządzonych z mieszaniny fosfatydylocholiny
i cholesterolu (9:1) metodą depolaryzacji fluorescencji
z użyciem difenyloheksatrienu
Postępowanie:
• Z fosfatydylocholiny i cholesterolu (9:1) sporządzić małe jednowarstwowe
pęcherzyki jak w ćwiczeniu na s. 94 stosując do hydratacji 12.5 mM bufor
boranowy, pH 8.0.
• Roztwór DPH (2 x 10
-3
M) w tetrahydrofuranie zmieszać przed użyciem z po-
wyższym buforem w stosunku l:l, a ten z kolei roztwór w stosunku 1:1 z za-
wiesiną błon i inkubować l godzinę w temperaturze pokojowej. Pomiary wy-
konywać w ustabilizowanej temperaturze przy długości fali wzbudzającej
360, a emisji 430 nm. Każdy pomiar wykonywać przy polaryzatorach usta-
wionych równolegle i prostopadle.
• Wyznaczyć zależność parametru r od temperatury dla temperatur 20, 25, 30,
35 i 40
0
C.
2.4. Liposomy jako modele
w badaniach procesu fuzji
błon biologicznych
Fuzja błon jest procesem kluczowym dla funkcjonowania wielu typów komórek.
Proces ten jest kluczowy nie tylko w zjawisku fuzji dwóch komórek, np. w czasie
zapłodnienia, lecz również w transporcie wewnątrzkomórkowym na drodze pę-
cherzykowej. Fuzja pęcherzyków transportowych wydaje się głównym mechani-
zmem takich procesów, jak biogeneza błon, wydzielanie na drodze egzocytozy
oraz wchłanianie na drodze endocytozy. Fuzja błon jest również kluczowym
etapem procesu wnikania opłaszczonych wirusów do komórek.
W badaniach molekularnych mechanizmów procesu fuzji niezastąpione oka-
zały się liposomy. Zalety tych systemów modelowych są oczywiste.
1. Liposomy tworzą stabilne, dobrze zdefiniowane struktury o małej prze-
puszczalności dla rozpuszczalnych w wodzie solutów, tak że mieszanie się prze-
strzeni wodnych w czasie procesu można łatwo śledzić.

2. Błony biologiczne 86
2. Skład lipidowy (typy fosfolipidów, długości reszt acylowych, obecność
cholesterolu czy glikolipidów) błony liposomalnej można łatwo zmieniać, co
umożliwia badania roli grup polarnych oraz takich m.in. czynników, jak separacja
faz w płaszczyźnie błony czy przejścia fazowe w procesie fuzji.
3. Układy liposomowe zapewniają również badanie roli tak białek błono-
wych, jak i cytoplazmatycznych w przebiegu fuzji.
4. Modelowe układy liposomowe umożliwiają kinetyczne śledzenie zacho-
dzącego procesu fuzji.
Dzięki tym badaniom stwierdzono, że aby proces fuzji dwu błon mógł nastą-
pić, muszą zostać pokonane dwie istotne bariery - odpychania elektrostatycznego
oraz odpychania, niedopuszczania do kontaktu dwu błon przez warstewki wody
hydratacyjnej. Coraz więcej danych wskazuje na udział niewarstwowych struktur
pośrednich w procesie tworzenia jednej wspólnej błony z dwóch oddzielnych. Na
Rys. 21 przedstawiono tworzenie, możliwe przekształcanie i udział tzw. we-
wnątrzbłonowych micelarnych intermediatów (IMI) we wczesnym etapie procesu
fuzji - tworzenia tzw. połączeń pomiędzywarstwowych (ILA).
Jak można z przedstawionego schematu zauważyć, IMI są intermediatami,
które mogą prowadzić do procesu zarówno fuzji, jak i destrukcji błony poprzez
rozbudowanie w struktury niewarstwowe typu H
II
. Badania wykazały, że o kie-
runku przekształceń decyduje m.in. struktura grup polarnych cząsteczek lipidów
uczestniczących w procesie. Stwierdzono, że zmiana parametru Z, czyli stosunku
powierzchni grupy polarnej lipidu wykazywanej w fazie warstwowej L
a
do po-
wierzchni tej grupy wykazywanej w fazie H
II
o 10% powoduje zmianę wartości
stałej tworzenia ILA o dwa rzędy wielkości.
Do czynników, które poprzez oddziaływanie na fosfolipidy błony biologicznej
umożliwiają przełamanie wymienionych barier spontanicznej fuzji, czyli oddzia-
łują fuzjogennie, należą białka i peptydy, szczególnie białka otoczek wirusowych,
wysokie pH i stężenie jonów wapniowych oraz tzw. chemiczne fuzjogeny, z któ-
rych najczęściej stosowany jest glikol polietylenowy.
Do badań procesów interakcji pomiędzy pęcherzykami liposomowymi i ich
fuzji używa się wielu różnych metod, takich jak spektroskopie NMR i ESR,
różnicowa kalorymetria, mikroskopia elektronowa, filtracja żelowa, a także badań
turbidymetrycznych oraz technik fluorescencyjnych. Wiele z tych technik umo-
żliwia jedynie przypuszczalne określenie, czy proces fuzji zachodzi, czy też nie,
gdyż opierają się one na pomiarach agregacji pęcherzyków, powiększania ich
rozmiarów, mieszania się lipidów oddzielnych błon czy też wycieku zawartości
pęcherzyków. Jedynie techniki, które umożliwiają wykazanie mieszania się za-
wartości przestrzeni wodnych pęcherzyków ulegających fuzji, spełniają naj-
ostrzejsze kryteria detekcji procesu fuzji.
Opracowano kilka technik umożliwiających śledzenie procesu mieszania się
zawartości pęcherzyków. Są to techniki fluorescencyjne, których zasady przedsta-
wiono schematycznie na Rys. 22.
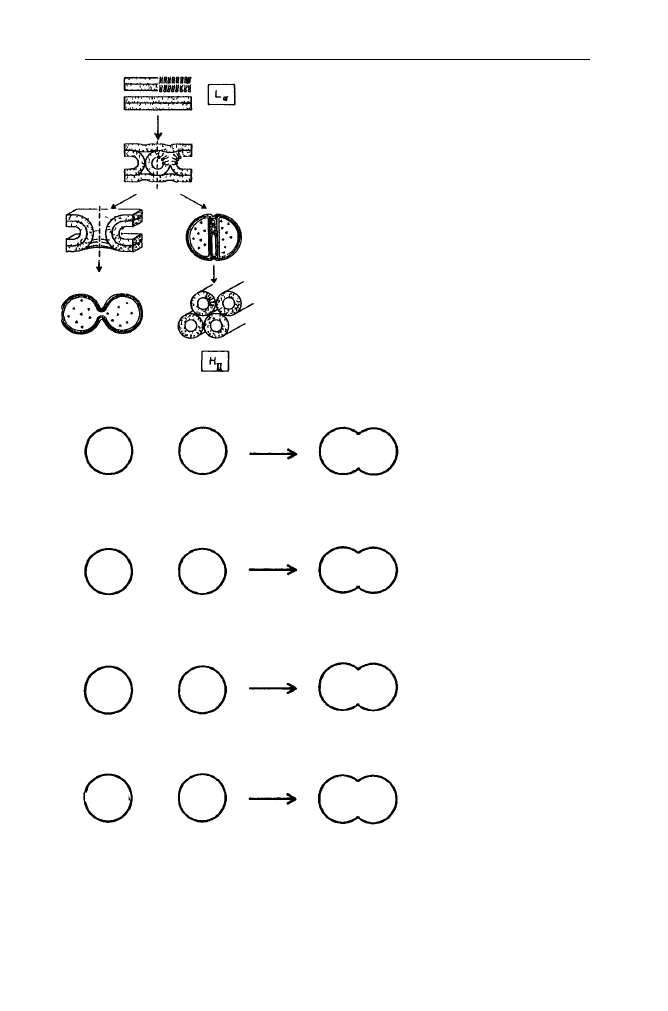
Rys. 21. Tworzenie IMI i możliwe ich przekształcanie
w ILA - prekursory fuzji lub prekursory fazy
niewarstwowej H
II
2.4. Liposomy jako modele w badaniach procesu fuzji błon biologicznych 87
Tb-terb
DPA-kwas dipikolinowy
ANTS-kwas amino-
naftalenotrójsulfonowy
DPX-bromek
p-ksylenobispirydyny
CF-karboksyfluoresoeina
NISKA FLUORESCENCJA WZROST FLUORESCENCJI
NISKA FLUORESCENCJA WYSOKA FLUORESCENCJA
FLUORESCENCJA WYGASZASZ NISKA FLUORESCENCJA
FUZJA
FUZJA
FUZJA
CF
BRAK FLUORESCENCJI WZROST FLUORESCENCJI
Rys. 22. Zasady metod fluorymetrycznych badania procesu fuzji poprzez śledzenie mieszania się
zawartości liposomów
FUZJA
Tb
DPA
Tb/DPA
ANTS
DPX
ANTS/DPX
CF
CF
EDTA
EDTA-Co
Kalceina
fuzja
IMI
ILA
Kalceina
-Co
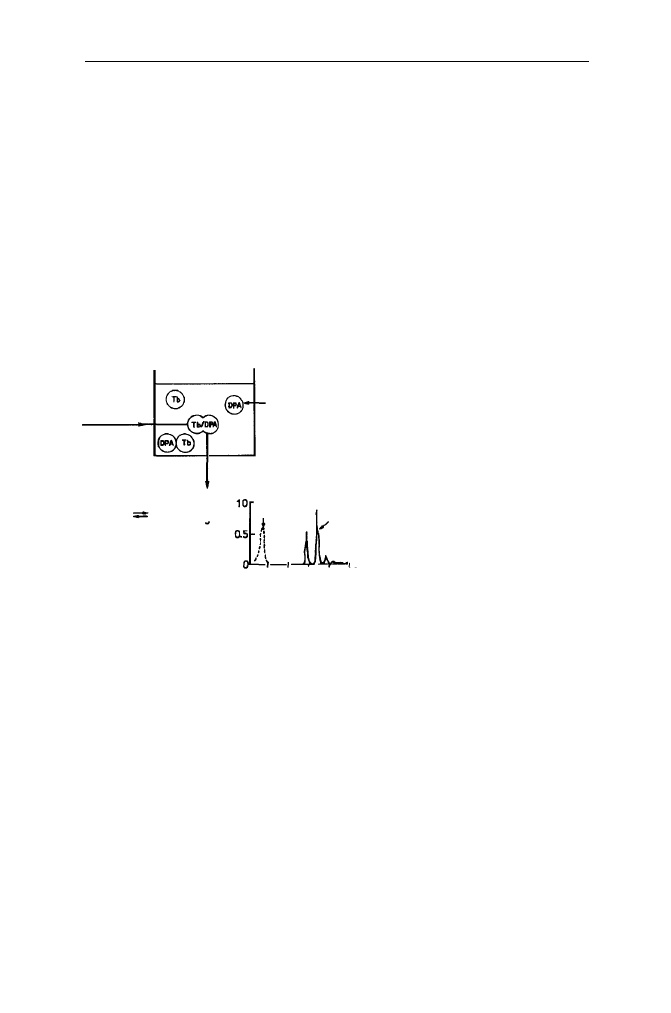
2. Błony biologiczne 88
2.4.1. Badanie fuzji liposomów
poprzez śledzenie mieszania zawartości
ich przestrzeni wodnych
(wg Wilschut J., Papahadjopoulos D., Nature 281 (1979) 690-692)
Jedną z najczęściej używanych metod badania mieszania zawartości liposomów
podczas ich fuzji jest technika Tb/DPA. Postępowanie polega na zamknięciu
w jednej populacji pęcherzyków liposomowych jonów terbu Tb
+3
, a w drugiej -
anionu kwasu dipikolinowego (DPA). W wyniku fuzji zachodzącej pomiędzy
zawierającymi Tb i DPA liposomami następuje zlanie ich zawartości i wytworze-
nie w jego wyniku fluoryzującego kompleksu Tb(DPA)
3+3
. Tworzenie tego kom-
pleksu jest bardzo szybkie, co umożliwia pomiary kinetyczne procesu fuzji.
Zasadę metody ilustruje Rys. 23.
Materiały i roztwory:
chloroformowe roztwory fosfatydyloseryny, fosfatydyloetanolaminy, cholestero-
lu (30 μmoli/ml);
roztwór 5 mM TbCl
3
, 50 mM cytrynian sodowy, 5 mM bufor Hepes pH 7.4;
roztwór 50 mM dipikolinianu sodowego (kwas dipikolinowy zobojętniony
NaOH) w 5 mM buforze Hepes pH 7.4;
100 mM NaCl - 0.5% dezoksycholan sodowy - 5 mM bufor Hepes pH 7.4;
100 mM NaCl - l mM EDTA - 5 mM bufor Hepes pH 7.4;
100 mM NaCl - 0.1 mM EDTA - 5 mM bufor Hepes pH 7.4;
20 mM CaCl
2
w 5 mM buforze Hepes pH 7.4;
żel Sephadex G-75 napęczniały w 100 mM NaCl - l mM EDTA - 5 mM bufor
Hepes pH 7.4.
Postępowanie:
Przygotowanie pęcherzyków liposomalnych
• W dwóch kolbach okrągłodennych do wyparki umieścić po 30 μmoli fosfoli-
Tb+3DPA Tb(DPA)
3-
pęcherzyk lipidowy
wzbudzanie
272 nm
emisja
492 nm
widmo
emisyjne
wzbudzenie
długość fali (nm)
300 500 700
Rys. 23. Zasada metody
Tb/DPA badania procesu fuzji
fluorescencj
a

2 4 Liposomy jako modele w badaniach procesu fuzji błon biologicznych 89
pidów lub ich mieszanin, odparować rozpuszczalnik w wyparce, a następnie
umieścić je w eksykatorze próżniowym na 12 godz
• Do jednej z kolbek dodać l ml roztworu TBC13, a do drugiej l ml roztworu
kwasu dipikolinowego Przygotować FAT-MLV, które dodatkowo należy
„wykalibrować" przez przeciskanie przez filtr membranowy 200, a następnie
l00 μm (Extruder)
• Uzyskane liposomy
oddzielić od nie zamkniętych w nich solutów przez sącze-
nie molekularne na kolumnach wypełnionych żelem Sephadex G-75 stosując
do elucji l00 mM NaCl - l mM EDTA - 5 mM bufor Hepes pH 7 4 Oczysz-
czone liposomy używać do dalszych doświadczeń Fosfor lipidowy oznaczyć
w zawiesinach liposomalnych metodą Rousera
Pomiary fuzji liposomów
Postępowanie:
• Na spektrofluorymetrze połączonym z rejestratorem ustawić długość fali
wzbudzenia 276 nm oraz długość fali emisji 540-545 nm Pomiędzy kuwetę
pomiarową a monochromator emisji można wstawić filtr odcinający fale
o długości poniżej 525 nm Szczeliny wzbudzenia i emisji ustawić na 10 nm
Kuwetę pomiarową umieścić w przystawce zapewniającej mieszanie jej za-
wartości oraz termostatowanie Pomiary prowadzić w 25
0
C
• W kuwecie umieścić 2 ml 100 mM NaCl - 0.l mM EDTA - 5 mM bufor
Hepes pH 7 4 i wstrzyknąć l00 μl mieszaniny obu rodzajów pęcherzyków
w stosunku l l Włączyć mieszanie i odczekać l min dla wyrównania tempe-
ratury Śledzić zmiany fluorescencji od chwili wstrzyknięcia liposomów
• Proces fuzji zainicjować przez wstrzyknięcie do kuwety roztworu CaCl
2
do
uzyskania stężenia końcowego 5 mM
• Zmiany fluorescencji śledzić przez kilka minut
• Wartość maksymalnej, możliwej do uzyskania dla danego układu fluorescen-
cji określić przez dodanie 100 μl mieszaniny liposomów do 2 ml 100 mM
NaCl - 0.5% dezoksycholan sodowy - 5 mM bufor Hepes pH 7 4
• Z początkowego nachylenia krzywej narastania fluorescencji obliczyć szyb-
kość początkową procesu fuzji, jako mieszania się zawartości pęcherzyków
liposomowych Stosując opisane postępowanie zbadać wpływ stężenia jonów
wapniowych (0.5-10 mM) na proces fuzji, a także modulację tego procesu
przez związki amfifilowe
Wyszukiwarka
Podobne podstrony:
czynniki biologiczne 2 id 66725 Nieznany
BiologiaVIaObUkNerw id 88384 Nieznany
Biologia 9 id 87703 Nieznany
czynniki biologiczne 4 id 66726 Nieznany
biologia 6 id 87698 Nieznany (2)
ODN BIOLOG id 331882 Nieznany
czynniki biologiczne 3 id 66726 Nieznany
biologia 1 id 87669 Nieznany (2)
czynniki biologiczne 1 id 66725 Nieznany
Biologia 8 id 87702 Nieznany
odpowiedzi biologia id 85891 Nieznany
czynniki biologiczne 6 id 66726 Nieznany
czynniki biologiczne 8 id 66726 Nieznany
czynniki biologiczne 5 id 66726 Nieznany
29 38 egzamin biologia!! id 32 Nieznany
ODNOWA BIOLOGICZNA id 331919 Nieznany
czynniki biologiczne 7 id 66726 Nieznany
Funkcja blony otrzewnowej id 18 Nieznany
Blony plodowe id 90529 Nieznany (2)
więcej podobnych podstron