
598
CHROMATOGRAPHY, AFFINITY
Vol. 1
CHROMATOGRAPHY, HPLC
Introduction
High performance liquid chromatography (hplc) is a powerful tool for character-
ization of synthetic and natural polymers by separating individual fractions by
molecular weight, chemical composition, functional groups, etc. It is also used
for isolation and purification of biopolymers and analysis of additives in complex
polymer formulations. As in any chromatographic technique, separation occurs
due to thermodynamic partitioning between the sample components in the mo-
bile and stationary phases. In hplc, this process takes place in solution inside a
chromatographic column packed with inorganic (usually, silica-based) or organic
(synthetic resin, such as polystyrene–divinyl benzene and acrylic-based) porous
particles, capable of resisting the high pressure created by a moving liquid (mo-
bile phase) mechanically pumped through the column. Depending on application,
isocratic (constant mobile phase composition) or gradient (variable composition)
modes of separation can be employed, which significantly extends the capabilities
of the technique.
Column liquid chromatography has been used for more than 60 years for an-
alytical and preparative separations of polymers. Baker–Williams Fractionation,
introduced in the mid-1950s, employed both temperature and solvent gradients to
separate polymer fractions according to solubility through multiple precipitation–
redissolution steps. This technique has been significantly improved in the last two
decades and is still used for fractionation of synthetic copolymers and polymer
blends under the term high performance precipitation liquid chromatography
(hpplc). Dramatic changes in instrumentation and column preparation for
liquid chromatography in 1970s, as well as better understanding of the mecha-
nisms of separation, had a dramatic impact on polymer separation approaches.
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.
Vol. 1
CHROMATOGRAPHY, HPLC
599
Thus, a new technique Chromatography, SEC (a.k.a. gel-permeation chromatog-
raphy for organic solutions or gel-filtration chromatography for aqueous mobile
phases) emerged at that time as a premier method for molecular weight distribu-
tion (MWD) determination of synthetic and natural polymers. Since the early
1970s, sec as well as traditional types of hplc (reversed-phase, ion-exchange,
hydrophobic-interaction, and affinity chromatography) have been widely used for
analysis of proteins and other biopolymers. For two decades, normal- and reversed-
phase chromatography have attracted significant interest as methods of separa-
tion of synthetic copolymers and polymer blends by chemical composition. Two-
dimensional hplc techniques (also called chromatographic cross-fractionation)
have been used for most complete polymer characterization in recent years. Thus,
consequently coupling two different modes of chromatographic separation can
result in determination of both molecular weight and chemical composition dis-
tributions of synthetic copolymers.
Instrumentation for HPLC
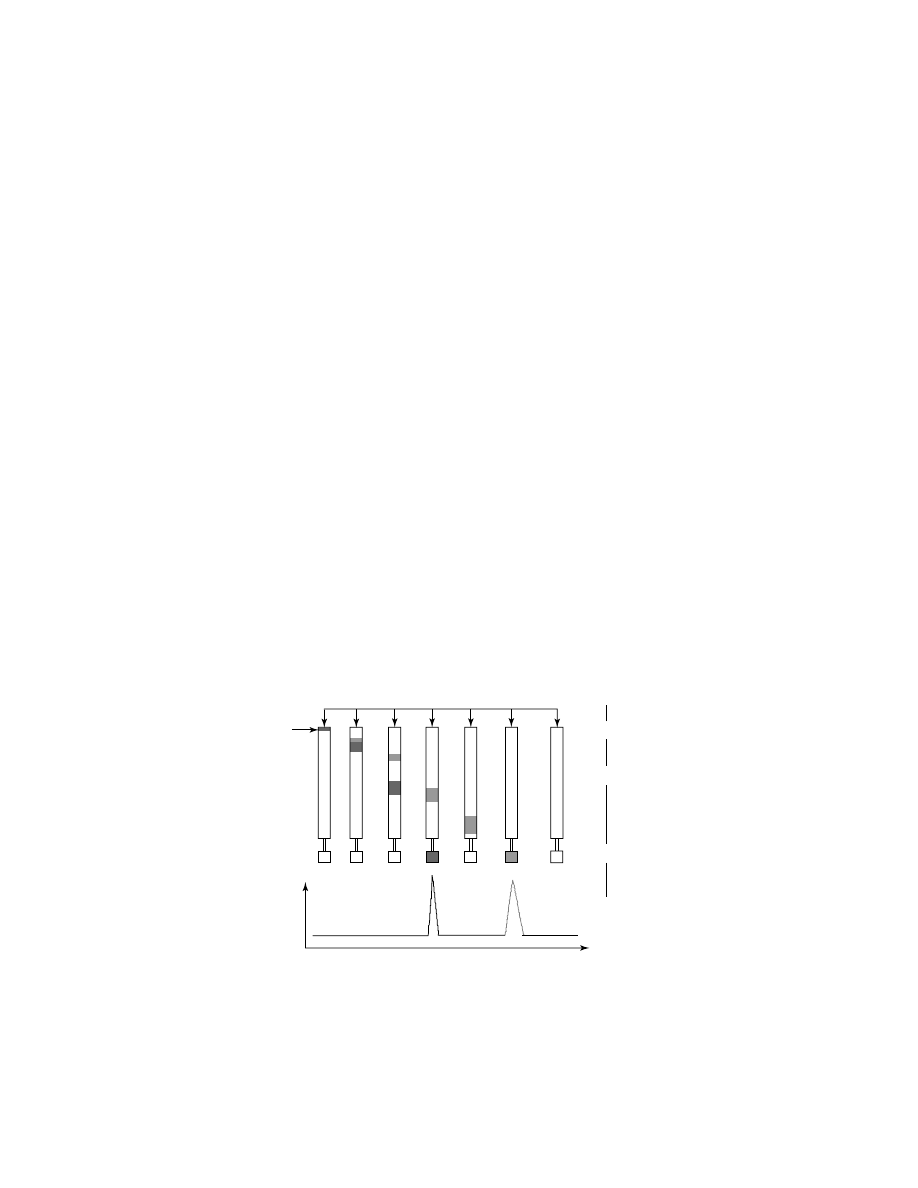
Each contemporary hplc instrument consists of a pump for solvent delivery, an
injector device for sample introduction, column(s) for sample separation, detec-
tor(s) for visualization of the separated components (solutes), and a computer for
system control and data acquisition and reduction, as depicted in Figure 1. Pre-
cise temperature control of columns and some other parts of a chromatographic
system becomes an important prerequisite for a successful separation.
Solvent Delivery.
The function of the solvent delivery system is to de-
liver the mobile phase (eluent) through the chromatograph, accurately and re-
producibly. The most popular are reciprocating-piston pumps, usually with two
Eluent
Sample
A
+ B
Detector Signal
B
A
Time (Volume)
Pump
Injector
Column
Detector
Computer
Fig. 1.
Schematic presentation of separation of two components, A and B. Reprinted from
Ref. 1 with permission.

600
CHROMATOGRAPHY, HPLC
Vol. 1
pump heads containing two sets of moving parts: the check valves and seal-piston
assembly. For analytical chromatography, such pumps can deliver liquid at a flow
rate from 100
µL/min up to 10 mL/min. Microscale capillary chromatography
needs syringe (positive displacement) pumps with capability to produce a flow rate
as low as 1
µL/min, while preparative chromatography utilizes flow rates up to
50 mL/min. Gradient pumping system is capable of delivering more than one sol-
vent during an analysis with variable mobile phase composition. The blending of
the solvents can occur in one of two ways: high pressure mixing or low pressure
mixing. In the former case the solvents are mixing on the injector side, while in
the latter case the mixing takes place at atmospheric pressure and a single high
pressure solvent delivery system is used to pump the mixture.
Accurate and reproducible eluent flow rate and composition are important
prerequisites for successful separation and detection. This is achieved in contem-
porary “intelligent” pumps through completely independent, digitally controlled
piston drives. The sophisticated, software-driven mechanism produces pulse-free
eluent delivery, compensates for changes in eluent viscosity, and automatically
purges any gaseous mobile phase, thereby enhancing performance for both iso-
cratic and gradient applications (2). The quality of solvents affects the accuracy
of flow and baseline noise. On-line solvent filtering and degassing become the
necessary elements of any commercial hplc system.
Sample Introduction.
To minimize the dispersion and broadening of
peaks, sample solution must be injected as a sharp plug with minimal interruption
of flow. Typically, two-position six-port valves are used for sample injection, which
may be operated manually or automatically (such as in an autosampler, where the
sample is introduced from a sample vial held in an injection station). While the
general intention is to keep the injection volume as small as possible, the limits
are imposed by column dimensions, the sensitivity of detectors, and the nature
of separation. An additional factor is the concentration of the analyte, which for
high molecular weight species may often be selected very low to reduce solution
viscosity and avoid intermolecular interaction.
Stationary and Mobile Phases.
Chromatographic columns provide the
means for polymer fractionation or separating mixtures into components. The se-
lectivity, capacity, and efficiency of the columns are all affected by the nature of
the packing material. Most modern hplc packings are porous spherical micropar-
ticles with 3- to 10-
µm size. The efficiency of a column increases with decreasing
particle size, but so does the backpressure, which usually cannot exceed 27.2 MPa
(4000 psi) on traditional instruments. The use of new generation of ultra-high
pressure chromatographs with backpressure up to 690 MPa (100,000 psi) can po-
tentially reduce the particle size up to 1
µm or lower. In general, silica-based
columns can withstand higher pressure as compared with resin-based columns.
Pores (mean diameter usually from 6 to 100 nm) play a crucial role in hplc, as
they provide the surface where retention and hence separation occur. The chemi-
cal structure of the surface determines the thermodynamic “environment” for the
analyte in thin surface layer (stationary phase) and in such a way determines
the mechanism of retention. Flexible macromolecules approaching the internal
surface of a solid particle change their spatial conformations because of steric in-
teraction, which plays an important role in any mode of polymer chromatography.
This interaction restricts the fluctuation motion of a macromolecule, decreases its

Vol. 1
CHROMATOGRAPHY, HPLC
601
conformation entropy, and effectively reduces the retention. In sec, where parti-
cles with inert surface are used and the size of macromolecules is comparable with
the internal pore diameter, steric interaction is dominant and separation occurs
according to size of macromolecules. All other modes of polymer chromatography
also exploit enthalpic (attraction) specific interactions between macromolecules
and the functional groups on the surface. For this reason they are often lumped
together as interaction polymer chromatography (ipc). The name “adsorption poly-
mer chromatography” is also used because the size of macromolecules usually far
exceeds the size of the surface functional groups participating in interaction, and
retention has the adsorption mechanism.
The nonsteric interactions in ipc depend on the chemical structure of the an-
alyte, and also on nature of stationary and mobile phases. In normal- or reversed-
phase hplc, neutral solutes are separated on the basis of their polarity. In the for-
mer case, polar stationary phases are employed (eg, bare silica with polar silanol
groups) and less polar mobile phases based on nonpolar hydrocarbons are used for
elution of the analytes. Solvent selectivity is controlled by adding a small amount
of a more polar solvent, such as 2-propanol or acetonitrile or other additives with
large dipole moments (methylene chloride and 1,2-dichloroethane), proton donors
(chloroform, ethyl acetate, and water), or proton acceptors (alcohols, ethers, and
amines). Correspondingly, the more polar the solute, the greater is its retention
on the column, yet increasing the polarity of the mobile phase results in decreased
solute retention.
The opposite situation takes place for reversed-phase separation, when non-
polar stationary phase is accompanied by a polar mobile phase, typically water, to
which a less polar solvent such as acetonitrile or methanol is added, and solvent
selectivity is controlled by the nature and amount of the added solvent. As a result,
a decrease in the polarity of the mobile phase leads to a decrease in solute reten-
tion. Many organic polymers are not soluble in water, and other polar solvents can
be used. Thus, the combination of acetonitrile as an initial solvent with addition of
tetrahydrofuran (THF) can be used in reversed-phase separation of many styrene-
based copolymers and acrylic polymers. Modern reversed-phase chromatography
typically refers to the use of chemically bonded stationary phases, where func-
tional groups, such as alkyl ( CH
3
, C
4
H
9
, C
8
H
17
, and C
18
H
37
) groups, phenyl
( C
6
H
5
) groups, cyano [ (CH
2
)
3
CN] groups, and amino [ (CH
2
)
3
NH
2
)] groups, are
bonded to the silica. Note that some of these bonded packings, such as those with
cyano- and amino-groups, as well as diol groups [ (CH
2
)
3
OCH
2
CH(OH)CH
2
OH],
can also be used in normal-phase separations, as they contain moderately polar
moieties. The same stationary phases are also used for separation of biomolecules
in hydrophobic-interaction chromatography. Weak nonionic interactions between
hydrophobic portions of proteins and nonpolar moieties of the stationary phase
have an entropic nature and are highly sensitive to salt concentration in a totally
aqueous, buffered mobile phase of high ionic strength.
In ion-exchange chromatography, the ionic species, such as proteins or syn-
thetic polyelectrolytes, are separated on the basis of differences in electric charge.
The stationary phases with ionic groups (fixed ions) are employed, and the mech-
anism of retention is the electrostatic attraction of ionic solutes in solution to
opposite charged ions (anions or cations) on the stationary phase support. The
stationary phase for ion-exchange chromatography is called ion exchanger and

602
CHROMATOGRAPHY, HPLC
Vol. 1
is classified as an anion-exchange material when the fixed ion carries a positive
charge, and as a cation exchanger in the opposite case. There are strong and weak
ion exchangers depending on the pH range over which the stationary phase can
retain the charge on the fixed ion. Acrylic-based copolymers treated with an ap-
propriate reagent to produce the desired functional group are the most used ion
exchanger because of wider pH range (pH 1–13) and higher ion-exchange capacity
as compared with silica-based columns (pH 2–8). Eluents for ion-exchange chro-
matography are aqueous solutions of salts with small addition of organic solvent.
Selectivity in the separation of polyelectrolytes may be varied by changing ei-
ther the pH of the mobile phase or the nature or concentration of the displacing
(competing) ions.
Chromatography, SEC is used for the separation of biomolecules on the basis
of the lock-and-key mechanism prevalent in biological systems.
Detectors.
When separated macromolecules leave the column, they are
detected by one or more on-line electrical devices with signals proportional to the
concentration of the analyte. In all hplc detectors, the eluent flows through a mea-
suring cell, where the change of a physical or chemical property with elution time
is detected. Each monitored trace represents a chromatogram: a set of chromato-
graphic peaks separated by baseline regions. The sensitivity (the ratio of peak
height to the sample concentration), signal-to-noise ratio (the amount of signal
visible above the baseline noise), as well as linear dynamic range (maximum lin-
ear response divided by the detector noise) are the most important characteristics
of any detector.
Ultraviolet (uv) absorption photometers capable of absorbing light at a se-
lected wavelength (in the range 190–350 nm for uv detectors, or 190–700 nm for
uv/Visible detectors) are the most popular detectors in hplc of both synthetic and
biological polymers. In case of proteins, wavelengths that are typically monitored
include the regions of absorbance of peptide bonds (210–220 nm), and tyrosine and
tryptophan side chains (ca 280 nm). Many synthetic polymers also contain chro-
mophoric groups. Variable-wavelength and photodiode array detectors have the
advantage that more than one wavelength can be monitored during a single run.
Photodiode array detectors allow for simultaneous measurement of an entire uv
spectrum at any point of the chromatogram, which can aid in peak identification.
Among other selective (solute property) detectors used in hplc of polymers are
fluorescence and IR photometers. For example, fluorescence detectors can provide
higher sensitivity, as well as greater selectivity, in the detection of proteins con-
taining tyrosine and tryptophan residues that have intrinsic fluorescence. Both
types of detectors are limited to certain mobile phases. A good alternative is off-
line coupling of ftir to hplc system using an evaporative interface (3). In such a
system, the eluent is sprayed onto a Germanium disc, which can be transferred
to any ftir spectrometer to yield the full spectrum information over any peak of
the chromatogram. The same principle is applied also to the matrix-assisted laser
desorption/ionization mass spectrometer, which otherwise is difficult to couple to
the hplc system (4).
A few universal (bulk property) detectors are of frequent use in hplc of
polymers. Refractive index detector is limited to isocratic elution and is utilized
primarily in sec. The same is true for the recently introduced density detector
based on mechanical oscillator principle (B. Trathnigg in Ref. 4). Evaporative light

Vol. 1
CHROMATOGRAPHY, HPLC
603
scattering detector (ELSD) becomes very popular in nonaqueous gradient elution
of synthetic copolymers and polymer blends (3). The eluent is nebulized, and the
solvent is evaporated from the droplets. Each droplet containing nonvolatile ma-
terial will form a particle, which scatters the light while crossing a light beam.
This detector has significant advantages over the uv detector for ability to work
with aliphatic polymers without chromophore groups, and also for its applicabil-
ity to practically any mobile phase which does not contain a nonvolatile buffer.
Despite these obvious merits, ELSD cannot be considered as an ultimate choice
for quantitation because of nonlinear concentration response, which depends on
numerous factors such as chemical nature of the sample including its molar mass
and chemical composition, the eluent composition, viscosity and surface tension,
and operating conditions. Coupling ELSD to another detector can help to solve
these problems with quantitation.
The quadrupole mass spectrometers, single-quadrupole (MS) or triple-
quadrupole (MS–MS), have proved to be the detectors of choice for interfacing
with chromatographs. The resulting combinations LC–MS or LC–MS–MS have
become very popular among protein scientists. Soft ionization methods, such as
electrospray ionization and matrix-assisted laser desorption, are used extensively
for biopolymers and some ionic synthetic polymers (5). The interface usually re-
quires the use of very low flow rates (below 10
µL/min) and therefore necessitates
capillary chromatographic equipment, or a sample splitter. Miniaturization of sep-
aration with laboratory-on-a-chip technology and microfluidics with nanoscale
columns can additionally improve the compatibility of column chromatography
with mass spectrometry detection.
In addition to the widespread use of mass spectrometry as a detection tech-
nique, scientists report investigations of other spectroscopic techniques such as
nuclear magnetic resonance (nmr) and inductively coupled plasma mass spectrom-
etry. These emerging technologies in a few years may have a major impact as on-
line detection methods for characterizing both synthetic and biological polymers.
Miniaturization of separation makes these detectors more feasible for on-line cou-
pling. Using internal reflectance spectroscopy to study molecular interactions at
phase surfaces has helped researchers to understand retention mechanisms and
to confirm or expand chromatographic data.
Data Treatment.
The traditional hplc method involves two types of data
analysis: qualitative (peak identification) and quantitative (determining the an-
alyte concentration). Both analyses require specific data manipulations, such as
matching retention times, peak integration and peak calibration. The use of spec-
troscopic detectors (photodiode array, ftir, and mass spectrometry) suggests addi-
tional mathematical procedures because of the necessity to manipulate with uv-,
ftir-, and MS-spectra and corresponding library matching (6). Most commercial
chromatographic software packages contain necessary functionalities and can be
applied to hplc of proteins, peptides, and DNAs. The characterization of other
natural and all synthetic polymers is a much more difficult problem, which in-
cludes calculating the distributions of different macromolecular parameters, such
as molecular mass, chemical composition, branching frequency and end group
functionality, from raw chromatograms. This involves additional calibration pro-
cedures (column calibration), calculation of statistical moments of distributions,
etc. Two-dimensional chromatographic cross-fractionation of synthetic copolymers
604
CHROMATOGRAPHY, HPLC
Vol. 1
requires the construction of two-dimensional distributions to characterize the
copolymer heterogeneity (7).
Retention Mechanism and Modes of Separation
Isocratic Elution.
Contrary to sec, where any nonsteric effects are unde-
sirable and suppressed, week nonsteric adsorption interactions between a solute
and a stationary phase play a dominant role in ipc. The best way to demonstrate
the difference between size-exclusion and interaction (adsorption) modes of poly-
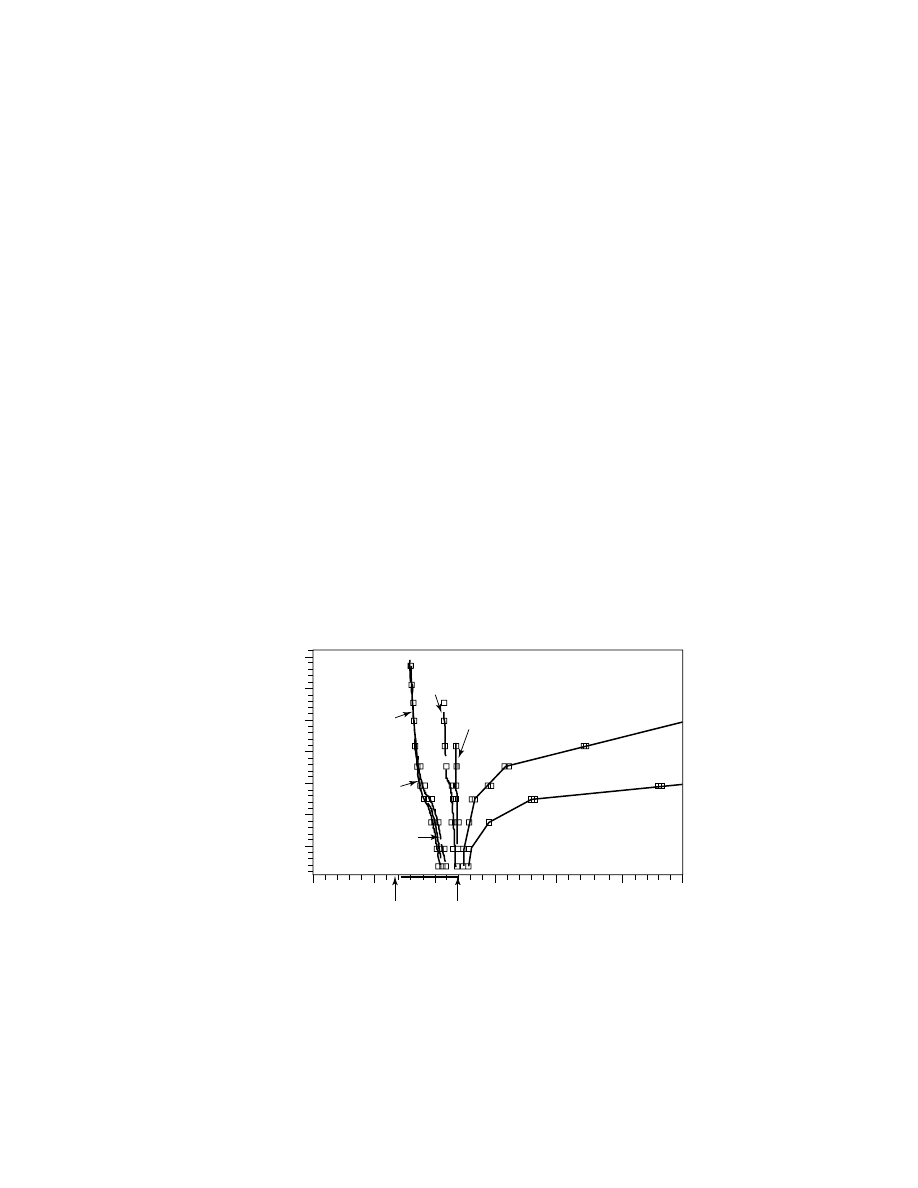
mer separation is to use the so-called molecular weight calibration curves (log M
vs V, where M is the molecular weight, and V is the elution volume) depicted in
Figure 2. The set of narrow polydispersity polystyrene samples with molecular
weights from several hundred up to million were subjected to isocratic elution on
the reversed-phase column (silica bonded with octadecyl chains) with different
acetonitrile/tetrahydrofuran (ACN/THF) mixtures as eluents. THF is less polar
than ACN (polarity parameters 4.0 and 5.8, respectively). Pure THF completely
suppresses nonpolar interactions between polystyrene and alkyl chains on silica,
and conventional sec behavior is obtained, that is, elution time increases with de-
creasing molar mass of the sample. The separation occurs within pore volume V
P
= V
T
− V
0
, where V
T
is the liquid volume inside the column and V
0
is the inter-
stitial volume between particles. The steric interactions prevail until the amount
of ACN in the mobile phase reaches ca. 50%. After that, a completely reversed
retention behavior is obtained, which is typical for the adsorption mode of sepa-
ration, when retention volume V
R
exceeds V
T
. At 45 and lower % THF, retention
6.0
5.5
5.0
4.5
4.0
3.5
3.0
0.0
1.0
2.0
3.0
4.0
5.0
6.0
48
50
45
40
100
80
70
V
0
V
T
V, mL
log
M
Fig. 2.
Transition from size-exclusion to adsorption mode for isocratic elution of
polystyrene samples with low polydispersity. Column: Nova-Pak C
18
(Waters Corporation,
USA), mobile phase: THF/ACN mixtures (numbers on the graph represent vol% THF), 1
mL/min, detector: ELSD Model 500 (Alltech Corp.).

Vol. 1
CHROMATOGRAPHY, HPLC
605
dramatically increases with molecular weight and becomes extremely sensitive
to the eluent composition, so that high molecular weight fractions are practically
irreversibly retained by the column. For this reason, the IPC separation of high
molecular weight polymers is usually performed in gradient mode.
It is important to note the elution at 48% THF: Retention becomes com-
pletely independent of molecular mass, and all fractions elute with the column’s
liquid volume V
T
. At this transition point, which is termed critical point of adsorp-
tion (CPA), entropy driven size-exclusion effects are completely compensated by
enthalpic adsorption interactions. Although both types of interaction strongly de-
pend on molecular weight, this mutual compensation phenomenon leads to molec-
ular weight independent retention at CPA. This phenomenon allows for practical
implementation of isocratic polymer separation other than sec. Thus, because of
chromatographic “invisibility” of a main polymer chain, effective separation of
telechelic polymers based on types of end-groups can be performed (Ref. 7, p. 125).
The same principle is applied for isocratic separation of some block copolymers
(Ref. 7, p. 151).
The isocratic separation at CPA requires very careful control of chromato-
graphic conditions, such as the quality of stationary phase, mobile phase compo-
sition, and temperature, because of high sensitivity of retention to these param-
eters, especially for polymers with high molecular weight. Another limitation of
the techniques is associated with the dynamic effects. The success of separation
depends on the ability of all macromolecules to penetrate all pores (the same way
as the solvent molecules). But small diffusion coefficients and steric restrictions
make the equilibration time for large macromolecules and narrow pores unrea-
sonably long. Because of this, the use of isocratic separation at CPA is practica-
ble only when the internal pore diameter exceeds the size of macromolecules in
solution.
Gradient Elution.
In gradient mode, the elution strength of the mobile
phase is gradually increased within the chromatographic run to facilitate the elu-
tion of strongly retained compounds. This is especially important for high molec-
ular weight polymers that otherwise are practically irreversibly retained by the
column in the adsorption mode. Such gradient can be performed by changing the
chemical composition, ionic strength or pH of the eluent, or even temperature.
In protein separation, the salt gradient is used in both hydrophobic-interaction
and ion-exchange chromatographies. During the chromatographic run, the salt
concentration is decreased in the former case and increased in the latter one.
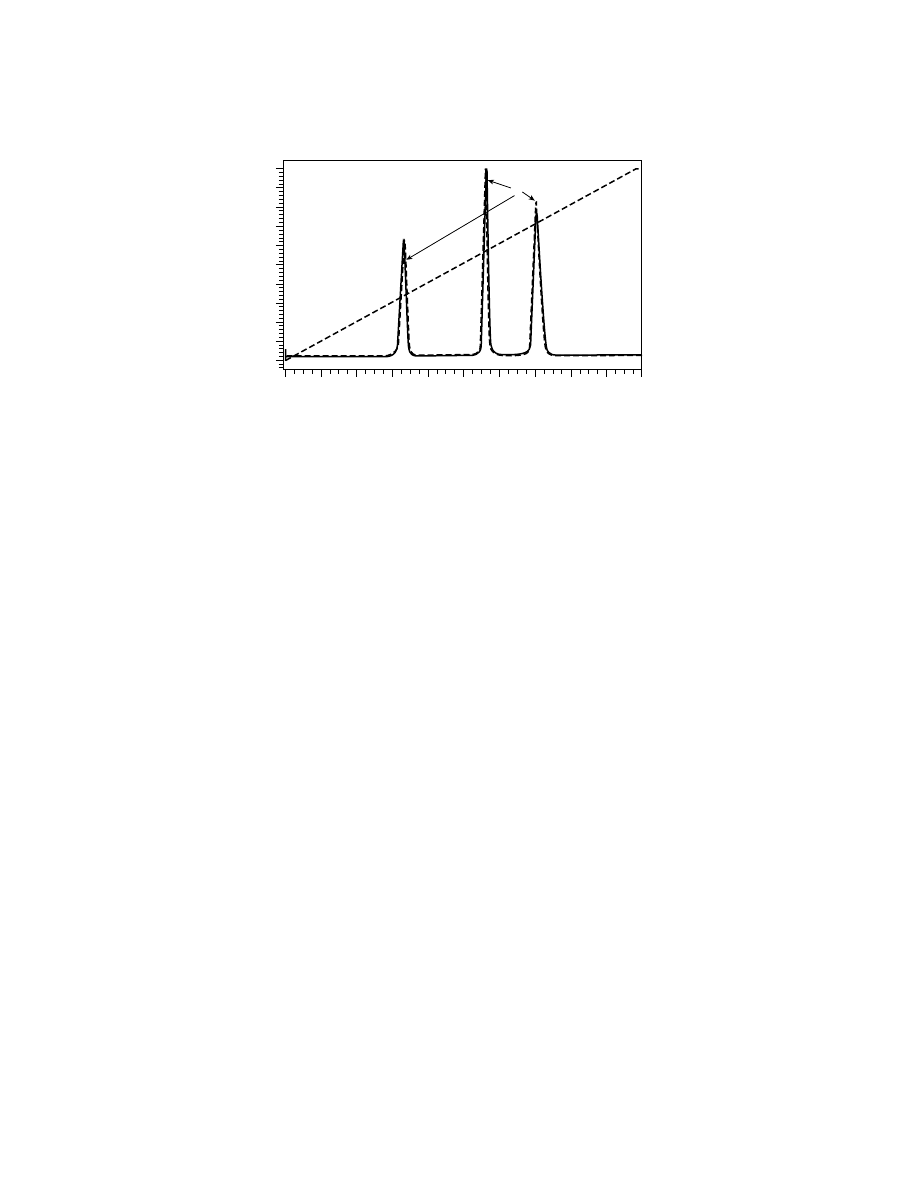
Typical reversed-phase gradient elution of synthetic polymers is demon-
strated in Figure 3 for the same set of polystyrene samples as in Figure 2. The in-
crease in eluent strength is achieved by linear increase of the concentration of less
polar THF in the mobile phase that allows the elution of all polystyrenes within
8 min. The elution of lower molecular weight samples (below 10,000 Da) occurs
in the adsorption mode at low content of THF in the mobile phase, and strongly
depends on molecular weight. Excellent resolution for polystyrene oligomers in
this mode allows one to distinguish between fractions with different end groups
(double peaks on the chromatograms). In contrast, all higher molecular weight
polystyrenes have practically the same elution volume, which coincides with the
volume where eluent reaches its CPA (48% THF). The principle that gradient
elution of high molecular weight polymers occurs at CPA has been introduced
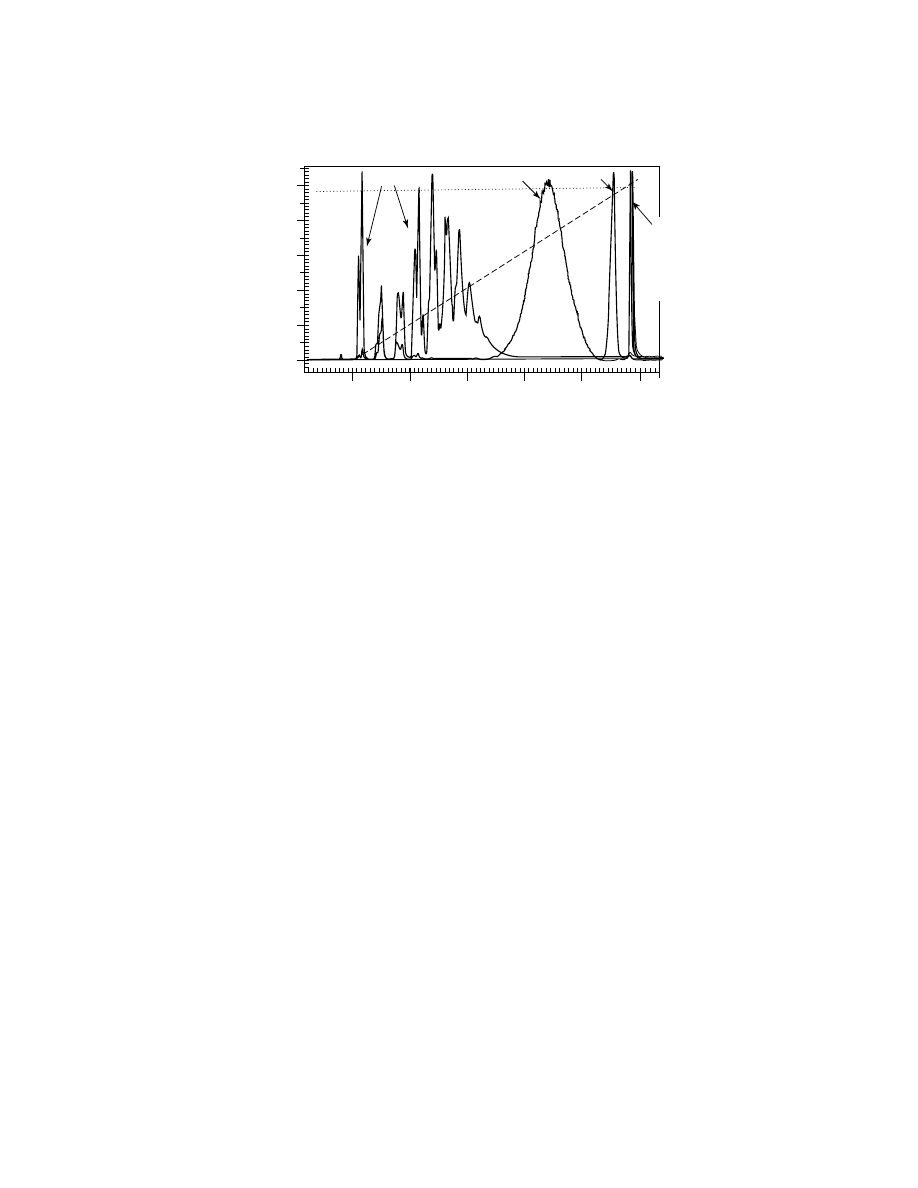
606
CHROMATOGRAPHY, HPLC
Vol. 1
50
40
30
20
10
0
3
4
5
6
7
8
Styrene Oligomers
2,890
43,900
190,000
355,000
706,000
1,090,000
V, mL
THF
, %
9,100
Fig. 3.
Gradient elution of polystyrene samples from Figure 1. The same chromatographic
system and conditions as in Figure 1, except eluent gradient: 100% ACN to 100% THF over
10 min (dashed line). Numbers on the graph: molecular weights of individual polystyrenes.
Broken line: critical point of adsorption (48% THF).
recently (8). It provides the basis for gradient separation of copolymers and poly-
mer blends by chemical composition. Each compositionally homogeneous fraction
of a copolymer or a polymer blend elutes at its own CPA independently of molec-
ular weight. By this means, gradient elution produces the chemical composition
distribution of copolymers and polymer blends.
In general, gradient elution is a much more versatile technique for polymer
separation as compared with isocratic elution at CPA Pore and particle sizes and
hence dynamic effects do not impose such significant restrictions. Macromolecules
do not need to penetrate the entire pore volume, and elution at CPA is achieved
when their size exceeds the pore diameter. Gradient run allows for a sample pre-
cipitation on the frit or inlet of the column, when starting eluent is a nonsolvent
for the polymer. This is a rather common situation because the solubility win-
dow for many polymers is relatively narrow, and the eluent with a poor elution
strength often has a poor thermodynamic quality. This is true even for some pro-
teins (membrane proteins or collagen), which can precipitate at the intermediate
stage of the gradient separation. However, the elution of different components
of a polymer sample still follows the rules of gradient separation described in
the previous paragraph as soon as all polymer fractions are redissolved and ad-
sorbed by the column during the chromatographic run. Only if macromolecules
elute without any adsorption interactions immediately after redissolution, as in
hpplc, the separation based solely on polymer solubility occurs. In this case, re-
tention always strongly depends on molecular weight and concentration of the
sample (9).
Multidimensional Separation.
In some cases, a single separation step
is not enough to complete the analysis of biological or synthetic polymers.
In this case, coupling two or more different modes of separation involv-
ing column-switching mechanism is widely used in multidimensional polymer

Vol. 1
CHROMATOGRAPHY, HPLC
607
chromatography. This approach provides the opportunity to simplify complex mix-
tures before their characterization by spectroscopic techniques, especially LC–MS
and LC–MS–MS. A typical example is the combination of ion-exchange or size-
exclusion separation with reversed-phase chromatography in protein purification
(10). Gradient or isocratic elution at CPA followed or preceded by size-exclusion
separation is often used in chromatographic cross-fractionation of synthetic poly-
mers (11). The use of ipc as a first step is a more feasible way to achieve the
true two-dimensional characterization of copolymers or polymers with functional
groups, because it allows for molecular weight independent separation by chemical
composition or by other structural differences in macromolecules. Size of macro-
molecules in each compositionally homogeneous fraction obtained after the first
separation, is directly related to molecular weight thereof, and the following size-
exclusion separation of all these fractions can complete the cross-fractionation.
The coupling of two modes of polymer separation can be achieved off-line
or on-line. In the former case fractions from one chromatographic system are
transferred to another system manually, in the second one, automatically, when
the eluent from the first instrument flows through the injection valve of the second
instrument. The most sophisticated automatic two-dimensional chromatographic
systems employ two six-port valves or one eight- or even ten-port injection valve
(10). Two storage loops make it possible to collect fractions continuously without
losses. The proper coordination of the flow rates in consecutive chromatographic
steps is an important feature of any automated system. Thus, in chromatographic
cross-fractionation, the collection time of one hplc fraction should coincide with
the analysis time in the following sec step (Ref. 7, p. 198).
Theory of Polymer Chromatography
Thermodynamic Treatment.
Each component of a polymer sample in-
jected in the chromatographic system can be characterized by its retention vol-
ume V
R
, ie, the volume of solvent that passes through the column from the time
of sample injection to the detection of the corresponding peak. To eliminate the
variability of retention caused by operational variance of the flow rate, mobile
phase, or other parameters, the normalized quantity k
= (V
R
− V
T
)/V
T
(capacity
factor) is often used instead of V
R
. The resolution of the chromatographic system,
ie, its ability to discriminate between individual macromolecules, depends on the
selectivity of separation, which is described by the ratio of corresponding capacity
factors.
The basic assumption in any chromatographic theory is that retention is
determined by the thermodynamic factors. In such a way, mobile and station-
ary phases are interpreted as true thermodynamic phases with volumes V
M
and
V
S
, respectively, so that retention volume depends on the partition (distribution)
equilibrium coefficient K of the solute in these two phases: V
R
= V
M
+ KV
S
. By def-
inition, all enthalpic and entropic interactions between the macromolecules and
the chromatographic surface occur in the stationary phase. If the size of macro-
molecules in solution is comparable with the internal diameter of pores, the entire
pore volume represents the stationary phase, V
S
= V
P
, yet the mobile phase is
formed by the interstitial volume only, V
M
= V
0
. This is not always the case for

608
CHROMATOGRAPHY, HPLC
Vol. 1
gradient separation of peptide or proteins, when the pore diameter usually far
exceeds the size of compact macromolecules, and practically the entire liquid vol-
ume, V
T
= V
0
+ V
P
, represents the mobile phase.
The role of thermodynamic theory is to relate the distribution coefficient K
(or free energy change due to solute sorption on the stationary phase) to the pa-
rameters describing the configurational and conformational structure of a macro-
molecule (such as molecular weight, chemical composition and shape), as well as
temperature, pore geometry, mobile phase composition, etc. These parameters de-
termine the entropy of steric interaction and the energy of multipoint nonsteric
interactions between the macromolecule and the surface, and in this way affect
the retention.
Construction of a universal thermodynamic model for chromatographic sep-
aration of proteins and other biological polymers is hardly probable because of
the complexity of chemical and spatial structure of such macromolecules, which
can undergo a diversity of conformational transitions during separation. Even for
small peptides composed of 5–15 amino acids, which are assumed to exist in solu-
tion as a random coil, amino acid identity and position, as well as peptide sequence,
all play an extremely important role in retention. Nevertheless, semiquantitative
approaches applied to specific modes of separation often allow for understanding
the mechanism of separation, as well as the effect of operating parameters such
as salt type and concentration, temperature, nature of packing material, organic
modifiers, and pH. Examples are the solvophobic (12) and preferential interaction
(13) theories applied to hydrophobic interaction and reversed-phase chromatogra-
phy of proteins (14). Several useful retention models exist in ion-chromatography,
where the equilibria, which govern ion-exchange separations, are generally well
understood (1).
In the case of synthetic polymers, a simple continuous random-flight model
of a flexible polymer chain allows for quantitative consideration of the chromato-
graphic process in a rather general way (15). The theory provides a simple ana-
lytical expression for the distribution constant K of a macromolecule penetrating
the slit-like or cylindrical pore and interacting with its walls, in terms of macro-
molecule and pore sizes, and a segment energy of nonsteric interactions between
the macromolecule and stationary phase. Depending on the mobile phase com-
position, the competition between steric (repulsive) and nonsteric (attractive) in-
teractions may produce either size-exclusion, K
<1, or adsorption, K>1, modes of
separations, yet the intermediate condition, K
=1, represents the critical mode of
adsorption, where both types of interaction counterbalance each other (see Fig. 2).
Present models of gradient elution (16) assume that migration in gradient
mode occurs in the same way as for isocratic elution. That is, given the dependence
of K on eluent composition, which changes during the mobile phase gradient, one
can calculate the gradient elution volume for gradients with various slopes and
shapes using the well-known mass-balance integral equation (17). The results
of such calculations applied to gradient separation of polymer homologous se-
ries can explain (8) the peculiarities depicted in Figure 3. Thus, the combination
of molecular–statistical theory of dilute polymer solutions in confined (porous)
media with conventional chromatographic theory produces a valuable tool in
elucidating the polymer chromatography (8). Recent extension (18) of the concept

Vol. 1
CHROMATOGRAPHY, HPLC
609
on copolymers and stationary phases with heterogeneous surface (which actu-
ally is inherent in most if not all of available sorbents) significantly improves the
feasibility of the theory for separation of complex polymer formulations.
Dynamic effects.
Besides the thermodynamically driven selectivity, the
resolution of the chromatographic system depends also on dynamic (kinetic) fac-
tors that determine the efficiency of separation. These factors cause the broaden-
ing of the chromatographic peaks so that even peaks from individual compounds
such as proteins have a finite width. For polydisperse polymers this chromato-
graphic peak broadening (or bandspreading) is usually hidden by the overall poly-
mer distribution, but still can produce erroneous results if no correction has been
made to compensate for this effect.
The major contributor to peak broadening in polymer chromatography is a
“stagnant” mobile phase mass transfer, that is, kinetics hindrance due to pene-
tration of macromolecules into the pores of the support packings. The molecular
diffusion is the only way for macromolecules to reach the internal surface of pores,
whereas the migration in interstitial volume is governed by the convection mecha-
nism. The effect of bandspreading tends to diminish with the ratio of characteristic
times of these two processes, t
D
= L
2
/D (L is the average length of pores, D is a
solute diffusion coefficient), and t
0
= V
0
/F (F is the flow rate). The diffusion coeffi-
cient of macromolecules inside pores drops rapidly as molecular weight increases,
and so does peak broadening. As the length of an individual pore is usually com-
parable with the size of a particle [exception is perfusive particles, which contain
very broad flow-through pores with only shallow diffusive pores and are mostly
used in preparative chromatography 19], the most popular solution to date for
minimizing the problem in biological applications is the use of small porous par-
ticles of 2–5
µm so that the depth of the stagnant pools is not excessive (20). It is
significant that in gradient separation of synthetic polymers the internal surface
area is not so important, and separation usually takes place only at the inlet of
very narrow pores, which significantly reduces the dynamic effects.
Applications
Separation of Biological Polymers.
The development of wide-pore
bonded-phase packings has dramatically affected the separation of biopolymers
in the life sciences and biotechnology (21). Nevertheless, several factors should
be taken into account in selecting the optimum mechanism of separation for a
specific application. Reversed-phase chromatography is the most popular mode
for separation of peptides because of exceptional selectivity and resolution. But
in case of proteins, hydrophobic interaction is often the method of choice because
of the much milder effect on solute conformation. Many proteins are denatured
by acids or organic solvents, as well as by the alkyl chain of the bonded phase,
the usual constituents of the reversed-phase separation (22). These conforma-
tional transitions may cause additional chromatographic problems, such as peak
broadening, multiple peaks, poor sample recovery, and ghosting, if the conforma-
tional transition time is of the same order as the migration time (23). Hydrophobic
interaction chromatography as well as ion-exchange chromatography have been
610
CHROMATOGRAPHY, HPLC
Vol. 1
predominantly used to analyze proteins, nucleic acids, and other biological macro-
molecules when maintenance of the three-dimensional structure is a primary
concern (24).
With the human genome now sequenced (primarily by capillary electrophore-
sis technology), scientists are turning their attention to proteomics. The term pro-
teome has been introduced to define the full complement of proteins in a cell. It also
requires a description of the localization, concentration, and multisubunit asso-
ciation of each of these proteins (25). The multidimensional hplc with automated
column switching technology can successfully complement the more conventional
approaches to protein separation, such as two-dimensional gel electrophoresis,
which usually involve many manual steps and are not conducive to high through-
put applications.
Separation of Synthetic Polymers.
Interest in interaction polymer
chromatography as a technique complementary to sec for characterization of syn-
thetic polymers and polymer blends has quickened in recent years. The princi-
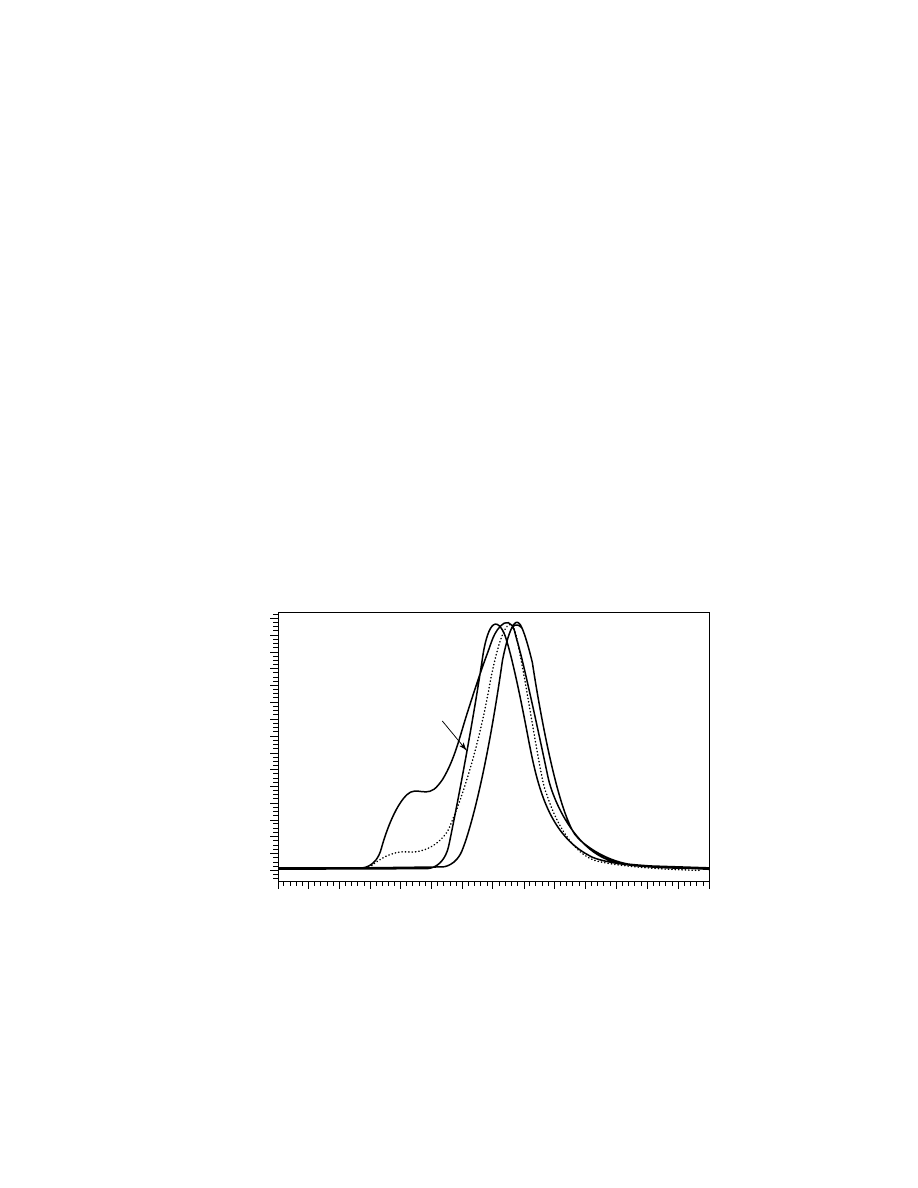
ple difference between these two methods is depicted in Figures 4 and 5, where
three polymers with close average molecular weights, as well as the equimolar
blend of these polymers were subjected to isocratic size-exclusion and gradient
reversed-phase separations, respectively. In the first case (Fig. 4), only molecu-
lar weight information is available from the chromatograms, the result of sec-
ond separation (Fig. 5) reveals the individual components, ie, chemical compo-
sition of the blend. This way, many statistical, block and graft copolymers and
blends were fractionated by chemical compositions in gradient mode on both
15
20
25
V, mL
Refr
activ
e Inde
x Response
3
2
4
1
Fig. 4.
Size-exclusion chromatograms of polystyrene (1), styrene–butadiene statistical
copolymer (2), styrene–acrylonitrile statistical copolymer (3), and equimolar mixture of
them (4, dotted line). Columns: 3 HR Styragel (Waters Corp.), mobile phase: THF, 1 mL/min,
detector: refractometer model 2410 (Waters Corp.).
Vol. 1
CHROMATOGRAPHY, HPLC
611
60
100
80
40
20
0
20
10
0
V, mL
THF
, %
3
1
4
2
Fig. 5.
Gradient reversed-phase elution of the same polymers as in Figure 3. Column:
SymmetryShield C
8
(Waters Corp.), mobile phase: 100% ACN to 100% THF over 20 min
(dashed line), 1 mL/min, detector: ELSD Model 500 (Alltech Corp.).
normal- and reversed-phase columns, without any practical limitations on the
molecular weight of polymers [see reviews in Refs. 7, 11 (chapt. 9), and 26–29].
On the other hand, several reported (7) isocratic separations of block copolymers
at CPA were limited by the oligomeric range of molecular weights. Many recent
publications deal with two-dimensional characterization of copolymers, where
the molecular weight independent separation is achieved by ipc as the first step
(7,30,31).
To summarize briefly, in spite of solid age, hplc of polymers remains the
workhorse of the biotechnology and chemical industries, and at the same time
presents a fruitful field for future development and improvement.
BIBLIOGRAPHY
“Chromatography” in EPSE 1st ed., Vol. 3, pp. 731–745, by P. M. Kamath and L. Wild, U.S.
Industrial Chemicals Co.; “Chromatography” in EPSE 2nd ed., Vol. 3, pp. 491–531, by J. F.
Johnson, The University of Connecticut at Storrs.
1. P. R. Haddad and P. E. Jackson, Ion Chromatography: Principles and Applica-
tions, Elsevier, New York, 1990, Chapt. “5”. Journal of Chromatography Library,
Vol. 46.
2. J. B. Li, J. Morawski, Liq. Chromatogr./Gas Chromatogr. 16, 468 (1998).
3. P. C. Cheung, S. T. Balke, and T. C. Schunk, in T. Provder, H. G. Barth, and M. W.
Urban, eds., Chromatographic Characterization of Polymers: Hyphenated and Multi-
dimensional Techniques, American Chemical Society, Washington, D.C., 1995.
4. J. L. Dwyer, in T. Provder, ed., Chromatography of Polymers: Hyphenated and Mul-
tidimensional Techniques, American Chemical Society, Washington, D.C., 1999. ACS
Symposium Series 731.
5. S. A. Carr and co-workers, Anal. Chem. 63, 2802 (1991).

612
CHROMATOGRAPHY, HPLC
Vol. 1
6. A. Weston and P. R. Brown, hplc and CE: Principles and Practice, Academic Press, San
Diego, Calif., 1997, p. 220.
7. H. Pasch and B. Trathnigg, hplc of Polymers, Springer, Berlin, 1998, Chapt. “7”.
8. Y. Brun, J. Liq. Chromatogr. Relat. Technol. 22, 3067–3090 (1999).
9. D. W. Armstrong and R. E. Boehm, J. Chromatogr. Sci. 22, 378 (1984).
10. H. J. Cortes and L. D. Rothman, in H. J. Cortes, ed., Multidimensional Chromatography:
Techniques and Applications, Marcel Dekker, Inc., New York, 1990. Chromatographic
Science Series 50.
11. G. Gl¨ockner, Gradient hplc of Copolymers and Chromatographic Cross-Fractionation,
Springer, Berlin, 1991, p. 148.
12. C. F. Poole and S. K. Poole, Chromatography Today, Elsevier, New York, 1991.
13. S. L. Wu, K. Benedek, and B. L. Karger, J. Chromatogr. 359, 3 (1986).
14. M. I. Aguilar and M. T. W. Hearn, in M. T. W. Hearn, ed., hplc of Proteins, Peptides and
Polynucleotides, VCH Publishers, New York, 1991.
15. A. A. Gorbunov and A. M. Skvortsov, Adv. Colloid Interface Sci. 62, 31–108
(1995).
16. J. Jandera and J. Churacek, Gradient Elution in Column Liquid Chromatography—
Theory and Practice, Elsevier, Amsterdam, 1985.
17. L. R. Snyder and M. A. Stadalius, in C. Horvath, ed., High-Performance Liquid Chro-
matography: Advances and Perspective, Vol. 4, Academic Press, Inc., Orlando, Fla.,
1986, p. 195.
18. Y. Brun, J. Liq. Chromatogr. Relat. Technol. 22, 3027–3065 (1999).
19. N. B. Afeyan and co-workers, J. Chromatogr. 519, 1 (1990).
20. R. L. Cunico, K. M. Gooding, and T. Wehr, Basic hplc and CE of Biomolecules, Bay
Bioanalytical Laboratory, Richmond, Calif., 1998, p. 23.
21. K. M. Gooding and F. E. Regnier, hplc of Biological Macromolecules: Methods and
Applications, Marcel Dekker, Inc., New York, 1990.
22. F. E. Regnier, Science 238, 319 (1987).
23. A. Sadana, Bioseparation of Proteins: Unfolding/Folding and Validations, Academic
Press, Inc., San Diego, Calif., 1998, Chapt. “5”.
24. M. T. W. Hearn, in S. Ahuja, ed., Handbook of Bioseparations, Vol. 2, Academic Press,
Inc., San Diego, 2000.
25. M. R. Wilkins, K. I. Williams, R. D. Appel, and D. F. Hochstrasser, eds., Pro-
teome Research: New Frontier in Functional Genomics, Springer-Verlag, New York,
1997.
26. S. Mori, in Ref. 3.
27. T. C. Chunk, J. Chromatogr. A 656, 591–615 (1993).
28. S. Teramachi, in D. Berek, ed., Chromatography of Polymers (special issue), Macromol.
Symp. 110, 217 (1996).
29. H. Sato, K. Ogino, T. Darwint, and I. Kiyokawa, in Ref. 28.
30. P. Kilz, R.-P. Kruger, H. Much, and G. Schulz, in Ref. 3.
31. B. Trathnigg, M. Kollroser, M. Parth, and S. Roblreiter, in T. Provder, ed., Chromatog-
raphy of Polymers: Hyphenated and Multidimensional Techniques, American Chemical
Society, Washington, D.C., 1999.
GENERAL REFERENCES
Refs. 6 and 19 are recommended as introductory material to hplc of biopolymers.
Refs. 14 and 20 deal critically with all aspects of hplc of biological molecules.

Vol. 1
CHROMATOGRAPHY, SIZE EXCLUSION
613
Refs. 23 and M. Kastner, ed., Protein Liquid Chromatography, Elsevier, Amsterdam, 2000,
J. Chrom. Library, Vol 61, reflect the current state of the art in protein chromatography.
U. D, Neue, hplc Columns: Theory, Technology, and Practice, Wiley-VCH, Inc., New York,
1997, in-depth review of column technology.
Refs. 7 and 11 reflect the current state of hplc of synthetic polymers.
Refs. 1, 8, and 17 deal with theoretical aspects of polymer chromatography.
Y
EFIM
B
RUN
Waters Corporation
Wyszukiwarka
Podobne podstrony:
Cz 11 Instrumentalne metody analizy ilościowej Wysokosprawna chromatografia cieczowa (HPLC)
chromanie przestankowe 2
192Preparatywna i procesowa chromatografia cieczowa
6Hydrophobic Interaction Chromatography
Chromatografia id 116057 Nieznany
chromatografia jonowymienna 2, Rok I, chemia fizyczna, chemia fizyczna-protokoły
CHROMATOGRAFIA CIECZOWA, I MU, Zaawansowana analiza
Chromatografia, Technologia chemiczna, Analiza instrumentalna
SPEKTOMETRIA MASS W POŁĄCZENIU Z CHROMATOGRAFIĄ GAZOWĄ
Oczyszczanie ludzkiego białka P2 na drodze chromatografii powinowactwa
chromanie przestankowe
Chromatografia gazowa
Machowski chromatografia flawonoidów i saponin
cw 6 chromatografia
chroma prezentacja
Chromatografia Powinowactwa
4Covalent Chromatography
CHROMATOGRAFIA PODSTAWY MK
więcej podobnych podstron