
FILOGENETYKA
Bioinformatyka, wyk
Bioinformatyka, wyk
ł
ł
ad 7
ad 7
(2
(2
4
4
.XI.200
.XI.200
8
8
)
)
krzysztof_pawlowski@sggw.pl
krzysztof_pawlowski@sggw.pl

Filogenetyka
Cel
–
rekonstrukcja historii ewolucji wszystkich
organizmów.
Klasyczne podejście:
historia ewolucji jest odtwarzana na podstawie
porównań
cech morfologicznych i fizjologicznych
badanych organizmów.

•
zadaniem
filogenetyki molekularnej
jest
zrekonstruowanie związków filogenetycznych
między badanymi sekwencjami
sekwencje przodka mutuj
sekwencje przodka mutuj
ą
ą
w sekwencje potomk
w sekwencje potomk
ó
ó
w
w
podobne gatunki s
podobne gatunki s
ą
ą
genetycznie blisko spokrewnione
genetycznie blisko spokrewnione
•
•
podstawowe za
podstawowe za
ł
ł
o
o
ż
ż
enia w filogenetyce molekularnej:
enia w filogenetyce molekularnej:
•
wyrazem analiz filogenetycznych są
drzewa filogenetyczne
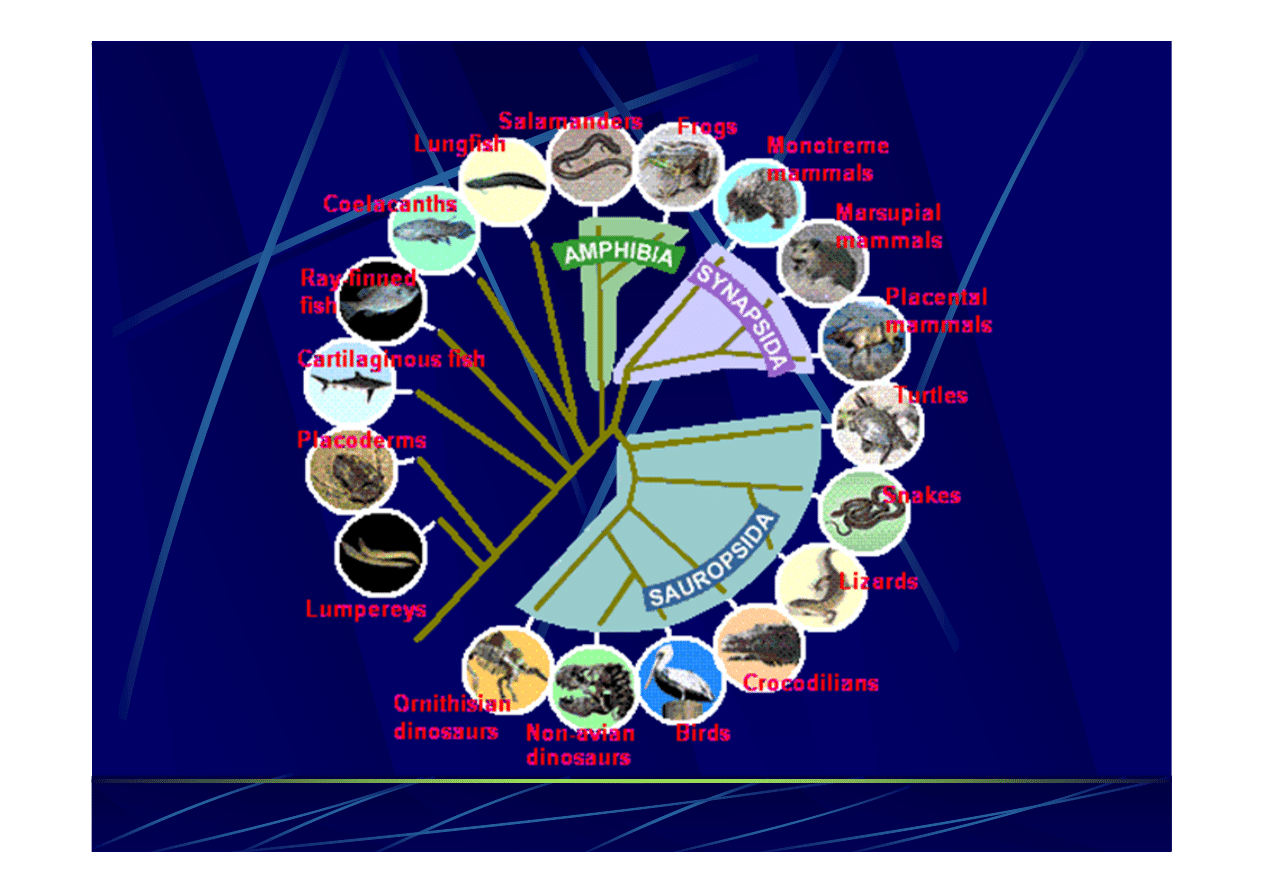

Tree
of
life
(Darwin)

eukarionty
archea
bakterie
Tree
of
life
(dziś)
kręgowce
grzyby
rośliny
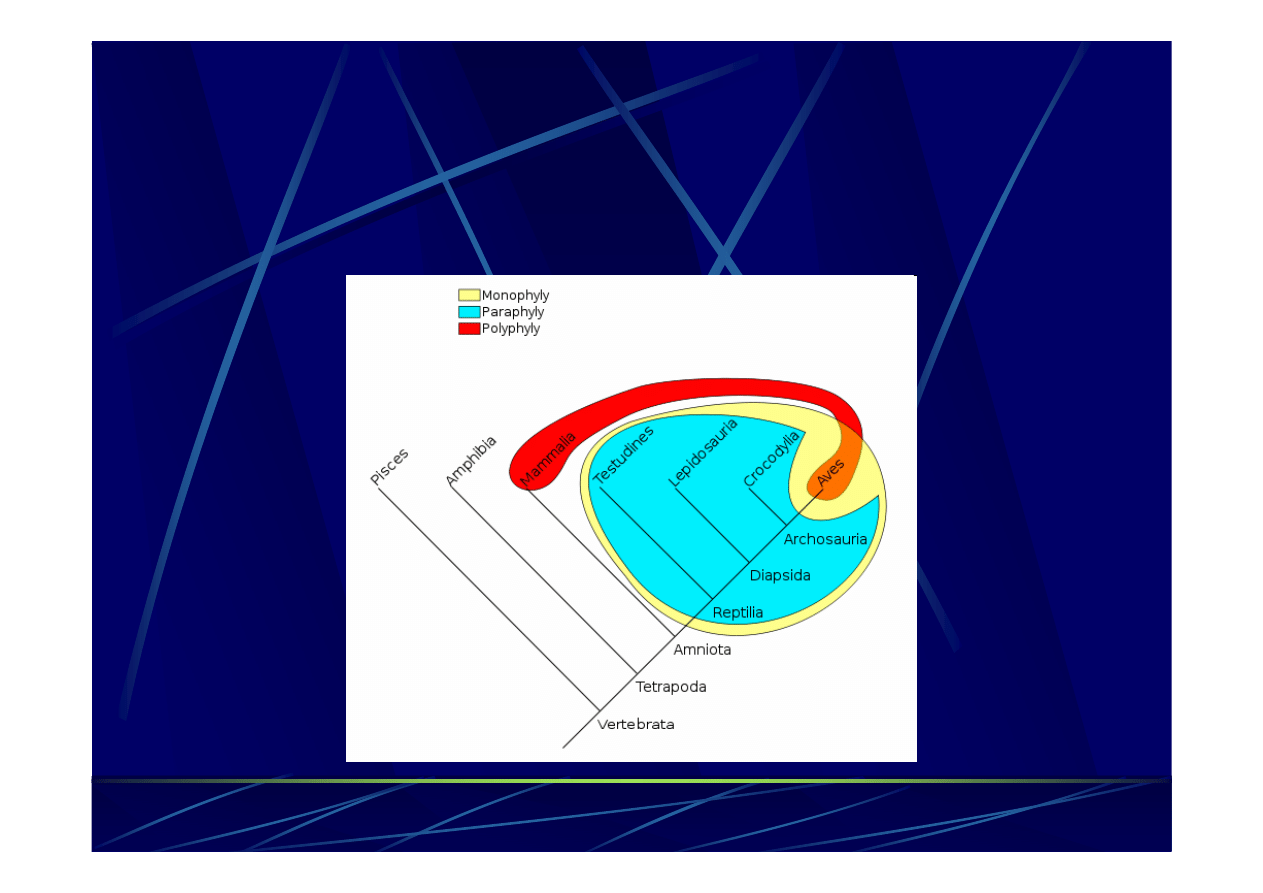
Taksony mono-
i polifiletyczne
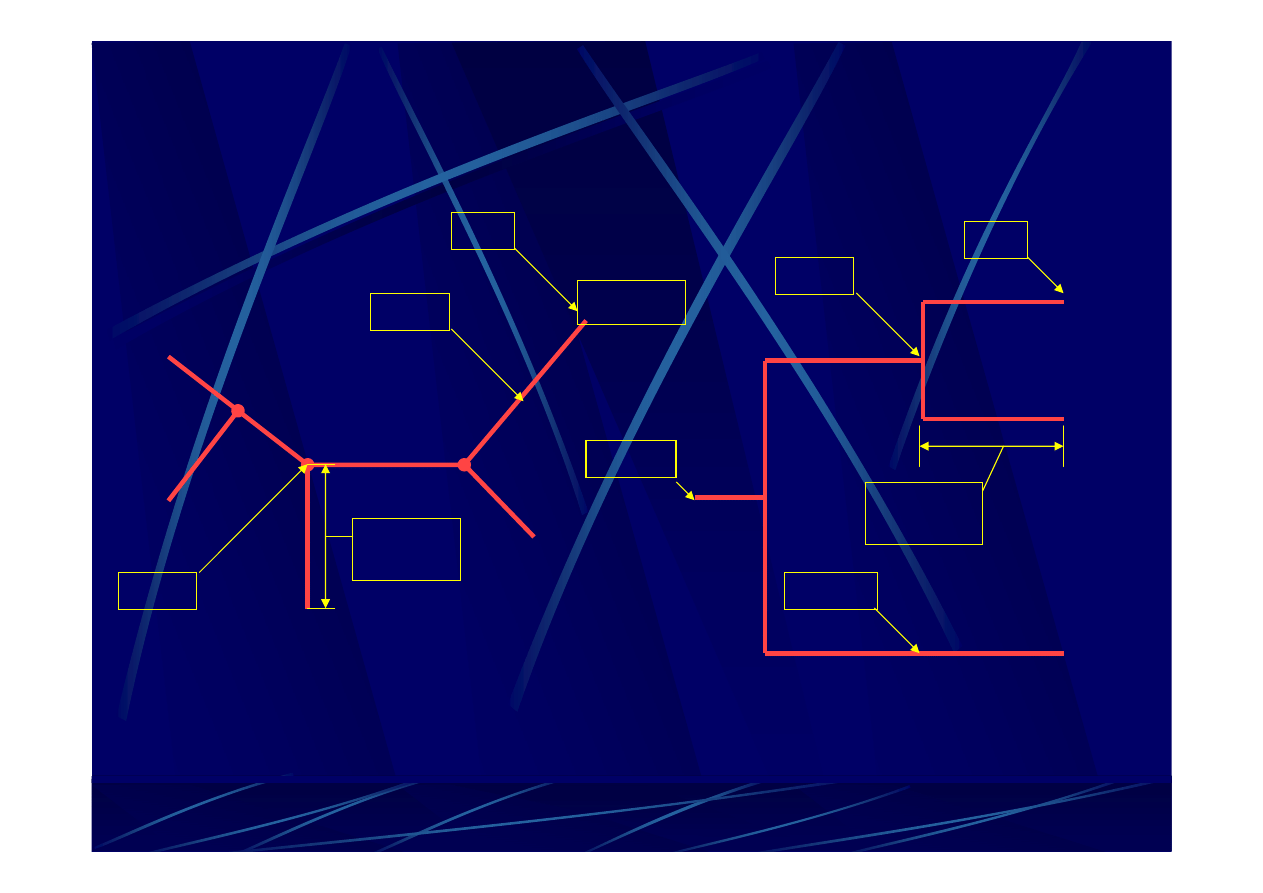
gatunek A
gatunek A
gatunek B
gatunek B
gatunek C
gatunek C
gatunek D
gatunek D
gatunek E
gatunek E
ga
ga
łąź
łąź
w
w
ę
ę
ze
ze
ł
ł
korze
korze
ń
ń
przykładowe
nieukorzenione
drzewo filogenetyczne
przykładowe
ukorzenione
drzewo filogenetyczne
d
d
ł
ł
ugo
ugo
ść
ść
ga
ga
łę
łę
zi
zi
gatunek A
gatunek A
gatunek B
gatunek B
gatunek C
gatunek C
w
w
ę
ę
ze
ze
ł
ł
ga
ga
łąź
łąź
d
d
ł
ł
ugo
ugo
ść
ść
ga
ga
łę
łę
zi
zi
li
li
ść
ść
li
li
ść
ść

Ga
Ga
łąź
łąź
-
-
obrazuje związki ewolucyjne między porównywanymi
jednostkami taksonomicznymi.
D
D
ł
ł
ugo
ugo
ść
ść
ga
ga
łę
łę
zi
zi
-
-
zazwyczaj reprezentuje liczbę
zmian, które się
zdarzyły
w danej linii ewolucyjnej.
Korze
Korze
ń
ń
-
-
wspólny przodek dla wszystkich taksonów.
Li
Li
ść
ść
-
-
reprezentuje aktualnie analizowaną
jednostkę
taksonomiczną.
W
W
ę
ę
ze
ze
ł
ł
-
-
reprezentuje jednostkę
taksonomiczną
(populację, organizm, gen).
Może przedstawiać
współcześnie istniejący takson, jak i jego przodka.

Mechanizmy ewolucji
-
Mutacje w genach.
Mutacje są
rozprzestrzeniane w populacji
poprzez dryf genetyczny
lub/i
selekcję
naturalną
-
Duplikacja i rekombinacja genów.

Etapy analizy filogenetycznej
Dobór i dopasowane sekwencji
Wybór modelu substytucji
Wybór metody oceny odległości ewolucyjnej
Konstrukcja drzewka
Ocena i analiza skonstruowanego drzewka
16S
16S
rRNA
rRNA
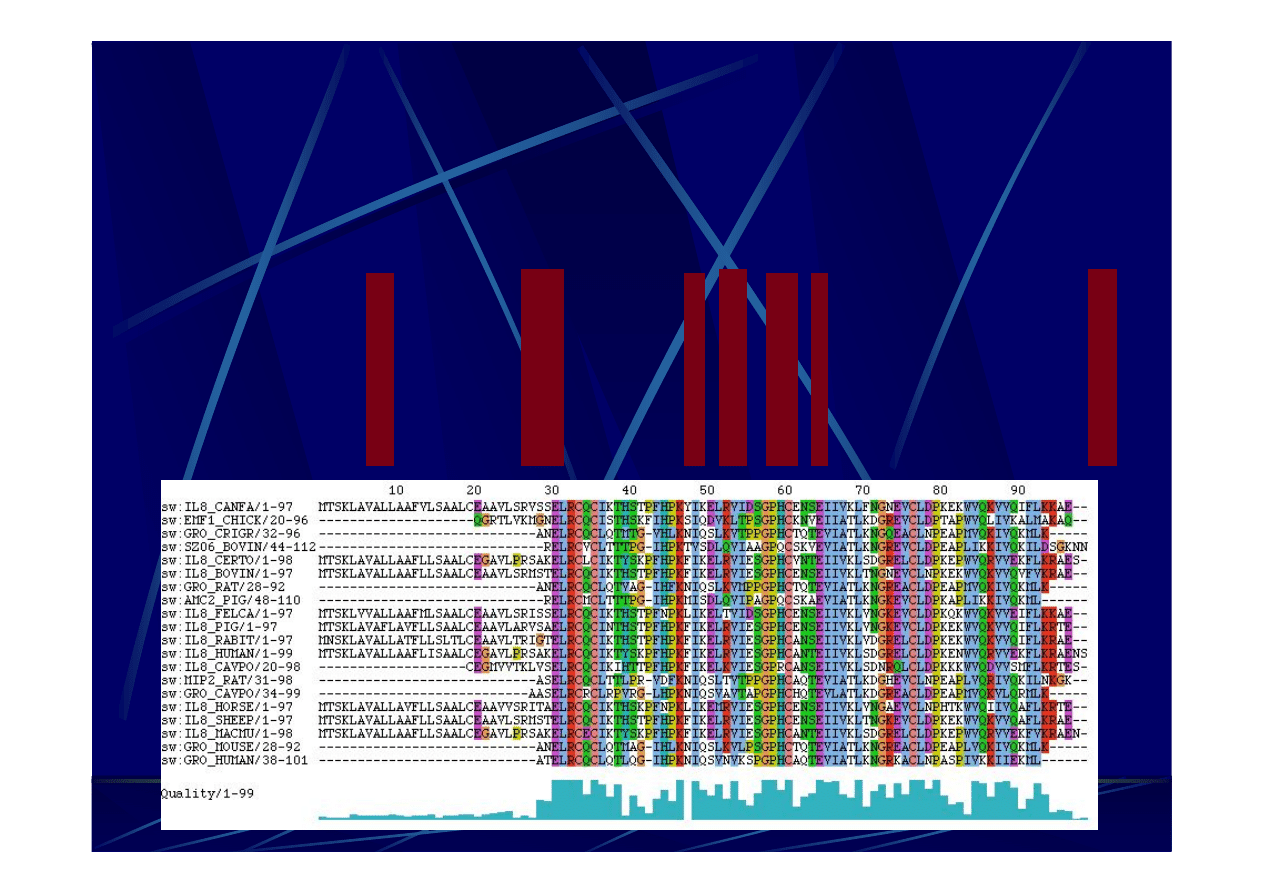
Dopasowanie wielu sekwencji
Dopasowanie wielu sekwencji
–
–
M
Multiple
sequence alignment (MSA)
E.coli
UCAGAUGU-GAAAUC-CCCGGG=CUCAA=CCUGGG=AACU=GCAUCUGA
Th. thermophilus UCCCAUGU-GAAAGA-CCACGG=CUCAA=CCGUGG=GGGA=GCGUGGGA
B.subtilis
UCUGAUGU-GAAAGC-CCCCGG=CUCAA=CCGGGG=AGGG=UCAUUGGA
Ancyst.nidulans UCUGUUGU-CAAAGC-GUGGGG=CUCAA=CCUCAU=ACAG=GCAAUGGA
Chl.aurantiacus UCGGCGCU-GAAAGC-GCCCCG=CUUAA=CGGGGC=GAGG=CGCGCCGA
match
** *** * ** ** * **
Thermus ruber
UCCGAUGC-UAAAGA-CCGAAG=CUCAA=CUUCGG=GGGU=GCGUUGGA
Grupa sekwencji
homologicznych
Dopasowanie
wielu sekwencji
Silne
podobieństwo
sekwencji?
Metoda
maksymalnej
parsymoni
-
MP
Rozpoznawalne
podobieństwo
sekwencji?
Metody oparte
na
odległościach
(dystansowe)
Metoda
maksymalnej
wiarygodności
-ML
Sprawdzanie
poprawności
rekonstrukcji
tak
tak
nie
nie
tak
tak
nie
nie
Metody tworzenia drzewek filogenetycznych

Metoda maksymalnej
Metoda maksymalnej
parsymonii
parsymonii
-
-
MP
MP
Drzewko filogenetyczne skonstruowane
metodą
MP
to takie, które wymaga
najmniejszej liczby zmian aby wyjaśnić
obserwowane różnice w analizowanych
sekwencjach
Metoda
Metoda
MP
MP
Seq1
Seq1
A
A
A G A G
A G A G
T
T
G
G
C
C
A
A
Seq2
Seq2
A
A
G C C G
G C C G
T
T
G
G
C
C
G
G
Seq3
Seq3
A
A
G A T A
G A T A
T
T
C
C
C
C
A
A
Seq4
Seq4
A
A
G A G A
G A G A
T
T
C
C
C
C
G
G
1
1
1
1
1
1
2
2
2
2
2
2
3
3
3
3
3
3
4
4
4
4
4
4
Miejsce „informatywne”
dla
sekwencji nukleotydowych to
takie, w którym obserwuje się
przynajmniej dwa różne
nukleotydy i są
one
prezentowane przynajmniej
w dwóch sekwencjach.

1 2
1 2
3 4
3 4
1 3
1 3
2 4
2 4
1 2
1 2
4 3
4 3
Position 2
Position 2
Position 3
Position 3
Position 4
Position 4
Position 5
Position 5
Position 7
Position 7
Position 8
Position 8
Seq1
Seq1
A
A
A G A G
A G A G
T
T
G
G
C
C
A
A
Seq2
Seq2
A
A
G C C G
G C C G
T
T
G
G
C
C
G
G
Seq3
Seq3
A
A
G A T A
G A T A
T
T
C
C
C
C
A
A
Seq4
Seq4
A
A
G A G A
G A G A
T
T
C
C
C
C
G
G
Sum
Sum
11
11
10
10
12
12
Position of
Position of
sequences on the
sequences on the
tree
tree
mutacja

Metoda maksymalnej wiarygodności
–
Maksimum likelihood
(ML)
Drzewko filogenetyczne
skonstruowane metodą
ML
to takie,
które z największym
prawdopodobieńswtem
odtwarza
obserwowane dane

Maximum
likelihood
method
(ML)
1. Wyliczana jest
wiarygodność
(prawdopodobieństwo -
L)
dla każdego
informatywnego
miejsca
2. Następnie sumowane są
wszystkie wartości L dla
każdego możliwego drzewa
3. Porównywane są
ze
sobą
wartości L dla
każdego możliwego drzewa
i wybierane jest to, które
ma najwyższą
wartość
L -
całościowe
czyli
Wybierane jest to drzewo,
które przy danym modelu
najbardziej pasuje do
analizowanych danych
Sekwencja 1: ACGCGTTGGG
Sekwencja 2: ACGCGTTGGG
Sekwencja 3: ACGCAATGAA
Sekwencja 4: AGACAGGGAA
1
2
3
4
T
T
A
G
?
ATGC
?
ATGC
?
ATGC
T
T
A
G
T
G
T
Rekonstrukcja drzewa metod
Rekonstrukcja drzewa metod
ą
ą
ML
ML
Analizujemy kolumnę
Proponujemy układ drzewa
Proponujemy układ nukleotydów
Przydzielenie
nukleotydów
Prawd = P(T) * P(T
G) * P(G
A) = 0.25*10
-6
*10
-6
Likelihood
konkretnej pozycji jest sumą
prawdopodobieństw wszystkich możliwych
rekonstrukcji przodków dla wybranego modelu .
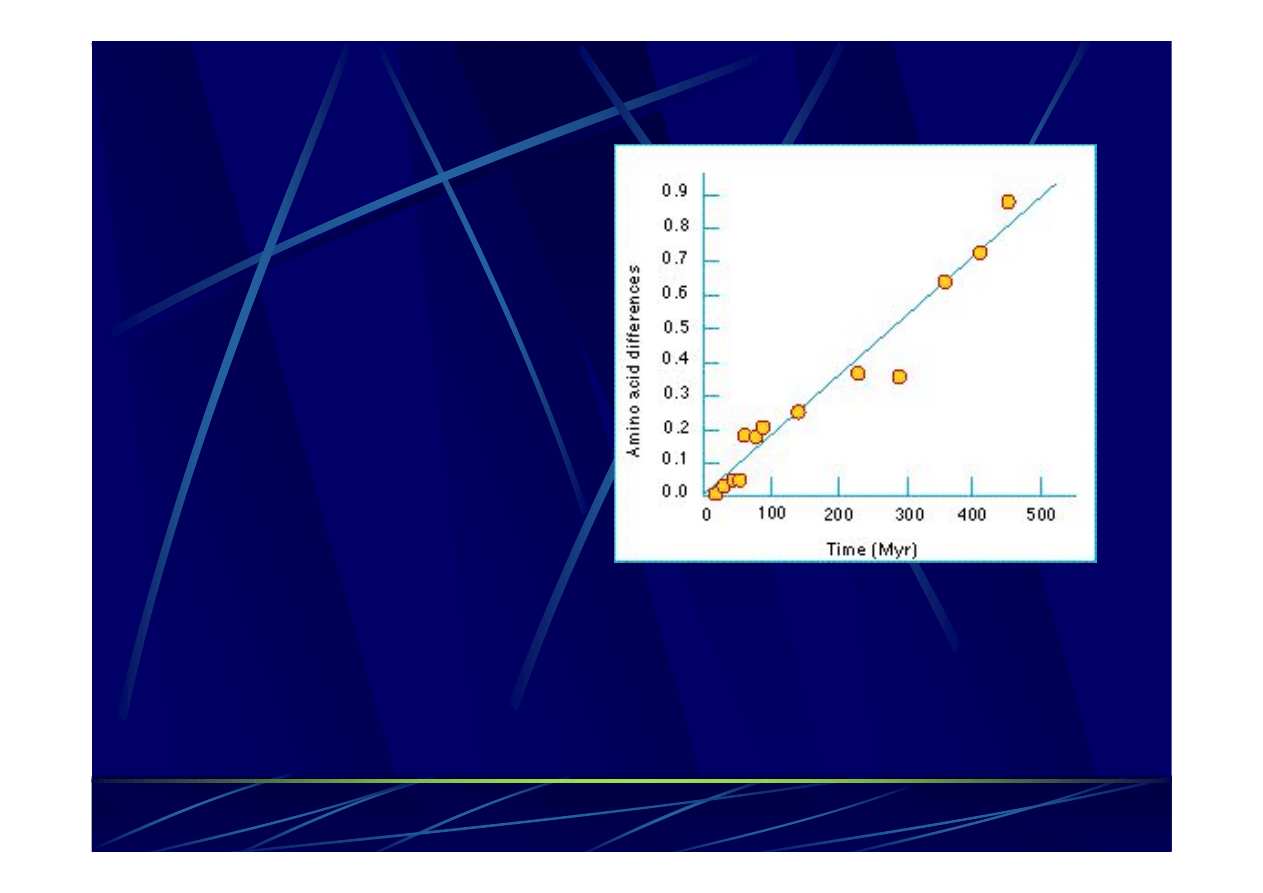
Czyli różnice między sekwencjami dwóch gatunków są
proporcjonalne do czasu jaki upłynął
od momentu gdy oba
gatunki miały wspólnego przodka.
Hipoteza zegara molekularnego (MC)
Hipoteza zegara molekularnego (MC)
Zaproponowana przez
Zuckerkandla
i Paulinga
w
roku 1962.
Opiera się
na założeniu, że
tempo ewolucji (akumulacja
mutacji) sekwencji
nukleotydowej czy
aminokwasowej
jest w
przybliżeniu stałe.

•
•
mo
mo
ż
ż
liwo
liwo
ść
ść
wyst
wyst
ą
ą
pienia wielokrotnych podstawie
pienia wielokrotnych podstawie
ń
ń
•
•
rewersja
rewersja
•
•
rzadko obserwuje si
rzadko obserwuje si
ę
ę
podstawienia mi
podstawienia mi
ę
ę
dzy a
dzy a
minokwasami pe
minokwasami pe
ł
ł
ni
ni
ą
ą
cymi
cymi
wa
wa
ż
ż
ne role w bia
ne role w bia
ł
ł
kach, jak
kach, jak
:
:
cysteina
cysteina
(C)
(C)
czy
czy
tryptofan
tryptofan
(W)
(W)
•
•
cz
cz
ęś
ęś
ciej obserwuje si
ciej obserwuje si
ę
ę
podstawienia mi
podstawienia mi
ę
ę
dzy a
dzy a
minokwasami podobnymi do
minokwasami podobnymi do
siebie, ze wzgl
siebie, ze wzgl
ę
ę
du na swoje w
du na swoje w
ł
ł
a
a
ś
ś
ciwo
ciwo
ś
ś
ci biochemiczne,
ci biochemiczne,
biofizyczne
biofizyczne
np
np
.
.
:
:
izoleucyna
izoleucyna
(I)
(I)
leucyna
leucyna
(L),
(L),
valina
valina
(V)
(V)
izoleucyna
izoleucyna
(I),
(I),
kwas asparaginowy
kwas asparaginowy
(D)
(D)
kwas glutaminowy
kwas glutaminowy
(
(
E),
E),
•
•
rzadko obserwuje si
rzadko obserwuje si
ę
ę
podstawienia mi
podstawienia mi
ę
ę
dzy aminokwasami bardzo
dzy aminokwasami bardzo
r
r
ó
ó
ż
ż
ni
ni
ą
ą
cymi si
cymi si
ę
ę
swoimi w
swoimi w
ł
ł
asno
asno
ś
ś
ciami
ciami
tryptofan
tryptofan
(W)
(W)
izoleucyna
izoleucyna
(I)
(I)
•
•
niekt
niekt
ó
ó
re aminokwasy, takie jak:
re aminokwasy, takie jak:
asparagina
asparagina
(N)
(N)
, kwas asparaginowy
, kwas asparaginowy
(D)
(D)
,
,
seryna
seryna
(S)
(S)
mutuj
mutuj
ą
ą
cz
cz
ęś
ęś
ciej ni
ciej ni
ż
ż
inne
inne
•
•
cz
cz
ęś
ęś
ciej obserwuje si
ciej obserwuje si
ę
ę
podstawienia typu
podstawienia typu
tranzycji
tranzycji
ni
ni
ż
ż
transwersji
transwersji
•
•
cz
cz
ęś
ęś
ciej obserwuje si
ciej obserwuje si
ę
ę
podstawienia w III pozycjach kodon
podstawienia w III pozycjach kodon
ó
ó
w
w
•
•
tempo mutacji zale
tempo mutacji zale
ż
ż
y od regionu w genomie, genie, rodzaju genu,
y od regionu w genomie, genie, rodzaju genu,

Protein
Rate (mean replacements per site
per 10
9
years)
Fibrinopeptides
8.3
Insulin C
2.4
Ribonuclease
2.1
Haemoglobins
1.0
Cytochrome C
0.3
Histone H4
0.01
przodek
rzeczywista liczba podstawie
rzeczywista liczba podstawie
ń
ń
zaobserwowana
zaobserwowana
liczba r
liczba r
ó
ó
ż
ż
nic
nic
MELSK
L
TGDPAR
Q
KEL
S
ML
WK
LSKLTGD
R
APFVYRV
L
KRL
MELSK
L
TGDPAR
Q
KEL
S
MLM
K
LSKLTGDPAPF
V
YRV
G
KRL
MELSK
T
TGDPAR
Q
KEL
S
MLM
K
LSKLTGDPAPF
Y
YRV
G
KRL
MELSK
T
TGDPAR
R
KEL
S
MLM
K
LSKLTGDPAPFVYRV
G
KRL
MELSK
T
TGDPAR
R
KELKMLMELSKLTGDPAPFVYRVLKRL
MELSKLTGDPAREKELKMLMELSKLTGDPAPFVYRVLKRL
potomek
3 zmiany
3 zmiany
2 zmiany
2 zmiana
2 zmiany
= 12 zmian
2 zmiany w stosunku do przodka
5 zmian w stosunku do przodka
6 zmian w stosunku do przodka
4 zmian w stosunku do przodka
5 zmian w stosunku do przodka
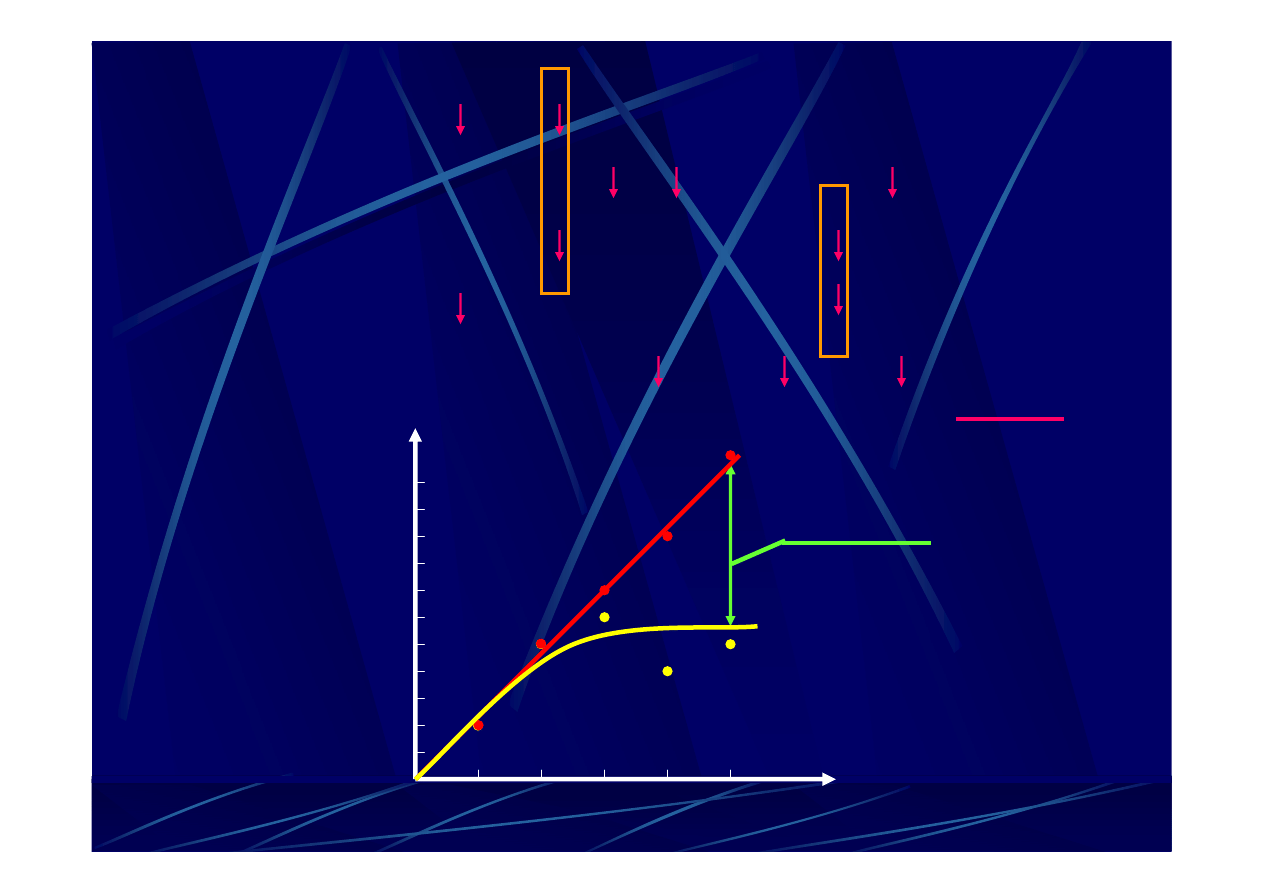
czas ewolucji
czas ewolucji
r
r
ó
ó
ż
ż
nice mi
nice mi
ę
ę
dz
y sek
w
encjami
dz
y sek
w
encjami
niedoszacowanie
niedoszacowanie

tranzycje
i transwersje

Juckes-Cantor
K80
–
(Kimura)
TN93 –
rozróżnia
tranzycje
i transwersje,
oraz typ tranzycji:
czy
zaszła ona między
purynami czy
pirymidynami
TN93
(Tamura-Nei, 93)
Macierze
Macierze
substutucji
substutucji
nukleotyd
nukleotyd
ó
ó
w
w

Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys Met Phe Pro Ser Thr Trp Tyr Val
A
R
N
D
C
Q
E
G
H
I
L
K
M
F
P
S
T
W
Y
V
Ala A 9867
2
9
10
3
8
17
21
2
6
4
2
6
2
22
35
32
0
2
18
Arg R
1
9913
1
0
1
10
0
0
10
3
1
19
4
1
4
6
1
8
0
1
Asn N
4
1
9822
36
0
4
6
6
21
3
1
13
0
1
2
20
9
1
4
1
Asp D
6
0
42
9859
0
6
53
6
4
1
0
3
0
0
1
5
3
0
0
1
Cys C
1
1
0
0
9973
0
0
0
1
1
0
0
0
0
1
5
1
0
3
2
Gln Q
3
9
4
5
0
9876
27
1
23
1
3
6
4
0
6
2
2
0
0
1
Glu E
10
0
7
56
0
35
9865
4
2
3
1
4
1
0
3
4
2
0
1
2
Gly G
21
1
12
11
1
3
7
9935
1
0
1
2
1
1
3
21
3
0
0
5
His H
1
8
18
3
1
20
1
0
9912
0
1
1
0
2
3
1
1
1
4
1
Ile I
2
2
3
1
2
1
2
0
0
9872
9
2
12
7
0
1
7
0
1
33
Leu L
3
1
3
0
0
6
1
1
4
22
9947
2
45
13
3
1
3
4
2
15
Lys K
2
37
25
6
0
12
7
2
2
4
1
9926
20
0
3
8
11
0
1
1
Met M
1
1
0
0
0
2
0
0
0
5
8
4
9874
1
0
1
2
0
0
4
Phe F
1
1
1
0
0
0
0
1
2
8
6
0
4
9946
0
2
1
3
28
0
Pro P
13
5
2
1
1
8
3
2
5
1
2
2
1
1
9926
12
4
0
0
2
Ser S
28
11
34
7
11
4
6
16
2
2
1
7
4
3
17
9840
38
5
2
2
Thr T
22
2
13
4
1
3
2
2
1
11
2
8
6
1
5
32
9871
0
2
9
Trp W
0
2
0
0
0
0
0
0
0
0
0
0
0
1
0
1
0
9976
1
0
Tyr T
1
0
3
0
3
0
1
0
4
1
1
0
0
21
0
1
1
2
9945
1
Val V
13
2
1
1
3
2
2
3
3
57
11
1
17
1
3
2
10
0
2
9901
Elementy pomnożone zostały przez 10 000
P
ercent
A
ccepted
M
utation
PAM1 -
M.
M.
Dayhoff
Dayhoff
1978r.
1978r.
element
M
IJ
tej macierzy reprezentuje
prawdopodobieństwo z jakim aminokwas
w kolumnie
j
zostanie podstawiony przez
aminokwas z wiersza
i
w czasie
ewolucyjnym 1 PAM
element diagonalny
M
ii
określa
prawdopodobieństwo, że dany
aminokwas nie ulegnie
substytucji w tym czasie

JEDNOSTKA PAM
JEDNOSTKA PAM
(
(
P
P
ercent
ercent
A
A
ccepted
ccepted
M
M
utation
utation
)
)
–
–
miara odleg
miara odleg
ł
ł
o
o
ś
ś
ci ewolucyjnej mi
ci ewolucyjnej mi
ę
ę
dzy sekwencjami
dzy sekwencjami
.
.
M.
M.
Dayhoff
Dayhoff
i wsp
i wsp
ó
ó
ł
ł
pracownicy
pracownicy
–
–
1978r.
1978r.
1 PAM
1 PAM
–
–
odpowiada takiemu czasowi ewolucyjnemu,
odpowiada takiemu czasowi ewolucyjnemu,
podczas kt
podczas kt
ó
ó
rego, w por
rego, w por
ó
ó
wnywanych sekwencjach,
wnywanych sekwencjach,
zmianie ulegnie 1 aminokwas na 100 (ok. 1
zmianie ulegnie 1 aminokwas na 100 (ok. 1
mln
mln
lat)
lat)
MELSKLT
G
DPAPFVYR
V
LKR ..... SKLT
G
DPAP ..... KVVFRIS
E
SP
M
IFKA
Y
PLDI ..... MELSK
L
TGDPA ... REKEL
K
MLMELSKLTGDPAPFVYRV
L
KRL ..... LDIVLSSLIHER
E
KELKML
MELSKLT
D
DPAPFVYR
Y
LKR ..... SKLT
Q
DPAP ..... KVVFRIS
R
SP
W
IFKA
V
PLDI ..... MELSK
T
TGDPA ... REKEL
D
MLMELSKLTGDPAPFVYRV
F
KRL ..... LDIVLSSLIHER
R
KELKML
1000 aminokwasów
10
zmienionych aminokwasów
Zmianie uległo 10/1000 = 1/100 aminokwasów, czyli 1%

Ewolucyjna macierz PAM
Ewolucyjna macierz PAM
Macierz PAM
Macierz PAM
-
-
Percent
Percent
Accepted
Accepted
Mutations
Mutations
(
(
Dayhoff
Dayhoff
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1978)
. 1978)
Utworzona przez por
Utworzona przez por
ó
ó
wnanie blisko spokrewnionych sekwencji bia
wnanie blisko spokrewnionych sekwencji bia
ł
ł
ek (ponad
ek (ponad
85% identyczno
85% identyczno
ś
ś
ci) o znanych powi
ci) o znanych powi
ą
ą
zaniach filogenetycznych; naliczenie 1572
zaniach filogenetycznych; naliczenie 1572
zmian zaakceptowanych (przez selekcj
zmian zaakceptowanych (przez selekcj
ę
ę
) w 71 grupach bia
) w 71 grupach bia
ł
ł
ek.
ek.
Uwzgl
Uwzgl
ę
ę
dnia
dnia
mutabilno
mutabilno
ś
ś
ci
ci
poszczeg
poszczeg
ó
ó
lnych aminokwas
lnych aminokwas
ó
ó
w
w
MWT
MWT
V
V
SALV
SALV
G
G
Q
Q
MWT
MWT
A
A
SALV
SALV
G
G
Q
Q
MWT
MWT
V
V
SALV
SALV
L
L
Q
Q
MWT
MWT
V
V
SALV
SALV
G
G
Q
Q
MWT
MWT
A
A
SALV
SALV
G
G
Q
Q
MWT
MWT
V
V
SALV
SALV
L
L
Q
Q
V
V
-
-
>
>
A
A
G
G
-
-
>
>
L
L

Macierz PAM
Macierz PAM
–
–
log
log
odds
odds
je
je
ż
ż
eli
eli
log
log
odds
odds
<
<
0
0
:
:
dana substytucja zachodzi rzadziej ni
dana substytucja zachodzi rzadziej ni
ż
ż
nale
nale
ż
ż
a
a
ł
ł
o si
o si
ę
ę
spodziewa
spodziewa
ć
ć
je
je
ż
ż
eli
eli
log
log
odds
odds
>
>
0
0
:
:
dana substytucja zachodzi cz
dana substytucja zachodzi cz
ęś
ęś
ciej ni
ciej ni
ż
ż
nale
nale
ż
ż
a
a
ł
ł
o si
o si
ę
ę
spodziewa
spodziewa
ć
ć
(
(
np
np
. +1 oznacza,
. +1 oznacza,
ż
ż
e dana substytucja jest obserwowana 10 razy cz
e dana substytucja jest obserwowana 10 razy cz
ęś
ęś
ciej ni
ciej ni
ż
ż
nale
nale
ż
ż
a
a
ł
ł
o si
o si
ę
ę
spodziewa
spodziewa
ć
ć
)
)
je
je
ż
ż
eli
eli
log
log
odds
odds
=
=
0
0
:
:
dana substytucja zachodzi z tak
dana substytucja zachodzi z tak
ą
ą
sam
sam
ą
ą
cz
cz
ę
ę
sto
sto
ś
ś
ci
ci
ą
ą
jak w sekwencji
jak w sekwencji
losowej
losowej
Wyliczenie warto
Wyliczenie warto
ś
ś
ci log
ci log
odds
odds
:
:
P
P
o
o
–
–
obserwowana cz
obserwowana cz
ę
ę
stotliwo
stotliwo
ść
ść
wyst
wyst
ę
ę
powania mutacji
powania mutacji
P
P
e
e
–
–
oczekiwana cz
oczekiwana cz
ę
ę
stotliwo
stotliwo
ść
ść
wyst
wyst
ę
ę
powania mutacji
powania mutacji
(losow
(losow
a
a
)
)
log
log
odds
odds
= log (P
= log (P
o/
o/
P
P
e
e
)
)

A R N D C Q E G H I L K M F P S T W Y V
A R N D C Q E G H I L K M F P S T W Y V
A
A
2
2
R
R
-
-
2 6
2 6
N 0 0 2
N 0 0 2
D 0
D 0
-
-
1 2 4
1 2 4
C
C
-
-
2
2
-
-
4
4
-
-
4
4
-
-
5 12
5 12
Q 0 1 1 2
Q 0 1 1 2
-
-
5 4
5 4
E 0
E 0
-
-
1 1 3
1 1 3
-
-
5 2 4
5 2 4
G 1
G 1
-
-
3 0 1
3 0 1
-
-
3
3
-
-
1 0 5
1 0 5
H
H
-
-
1 2 2 1
1 2 2 1
-
-
3 3 1
3 3 1
-
-
2 6
2 6
I
I
-
-
1
1
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
3
3
-
-
2 5
2 5
L
L
-
-
2
2
-
-
3
3
-
-
3
3
-
-
4
4
-
-
6
6
-
-
2
2
-
-
3
3
-
-
4
4
-
-
2 2 6
2 2 6
K
K
-
-
1 3 1 0
1 3 1 0
-
-
5 1 0
5 1 0
-
-
2 0
2 0
-
-
2
2
-
-
3 5
3 5
M
M
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
5
5
-
-
1
1
-
-
2
2
-
-
3
3
-
-
2 2 4 0 6
2 2 4 0 6
F
F
-
-
4
4
-
-
4
4
-
-
4
4
-
-
6
6
-
-
4
4
-
-
5
5
-
-
5
5
-
-
5
5
-
-
2 1 2
2 1 2
-
-
5 0 9
5 0 9
P 1 0
P 1 0
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
1
1
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
1
1
-
-
2
2
-
-
5 6
5 6
S 1 0 1 0 0
S 1 0 1 0 0
-
-
1 0 1
1 0 1
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
2
2
-
-
3 1 2
3 1 2
T 1
T 1
-
-
1 0 0
1 0 0
-
-
2
2
-
-
1 0 0
1 0 0
-
-
1 0
1 0
-
-
2 0
2 0
-
-
1
1
-
-
3 0 1 3
3 0 1 3
W
W
-
-
6 2
6 2
-
-
4
4
-
-
7
7
-
-
8
8
-
-
5
5
-
-
7
7
-
-
7
7
-
-
3
3
-
-
5
5
-
-
2
2
-
-
3
3
-
-
4 0
4 0
-
-
6
6
-
-
2
2
-
-
5 17
5 17
Y
Y
-
-
3
3
-
-
4
4
-
-
2
2
-
-
4 0
4 0
-
-
4
4
-
-
4
4
-
-
5 0
5 0
-
-
1
1
-
-
1
1
-
-
4
4
-
-
2 7
2 7
-
-
5
5
-
-
3
3
-
-
3 0 10
3 0 10
V 0
V 0
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
1
1
-
-
2 4 2
2 4 2
-
-
2 2
2 2
-
-
1
1
-
-
1
1
-
-
1 0
1 0
-
-
6
6
-
-
2 4
2 4
Rzadkie aminokwasy maj
Rzadkie aminokwasy maj
ą
ą
du
du
ż
ż
e wagi
e wagi
Pospolite
Pospolite
aminokwasy maj
aminokwasy maj
ą
ą
ma
ma
ł
ł
e wagi
e wagi
A R N D C Q E G H I L K M F P S T W Y V
A R N D C Q E G H I L K M F P S T W Y V
A
A
2
2
R
R
-
-
2 6
2 6
N 0 0 2
N 0 0 2
D 0
D 0
-
-
1 2 4
1 2 4
C
C
-
-
2
2
-
-
4
4
-
-
4
4
-
-
5 12
5 12
Q 0 1 1 2
Q 0 1 1 2
-
-
5 4
5 4
E 0
E 0
-
-
1 1 3
1 1 3
-
-
5 2 4
5 2 4
G 1
G 1
-
-
3 0 1
3 0 1
-
-
3
3
-
-
1 0 5
1 0 5
H
H
-
-
1 2 2 1
1 2 2 1
-
-
3 3 1
3 3 1
-
-
2 6
2 6
I
I
-
-
1
1
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
3
3
-
-
2 5
2 5
L
L
-
-
2
2
-
-
3
3
-
-
3
3
-
-
4
4
-
-
6
6
-
-
2
2
-
-
3
3
-
-
4
4
-
-
2 2 6
2 2 6
K
K
-
-
1 3 1 0
1 3 1 0
-
-
5 1 0
5 1 0
-
-
2 0
2 0
-
-
2
2
-
-
3 5
3 5
M
M
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
5
5
-
-
1
1
-
-
2
2
-
-
3
3
-
-
2 2 4 0 6
2 2 4 0 6
F
F
-
-
4
4
-
-
4
4
-
-
4
4
-
-
6
6
-
-
4
4
-
-
5
5
-
-
5
5
-
-
5
5
-
-
2 1 2
2 1 2
-
-
5 0 9
5 0 9
P 1 0
P 1 0
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
1
1
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
1
1
-
-
2
2
-
-
5 6
5 6
S 1 0 1 0 0
S 1 0 1 0 0
-
-
1 0 1
1 0 1
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
2
2
-
-
3 1 2
3 1 2
T 1
T 1
-
-
1 0 0
1 0 0
-
-
2
2
-
-
1 0 0
1 0 0
-
-
1 0
1 0
-
-
2 0
2 0
-
-
1
1
-
-
3 0 1 3
3 0 1 3
W
W
-
-
6 2
6 2
-
-
4
4
-
-
7
7
-
-
8
8
-
-
5
5
-
-
7
7
-
-
7
7
-
-
3
3
-
-
5
5
-
-
2
2
-
-
3
3
-
-
4 0
4 0
-
-
6
6
-
-
2
2
-
-
5 17
5 17
Y
Y
-
-
3
3
-
-
4
4
-
-
2
2
-
-
4 0
4 0
-
-
4
4
-
-
4
4
-
-
5 0
5 0
-
-
1
1
-
-
1
1
-
-
4
4
-
-
2 7
2 7
-
-
5
5
-
-
3
3
-
-
3 0 10
3 0 10
V 0
V 0
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
1
1
-
-
2 4 2
2 4 2
-
-
2 2
2 2
-
-
1
1
-
-
1
1
-
-
1 0
1 0
-
-
6
6
-
-
2 4
2 4
Dodatnie warto
Dodatnie warto
ś
ś
ci dla cz
ci dla cz
ę
ę
stszych
stszych
podstawie
podstawie
ń
ń
A R N D C Q E G H I L K M F P S T W Y V
A R N D C Q E G H I L K M F P S T W Y V
A
A
2
2
R
R
-
-
2 6
2 6
N 0 0 2
N 0 0 2
D 0
D 0
-
-
1 2 4
1 2 4
C
C
-
-
2
2
-
-
4
4
-
-
4
4
-
-
5 12
5 12
Q 0 1 1 2
Q 0 1 1 2
-
-
5 4
5 4
E 0
E 0
-
-
1 1 3
1 1 3
-
-
5 2 4
5 2 4
G 1
G 1
-
-
3 0 1
3 0 1
-
-
3
3
-
-
1 0 5
1 0 5
H
H
-
-
1 2 2 1
1 2 2 1
-
-
3 3 1
3 3 1
-
-
2 6
2 6
I
I
-
-
1
1
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
3
3
-
-
2 5
2 5
L
L
-
-
2
2
-
-
3
3
-
-
3
3
-
-
4
4
-
-
6
6
-
-
2
2
-
-
3
3
-
-
4
4
-
-
2 2 6
2 2 6
K
K
-
-
1 3 1 0
1 3 1 0
-
-
5 1 0
5 1 0
-
-
2 0
2 0
-
-
2
2
-
-
3 5
3 5
M
M
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
5
5
-
-
1
1
-
-
2
2
-
-
3
3
-
-
2 2 4 0 6
2 2 4 0 6
F
F
-
-
4
4
-
-
4
4
-
-
4
4
-
-
6
6
-
-
4
4
-
-
5
5
-
-
5
5
-
-
5
5
-
-
2 1 2
2 1 2
-
-
5 0 9
5 0 9
P 1 0
P 1 0
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
1
1
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
1
1
-
-
2
2
-
-
5 6
5 6
S 1 0 1 0 0
S 1 0 1 0 0
-
-
1 0 1
1 0 1
-
-
1
1
-
-
1
1
-
-
3 0
3 0
-
-
2
2
-
-
3 1 2
3 1 2
T 1
T 1
-
-
1 0 0
1 0 0
-
-
2
2
-
-
1 0 0
1 0 0
-
-
1 0
1 0
-
-
2 0
2 0
-
-
1
1
-
-
3 0 1 3
3 0 1 3
W
W
-
-
6 2
6 2
-
-
4
4
-
-
7
7
-
-
8
8
-
-
5
5
-
-
7
7
-
-
7
7
-
-
3
3
-
-
5
5
-
-
2
2
-
-
3
3
-
-
4 0
4 0
-
-
6
6
-
-
2
2
-
-
5 17
5 17
Y
Y
-
-
3
3
-
-
4
4
-
-
2
2
-
-
4 0
4 0
-
-
4
4
-
-
4
4
-
-
5 0
5 0
-
-
1
1
-
-
1
1
-
-
4
4
-
-
2 7
2 7
-
-
5
5
-
-
3
3
-
-
3 0 10
3 0 10
V 0
V 0
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
2
2
-
-
1
1
-
-
2 4 2
2 4 2
-
-
2 2
2 2
-
-
1
1
-
-
1
1
-
-
1 0
1 0
-
-
6
6
-
-
2 4
2 4
Ujemne warto
Ujemne warto
ś
ś
ci dla rzadkich
ci dla rzadkich
podstawie
podstawie
ń
ń
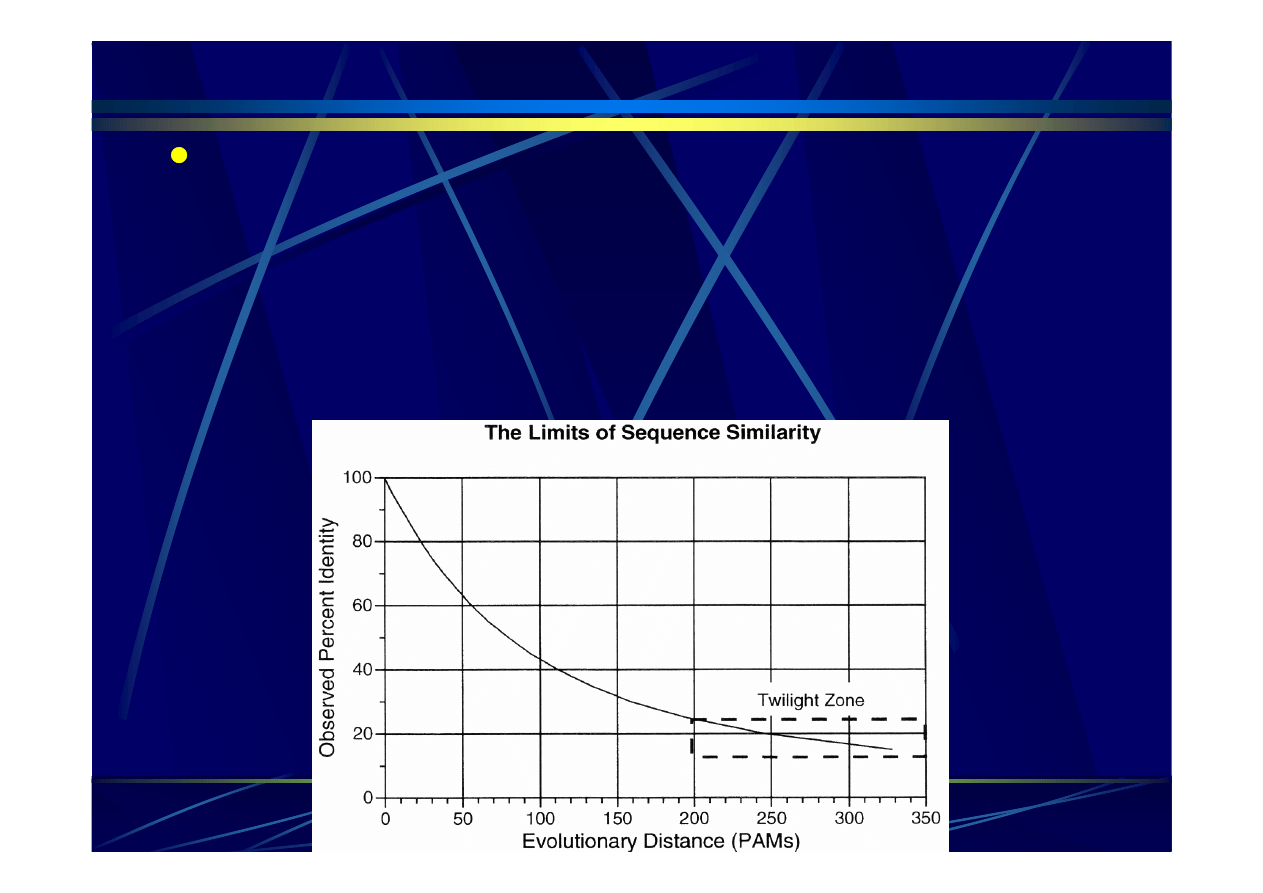
Ewolucyjna macierz PAM
Ewolucyjna macierz PAM
Ekstrapolowanie cz
Ekstrapolowanie cz
ę
ę
sto
sto
ś
ś
ci
ci
podstawie
podstawie
ń
ń
zaobserwowanych na kr
zaobserwowanych na kr
ó
ó
tkich
tkich
dystansach na d
dystansach na d
ł
ł
u
u
ż
ż
sze
sze
dystansy
dystansy
ewolucyjne
ewolucyjne
–
–
mno
mno
ż
ż
enie macierzy przez siebie
enie macierzy przez siebie
–
–
uzyskanie serii tablic PAM:
uzyskanie serii tablic PAM:
PAM1
PAM1
-
-
> PAM60, PAM80, PAM120,
> PAM60, PAM80, PAM120,
PAM250
PAM250
Podobie
Podobie
ń
ń
stwo:
stwo:
99%
99%
60%
60%
50%
50%
40%
40%
20%
20%
Liczba podstawie
Liczba podstawie
ń
ń
na miejsce:
na miejsce:
0.01
0.01
0.6
0.6
0.8
0.8
1.2
1.2
2.5
2.5

Macierz PAM
Macierz PAM
–
–
wady z powodu za
wady z powodu za
ł
ł
o
o
ż
ż
e
e
ń
ń
:
:
Podstawienia
Podstawienia
aminokwas
aminokwas
ó
ó
w zachodz
w zachodz
ą
ą
niezale
niezale
ż
ż
nie od
nie od
siebie. W rzeczywisto
siebie. W rzeczywisto
ś
ś
ci zmiany w r
ci zmiany w r
ó
ó
ż
ż
nych regionach
nych regionach
sekwencji s
sekwencji s
ą
ą
ze sob
ze sob
ą
ą
skorelowane.
skorelowane.
Te same tempo podstawie
Te same tempo podstawie
ń
ń
w r
w r
ó
ó
ż
ż
nych regionach
nych regionach
sekwencji. W rzeczywisto
sekwencji. W rzeczywisto
ś
ś
ci r
ci r
ó
ó
ż
ż
ne regiony wykazuj
ne regiony wykazuj
ą
ą
r
r
ó
ó
ż
ż
ny
ny
stopie
stopie
ń
ń
konserwatywno
konserwatywno
ś
ś
ci i ewoluuj
ci i ewoluuj
ą
ą
z r
z r
ó
ó
ż
ż
n
n
ą
ą
pr
pr
ę
ę
dko
dko
ś
ś
ci
ci
ą
ą
.
.
W r
W r
ó
ó
ż
ż
nych regionach r
nych regionach r
ó
ó
ż
ż
ne
ne
podstawienia
podstawienia
zdarzaj
zdarzaj
ą
ą
si
si
ę
ę
z r
z r
ó
ó
ż
ż
n
n
ą
ą
cz
cz
ę
ę
sto
sto
ś
ś
ci
ci
ą
ą
.
.
Cz
Cz
ę
ę
sto
sto
ść
ść
poszczeg
poszczeg
ó
ó
lnych podstawie
lnych podstawie
ń
ń
nie zmieniaj
nie zmieniaj
ą
ą
si
si
ę
ę
w czasie. W rzeczywisto
w czasie. W rzeczywisto
ś
ś
ci cz
ci cz
ę
ę
sto
sto
ś
ś
ci podstawie
ci podstawie
ń
ń
mog
mog
ą
ą
si
si
ę
ę
zmienia
zmienia
ć
ć
w czasie.
w czasie.

Macierz BLOSUM
Macierz BLOSUM
Macierz BLOSUM
Macierz BLOSUM
–
–
BLOcks
BLOcks
Substitution
Substitution
Matrix
Matrix
(
(
Henikoff
Henikoff
i
i
Henikoff
Henikoff
1992)
1992)
Utworzona przez por
Utworzona przez por
ó
ó
wnanie oko
wnanie oko
ł
ł
o 2000
o 2000
zachowanych
zachowanych
blok
blok
ó
ó
w (region
w (region
ó
ó
w
w
sekwencji) w ponad 500 rodzinach bia
sekwencji) w ponad 500 rodzinach bia
ł
ł
ek o r
ek o r
ó
ó
ż
ż
nej odleg
nej odleg
ł
ł
o
o
ś
ś
ci ewolucyjnej. Bloki s
ci ewolucyjnej. Bloki s
ą
ą
regionami sekwencji odpowiedzialnymi za podobn
regionami sekwencji odpowiedzialnymi za podobn
ą
ą
funkcj
funkcj
ę
ę
biochemiczn
biochemiczn
ą
ą
lub
lub
struktur
struktur
ę
ę
.
.
Macierze dla r
Macierze dla r
ó
ó
ż
ż
nych odleg
nych odleg
ł
ł
o
o
ś
ś
ci ewolucyjnych zosta
ci ewolucyjnych zosta
ł
ł
y wyliczone z por
y wyliczone z por
ó
ó
wnania
wnania
sekwencji odpowiednio odleg
sekwencji odpowiednio odleg
ł
ł
ych:
ych:
BLOSUM30
BLOSUM30
–
–
bloki sekwencji o co najmniej 30% identyczno
bloki sekwencji o co najmniej 30% identyczno
ś
ś
ci reszt aminokwasowych
ci reszt aminokwasowych
BLOSUM62
BLOSUM62
–
–
bloki sekwencji o co najmniej 62% identyczno
bloki sekwencji o co najmniej 62% identyczno
ś
ś
ci reszt aminokwasowych
ci reszt aminokwasowych
BLOSUM80
BLOSUM80
–
–
bloki sekwencji o co najmniej 80% identyczno
bloki sekwencji o co najmniej 80% identyczno
ś
ś
ci reszt aminokwasowych
ci reszt aminokwasowych
Macierz BLOSUM
Macierz BLOSUM
–
–
BLOcks
BLOcks
Substitution
Substitution
Matrix
Matrix
bloki
bloki

A R N D C Q E G H I L K M F P S T W Y V
A R N D C Q E G H I L K M F P S T W Y V
A 4
A 4
R
R
-
-
1 5
1 5
N
N
-
-
2 0 6
2 0 6
D
D
-
-
2
2
-
-
2 1 6
2 1 6
C 0
C 0
-
-
3
3
-
-
3
3
-
-
3 9
3 9
Q
Q
-
-
1 1 0 0
1 1 0 0
-
-
3 5
3 5
E
E
-
-
1 0 0 2
1 0 0 2
-
-
4 2 5
4 2 5
G 0
G 0
-
-
2 0
2 0
-
-
1
1
-
-
3
3
-
-
2
2
-
-
2 6
2 6
H
H
-
-
2 0 1
2 0 1
-
-
1
1
-
-
3 0 0
3 0 0
-
-
2 8
2 8
I
I
-
-
1
1
-
-
3
3
-
-
3
3
-
-
3
3
-
-
1
1
-
-
3
3
-
-
3
3
-
-
4
4
-
-
3 4
3 4
L
L
-
-
1
1
-
-
2
2
-
-
3
3
-
-
4
4
-
-
1
1
-
-
2
2
-
-
3
3
-
-
4
4
-
-
3 2 4
3 2 4
K
K
-
-
1 2 0
1 2 0
-
-
1
1
-
-
3 1 1
3 1 1
-
-
2
2
-
-
1
1
-
-
3
3
-
-
2 5
2 5
M
M
-
-
1
1
-
-
1
1
-
-
2
2
-
-
3
3
-
-
1 0
1 0
-
-
2
2
-
-
3
3
-
-
2 1 2
2 1 2
-
-
1 5
1 5
F
F
-
-
2
2
-
-
3
3
-
-
3
3
-
-
3
3
-
-
2
2
-
-
3
3
-
-
3
3
-
-
3
3
-
-
1 0 0
1 0 0
-
-
3 0 6
3 0 6
P
P
-
-
1
1
-
-
2
2
-
-
2
2
-
-
1
1
-
-
3
3
-
-
1
1
-
-
1
1
-
-
2
2
-
-
2
2
-
-
3
3
-
-
3
3
-
-
1
1
-
-
2
2
-
-
4 7
4 7
S 1
S 1
-
-
1 1 0
1 1 0
-
-
1 0 0 0
1 0 0 0
-
-
1
1
-
-
2
2
-
-
2 0
2 0
-
-
1
1
-
-
2
2
-
-
1 4
1 4
Macierz BLOSUM62
Macierz BLOSUM62
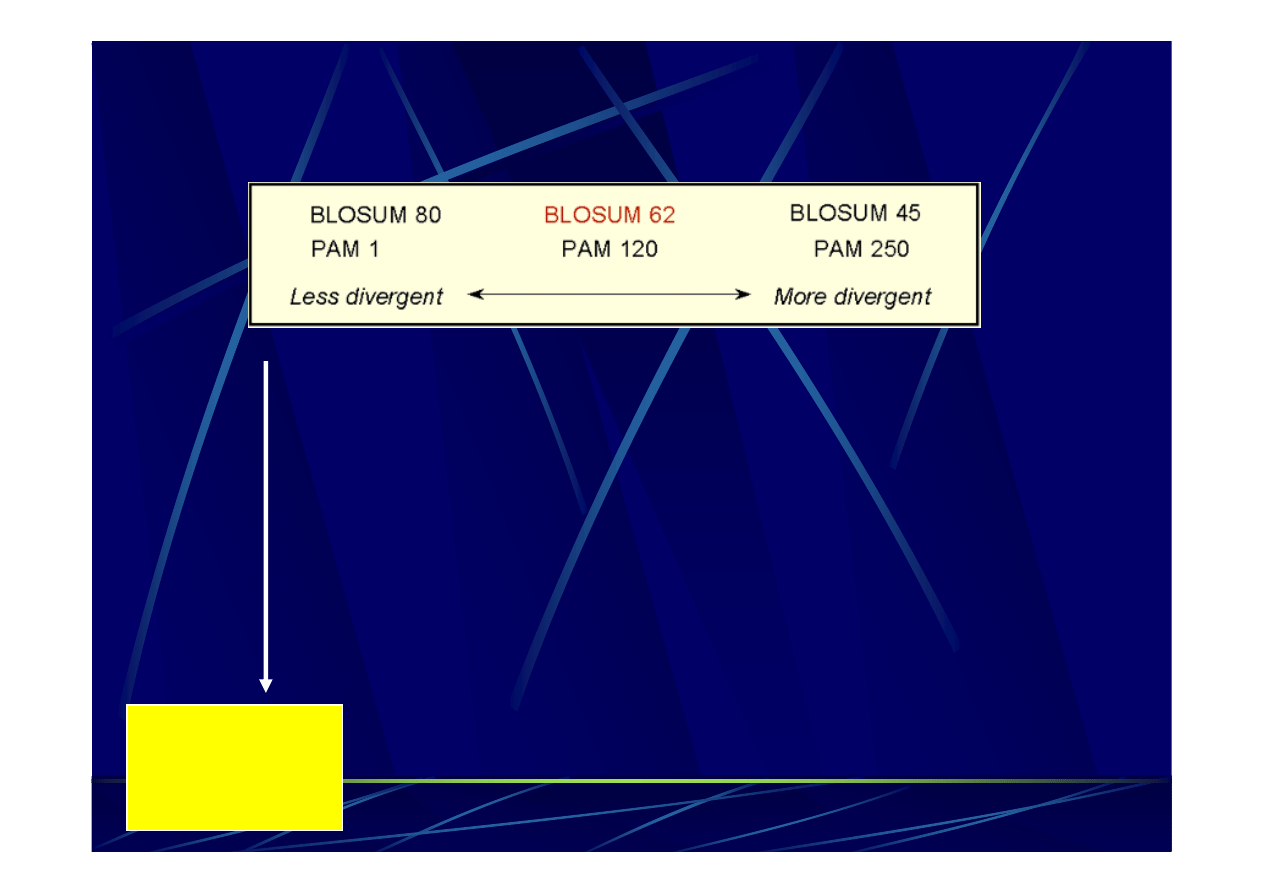
PAM Vs. BLOSUM
PAM100 =~ BLOSUM90
PAM120 =~ BLOSUM80
PAM160 =~ BLOSUM60
PAM200 =~ BLOSUM52
PAM250 =~ BLOSUM45
Bardziej odległe
sekwencje

Inne macierze
Inne macierze
substutucji
substutucji
aminokwasow
aminokwasow
ó
ó
w
w
Oparte na kodzie genetycznym
Oparte na kodzie genetycznym
-
-
zwi
zwi
ą
ą
zane z kodowaniem aminokwas
zane z kodowaniem aminokwas
ó
ó
w przez
w przez
kodony (
kodony (
Fitch
Fitch
1966;
1966;
Benner
Benner
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1994)
. 1994)
Uwzgl
Uwzgl
ę
ę
dniaj
dniaj
ą
ą
ce w
ce w
ł
ł
a
a
ś
ś
ciwo
ciwo
ś
ś
ci fizyko
ci fizyko
-
-
chemiczne aminokwas
chemiczne aminokwas
ó
ó
w (
w (
Vogt
Vogt
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1995)
. 1995)
i podobie
i podobie
ń
ń
stwo strukturalne
stwo strukturalne
ł
ł
a
a
ń
ń
cuch
cuch
ó
ó
w bocznych (
w bocznych (
Feng
Feng
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1985)
. 1985)
Uwzgl
Uwzgl
ę
ę
dniaj
dniaj
ą
ą
ce struktur
ce struktur
ę
ę
trzeciorz
trzeciorz
ę
ę
dow
dow
ą
ą
(
(
Risler
Risler
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1988; Johnson i
. 1988; Johnson i
Overington
Overington
1993;
1993;
Henikoff
Henikoff
i
i
Henikoff
Henikoff
1993;
1993;
Sander
Sander
i Schneider 1991)
i Schneider 1991)
Macierz
Macierz
dwupeptyd
dwupeptyd
ó
ó
w
w
(
(
Gonnet
Gonnet
i
i
wsp
wsp
ó
ó
ł
ł
pr
pr
. 1994)
. 1994)
-
-
400 x 400, uwzgl
400 x 400, uwzgl
ę
ę
dnia wp
dnia wp
ł
ł
yw
yw
przyleg
przyleg
ł
ł
ych aminokwas
ych aminokwas
ó
ó
w na cz
w na cz
ę
ę
sto
sto
ść
ść
substytucji
substytucji
Macierz PAM z uwzgl
Macierz PAM z uwzgl
ę
ę
dnieniem bia
dnieniem bia
ł
ł
ek transmembranowych (Jones i
ek transmembranowych (Jones i
wsp
wsp
ó
ó
ł
ł
pr
pr
.
.
1994)
1994)

Etapy analizy filogenetycznej
Dobór i dopasowane sekwencji
Wybór modelu substytucji
Wybór metody oceny odległości ewolucyjnej
Konstrukcja drzewka
Ocena i analiza skonstruowanego drzewka
FM
–
Fitch
-
Margoliash
0.047
Human
Chimpanzee
Gorilla
0.056
0.047
Orangutan
0. 094
0.108
Gibbon
UPMGA –
Unweighted
Pair
Group
Method
with
Arithmetic
Mean

A
B
C
D
E
A
B
E
D
C
A
B
NJ -
Neighbour
joining

Ocena poprawności rekonstrukcji
filogenetycznej –
metoda bootstrap
Site
OTU 1
2
3 4 5 6 7 8
9
10
1
2
3
4
A
A
C
C
Site
OTU
1
2
3
4 5 6 7 8 9 10
1
2
3
4
T
C
A
G A T C T A G
T
T
A
G A A C T A G
T
T
C
G A T C G A G
T
T
C
T A A G G A C
C
T
T
T
T
A
T
A
A
A
A
A
G
G
G
C
C
C
C
G
A
A
A
A
T
A
T
A
T
T
T
T
A
A
A
A
oryginalne
dopasowanie
powtórne dopasowanie
1
2
3
4
1
3
2
4
Losujemy nowe kolumny dopasowania (z powtórzeniami!)
Powtarzamy 100 -
1000 i tworzymy drzewko konsensusowe
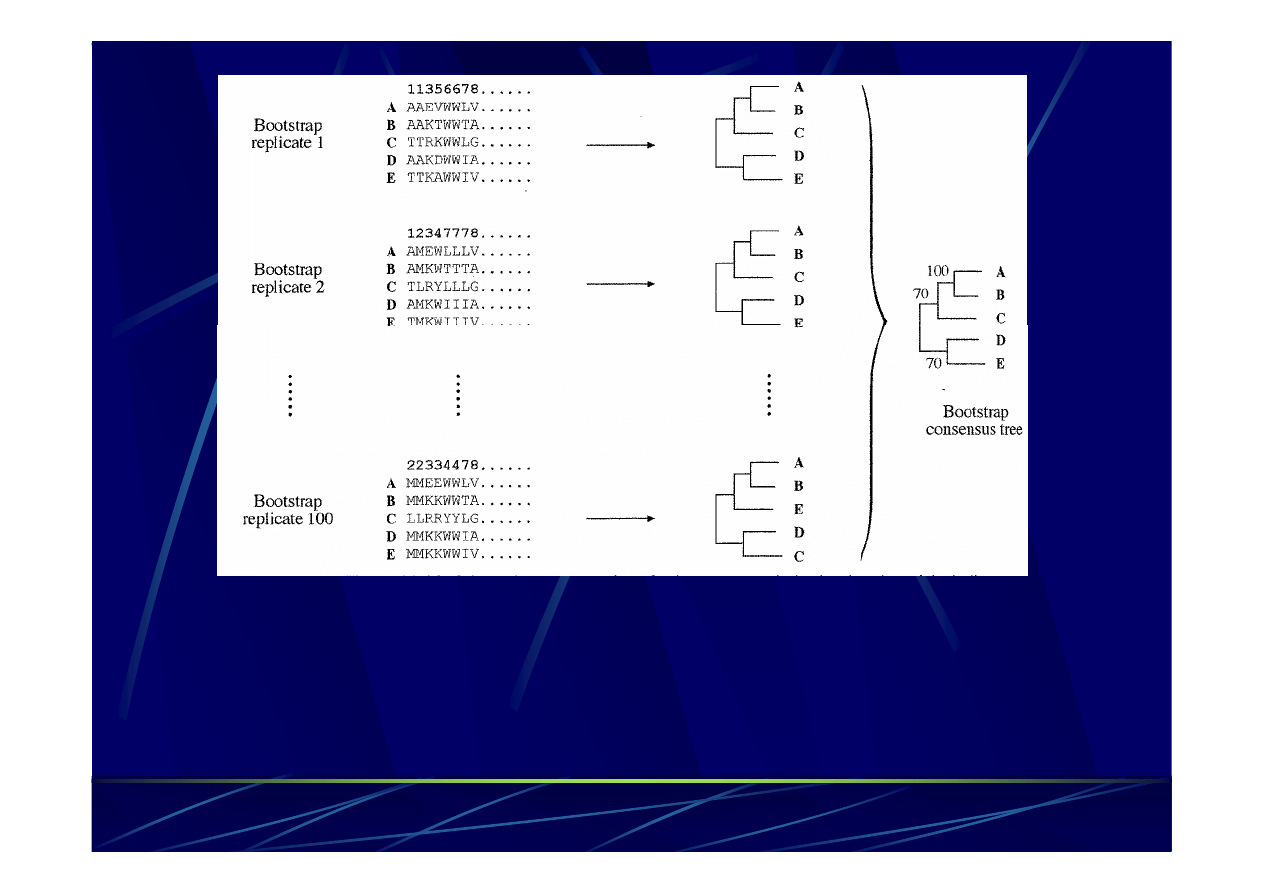
Wartości bootstrap:
> 95% topologia drzewka bardzo prawdopodobna
< 75% nie ma wystarczająco silnych dowodów potwierdzających
taką
topologię
drzewka co wcale nie oznacza, że nie jest ona
prawidłowa!!!
Document Outline
- FILOGENETYKA
- Slide Number 2
- Slide Number 3
- Slide Number 4
- Tree of life (Darwin)
- Slide Number 6
- Slide Number 7
- Taksony mono- i polifiletyczne
- Slide Number 9
- Slide Number 10
- Mechanizmy ewolucji
- Etapy analizy filogenetycznej
- Slide Number 13
- Slide Number 14
- Metoda maksymalnej parsymonii - MP
- Slide Number 16
- Slide Number 17
- Metoda maksymalnej wiarygodności – Maksimum likelihood (ML)
- Maximum likelihood method (ML)
- Slide Number 20
- Slide Number 21
- Slide Number 22
- Slide Number 23
- Slide Number 24
- Slide Number 25
- Slide Number 26
- Slide Number 27
- Slide Number 28
- Slide Number 29
- Slide Number 30
- Slide Number 31
- Slide Number 32
- Slide Number 33
- Slide Number 34
- Slide Number 35
- Slide Number 36
- Slide Number 37
- Slide Number 38
- Slide Number 39
- Slide Number 40
- Etapy analizy filogenetycznej
- Slide Number 42
- Slide Number 43
- Slide Number 44
- Slide Number 45
Wyszukiwarka
Podobne podstrony:
bioinformatyka w13 2008 9 web
bioinformatyka w2 2008 web
bioinformatyka w9 2008 web
bioinformatyka w6 2008 web
bioinformatyka w11 2008 web
bioinformatyka w4 2008 web
bioinformatyka w10 2008 web
bioinformatyka w12 2008 9 web
bioinformatyka w3 2008 web
bioinformatyka w1 2008 web
bioinformatyka w8 2008 web
bioinformatyka w5 2008 web
bioinformatyka w13 2008 9 web
bioinformatyka w2 2008 web
bioinformatyka w9 2008 web
więcej podobnych podstron