
Katowice, 26.01.2015 r.
Sprawozdanie z chemii teoretycznej
Analiza cząsteczki wody przy pomocy programu GAMESS
1. Wstęp teoretyczny:
GAMESS (General Atomic and Molecular Electronic Structure System) jest to program służący do
wykonywania obliczeń z zakresu chemii kwantowej. Do możliwości pakietu należy dokonywanie
obliczeń z użyciem hamiltonianu ab initio (m.in. metoda Hartree – Focke'a, metoda pełnego
mieszania konfiguracji (Full Configuration Interaction, FCI)) z wykorzystaniem wszystkich
powszechnie stosowanych baz funkcyjnych, włączając w to efektywne potencjały rdzenia (Effective
Core Potential, ECP), a także półempiryczne hamiltoniany MNDO, AM1, PM3.
Do pozostałych metod ab initio stosowanych w tym programie należą:
a) metoda perturbacyjna Moellera - Plesseta MP2,
b) metoda sprzężonych klasterów (CC): CCSD, CCSD[T], CCSD(T), CCSD(TQf), renormalized
(R) i complete renormalized (CR) CC: np. R-CCSD(T), R-CCSD(TQ), CR-CCSD(T).
Oprócz tego obliczeń można dokonywać z zastosowaniem metod bazujących na gęstości
elektronowej – DFT (Density Functional Theory).
Pakiet GAMESS posiada również możliwość uwzględnienia sprzężenia spinowo-orbitalnego.
Funkcje falowe jakie oferuje program to te uzyskane w przybliżeniu Hartree – Focka: RHF, ROHF,
UHF oraz za pomocą uogólnionej metody wiązań walencyjnych (GVB) albo też w przybliżeniu
wielokonfiguracyjnym (CI, MCSCF).
GAMESS ma szeroki zakres zastosowań, służy m.in. do:
–
obliczania własności molekularnych np. ładunku populacyjnego, potencjałów
elektrostatycznych, momentów dipolowych, polaryzowalności, hiperpolaryzowalności,
gęstości spinowej,
–
obliczania orbitali zlokalizowanych, optymalizacji geometrii układu, lokalizacji stanów
przejściowych, obliczania dróg reakcji, prawdopodobieństw przejść.
2. Część 1 sprawozdania:
Za pomocą programu, dla analizowanej na zajęciach cząsteczki wody, udało się otrzymać:
–
bazę minimalną (STO), liczbę orbitali zajętych i niezajętych
–
całkowitą energię stanu związanego
–
energię wszystkich orbitali molekularnych
–
energię przyciągania elektron – jądro oraz energie odpychania elektron – elektron,
jądro – jądro
–
wartość momentu dipolowego
–
długość wiązania O – H i odległość między atomami wodoru w H
2
O
a) Baza Slatera (STO) - typ bazy minimalnej składającej się z orbitali typu Slatera.
Obliczenia prowadzone są za pomocą metody SCF, wariantem RHF.
Dzięki procedurze wariacyjnej HF (Hartree – Focke'a) utworzono 7 orbitali molekularnych.
Baza minimalna (STO): 7 orbitali (7 funkcji bazy)
liczba orbitali zajętych – 5 orbitali
liczba orbitali niezajętych – 2 orbitale
b) Całkowita energia stanu związanego: -74,962039 j.at = -196740,8 kJ/mol
Przeliczenia jednostek:
(1 j.at to 6,2773 ∙ 10
2
kcal/mol)
1 j.at
–
6,2773 ∙ 10
2
kcal/mol
-74,962039 j.at
–
x kcal/ mol
x = -47055,920741 kcal/mol
(1 kcal to 4184 J)
-47055,920741 kcal/mol
–
x J/mol
1 kcal
–
4184 J
x = -196740,8046 kJ/ mol
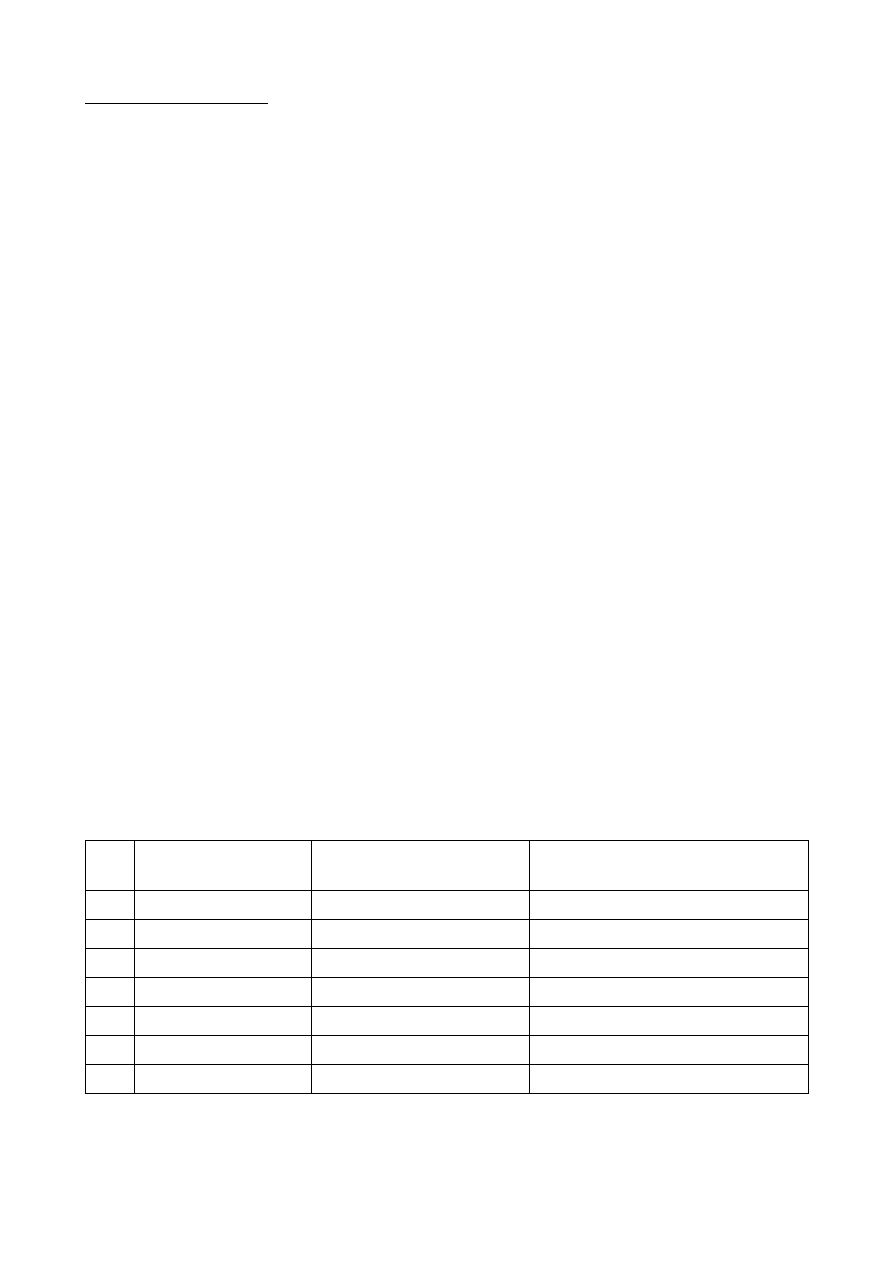
c) Wektory własne wiązania (orbitale molekulane):
nr
Typ reprezentacji:
Energia orbitalu
molekularnego [j.at.]:
Orbital atomowy wnoszący główny
udział do orbitalu molekularnego:
1
A
1
-20,2405
1s, O
2
A
1
-1,2702
2s, O
3
B
2
-0,6212
-
4
A
1
-0,4524
-
5
B
1
-0,3912
-
6
A
1
0,6095
-
7
B
2
0,7494
-
Energia orbitali zajętych ma wartość ujemną, a orbitali niezajętych wartość dodatnią. Orbital nr 5
(2p
x
, O) nie bierze udziału w tworzeniu wiązania, ponieważ nie ma odpowiedniej symetrii, jest
skierowany prostopadle do płaszczyzny wiązania. Na orbitalu nr 5 (2p
x
, O) występuje wolna para

elektronowa. Orbital 2p
y
tlenu (nr 3, reprezentacja B
2
) i orbital 2p
z
tlenu (nr 4, reprezentacja A
1
) są
orbitalami wirtualnymi (niezajętymi).
d) Energia przyciągania elektron – jądro: -197,051 j.at.
Energia odpychania elektron – eletron: 38,249 j.at.
Energia odpychania jądro – jądro: 9,239 j.at.
Największy wkład do całkowitej energii ma energia przyciągania elektron – jądro.
e) Wartość momentu dipolowego: 1,728 D (Debye'a)
DX
DY
DZ
-0
0
1,728
Moment dipolowy występuje w jednym kierunku, wzdłuż osi Z, pokrywającej się z osią obrotu dla
grupy C
2V .
Składowa Z należy do reprezentacji pełnosymetrycznej.
Tablicowa wartość momentu dipolowego wody: 1,84 D
Błąd względny między wartością wyznaczoną, a tablicową: 6,09%
f) Długość wiązania O – H: 0,9525 Å
Odległość między atomami wodoru: 1,51136 Å
(powyższe dane zostały przepisane z przykładu podanego przez program GAMESS)
sin α = 0,775568/0,9525
sin α = 0,7933648
α = 52,5
0
kąt wiązania H-O-H: 2α = 105
0
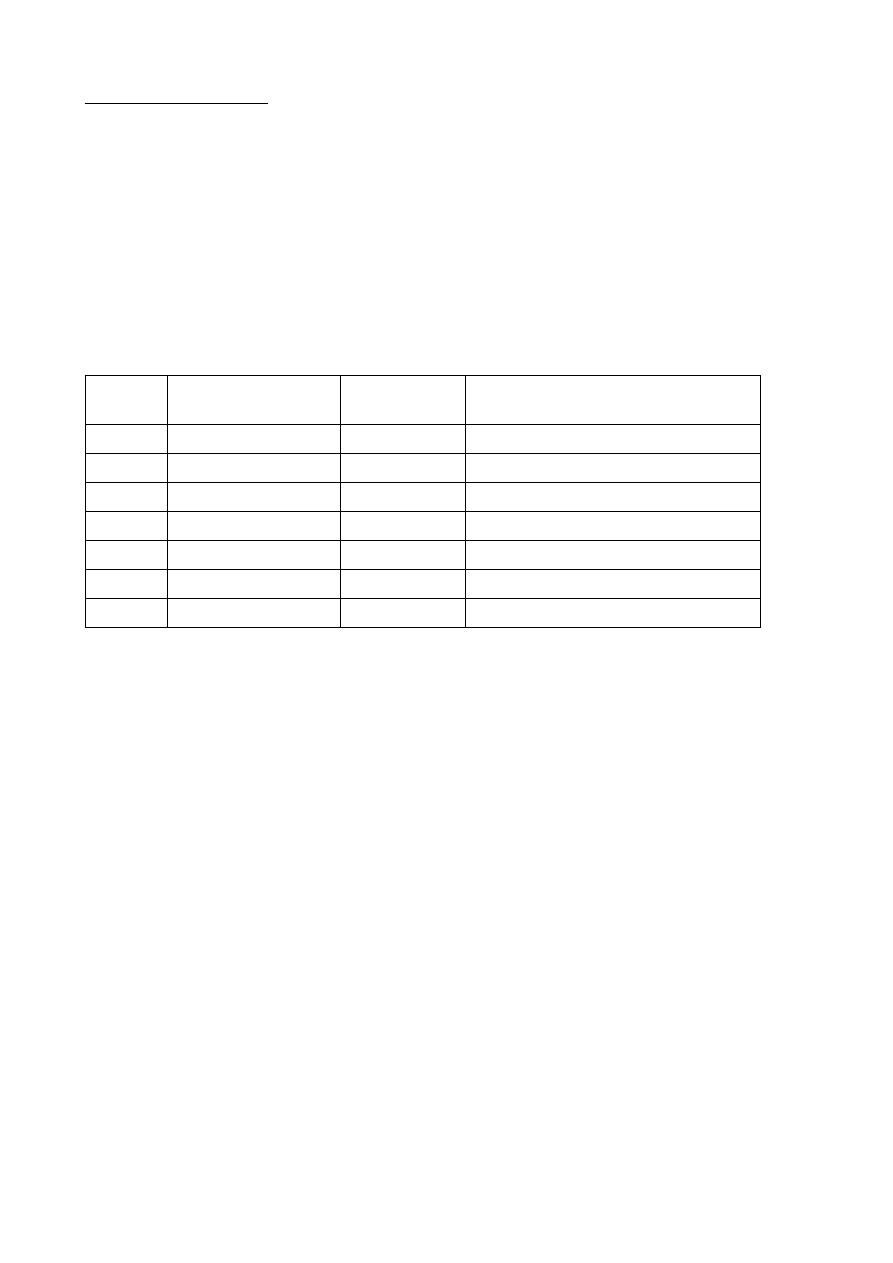
3. Część 2 sprawozdania:
Baza: ACCD (aug-cc-pVDZ) jest rozszerzoną bazą korelacyjno – konsystentną Dunninga,
powstającą przez dodanie jednej dodatkowej funkcji o małym wykładniku do każdego orbitalu
obecnego w bazie. Dla atomu tlenu przypisuje ona: 4s 3p 2d – w sumie 23 funkcje, a dla atomu
wodoru: 3s 2p – w sumie 9 funkcji.
Obliczenia prowadzi się metodą MP2 (metodą perturbacyjną Mollera – Plesseta II rzędu), która
pozwala opisać znaczną część energii korelacji kulombowskiej. Pozwala ona wyznaczyć poprawkę
zarówno do energii elektronowej, jak i funkcji falowej z obliczeń HF.
a) Całkowita energia (11 iteracji): -76,0416632123 j.at. = -199717,5 kJ/mol
b) Wektory własne wiązania (orbitale molekularne):
nr
Energia orbitalu
molekularnego [j.at]:
Typ
reprezentacji:
Orbital atomowy wnoszący główny
udział do orbitalu molekularnego:
1
-20,5762
A
1
1s, O
2
-1,3587
A
1
2s, O
3
-0,7229
B
2
-
4
-0,5856
A
1
-
5
-0,5097
B
1
-
6
0,0356
A
1
-
7
0,0579
B
2
-
W sumie występuje 41 wektorów własnych wiązania, 34 wektory własne zostały pominięte
w powyższej tabelce.
Energia orbitali zajętych ma wartość ujemną, a orbitali niezajętych wartość dodatnią. Orbital nr 5
(2p
x
, O) nie bierze udziału w tworzeniu wiązania, ponieważ nie ma odpowiedniej symetrii, jest
skierowany prostopadle do płaszczyzny wiązania. Na orbitalu nr 5 (2p
x
, O) występuje wolna para
elektronowa.
Orbital HOMO – najwyższy zajęty orbital molekularny, czyli orbital o najwyższej energii wśród
zajętych.
Orbital LUMO – najniższy niezajęty orbital molekularny, czyli orbital o najniżej energii wśród
niezajętych.
HOMO: -0,5097 j.at.
LUMO: 0,0356 j.at.
Różnica energii pomiędzy orbitalem HOMO i LUMO: -0,5097 – 0,0356 = -0,5453 j.at.
Wartość momentu dipolowego: 1,986948 D (Debye'a)
DX
DY
DZ
-0
0
1,986948
Moment dipolowy występuje w jednym kierunku, wzdłuż osi Z, pokrywającej się z osią obrotu dla
grupy C
2V .
Składowa Z należy do reprezentacji pełnosymetrycznej.
Tablicowa wartość momentu dipolowego wody: 1,84 D
Błąd względny między wartością wyznaczoną, a tablicową: 7,98%

4. Część 3 sprawozdania:
Baza: cc-pVTZ należy do baz korelacyjno - konsystentnych Dunninga. Dla atomu tlenu przypisuje
ona: 4s 3p 2d 1f – w sumie 30 funkcji, a dla atomu wodoru: 3s 2p 1d – w sumie 14 funkcji.
Obliczenia prowadzi się metodą MP2 (metodą perturbacyjną Mollera – Plesseta II rzędu), która
opisuje znaczną część energii korelacji kulombowskiej. Pozwala ona wyznaczyć poprawkę zarówno
do energii elektronowej, jak i funkcji falowej z obliczeń HF.
a) Całkowita energia (11 iteracji): -76,0574586573 j.at. = -199759,0 kJ/mol
b) Wektory własne wiązania (orbitale molekularne):
nr
Energia orbitalu
molekularnego [j.at]:
Typ
reprezentacji:
Orbital atomowy wnoszący główny
udział do orbitalu molekularnego:
1
-20,5537
A
1
1s, O
2
-1,3477
A
1
2s, O
3
-0,7133
B
2
-
4
-0,5776
A
1
-
5
-0,5047
B
1
-
6
0,1428
A
1
-
7
0,2044
B
2
-
W sumie występuje 58 wektorów własnych wiązania, 51 wektory własne zostały pominięte
w powyższej tabelce.
Energia orbitali zajętych ma wartość ujemną, a orbitali niezajętych wartość dodatnią. Orbital nr 5
(2p
x
, O) nie bierze udziału w tworzeniu wiązania, ponieważ nie ma odpowiedniej symetrii, jest
skierowany prostopadle do płaszczyzny wiązania. Na orbitalu nr 5 (2p
x
, O) występuje wolna para
elektronowa.
Orbital HOMO – najwyższy zajęty orbital molekularny, czyli orbital o najwyższej energii wśród
zajętych.
Orbital LUMO – najniższy niezajęty orbital molekularny, czyli orbital o najniżej energii wśród
niezajętych.
HOMO: -0,5047 j.at.
LUMO: 0,1428 j.at
Różnica energii pomiędzy orbitalem HOMO i LUMO: -0,5047 – 0,1428 = -0,6475 j.at.
Wartość momentu dipolowego: 2,013474 D (Debye'a)
DX
DY
DZ
0
0
2,013474
Moment dipolowy występuje w jednym kierunku, wzdłuż osi Z, pokrywającej się z osią obrotu dla
grupy C
2V .
Składowa Z należy do reprezentacji pełnosymetrycznej.
Tablicowa wartość momentu dipolowego wody: 1,84 D
Błąd względny między wartością wyznaczoną, a tablicową: 9,43%

Wnioski:
Najlepszą metodą jest metoda omówiona w 3 części sprawozdania (baza cc-pVTZ, metoda MP2),
ponieważ uzyskuje się dzięki niej najniższą wartość energii całkowitej.
Wyszukiwarka
Podobne podstrony:
10.6 poprawione, semestr 4, chemia fizyczna, sprawka laborki, 10.6
28fizyczna, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyc
2 chemia teoretyczna
chemia nr 4-sik, Studia budownictwo pierwszy rok, Chemia budowlana, sprawka z chemii
spr 8.5 obliczenia1, semestr 4, chemia fizyczna, sprawka laborki, 8.5
SprawozdanieNr2Kevcio, Studia budownictwo pierwszy rok, Chemia budowlana, sprawka z chemii
sprawozdaniewapno2, Studia budownictwo pierwszy rok, Chemia budowlana, sprawka z chemii
sprawozdanie chemia 10, sprawka z chemi utp rok I
41, CHEMIA FIZYCZNA SPRAWKA 4 SEM
chemia fizyczna 21, CHEMIA FIZYCZNA SPRAWKA 4 SEM
6, semestr 4, chemia fizyczna, sprawka laborki, 6.11
moje 4, chemia w nauce i gospodarce Uł, semestr V, sprawozdania chemia fizyczna i analityczna uł, Ch
Chemia ostatnie sprawko
Sprawozdanie z ćw nr1 - chemia bud, Studia budownictwo pierwszy rok, Chemia budowlana, sprawka z che
chemia nr3-sik, Studia budownictwo pierwszy rok, Chemia budowlana, sprawka z chemii
sekuła, inżynieria materiałowa - semestr 4, Inżynieria Materiałowa pwr - semestr 4, Chemia Fizyczna,
więcej podobnych podstron