
349
Streszczenie
Wprowadzenie: zespół Crouzona jest rzadkim ze-
społem wad wrodzonych o podłożu genetycznym.
Jego występowanie jest związane z mutacjami genu
FGFR (receptora czynnika wzrostu fibroblastów).
Dominującym objawem jest przedwczesne zarośnię-
cie szwów czaszki, prowadzące do wzrostu ciśnie-
nia śródczaszkowego. Zaburzenia rozwojowe części
twarzowej czaszki obejmują hipoplazję szczęki i
zwężenia łuków zębowych. W badaniu okulistycznym
stwierdza się: hiperteloryzm, wytrzeszcz, zanik ner-
wu wzrokowego. Objawy zwykle występują w okre-
sie okołoporodowym. Szczególnie niebezpieczna jest
progresywna postać zespołu, której symptomy mogą
być początkowo słabo zaznaczone.
Materiał i metody: omówiono epidemiologię, pato-
genezę, objawy kliniczne i radiologiczne, ze szcze-
gólnym uwzględnieniem zmian w obrębie narządu
żucia. Przedstawiono metody diagnostyki dysostosis
craniofacialis oraz diagnostykę różnicową. Zapre-
zentowano objawy zespołu Crouzona u pacjentów,
którzy zgłosili się do Zakładu Ortodoncji IS AM w
Warszawie w celu rozpoczęcia zespołowego leczenia
ortodontyczno– chirurgicznego.
Podsumowanie: leczenie pacjentów z zespołem Cro-
uzona jest wieloletnie i wymaga współpracy specja-
listów wielu dziedzin.
Zespół Crouzona na podstawie piśmiennictwa
i obserwacji własnych
Crouzon’s syndrome – review of literature and own observations
Małgorzata Zadurska
1
, Barbara Siemińska-Piekarczyk
1
,
Konrad Perkowski
1
, Janusz Piekarczyk
2
, Maciej Jagielak
2
Z Zakładu Ortodoncji IS AM w Warszawie
1
p. o. Kierownika: dr n. med. B. Siemińska-Piekarczyk
Z II Kliniki Chirurgii Szczękowo-Twarzowej IS AM w Warszawie
2
Kierownik: prof. dr hab. n. med. J. Piekarczyk
Summary
Introduction: Crouzon’s syndrome is a rare genetic
disorder. Congenital defects are caused by mutations
in FGFR gene (fibroblast growth factor receptor).
The main symptoms include craniosynostosis
leading to increased intracranial pressure.
Impaired development of craniofacial bones
includes hypoplastic maxilla and the narrowing of
dental arches. Ophthalmic examination reveals:
hypertelorism, exophthalmos, atrophy of the optic
nerve. Symptoms occur at birth. The progressive
subtype of the syndrome, with initially poorly
accented symptoms, is especially dangerous.
Material and methods: Epidemiology, pathogenesis,
clinical and radiological manifestations have been
discussed with particular emphasis on masticatory
changes. Diagnostics of dysostosis craniofacialis
and differential diagnosis were discussed. The
paper illustrates Crouzon’s syndrome as manifested
by patients who were admitted to the Department of
Orthodontics of the Medical University in Warsaw
to start comprehensive orthodontic and surgical
treatment.
Conclusion: The management of patients with
Crouzon’s syndrome takes years and requires the
co-operation of many specialists.
HASŁA INDEKSOWE:
zespół Crouzona, craniosynostosis, objawy klinicz-
ne, objawy radiologiczne, zmiany w narządzie żu-
cia
KEYWORDS:
Crouzon’s syndrome, craniosynostosis, clinical
signs, radiological symptoms, changes in masticatory
system
Czas. Stomatol., 2006, LIX, 5, 349-360
Organ Polskiego Towarzystwa Stomatologicznego
http://www.czas.stomat.net

350
M. Zadurska i in.
Czas. Stomatol.,
Zespół Crouzona jest rzadkim zespołem wad
wrodzonych, po raz pierwszy opisanym przez
francuskiego neurologa Octave’a Crouzona
w 1912 roku (10). Innymi określeniami tego
zespołu są: dysostosis craniofacialis, dysto-
nia craniofacialis, „zespół papuziej głowy”.
Jest zaliczany do symetrycznych zaburzeń I
i II łuku skrzelowego. Podstawową jego ce-
chą jest przedwczesne zarośnięcie szwów
czaszki, czyli kraniosynostoza. Po raz pierw-
szy skutki przedwczesnego zarośnięcia szwów
czaszki opisał Sömmering w 1800 roku (31).
Termin „craniostenosis” został wprowadzony
w 1851 roku przez Virchowa (35), który wy-
jaśnił wpływ zaburzenia wzrostu w obrębie
szwów na kształt czaszki i dokonał podziału
tych zniekształceń (3, 26, 19).
Kraniosynostozy występują z częstością 1
na 2000 do 3000 urodzeń. Zespół Crouzona
stanowi około 4,5-5% schorzeń z tej grupy (2,
14, 16), co odpowiada 15 do 16,5 przypadków
na 1000 000 urodzeń (2, 18, 27). Według in-
nych autorów (5, 16) występuje częściej – 1 na
25 000 urodzeń (40 na 1000 000).
Zespół Crouzona dziedziczy się w sposób
autosomalny dominujący, z różną penetracją
genu (2). U około 30 do 60% pacjentów wy-
wiad rodzinny jest ujemny – choroba nie wy-
stępowała u wcześniejszych pokoleń i jest wy-
nikiem nowej mutacji (11). Wykazano związek
między zaawansowanym wiekiem ojca dziec-
ka (powyżej 35 roku życia), a częstością po-
wstawania mutacji genów prowadzących do
wystąpienia zespołu (2, 11, 16).
Kraniostenozy występują w ponad 100 ze-
społach wad wrodzonych (2, 14, 18).W gru-
pie tej stwierdza się mutacje 3 genów kodu-
jących receptory czynnika wzrostu fibrobla-
stów, tzw. FGF (15). Dysostosis craniofacialis
jest skutkiem mutacji genu FGFR 2 (2, 11, 13,
14). Bierze on udział w procesie proliferacji
i dojrzewania chondrocytów. Jest to proces
złożony, podlegający wieloczynnikowej re-
gulacji. Ocenia się, że mutacje genu kodu-
jącego FGFR 2 powodują co najmniej 5 ze-
społów wad wrodzonych w grupie kraniosy-
nostoz. Klinicznie różne zespoły: Crouzona,
Pfeiffera, Aperta, Jacksona-Weissa, Baere-
Stevensona mają zbliżone podłoże genetycz-
ne (13, 14, 15). Mutacje genu FGFR 2 powo-
dują aktywację receptora i zahamowanie pro-
liferacji chondrocytów. Równowaga wzrostu
w obrębie szwów zostaje zachwiana, doj-
rzewające osteoblasty powodują ich zarasta-
nie (3, 13, 14). Do chwili obecnej opisano
około 30 mutacji w genomie FGFR 2, któ-
re mogą spowodować zespół Crouzona (11).
Prowadzone obecnie badania mają na celu
ustalenie, które z tych mutacji odpowiadają
za wystąpienie określonych cech fenotypo-
wych (14). Mutacja genu FGFR 3 odpowia-
da za występowanie dyzostozy czaszkowo-
-twarzowej z towarzyszącym zrogowaceniem
ciemnym (2, 13, 34). Jest to rodzaj hiperkera-
tozy z przebarwieniem skóry o brodawkowa-
tej powierzchni, czasem określanej jako „ak-
samitna”. Klinicznie zrogowacenie ciemne
przybiera różne formy, najczęściej występuje
w okolicy zgięć kończyn i na szyi. Dla zespo-
łu Crouzona charakterystyczne jest wczesne
pojawianie się zmian i ich występowanie w
okolicy ust (36).
Zespół Crouzona należy różnicować z
zespołami: rzekomym Crouzona, Aperta,
Franceschettiego, Griega, Gorlina-Cohena,
Waardenburga, Lowry’ego (19). Objawy ze-
społu Crouzona, mogą wystąpić z różnym na-
sileniem, w zależności od przypadku. Zmiany
zwykle są widoczne już u noworodka lub po-
jawiają się w okresie niemowlęcym. Na szcze-
gólną uwagę zasługuje tzw. progresywna po-
urodzeniowa postać zespołu (9, 12). U pacjen-
tów dotkniętych tą postacią choroby, kształt
czaszki w okresie niemowlęcym jest prawidło-

351
2006, LIX, 5
Zespół Crouzona
wy. Kraniosynostoza rozpoczyna się w okre-
sie pourodzeniowym, prowadząc do wzrostu
ciśnienia śródczaszkowego. Ze względu na
nietypowy przebieg, może zostać rozpoznana
zbyt późno. Nieleczona chirurgicznie, dopro-
wadza do nieodwracalnych uszkodzeń mózgu
i narządu wzroku. Możliwość wystąpienia do-
datkowych zaburzeń i wad rozwojowych in-
nych niż typowe, a także późnego wystąpienia
objawów wskazuje na konieczność prowadze-
nia kompleksowych badań i obserwacji rozwo-
ju pacjentów (8, 27).
Podstawowe objawy są związane z przed-
wczesnym zarośnięciem szwu wieńcowego,
strzałkowego i węgłowego. Zarastają rów-
nież chrząstkozrosty podstawy czaszki, szwy
oczodołów i szczęki. Na skutek przedwcze-
snego zarośnięcia szwów czaszki, zmienia
się jej kształt, co ujawnia się zwykle w okre-
sie okołoporodowym. W celu odróżnienia od
zniekształceń czaszki związanych z porodem,
prowadzona jest diagnostyka radiograficzna.
U noworodków z zespołem Crouzona szwy
kostne zwykle nie są zarośnięte. Synostoza
postępuje z wiekiem, szwy wieńcowy i strzał-
kowy zarastają około 1 roku życia, węgłowy
później (25). Kształt czaszki pacjentów zale-
ży od zakresu stenozy, czasu jej wystąpienia
oraz kolejności i tempa zarastania szwów (7,
13). Wynika on z kierunku kompensacyjnego
wzrostu pozostałych kości. Kierunek wzrostu
czaszki zależy z kolei od położenia szwów ak-
tywnych względem szwów zarośniętych (3).
Spośród opisanych przez Virchowa kształtów
czaszek w kraniostenozach, najczęściej spo-
tykana jest brachycefalia. Jest ona skutkiem
obustronnego zarośnięcia szwu wieńcowego.
Czaszka jest skrócona w wymiarze przed-
nio-tylnym. Czoło jest cofnięte, ustawione
pionowo, z wydatną wypukłością. Nasada
nosa i okolica łuków brwiowych są cofnię-
te. Okolica kości skroniowych jest wyraź-
nie uwypuklona. Poprzeczny wymiar czaszki
jest zwiększony (1, 6, 26).Wzrost w obrębie
szwów podstawy czaszki: klinowo-sitowe-
go, czołowo-sitowego i potyliczno-klinowe-
go jest zahamowany (5, 30). Przedni i tylny
dół czaszki są skrócone. Częstą cechą dyso-
stosis craniofacialis jest występowanie zro-
śniętych kręgów szyjnych, zwykle C2 i C3.
Część autorów wiąże proces zrastania krę-
gów szyjnych ze zrastaniem szwów podsta-
wy czaszki (mają one wspólne pochodzenie
filogenetyczne). Histologicznie szwy podsta-
wy czaszki są bowiem chrząstkozrostami, na-
tomiast kości sklepienia czaszki łączą wię-
zozrosty. Carinci (5) uważa, że punktem, od
którego rozprzestrzenia się stenoza czaszki
są właśnie chrząstkozrosty podstawy, dopie-
ro później proces obejmuje więzozrosty kości
sklepienia. Inną cechą charakterystyczną dla
zespołu Crouzona jest kostnienie w obrębie
więzadła rylcowo-gnykowego (5).
Zmiany morfologiczne w dysostosis cranio-
facialis pociągają za sobą występowanie kom-
plikacji neurologicznych. Powszechnie stwier-
dzane jest postępujące wodogłowie i posze-
rzenie układu komór mózgu. Wzrost ciśnienia
śródczaszkowego jest największym zagroże-
niem. Nieleczony, prowadzi do ślepoty, upo-
śledzenia umysłowego lub śmierci pacjenta.
Występuje u około 60% pacjentów z zespołem
Crouzona (25, 33). U około 30% pacjentów sto-
sowane są zastawki komorowo-otrzewnowe w
celu zmniejszenia ciśnienia śródczaszkowego
(25). Uważa się, że ciśnienie wzrasta do oko-
ło 6 roku życia, a potem utrzymuje się na sta-
łym poziomie lub spada (32).Wzrost ciśnienia
śródczaszkowego jest wypadkową wielu czyn-
ników. Pierwotną przyczyną jest przedwcze-
sne zarośnięcie szwów czaszki i zmniejsze-
nie jej objętości. Wtórnie sytuację pogarszają:
przewlekłe wklinowanie migdałków móżdżku,
zmniejszenie odpływu żylnego, wodogłowie i

352
M. Zadurska i in.
Czas. Stomatol.,
zwężenie dróg oddechowych (33). Badane są
przyczyny wzrostu ciśnienia żylnego w żyłach
szyjnych wewnętrznych. Podkreśla się rolę
zwężenia otworu żyły szyjnej wewnętrznej i
przerostu opony twardej w tej okolicy (28).
Przewlekłe wklinowanie migdałków móżdż-
ku występuje u większości (około 70-75%)
pacjentów z zespołem Crouzona (16). Uważa
się, że jest ono spowodowane wczesnym (do
2 roku życia) zarośnięciem szwu węgłowego
i zmniejszeniem objętości tylnego dołu czasz-
ki (16, 28). Upośledzenie umysłowe nie jest
cechą charakterystyczną dyzostozy czaszko-
wo-twarzowej. Pojawia się wtórnie, jako kon-
sekwencja utrzymującego się zwiększonego
ciśnienia śródczaszkowego (20).Występuje u
około 3% pacjentów (7, 29).
Nie ma obecnie bezpiecznych, bezinwa-
zyjnych metod, którymi można byłoby bez-
pośrednio stwierdzić wzrost ciśnienia śród-
czaszkowego. Rozpoznanie jest najczęściej
stawiane na podstawie danych pośrednich:
obecności wycisków palczastych, powiększe-
nia układu komór mózgu i zmiany ich kształ-
tu, obrzęku tarczy nerwu wzrokowego, zani-
ku nerwu wzrokowego, zniekształceń czasz-
ki, wymiotów oraz zmian w zachowaniu pa-
cjenta. Są to metody mało precyzyjne (21,
32). Nawet niewielkie, przewlekłe podnie-
sienie ciśnienia, przebiegające bez ostrych
objawów, może spowodować upośledzenie
umysłowe pacjenta (25). Badania prowadzo-
ne na pacjentach z niezespołowymi synosto-
zami wykazały, że stosunkowo często (do
47%), pojawiają się upośledzenia lżejszego
stopnia określane jako zaburzenia uczenia się
(oceniane zdolności to czytanie, pisanie, li-
czenie, pamięć wzrokowa, koncentracja). Nie
wykazano związku między występowaniem
zaburzeń umysłowych a wykonanymi zabie-
gami chirurgicznymi odbarczającymi mózg.
Częstość występowania zaburzeń była po-
dobna u pacjentów nieleczonych chirurgicz-
nie, jak i u pacjentów leczonych, niezależnie
od wieku (17, 21, 32).
Najczęstszą i najbardziej wyraźną cechą ze-
społu Crouzona, ujawniającą się często tuż
po urodzeniu, jest wytrzeszcz spowodowa-
ny spłyceniem oczodołów (1, 6). Spłycenie
oczodołów jest niejednokrotnie tak znacz-
ne, że powoduje zwichnięcia gałek ocznych.
Cechą charakterystyczną jest także hiperte-
loryzm i powiększone wejście do oczodołów
(5, 6). Wytrzeszcz, powodujący niedomykanie
szpary powiekowej może prowadzić do kera-
topatii ekspozycyjnej (obrzęki, owrzodzenia
rogówki). Zaburzenia budowy przewodów no-
sowo-łzowych mogą dodatkowo pogarszać ten
stan (1, 32). Obrzęk tarczy nerwu wzrokowe-
go, występujący u około 35% pacjentów, jest
skutkiem wzrostu ciśnienia śródczaszkowego.
Proces degeneracji postępuje, prowadząc do
zaniku nerwu wzrokowego, co stwierdza się u
około 10% pacjentów (25). Innymi opisywa-
nymi objawami są: ograniczenie pola widze-
nia, krótkowzroczność, astygmatyzm, osłabie-
nie ostrości wzroku, oczopląs, zezy rozbieżne,
ślepota (16, 18). Ograniczenie pola widzenia
jest zwykle jednym z pierwszych objawów za-
grożenia ślepotą (32). Rzadko spotykaną ano-
malią jest niedorozwój powiek (1).
Dla rysów twarzy charakterystyczne są:
skrócenie górnej wargi, zahamowanie wzro-
stu środkowego piętra twarzy (retrognacja),
wklęsły profil. W obrębie narządu żucia naj-
częściej obserwowanymi zaburzeniami są:
hipoplastyczna, wąska szczęka, zwężenie łu-
ków zębowych, podniebienie gotyckie, stło-
czenia i zaburzenia liczby zębów (hipodoncja
lub nadliczbowość), makrodoncja, wady z gru-
py zgryzów otwartych i przodozgryzów (7).
Występowanie rozszczepu podniebienia pier-
wotnego i/lub wtórnego nie jest cechą charak-
terystyczną zespołu.

353
2006, LIX, 5
Zespół Crouzona
W analizie cefalometrycznej obserwuje się:
skrócenie szczęki, retrognację (zmniejszenie
kąta SNA), wklęsły profil twarzy, zwiększo-
ną szerokość żuchwy (wymiar Go-Go), pra-
widłową długość gałęzi i podstawy żuchwy,
prawidłową wartość kąta żuchwy, kąt SNB w
granicach normy lub zmniejszony, zmniejszo-
ną szerokość twarzy (rozmiar Zy-Zy) (6,24).
Inni autorzy wskazują na zwiększenie długo-
ści gałęzi żuchwy (23, 24).
Wyniki badań stanu higieny jamy ustnej do-
konane w Wielkiej Brytanii są zaskakujące.
Higiena jamy ustnej, gorsza u pacjentów z
zespołem Crouzona, niż w grupie kontrolnej
(wyższe wskaźniki płytki, zapalenia dziąseł)
jest spowodowana stłoczeniami i trudnością z
oczyszczaniem zębów. Jednocześnie, wartości
PUW/PUWP są niższe niż w grupie kontrol-
nej. Wyniki te są tłumaczone regularnymi wi-
zytami kontrolnymi i większą świadomością
stomatologiczną pacjentów z dysostosis cra-
niofacialis (22).
Często opisywane są zaburzenia dotyczą-
ce drożności dróg oddechowych w zespole
Crouzona. Zwężenia górnych i dolnych dróg
oddechowych występują u około 40% pacjen-
tów. Spowodowane są one skróceniem pod-
stawy czaszki i szczęki, zwężeniem lub nawet
zarośnięciem nozdrzy, jam nosa i nosogardła.
Wymiary nosogardła (wysokość i głębokość)
oraz jamy nosa są zmniejszone. Zmiany te,
wraz z przerostem tkanki chłonnej nosogardła,
wymuszają ustny tor oddychania. Zwężenie
dróg oddechowych czasem wymaga wykona-
nia zabiegu tracheotomii (29). Często wystę-
pują bezdechy senne o różnym stopniu na-
silenia. Przypadki występowania chrzęstnego
mankietu w tchawicy są opisywane wyłącznie
u pacjentów z kraniosynostozami, w tym dyzo-
stozie czaszkowo-twarzowej. Mogą być ogra-
niczone do kilku łuków, a w najcięższych po-
staciach obejmować tchawicę, jej rozwidlenie
i oskrzela (5, 29). Nieleczone zwężenie dróg
oddechowych może prowadzić do powstania
serca płucnego (29).
W obrębie ucha środkowego spotykane są
deformacje różnego stopnia, najczęściej wy-
stępuje niedosłuch przewodzeniowy. U około
55% pacjentów obserwuje się zwężenie prze-
wodów słuchowych, u około 13 % pacjentów
są one niewykształcone. Pojawiające się wtór-
nie przewlekłe stany zapalne ucha środkowego
pogłębiają istniejące zaburzenia słuchu.
Innym spotykanym zaburzeniem towarzy-
szącym zespołowi Crouzona jest choroba
Meniera. Poza tym opisano przypadki braku
błony bębenkowej, ankylozy młoteczka, de-
formacji strzemiączka, błędnika i ucha środ-
kowego (16, 29).
W przeszłości, cechami różnicującymi ze-
spół Crouzona z innymi zespołami wad wro-
dzonych, m in. z zespołem Aperta, były prawi-
dłowo rozwinięte kończyny (2, 4). Klinicznie
zauważalne zaburzenia rozwojowe dłoni i stóp
są rzadko spotykane. Badania radiologiczne
wskazują jednak na występowanie niewiel-
kich, subklinicznych zmian, m. in. skrócenia
lub hipoplazji paliczków oraz zrostów między
kościami nadgarstka (4). Zmiany te nie mają
wpływu na powstawanie i dojrzewanie jąder
kostnienia w obrębie nadgarstka. Świadczy to
o możliwości właściwej oceny wieku kostne-
go u pacjentów z dysostosis craniofacialis na
podstawie zdjęć nadgarstka. Opóźnienie wie-
ku kostnego nie jest cechą charakterystyczną
zespołu.
Dla określenia ekspresji klinicznej, pod-
stawowe znaczenie mają zdjęcia rentgenow-
skie czaszki (ocena szwów, asymetrii, zabu-
rzeń kształtu kości). Tomografia komputerowa
umożliwia ponadto ocenę mózgowia i innych
tkanek miękkich. W niemowlęctwie propono-
wane jest wykonywanie pomiarów kraniome-
trycznych na zdjęciach TK, zamiast na stan-

354
M. Zadurska i in.
Czas. Stomatol.,
dardowych odległościowych zdjęciach bocz-
nych głowy, co jest uwarunkowane między
innymi trudnościami technicznymi przy wy-
konywaniu zdjęć rentgenowskich głowy u nie-
mowląt (6).
Zespół Crouzona może być rozpoznany u
płodu, za pomocą rezonansu magnetycznego
i badania ultrasonograficznego. W przypad-
ku dodatniego wywiadu rodzinnego wskaza-
ne jest poradnictwo genetyczne i badania pre-
natalne. Opisano badania genetyczne blasto-
cyst przed implantacją, u rodziców z zespołem
Crouzona planujących zabieg zapłodnienia in
vitro. Tylko te blastocysty, w których wyklu-
czono wystąpienie zespołu były implantowa-
ne (2).
Obserwacje
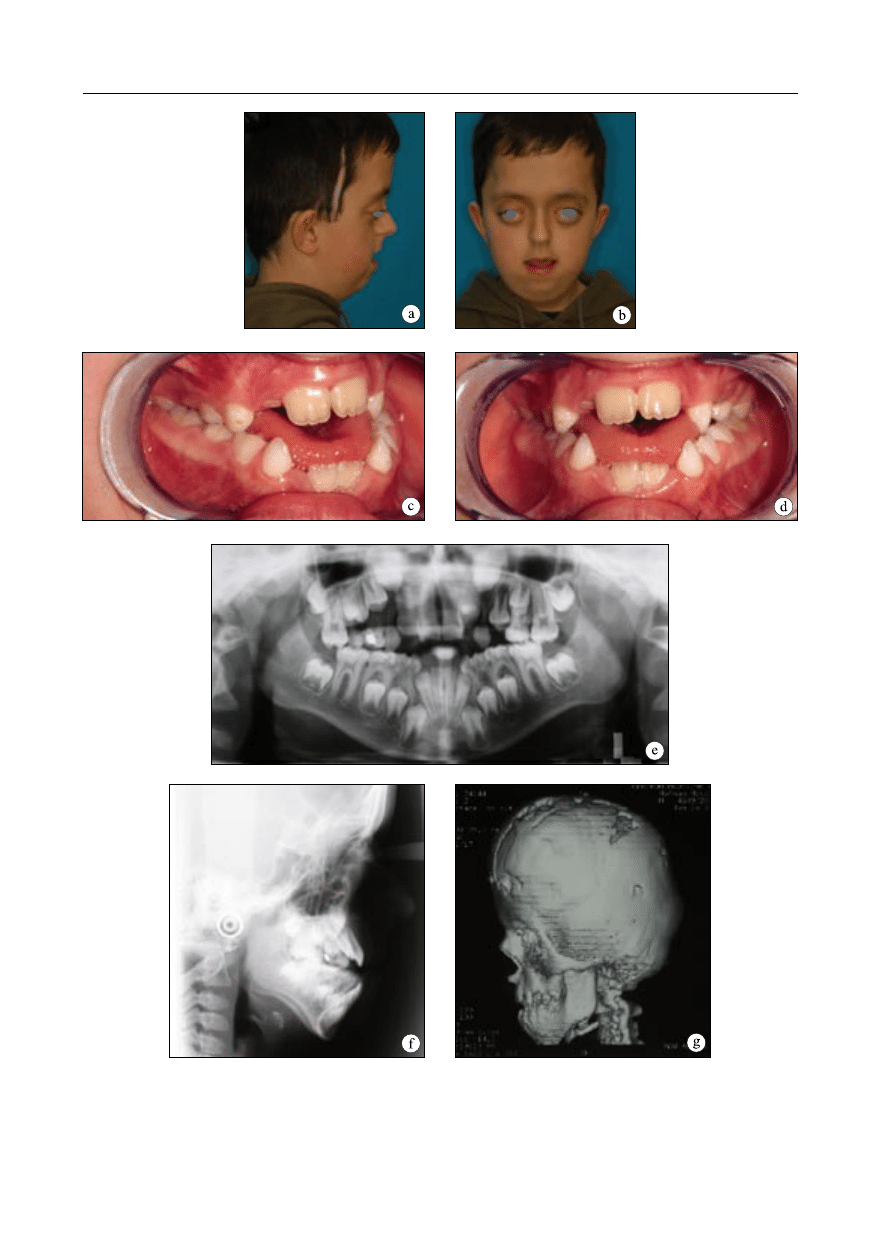
Obserwacja 1
Chłopiec zgłosił się do leczenia w Zakładzie
Ortodoncji IS AM w Warszawie w wieku 8 lat
i 3 miesięcy. Zespół Crouzona nie występował
w rodzinie pacjenta. Schorzenie rozpoznano w
niemowlęctwie.
Wykonano badania kliniczne ogólne oraz
miejscowe – zewnątrzustne, wewnątrzustne i
czynnościowe. Rozwój fizyczny chłopca prze-
biegał na poziomie 90 centyla. Rysy twarzy
były charakterystyczne dla zespołu Crouzona:
czaszka skrócona w wymiarze przednio-tyl-
nym, nieco zwiększony poprzeczny wymiar
czaszki, wysokie, pionowo ustawione czoło z
wydatną wypukłością, zahamowanie wzrostu
środkowego piętra twarzy, spłaszczenie oko-
licy podoczodołowej i wklęsły profil, wąski,
„papuzi” nos, skrócenie górnej wargi. Badanie
układu wzrokowego wykazało: spłycenie
oczodołów, hiperteloryzm, wytrzeszcz i zez
rozbieżny (ryc. 1a, b).
W badaniu wewnątrzustnym stwierdzono:
uzębienie mieszane, przodozgryz całkowity,
zgryz otwarty całkowity, nieprawidłowości zę-
bowe, wysoko wysklepione podniebienie, łuki
zębowe zwężone, kształtu zbliżonego do trój-
kąta (ryc. 1c, d).
Zaburzeniom morfologicznym towarzyszy-
ły zaburzenia czynności narządu żucia: prze-
trwały niemowlęcy typ połykania, ustny tor
oddychania, upośledzone żucie, wada wymo-
wy (seplenienie).
Wykonano analizę modeli diagnostycznych
i zdjęcia rentgenowskie (pantomogram, tele-
rentgenogram boczny głowy – ryc. 1e, f).Wcze-
śniej, w związku z prowadzonym leczeniem
neurochirurgicznym, wykonano tomografię
komputerową (ryc. 1g). Na zdjęciu pantomo-
graficznym stwierdzono obecność wszystkich
zawiązków zębów stałych. Badanie TK wy-
kazało przewlekłe wklinowanie migdałków
móżdżku i obecność wycisków palczastych,
które są pośrednim objawem podwyższonego
ciśnienia śródczaszkowego. W związku z po-
stępującym wodogłowiem u pacjenta w wieku
1,5 roku wykonano zabieg wszczepienia za-
stawki komorowo-otrzewnowej w celu zmniej-
szenia ciśnienia śródczaszkowego. Została ona
usunięta po dwóch miesiącach. Rozwój umy-
słowy pacjenta jest prawidłowy.
Analiza cefalometryczna telerentgenogramu
bocznego głowy wykazała retrognatyczny typ
twarzy, neutralną sagitalną relację podstaw.
Kąt SNA wynosił 65,1°, SNB = 67,8°. Kąt
ANB wskazywał na III klasę szkieletową i wy-
nosił -2,8°. Pomiar Wits -9,9 mm. Kąt żuchwy
wykazywał prawidłową wielkość (GntGoAr
= 131,1°). Długość trzonu i gałęzi żuchwy
były prawidłowe (odpowiednio 62,3 mm i 45,7
mm). Długość szczęki była prawidłowa i wy-
nosiła 40,4 mm. Żuchwa wykazywała znacz-
ną posteriorotację, SGo/NMe = 54,5%. Kąt
nosowo-wargowy był powiększony i wynosił
125,6°. Kąt NS – ML wynosił 60,2°.
355
2006, LIX, 5
Zespół Crouzona
Ryc. 1. Obserwacja 1. Rysy twarzy 8-letniego pacjenta z zespołem Crouzona; a – profil prawy, b – en face.
Warunki zgryzowe; c – strona prawa, d – en face. Badania radiologiczne; e – pantomogram, f – teleradio-
gram boczny głowy, g – tomografia komputerowa, rekonstrukcja 3D.
356
M. Zadurska i in.
Czas. Stomatol.,
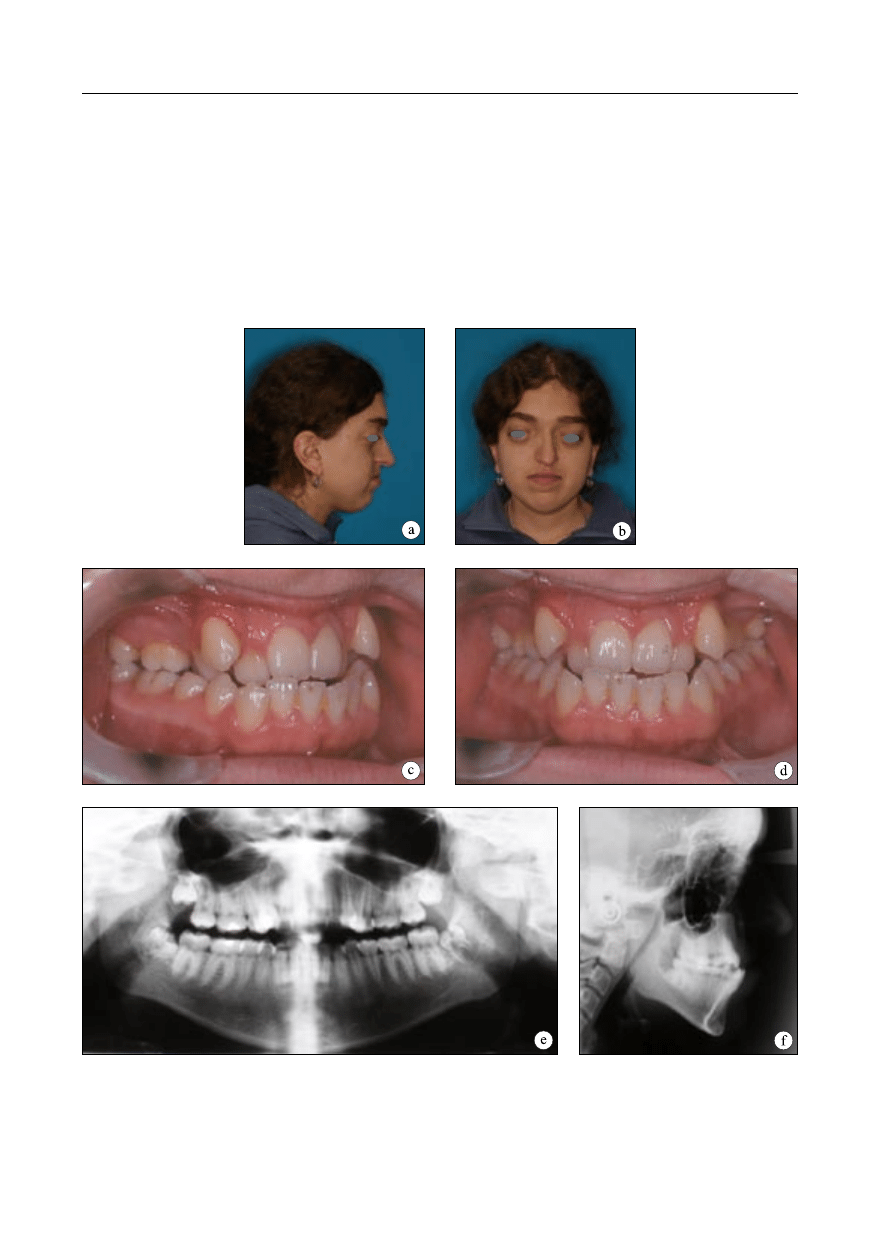
Obserwacja 2
Pacjentka została skierowana z Kliniki
Chirurgii Szczękowo-Twarzowej do leczenia
w Zakładzie Ortodoncji IS AM w Warszawie
w wieku lat 18. Zespół Crouzona nie wystę-
pował w rodzinie. Schorzenie rozpoznano w
niemowlęctwie. Przeprowadzono badania kli-
niczne ogólne oraz miejscowe – zewnątrzust-
ne, wewnątrzustne i czynnościowe. Rysy twa-
rzy pacjentki były charakterystyczne dla ze-
społu Crouzona: czaszka skrócona w wymia-
rze przednio-tylnym, zwiększony poprzecz-
ny wymiar czaszki, zahamowanie wzrostu
środkowego piętra części twarzowej czaszki,
Ryc. 2. Obserwacja 2. Rysy twarzy 18-letniej pacjentki z zespołem Crouzona; a – profil prawy, b – en face.
Warunki zgryzowe; c – strona prawa, d – en face. Badania radiologiczne; e – pantomogram, f – teleradio-
gram boczny głowy

357
2006, LIX, 5
Zespół Crouzona
spłaszczenie okolicy podoczodołowej i wklę-
sły profil, skrócenie wargi górnej. Badaniem
okulistycznym rozpoznano: spłycenie oczodo-
łów, wytrzeszcz, hiperteloryzm, zez rozbieżny
i oczopląs (ryc. 2a, b). U pacjentki występowa-
ło wodogłowie, niewymagające wszczepienia
zastawki. Nie wykazuje ona żadnych niepra-
widłowości rozwoju umysłowego.
W badaniu wewnątrzustnym stwierdzono:
uzębienie stałe, przodozgryz całkowity, zgryz
krzyżowy częściowy boczny obustronny, nie-
prawidłowości zębowe – stłoczenia, wysoko
wysklepione gotyckie podniebienie, łuki zę-
bowe zwężone, górny w kształcie zbliżonym
do trójkąta, dolny trapezoidalny (ryc. 2c, d).
Zaburzeniom morfologicznym w obrębie na-
rządu żucia towarzyszyły zaburzenia czynno-
ści: ustny tor oddychania, upośledzone żucie,
wada wymowy (seplenienie).
Wykonano analizę modeli diagnostycz-
nych i zdjęcia rentgenowskie (pantomogram
i telerentgenogram boczny głowy– ryc. 2e, f).
Pantomogram wykazał obecność zawiązków
wszystkich zębów stałych, zdjęcie teleradio-
graficzne – obecność charakterystycznych dla
zespołu Crouzona wycisków palczastych. W
analizie cefalometrycznej telerentgenogramu
bocznego głowy stwierdzono retrognatyczny
profil twarzy i doprzednią sagitalną relację
podstaw. Kąt SNA = 68,6°, SNB = 72,9°. Kąt
ANB = -4,4°, wskazywał na III klasę szkieleto-
wą. Pomiar Wits wynosił -3,9 mm. Kąt żuchwy
wykazywał prawidłową wielkość (GntGo Ar
= 122,2°).
Długość trzonu żuchwy była prawidłowa
(72,7 mm).
Długość gałęzi żuchwy była zwiększona
i wynosiła 67,9 mm. Długość szczęki była
zmniejszona i wynosiła 45,4 mm. Żuchwa wy-
kazywała anteriorotację (SGo/NMe 66,2 %).
Kąt NS – ML wynosił 41,8°, kąt nosowo-war-
gowy był w normie i wynosił 104,4°.
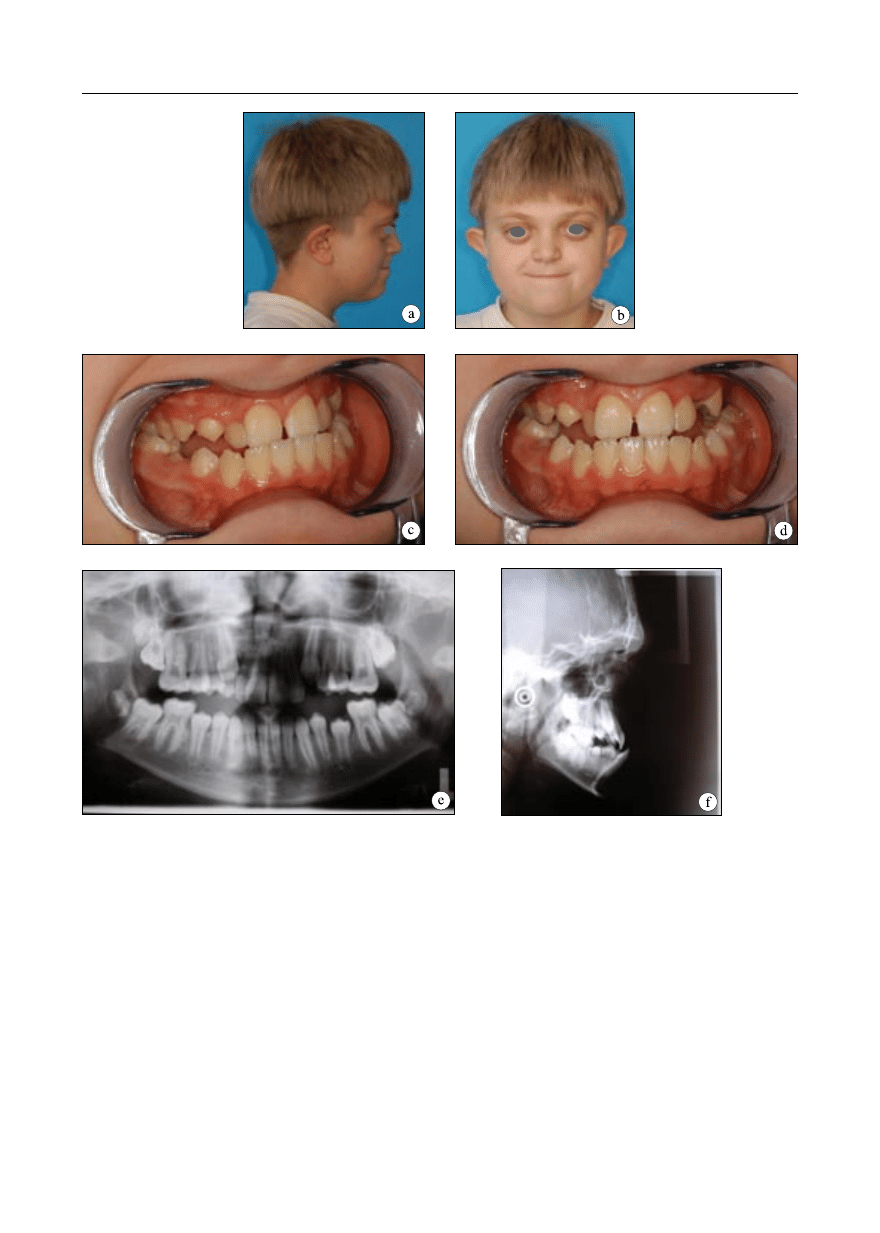
Obserwacja 3
Pacjent zgłosił się do leczenia w Zakładzie
Ortodoncji IS AM w Warszawie w wieku lat
12. Zespół Crouzona nie występował w ro-
dzinie. Schorzenie rozpoznano w niemow-
lęctwie. Przeprowadzono badania klinicz-
ne ogólne oraz miejscowe – zewnątrzustne,
wewnątrzustne i czynnościowe. Rysy twarzy
pacjenta były charakterystyczne dla zespo-
łu Crouzona: czaszka skrócona w wymia-
rze przednio-tylnym, zwiększony poprzeczny
wymiar czaszki, zahamowanie wzrostu środ-
kowego piętra twarzy, spłaszczenie okolicy
podoczodołowej, wklęsły profil i skrócenie
górnej wargi. Małżowiny uszne odstające.
Z objawów ocznych stwierdzono: spłycenie
oczodołów, wytrzeszcz, hiperteloryzm (ryc.
3a, b). W badaniu okulistycznym stwierdzo-
no zanik lewego nerwu wzrokowego i wadę
wzroku. Pacjent nie wykazuje żadnych nie-
prawidłowości rozwoju umysłowego.
W badaniu wewnątrzustnym stwierdzono:
uzębienie mieszane, przodozgryz całkowity,
zgryz krzyżowy częściowy boczny obustron-
ny, nieprawidłowości zębowe – stłoczenia zę-
bów, wysoko wysklepione gotyckie podnie-
bienie, zwężony górny łuk zębowy, w kształ-
cie zbliżonym do trójkąta, dolny prawidłowej
szerokości (ryc. 3c, d). Zaburzeniom morfolo-
gicznym w obrębie narządu żucia towarzyszy-
ły zaburzenia czynności: ustny tor oddychania
i upośledzone żucie.
Wykonano analizę modeli diagnostycz-
nych i zdjęcia rentgenowskie: pantomogram
i telerentgenogram boczny głowy (ryc. 3e, f).
Telerentgenogram wykazał obecność wycisków
palczastych, pantomogram – obecność zawiąz-
ków wszystkich zębów stałych. W analizie cefa-
lometrycznej telerentgenogramu bocznego gło-
wy stwierdzono retrognatyczny profil twarzy i
doprzednią sagitalną relację podstaw. Kąt SNA
= 75,2°, SNB = 81,3°. Kąt ANB = -6,1° wska-
358
M. Zadurska i in.
Czas. Stomatol.,
zywał na III klasę szkieletową. Pomiar Wits wy-
nosił -9,9 mm. Kąt żuchwy wykazywał prawi-
dłową wielkość (GntGoAr = 119,1°).
Długość trzonu żuchwy była prawidłowa
(70,7 mm). Długość gałęzi żuchwy była zwięk-
szona i wynosiła 59,3 mm. Długość szczęki
wynosiła 45,8 mm. Żuchwa wykazywała ante-
riorotację (SGo/NMe = 69,3%). Kąt NS – ML
wynosił 32°, kąt nosowo-wargowy był zwięk-
szony i wynosił 124,5°.
Obserwowani pacjenci wykazują charakte-
rystyczne dla zespołu Crouzona zniekształ-
cenia mózgowej i twarzowej części czasz-
ki: skrócenie w wymiarze przednio-tylnym,
wklęsły profil, zahamowanie wzrostu środko-
wego piętra twarzy, skrócenie górnej wargi.
Ryc. 3. Obserwacja 3. Rysy twarzy 12 – letniego pacjenta z zespołem Crouzona; a– profil prawy, b– en face.
Warunki zgryzowe; c – strona prawa, d – en face. Badania radiologiczne; e – patomogram, f– teleradiogram
boczny głowy

359
2006, LIX, 5
Zespół Crouzona
Wspólnymi cechami w obrębie narządu wzro-
ku były: hiperteloryzm, wytrzeszcz i zez roz-
bieżny. Stwierdzono u nich wzrost ciśnienia
śródczaszkowego, którego pośrednim obja-
wem była obecność wycisków palczastych na
zdjęciach rentgenowskich czaszki.
W obrębie narządu żucia obserwowano:
przodozgryzy całkowite, stłoczenia zębów,
wysoko wysklepione podniebienia. Wadom
zgryzu towarzyszyły zaburzenia czynności
narządu żucia.
Analiza cefalometryczna zdjęć bocznych
głowy wykazała zmniejszenie kąta SNA u troj-
ga pacjentów. Budowa żuchwy pierwszego,
rozwijającego się pacjenta jest prawidłowa i
zgodna z wynikami badań Carinci (5). U po-
zostałych pacjentów stwierdzono zwiększoną
długość gałęzi żuchwy, co potwierdza badania
Perzyny i Ostrowskiego (30).
Leczenie pacjentów z zespołem Crouzona
jest wieloletnie i wymaga współpracy specja-
listów wielu dziedzin. Obserwowani pacjen-
ci rozpoczęli obecnie leczenie ortodontyczne,
które jest przygotowaniem do właściwego le-
czenia chirurgicznego.
Piśmiennictwo
1. Abdallah A. M., Nelson L. B.: A severe form
of Crouzon’s disease: Clinical and radiologi-
cal correlation. J.Pediatr. Ophthalmology and
Strabismus, 1998, 35, 4, 230-231. – 2. Abou-
Sleiman P. M., Apessos A., Harper J. C., Serhal
P., Delhanty J. D. A.:Pregnancy following preim-
plantation genetic diagnosis for Crouzon syndro-
me. Mol. Hum. Reprod., 2002, 8, 3, 304-309. – 3.
Alden T. D., Lin K. Y., Jane A.: Mechanisms of
premature closure of cranial sutures. Child’s Nerv.
Syst., 1999, 15, 670-675. – 4. Anderson P. J., Hall
C. M., Evans R. D., Jones B. M., Hayward R. D.:
Hand anomalies in Crouzon syndrome. Skeletal
Radiol., 1997, 26, 113-115. – 5. Carinci F.,
Avantaggiato A., Curioni C.: Crouzon Syndrome:
Cephalometric analysis and evaluation of patho-
genesis. Cleft Palate Craniofac. J., 1994, 31, 3,
201-209. – 6. Carr M., Posnick J. C., Pron G.,
Armstrong D.: Cranio-orbito-zygomatic mea-
surments from standard CT scans in unoperated
Crouzon and Apert infants: comparison with nor-
mal controls. Cleft Palate Craniofac. J., 1992, 29,
2, 129-136. – 7. Charazińska Z.: Z problematyki
leczenia szczękowo-ortopedycznego pacjentów z
dyzostozą czaszkowo-twarzową. Czas. Stomatol.,
1977, XXX, 10, 831-840. – 8. Cohen S.R., Dauser
R.C., Górski J.L.: Insidious onset of familial cra-
niosynostosis. Cleft Palate Craniofac. J., 1993, 30,
4, 401-405. – 9. Conolly J.P., Gruss J., Seto M.L.,
Whelan M.F., Ellenbogen R., Weiss A., Buchman
S.R., Cunningham M.L.: Progressive postnatal
craniosynostosis and increased intracranial pres-
sure. Plast. Reconstr. Surg., 2004, 15, 113, 1313-
1323. – 10. Crouzon O.: Dysostose cranio– faciale
hereditaire. Bull. Soc. Med. Hop. Paris, 1912, 33,
545– 555.
11. Glaser R. L., Jiang W., Boyadjiev S. A., Tran
A.K., Zachary A.A., Van Maldergem L., Johnson
D., Walsh S., Oldridge M., Wall S. A., Wilkie O.
M., Wang Jabs E.:Paternal origin of FGFR2 muta-
tions in sporadic cases of Crouzon syndrome and
Pfeiffer syndrome. Am. J. Hum. Genet., 2000, 66,
768-777. – 12. Hoefkens M. F., Vermeij-Keers C.,
Vaandrager J. M.: Crouzon syndrome: phenotypic
signs and symptoms of the postnatally expressed
subtype. J. Craniofac. Surg., 2004, 15, 233-240.
– 13. Hollway G. E., Suthers G. K., Haan E. A.,
Thompson E., David D. J., Gecz J., Mulley J. C.:
Mutation detection in FGFR 2 craniosynostosis
syndromes. Hum. Genet., 1997, 99, 251-255. – 14.
Ibrahimi O. A., Zhang F., Eliseenkowa A. V., Itoh
N., Linhardt R. J., Mohamadi M.: Biochemical
analysis of pathogenic ligand-dependent FGFR2
mutations suggests distinct pathophysiologi-
cal mechanisms for craniofacial and limb abnor-
malities. Hum. Mol. Genet., 2004, 13, 19, 2313-
2324. – 15. Ingersoll R. G., Paznekas W. A., Tran
A. K., Scott A. F.: Fibroblast growth factor recep-
tor 2 (FGFR2): genomic sequence and variations.
Cytogenet. Cell Genet., 2001, 94, 121-126. – 16.
Kabbani H., Raghuveer T. S.: Craniosynostosis.
Am. Family Physician, 2004, 69, 12, 2863-2869.
– 17. Kapp-Simon K. A.:Mental development and

360
M. Zadurska i in.
Czas. Stomatol.,
learning disorders in children with single suture
craniosynostosis. Cleft Palate Craniof. J., 1998,
35, 3, 197-203. – 18. Khan S. H., Nischal K. K.,
Dean F., Hayward R. D., Walker J.: Visual out-
comes and amblyogenic risk factors in cranio-
synostotic syndromes: a review of 141 cases. Br.
J. Opht., 2003, 87, 999-1003. – 19. Kopyść Z.
(red.): Kompendium zespołów i rzadkich cho-
rób dziecięcych. Wydawnictwo Lekarskie PZWL,
Warszawa, 1984, 130-131. – 20. Manikoglu B.,
Manikoglu A.: A father and son with nonsevere
form of Crouzon’s syndrome. Ear, Nose Throat J.,
2000, 79, 5, 368-369.
21. Mouradian W. E.: Controversies in the di-
agnosis and management of craniosysnostosis. A
panel discussion.Cleft Palate Craniof. J., 1998, 35,
3, 190-193. – 22. Mustafa D., Lucas V. S., Junod
P., Evans R., Mason C., Roberts G. J.: The den-
tal health and caries-related microflora in children
with craniosynostosis.Cleft Palate-Craniofac. J.,
2001, 38, 6, 629-635. – 23. Perzyna B., Ostrowski
J.: Wielkość żuchwy w zespole Crouzona.
Czas. Stomatol., 1988, XLI, 9, 565-573. – 24.
Pospieszyńska M.: Ocena cefalometryczna zmian
twarzowej części czaszki w zespole Crouzona.
Czas. Stomatol., 2005, LVIII, 2, 124-128. – 25.
Renier D., Lajeunie E., Arnaud E., Marchac D.:
Management of craniosynostoses. Child’s Nerv.
Syst., 2000, 16, 645-658. – 26. Richtsmeier J. T.,
Grausz H. M., Morris G. R., Marsh J. L., Vannier
M. W.: Growth of the cranial base in craniosynos-
tosis. Cleft Palate Craniofac. J. 1991, 28, 1, 55-
-67. – 27. Rokicki W., Rokicka A.: Współistnienie
zespołu Crouzona z ubytkiem przegrody między-
komorowej serca. Wiad. Lek., 2003, LVI, 5-6. –
28. Rollins N., Booth T., Shapiro K.: MR venog-
raphy in children with complex craniosynosto-
sis. Pediatr. Neurosurg., 2000, 32, 308-315. – 29.
Scheid S. C., Spector A. R., Luft J. D.: Tracheal
cartilaginous sleeve in Crouzon syndrome. Int.
J. Pediatr. Otorhinolaryngol., 2002, 65, 147-152.
– 30. Sgouros S., Natarajan K., Hockley A. D.,
Goldin J. H., Wake M.: Skull base growth in cra-
niosynostosis. Pediatr. Neurosurg., 1999, 31, 281-
293.
31. Sommering S. T.: Vom baue des mensch-
lichen korpers. 1st ed., Leipzig, Germany, Voss
1800. – 32. Stavrou P., Sgouros S., Willshaw H.
E., Goldin J. H., Hockley A. D., Wake M. J. C.:
Visual failure caused by raised intracranial pres-
sure in craniosynostosis. Child’s Nerv. Syst.,
1997, 13, 64-67. – 33. Tamburrini G., Di Rocco
C., Velardi F., Santini P.: Prolonged intracranial
pressure (ICP) monitoring in non-traumatic pe-
diatric neurosurgical diseases. Med. Sci. Monit.,
2004, 10, 4, 53-63. – 34. Torley D., Bellus G. A.,
Munro C. S.: Genes, growth factors and acantho-
sis nigricans. Br. J. Dermatol., 2002, 147, 1096-
1101. – 35. Virchow H. R.: Ueber den Cretinismus,
namentlich in Franken, und ueber pathologi-
sche Schaedelforamen. Verh. Phys. Med. Ges.
Wuerzburg, 1852, 2, 230– 271. – 36. Wilkes D.,
Rutland P., Pulleyn L. J., Reardon W., Moss C. J.,
Ellis P., Winter R. M., Malcolm S.: A recurrent mu-
tation, ala391glu, in the transmembrane region of
FGFR3 causes Crouzon syndrome and acanthosis
nigricans. J. Med. Genet., 1996, 33, 744-748.
Otrzymano: dnia 4.I.2005 r.
Adres autorów: 02-005 Warszawa, ul. Nowogrodzka 59.
Wyszukiwarka
Podobne podstrony:
Ogólne założenia promocji zdrowia na podstawie piśmiennictwa
PDW na podstawie obserwacji pedagogicznej
8 Wyznaczenie częstości generatora na podstawie obserwacji dudnień i krzywych Lissajous2012
18 Uczenie się na podstawie obserwacji
Temat Problemy oceny pracowników ( na podstawie własnych doświadczeń )
Na podstawie obserwacji pracy studenta oraz rozmow pohospitacyjnych stwierdza sie ze przygotowani
Ćwiczenia na wyskok, Ćwiczenia na wyskok, Opisany tu trening powstał na podstawie własnych doświadcz
OII08 Wyznaczanie czestosci generatora na podstawie obserwacji dudnień i krzywych Lissajous
8 Wyznaczenie częstości generatora na podstawie obserwacji dudnień i krzywych Lissajou
Badanie ukł. wyświetlania informacji na podstawie prostego woltomierza, Zespół Szkół Elektrycznych n
4 Wyznaczanie czestosci generatora na podstawie obserwacji dudnien i krzywych Lissajous, Fizyka spra
PDW na podstawie obserwacji pedagogicznej
Malaczewski, Maciej Prace zespołowe na przedmiotach ilościowych w świetle własnych doświadczeń i op
Praca zespolonych słupów stalowo betonowych na podstawie badań i analizy metodą MES
18 Uczenie się na podstawie obserwacji
Człowiek nie może żyć bez miłości rozwiń myśl Jana Pawła II na podstawie literatury i własnych prze
ING Lojalność wobec klientów na podstawie ING Banku Śląskiego S A
więcej podobnych podstron