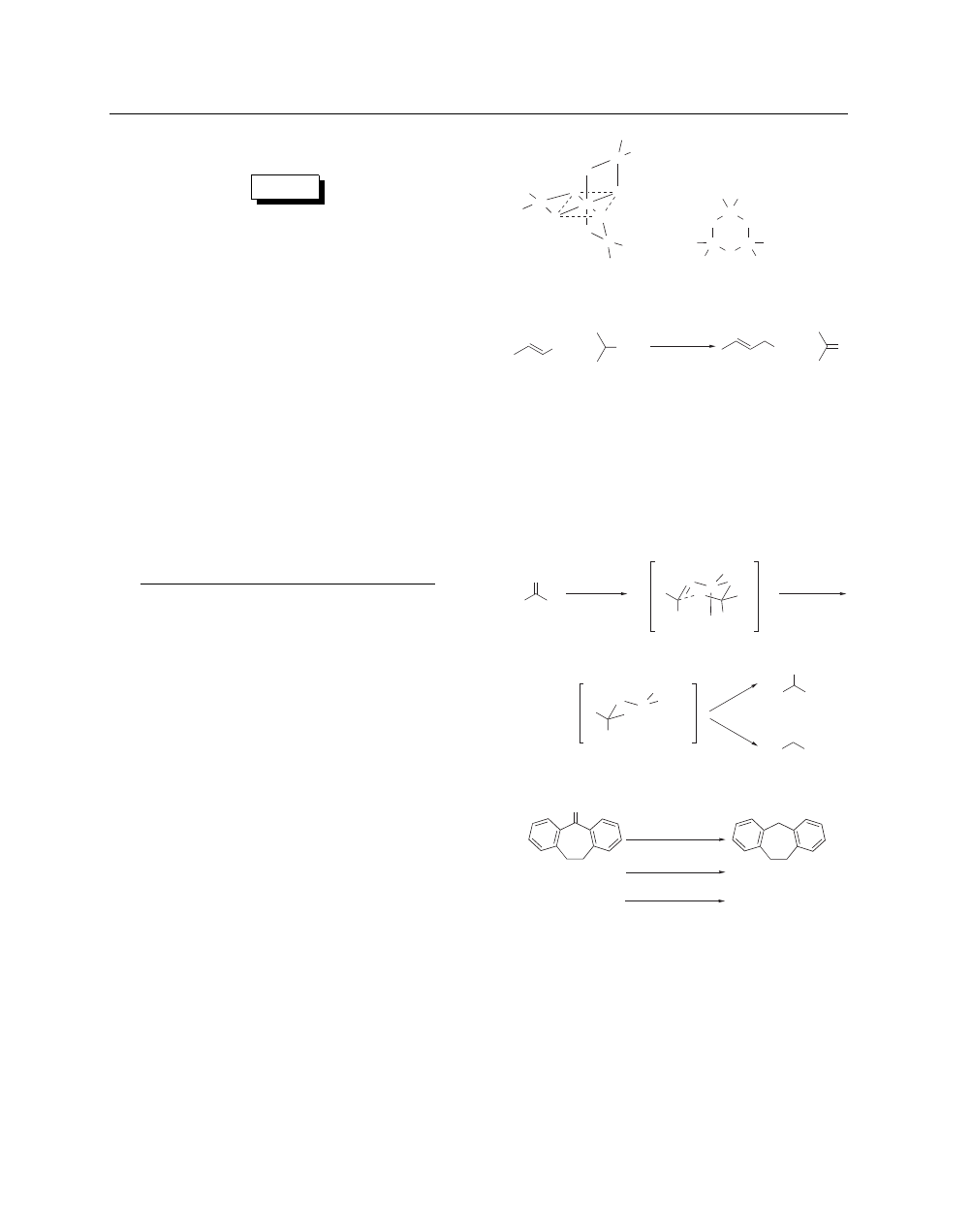
ALUMINUM ISOPROPOXIDE
1
Aluminum Isopropoxide
1
Al(O-i-Pr)
3
[555-31-7]
C
9
H
21
AlO
3
(MW 204.25)
InChI = 1/3C3H7O.Al/c3*1-3(2)4;/h3*3H,1-2H3;/q3*-1;+3/
rC9H21AlO3/c1-7(2)11-10(12-8(3)4)13-9(5)6/h7-9H,
1-6H3
InChIKey = SMZOGRDCAXLAAR-UYRFGFQLAJ
(mild
reagent
for
Meerwein–Ponndorf–Verley
reduction;
1
Oppenauer oxidation;
13
hydrolysis of oximes;
16
rearrangement
of epoxides to allylic alcohols;
17
regio- and chemoselective ring
opening of epoxides;
20
preparation of ethers
21
)
Alternate Name:
triisopropoxyaluminum.
Physical Data:
mp 138–142
◦
C (99.99+%), 118
◦
C (98+%); bp
140.5
◦
C; d 1.035 g cm
−3
.
Solubility:
sol benzene; less sol alcohols.
Form Supplied in:
white solid (99.99+% or 98+% purity based
on metals analysis).
Preparative Methods:
see example below.
Handling, Storage, and Precautions:
the dry solid is corrosive,
moisture sensitive, flammable, and an irritant. Use in a fume
hood.
Original Commentary
Kazuaki Ishihara & Hisashi Yamamoto
Nagoya University, Nagoya, Japan
NMR Analysis of Aluminum Isopropoxide. Evidence from
molecular weight determinations indicating that aluminum iso-
propoxide aged in benzene solution consists largely of the tetramer
(1), whereas freshly distilled molten material is trimeric (2),
2
is
fully confirmed by NMR spectroscopy.
3
Meerwein–Ponndorf–Verley Reduction.
One use of the
reagent is for the reduction of carbonyl compounds, particularly
of unsaturated aldehydes and ketones, for the reagent attacks only
carbonyl compounds. An example is the reduction of crotonalde-
hyde to crotyl alcohol (eq 1).
1
A mixture of 27 g of cleaned Alu-
minum foil, 300 mL of isopropanol, and 0.5 g of Mercury(II)
Chloride is heated to boiling, 2 mL of carbon tetrachloride is
added as catalyst, and heating is continued. The mixture turns
gray, and vigorous evolution of hydrogen begins. Refluxing is
continued until gas evolution has largely subsided (6–12 h). The
solution, which is black from the presence of suspended solid,
can be concentrated and the aluminum isopropoxide distilled in
vacuum (colorless liquid) or used as such. Thus the undistilled
solution prepared as described from 1.74 mol of aluminum and
500 mL of isopropanol is treated with 3 mol of crotonaldehyde
and 1 L of isopropanol. On reflux at a bath temperature of 110
◦
C,
acetone slowly distills at 60–70
◦
C. After 8–9 h, when the distil-
late no longer gives a test for acetone, most of the remaining iso-
propanol is distilled at reduced pressure and the residue is cooled
and hydrolyzed with 6 N sulfuric acid to liberate crotyl alcohol
from its aluminum derivative.
RO
Al
O
R
Al
OR
Al
OR
OR
RO
RO
OR
RO
R
O
OR
O
R
OR
Al
RO
Al
Al
RO
Al
OR
OR
OR
OR
RO
RO
(1)
(2)
CHO
OH
O
OH
+
Al(O-i-Pr)
3
60%
+
(1)
The Meerwein–Ponndorf–Verley reduction of the ketone (3)
involves formation of a cyclic coordination complex (4) which,
by hydrogen transfer, affords the mixed alkoxide (5), hydrolyzed
to the alcohol (6) (eq 2).
4
Further reflection suggests that under
forcing conditions it might be possible to effect repetition of the
hydrogen transfer and produce the hydrocarbon (7). Trial indeed
shows that reduction of diaryl ketones can be effected efficiently
by heating with excess reagent at 250
◦
C (eq 3).
5
O-i-Pr
Ar
Ar
O
O
Al
+
O
H
Ar
Ar
O-i-Pr
O-i-Pr
Al
O
H
Ar
Ar
O-i-Pr
OH
Ar
Ar
Ar
Ar
–
Al(O-i-Pr)
3
(3)
(4)
H
+
250 °C
(5)
(6)
(7)
(2)
(3)
O
Al(O-i-Pr)
3
, 250 °C
95%
anthraquinone
anthrone
anthracene
anthracene
75%
92%
A study
6
of this reduction of mono- and bicyclic ketones shows
that, contrary to commonly held views, the reduction proceeds
at a relatively high rate. The reduction of cyclohexanone and of
2-methylcyclohexanone is immeasurably rapid. Even menthone
is reduced almost completely in 2 h. The stereochemistry of the
reduction of 3-isothujone (8) and of 3-thujone (11) has been ex-
amined (eqs 4 and 5). The ketone (8) produces a preponderance
of the cis-alcohol (9). The stereoselectivity is less pronounced in
the case of 3-thujone (11), although again the cis-alcohol (12)
predominates. The preponderance of the cis-alcohols can be in-
creased by decreasing the concentration of ketone and alkoxide.
Avoid Skin Contact with All Reagents

2
ALUMINUM ISOPROPOXIDE
(4)
7:1
(10)
(9)
(8)
+
O
i
-Pr
i
-Pr
OH
OH
i
-Pr
(5)
3.2:1
(13)
(12)
(11)
+
O
i
-Pr
i
-Pr
OH
OH
i
-Pr
This reducing agent is the reagent of choice for reduction
of enones of type (14) to the α,β-unsaturated alcohols (15)
(eq 6). Usual reducing agents favor 1,4-reduction to the saturated
alcohol.
7
(6)
(15)
20%, each isomer
(14)
Al(O-i-Pr)
3
O
C
5
H
11
O
O
BPCO
BPCO
O
OH
C
5
H
11
O
BPC = biphenylcarbonyl
The Meerwein–Ponndorf–Verley reduction of pyrimidin-
2(1H)-ones using Zirconium Tetraisopropoxide or aluminum iso-
propoxide leads to exclusive formation of the 3,4-dihydro isomer
(eq 7).
8
The former reducing agent is found to be more effective.
(7)
Zr(O-i-Pr)
4
or Al(O-i-Pr)
3
i
-PrOH, 90 °C, 2 days
20–91%
R = H, halide
N
N
O
R
Bn
N
Bn
R
O
HN
Reductions with Chiral Aluminum Alkoxides.
The re-
duction of cyclohexyl methyl ketone with catalytic amounts of
aluminum alkoxide and excess chiral alcohol gives (S)-1-cyclo-
hexylethanol in 22% ee (eq 8).
9
(8)
Al(OR)
3
22% ee
O
OH
OH
Isobornyloxyaluminum dichloride is a good reagent for reduc-
ing ketones to alcohols. The reduction is irreversible and subject
to marked steric approach control (eq 9).
10
(9)
70% ee
Ph
O
OAlCl
2
OH
Ph
Diastereoselective Reductions of Chiral Acetals. Recently,
it has been reported that Pentafluorophenol is an effective accel-
erator for Meerwein–Ponndorf–Verley reduction.
11
Reduction of
4-t-butylcyclohexanone with aluminum isopropoxide (3 equiv) in
dichloromethane, for example, is very slow at 0
◦
C (<5% yield for
5 h), but in the presence of pentafluorophenol (1 equiv), the reduc-
tion is cleanly completed within 4 h at 0
◦
C (eq 10). The question
of why this reagent retains sufficient nucleophilicity is still open.
It is possible that the o-halo substituents of the phenoxide lig-
and may coordinate with the aluminum atom, thus increasing the
nucleophilicity of the reagent.
(10)
Al(O-i-Pr)
3
(3 equiv)
C
6
F
5
OH (1 equiv)
CH
2
Cl
2
, 0 °C
O
t
-Bu
t
-Bu
OH
Chiral acetals derived from (−)-(2R,4R)-2,4-Pentanediol and
ketone are reductively cleaved with high diastereoselectivity by a
1:2 mixture of diethylaluminum fluoride and pentafluorophenol.
11
Furthermore, aluminum pentafluorophenoxide is a very powerful
Lewis acid catalyst for the present reaction.
12
The reductive cleav-
age in the presence of 5 mol % of Al(OC
6
F
5
)
3
affords stereose-
lectively retentive reduced β-alkoxy ketones. The reaction is an
intramolecular Meerwein–Ponndorf–Verley reductive and Oppe-
nauer oxidative reaction on an acetal template (eq 11).
(11)
K
2
CO
3
Et
2
AlF–2C
6
F
5
OH
(1.2 equiv)
or
Al(OC
6
F
5
)
3
(5 mol %)
CH
2
Cl
2
or toluene
R
1
> R
2
O
O
R
1
R
2
R
1
R
2
O
O
OH
R
2
R
1
The direct formation of α,β-alkoxy ketones is quite useful.
Removal of the chiral auxiliary, followed by base-catalyzed β-
elimination of the resulting β-alkoxy ketone, easily gives an opti-
cally pure alcohol in good yield. Several examples of the reaction
are summarized in Table 1.
(trans:cis)
82:18
73:27
94:6
99:1
92:8
95:5
81:19
83
61
90
71
78
89
67
Ratio
(S:R)
Yield (%)
R
2
R
1
Al(OC
6
F
5
)
3
(5 mol %)
CH
2
Cl
2
Table 1 Reductive cleavages of acetals using Al(OC
6
F
5
)
3
catalyst
O
O
R
2
R
1
R
2
R
1
O
O
CH
2
CH
2
t
-Bu
>
C
5
H
11
i
-Bu
i
-Pr
Ph
Ph
c
-Hex
Me
Me
Me
Me
Et
Me
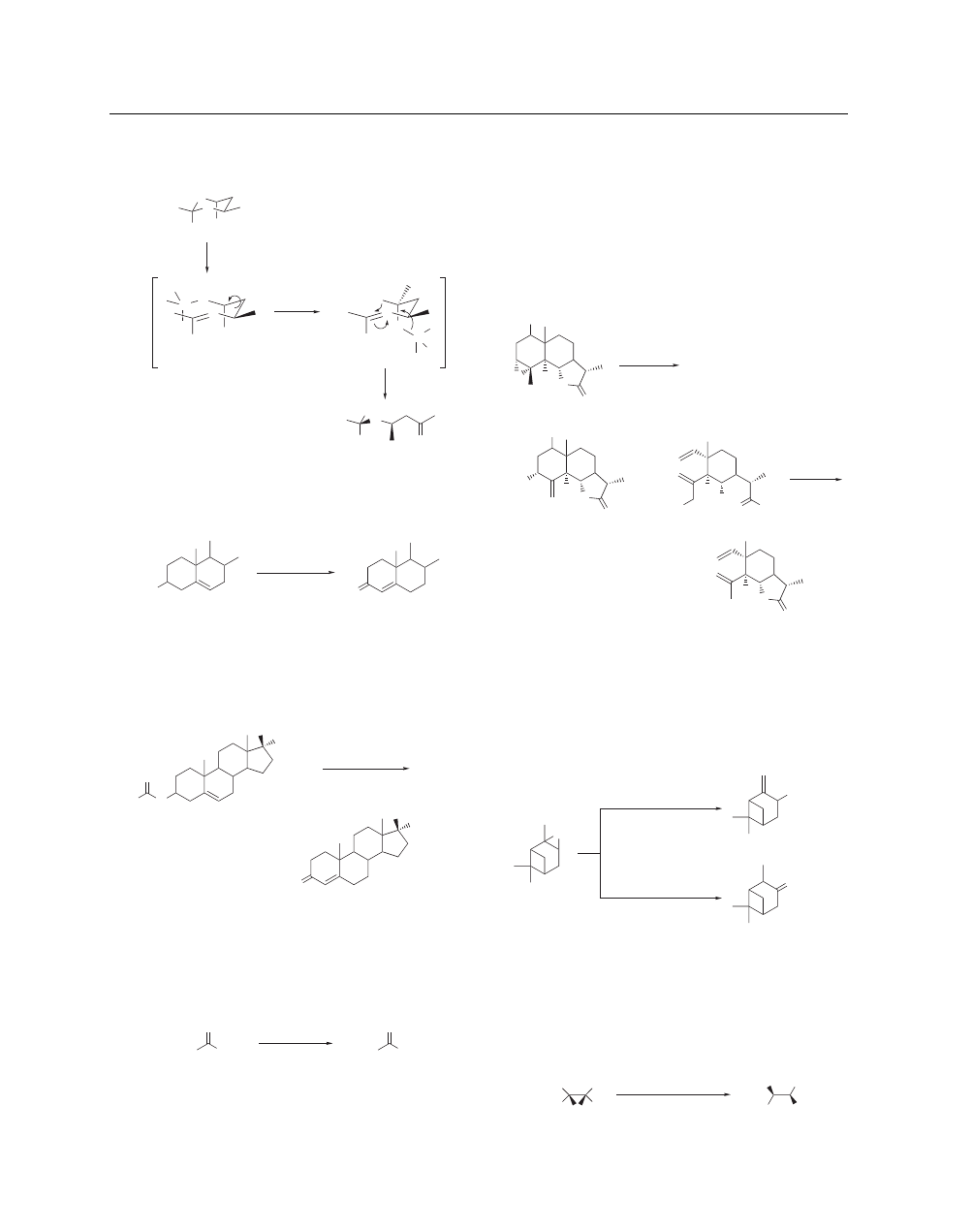
Although the detailed mechanism is not yet clear, it is assumed
that an energetically stable tight ion-paired intermediate is gener-
ated by stereoselective coordination of Al(OC
6
F
5
)
3
to one of the
oxygens of the acetal; the hydrogen atom of the alkoxide is then
A list of General Abbreviations appears on the front Endpapers
ALUMINUM ISOPROPOXIDE
3
transferred as a hydride from the retentive direction to this depart-
ing oxygen, which leads to the (S) configuration at the resulting
ether carbon, as described (eq 12).
(12)
R
1
> R
2
L = OC
6
F
5
Al(OC
6
F
5
)
3
rotation
O
O
R
1
R
2
R
2
R
1
O
O
H
Al
L
O
+
O
R
1
R
2
L
L
O
+
O
H
R
1
R
2
Al
L
L
L
–
–
Oppenauer Oxidation.
13
Cholestenone is prepared by oxida-
tion of cholesterol in toluene solution with aluminum isopropox-
ide as catalyst and cyclohexanone as hydrogen acceptor (eq 13).
14
(13)
HO
O
Al(O-i-Pr)
3
cyclohexanone
toluene
72–74%
A formate, unlike an acetate, is easily oxidized and gives the
same product as the free alcohol.
15
For oxidation of (16) to (17) the
combination of cyclohexanone and aluminum isopropoxide and a
hydrocarbon solvent is used: xylene (bp 140
◦
C at 760 mmHg) or
toluene (bp 111
◦
C at 760 mmHg) (eq 14).
(14)
COMe
OCOMe
O
O
OCOMe
COMe
Al(O-i-Pr)
3
cyclohexanone
xylene or toluene
86%
(16)
(17)
O
H
Hydrolysis of Oximes.
16
Oximes can be converted into parent
carbonyl compounds by aluminum isopropoxide followed by acid
hydrolysis (2N HCl) (eq 15). Yields are generally high in the case
of ketones, but are lower for regeneration of aldehydes.
(15)
1. Al(O-i-Pr)
3
2. HCl, H
2
O
R
1
R
2
R
1
R
2
NOH
O
Rearrangement of Epoxides to Allylic Alcohols. The key
step in the synthesis of the sesquiterpene lactone saussurea lactone
(21) involved fragmentation of the epoxymesylate (18), obtained
from α-santonin by several steps (eq 16).
17
When treated with
aluminum isopropoxide in boiling toluene (N
2
, 72 h), (18) is con-
verted mainly into (20). The minor product (19) is the only product
when the fragmentation is quenched after 12 h. Other bases such
as potassium t-butoxide, LDA, and lithium diethylamide cannot
be used. Aluminum isopropoxide is effective probably because
aluminum has a marked affinity for oxygen and effects cleav-
age of the epoxide ring. Meerwein–Ponndorf–Verley reduction is
probably involved in one step.
(16)
Al(O-i-Pr)
3
68%
O
O
MsO
H
O
O
H
MsO
HO
O
HO
O
O-i-Pr
HO
H
H
O
O
+
5 steps
(18)
(19) 9%
(20) 68%
(21)
α
-Pinene oxide (22) rearranges to pinocarvenol (23) in the pres-
ence of 1 mol % of aluminum isopropoxide at 100–120
◦
C for
1 h.
18
The oxide (22) rearranges to pinanone (24) in the presence
of 5 mol % of the alkoxide at 140–170
◦
C for 2 h. Aluminum iso-
propoxide has been used to rearrange (23) to (24) (200
◦
C, 3 h,
80% yield) (eq 17).
19
(17)
OH
O
O
(22)
(23)
(24)
Al(O-i-Pr)
3
, 100 °C
~85%
Al(O-i-Pr)
3
, 150 °C
69%
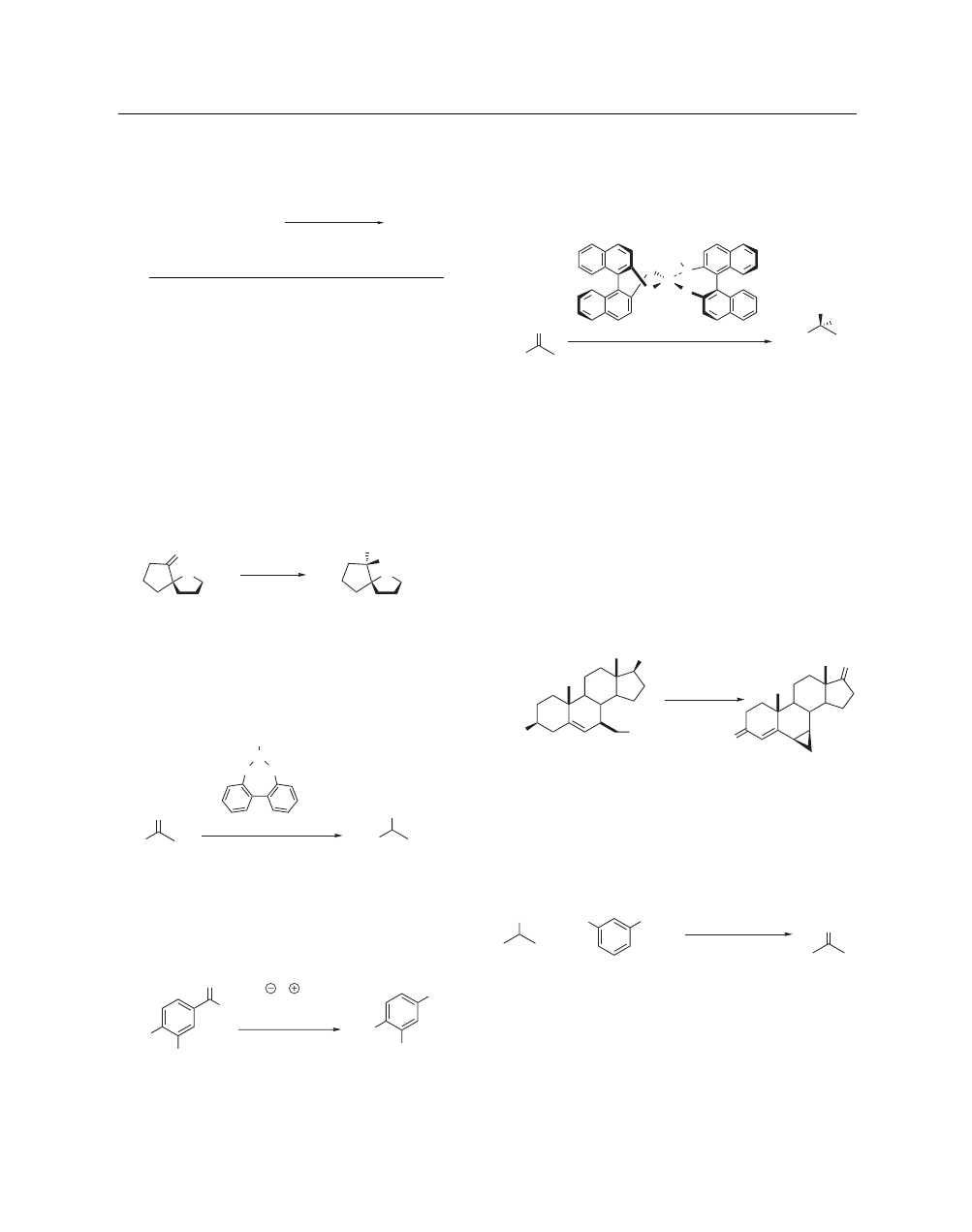
Regio- and Chemoselective Ring Opening of Epoxides.
Functionalized epoxides are regioselectively opened using
trimethylsilyl azide/aluminum isopropoxide, giving 2-trimethyl-
siloxy azides by attack on the less substituted carbon (eq 18).
20
(18)
Me
3
SiN
3
(1.5 equiv)
Al(O-i-Pr)
3
(0.1 equiv)
CH
2
Cl
2
59–93%
O
R
1
H
R
2
H
N
3
TMSO
R
1
R
2
Avoid Skin Contact with All Reagents
4
ALUMINUM ISOPROPOXIDE
Preparation of Ethers.
Ethers ROR
′
are prepared from
aluminum alkoxides, Al(OR)
3
, and alkyl halides, R
′
X. Thus
EtCHMeOH is treated with Al, HgBr
2
, and MeI in DMF to give
EtCHMeOMe (eq 19).
21
(19)
Al(OR)
3
+
R
1
X
DMF, reflux, 2 days
20–80%
ROR
1
R, R
1
= alkyl; X = halide
First Update
David Crich
Wayne State University, Detroit, MI, USA
Meerwein–Ponndorf–Verley and Related Reductions. The
classic MPV reaction,
22
for which computational support has now
been advanced in support of the concerted six-membered cyclic
transition state (eq 2),
23
continues to be a major use of this reagent.
For example, aluminum isopropoxide was found to be particularly
well suited to the reduction of the spirocyclic ketone 25, as no
other reagents examined gave a useful amount of the β-carbinol
26 (eq 20).
24
O
O
Al(O-i-Pr)
3
O
OH
H
i
-PrOH,
∆
70%,
β :α = 85:15
(20)
(25)
(26)
It was found that a complex 27, derived in situ from aluminum
isopropoxide and a chelating hydroxyl biphenylsulfonamide, was
an effective catalyst for the reduction of acetophenone by iso-
propanol (eq 21).
25
Ph
O
NSO
2
(CF
2
)
7
CF
3
Al
O
O-i-Pr
Ph
OH
(27) (5 mol %)
i
-PrOH, CH
2
Cl
2
, rt
82%
(21)
Reaction of aluminum isopropoxide with trifluoroacetic acid in
dichloromethane at room temperature affords a white solid for-
mulated as the salt 28, which is capable of reducing a wide range
of alkyl and aryl aldehydes and ketones at room temperature in
dichloromethane (eq 22).
26
O
H
OMe
MeO
CH
2
OH
OMe
MeO
CF
3
CO
2
Al(i-O-Pr)
2
(28)
CH
2
Cl
2
, rt
(22)
97%
The catalytic asymmetric MPV reaction has been much exam-
ined and recently reviewed,
27
but the catalyst precursor of choice
is more usually a trialkylaluminum than aluminum isopropoxide.
Complexes such as that derived from aluminum isopropoxide with
2 equiv of enantiomerically pure BINOL (29) catalyze the enan-
tioselective reduction of aromatic ketones by borane-dimethyl
sulfide, through binding of both the borane and the ketone
(eq 23).
28
Al
O
O
O
O
H
Ph
O
Ph
OH
H
(29) (10 mol %)
96%, 74% ee
BH
3
·
SMe
2
, CH
2
Cl
2
, 40
°C
(23)
Aluminum BINOL complexes have also been used to promote
the reduction of N-diphenylphosphinoyl ketimines to the corre-
sponding amines by isopropanol with excellent ee’s. However,
again, the preferred catalyst precursor is trimethylaluminum.
29
Diasteroselective reductive amination of α-methylbenzylamine-
derived imines has also been achieved with hydrogen over a cata-
lyst combination of Raney nickel and aluminum isopropoxide, but
titanium tetra(isopropoxide) was the metal alkoxide of choice.
30
Oppenauer Oxidation. Oppenauer oxidation of the mesy-
loxymethyl steroidal diol 30 took place with concomitant alu-
minum isopropoxide promoted dienolate alkylation, leading to
the formation of a cyclopropa-derivative 31 (eq 24).
31
OH
HO
OMs
O
O
78%
(30)
(31)
(24)
cyclohexanone
toluene,
∆
Al(O-i-Pr)
3
The Oppenauer oxidation has also been rendered catalytic
through the use of electron-deficient aldehydes as hydride accep-
tors. Optimal yields were obtained with trimethylaluminum as
catalyst, but the process also functioned in a respectable manner
with aluminum isopropoxide (eq 25).
32
OH
O
2
N
CHO
O
+
(3 equiv)
10 mol % Al(O-i-Pr)
3
(25)
73%
toluene, rt
Rearrangement of Epoxides to Allylic Alcohols and Open-
ing of Lactones and Anhydrides. Treatment of the epoxylac-
tone 32 with aluminum isopropoxide in toluene at reflux resulted
in cleavage to the allylic alcohol in tandem with nucleophilic open-
ing of the lactone (eq 26). The diastereomeric epoxylactone 33
A list of General Abbreviations appears on the front Endpapers
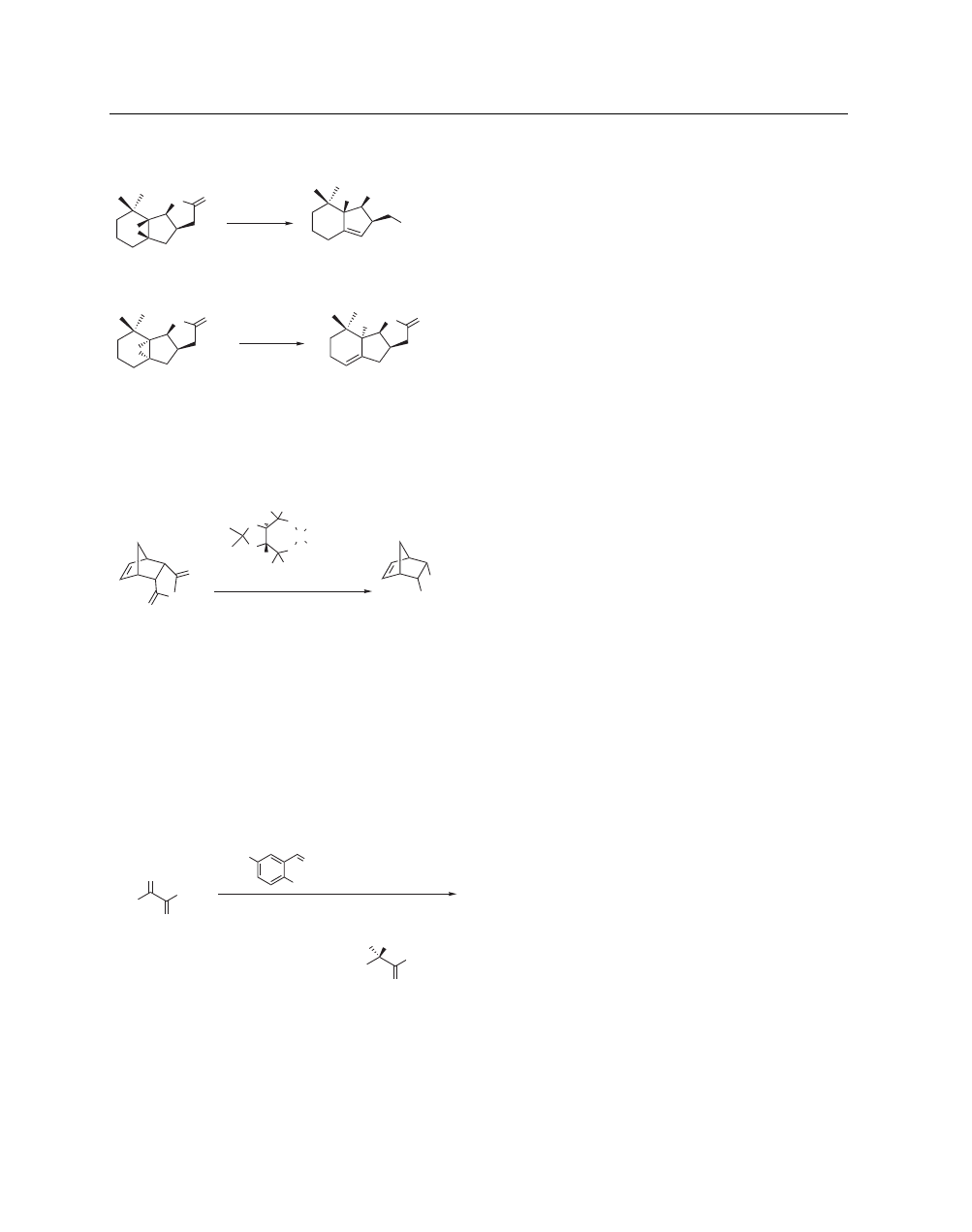
ALUMINUM ISOPROPOXIDE
5
did not suffer opening of the lactone under the same conditions
(eq 27).
33
O
O
O
OH
OH
CO
2
-i-Pr
(32)
Al(O-i-Pr)
3
66%
(26)
toluene,
∆
O
O
O
O
OH
O
(33)
Al(O-i-Pr)
3
(27)
66%
toluene,
∆
The meso-anhydride 34 undergoes highly enantioselective ring
opening with aluminum isopropoxide and catalytic titanium TAD-
DOLate 35 at, −34
◦
C in good yield, albeit very slowly (eq 28).
34
O
O
O
CO
2
H
CO
2
-i-Pr
O
Ti
O
O
O
Ar
Ar
Ar
Ar
H
H
O-i-Pr
O-i-Pr
(34)
(35) (Ar =
β-naphthyl) (20 mol %)
80 mol % Al(O-i-Pr)
3
, Et
2
O
–34
°C, 24 days
74%, 98:2 er
(28)
Enantioselective Alkylation of α
α
α
-Ketoesters.
The combi-
nation of a dipeptide-derived ligand and 15 mol % aluminum
isopropoxide promotes the efficient enantioselective addition of
dimethyl and diethylzinc to alkyl, aryl, and α,β-unsaturated α-
ketoesters in good to excellent yield and ee. The ee’s were gen-
erally higher in the presence of 50 mol % diethylphosphoramide,
which is thought to serve as an additional ligand to the intermedi-
ate organozinc reagent, and with aryl α-ketoesters (eq 29).
35
Et
O
O
OMe
MeO
OH
N-L-Thr(Trt)-L-(Thr(Trt)-NHBu
Et
O
OMe
Me
H
(15 mol %)
10 equiv Me
2
Zn, 15 mol % Al(O-i-Pr)
3
50 mol % (EtO)
2
P(=O)NH
2
, toluene, –78
°C
44%
(98% conversion)
92% ee
(29)
1.
Wilds, A. L., Org. React. 1944, 2, 178.
2.
Shiner, V. J.; Whittaker, D.; Fernandez, V. P., J. Am. Chem. Soc. 1963,
85
, 2318.
3.
Worrall, I. J., J. Chem. Educ. 1969, 46, 510.
4.
Woodward, R. B.; Wendler, N. L.; Brutschy, F. J., J. Am. Chem. Soc.
1945, 67, 1425.
5.
Hoffsommer, R. D.; Taub, D.; Wendler, N. L., Chem. Ind. (London) 1964,
482.
6.
Hach, V., J. Org. Chem. 1973, 38, 293.
7.
Picker, D. H.; Andersen, N. H.; Leovey, E. M. K., Synth. Commun. 1975,
5
, 451.
8.
Høseggen, T.; Rise, F.; Undheim, K., J. Chem. Soc., Perkin Trans. 1 1986,
849.
9.
Doering, W.; von, E.; Young, R. W., J. Am. Chem. Soc. 1950, 72,
631.
10.
Nasipuri, D.; Sarker, G., J. Indian Chem. Soc. 1967, 44, 165.
11.
Ishihara, K.; Hanaki, N.; Yamamoto, H., J. Am. Chem. Soc. 1991, 113,
7074.
12.
Ishihara, K.; Hanaki, N.; Yamamoto, H., Synlett 1993, 127; J. Am. Chem.
Soc.
, 1993, 115, 10 695.
13.
Djerassi, C., Org. React. 1951, 6, 207.
14.
Eastham, J. F.; Teranishi, R., Org. Synth., Coll. Vol 1963, 4, 192.
15.
Ringold, H. J.; Löken, B.; Rosenkranz, G.; Sondheimer, F., J. Am. Chem.
Soc. 1956
, 78, 816.
16.
Sugden, J. K., Chem. Ind. (London) 1972, 680.
17.
Ando, M.; Tajima, K.; Takase, K., Chem. Lett. 1978, 617.
18.
Scheidl, F., Synthesis 1982, 728.
19.
Schmidt, H., Chem. Ber. 1929, 62, 104.
20.
Emziane, M.; Lhoste, P.; Sinou, D., Synthesis 1988, 541.
21.
Lompa- Krzymien, L.; Leitch, L. C., Pol. J. Chem. 1983, 57, 629.
22.
De Graauw, C. F.; Peters, J. A.; van Bekkum, H.; Huskens, J., Synthesis
1994, 1007.
23.
Cohen, R.; Graves, C. R.; Nguyen, S. T.; Martin, J. M. L.; Ratner, M. A.,
J. Am. Chem. Soc. 2004
, 126, 14796.
24.
Paquette, L. A.; Owen, D. R.; Bibart, R. T.; Seekamp, C. K.; Kahane, A.
L.; Lanter, J. C.; Corral, M. A., J. Org. Chem. 2001, 66, 2828.
25.
Ooi, T.; Ichikawa, H.; Maruoka, K., Angew. Chem. Int. Ed. 2001, 40,
3610.
26.
(a) Akamanchi, K. G.; Varalakshmy, N. R., Tetrahedron Lett. 1995, 36,
3571. (b) Akamanchi, K. G.; Varalakshmy, N. R.; Chaudhari, B. A.,
Synlett 1997
, 371.
27.
Graves, C. R.; Campbell, E. J.; Nguyen, S. T., Tetrahedron: Asymmetry
2005, 16, 3460.
28.
Fu, I. -P.; Uang, B. -J., Tetrahedron: Asymmetry 2001, 12, 45.
29.
Graves, C. R.; Scheidt, K. A.; Nguyen, S. T., Org. Lett. 2006, 8,
1229.
30.
Nugent, T. C.; Ghosh, A. K.; Wakchaure, V. N.; Mohanty, R. R., Adv.
Synth. Catal. 2006
, 348, 1289.
31.
Deng, G.; Li, Z.; Peng, S. -Y.; Fang, L.; Li, Y. -C., Tetrahedron 2007,
63
, 4630.
32.
Graves, C. R.; Zeng, B. -S.; Nguyen, S. T., J. Am. Chem. Soc. 2006, 128,
12596.
33.
Reizelman, A.; Zwanenburg, B., Synthesis 2000, 1952.
34.
Jaeschke, G.; Seebach, D., J. Org. Chem. 1998, 63, 1190.
35.
Wieland, L. C.; Deng, H.; Snapper, M. L.; Hovedya, A. H., J. Am. Chem.
Soc. 2005
, 127, 15453.
Avoid Skin Contact with All Reagents
Wyszukiwarka
Podobne podstrony:
aluminum chloride eros ra079
aluminium ethoxide eros ra081
aluminium amalgam eros ra076
aluminum eros ra075
potassium on alumina eros rp192
alumina eros ra074
Aluminum i miedź Mateusz Bednarski
benzyl chloride eros rb050
hydrobromic acid eros rh031
chloroform eros rc105
magnesium eros rm001
więcej podobnych podstron