
Pathophysiology of the
liver

Mechanisms of cholestasis
Stasis of bile flow may result from three processes:
a) failure to secrete bile into the canaliculi, due to injury to
hepatocytes and/or the canalicular membrane or altered
function of the canalicular bile salt transporter;
b) increased permeability of the canaliculi, cholangiocytes
and/or their tight junctions; and
c) mechanical obstruction of the biliary tree at any level.
Diffuse injury to canalicular membranes causes loss of
microvilli in dilated canaliculi which can be plugged by
inspissated bile. There is impaired secretion of all
components of bile.

Classification of cholestasis
1. Canalicular Cholestasis
a. In drug-induced cholestasis, (phenothiazines, sulfonylureas)
b. In steroid- induced cholestasis (normal pregnancy, or treatment with
synthetic estrogens, or with 17-a-alkyl anabolic steroids),
c. Postoperative cholestasis.
d. Other causes of canalicular cholestasis (severe infections with
endotoxemia and/or septicaemia, total parenteral nutrition, and sickle cell
anaemia crises).
2. Obstructive cholestasis
a. Obstructive intrahepatic cholestasis (primary biliary cirrhosis, sclerosing
cholangitis and intrahepatic lithiasis; granulomas, lymphomas, metastatic
nodules obstructing interlobular and larger intrahepatic bile ducts).
b. Obstructive extrahepatic cholestasis is most often caused by local lesions
such as carcinoma of the pancreas, or by common bile duct stones or
structures.

HYPERBILIRUBINEMIA
• Hyperbilirubinemia is defined as a total bilirubin level greater
than 1.5 mg/dL, an unconjugated level greater than 1 mg/dL,
or a conjugated bilirubin level greater than 0.3 mg/dL.
• The causes of hyperbilirubinemia can be divided into
problems of excess production and abnormal clearance. The
concentration of bilirubin in plasma is directly related to the
production rate of bilirubin and inversely related to hepatic
and renal removal rates.
• In general, hyperbilirubinemia is separated into conjugated
and unconjugated bilirubin excess.
• This distinction is particularly helpful at low bilirubin
concentrations and assists in determining which set of
differential diagnoses should be sought.

Unconjugated versus Conjugated
Hyperbilirubinemia

1. Pathophysiology of Unconjugated
Hyperbilirubinemia
• Unconjugated hyperbilirubinemia, characterized by
retention exclusively of UCB in the serum, without
bilirubinuria, results from insufficient hepatic
clearance and/or conjugation of the load of the UCB
produced each day, and is subclassified according
to the step(s) in bilirubin metabolism that are
deranged.
• The most common aetiologies are:
overproduction of unconjugated bilirubin due to disorders
of red blood cells;
impaired delivery of unconjugated bilirubin due to
disturbances in hepatic circulation; and
functional or hereditary defects in uptake, storage, or
conjugation of bilirubin.

Pathophysiology of Unconjugated
Hyperbilirubinemia continued
a. Overproduction of bilirubin due to accelerated heme catabolism is a very
common cause of unconjugated hyperbilirubinemia and often augments
jaundice of other causes. Most often caused by hemolytic anemias. Diagnosis
rests mainly on hematological studies.
b. Decreased delivery of bilirubin and other substances in plasma to the liver
cells is the most common form of unconjugated jaundice. It is most often due
to right-sided congestive heart failure and clears as the heart failure is
controlled. Another major cause is portosystemic shunting, due to either
cirrhosis or surgical anastomosis, which diverts the bilirubin formed in the
spleen past the liver directly into the systemic circulation.
c&d. Diminished clearance (uptake and storage) of UCB is often due to
competitive inhibition of these processes by drugs (e.g., rifampicin). The
jaundice usually resolves within 2-3 days after the drug is discontinued.
Hypothyroidism and febrile illnesses may impair hepatic storage capacity for
UCB. Impaired uptake of UCB and other organic anions is also present in most
patients with the hereditary disorder of conjugation, Gilbert's syndrome.
e. Impaired conjugation of bilirubin is usually due to hereditary defects, since
the activity of UGT1A1 is preserved in hepatobiliary diseases, except in end-
stage cirrhosis or acute hepatic failure.

Hereditary autosomal recessive
disorders of UCB conjugation
Gilbert's syndrome is characterized by a chronic, mild, fluctuating
unconjugated hyperbilirubinemia, caused by a polymorphism in the
promoter of both gene alleles encoding UGT1A1. This very common
hereditary defect leads to a 70-80% decrease in the expression
and activity of UGT1A1. This, by itself, does not cause
hyperbilirubinemia unless combined with overproduction and/or
impaired uptake of bilirubin. Bilirubin production and/or clearance
are further (reversibly) altered during fasting and stress, often
unmasking or augmenting the severity of jaundice.
The Crigler-Najjar syndromes are two more severe, rare, hereditary,
recessive deficiencies of UGT1A1.
In Type I, there is no detectable activity of UGT1A1 in the liver, and
glucuronide conjugation of many drugs may be impaired also.
Jaundice is severe from the neonatal period, and bilirubin
encephalopathy is the rule if the patients are untreated.
In Type II, UGT1A1 activity is detectable but at less than 10% of
normal levels. Jaundice begins usually in late childhood, is less
severe, and seldom causes bilirubin encephalopathy.

2. Pathophysiology of Conjugated
Hyperbilirubinemia
Conjugated hyperbilirubinemia is characterized by retention principally of
conjugated bilirubin in the serum.
Plasma UCB concentrations are elevated also, due in part to hydrolysis of retained
conjugated bilirubins by tissue ß-glucuronidases, as well as by contributions
from associated haemolysis and/or impairment of delivery, uptake, and storage
of UCB.
Conjugated hyperbilirubinemia results from impairment of the canalicular secretion
or biliary flow of conjugated bilirubins. Other organic anions, which share the
same transport system as conjugated bilirubin, are also excreted poorly.
The retained conjugated bilirubins regurgitate through the hepatocytes,
cholangiocytes and their weakened tight junctions into the space of Disse and
thence via the lymph to the plasma.
The small fraction of retained conjugated bilirubins that is not bound to plasma
albumin filters at the glomerulus, producing bilirubinuria; this is not only
diagnostic of conjugated hyperbilirubinemia, but also constitutes the major
alternate pathway for excretion of conjugated bilirubin and other organic anions
in the face of reduced hepatobiliary excretion.

2. Pathophysiology of Conjugated
Hyperbilirubinemia continued
• In contrast to unconjugated hyperbilirubinemia, jaundice with bilirubinuria
almost always results from significant hepatobiliary disease, and is
classified further according to whether canalicular secretion or biliary flow
is primarily impaired.
• Specific defects in canalicular secretion of bilirubin conjugates and other
organic anions secreted by MRP2 are characteristic of the hereditary
Dubin-Johnson and Rotor's syndromes, and common in hepatocellular
diseases.
• Generalized defects in canalicular secretion, or biliary flow, produce
cholestasis, in which there is also marked retention of bile salts.
• In all cases, the canalicular transporters, BSEP and MRP2, are dislocated
into subapical vesicles where they can no longer export bile salts and other
organic anions into bile.
• Dislocation of alkaline phosphatase to the basolateral membrane leads to
regurgitation of this enzyme into the space of Disse and thence to plasma.
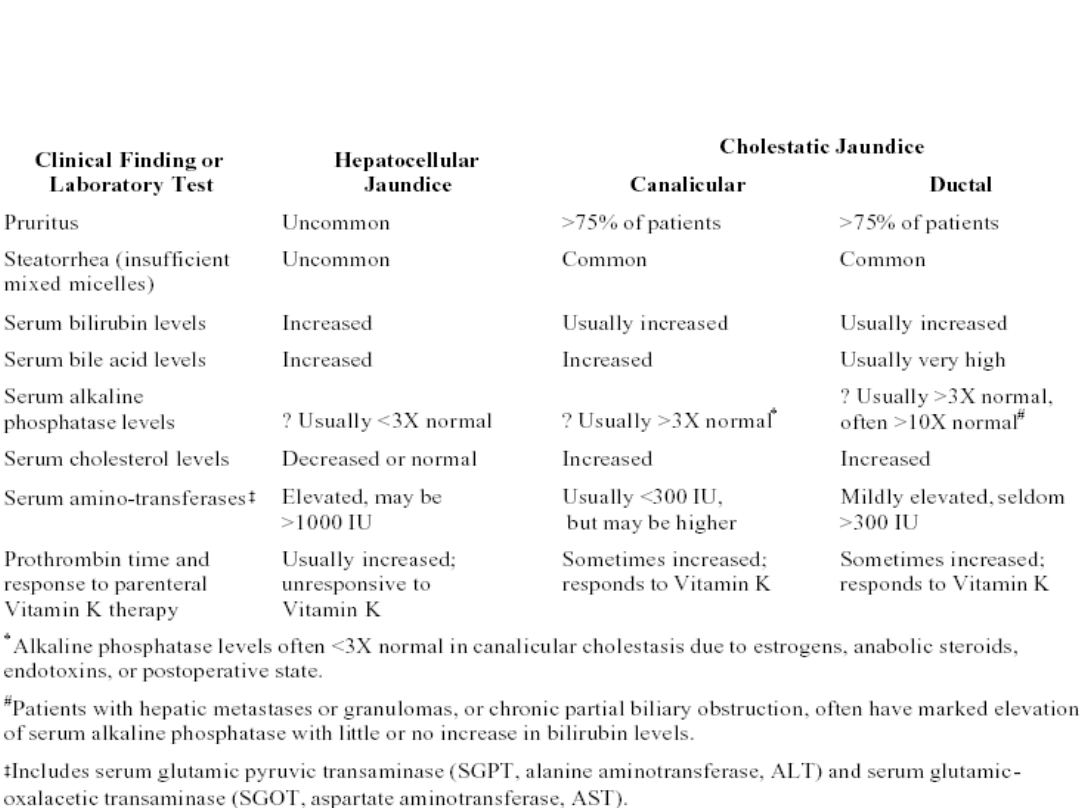
Differential Diagnosis of Conjugated
Hyperbilirubinemia –
Hepatocellular versus Cholestatic Jaundice

Effects of hyperbilirubinemia
• Kernicterus in infants
• Renal failure
• Increased postoperative mortality and morbidity
• Decreased cardiovascular response to vasopressors
• Decreased adsorption of fat-soluble vitamins
• Pigmented gallstones

Effects of cholestasis
• Impaired transport of bile salts, organic anions, and bile
components (e.g., lipids, bilirubin)
• Pruritus
• Decreased amounts of bile acids in the intestinal lumen
Decreased absorption of fat-soluble vitamins (A, D, E, and K)
Decreased calcium absorption
Osteoporosis
Coagulopathy
Steatorrhea
• Decreased secretion cholesterol into bile
Hypercholesterolemia, increased lipoprotein X
Xanthomas, increased phospholipids
Altered erythrocyte membrane with hemolysis
• Stimulation of secretion or synthesis of
Alkaline phosphatase
5´-Nucleotidase
gGlutamyltransferase

Cirrhosis of the Liver
• Cirrhosis of the liver is defined, pathologically, as
widely distributed, irregular hepatic fibrosis with
distortion of the lobular architecture and vasculature,
resulting from persistent inflammation and/or
parenchymal cell necrosis, combined with nodular
regeneration.
• Cirrhosis always has three basic features:
a) destruction of liver cells;
b) replacement of groups of lobules, entire lobules, or
parts of lobules with fibrous tissue;
c) nodular regeneration of the residual hepatic
parenchyma.

Cirrhosis of the Liver
• Cirrhosis is usually progressive, at a tempo which
may be variable and intermittent.
• In the earlier stages, this is due to persistent or
repeated damage to liver cells from the causative
agent, but resorption of fibrous tissue and
regeneration of hepatocytes may repair the
damage and reverse the process if the offending
agent is treated or removed.
• By contrast, in late stages of cirrhosis, the
secondary distortion of the hepatic circulation may
lead to chronic ischemia, with increasing fibrosis,
continued cell loss and eventually liver failure.

Clinical Consequences of Cirrhosis 1
a. Diminished hepatocytic synthetic capacity leads
to hypoalbuminemia, deficiency of clotting factors,
and (usually) hypocholesterolemia (except with
chronic cholestasis).
b. Impaired oestrogen metabolism causes palmar
erythema (red palms), spider angiomata,
amenorrhea in females, complicated by alcohol-
induced testicular atrophy and feminization in males.
c. Impaired detoxification/excretory function,
combined with shunting of blood around the liver,
causes jaundice, encephalopathy, and excessive
responses to administered drugs.

Clinical Consequences of Cirrhosis
2
d. Altered metabolism of vasoactive substances leads to
splanchnic vasodilatation, activation of the renin-
angiotensin-catecholamine system, sodium retention,
ascites and oedema, and functional renal failure (hepato-
renal syndromes).
e. Portal hypertension and the compensatory development
of portosystemic collateral circulation cause oesophageal
varices (often with GI haemorrhage), splenomegaly with
pancytopenia, and ascites (hypoalbuminemia contributes).
f. Bacterial overgrowth, with translocation of bacteria and
toxins through the congested bowel wall into the portal
system, leads to systemic infections and spontaneous
bacterial peritonitis (infected ascites).

Haemochromatosis
• Haemochromatosis is the commonest inherited liver disease in
the United Kingdom.
• It affects about 1 in 200 of the population and is 10 times more
common than cystic fibrosis.
• Haemochromatosis produces iron overload, and patients
usually present with cirrhosis or diabetes due to excessive iron
deposits in the liver or pancreas.
• The genetic defect responsible is a single base change at a
locus of the HFE gene on chromosome 6, with this defect
responsible for over 90% of cases in the United Kingdom.
• Genetic analysis is now available both for confirming the
diagnosis and screening family members.
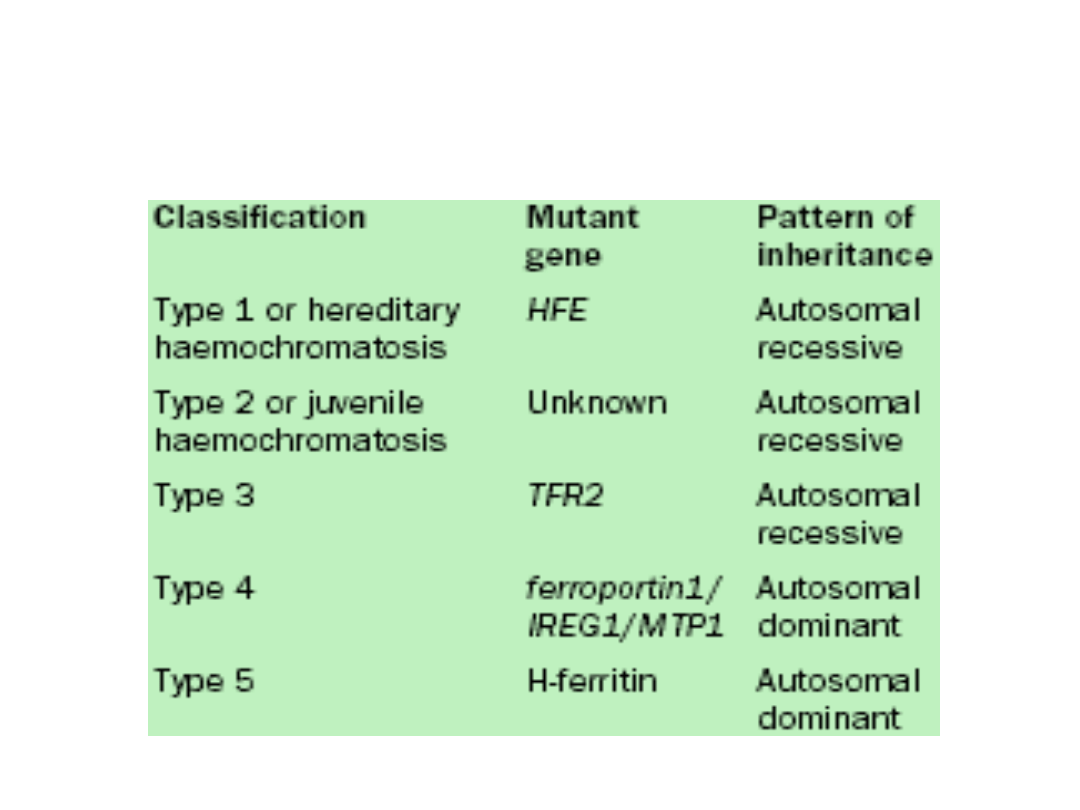
Genes mutated in hereditary
haemochromatosis

Haemochromatosis
• In haemochromatosis, iron accumulates first in the
transferrin pool, manifested by a rise in serum
transferrin saturation, and subsequently in tissue
stores—especially the hepatic parenchyma—which is
accompanied by a progressive increase in
concentrations of serum ferritin.
• The clinical features of the disease arise as a result of
the progressive accumulation of iron in the
parenchymal cells of the liver, pancreas, heart, and
anterior pituitary.
• In the absence of treatment to reduce iron
concentrations, a characteristic pattern of tissue injury
and organ failure can develop.

Haemochromatosis
• In its most extreme form, the disease manifests as cirrhosis,
hepatocellular cancer, diabetes mellitus, sexual dysfunction
due to hypogonadotropic hypogonadism, cardiomyopathy, a
destructive arthritis, and generalised skin pigmentation.
• This phenotype is now less frequently seen, however, than
previously because of:
increased awareness of iron overload and early diagnosis,
genotyping done for family studies and screening
programmes .
• There is an average delay of 10 years between onset of
symptoms (lethargy, arthralgia) and diagnosis of
haemochromatosis, because of the non-specific nature of
the symptoms and because of the unfounded belief among
health professionals that haemochromatosis is rare.

Haemochromatosis
• Excess iron damages the liver, and presumably
other parenchymal organs, by induction of
oxidative stress and expression of cytokines, such
as transforming growth factor (TGF), which
promote hepatic fibrosis.
• The chemical nature of the reactive iron species
in cells is not known, but the presence of iron in
the circulation in excess of that bound by plasma
transferrin is well documented and is assumed to
promote oxidative damage to the lipid component
of cell membranes and intracellular organelles.

Haemochromatosis
• The disease typically affects middle aged men.
Menstruation and pregnancy probably account for
the lower presentation in women.
• Patients who are homozygous for the mutation
should have regular venesection to prevent
further tissue damage. Heterozygotes are
asymptomatic and do not require treatment.
Cardiac function is often improved by venesection
but diabetes, arthritis, and hepatic fibrosis do not
improve.

Presenting conditions in
haemochromatosis
• Cirrhosis (70%)
• Diabetes (adult onset) (55%)
• Cardiac failure (20%)
• Arthropathy (45%)
• Skin pigmentation (80%)
• Sexual dysfunction (50%)

Wilson's disease
• Wilson's disease is a rare autosomal recessive cause
of liver disease due to excessive deposition of copper
within hepatocytes.
• Abnormal copper deposition also occurs in the basal
ganglia and eyes.
• The defect lies in a decrease in production of the
copper carrying enzyme ferroxidase.
• Unlike most other causes of liver disease, it is
treatable and the prognosis is excellent provided that
it is diagnosed before irreversible damage has
occurred.

Wilson’s disease
• Patients may have a family history of liver or
neurological disease and a greenish brown
corneal deposit of copper (a Kayser Fleischer
ring), which is often discernible only with a
slit lamp.
• Most patients have a low caeruloplasmin
level and low serum copper and high urinary
copper concentrations.
• Liver biopsy confirms excessive deposition
of copper.

Risk factors associated with formation
of cholesterol gallstones
• Age > 40 years
• Female sex (twice risk
in men)
• Genetic or ethnic
variation
• High fat, low fibre diet
• Obesity
• Pregnancy (risk
increases with number
of pregnancies)
• Hyperlipidaemia
• Bile salt loss (ileal
disease or resection)
• Diabetes mellitus
• Cystic fibrosis
• Antihyperlipidaemic
drugs (clofibrate)
• Gallbladder dysmotility
• Prolonged fasting
• Total parenteral
nutrition

Charcot's triad of symptoms in severe
cholangitis
• Pain in right upper quadrant
• Jaundice
• High swinging fever with rigors and
chills

HELLP
• HELLP (hemolysis, elevated liver tests, low platelets) syndrome and
acute fatty liver of pregnancy should be considered in the differential
diagnosis of abnormal liver tests in the second half of pregnancy,
usually in the third trimester.
• In HELLP syndrome, patients have signs of pre-eclampsia as well as
thrombocytopenia.
• Pre-eclampsia affects 3–10% of pregnancies, and HELLP syndrome
occurs in 20% of patients with severe pre-eclampsia.
• The most common symptom is abdominal pain, but it occurs in only
65% of affected patients.
• Many patients have no specific symptoms, and the condition is
diagnosed when laboratory tests are done on patients with pre-
eclampsia.
• Renal failure or seizures (eclampsia) may complicate the pre-
eclampsia.
• As many as 30% of patients with HELLP present, or are diagnosed,
after delivery.
• As with pre-eclampsia, the pathogenesis of this condition is unknown.

HELLP
• Aminotransferase elevations are the hallmark of this
syndrome, with AST elevations ranging from 70 to 6,000,
with a mean of 250 in a large series.
• This is not a true hepatic failure, and the prothrombin time is
normal except in the most severe cases complicated by
disseminated intravascular coagulation.
• The thrombocytopenia may be modest to very severe.
• Most patients are not jaundiced, and the hemolysis is
manifested as schistocytes and burr cells on peripheral
smear.
• The liver biopsy shows the findings typical of pre-eclampsia:
periportal hemorrhage and fibrin deposition. The severity of
the histological changes is not uniformly reflected in the
laboratory abnormalities, and biopsy is not usually needed
for diagnosis.
• The differential diagnosis includes viral hepatitis or, rarely,
ITP. Viral serologies are useful.

Summary of main viral hepatitis
characteristics

Common symptoms of acute viral
hepatitis
• Myalgia
• Nausea and vomiting
• Fatigue and malaise
• Change in sense of smell or taste
• Right upper abdominal pain
• Coryza, photophobia, headache
• Diarrhoea (may have pale stools and
dark urine)

Variations in the clinical course of
acute viral hepatitis
• Normal Convalescence: This is by far the most frequent
course in Hepatitis A, B, and E. Recovery occurs in only
about 15%-20% of patients with Hepatitis C.
• Acute, Fulminant, Massive Necrosis: Massive loss of
hepatocytes with collapse of residual structures.
• Submassive Necrosis
• Chronic Hepatitis : common with HCV, but not a sequela of
Hepatitis A or E. Occasionally, the acute hepatitis subsides
but never totally heals and may even show intermittent
acute relapses. Scattered focal liver cell death continues to
occur, especially in the periportal region (piecemeal
necrosis).

Other biochemical or haematological
abnormalities seen in acute hepatitis
• Leucopoenia is common ( < 5 x 10
9
/l
in 10% of patients)
• Anaemia and thrombocytopenia
• Immunoglobulin titres may be raised

Liver enzyme activity in liver disease
Hepatitis
Cholestasis or obstruction
“Mixed”
Alkaline
Phosphatase
Normal
Raised
Raised
γ glutamyl-
transferase
Normal
Raised
Raised
Alanine
transaminase Raised
Normal
Raised
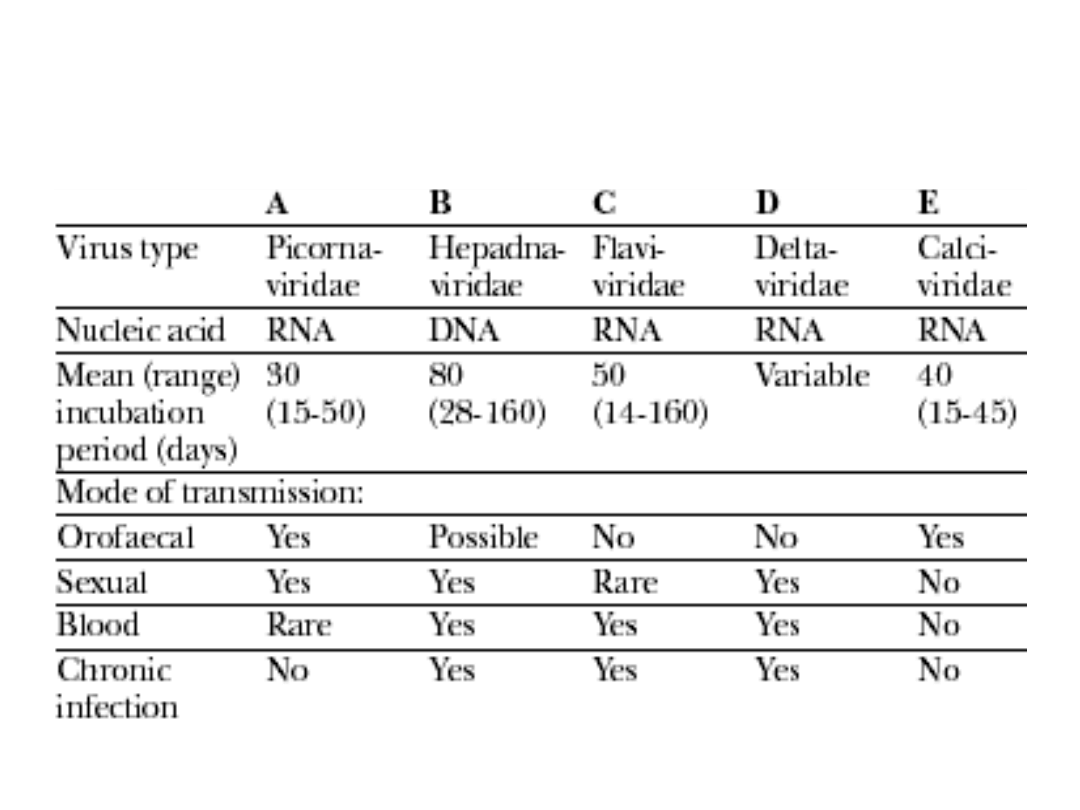
Types and modes of transmission of
human hepatitis viruses
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
Wyszukiwarka
Podobne podstrony:
ABC Transplantation of the liver and pancreas
Aspden THE CREATION OF THE PROTON (2005)
Investigating the Afterlife Concepts of the Norse Heathen A Reconstuctionist's Approach by Bil Linz
16 Changes in sea surface temperature of the South Baltic Sea (1854 2005)
0262033291 The MIT Press Paths to a Green World The Political Economy of the Global Environment Apr
2005 10 Dawn of the Uber Distro
Mordwa, Stanisław Religious Minorities of the Internet the Case of Lodz, Poland (2005)
Faculty of Theology operates on the basis of the Act of 27 July 2005 Law on Higher Education
Marilyn Yalom Birth of the Chess Queen A History (2005)
Publications of The Metropolitan Museum of Art 1964 2005 A Bibliography
Communist League Basic Principles of the Communist League (2005)
0791464539 State University of New York Press The Gathering Of Reason May 2005
148 Bitwa o brzuchy Battle of the Bulge, Jay Friedman, Jun 8, 2005
07 WoW War Of The Ancients Trilogy 03 The Sundering (2005 07)
Mansour Pasupathi The wisdom of experience Autobiographical narratives International Journal of Beha
BS EN ISO 1133 2005 Plastics Determination of the melt mass flow rate (MFR) and the melt volume flow
WarCraft (2005) War of the Ancients Trilogy 03 The Sundering Richard A Knaak
Falcon Aristotle and the Science of Nature (Cambridge, 2005)
The Modern Scholar David S Painter Cold War On the Brink of Apocalyps, Guidebook (2005)
więcej podobnych podstron