
ZASADY ZAPISU RODOWODU
– rysowanie rodowodów
Zakład Genetyki Medycznej
Warszawski Uniwersytet Medyczny
Ćwiczenia z propedeutyki genetyki klinicznej

Wywiad rodzinny
Prawidłow
e zebranie
wywiadu
rodzinneg
o
Gdy pytasz osobę o braci i
siostry, dowiedz się, czy miała
rodzeństwo, które zmarło:
często ludzie, nie pytani,
wymieniają tylko osoby żyjące.
Spytaj o przypadki śmierci
noworodków i poronienia.
Zwróć uwagę na to, czy
omawiane rodzeństwo ma tych
samych oboje rodziców, co
badana osoba oraz na
rodzeństwo adoptowane.
Spytaj o związki
krewniacze.
Spytaj o pochodzenie
etniczne różnych gałęzi
rodzinny.
Spróbuj uzyskać
dokumentację
(np. wyniki badań) istotną dla
wywiadu rodzinnego; często
członkowie rodziny na
podstawie powierzchownych
informacji,
które mogą być nieprawidłowe,
wnioskują, że krewni cierpią na
zaburzenia „przekazywane w
rodzinie”.
Spytaj o dzieci z zapłodnienia in vitro.
Spytaj o wczesne zgony.

Dokumentacja użyteczna
w poradnictwie genetycznym
• Dokumentacja medyczna z archiwów szpitalnych,
prywatnych gabinetów lekarskich oraz innych
zakładów leczniczych
• Odpowiednie instytucje i urzędy lokalne lub
regionalne
• Biura parafialne
• Do najbardziej przydatnych dokumentów należą:
wyniki badań sekcyjnych
informacje o zabiegach chirurgicznych
kompleksowy opis wszystkich badań
przeprowadzonych
u danej osoby
wyniki badań laboratoryjnych np. badania
cytogenetyczne
fotografie rodzinne

Różnorodność symboli
stosowanych przy
sporządzaniu rodowodu
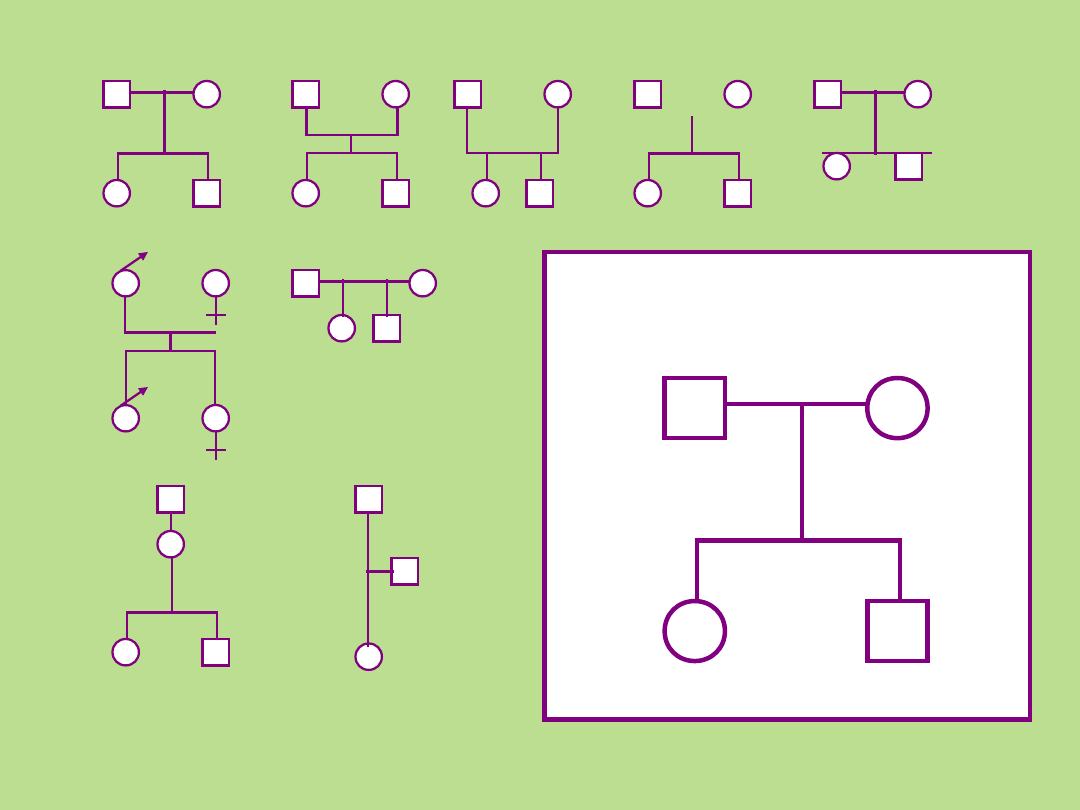
Biologiczni rodzice i ich
potomstwo
x
Rekomendowany przez
PSTF
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
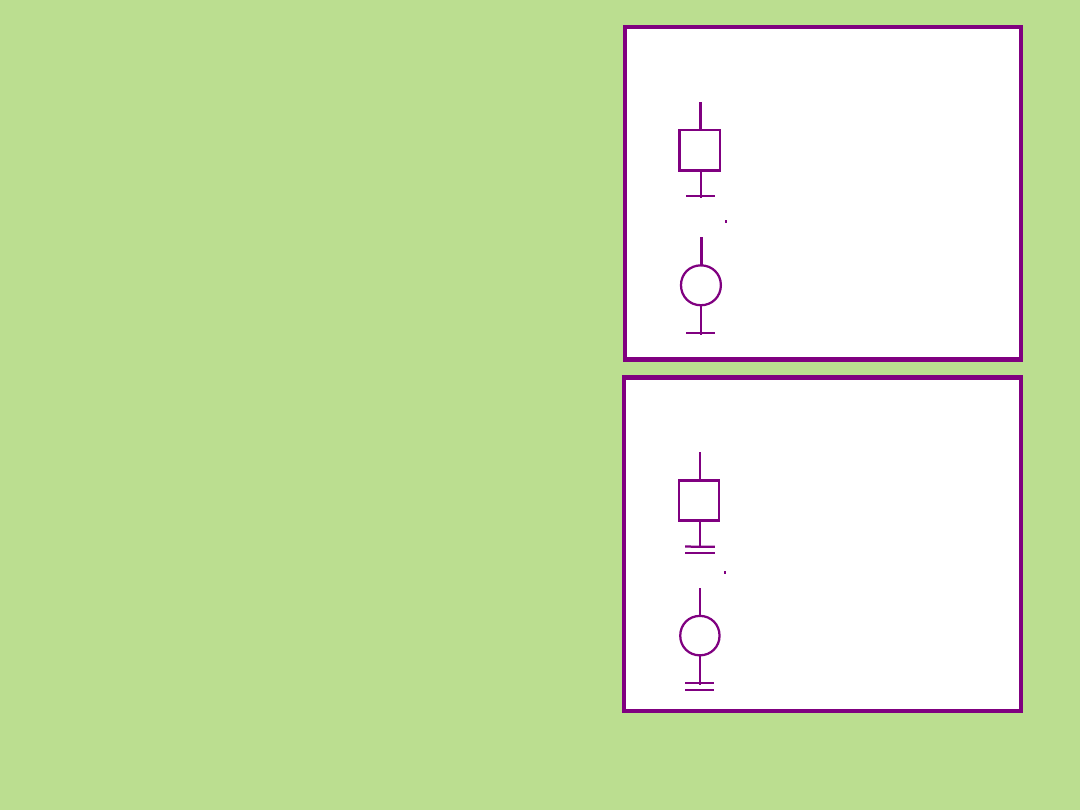
Osoba zdrowa
z wykluczoną chorobą –
udokumentowana ocena
lekarza
płeć męska
płeć
żeńska
Udokumentowane
*
*
płeć męska
płeć
żeńska
Nieudokumentowane
Osoba zdrowa
nieudokumentowana ocena
konsultanta, brak diagnozy
lekarskiej
– badanie podmiotowe
płeć
nieznana
płeć
nieznana
*
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Osoba w stadium
przedobjawowym
choroby
Duże prawdopodobieństwo
wystąpienia objawów w
przyszłości, por. pląsawica
Huntingtona
Nosiciel allela recesywnego
Niezależnie od typu dziedziczenia!
Rekomendowane przez
PSTF
płeć męska
płeć żeńska
Rekomendowane przez
PSTF
płeć męska
płeć żeńska
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
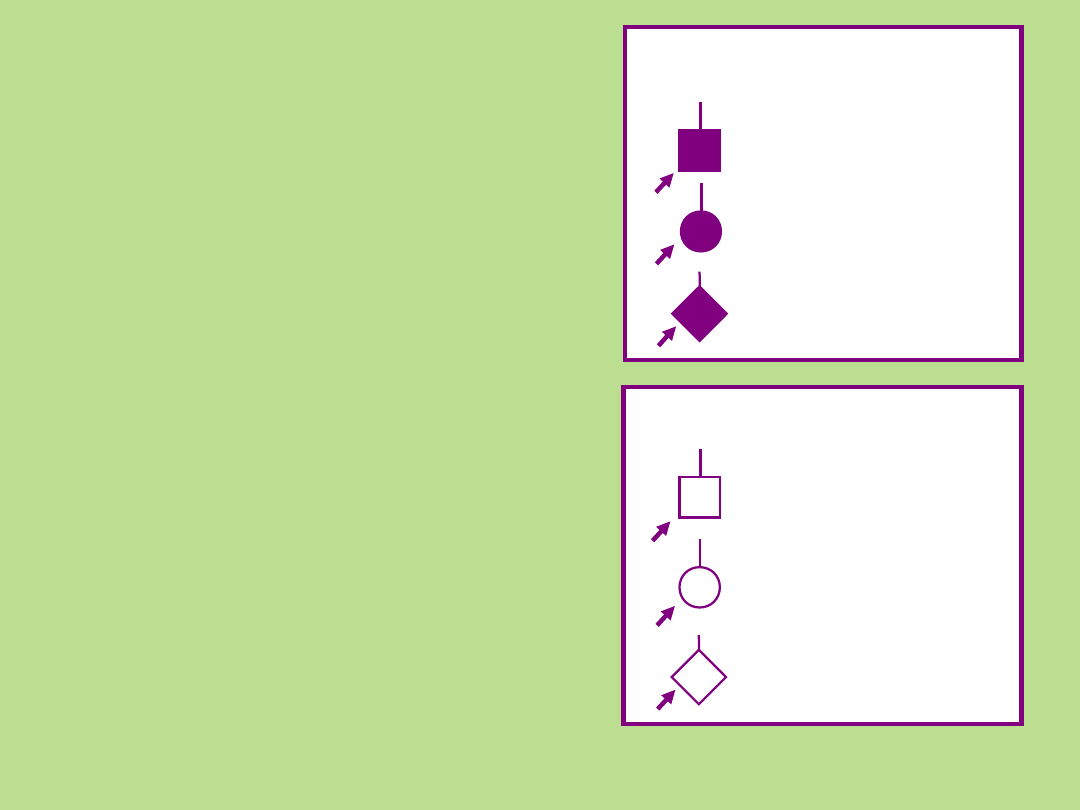
Osoba chora
płeć męska
płeć
żeńska
płeć
nieznana
*
#
c
Rekomendowane przez
PSTF
płeć męska
płeć żeńska
płeć nieznana
*
Rekomendowane przez
NSGC PSTF
Osoba
nieżyjąca
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Grupa osób (nieznana
liczba)
Rekomendowane przez
NGSC PSTF
płeć męska
płeć
żeńska
płeć
nieznana
Grupa osób (znana liczba)
Rekomendowane przez
PSTF
płeć męska
płeć żeńska
płeć nieznana,
nieokreślona
n
n
n
5
2
4
2
(3)
5
5
5
n
n
n
n
(n)
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Martwe urodzenie
wewnątrzmaciczne obumarcie
płodu, zgon płodu
(ang. still birth)
Rekomendowane przez
NGSC PSTF
płeć męska
płeć
żeńska
płeć
nieznana
Ciąża
Rekomendowane przez
NGSC
PSTF
płeć męska
płeć żeńska
płeć nieznana
P
P
?
P
P
P
SB
SB
SB
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Przerwanie ciąży
Poronienie spontaniczne
(Sp)
Rekomendowane przez NGSC PSTF
płeć męska
płeć
żeńska
płeć nieznana
Poronienia
spontaniczne
Poronienia
spontaniczne
- chory płód
Płeć męska
Płeć żeńska
Ciąża
pozamaciczna
Płeć męska
Płeć
żeńska
Rekomendowane przez
PSTF
płeć męska
płeć
żeńska
płeć nieznana
Przerwanie
ciąży
Przerwanie ciąży
- chory płód
Płeć męska
Płeć żeńska
Płeć męska
Płeć
żeńska
SA
T
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Niepłodność
Brak potomstwa
Rekomendowane przez
NGSC PSTF
płeć męska
płeć
żeńska
Rekomendowane przez
NGSC PSTF
płeć męska
płeć
żeńska
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
Konsultant (osoba badana)
Rekomendowane przez
NGSC PSTF
płeć męska
płeć
żeńska
Proband
Rekomendowane przez
PSTF
płeć męska
płeć żeńska
płeć nieznana
płeć
nieznana
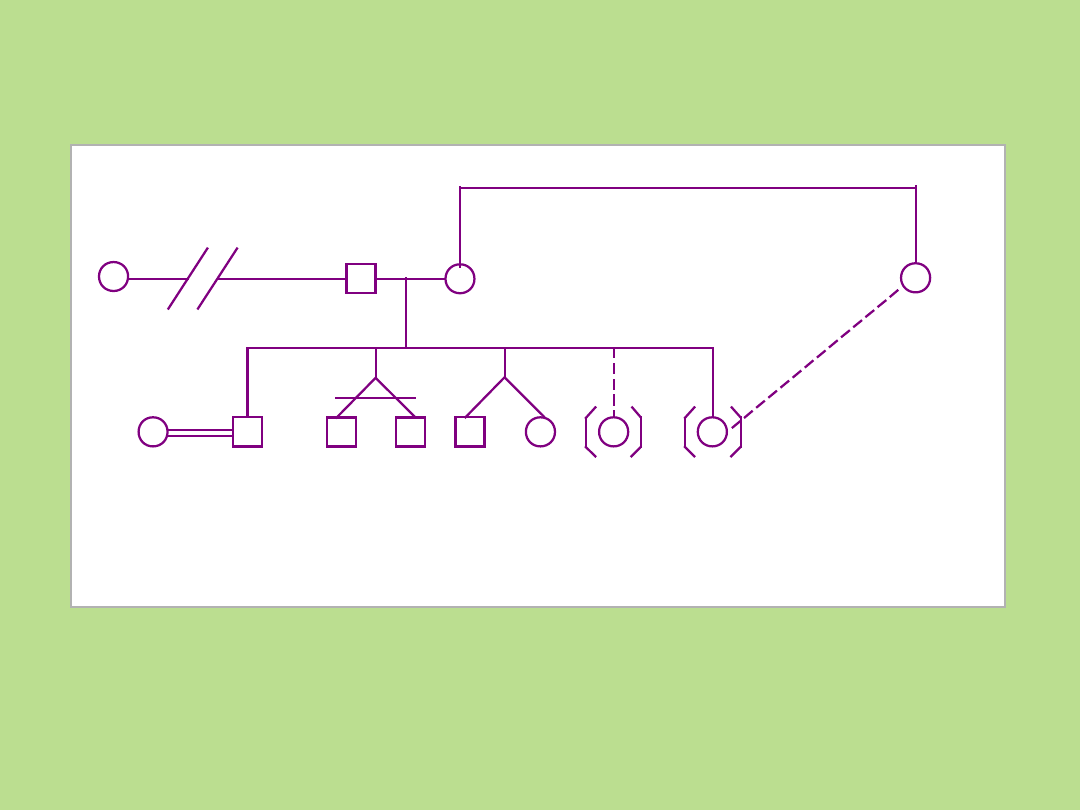
związek się
zakończył
związek
krewniacz
y
bliźnięta
jednojajo
we
bliźnięta
dwujajow
e
oddanie
do
adopcji
adopcj
a
adopcja przez
członka
rodziny
Bliźnięta, adopcje, związek
krewniaczy
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
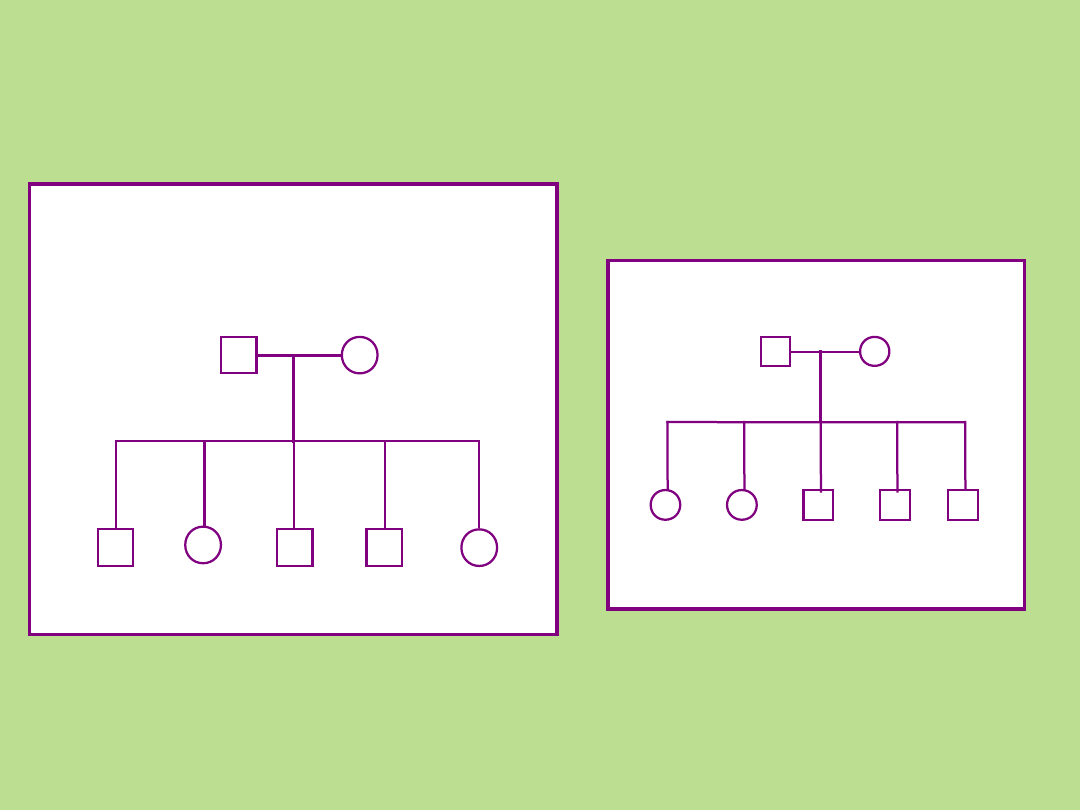
Potomstwo każdej pary rodziców zaznacza się w kolejności urodzeń,
umieszczając najstarsze po lewej stronie
1985 1990 1955 1960 1965
zachowana kolejność
urodzeń
niezachowania kolejność urodzeń
Bennett R.L et al.. J Genet Counsel (2008) 17:424-433 56: 745-752.
Stienhaus K.A.,et al.. Am. J. Hum. Genet. (1995) 56: 291-295
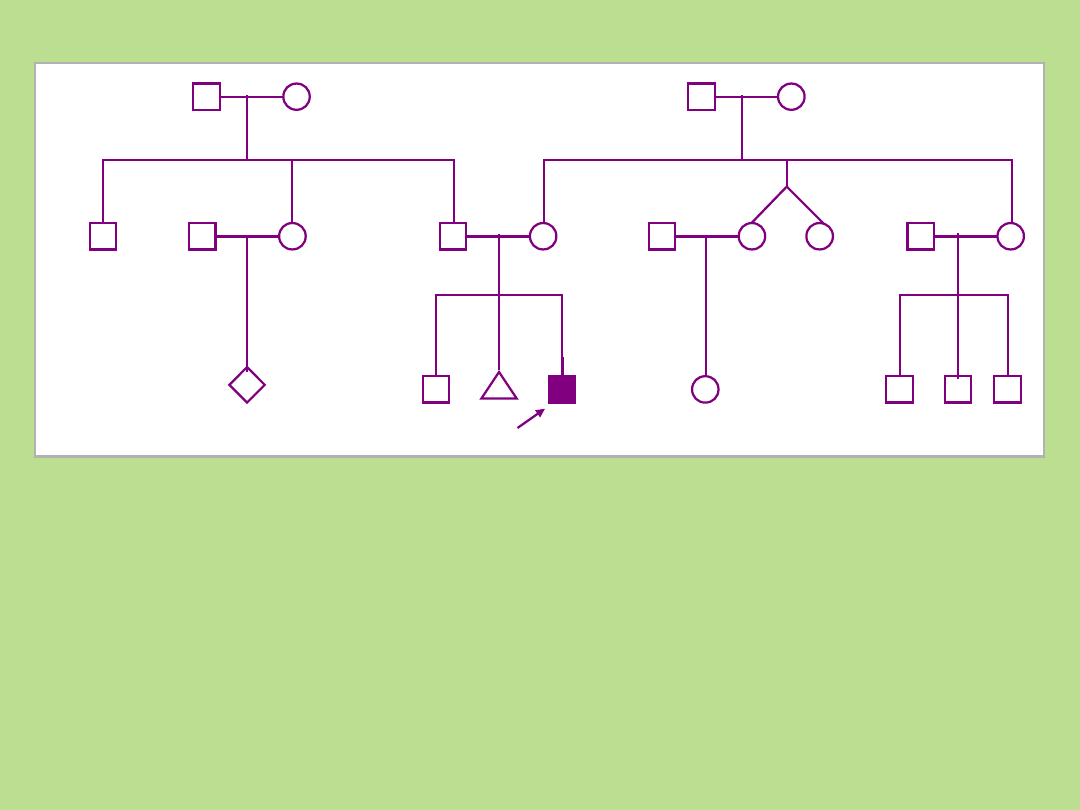
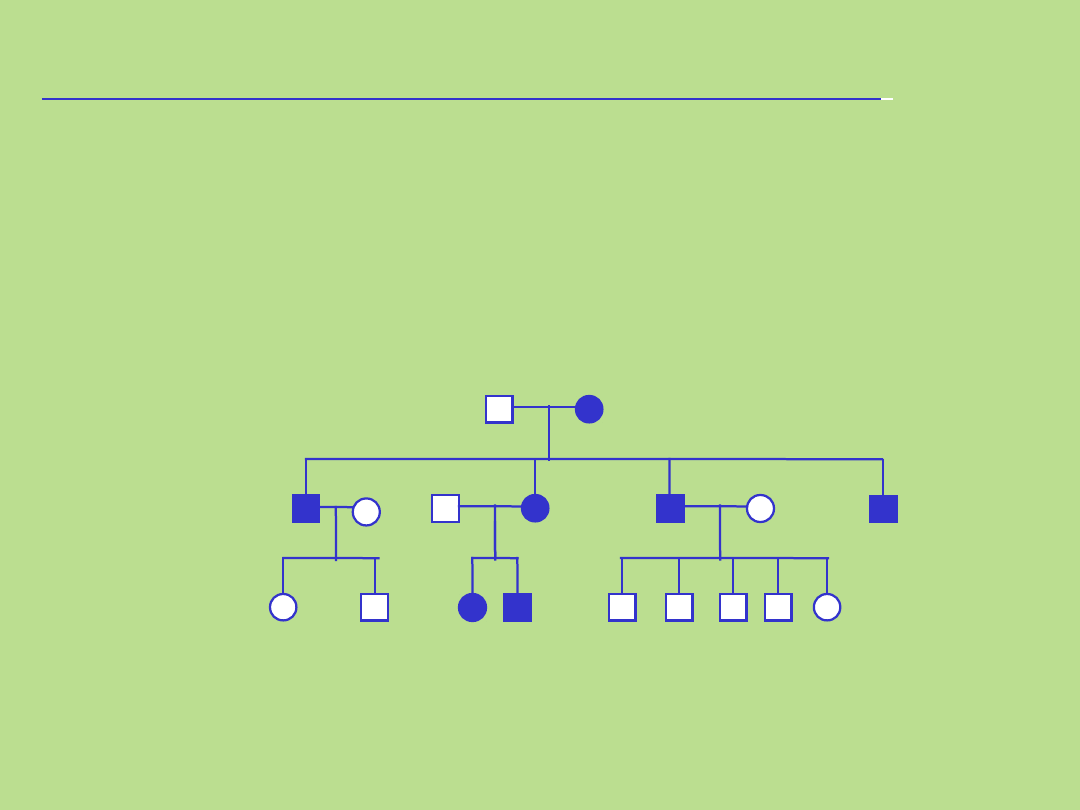
Przykład rodowodu
rodziny:
Podstawowe reguły:
• Linia męska po lewej stronie
• Wszyscy członkowie tego samego pokolenia w jednej linii poziomej
• Cyframi rzymskimi oznacza się kolejne pokolenia, poczynając od najwcześniejszego
• Cyframi arabskimi oznacza się kolejne osoby w pokoleniu (numery od strony lewej do
prawej)
• Potomstwo każdej pary rodziców zaznacza się w kolejności urodzeń,
umieszczając najstarsze po lewej stronie
• Zaleca się, aby rodowód rozpocząć od dołu,
zaznaczając najmłodszą generację, przesuwając się ku górze arkusza
Bennet L. et al. J Genet Counsel (2008) 17:424-
433
P
I
II
III
1
2
3
4
1
2
3
4
5
6
7
8
9
1
0
1
2
4
5
6
7
8
Płeć żeńska
3
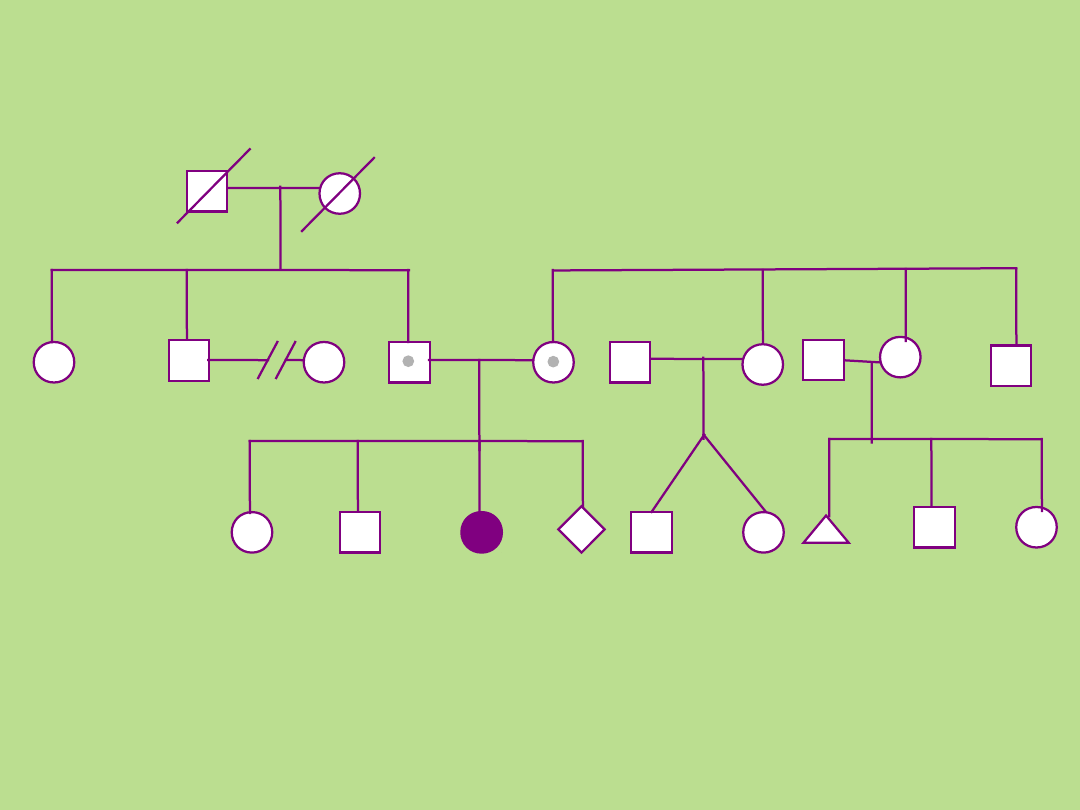
Kamila
Robert
Bartek
Lidka
Maria
Krysi
a
Kamil
Zuzia
Jarek
Kasia
Maciej
Płeć męska
Zbysze
k
Mart
a
Klaudia
Przykład 1
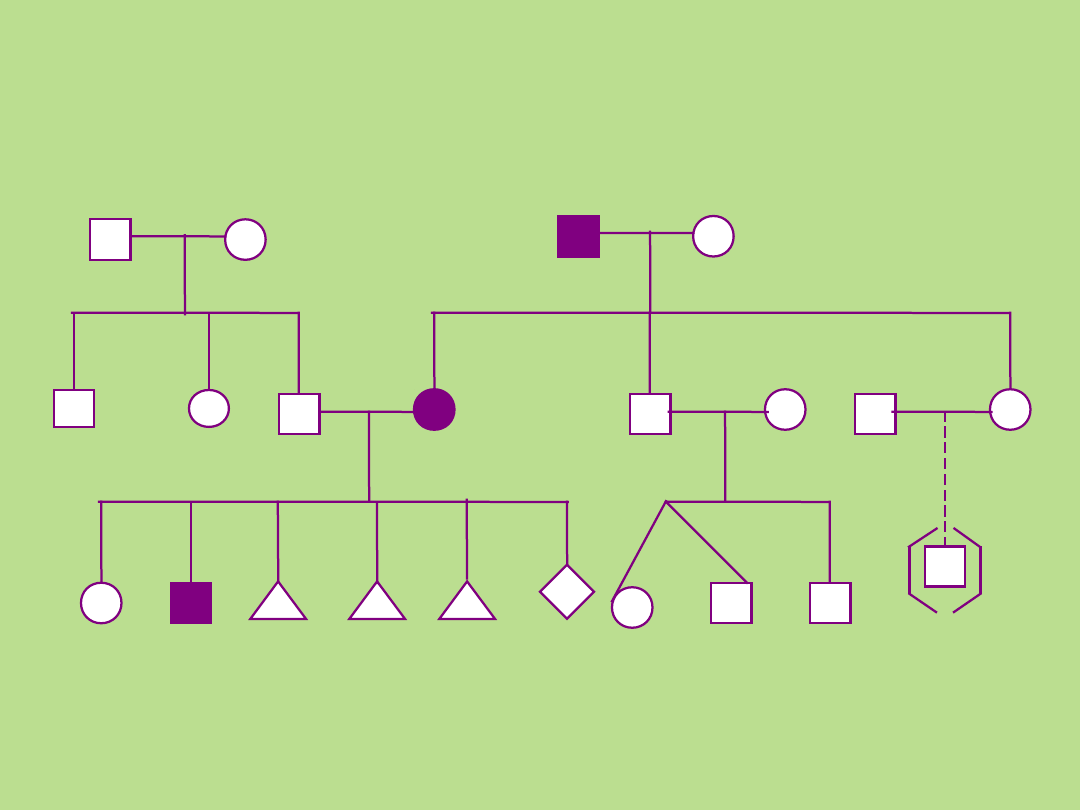
P
P
Kasia
Karol
Paweł
Karolina
Mateusz
Magda
Kamil
a
Przykład
2
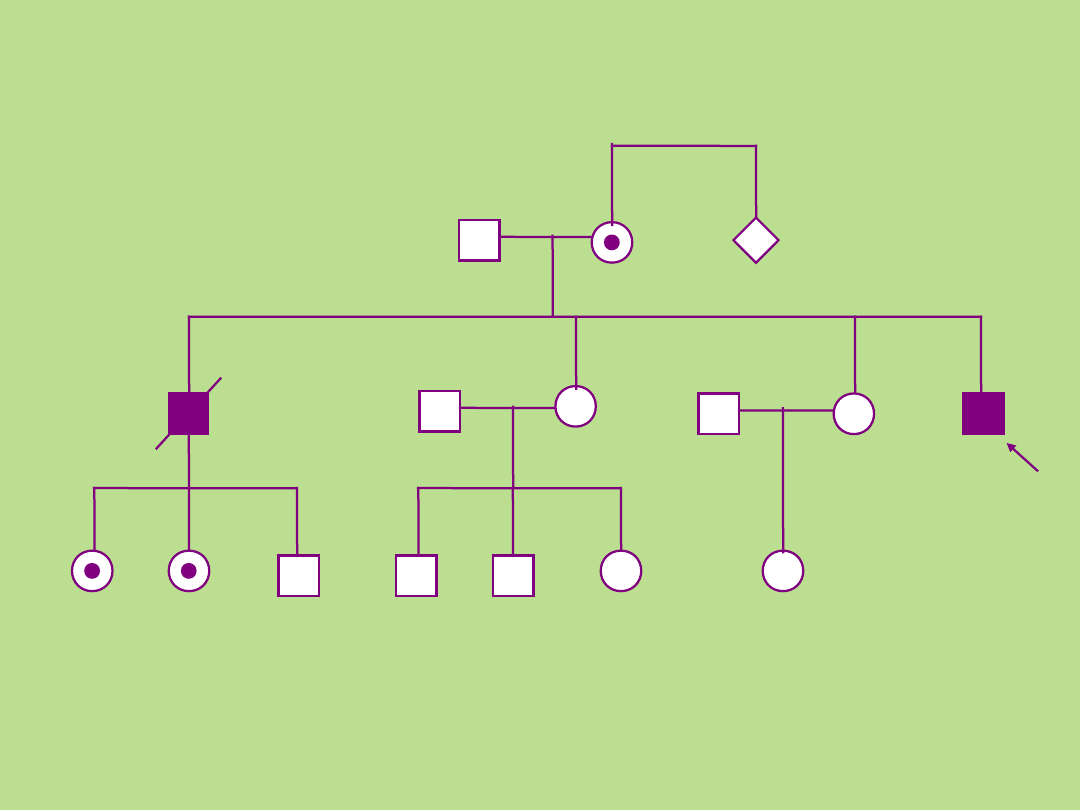
n
Marian
Maria
Kuba
Joanna
Ola
Mirek
Tomek
Marek
6 y
Michał
4 y
Zosia
Kinga
Przykład 3

Programy komputerowe
do rysowania rodowodów
• Haplopainter
http://haplopainter.sourceforge.net
• PEdigree Drawing software
( Za: Bennett R.L., Steinhaus K.A., Uhrich S.B., et al. Recomendations for standardized human pedigree nomenclature.
Pedigree Standarization Tack Force of National Society of Genetic Counselors. Am. J. Hum. Genet. 1995: 745-752.
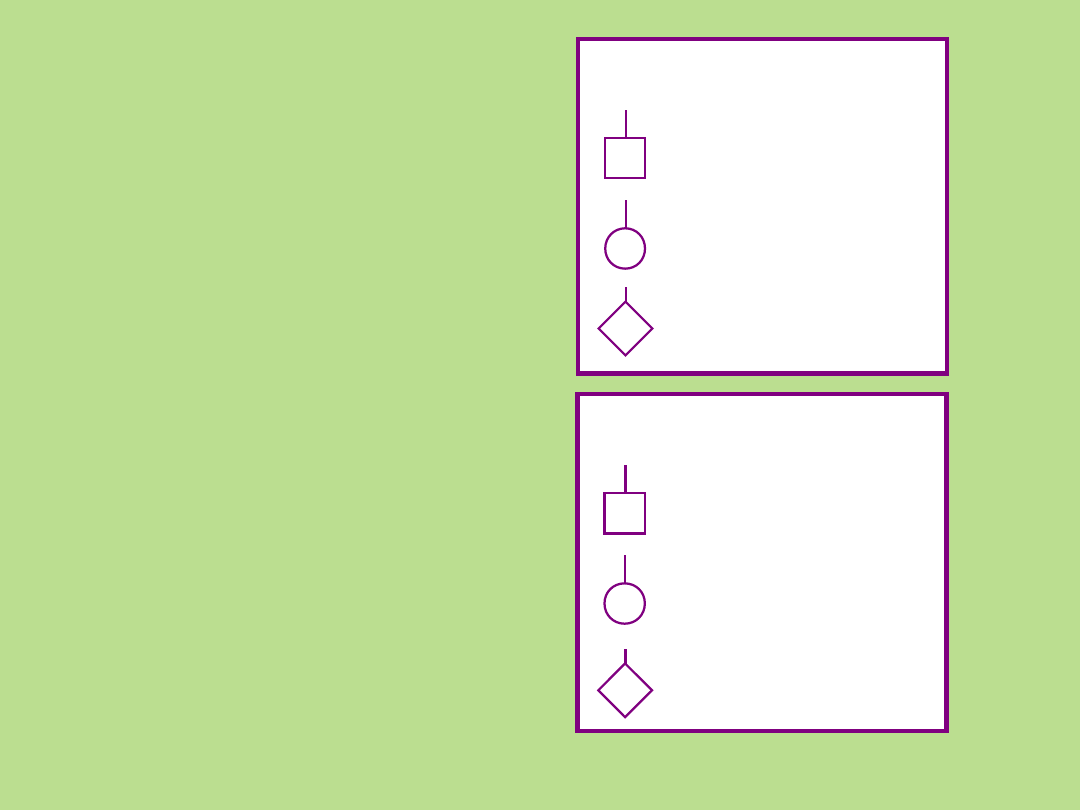
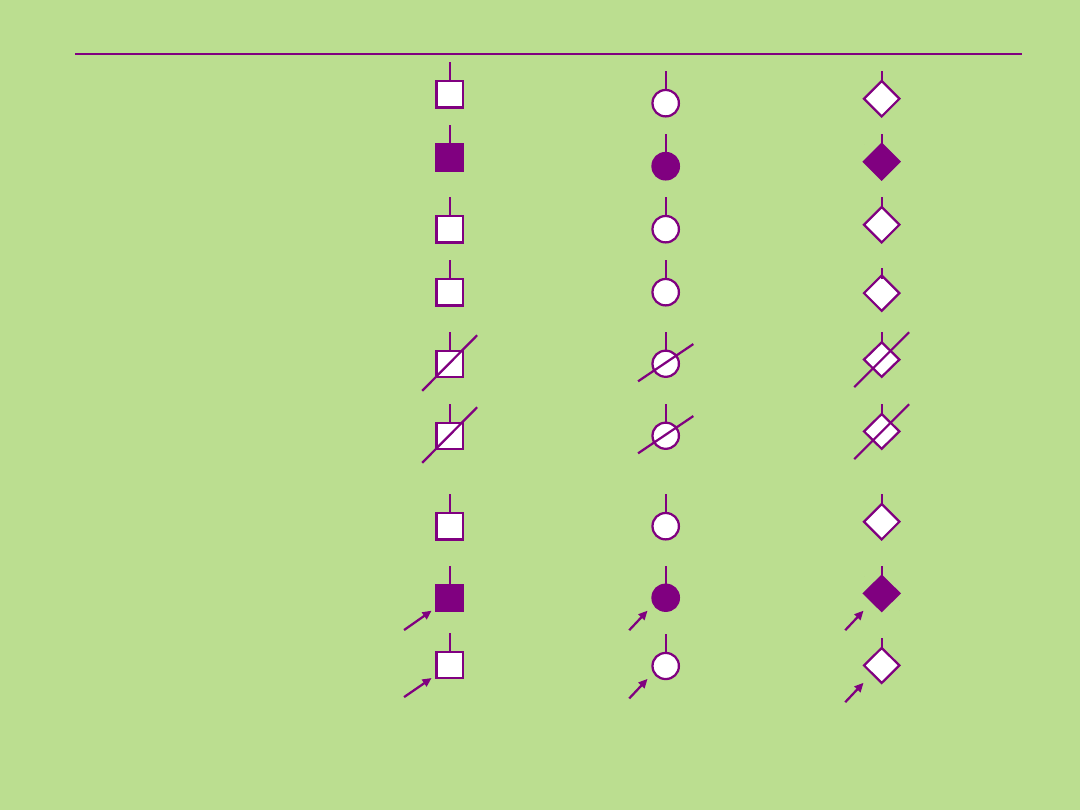
Symbole stosowane przy sporządzaniu
rodowodu
Płeć męska
Płeć żeńska
Płeć
nieznana
Osoba
Osoba chora
Grupa osób
(znana liczba)
Grupa osób
(nieznana liczba)
Osoba nieżyjąca
Martwe urodzenia
(ang. stillbirth, SB)
Ciąża
(ang. pregnancy, P)
Proband
Osoba badana
5
n
SB
P
5
n
SB
P
5
n
SB
P
( Za: Bennett R.L., Steinhaus K.A., Uhrich S.B., et al. Recomendations for standardized human pedigree nomenclature.
Pedigree Standarization Tack Force of National Society of Genetic Counselors. Am. J. Hum. Genet. 1995: 745-752.
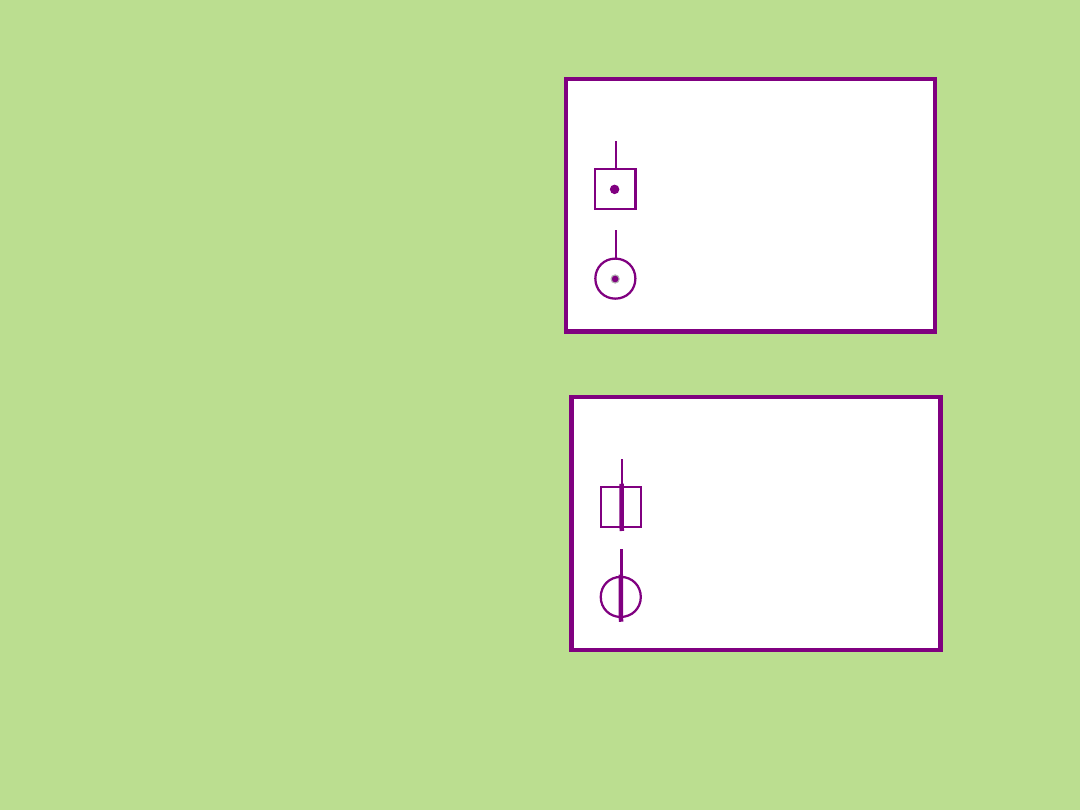
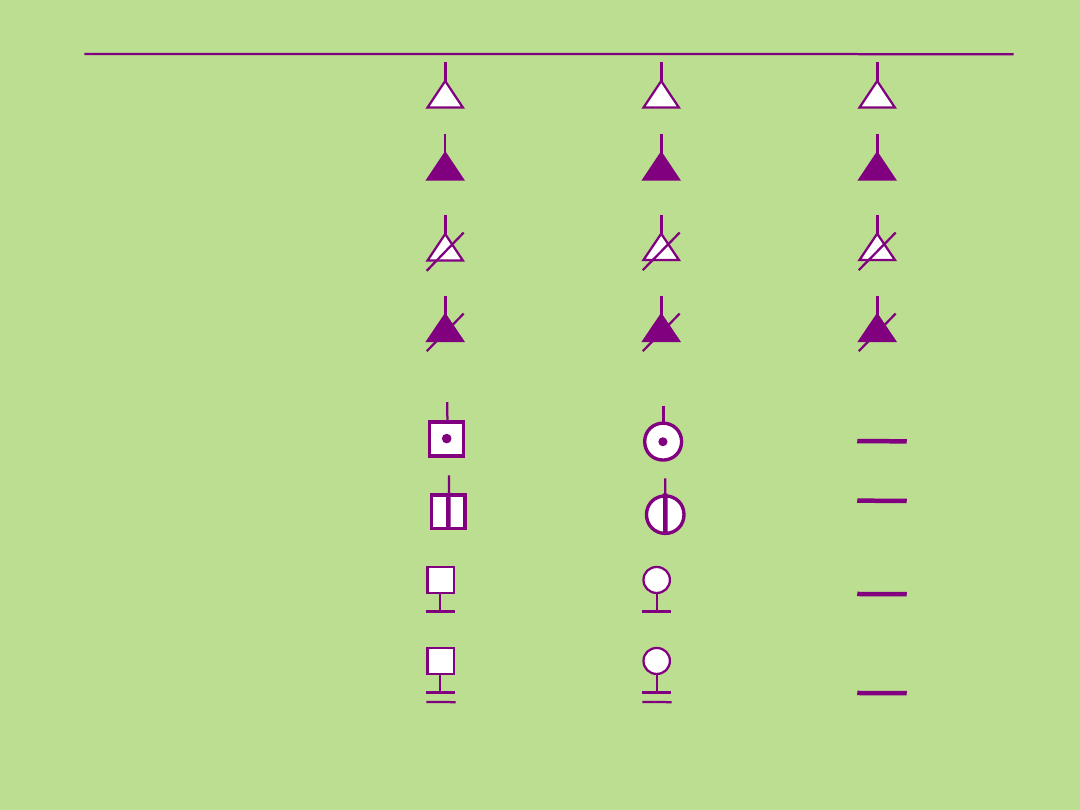
Symbole stosowane przy sporządzaniu
rodowodu
Płeć męska
Płeć żeńska
Płeć
nieznana
Poronienia
spontaniczne
Poronienia
spontaniczne
- chory płód
Przerwanie
ciąży
Przerwanie ciąży
- chory płód
Płeć męska
Płeć żeńska
Ciąża
pozamaciczna
Płeć męska
Płeć
żeńska
Płeć męska
Płeć żeńska
Płeć
żeńska
Płeć męska
Nosiciel allela recesywnego
Osoba w okresie
przedobjawowym choroby
Brak
potomstwa
Niepłodność

Rodzaje
dziedziczenia
Rodzaje
dziedziczenia
Dziedziczenie
mendlowskie
Dziedziczenie
wieloczynniko
we
Dziedziczenie
mitochondrial
ne
Dominujące
Autosomalne
Sprzężone z
chromosomem X
Recesywne
Piętnowanie (imprinting) genomowe
Dominujące
Recesywne
Dziedziczenie jednogenowe

Mechanizmy molekularne
• Nasilenie/Nabycie nowej funkcji
(gain of function/novel property)
• Efekt negatywny dominujący
(dominant negative)
(zadanie: jaka część
homotetramerów będzie prawidłowa u heterozygoty?)
• Niewydolność haplotypowa
(haplotype insufficiency)
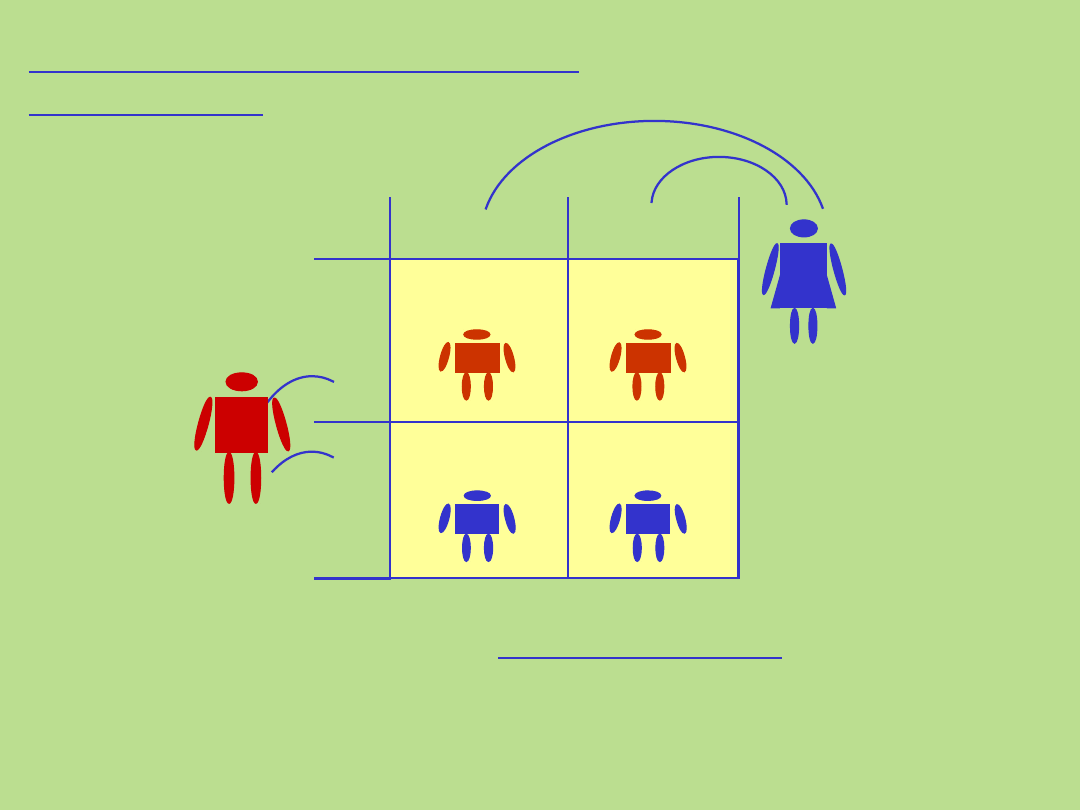
Dziedziczenie autosomalne
dominujące
50%
M
m
m
m
M
m
m m
m m
M
m
Ojcie
c
M
m
Matk
am m
Marfan Syndrome: How is it inherited?
Przykłady chorób:
•zespół Marfana
•choroba Huntingtona
Problemy diagnostyczne
•mozaikowość germinalna
•Mutacje de novo
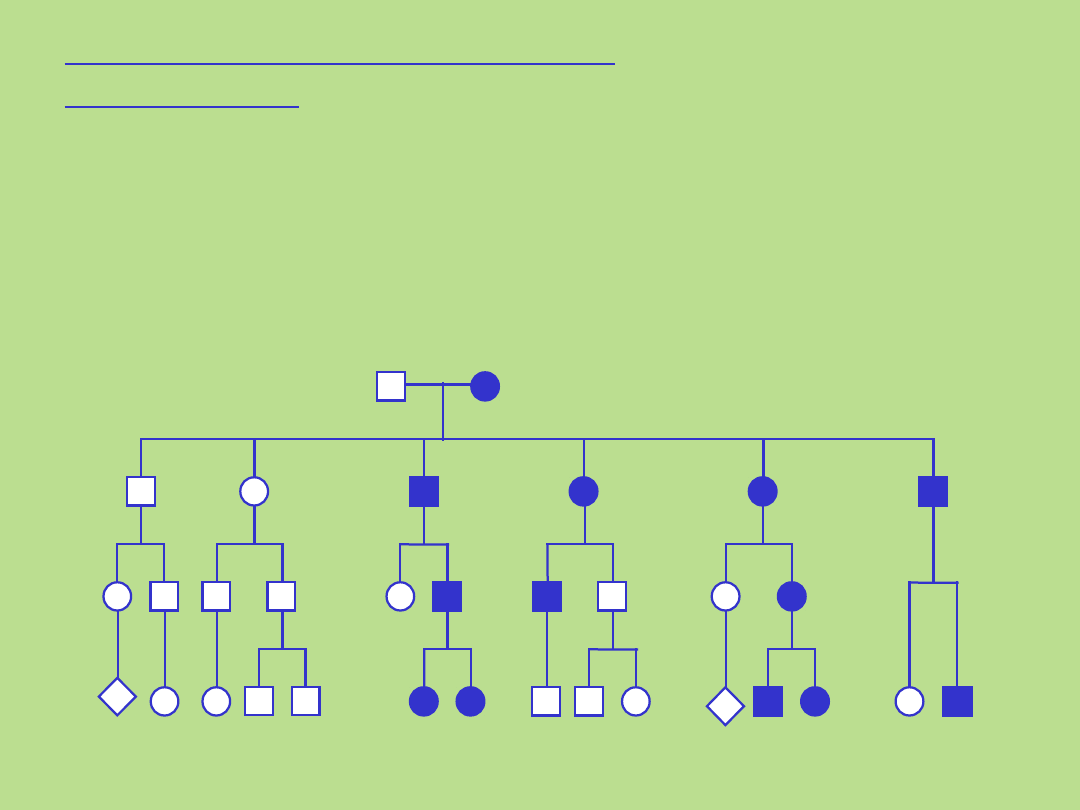
5
2
Pedigree typical of autosomal dominant
inheritance
Dziedziczenie autosomalne
dominujące
. Kryteria dla tego typu dziedziczenia:
• występowanie choroby z jednakową częstością u obu płci
• pionowy wzór dziedziczenia choroby w rodowodzie, bez przeskoków z
pokolenia na pokolenie
• każde chore dziecko ma chorego rodzica (?)
• prawdopodobieństwo przekazania nieprawidłowego genu
potomstwu wynosi 50% - zarówno ojciec jak i matka mogą przekazać
nieprawidłowy gen
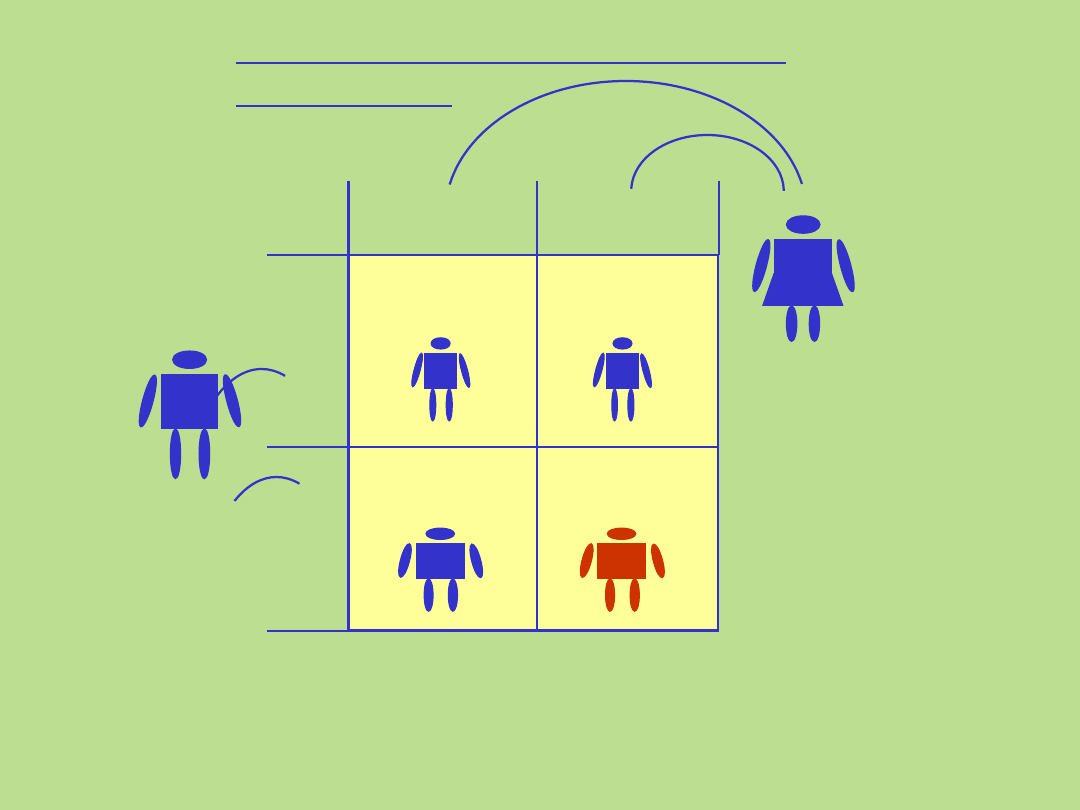
Dziedziczenie autosomalne
recesywne
25%
Matka
C
c
C
c
C
c
C
c
c c
C
c
C C
Ojciec
C
c
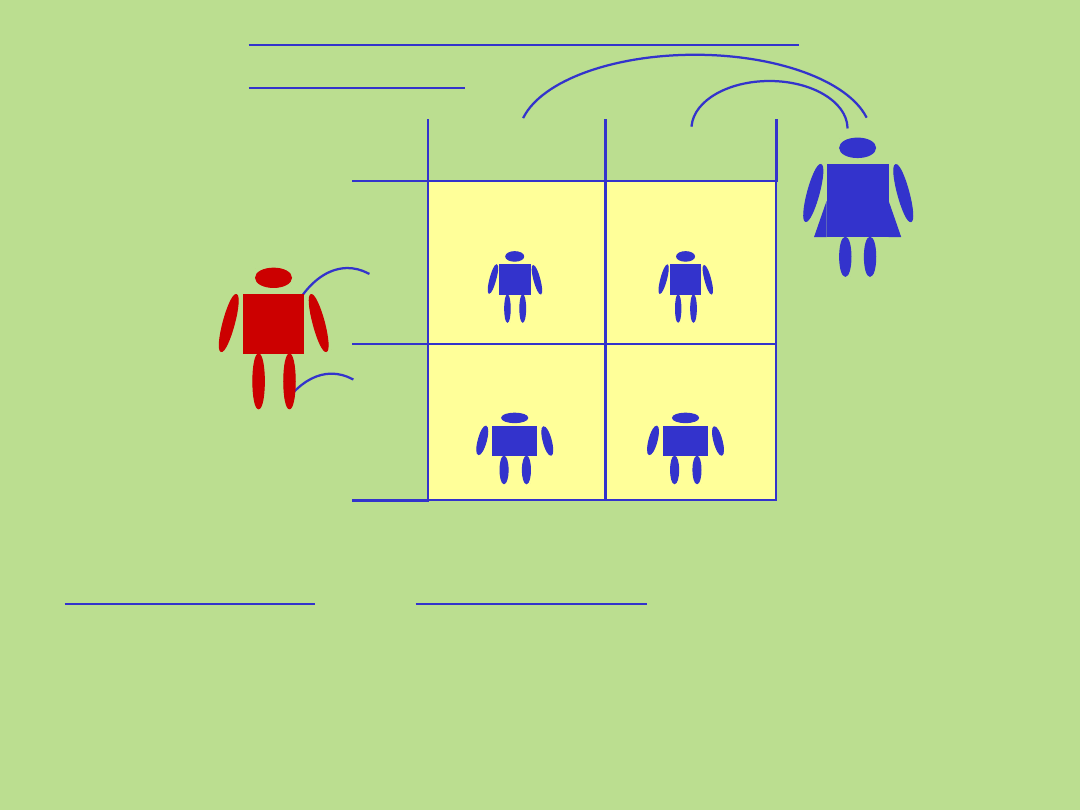
0
%
Ojciec
c
c
c
c
C
C
C
c
C
c
C
c
C
c
Matka
C C
Dziedziczenie autosomalne
recesywne
• nosicielstwo
• pokrewieństwo
Cechy specyficzne:
• mukowiscydoza
• anemia sierpowatokrwinkowa
Przykłady chorób
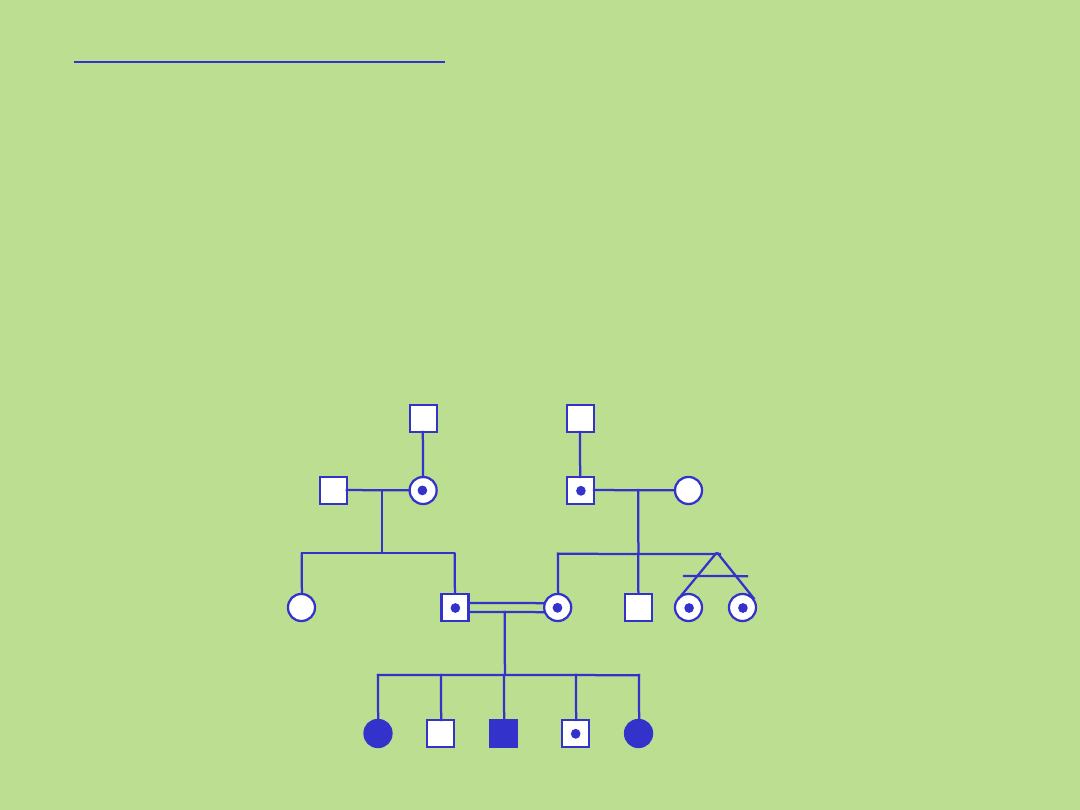
Autosomalne recesywne
Rodowód typowy dla dziedziczenia autosomalnego
recesywnego
1. Kryteria dla tego typu dziedziczenia:
• występowanie choroby z jednakową częstością u
obu płci
• poziomy wzór dziedziczenia choroby ponieważ choroba występuje zazwyczaj wśród
rodzeństwa
• ujawnienie się choroby u homozygot
• jeżeli choroba (lub cecha) występuje w populacji bardzo rzadko, istnieje duże
prawdopodobieństwo pokrewieństwa miedzy rodzicami chorego
• obydwoje rodzice chorego dziecka są bezobjawowymi klinicznie heterozygotami
(nosicielami) pod względem zmutowanego genu
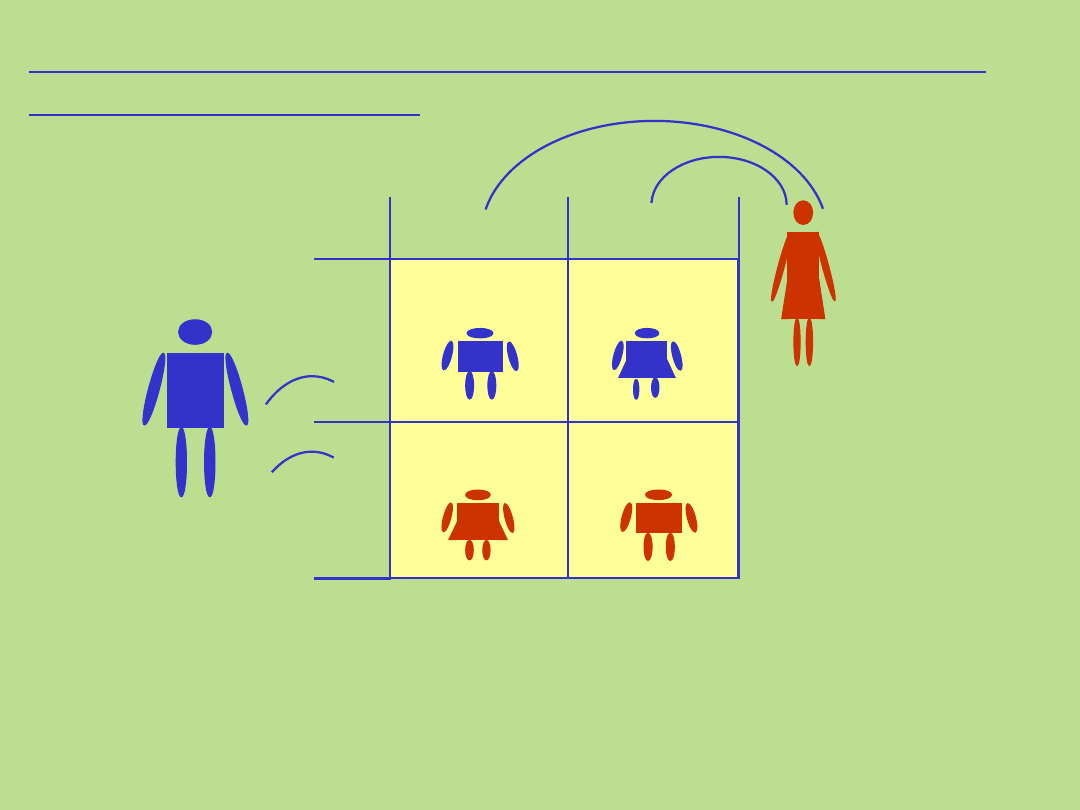
Dziedziczenie autosomalne pseudodominujące
(quasi-dominujące)
50%
M
m
m
m
M
m
m
m
m
m
M
m
Ojcie
c
M m
Matk
a
m
m
Marfan Syndrome: How is it inherited?
Dziedziczenie autosomalne recesywne naśladujące autosomalne
dominujące
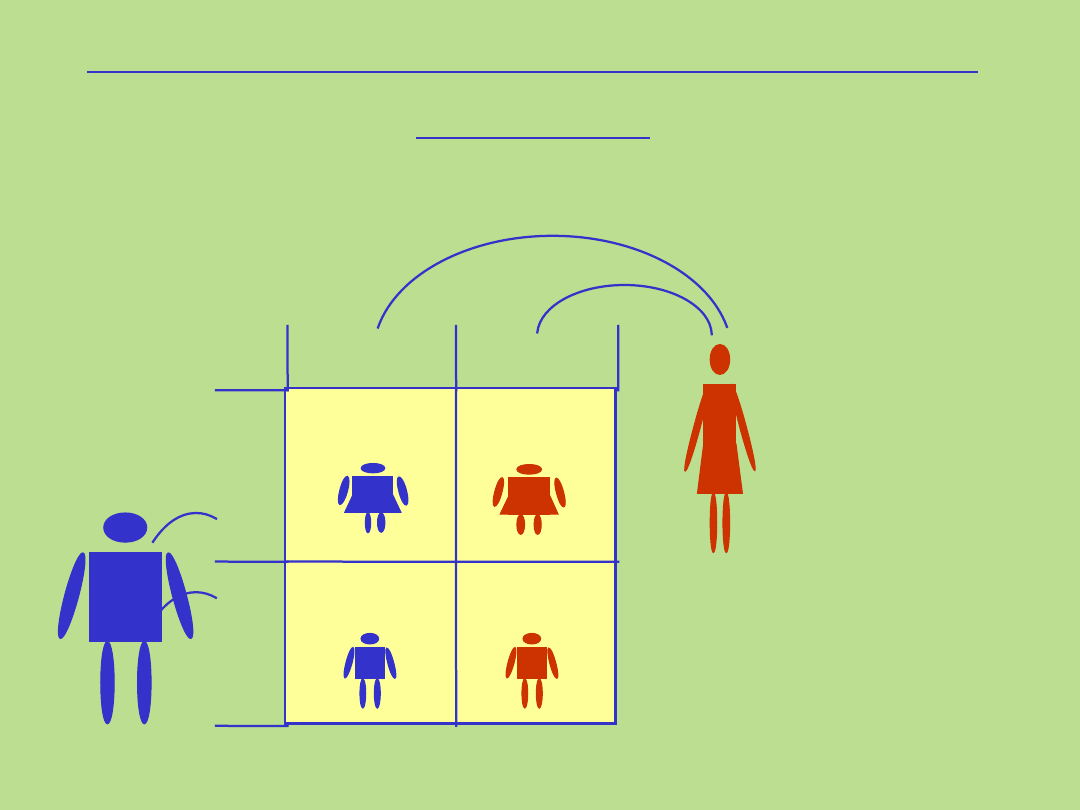
50%
X
Y
X
X*
X
X*
X*
Y
X Y
X X
Dziedziczenie sprzężone z chromosomem X
dominujące
gen przekazany przez matkę
Matka
X
X
*
chora
Ojciec
X Y
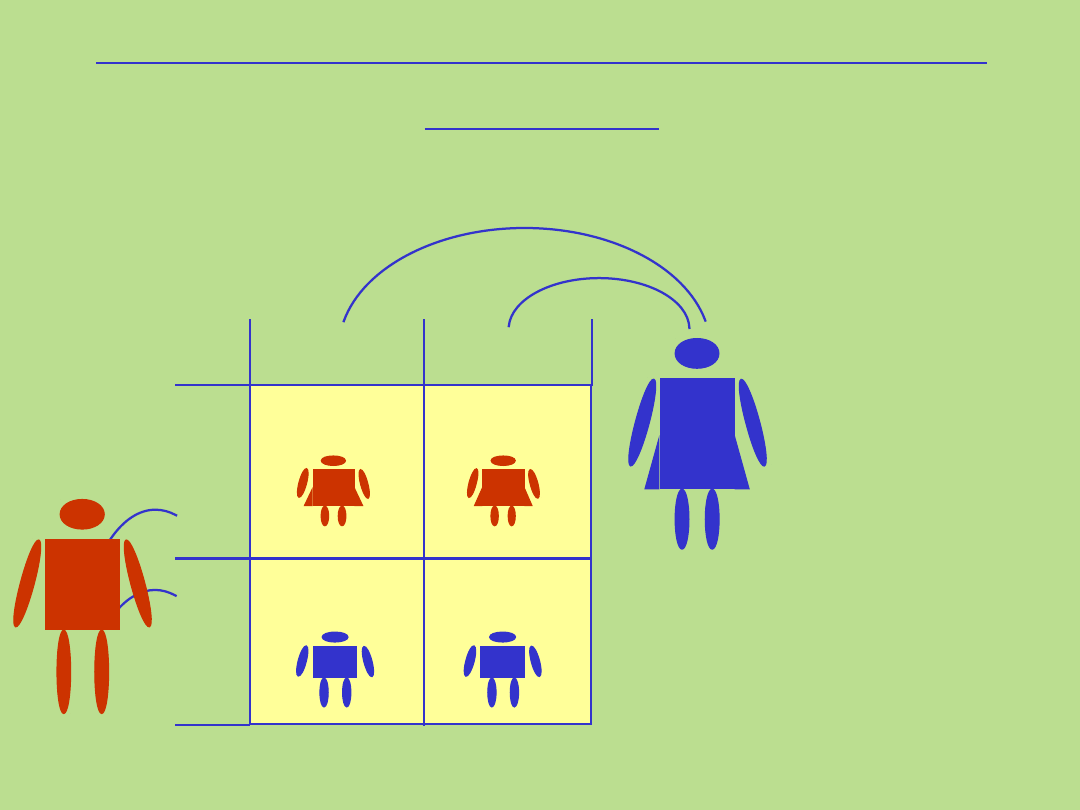
50%
(tylko córki)
X*
Y
X
X
X*
X
X Y
X Y
X*
X
Ojciec
X*
Y
Dziedziczenie sprzężone z chromosomem X
dominujące
gen przekazany przez ojca
Matka
X X
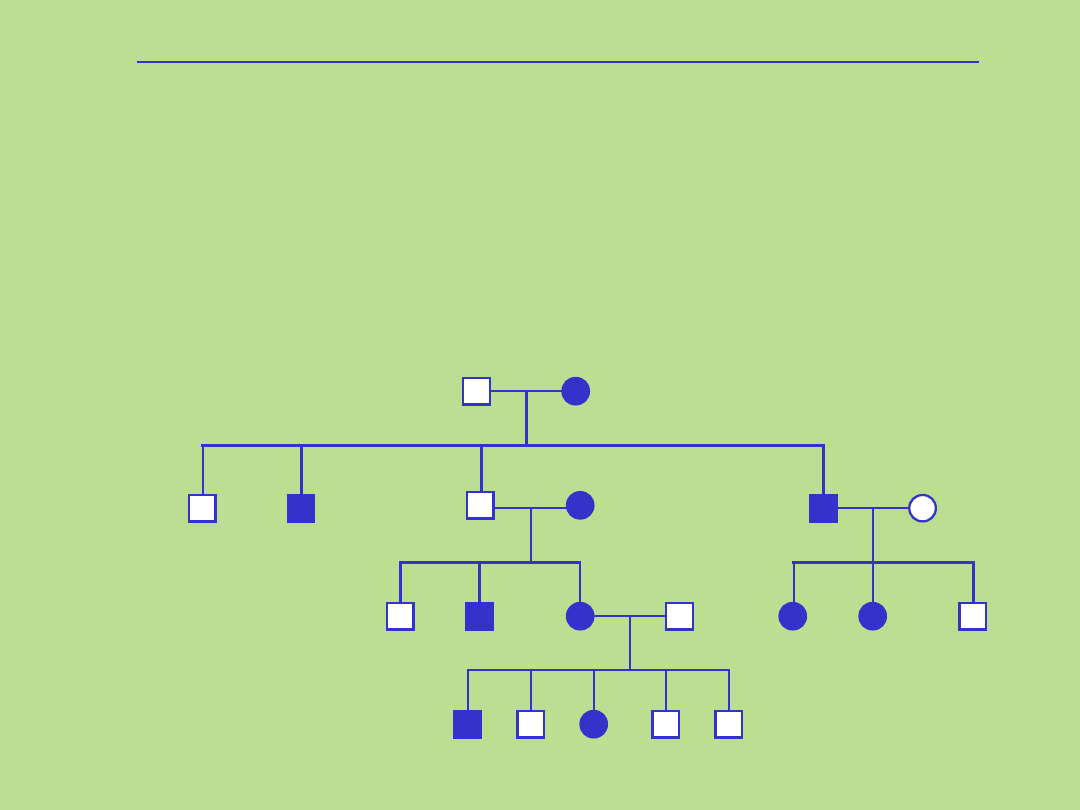
Dziedziczenie dominujące sprzężone z chromosomem X
Rodowód typowy dla dziedziczenia dominującego sprzężonego z
chromosomem X
1. Kryteria dla tego typu dziedziczenia:
• występowanie choroby u obu płci
• chorzy mężczyźni są hemizygotami
• chore kobiety maja prawidłowy allel na drugim chromosomie X
• kobiety przekazują zmutowany allel córkom i synom
• chorzy mężczyźni przekazują zmutowany allel wszystkim córkom , nigdy synom
• wszystkie córki chorego mężczyzny będą chore
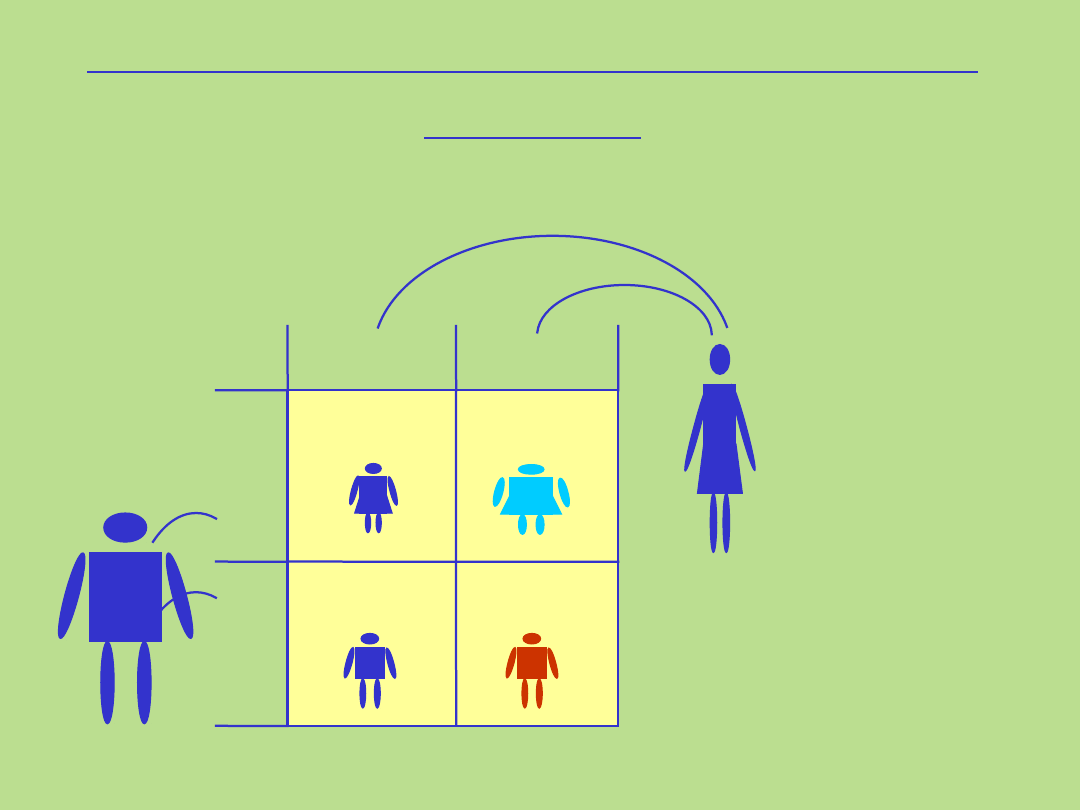
25%
(tylko
synowie)
X
Y
X
X*
X
X*
X*
Y
X Y
X X
Dziedziczenie sprzężone z chromosomem X
recesywne
gen przekazany przez matkę
Matka
X
X
*
nosicielka
Ojciec
X Y
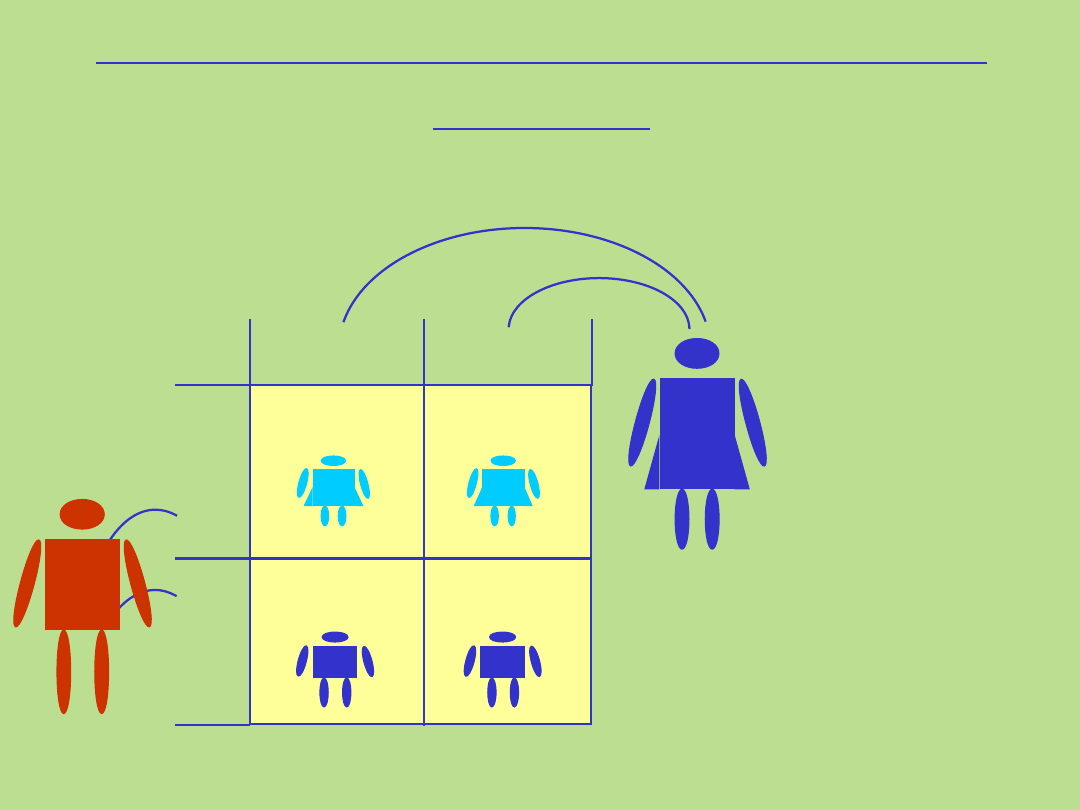
50%
(tylko córki)
X*
Y
X
X
X*
X
X Y
X Y
X*
X
Ojciec
X*
Y
Dziedziczenie sprzężone z chromosomem X
recesywne
gen przekazany przez ojca
Matka
X X
Recesywne sprzężone z chromosomem
X
Rodowód typowy dla dziedziczenia recesywnego sprzężonego z chromosomem
X
1. Kryteria dla tego typu dziedziczenia:
• występowanie choroby tylko u mężczyzn
• kobiety nosicielki mutacji genowej, w zasadzie nie wykazują cech
choroby
• hemizygotyczni chorzy mężczyźni przekazują zmutowany allel wszystkim
córkom, nigdy synom
• wszystkie córki chorego mężczyzny będą nosicielkami mutacji
• kobiety przekazują zmutowany allel córkom i synom
• matka chorego mężczyzny jest nosicielką mutacji genowej

Dziedziczenie dominujące i recesywne sprzężone z
chr. X
Cechy specyficzne:
• przypadki sporadyczne
•
heterogenność
Przykłady kliniczne:
• dystrofia mięśniowa Duchenne’a /Beckera
• zespół łamliwego chromosomu X
• hemofilia
• krzywica oporna na witaminę D

Dziedziczenie mitochondrialne
Cechy specyficzne:
- dziedziczenie w linii matczynej
- heteroplazmia, czyli występowanie zmutowanego i dzikiego mtDNA
jednocześnie
- niepełna/pełna penetracja
- najczęstsze defekty:
•
mutacje punktowe
•
rozległe delecje
•
deplecja, czyli redukcja liczby kopii mtDNA
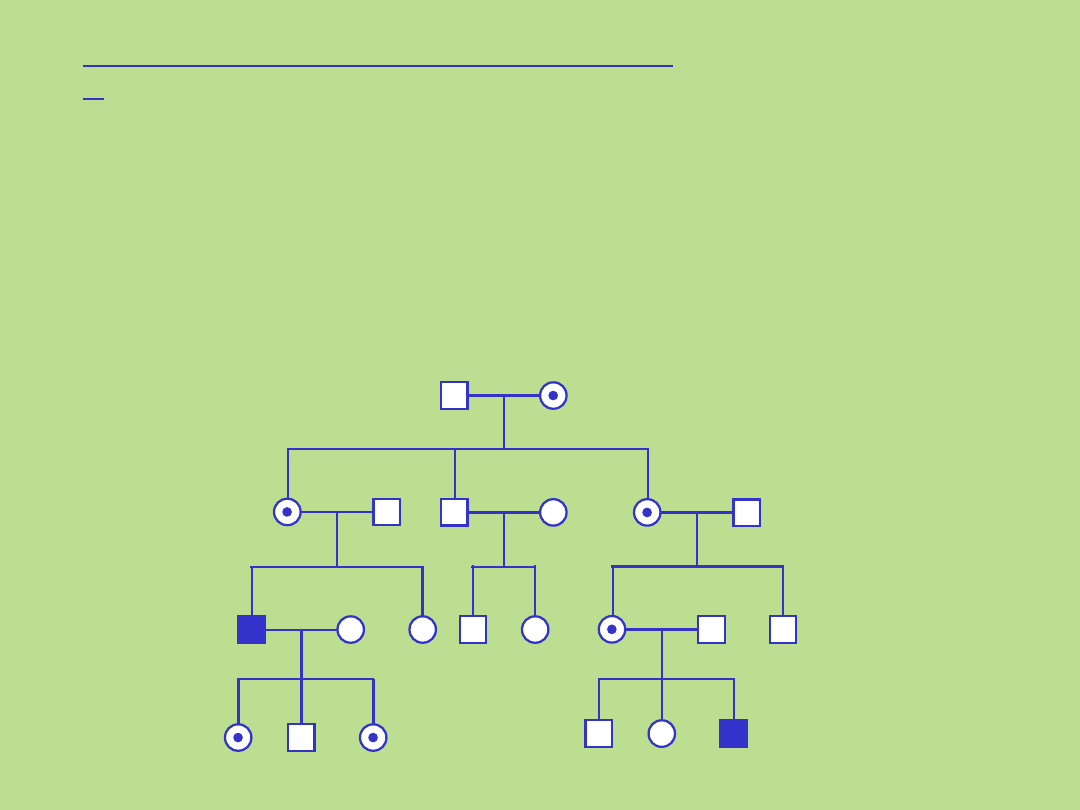
Dziedziczenie mitochondrialne – kryteria:
•matka przekazuje cechę wszystkim swoim potomkom
•tylko kobiety przenoszą tę cechę
•chorzy ojcowie nigdy nie przekazują choroby swoim
dzieciom
Rodowód typowy dla dziedziczenia mitochondrialnego

Imprinting
piętnowanie genomowe
Rodzaj mechanizmu regulacji ekspresji genów.
Efekt zależy od tego, czy imprinting występuje
na chromosomie ojcowskim, czy matczynym – zróżnicowana aktywacja genów.
Genomowe piętno rodzicielskie jest ustanawiane w trakcie gametogenezy -
– specyficzna płciowo modyfikacja genomu

Skutki imprintingu –
przykład rodzicielskiego,
zależnego od płci wpływu na
fenotyp
• klacz × osioł = muł,
• ogier × oślica = osłomuł - krótsze
uszy, gęstsza grzywa i ogon, nogi
silniejsze, niż u muła)
Imprinting rodzicielski – zróżnicowana ekspresja alleli w
zależności od płci rodzica
Matczyny – kopia genu od matki jest wyłączona, nie ulega
ekspresji.
Ojcowski – kopia genu od ojca jest wyłączona, nie ulega
ekspresji.
Geny takie są często zmetylowane!
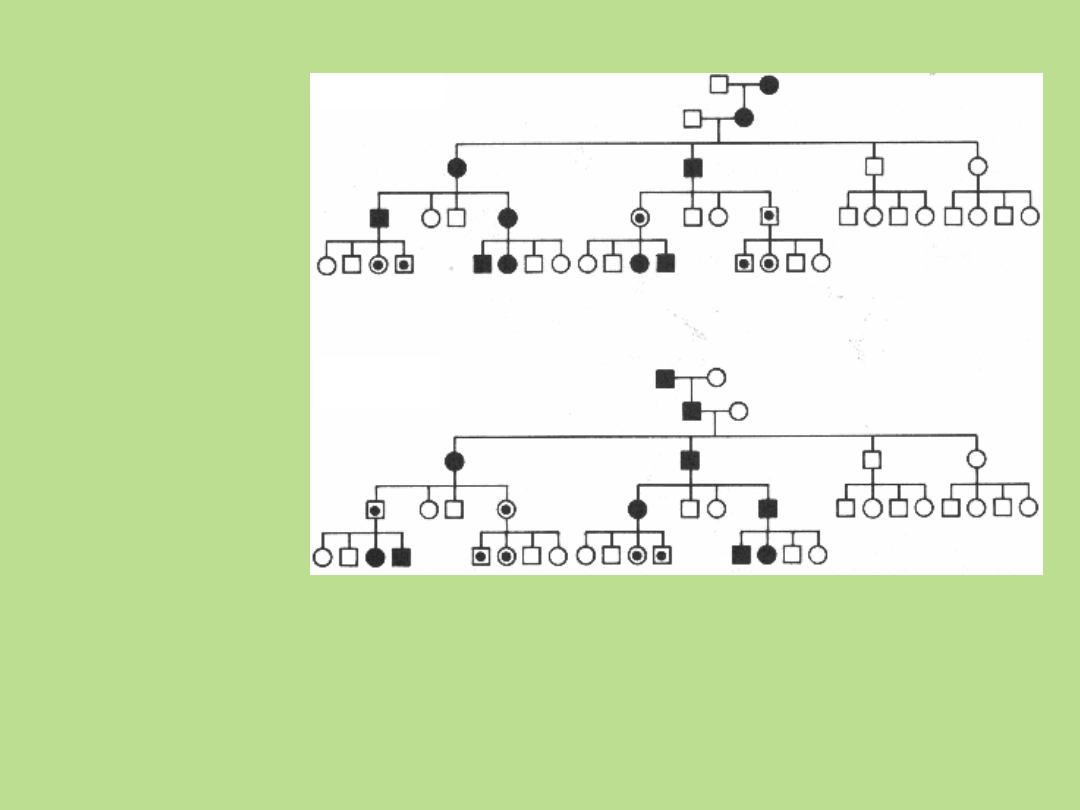
Imprinting rodzicielski - zasady
dziedziczenia
Przykładowe rodowody przedstawiające wpływ imprintingu matczynego i ojcowskiego na
ujawnienie się choroby u posiadacza zmutowanego genu. Termin "imprinting" wskazuje na
modyfikację ekspresji genu. Zmienione allele są dziedziczone w sposób mendlowski, lecz ich
ekspresja jest determinowana przez płeć rodzica, od którego pochodzą. Termin "imprinting
matczyny" oznacza brak ekspresji fenotypowej zmienionego allela w przypadku jego pochodzenia
od matki. Termin „imprinting ojcowski” oznacza brak ekspresji fenotypowej zmienionego allela w
przypadku jego pochodzenia od ojca. Efekt fenotypowy wystąpi tylko, gdy gen/segment
chromosomowy pochodzi od jednego, lub drugiego rodzica. Istnieje wielu bezobjawowych
nosicieli.
Imprinting ojcowski
Imprinting matczyny
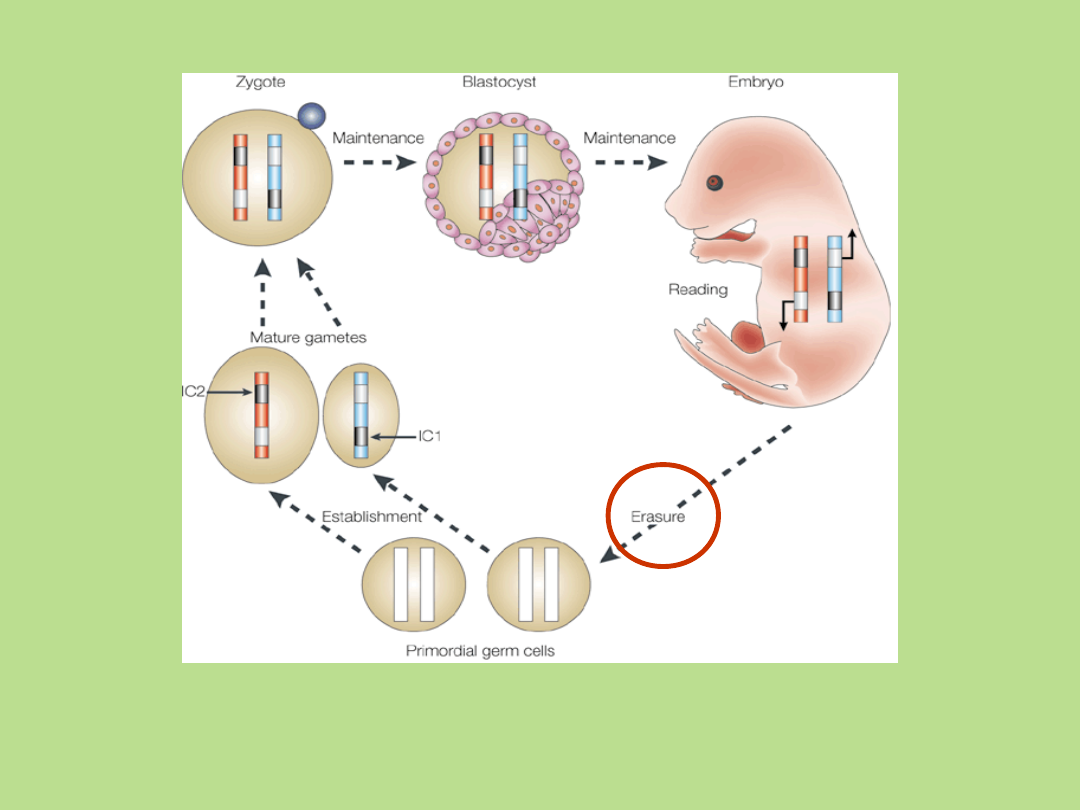
Cykl życiowy imprintów
metylacyjnych
Hum Mol Genet. 1992 Apr;1(1):7-10
IC – Element kontrolny imprintingu. Kolor szary - modyfikacja, kolor biały –
brak modyfikacji w odpowiadających sobie allelach. Chromosomy
rodzicielskie: kolor niebieski – ojcowskie, kolor czerwony - matczyne. Strzałki
wskazują sposób odczytu (transkrypcyjna interpretacja pierwotnych
imprintów) w rozwijającym się zarodku.
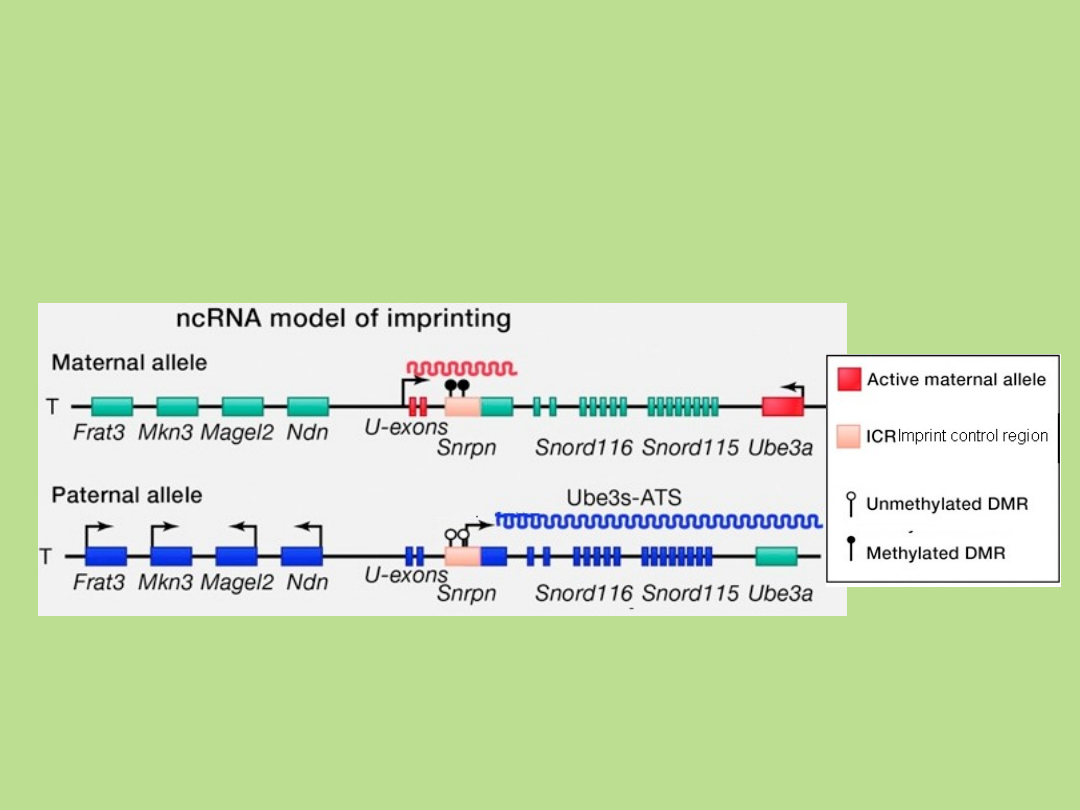
Imprinting – mechanizmy inaktywacji
• metylacja
• kondensacja chromatyny
• ncRNA
Cell 152, March 14,
2013
Allel ojcowski: niezmetylowane ICR → ekspresja
ncRNA → blokada ekspresji Ube3a
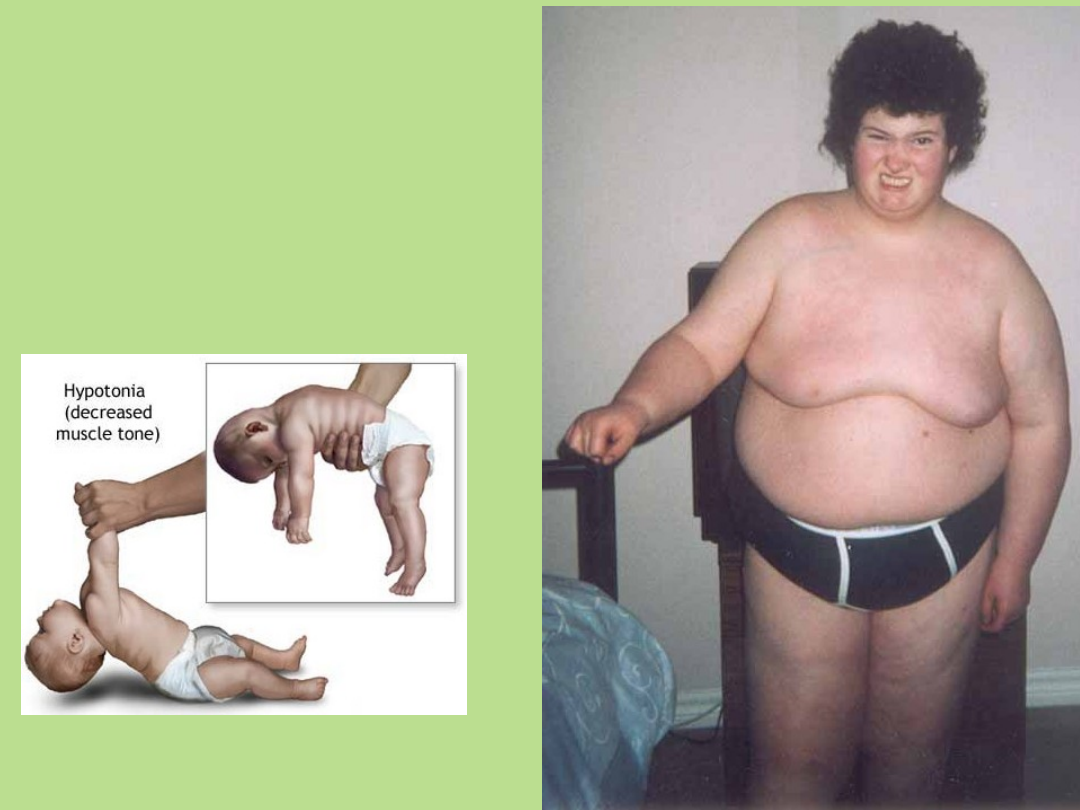
Fenotyp w zespole
Prader-Willi’ego:
U 75% pacjentów wykazano
duże delecje chromosomowe +/-
4 Mb w regionie 15q11-13.
Zawsze delecja występuje na
chromosomie przekazanym
przez ojca.
sou

Zespół Angelmana
• Znaczne funkcjonalne
opóźnienie rozwoju,
• Upośledzenie mowy,
nieużywanie, lub
minimalne używanie słów;
poziom poznawczej i
niewerbalnej zdolności
komunikacji wyższy, niż
webralnej,
• Zaburzenia ruchowe i
równowagi, zwykle ataksja
i nerwowe ruchy kończyn,
• Unikalność zachowania:
kombinacja częstego
śmiechu/uśmiechania się;
pozornie wesołe
usposobienie; łatwość
ekscytacji, z częstym
klaskaniem dłońmi;
zachowanie
hipermotoryczne; krótkie
okresy utrzymania uwagi
• Wygląd „szczęśliwej
kukiełki”
http://www.asclepius.com/
U 70% pacjentów wykazano
delecje +/- 4 Mb w regionie
15q11-13. Delecja występuje
zawsze na chromosomie
przekazanym przez matkę.
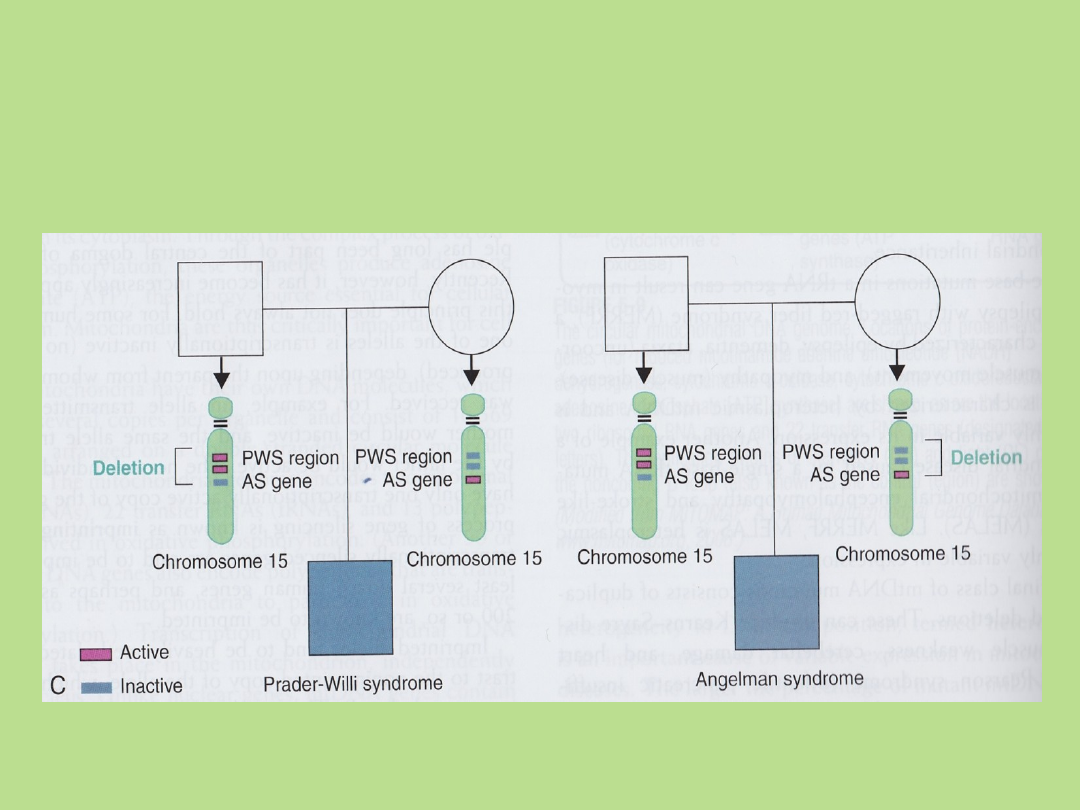
Zespół Pradera-Williego (PWS)
i zespół Angelmana (AS)
Medical genetics, Jorde, Carey, Bamshad, 4
th
edition
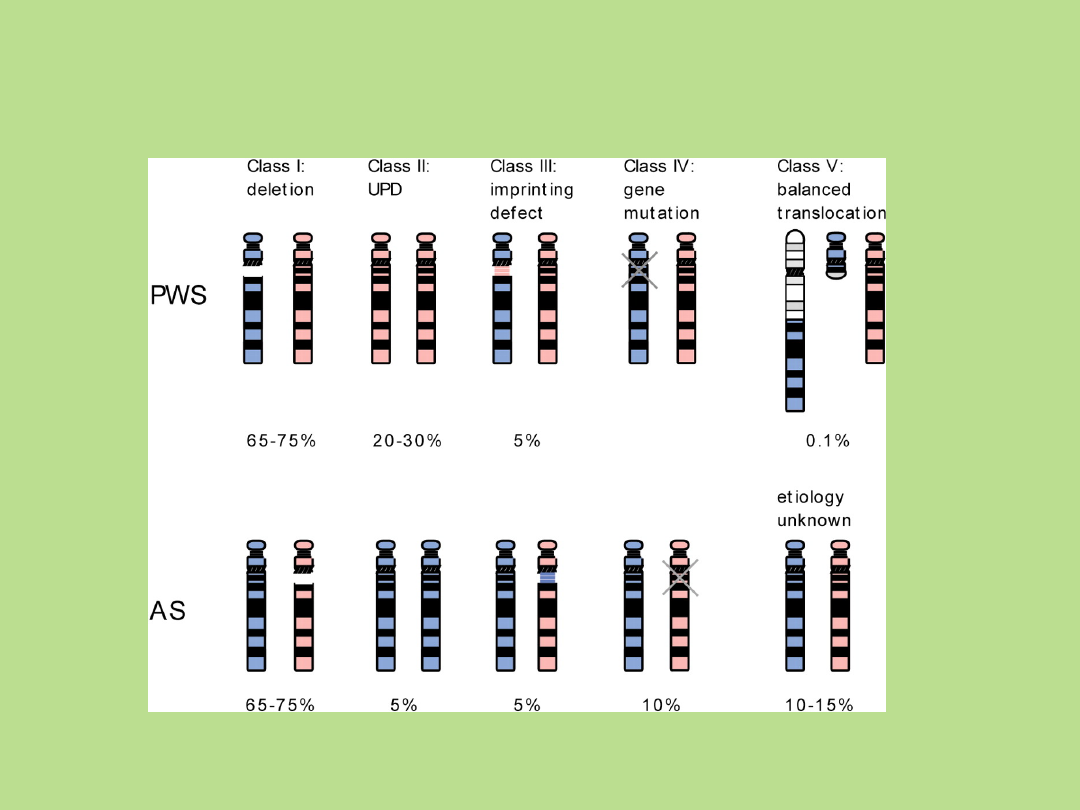
Przyczyny PWS i AS
Czerwone – chromosomy matczyne
Niebieskie –
chromosomy ojcowskie
Ann Rev 2001
No single gene muations.
A polygenic defect?
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
- Slide 23
- Slide 24
- Slide 25
- Slide 26
- Slide 27
- Slide 28
- Slide 29
- Slide 30
- Slide 31
- Slide 32
- Slide 33
- Slide 34
- Slide 35
- Slide 36
- Slide 37
- Slide 38
- Slide 39
- Slide 40
- Slide 41
- Slide 42
- Slide 43
- Slide 44
- Slide 45
- Slide 46
- Slide 47
- Slide 48
- Slide 49
- Slide 50
Wyszukiwarka
Podobne podstrony:
Rodowody, dziedziczenie, imprinting
Wzory rodowodów i rodzaje dziedziczenia
7 Dziedziczenie jednogenowe Analiza rodowodow 01
dziedziczenie chorob jednogenowych
dziedziny wychowania(1)
Kolonialne dziedzictwo
16 Dziedziczenie przeciwtestamentowe i obliczanie zachowkuid 16754 ppt
078c rozp zm rozp min gosp w spr szkolenia w dziedzinie bhp
Decyzja Rady 90 424 EWG z dnia 26 czerwca 1990 r w sprawie wydatków w dziedzinie weterynarii
więcej podobnych podstron