Dziedziczenie chorób
jednogenowych

• GENOTYP
– zespół wszystkich
genów organizmu warunkujący
jego właściwości dziedziczne
(genetyczny skład organizmu).
• FENOTYP
– zespół cech
morfologicznych; jest efektem
współdziałania genów i czynników
środowiskowych.

I i II prawo Mendla
• I prawo Mendla
– (prawo czystości
gamet) mówi, że do gamety przechodzi
tylko jeden allel danego genu.
• II prawo Mendla
– (
niezależnego
dziedziczenia się cech) stwierdza, że
przy krzyżowaniu 2 organizmów
różniących się więcej niż 1 cechą —
każda z cech dziedziczy się niezależnie
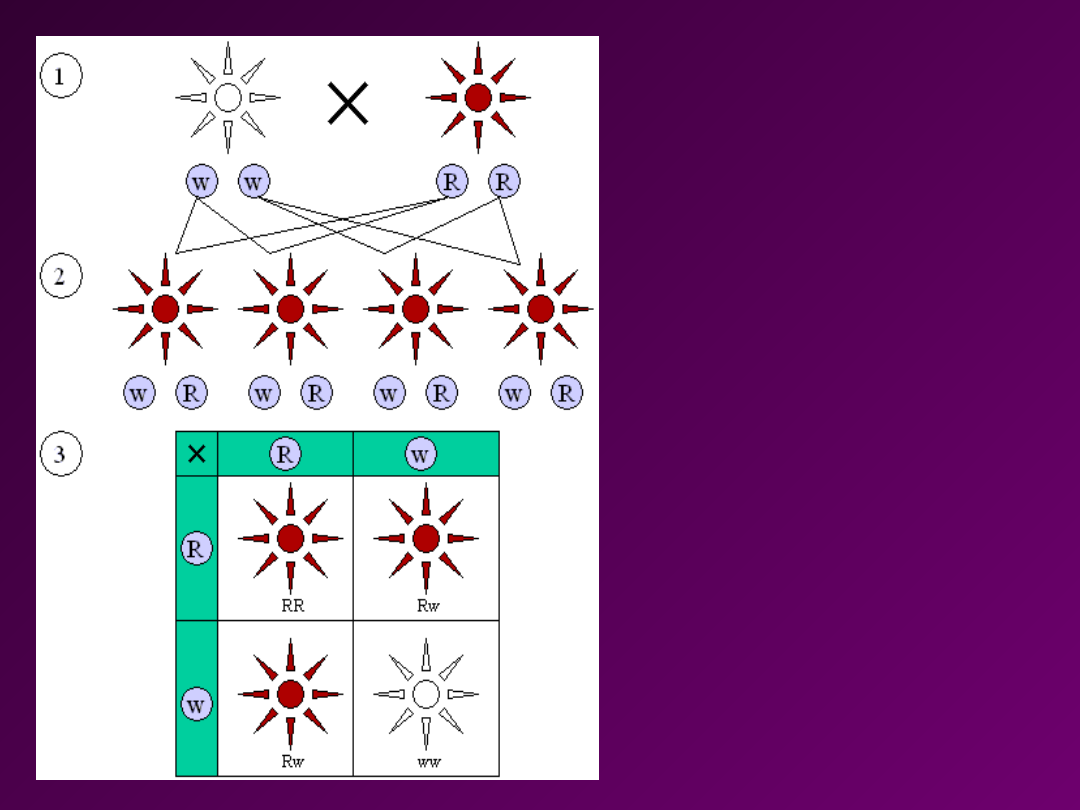
Rozkład cechy
3:1
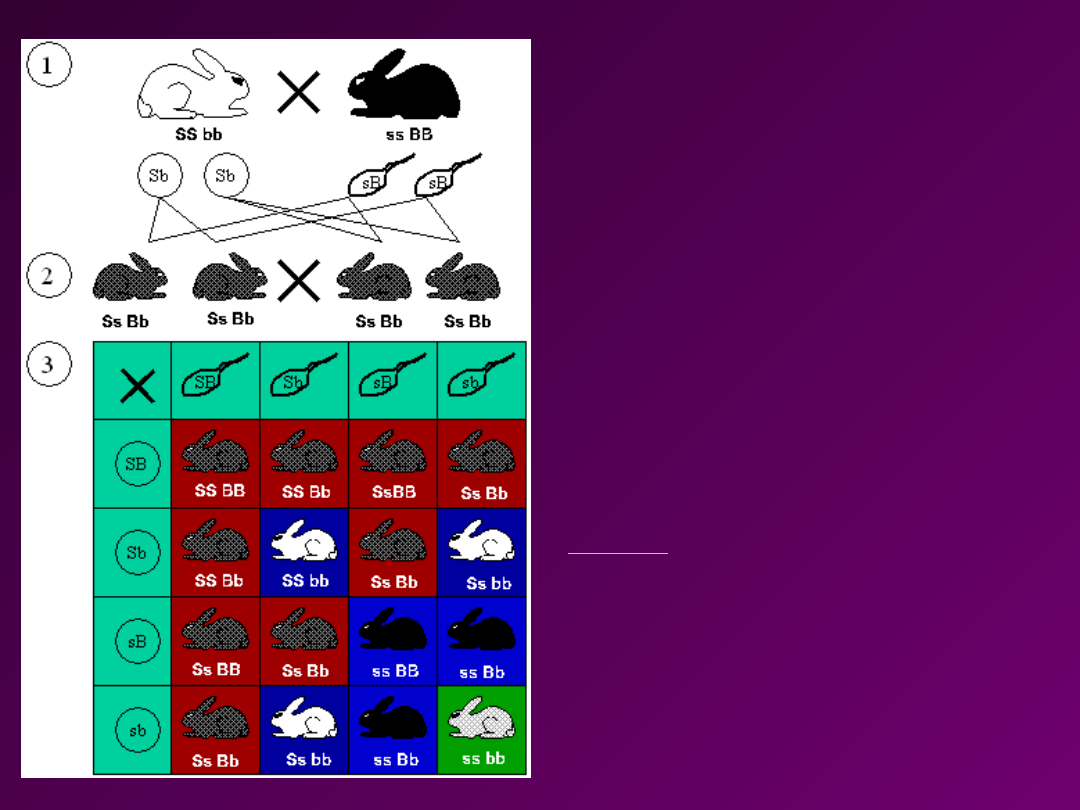
Dwie cechy
Czarne zabarwienie / Białe
zabarwienie
Krótka sierść / Długa sierść
Rozkład fenotypów w pokoleniu
F2
9:3:3:1
S=krótka, s=długa,
B=czarna, b=biała
Wynik:
9x krótkie czarne futro,
3x długie czarne futro,
3x krótkie białe futro,
1x długie białe futro
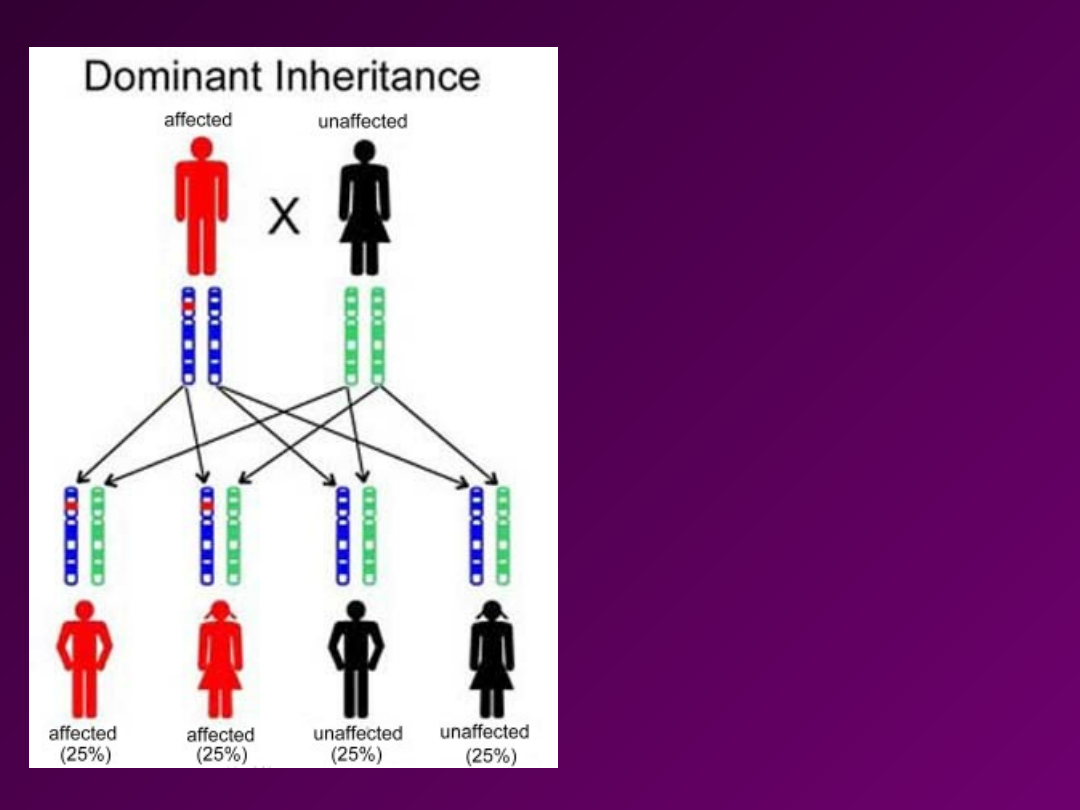
KRYTERIA
• Cecha jest przekazywana z
pokolenia na pokolenie bez
przeskoku (występuje w
każdym pokoleniu)
• Choroba występuje z tą
samą częstością u obu płci.
• Ze związku chorej
heterozygoty (Aa) i osoby
zdrowej (aa), 50%
potomstwa jest chore, a 50%
jest zdrowe.

• PENETRACJA
– zjawisko „wszystko albo
nic”; pojawianie się określonego
fenotypu
wśród osobników o tym samym
genotypie
.
Penetracja może być całkowita, gdy
wszystkie osobniki o tym samym
genotypie przejawiają cechę fenotypową,
wywołaną tym samym genem, lub
częściowa, gdy tylko część osobników
wykazuje taką cechę.
• EKSPRESJA
– nasilenie cechy

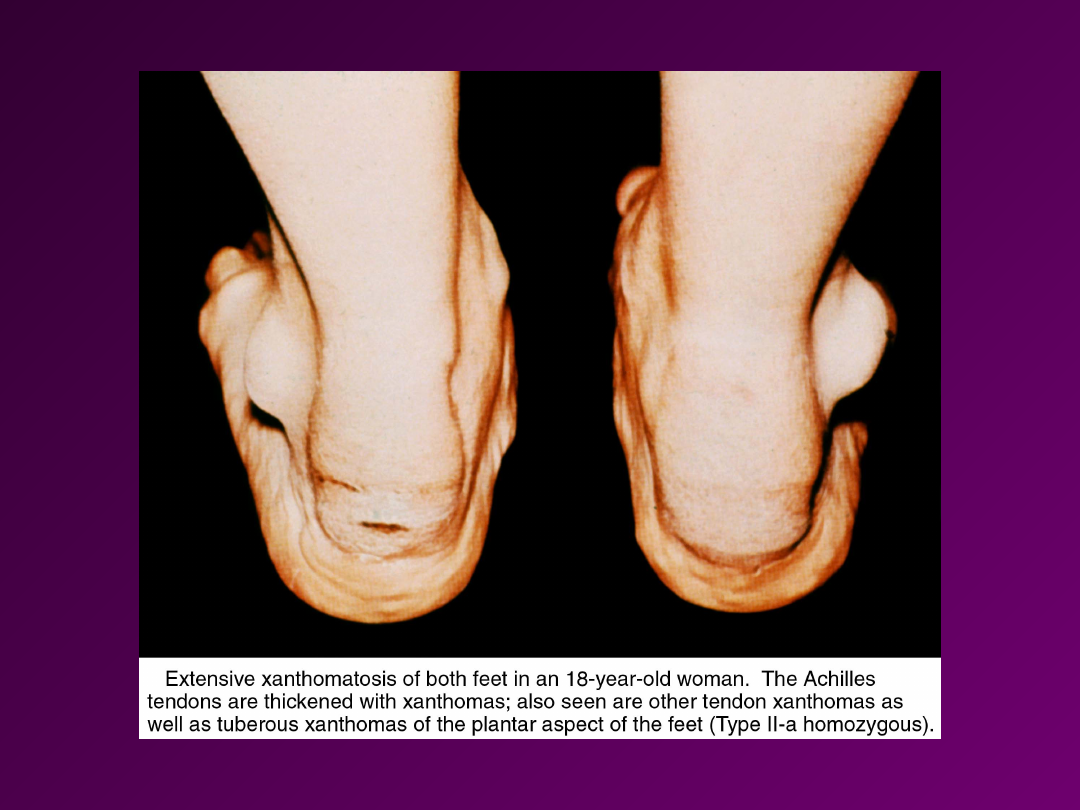
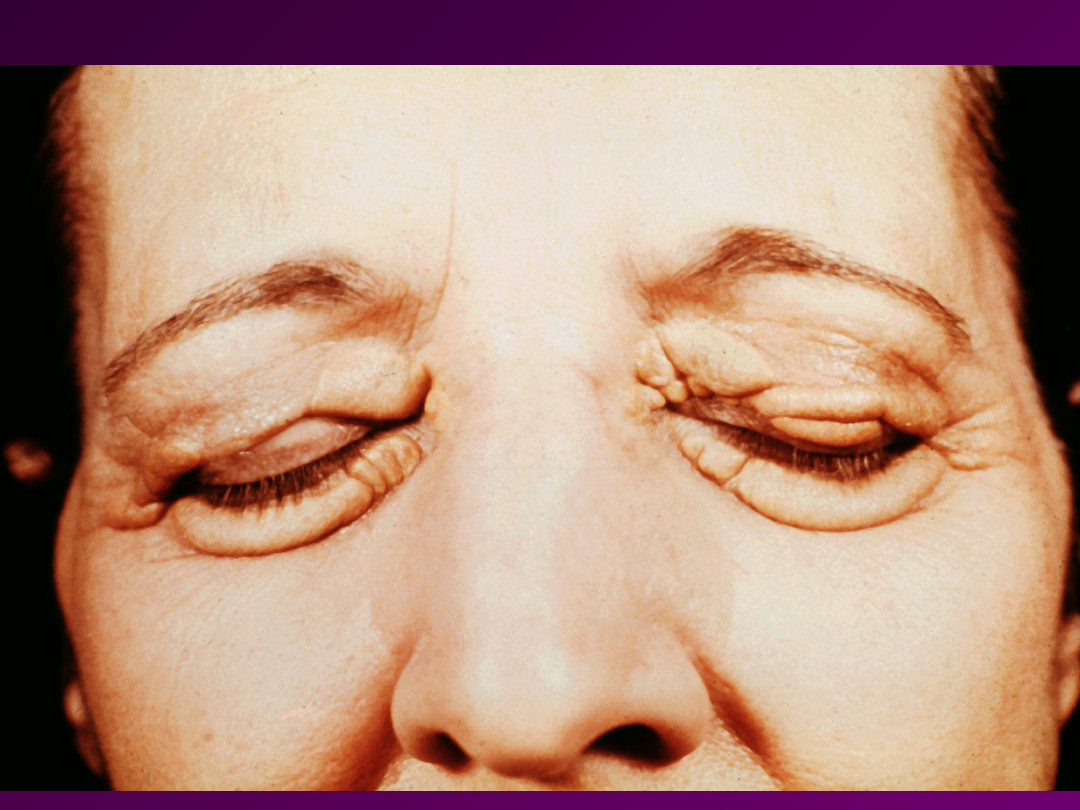
• HIPERCHOLESTEROLEMIA
– choroba
polega na znacznym wzroście poziomu
cholesterolu we krwi (efekt zaburzenia
działania białka LDL) – nawet do
1000mg/100ml (górna granica normy
~200 mg/100 ml). Cholesterol odkłada
się w skórze i naczyniach krwionośnych.
Na skórze i ścięgnach pojawiają się tzw.
Kępki żółte. Dochodzi do
przedwczesnego rozwoju zmian
miażdzycowych.

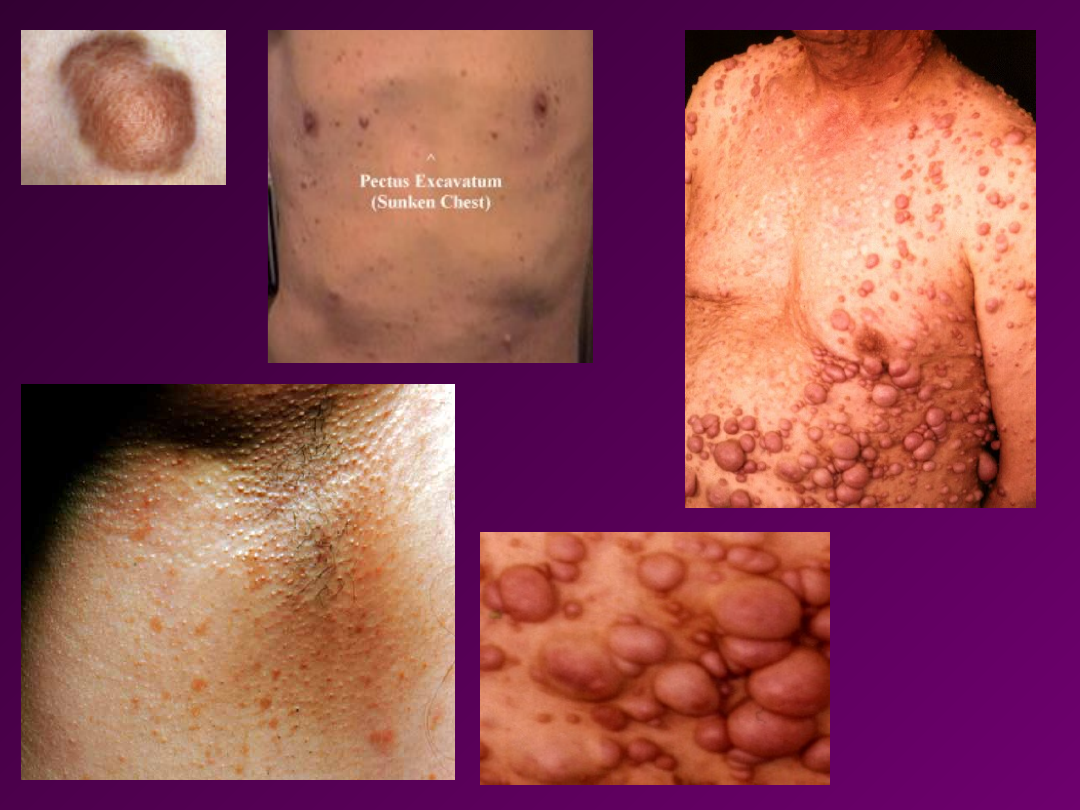
• NERWIAKOWŁÓKNIAKOWATOŚĆ
–
(choroba Recklinhausena).
Cecha występuje z pełną penetracją, ale
zmienną ekspresją. Na skórze mogą
występować zmiany barwnikowe w
kolorze kawy z mlekiem tzw. Cafe au-lait
lub guzki wyrastające z nerwów
obwodowych (włókniaki,
nerwiakowłókniaki). Guzki są zmianami
łagodnymi, jednak w 3-13 % ulegają
transformacji nowotworowej.
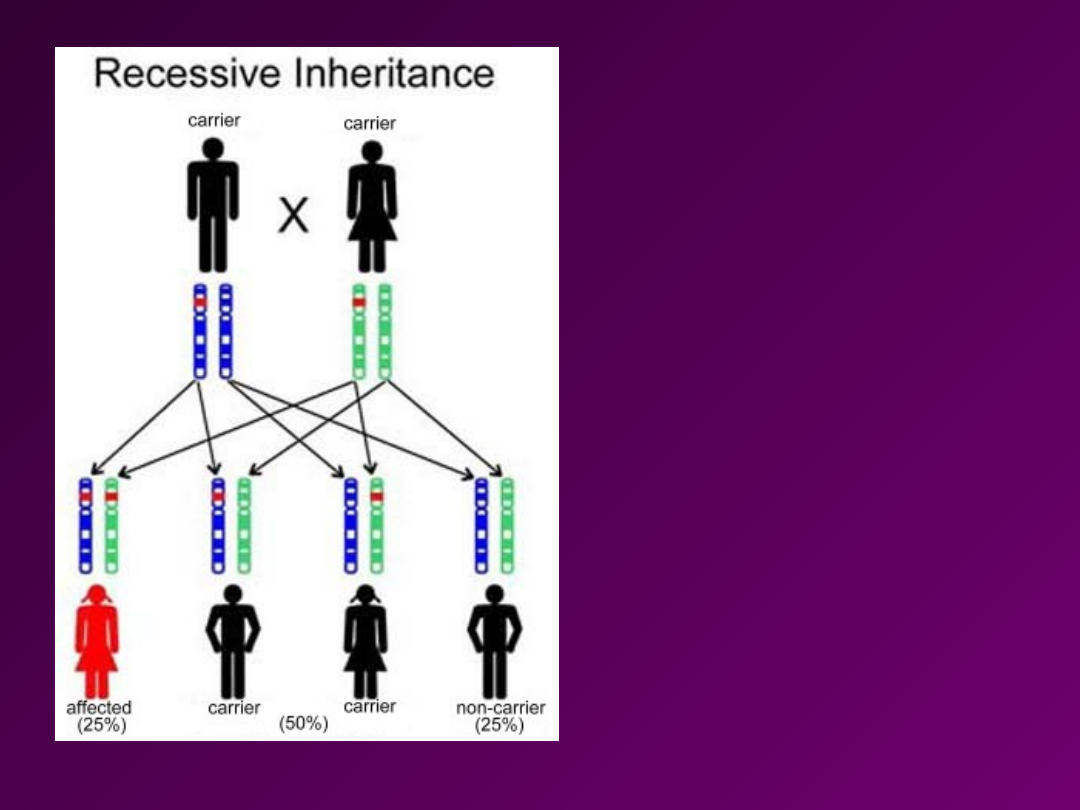
KRYTERIA
• Choroba ujawnia się tylko u
Homozygot recesywnych
(niezależnie od płci)
• Rodzice i krewni probanta są
zwykle zdrowi.
• Jeśli oboje rodziców jest
heterozygotami to szansa
urodzenia dziecka chorego
jest 25%.

MUKOWISCYDOZA
– Przyczyną choroby jest wrodzony
defekt metaboliczny prowadzący
do zalegania gęstego śluzu – głównie w obrębie
układu oddechowego (płuca) i pokarmowego
(trzustka). W płucach dochodzi do zalegania wydzieliny
oskrzelowej, co prowadzi do wzrostu częstości zakażeń
bakteryjnych.
W trzustce zatkaniu ulegają przewody, którymi do
dwunastnicy dostarczane są enzymy trawienne (m.in.
trawiące tłuszcz).
Dolegliwości i objawy:
przewlekły i napadowy kaszel (z
trudną do odkrztuszenia wydzieliną), przewlekłe
zapalenie oskrzeli i/lub płuc, nawracające zapalenie
zatok, przewlekła biegunka i kłopoty z tolerancją
pokarmów (wzdęcia, bóle brzucha, biegunki), niedobór
masy ciała i/lub wzrostu
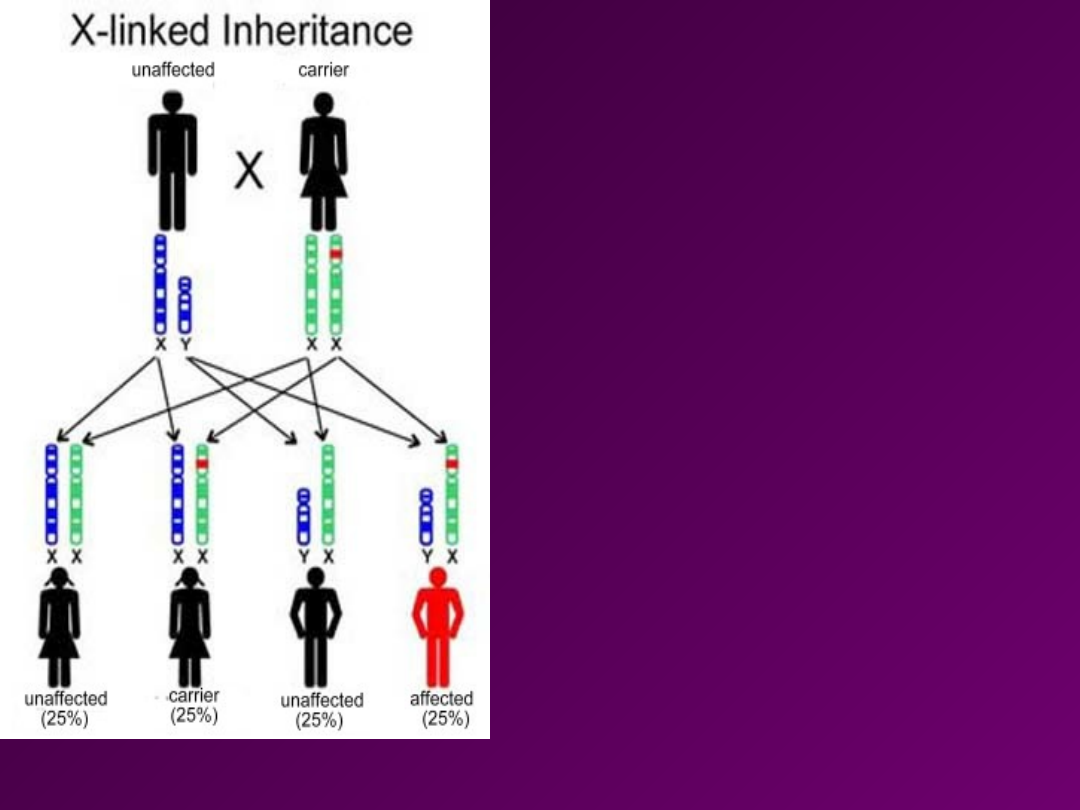
KRYTERIA
XD
• Chory mężczyzna ma wyłącznie chore
córki i zdrowych synów
• Chore kobiety (heterozygoty)
przekazują cechę 50% swego
potomstwa.
• Chore kobiety (homozygoty)
przekazują cechę 100% swego
potomstwa.
Krzywica hipofosfatemiczna oporna na
leczenie witaminą D
XR
• Choroba występuje znacznie częściej
u mężczyzn niż u kobiet.
• Chory mężczyzna ma zdrowych synów
i córki nosicielki (bezobjawowe).
Dystrofia mięśniowa Duchenne’a

• KRZYWICA HIPOFOSFATEMICZNA OPORNA NA LECZENIE
WITAMINĄ D
XLH występuje z częstością 1:20000 żywo urodzonych dzieci
i manifestuje się hipofosfatemią, niedoborem wzrostu i
krzywicą, szczególnie w obrębie kończyn dolnych, a w
starszym wieku wzmożoną osteoporozą.
Pierwotny defekt dotyczy uszkodzenia genu, którego
ekspresja zachodzi w osteoblaście - miejscu czynnej
mineralizacji kości. Pod koniec lat 80-tych zlokalizowano gen
choroby na chromosomie X w rejonie p22.2 a w latach 1995-
97 poznano sekwencję i strukturę genu PHEX. Gen koduje
transmembranowe białko odpowiedzialne za aktywację lub
degradację hormonów peptydowych. Białko PHEX bierze
prawdopodobnie udział w potranslacyjnej obróbce
krążącego czynnika fosfaturycznego, MEPE, który kontroluje
proces mineralizacji oraz reguluje reabsopcję fosforanów i
metabolizm 1,25-dihydroksy-witaminy D3 w nerkach.

• Dystrofia mięśniowa Duchenne'a (DMD)
-
występuje z częstością 1 : 3500 żywo urodzonych
chłopców; polega na stopniowym i nieodwracalnym
zaniku mięśni wywołanym uszkodzeniem białka -
dystrofiny. Choroba dotyka chłopców, kobiety są
zazwyczaj bezobjawowymi jej nosicielkami.
Opisano dwie postacie kliniczne: z ostrym jej
przebiegiem typ Duchenne'a (DMD) i z łagodnym typ
Beckera (BMD). W ostrej postaci pacjenci przestają
samodzielnie chodzić w wieku 8-10 lat, a śmierć na
skutek niewydolności oddechowej następuje około 20
roku życia, w postaci łagodnej spektrum objawów
jest zróżnicowane od objawów nieco łagodniejszych
niż w postaci Duchenne'a poprzez kardiomiopatie do
przypadków bezobjawowych.

Podłożem molekularnym choroby są mutacje w genie
DMD - delecje (60%), duplikacje (5-10%) i mutacje
punktowe (30-35%) uszkadzające syntezę dystrofiny.
Mutacje naruszające ramkę odczytu wywołują
całkowity brak białka i ostrą postać choroby - DMD,
mutacje zachowujące ramkę odczytu powodują
powstanie skróconej (truncated) formy białka i w
zależności od ilości i stopnia zmian cząsteczki białka
powodują różny stopień nasilenia objawów BMD.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
- Slide 14
- Slide 15
- Slide 16
- Slide 17
- Slide 18
- Slide 19
- Slide 20
- Slide 21
- Slide 22
Wyszukiwarka
Podobne podstrony:
Ćw 12 Dziedziczenie chorob jednogenowych
Choroby jednogenowe
Biologia Choroby jednogenne, chromosomalne i wieloczynnikowe
7 Choroby jednogenowe 01
W8 Choroby jednogenowe
Choroby jednogenowe
Choroby jednogenowe
Choroby jednogenowe –
Dziedziczenie chorób sprzężonych z chromosomem X
Choroby dziedziczne, Szkoła, przydatne w szkole
Choroby dziedziczne
Mechanizmy chorób dziedzicznych 1
Wykłady z genetyki (Choroby, dziedziczność) by Kusy
CHOROBA ZWYRODNIENIOWA STAWÓW, Materiały naukowe z różnych dziedzin, Reumatologia
więcej podobnych podstron