W 1934 roku Asbjőrn Főlling poszukując
przyczyny upośledzenia umysłowego u dwojga
rodzeństwa zaobserwował, ze ich mocz po dodaniu
FeCl3 przybrał charakterystyczne oliwkowozielone
zabarwienie.
Taką samą reakcję barwną wykazał mocz 8
innych osób z grupy 430 pacjentów opóźnionych w
rozwoju umysłowym. Występowanie aż 4 par
bliźniąt wśród 10 pacjentów, których mocz dał
pozytywną reakcję barwną z FeCl3 sugerowało, ze
przyczyna choroby może być wrodzone zaburzenie
metaboliczne.
Okazało się, że za reakcję barwną moczu
odpowiedzialny jest fenylopirogronian, skąd
chorobę nazwano fenyloketonurią.
Folling wysunął hipotezę, ze przyczyną
zahamowania rozwoju umysłowego mogą być
wrodzone wady metabolizmu fenyloalaniny.
Rozpoznanie choroby

Rzeczywiscie fenyloketonuria, jest spowodowana genetycznie
uwarunkowanym
niedoborem enzymu hydroksylazy fenyloalaniny
lub (znacznie rzadziej) jej kofaktora – tetrahydrobiopteryny
wskutek czego fenyloalanina nie może zostac przekształcona w
tyrozynę i gromadzi się we wszystkich płynach ustrojowych.
Fenyloketonuria jest chorobą genetyczną spowodowaną
mutacją, położonego na chromosomie 12 (12q22-q24.1)
genu kodującego enzym hydroksylazę fenyloalaninową
(PAH, EC 1.14.16.1).
U podłoża choroby leży mutacja genu odpowiedzialnego za aktywność tego
enzymu
Chromosom 12 – jeden z 23 parzystych chromosomów
człowieka. DNA chromosomu 12 liczy ponad 132 milionów
par nukleotydów, co stanowi około 4-4,5% materiału
genetycznego ludzkiej komórki.
Liczbę genów mających swoje locus na chromosomie 12
szacowana jest na 1.000-1.300
.
Niektóre geny mające swoje locus na chromosomie 12:
•ACVRL1
•COL2A1
•HPD
•MMAB
•MYO1A (kodujący miozynę IA)
•NANOG
•PAH (kodujący hydroksylazę
fenyloalaninową)
11
12
GT
13
EKSON
EKSON
EKSON
INTRON
INTRON
Transkrypt pierwotny
splicing
11
12
13
11
12
AT
13
EKSON
EKSON
EKSON
mRNA
A
B
Transkrypt pierwotny
Nieprawidłowy splicing
11
13
mRNA z
delecją
INTRON
INTRON
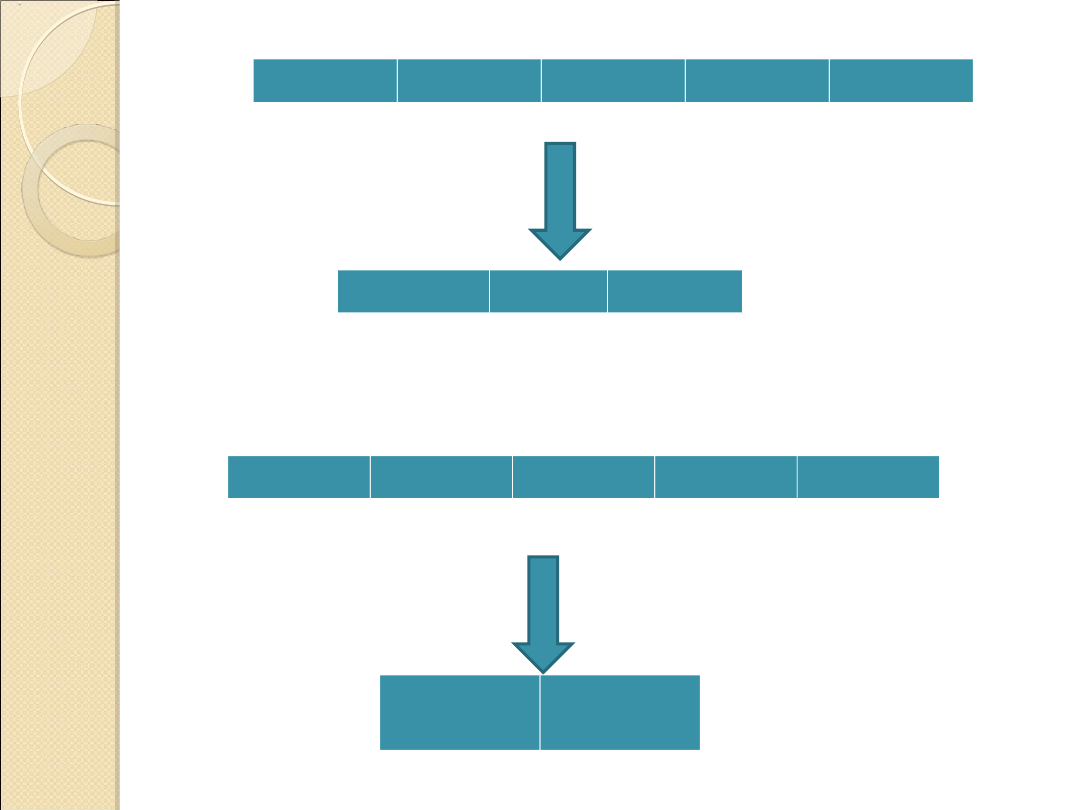
Przyczyną
fenyloketonurii
mogą być
mutacje w
intronie
zmieniające
splicing.
A -normalny
transkrypt
pierwotny i
mRNA
B- Mutacja G do
A w intronie 12
powoduje utratę
eksonu.
Większość
mutacji
odpowiedzialnyc
h za wystąpienie
fenyloketonurii
występuje w
rejonie
kodujacym

W Polsce rocznie rodzi się 60-70 chorych dzieci.
Jest to choroba o podłożu genetycznym,
przekazywana dziecku przez oboje z rodziców.
Dziedziczy się w sposób autosomalny recesywny co
oznacza, że chore dziecko musi otrzymać po 1
nieprawidłowym genie wywołującym PKU od
każdego z rodziców.
Do pełnego wystąpienia choroby potrzebne są dwa
„chore” geny.
Rodzice są najczęściej tylko bezobjawowymi
nosicielami nieprawidłowego genu i zwykle nie
zdają sobie sprawy z tego, że mogą go przekazać
swojemu potomstwu.
Są oni zdrowi, ponieważ drugi obecny u nich gen
jest „zdrowy”. W populacji polskiej co 46 osoba jest
zdrowym nosicielem nieprawidłowego genu PKU.
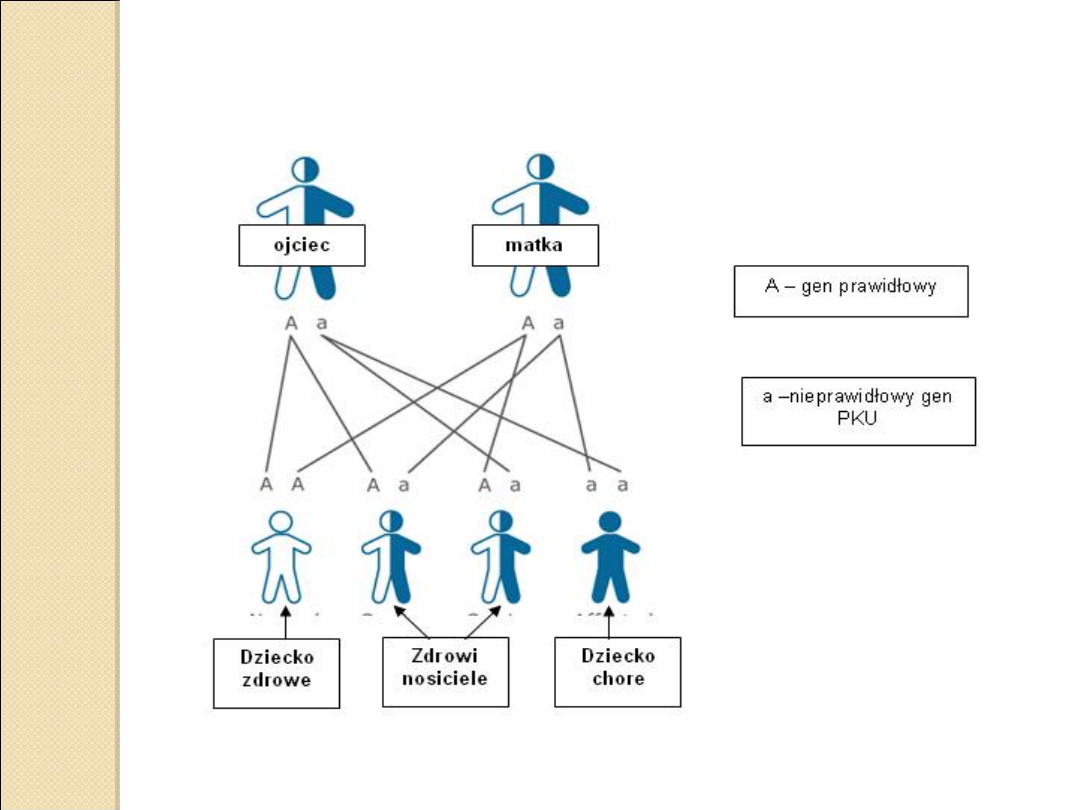
Dziedziczy się w sposób autosomalny recesywny co oznacza,
że chore dziecko musi otrzymać po 1 nieprawidłowym genie
wywołującym PKU od każdego z rodziców.
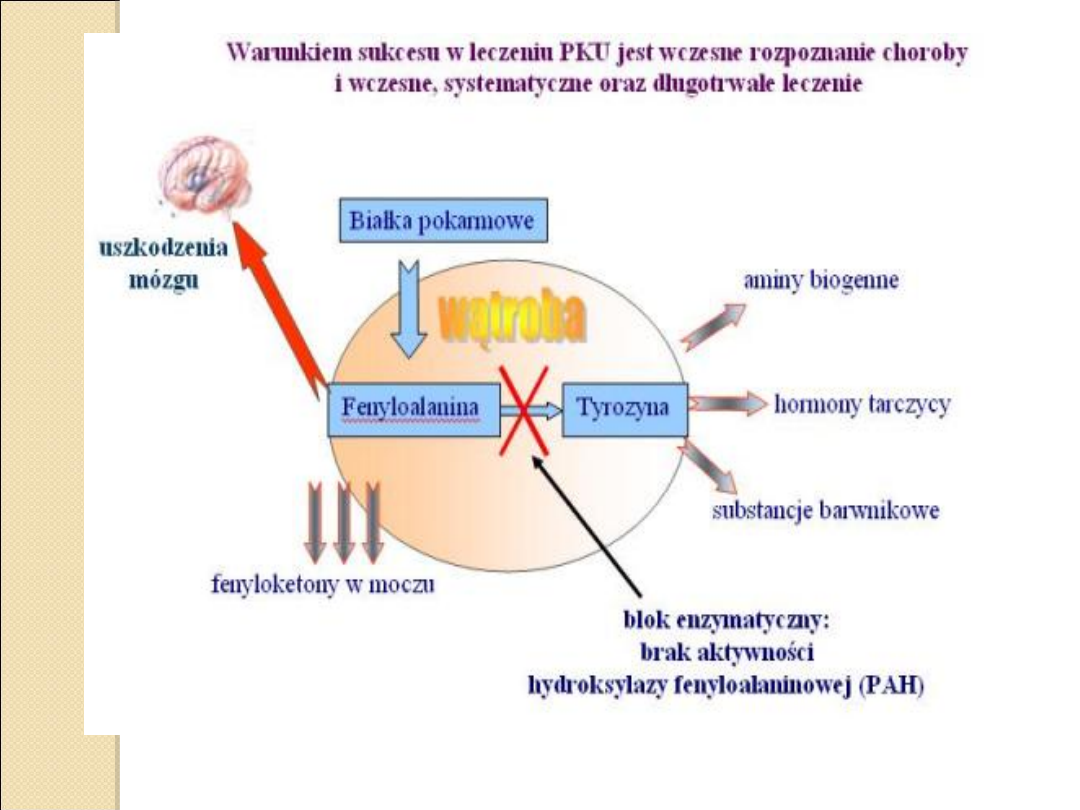

We krwi dziecka chorego na fenyloketonurię zaczyna
gromadzić się fenyloalanina i produkty jej
metabolizmu (np. kwasu fenylopirogronowego), przy
względnym niedoborze tyrozyny.
Wraz z krwią fenyloalanina dostaje się do
ośrodkowego układu nerwowego nerwowego (OUN),
gdzie wywołuje nieodwracalne uszkodzenia.
Powoduje to różne formy niepełnosprawności
intelektualnej i zaburzenia neurologiczne, które
stanowią główne objawy nieleczonej lub leczonej
niewystarczająco PKU.
Chociaż dokładny mechanizm jej działania nie jest
jeszcze ostatecznie poznany wiadomo, że
fenyloalanina:
• zaburza syntezę białek tkanki nerwowej
• powoduje zaburzenia w syntezie mieliny, substancji
niezbędnej dla prawidłowego funkcjonowania
komórek nerwowych .

Mózg chorych pacjentów wykazuje mase mniejszą od
prawidłowe, otoczka mielinowa ich nerwów jest źle
wykształcona, chorzy wykazują nadpobudliwosć.
Nieleczona fenyloketonuria drastycznie skraca długosc
życia, połowa chorych umiera przed osiągnięciem
dwudziestu lat, ¾ przed osiągnięciem 30 lat.
• zaburza metabolizm tzw. neurotransmiterów, czyli
związków zaangażowanych w procesy przekazywania
impulsów nerwowych

Objawy
Objawami nieleczonej choroby są m.in. :
• ciężka niepełnosprawność intelektualna
• padaczka
• zaburzenia mowy – niektórzy chorzy w ogóle nie
potrafią się porozumiewać
• zaburzenia chodu – najciężej dotknięci pacjenci,
szczególnie w późniejszych latach życia, poruszają się
jedynie z pomocą osób trzecich lub są na trwale
unieruchomieni w łóżku
• postępujące wraz z wiekiem zaburzenia
neurologiczne
• małogłowie
• zmiany wypryskowe na skórze
• nieprawidłowy „mysi” zapach potu.
Wielu chorych prezentuje ciężkie i utrudniające
codzienne życie zaburzenia zachowania, takie jak:
• nadpobudliwość z niekontrolowanymi napadami
złości
• napady agresji wobec innych oraz wobec siebie - te
ostatnie mogą doprowadzić do dotkliwych
samookaleczeń

• zachowania destrukcyjne
• zaburzenia snu
• deficyty uwagi i zaburzenia koncentracji
• zachowania autystyczne, m.in. lęk wobec osób obcych i
wobec zmiany otoczenia
• psychozy
Dzieci, u których choroba zostaje wykryta w pierwszych
dniach życia i które następnie są wcześnie, systematycznie i
długotrwale (optymalnie do końca życia) leczone, rozwijają się
prawidłowo, wykorzystując w pełni swój potencjał
intelektualny.

Wykrywanie fenyloketonurii
Diagnostyka PKU opiera się o tzw. badania
przesiewowe noworodków, które pozwalają
na identyfikację chorych dzieci już w
pierwszych dniach ich życia.
Dokonuje się tego poprzez pobranie kilku
kropli krwi z pięty każdego noworodka po
ukończeniu przez niego 72 godzin życia i
oznaczenia w niej stężenia fenyloalaniny.
Aktualnie w Polsce badania wykonuje się
przy użyciu metody kolorymetrycznej.
Warunkiem prawidłowego wykonania testu
jest wcześniejsze karmienie dziecka
mlekiem matki lub mlekiem
modyfikowanym.
. U dzieci zdrowych stężenia fenyloalaniny
mieszczą się na ogół poniżej 2 mg%, u
noworodków chorych są wielokrotnie
wyższe, przekraczając często wartość 20
mg%.

Inną metodą stosowaną niekiedy w diagnostyce
fenyloketonurii jest badanie metabolitów fenyloalaniny w
moczu z zastosowaniem FeCl
3
.
Zwiększone stężenie fenyloalaniny we krwi może nie
pojawić się przed 3 dniem życia.
Dodatkowo u wcześniaków stwierdza się występowanie
wyników fałszywie dodatnich (ze względu na
niedojrzałość układów enzymatycznych).

LECZENIE
Terapia w przypadku fenylokeronurii polega na
stosowaniu diety ubogiej w fenyloalaninę, czyli
zawierającej akurat tyle fenyloalaniny, ile potrzeba jej
do wzrostu organizmu i wymiany w białkach.
Kontrolowaną zawartość fenyloalaniny w diecie można
uzyskać, hydrolizując białka ubogie w ten aminokwas,
takie jak kazeina i usuwając fenyloalaninę przez
adsorpcję na odpowiednich sorbentach.
Aby uniknąć nieodwracalnych zmian w mózgu,
podawanie diety ubogiej w fenyloalaninę należy w
przypadkach fenyloketonurii rozpocząć wkrótce po
urodzeniu .
Przyjęty współczynnik inteligencji chorych, których
leczenie podjęto wkrótce po urodzeniu, wynosił 93,
podczas gdy w grupie kontrolnej, której leczenie
rozpoczęto po roku współczynnik ten wynosił 53.
Document Outline
- Slide 1
- Slide 2
- Slide 3
- Slide 4
- Slide 5
- Slide 6
- Slide 7
- Slide 8
- Slide 9
- Slide 10
- Slide 11
- Slide 12
- Slide 13
Wyszukiwarka
Podobne podstrony:
prezentacja finanse ludnosci
prezentacja mikro Kubska 2
Religia Mezopotamii prezentacja
Prezentacja konsument ostateczna
Strategie marketingowe prezentacje wykład
motumbo www prezentacje org
lab5 prezentacja
Prezentacja 18
Materialy pomocnicze prezentacja maturalna
Prezentacja na seminarium
Lato prezentacja 3
Prezentacja1
Prezentacja 2 analiza akcji zadania dla studentow
prezentacja soc rodziny
więcej podobnych podstron