
CHEMIA 1

Kinetyka
◦
Teoria kompleksu aktywnego i zderzeń aktywnych, energia
aktywacji
◦
Katalizatory
Wykład 14

Teoria kompleksu aktywnego
i zderzeń aktywnych, energia
aktywacji
Wprowadzenie
Na przełomie XIX i XX wieku stwierdzono (Van’t Hoff), że
szybkość reakcji chemicznych, przy wzroście temperatury o 10°C
rośnie ok. 2-4 razy
. Reguła Van’t Hoffa spełniona jest dla dużej
grupy reakcji homogenicznych do temperatury ok. 500°C.
Ponieważ ogólne równanie kinetyczne reakcji ma postać:
a stężenia reagentów b. słabo zależą od temperatury, główną
przyczyną zmiany szybkości musi być temperaturowa zależność
stałej szybkości reakcji
k
.
Arrhenius ustalił, że stała szybkości spełnia równanie:
gdzie A i B to dodatnie stałe zależne od reagujących substancji.
Zależność tą można uzasadnić w oparciu o teorię zderzeń
aktywnych.
...
c
c
c
k
v
γ
C
β
B
α
A
T
B
A
lnk
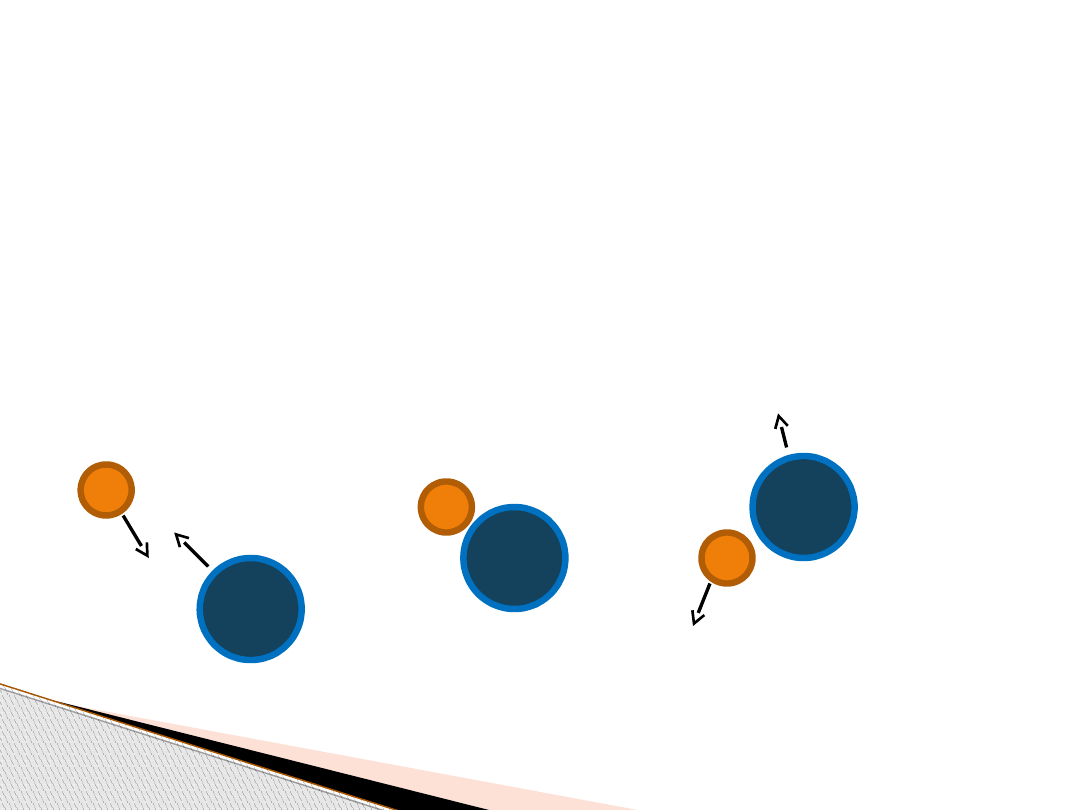
Teoria zderzeń aktywnych
Szybkość reakcji chemicznej powinna być proporcjonalna do
ilości zderzeń pomiędzy cząsteczkami reagujących substratów,
ponieważ atomy (cząsteczki) reagujących substancji muszą
„wejść w kontakt” (zderzyć się).
Nie każde takie zderzenie prowadzić będzie do zajścia reakcji:
zd
*
N
k
v
A
B
A
B
A
B
zderzenie sprężyste, reakcja nie zachodzi
E
A
+ E
B
< E
a
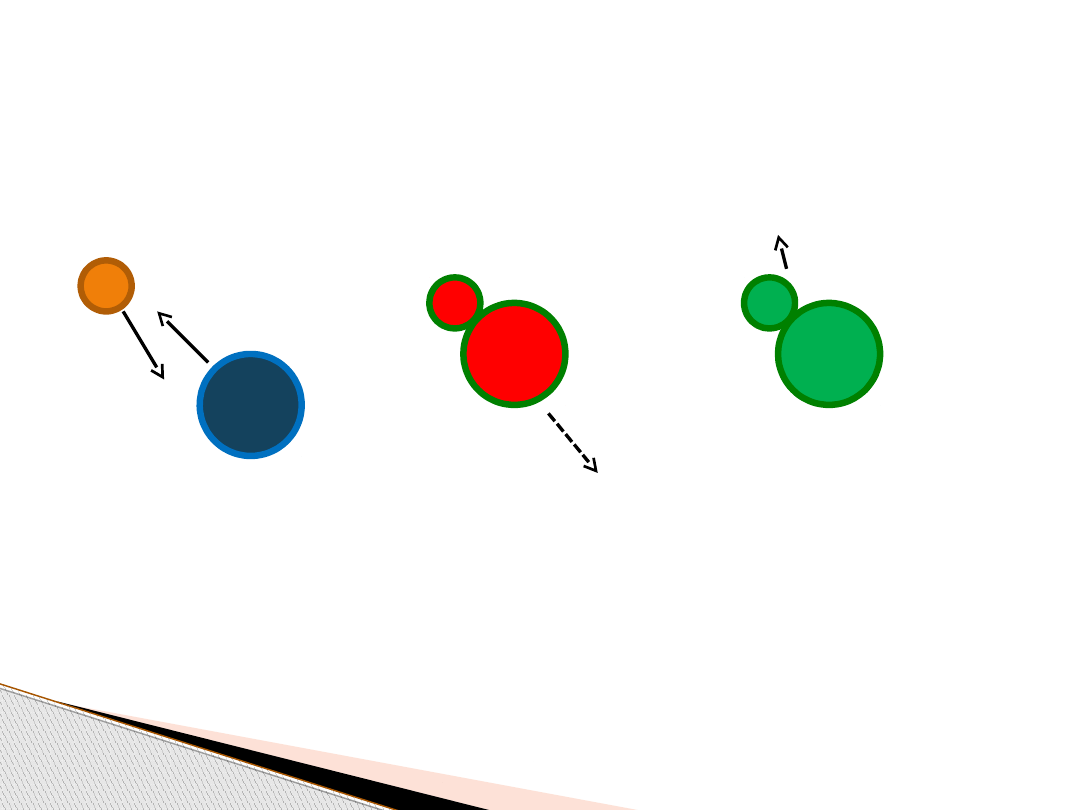
Teoria zderzeń aktywnych
Aby reakcja zaszła, konieczne jest utworzenie kompleksu
aktywnego, czyli zderzenie musi być aktywne.
A
B
zderzenie aktywne, reakcja zachodzi
E
A
+ E
B
≥ E
a
C
kompleks aktywny
wydziela się
nadmiar energii
C = AB

Teoria zderzeń aktywnych
Szybkość reakcji chemicznej będzie wobec tego zależna od ilości
zderzeń aktywnych, przeliczonych na jednostkę objętości,
zachodzących w danym czasie.
Można wykazać, że w mieszaninie gazów A i B liczba zderzeń
pomiędzy
cząsteczkami
A i B dana jest zależnością:
Aby zderzenie było aktywne, zderzające cząstki muszą mieć
wystarczająco wysoką energię (równą lub przekraczającą
wartość E
A
i E
B
). Ilość takich cząstek dana jest zależnością:
zd(akt)
N
k
v
B
A
#
zd
c
c
T
k
N
T
R
E
A
)
E
A(E
A
A
e
N
n
T
R
E
B
)
E
B(E
B
B
e
N
n

Teoria zderzeń aktywnych
Można zatem wyrazić liczbę zderzeń aktywnych zależnością:
Korzystając ze wzorów:
otrzymujemy:
T
R
E
zd
T
R
E
E
zd
T
R
E
T
R
E
zd
zd(akt)
a
B
A
B
A
e
N
k
e
N
k
e
e
N
k
N
B
A
#
zd
c
c
T
k
N
zd(akt)
N
k
v
T
R
E
B
A
T
R
E
B
A
#
T
R
E
zd
a
a
a
e
c
c
T
k
v
e
c
c
T
k
k
k
e
N
k
k
v
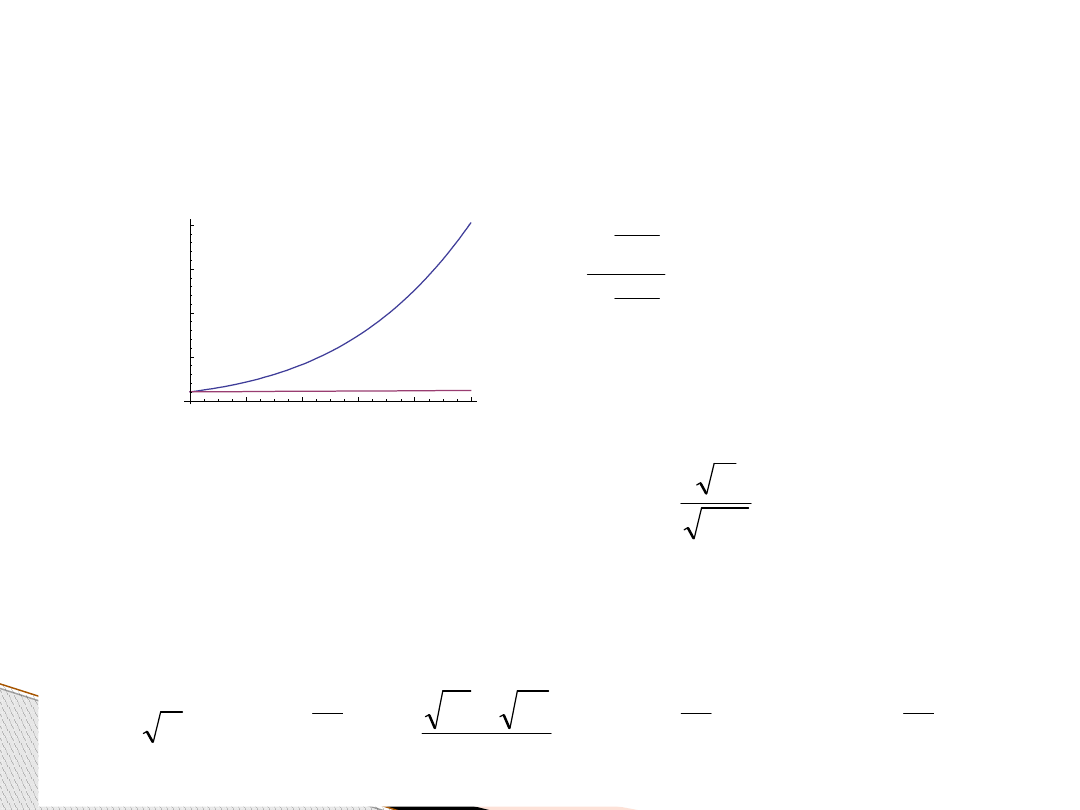
Teoria zderzeń aktywnych
Zmiany temperaturowe związane z czynnikiem eksponencjalnym
są jednak nieporównywalnie większe, w porównaniu do czynnika
T
0,5
.
Można więc zastosować przybliżenie:
3 2 0
3 4 0
3 6 0
3 8 0
4 0 0
5
1 0
1 5
2 0
T [K]
p
rz
y
ro
s
t
c
zy
n
n
ik
a
e
k
s
p
o
n
e
n
c
ja
ln
e
g
o
i
p
ie
rw
ia
s
tk
o
w
e
g
o
00
3
R
30000
T
R
30000
e
e
300
T
T
R
E
B
A
0
T
R
E
B
A
2
1
T
R
E
B
A
a
a
a
e
c
c
k
e
c
c
2
T
T
k
e
c
c
T
k
v

Teoria zderzeń aktywnych
W ogólnym przypadku, dla reakcji zachodzących w układzie
homogenicznym można stosować wyrażenie na szybkość reakcji:
Stałe w równaniu Arrheniusa wynoszą:
T
R
E
0
γ
C
β
B
α
A
γ
C
β
B
α
A
T
R
E
0
a
a
e
k
k
...
c
c
c
k
...
c
c
c
e
k
v
T
B
A
lnk
0
lnk
A
R
E
B
a

Energia aktywacji reakcji
A + B → AB
e
n
e
rg
ia
droga reakcji
energia substratów
energia produktu
energia kompleksu
aktywnego
A + B
(A+B)*
AB = C
E
a
Q
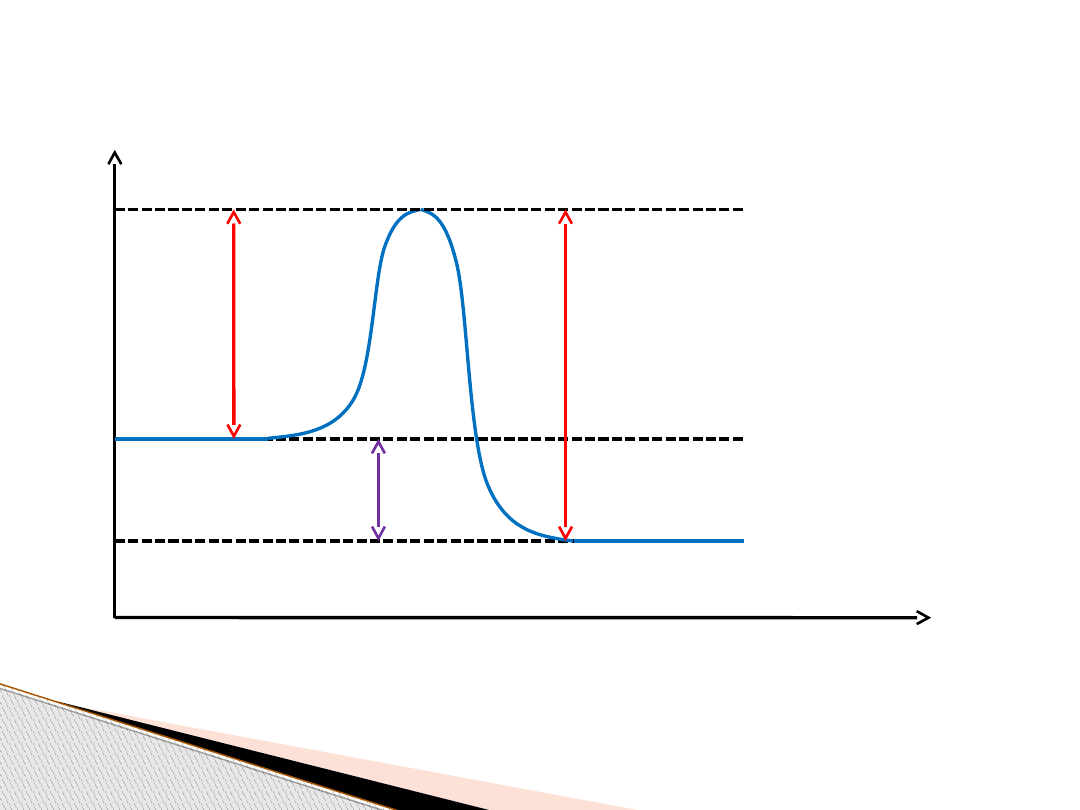
Energia aktywacji w reakcji
odwracalnej
A + B ↔ AB
e
n
e
rg
ia
droga reakcji
energia substratów
energia produktu
energia kompleksu
aktywnego
A + B
(A+B)*
AB = C
E
a L→P
Q
E
a P→L
Q = E
a P→L
– E
a P→L

Kompleks aktywny i teoria stanu
przejściowego
W trakcie zderzenia aktywnego powstaje kompleks aktywny.
Proces tez zachodzi zgodnie z równaniem
A + B → (A+B)*
. Można
zapisać wyrażenie na stałą stężeniową:
Analizując drgania w kompleksie aktywnym, można określić
szybkość zachodzenia reakcji:
[B]
[A]
B)*]
[(A
K
A
B
(A+B)*

Kompleks aktywny i teoria stanu
przejściowego
Średnia częstotliwość drgań w kompleksie aktywnym dana jest
zależnością:
gdzie k – stała Boltzmanna, h – stała Plancka.
Jeśli kompleks aktywny ma wyższą częstotliwość drgań to
częściej przybierze konfigurację umożliwiająca rozpad na
produkty
reakcji.
A
więc
szybkość
reakcji
v
będzie
proporcjonalna do v
śr
. Z drugiej strony, szybkość reakcji będzie
też proporcjonalna do stężenia kompleksów aktywnych, wobec
czego:
Z termodynamiki można wyprowadzić zależność:
h
T
k
v
śr
[A][B]
K
h
T
k
k
B)*]
[(A
v
k
v
$
śr
$
RT
ΔH
R
ΔS
0
0
e
e
K

Kompleks aktywny i teoria stanu
przejściowego
Otrzymuje się zatem równanie:
ΔH
0
dla kompleksu aktywnego ma więc sens energii aktywacji.
R
ΔS
$
0
t
RT
ΔH
0
t
RT
ΔH
R
ΔS
$
0
0
0
0
e
h
T
k
k
k
[A][B]
e
k
[A][B]
e
e
h
T
k
k
v

Katalizatory

Katalizatory
Energia aktywacji można traktować jako pewną barierę, którą
muszą pokonać cząsteczki substratów aby utworzyć produkty.
Obniżenie tej bariery spowodowało by zwiększenie szybkości
zachodzenia reakcji w danej temperaturze, dla tych samych
wyjściowych stężeń substratów .
Aby to osiągnąć stosuje się tzw. katalizatory.
Kataliza to
zjawisko zmiany szybkości reakcji chemicznych pod wpływem
katalizatorów.
Wyróżniamy
katalizę homogeniczną
: substraty i katalizator
tworzą jedną fazę, oraz
katalizę heterogeniczną
: substraty i
katalizator występują w różnych fazach.
Zapis reakcji homogenicznej z katalizatorem można przedstawić
jako:
A + B + K → (AK)* + B → (AKB)* → AB + K
lub
A + B + K → A + (BK)* → (ABK)* → AB + K
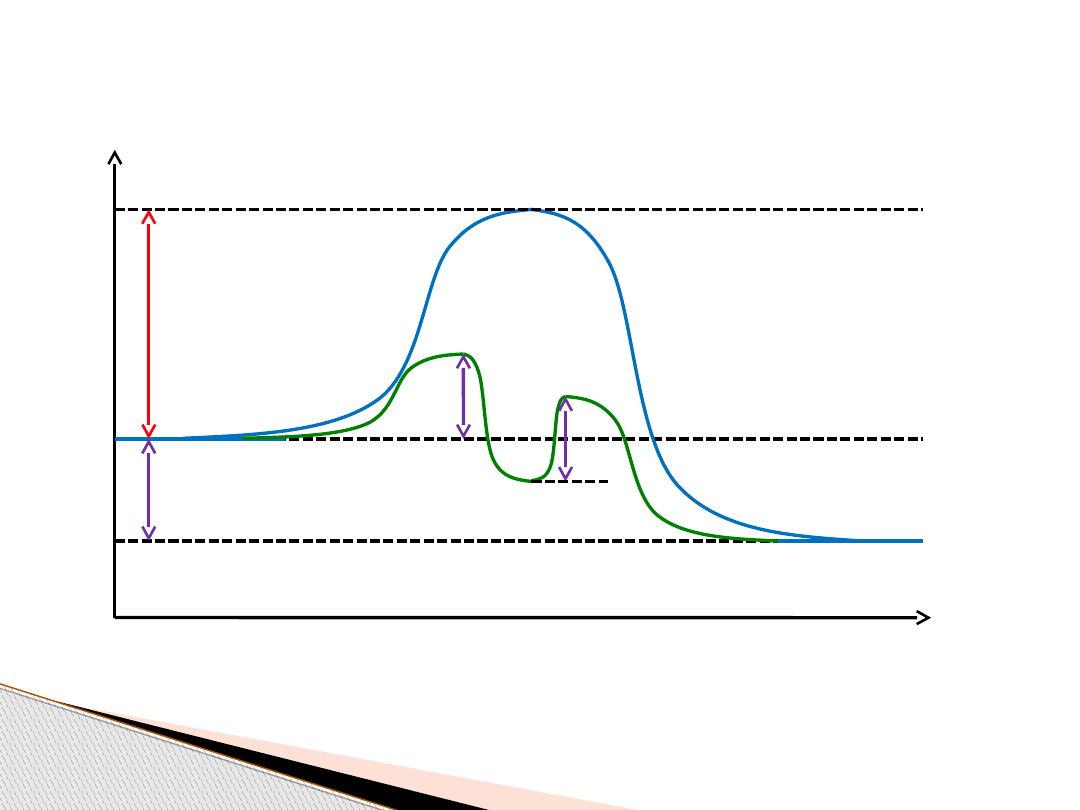
Przykład działania katalizatora:
A + B
+ K → AB + K
e
n
e
rg
ia
droga reakcji
A + B
+ K
(AK)* + B
AB = C
+ K
E
a
Q
E
a
> E
I
a
E
a
> E
II
a
(AK+B)*
E
I
a
E
II
a
AK + B
Document Outline
- Slide 1
- Wykład 14
- Slide 3
- Wprowadzenie
- Teoria zderzeń aktywnych
- Teoria zderzeń aktywnych
- Teoria zderzeń aktywnych
- Teoria zderzeń aktywnych
- Teoria zderzeń aktywnych
- Teoria zderzeń aktywnych
- Energia aktywacji reakcji A + B → AB
- Energia aktywacji w reakcji odwracalnej A + B ↔ AB
- Kompleks aktywny i teoria stanu przejściowego
- Kompleks aktywny i teoria stanu przejściowego
- Kompleks aktywny i teoria stanu przejściowego
- Slide 16
- Katalizatory
- Przykład działania katalizatora: A + B + K → AB + K
Wyszukiwarka
Podobne podstrony:
14 chemiaogolna wyklad 171109, Stała dysocjacji K zależy od temperatury
wykladChK-14, Chemia UŁ, teoretyczna wykład
Chemia organiczna wykład 14
wyklad 14
wyklad 14 15 2010
Wyklad 14 2010
Wyklad 14 PES TS ZPE
Wyklad 14
Chemia wyklad I i II (konfiguracja wiÄ…zania Pauling hybrydyzacja wiazania pi i sigma)
Wykład 14
Wykład 14
patomorfologia wyklad 2 14 10 2011 2
Chemia Wykład 6
IS wyklad 14 15 01 09 MDW id 22 Nieznany
pytania testowe i chemia budowlana -zestaw3, Szkoła, Pollub, SEMESTR II, chemia, wykład, testy
Wyklad z 14, szkoła
PiK wykład 14 10 16
więcej podobnych podstron