
Zasady diagnostyki
chorób mitochondrialnych
Dorota Piekutowska-Abramczuk
Zakład Genetyki Medycznej IPCZD
Warszawa, 12.01.12

Chorobą mitochondrialną
nazywamy stan chorobowy wywołany
uwarunkowaną genetycznie zmianą budowy
białka, pierwotnie zaburzającą przebieg
procesu fosforylacji oksydacyjnej w komórce.
45
7
38
4
4
11
1
10
13
3
10
16
2
14
Podjednostki
mtDNA
nDNA
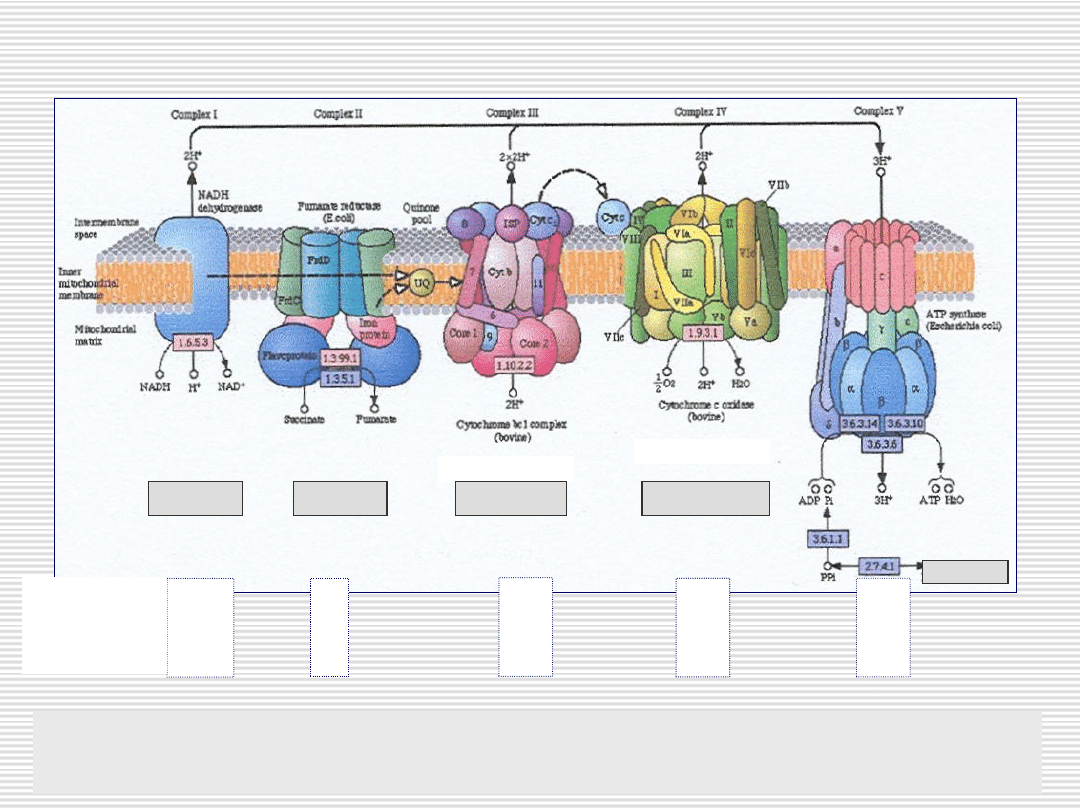
System fosforylacji oksydacyjnej (OXPHOS)
Dehydrogenaza
bursztynianowa
Dehydrogenaza
NADH
Kompleks
cytochromów bc1
Oksydaza
cytochromu c
Syntaza ATP
Zaburzenia aktywności kompleksów – izolowane lub złożone
najczęściej – KI i KIV

Choroby mitochondrialne
Częstość występowania 1:8500 - 1:5000
Prawdopodobnie wiele przypadków pozostaje nierozpoznanych
Heterogenność kliniczna, biochemiczna i genetyczna
Patomechanizm
obniżenie efektywności syntezy ATP
wzrost produkcji reaktywnych form tlenu
zaburzenia homeostazy wapnia

Choroby mitochondrialne
Uwarunkowane mutacjami w genach:
› mitochondrialnych
» strukturalne podjednostki OXPHOS
» tRNA
» rRNA
› jądrowych
» strukturalne podjednostki OXPHOS
» niestrukturalne
»› czynniki towarzyszące (
assembly factors
) (I-V)
»›
czynniki zaburzające replikację mtDNA
»› zaburzające transkrypcję i translację genów OXPHOS
»›
zaburzające import białek mitochondrialnych
»› biosynteza kofaktorów
»›
deficyt CoQ i in.
>50% wszystkich mutacji w mtDNA (gł.
MTTK, MTTL1, MTTI)

Dziedziczenie
matczyne
autosomalne recesywne
autosomalne dominujące
sprzężone z chromosomem X
Przypadki sporadyczne
Występuje w 10
3
– 10
5
kopii
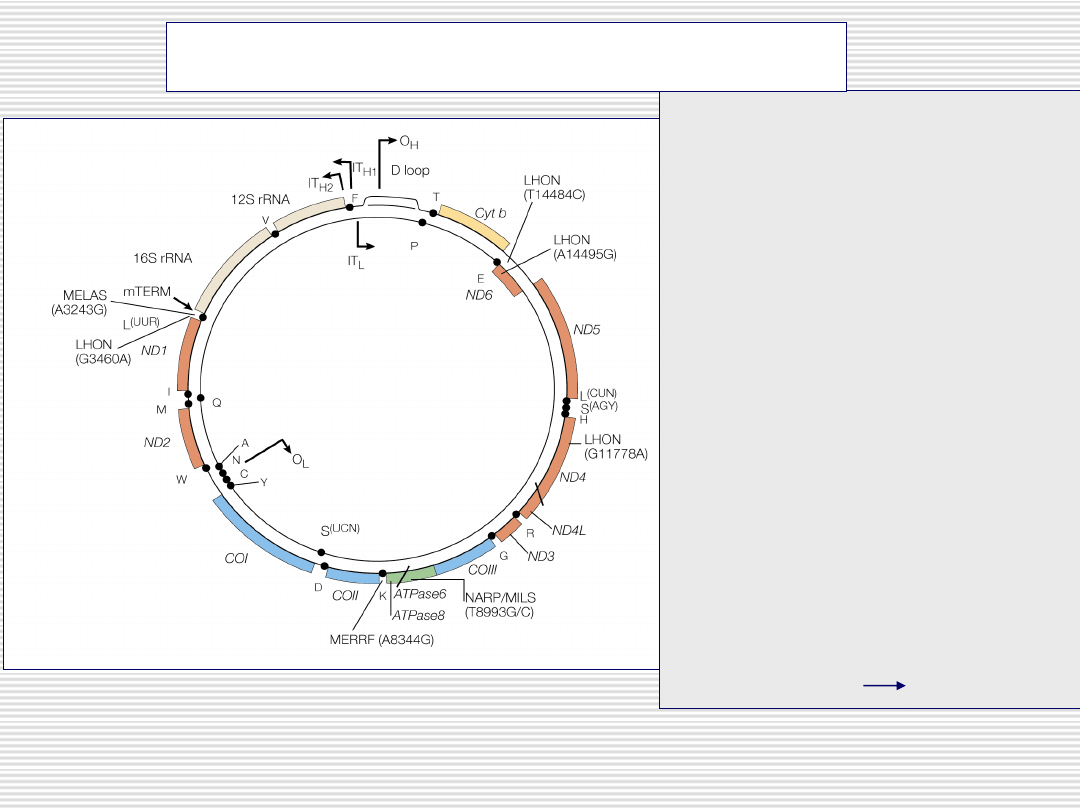
Kolista cząsteczka o wielkości
16 569 kpz (nić H i L)
Zawiera 37 genów (13
strukturalnych podjednostek
systemu OXPHOS, 2 rRNA, 22
tRNA)
Małe lub brak przestrzeni
międzygenowych
Brak intronów
2 małe obszary niekodujące:
pętla D – zawiera miejsca
inicjacji transkrypcji i replikacji
nici H i miejsce startu replikacji
nici L
Odstępstwa od uniwersalnego
kodu genetycznego
(np. TGA (Stop) Trp)
Mitochondrialne DNA

Genetyka mitochondrialna
Dziedziczenie matczyne
Tylko kobieta przekazuje zmutowany gen swemu potomstwu, zarówno córce
jak i synowi (nieliczne mtDNA plemnika są aktywnie usuwane we wczesnych
podziałach zygoty).
Poliploidalność mitochondriów (setki mitochondriów, a w każdym do 10
cząsteczek mtDNA)
Szybkie tempo ewolucji (mała wydajność systemów naprawy, brak
histonów, brak zjawiska rekombinacji, nadprodukcja ROS)
Heteroplazmia i wartość progowa
Prezentacja objawów zależy od poziomu heteroplazmii i wartości progowej
(
treshold effect
).
Segregacja mitotyczna (efekt
bottleneck
) w oogenezie tłumaczy częste
zmiany poziomu mutacji w kolejnych pokoleniach i znaczne zróżnicowanie
fenotypowe.
Ekspansja klonalna (preferencyjna amplifikacja mutacji mtDNA do
wysokiego poziomu w tkankach postmitotycznych)
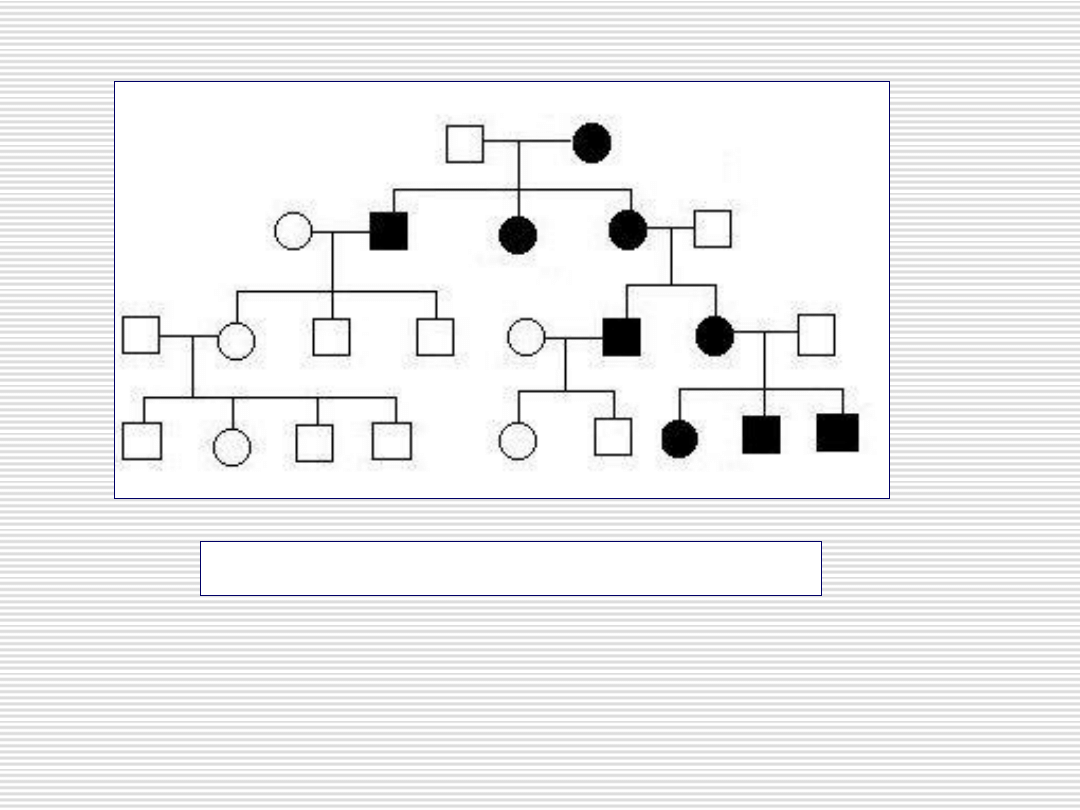
Dziedziczenie w linii matczynej
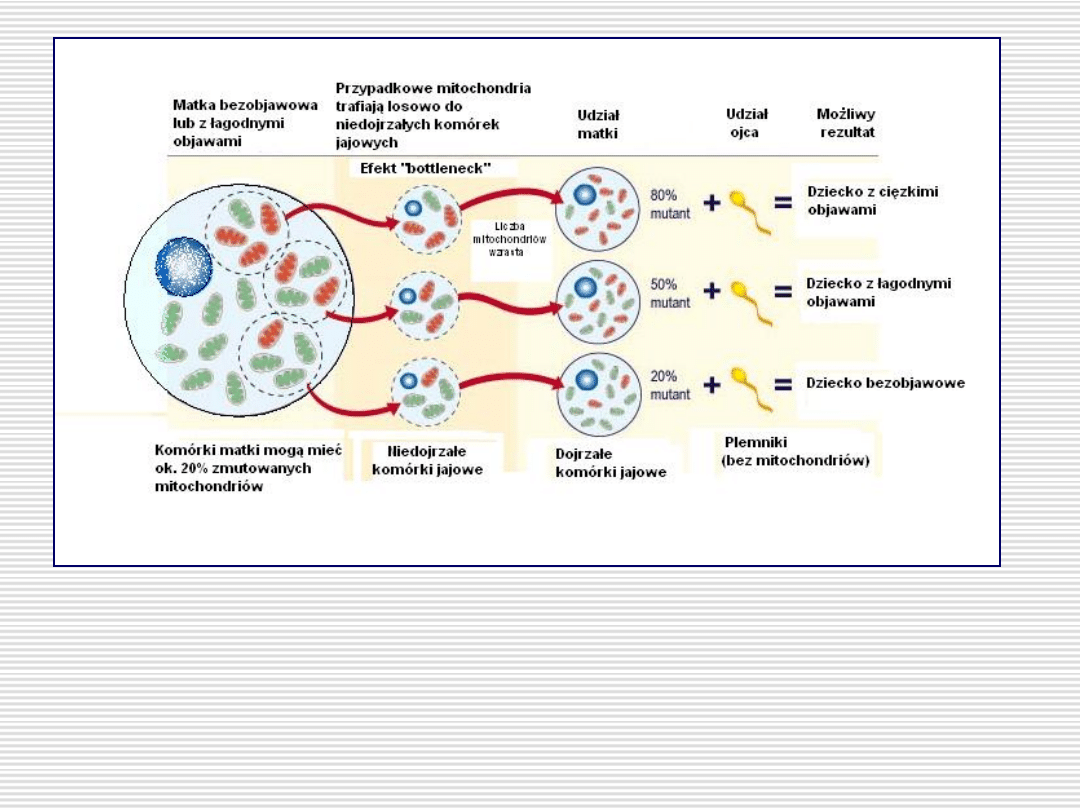
Schemat mitochondrialnego efektu szyjki butelki
(bottleneck) w dojrzewających komórkach jajowych

Cechy mutacji mtDNA
Heteroplazmatyczne
Homoplazmatyczne
Stabilne
• silna korelacja genotyp-fenotyp
• mała/brak zmienność poziomu mutacji w różnych tkankach
• mała/brak zmienność poziomu w czasie (podczas rozwoju płodowego i
po urodzeniu)
m.8993T>G, m.8993T>C
Niestabilne
• bardzo zróżnicowany obraz kliniczny
• niejednorodne tkankowe rozmieszczenie mutacji
• duża zmienność poziomu mutacji w czasie
m.3243A>G

Choroby mitochondrialne u dzieci
Częściej wynikają z mutacji w jądrowym DNA
Cięższe od objawiających się w wieku późniejszym i częściej
wielonarządowe;
Dotyczą dysfunkcji wątroby (MDS), choroby nerek (MDS, deficyt
KIII) i zaburzeń hemopoezy (zespół Pearsona)
Opóźnienie rozwoju w połączeniu z kwasicą mleczanową
Postępujący regres rozwojowy, utrata posiadanych umiejętności
(zespół Leigha)
Niespecyficzne objawy zaburzenia w odżywianiu, wzrastania,
drgawki, częste infekcje
Rzadko obecne RRF

Rozpoznanie choroby mitochondrialnej
Zdefiniowane, wysoce prawdopodobne
analiza mutacji
Prawdopodobne
biopsja mięśnia
Możliwe
obserwacja, biopsja mięśnia, zabezpieczenie
materiału w wypadku zgonu

Materiał biologiczny
mięsień
krew obwodowa
wymaz z nabłonka jamy ustnej
mocz
wątroba
fibroblasty
włosy
płyn owodniowy
kosmki kosmówki
Oragene DNA
www.dnagenotek.com

Biopsja mięśnia
kompleksowe potwierdzenie rozpoznania
Ocena morfologiczna (włókna RRF)
Badanie histochemiczne (oksydaza cytochromu c mozaikowość,
dehydrogenaza bursztynianowa)
Badanie proteomiczne (obecność i aktywność kompleksów łańcucha
oddechowego i ich poszczególnych podjednostek)
Analiza enzymatyczna (aktywność kompleksów łańcucha oddechowego i
syntazy cytrynianowej)
Analiza molekularna

Przekrój poprzeczny przez mięsień szkieletowy.
Włókna RRF (A) wykazują dodatnią reakcję w kierunku dehydrogenazy bursztynianowej (B) i
brak aktywności oksydazy cytochromowej (C). Uszkodzone włókna wykazują niewielki wzrost
aktywności kwaśnej fosfatazy (D)
D
C
A
B
(A)
(B)
(C)
(D)

Zespół mitochondrialny IPCZD
Klinika Chorób Metabolicznych
Klinika Neurologii i Epileptologii
Klinika Gastroenterologii, Hepatologii i Immunologii
Zakład Biochemii i Medycyny Doświadczalnej
Zakład Patologii
Zakład Genetyki
Współpraca zewnętrzna

Diagnostyka molekularna chorób
mitochondrialnych w ZGM IPCZD
1. Identyfikacja powszechnych mutacji
• m.8993T>C/G (
MTATP6
) - NARP/MILS
• m.3243A>G (
MTTL1
) - MELAS
• m.8344A>G (
MTTK
) - MERRF
• delecja 4977pz (m.8470_13446del) KSS i in. zespoły delecyjne
• m.11778G>A (
MTND4
), m.3460G>A (
MTND1
), m.14484T>C (
MTND6
) - LHON
• g.1541G>A (
SCO2
) - deficyt białka SCO2
• c.845_846delCT, c.311_312insAT312_321del10(
SURF1
) – LS
2. Poszukiwanie mutacji w innych, znanych genach związanych z fenotypem
•
MTND1-MTND6 -
LS
•
DGUOK, MPV17
- HC-MDS
3. W przyszłości dalsza analiza molekularna obejmująca geny nie badane lub nowo
odkryte.
podstawowy
skrining mtDNA

Izolacja DNA
Amplifikacja, w tym L-PCR (delecje)
Sekwencjonowanie
MLPA (mutacje punktowe, delecje)
Real-Time PCR (poziom heteroplazmii, deplecji)
Analiza restrykcyjna (poziom heteroplazmii)
Schemat postępowania

Negatywny wynik badania molekularnego zwykle nie
wyklucza rozpoznania.
Wyszukiwarka
Podobne podstrony:
IPN 16 2007 06 01
Podstawowe zasady diagnozy psychologicznej
06 01 88
1968stories?utsch 06 01 06
ZASADY DIAGNOSTYKI I LECZENIA BÓLU wyklady z fizjologii
1968stories english 06 01 06
2015 08 20 07 50 06 01
wyklad 06[1].01.2008, Zarządzanie studia licencjackie, Finanse publiczne
MPLP 302;303 06.01.2011;18.01.2011
Paczka Nutek Disco Polo Nowości (06 01 2010)
2015 08 20 08 04 06 01
0656PWsrT Rysunek 06 01
Incomings SCORE w Krakowie 02 06 01 2014
10 06 01 chkol3
Matematyka II (Ćw) 2012 06 01
11 01 06 01 xxx?hrrgln allg m L
GIge zal 06 01 04 Przekroj geo inz
06 01 Roboty w zbiornikach i komorach
więcej podobnych podstron