

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
MINISTERSTWO EDUKACJI
NARODOWEJ
Małgorzata Klimczak
Wykonywanie analizy jakościowej i ilościowej produktów
leczniczych 322[10].Z1.01
Poradnik dla ucznia
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy
Radom 2007

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
1
Recenzenci:
dr hab. n. farm. Anna Gumienniczek
dr n. farm. Dorota Kowalczuk
Opracowanie redakcyjne:
mgr Alina Krawczak
Konsultacja:
dr hab. inż. Henryk Budzeń
Poradnik stanowi obudowę dydaktyczną programu jednostki modułowej „Wykonywanie
analizy jakościowej i ilościowej produktów leczniczych” 322[10].Z1.01 zawartego
w modułowym programie nauczania dla zawodu technik farmaceutyczny.
Wydawca
Instytut Technologii Eksploatacji – Państwowy Instytut Badawczy, Radom 2007

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
2
SPIS TREŚCI
1. Wprowadzenie
3
2. Wymagania wstępne
5
3. Cele kształcenia
6
4. Materiał nauczania
7
4.1. Analiza jakościowa związków nieorganicznych i organicznych
7
4.1.1. Materiał nauczania
7
4.1.2. Pytania sprawdzające
12
4.1.3. Ćwiczenia
13
4.1.4. Sprawdzian postępów
16
4.2. Analiza ilościowa
17
4.2.1. Materiał nauczania
17
4.2.2. Pytania sprawdzające
22
4.2.3. Ćwiczenia
23
4.2.4. Sprawdzian postępów
28
5. Sprawdzian osiągnąć
29
6. Literatura
34

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
3
1. WPROWADZENIE
Poradnik będzie Ci pomocny w przyswojeniu wiedzy teoretycznej i nabyciu umiejętności
praktycznych związanych z wykonywaniem analizy jakościowej i ilościowej produktów
leczniczych.
W poradniku zamieszczono:
−
wymagania wstępne, czyli wykaz niezbędnych umiejętności i wiedzy, które powinieneś
mieć opanowane, aby przystąpić do realizacji tej jednostki modułowej,
−
cele kształcenia tej jednostki modułowej,
−
materiał nauczania (rozdział 4), który umożliwia samodzielne przygotowanie się do
wykonania ćwiczeń i zaliczenia sprawdzianów.
Wykorzystaj do poszerzenia wiedzy wskazaną literaturę oraz inne źródła informacji.
Obejmuje on również ćwiczenia, które zawierają:
−
treść ćwiczeń,
−
sposób ich wykonania,
−
wykaz materiałów i sprzętu potrzebnego do realizacji ćwiczenia,
−
sprawdzian postępów, który umożliwi Ci sprawdzenie opanowania zakresu materiału po
zrealizowaniu każdego podrozdziału – wykonując sprawdzian postępów powinieneś
odpowiadać na pytanie tak lub nie, co oznacza, że opanowałeś materiał albo nie,
−
sprawdzian osiągnięć, czyli zestaw zadań testowych sprawdzających Twoje opanowanie
wiedzy i umiejętności z zakresu całej jednostki. Zaliczenie tego ćwiczenia jest dowodem
osiągnięcia umiejętności praktycznych określonych w tej jednostce modułowej.
Jeżeli masz trudności ze zrozumieniem tematu lub ćwiczenia, to poproś nauczyciela
o wyjaśnienie i ewentualne sprawdzenie, czy dobrze wykonujesz daną czynność.
Po opracowaniu materiału spróbuj rozwiązać sprawdzian z zakresu jednostki modułowej.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
4
Schemat układu jednostek modułowych
322[10].Z1
Podstawy analizy i wytwarzania produktów
leczniczych
322[10].Z1.01
Wykonywanie analizy jakościowej
i ilościowej produktów leczniczych
322[10].Z1.02
Pozyskiwanie i przetwarzanie roślinnych
surowców leczniczych
322[10].Z1.03
Analizowanie procesów technologicznych
produkcji leków

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
5
2. WYMAGANIA WSTĘPNE
Przystępując do realizacji programu jednostki modułowej, powinieneś umieć:
−
korzystać z różnych źródeł informacji,
−
przestrzegać przepisów bezpieczeństwa i higieny pracy, ochrony przeciwpożarowej oraz
ochrony środowiska,
−
organizować stanowisko pracy zgodnie z wymaganiami ergonomii,
−
udzielać pierwszej pomocy w stanach zagrożenia życia i zdrowia.
−
zadbać o stan wyposażenia oraz ład i porządek w miejscu pracy,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
6
3. CELE KSZTAŁCENIA
W wyniku realizacji programu jednostki modułowej powinieneś umieć:
−
wykonać podstawowe czynności laboratoryjne,
−
obsłużyć sprzęt, aparaturę oraz urządzenia stosowane w laboratoriach,
−
wypełnić dokumentację laboratoryjną,
−
wykonać oznaczenia z zakresu analizy jakościowej związków nieorganicznych
i organicznych,
−
określić tożsamość surowców farmaceutycznych i środków leczniczych,
−
pobrać i przygotować próbki do badań,
−
przeprowadzić wybranymi metodami analizę jakościową i ilościową, klasyczną
i instrumentalną leków, surowców farmaceutycznych i produktów leczniczych,
−
obliczyć wyniki analizy ilościowej z wykorzystaniem metod matematycznych, graficznych i
statystycznych,
−
przechować zgodnie z zasadami próbki analityczne substancji leczniczych,
−
zastosować metody wagowe, miareczkowe i instrumentalne do badania czystości
i zawartości substancji leczniczych,
−
ocenić czystość surowców i produktów leczniczych,
−
dokonać rozdziału leków złożonych metodą: destylacji, ekstrakcji, wytrącania osadów,
sączenia, przemywania, wirowania, krystalizacji,
−
zinterpretować wyniki przeprowadzonych badań,
−
zastosować obowiązujące normy oceny jakości produktów leczniczych i surowców
różnego pochodzenia,
−
zastosować zasady Dobrej Praktyki Laboratoryjnej.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
7
4. MATERIAŁ NAUCZANIA
4.1. Analiza
jakościowa
związków
nieorganicznych
i organicznych
4.1.1. Materiał nauczania
Organizacja pracy i wyposażenie laboratorium.
W celu zapewnienia bezpieczeństwa w laboratorium należy przestrzegać następujących
zasad:
−
używaj zawsze fartucha, okularów ochronnych i rękawic ochronnych,
−
podczas pracy w laboratorium noś odpowiednie obuwie na podeszwie antypoślizgowej,
długie włosy powinny być krótko upięte,
−
przed przystąpieniem do pracy zapoznaj się z kartami charakterystyk substancji
niebezpiecznych, dotyczącymi danego ćwiczenia,
−
na stanowisku pracy gromadź wyłącznie sprzęt i odczynniki niezbędne do wykonywania
ćwiczenia,
−
prace z substancjami łatwopalnymi i żrącymi wykonuj pod sprawnym wyciągiem,
−
prace z substancjami łatwopalnymi (eter, chloroform) wykonuj z dala od ognia,
−
zapoznaj się z lokalizacją sprzętu ratunkowego oraz drogami ewakuacji,
−
w laboratorium nie wolno pić, jeść, żuć gumy,
−
nie prowadź głośnych rozmów w czasie ćwiczeń, nie używaj telefonu komórkowego,
−
nie pipetuj ustami, nie badaj smaku analizowanych substancji,
−
utrzymuj porządek na stanowisku w trakcie pracy,
−
odstawiaj wszystkie odczynniki na miejsce,
−
odpady chemiczne składaj do właściwych pojemników,
−
po zakończeniu ćwiczenia uprzątnij stanowisko pracy,
−
po zakończeniu ćwiczeń umyj dokładnie ręce.
Przepisy bezpieczeństwa i higieny pracy obowiązują na terenie laboratorium.
W laboratorium znajduje się sprzęt do usuwania skutków wypadków:
sprzęt ochrony
przeciwpożarowej (gaśnica śniegowa i proszkowa, koc gaśniczy),
zestaw adsorbentów
i środków do neutralizacji rozlanych chemikaliów, apteczka ze środkami pierwszej pomocy.
O każdym wypadku przy pracy należy natychmiast powiadomić nauczyciela.
Postępowanie w nagłych wypadkach
Skaleczenia
Ranę przemyć wodą utlenioną, obejrzeć, czy nie tkwią w niej odłamki szkła. Z rany
można usunąć, przy pomocy pęsety, wyłącznie luźno leżące szklane odłamki, nie wolno
penetrować wnętrza rany. Zranienie należy zabezpieczyć jałowym opatrunkiem.
Oparzenia termiczne
Pierwsza pomoc polega na ochładzaniu uszkodzonego miejsca dużą ilością bieżącej
zimnej wody.
Oparzenie chemiczne
Pierwsza pomoc polega na natychmiastowym zmyciu żrącej substancji dużą ilością
bieżącej zimnej wody. W przypadku oparzenia zasadą należy przemyć oparzone miejsce w celu
zobojętnienia 1% roztworem kwasu octowego lub borowego, przy oparzeniu kwasem

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
8
oparzone miejsce należy przemyć 1% roztworem wodorowęglanu sodu. Jeżeli oparzenie jest
rozległe, to należy skonsultować się z lekarzem.
Ciało obce w oku
Pierwsza pomoc polega na przemywaniu uszkodzonego oka dużą ilością bieżącej wody
przez co najmniej 10 minut, przy szeroko rozwartej powiece, pamiętając aby strumień wody
nie był zbyt silny.
Zatrucie
W przypadku zatrucia parami substancji należy wyprowadzić poszkodowanego
z laboratorium i ulokować go wygodnie w pobliżu otwartego okna. W cięższych przypadkach
należy wezwać pomoc lekarską. W przypadku połknięcia substancji toksycznej należy podać
do wypicia czystą wodę i wezwać lekarza.
Porażenie prądem
Należy wyłączyć instalację elektryczną. Udzielić pierwszej pomocy. Natychmiast wezwać
pomoc lekarską.
Postępowanie w razie pożaru
Należy natychmiast ewakuować ludzi z zagrożonego pomieszczenia, wyłączyć źródła
ognia (instalację gazową i elektryczną). Laboratorium wyposażone jest w różne środki
gaśnicze: gaśnice, koce gaśnicze, piasek i wodę. Dobór odpowiedniego sposobu gaszenia
zależy od rodzaju płonącego materiału i wielkości pożaru.
Postępowanie w przypadku awarii sieci gazowej
W przypadku awarii sieci gazowej, należy zamknąć główny zawór gazowy, wyłączyć
wszystkie źródła ognia, ewakuować ludzi z zagrożonego laboratorium i otworzyć szeroko
okna.
Wyposażenie laboratorium
Pomieszczenia laboratorium wyposażone są w instalację wodną, gazową, elektryczną
i wentylacyjną. Podstawowy sprzęt w laboratorium: stół laboratoryjny z szufladami, półkami
i szafkami, dygestorium z pełnym wyposażeniem. Podstawowy sprzęt laboratoryjny:
−
szklany (zlewki, kolbki stożkowe, lejki, cylindry miarowe, probówki, bagietki szklane,
szkiełka zegarkowe, kolby miarowe, pipety, biurety),
−
metalowy (statywy, łączniki, łapy, kółka, trójnogi, trójkąty, szczypce),
porcelanowy (parownice, łyżki, tygle),
−
gumowy (węże, korki),
−
z tworzyw sztucznych (łyżki, statywy do pipet, tace, koszyczki),
−
drewniany (łapy),
−
ponadto komory chromatograficzne, pipety automatyczne, aparat do wywoływania
chromatografów.
Urządzenia:
−
suszarka laboratoryjna,
−
łaźnia wodna, piaskowa,
−
wirówka,
−
piec do spalania,
−
destylarka do wody,
−
lampa kwarcowa laboratoryjna.
Sprzęt i aparatura pomiarowa:
−
wagi laboratoryjne i analityczne,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
9
−
areometr,
pehametr,
potencjometr,
kolorymetr,
refraktometr,
polarymetry,
spektrofotometry UV i VIS, chromatografy gazowy lub cieczowy HPLC, spektofotometr
płomieniowy,
−
odczynniki chemiczne do wykonania przewidzianych w programie zadań, butle z wodą
destylowaną.
Dokumentacja laboratoryjna
Każdy uczeń powinien posiadać odpowiedni zeszyt (dziennik laboratoryjny), w którym
podczas wykonywania ćwiczeń dokonuje zapisu: obserwacji, naważek, wyników.
W zeszycie przeprowadza obliczenia i analizę błędów.
Dobra Praktyka Laboratoryjna
Zasady
Dobrej
Praktyki
Laboratoryjnej
zostały
wprowadzone
formalnie
„Rozporządzeniem Ministra Zdrowia z dnia 4 czerwca 2003 r. w sprawie kryteriów, które
powinny spełniać jednostki organizacyjne wykonujące badania substancji i preparatów
chemicznych oraz kontroli spełnienia tych kryteriów” (Dz. U. Nr 116, poz.1103). Dobra
Praktyka Laboratoryjna jest systemem zapewnienia jakości badań, określającym zasady
organizacji jednostek badawczych wykonujących nie kliniczne badania z zakresu
bezpieczeństwa i zdrowia człowieka i środowiska, w szczególności badania substancji
i preparatów chemicznych wymagane ustawą i warunki, w jakich te badania są planowane,
przeprowadzane i monitorowane, a ich wyniki są zapisywane, przechowywane i podawane
w sprawozdaniu. Celem Dobrej Praktyki Laboratoryjnej jest promowanie jakości
i wiarygodności uzyskiwanych wyników badań, od momentu ich planowania, aż po właściwe
przechowywanie danych źródłowych i sprawozdań, tak aby możliwe było prześledzenie toku
badania lub jego całkowite odtworzenie. Zasady Dobrej Praktyki Laboratoryjnej dotyczą tak
całości działania laboratorium, jak i poszczególnych etapów procesu analitycznego.
Szczególnie istotne są Zasady Dobrej Praktyki Laboratoryjnej w następujących procesach:
−
pobierania i przygotowywania próbek,
−
walidacji metod,
−
kalibrowania aparatury,
−
gospodarki chemikaliami,
−
dokumentowania i nadzoru nad sporządzoną dokumentacją.
Pobranie próbki do analizy
Partia produktu to cała ilość materiału tej samej jakości, którą mamy ocenić na podstawie
analizy. Wielkość partii określają normy przedmiotowe.
Próbka pierwotna to porcja materiału pobrana jednorazowo z wielu miejsc wybranych
losowo za pomocą zgłębników, czerpaków.
Próbka jednostkowa – próbka pierwotna pobrana z mniejszego opakowania (worka,
skrzyni, beczki).
Próbka ogólna – połączone próbki pierwotne pobrane z partii materiału, stanowi 1‰
partii.
Średnia próbka laboratoryjna – zmniejszona, wymieszana próbka ogólna zamknięta
hermetycznie, wysłana do laboratorium.
Z tej próbki pobieramy próbkę do badań.
Metody oczyszczania i rozdziału związków chemicznych
Destylacja jest metodą rozdziału wieloskładnikowych mieszanin ciekłych czyli
oczyszczania substancji lotnych. Polega ona na odparowaniu najbardziej lotnego w danych
warunkach ciśnienia i temperatury składnika, następnie na skropleniu par i zebraniu skroplonej

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
10
cieczy (destylatu). W trakcie ogrzewania cieczy prężność pary wzrasta aż do chwili, gdy staje
się równa ciśnieniu atmosferycznemu i rozpoczyna się wrzenie czyli parowanie w całej
objętości cieczy. Rozróżnia się cztery typy destylacji: destylację prostą, destylację frakcyjną,
destylację z parą wodną i destylację pod zmniejszonym ciśnieniem.
W destylacji prostej pary cieczy poddaje się skropleniu przez bezpośrednie oziębianie, co
pozwala tylko na zagęszczenie składników mieszaniny w poszczególnych coraz wyżej
wrzących frakcjach. Stosujemy ją w przypadku, gdy temperatury wrzenia destylowanych
cieczy zawarte są w granicach 40-150
o
C.
W czasie destylacji frakcyjnej pary cieczy podążają ku górze kolumny destylacyjnej
ulegając w tym czasie częściowemu skropleniu. Utworzony kondensat spływa w dół kolumny i
spotyka się z dążącymi ku górze gorącymi parami. Dochodzi do wymiany cieplnej pomiędzy
dwiema fazami, a pary wzbogacają się w składnik bardziej lotny. Stosujemy ją w przypadku,
gdy rozdzielanie mieszaniny przez jednorazową prostą destylację jest niewystarczające,
zazwyczaj, gdy różnice temperatur wrzenia są mniejsze od 80
o
C.
Destylacja z parą wodną jest metodą oczyszczania substancji stałych i ciekłych, nie
mieszających się z wodą, lotnych zaś z parą wodną. Metodę tę stosuje się do destylacji cieczy
lub ciał stałych o wysokich temperaturach wrzenia lub do wydzielania lotnego z parą wodną
składnika ze złożonych mieszanin.
Destylacja pod zmniejszonym ciśnieniem służy do oczyszczania lub rozdzielania cieczy
o bardzo wysokich temperaturach wrzenia (znacznie powyżej 200
°C) lub takich, które ulegają
znacznemu
rozkładowi
przed
osiągnięciem
temperatury
wrzenia pod ciśnieniem
atmosferycznym.
Metody ekstrakcyjne
Ekstrakcja w układzie ciecz-ciecz to proces przenoszenia substancji rozpuszczonej
w jednej fazie ciekłej do drugiej fazy ciekłej, nie mieszającej się z pierwszą. Jedną fazą ciekłą
jest
zwykle
roztwór
wodny,
drugą
organiczny
rozpuszczalnik
lub
mieszanina
rozpuszczalników, które trudno mieszają się z wodą. Aby szybko osiągnąć równowagę
między fazami trzeba zwiększyć i odnawiać powierzchnię zetknięcia faz poprzez ręczne lub
mechaniczne wstrząsanie całości. Do ekstrakcji i rozdzielania warstw nie mieszających się ze
sobą cieczy używa się rozdzielaczy. Ekstrakcję analitu z fazy wodnej prowadzi się używając
rozpuszczalników o gęstości mniejszej lub większej niż gęstość wody. Ekstrakcję w układzie
ciało stałe – ciecz przeprowadza się w sytuacji gdy trzeba wyekstrahować z próbki stałej
składnik rozpuszczalny w użytym rozpuszczalniku.
Krystalizacja to sposób oczyszczania substancji stałych, opierający się na różnicy
rozpuszczalności danego związku w danym rozpuszczalniku w różnych temperaturach.
Substancja powinna rozpuszczać się bardzo dobrze na gorąco, a słabo po oziębieniu. Przy
krystalizacji stosuje się kolbę kulistą zaopatrzoną w chłodnicę zwrotną. Źródłem ciepła
najczęściej jest płaszcz grzejny ewentualnie łaźnia wodna.
Odwirowanie osadu to sposób oddzielenia osadu od roztworu. Przeprowadza się je
w probówkach stożkowych, przy użyciu wirówki.
Analiza chemiczna
Analiza chemiczna – zespół czynności prowadzących do ustalenia składu chemicznego,
jakościowego i ilościowego badanej substancji. Analizę chemiczną dzieli się tradycyjnie na dwa
zasadnicze działy – analizę jakościową i ilościową.
Analiza jakościowa ma za zadanie określić skład jakościowy badanego ciała tzn. ustalenie
z jakich pierwiastków, grup atomów lub związków chemicznych ciało to się składa. Analiza
jakościowa zajmuje się wykrywaniem i identyfikacją składników ciała.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
11
Zadaniem analizy ilościowej jest określenie ilości pojedynczych składników (pierwiastków,
jonów, itp.) w badanej substancji. Analiza chemiczna stanowi nierozerwalną całość i często te
same reakcje chemiczne, zjawiska fizyczne i fizykochemiczne mogą służyć zarówno do
wykrywania, identyfikacji jak i do oznaczania danego pierwiastka.
Metoda analityczna to określony sposób postępowania, według którego wykonuje się
analizę (wykrywanie lub oznaczanie).
Technika analityczna to grupa metod analitycznych opartych na tej samej zasadzie
fizycznej.
Klasyczna analiza jakościowa
Klasyczna, mokra analiza jakościowa polega na tym, że do badanego roztworu dodaje się
odczynnika chemicznego, pod wpływem którego zachodzi jakaś zauważalna,
charakterystyczna reakcja analityczna: roztwór zabarwia się, powstaje lub rozpuszcza się osad,
wydziela się gaz. Odczynnik i substancja badana znajdują się w roztworze wodnym, są
zdysocjowane, reakcja zachodzi między jonami.
Odczynniki stosowane w analizie jakościowej można podzielić na specyficzne, selektywne,
grupowe, charakterystyczne i maskujące.
Odczynniki specyficzne – w określonych warunkach dają reakcję z danym jonem,
pozwalają go wykryć w obecności innych jonów.
Odczynniki selektywne – dają podobną reakcję z pewną ograniczoną grupą jonów.
Odczynniki maskujące – łączą się z jonem ubocznym, wyłączają go z udziału w roztworze.
Odczynniki grupowe – wytrącają pewną określoną kategorię jonów z roztworu
w pewnych określonych warunkach; pozwalają na rozdzielenie jonów znajdujących się
w badanym roztworze na grupy analityczne.
Odczynniki charakterystyczne – służą do wykrywania poszczególnych jonów w obrębie
grupy analitycznej.
Jakościowa analiza kationów
Podstawą systematycznej analizy kationów jest wynik reakcji z odczynnikiem grupowym.
Systematyczna analiza jakościowa kationów opiera się na ich podziale na grupy analityczne.
Każda z nich posiada odpowiedni odczynnik grupowy.
Grupa I
Do I grupy kationów zaliczamy następujące jony:Ag
+
,Hg
2
2+
,Pb
2+
. Odczynnikiem
grupowym jest kwas solny HCl, z którym wymienione jony tworzą chlorki nierozpuszczalne w
wodzie i rozcieńczonych kwasach. PbCl
2
jest rozpuszczalny w gorącej wodzie.
Grupa II
Do II grupy kationów zaliczamy następujące jony: Hg
2+
, Bi
3+
, Cu
2+
, Cd
2+
, Sn
2+
, Sn
4+
,
As
3+
,As
5+
,Sb
3+
,Sb
5+
.Odczynnikiem grupowym jest roztwór AKT - amid kwasu tiooctowego,
CH3CSNH2 w 0,3 mol/l HCl. Iloczyny rozpuszczalności siarczków metali tej grupy są
bardzo małe, łatwo je przekroczyć przy małym stężeniu jonów S
2-
powstałych na skutek
dysocjacji siarkowodoru w środowisku kwaśnym.
Grupa III
Do III grupy kationów zaliczamy następujące jony:Al
3+
, Cr
3+
, Fe
2+
, Fe
3+
, Ni
2+
, Co
2+
,
Mn
2+
,Zn
2+
.Odczynnikiem grupowym jest roztwór AKT w obecności NH
3
.
H
2
O i NH
4
Cl.
Kationy tej grupy wytrącają się w postaci siarczków lub wodorotlenków(Al
3+
,Cr
3+
).
Grupa IV
Do IV grupy kationów zaliczamy następujące jony: Ba
2+
, Sr
2+
, Ca
2+
.Odczynnikiem
grupowym jest (NH
4
)
2
CO
3
w obecności NH
4
Cl i NH
3
.
H
2
O. Kationy wytrącają się w postaci
węglanów.
Grupa V

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
12
Do V grupy kationów zaliczamy następujące jony:Mg
2+
,NH
4
+
,K
+
,Na
+
. Grupa V nie
posiada odczynnika grupowego, który by je strącił jednocześnie. Sole ich w większości są
łatwo rozpuszczalne.
Jakościowa analiza anionów
Podział anionów na grupy analityczne wg Bunsena. Podział ten jest oparty na reakcji
z jonami Ag
+
i Ba
2+
.
Grupa I
Do grupy I anionów zaliczamy następujące jony: Cl-, Br-, I-, CN-, SCN
-
, Fe(CN)
6
4-
,
Fe(CN)
6
3-
, ClO
-
(jony Ag
+
wytrącają osady nierozpuszczalne w rozcieńczonym HNO3, jony
Ba
2+
nie tworzą osadów).
Grupa II
Do grupy II anionów zaliczamy następujące jony: S
2
-, NO
2
-, CH
3
COO- (jony Ag
+
wytrącają białe osady rozpuszczalne w rozcieńczonym HNO
3
, jony Ba
2+
nie tworzą osadów).
Grupa III
Do grupy III anionów zaliczamy następujące jony: BO
2
-, SO
3
2
-, CO
3
2
-, C
2
O
4
2
- C
4
H
4
O
6
2
-
(jony Ag
+
wytrącają białe osady rozpuszczalne w rozcieńczonym HNO3, jony Ba
2+
wytrącają
osady rozpuszczalne w rozcieńczonym HNO
3
Grupa IV
Do grupy IV anionów zaliczamy następujące jony: PO
4
3
-, AsO
4
-, AsO
3
3
-, S
2
O
3
2
-, CrO
4
2-
Cr
2
O
7
2-
(jony Ag
+
wytrącają barwne osady rozpuszczalne w rozcieńczonym HNO3, jony Ba
2+
wytrącają osady rozpuszczalne w rozcieńczonym HNO
3
)
Grupa V
Do grupy V anionów zaliczamy następujące jony: NO
3
-, ClO
3
-, Mno
4
- ClO
4
-, (jony Ag
+
nie wytrącają osadu, jony Ba
2+
nie wytrącają osadu).
Grupa VI
Do grupy VI anionów zaliczamy następujące jony: SO
4
2
-, F-, SiF
6
2
-, (jony Ag
+
nie
wytrącają osadu, jony Ba
2+
wytrącają osad).
Grupa VII
Do grupy VII anionów zaliczamy następujące jony: SiO
3
2
- (jony Ag
+
wytrącają żółty
osad rozpuszczalny w HNO
3
, jony Ba
2+
wytrącają biały osad rozpuszczalny w HNO
3
).
Analiza jakościowa związków organicznych
Identyfikację związków organicznych rozpoczynamy od przeprowadzenia próby spalenia,
badania rozpuszczalności, oznaczania temperatury topnienia i wrzenia. Kolejno przystępujemy
do wykrywania grup funkcyjnych charakterystycznych dla danej grupy związków.
4.1.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1. Czy znasz zasady bezpieczeństwa obowiązujące w laboratorium?
2. Czy znasz zasady postępowania w przypadku pożaru?
3. Czy umiesz pobrać próbkę do badań laboratoryjnych?
4. Czy wiesz czym zajmuje się analiza jakościowa?
5. Czy wiesz jak postępować w przypadku oparzenia chemicznego?
6. Czy potrafisz podzielić kationy i aniony na grupy analityczne?
7. Czy potrafisz wyliczyć odczynniki grupowe?
8. Czy znasz sposoby identyfikacji kationu?
9. Czy wiesz co to jest systematyczna analiza anionów i kationów?
10. Czy wiesz na czym polega Dobra Praktyka Laboratoryjna?
11. Czy potrafisz wyjaśnić pojęcia: ekstrakcja i krystalizacja?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
13
4.1.3. Ćwiczenia
Ćwiczenie 1
Zmierz gęstość alkoholu izopropylowego za pomocą areometru i piknometru.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) napełnić alkoholem cylinder o pojemności 50 ml,
4) zanurzyć w alkoholu areometr,
5) odczytać poziom zanurzenia na skali, zapisać wynik w zeszycie,
6) napełnić piknometr alkoholem izopropylowym,
7) zważyć napełniony piknometr na wadze analitycznej,
8) zważyć taką samą objętość wody,
9) obliczyć gęstość wg wzoru
m
D
20
= * 0,997 + 0,0012 [g/ml]
w
m – masa badanej substancji w g
w – masa takiej samej objętości wody w g
0,997 – gęstość wody
0,0012 – poprawka na ważenie w powietrzu
wszystkie dane dotyczą temp. otoczenia 20
o
C
10) zapisać obserwacje i efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
areometr, piknometr
−
szkło laboratoryjne,
−
waga analityczna,
−
tablice gęstości,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 2
Rozpoznaj kationy w otrzymanej mieszaninie kationów.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej
2) uważnie przeczytać treść zadania,
3) przenieść część otrzymanej analizy do probówek,
4) przeprowadzić próby wstępne w celu identyfikacji kationu NH
4
+
,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
14
5) przeprowadzić reakcje specyficzne jonów Fe
2+
, Fe
3+
, Mn
2+
,
6) dokonać rozdziału kationów na grupy analityczne dodając kolejne odczynniki grupowe,
7) po wytrąceniu grupy oddzielić osad przez wirowanie i przemyć wodą destylowaną,
8) przeprowadzić identyfikację kationu za pomocą reakcji charakterystycznych,
9) zapisać efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje,
−
wirówka,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 3
Rozpoznaj aniony w otrzymanej mieszaninie anionów.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) przenieść część otrzymanej analizy do kilku probówek,
4) dodać odczynniki grupowe AgNO
3
i BaCl
2
,
5) po otrzymaniu osadu sprawdzić jego rozpuszczalność w rozcieńczonym kwasie azotowym
(V),
6) po przeprowadzonych próbach zakwalifikować anion do jednej z grup analitycznych,
7) zidentyfikować anion w obrębie grupy za pomocą reakcji charakterystycznych,
8) przeprowadzić próbę z manganianem (VII) potasu, jeżeli anion należy do I, II, III i IV
grupy analitycznej anionów,
9) zapisać efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje,
−
wirówka,
−
zeszyty,
−
przybory do pisania
Ćwiczenie 4
Zbadaj tożsamość kwasu cytrynowego według Farmakopei Polskiej.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
15
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) sprawdzić rozpuszczalność kwasu cytrynowego w 95
o
etanolu i eterze etylowym,
4) rozpuścić 50 mg substancji w 1 ml wody i zmierzyć pH roztworu,
5) przeprowadzić ostrożne ogrzewanie substancji, dokonać obserwacji,
6) wrzucić substancję do tetrachlorku węgla i obserwować czy tonie,
7) rozpuścić 1,0 g substancji w 10 ml wody,
8) przenieść po 2 ml roztworu do dwóch probówek,
9) zobojętnić roztwór w pierwszej probówce roztworem wodorotlenku sodu (20 g/l) i dodać
2 ml roztworu chlorku wapnia (100 g/l),
10) dokonać obserwacji a następnie zagotować,
11) do probówki drugiej dodać 0,25 ml etanolowego roztworu waniliny (10 g/l), odparować
na łaźni wodnej do sucha,
12) do suchej pozostałości dodać 0,1 ml kwasu siarkowego (178 g/l), ogrzewać 15 min.,
obserwować zabarwienie,
13) dodać do analizy wody, obserwować zabarwienie,
14) następnie dodać do analizy wodorotlenku amonu (96 g/l), obserwować zabarwienie,
15) zapisać efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne,
−
pehametr,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
łaźnia wodna,
−
zeszyty,
−
przybory do pisania
Ćwiczenie 5
Zbadaj czystość azotanu srebra według Farmakopei Polskiej.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) rozpuścić 5 g substancji w 50 ml wody,
4) obserwować przezroczystość roztworu,
5) przenieść po 5 ml roztworu do 4 probówek,
6) do pierwszej probówki dodać 0,1 ml roztworu czerwieni fenolowej i obserwować
zabarwienie,
7) do drugiej probówki dodać 0,1 ml roztworu zieleni bromokrezolowej,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
16
8) do trzeciej probówki dodać 5 ml wody, 1 ml kwasu azotowego (287 g/l); 0,05 ml
roztworu manganianu (VII) potasu (0,02 mol/l),
9) do czwartej probówki dodać kroplami wodorotlenek amonowy (96 g/l) do rozpuszczenia
powstającego osadu,
10) do 10 ml roztworu wyjściowego dodać 10 ml wody, 4 ml kwasu solnego (105 g/l), silnie
wytrząsnąć, ogrzać 5 min. na łaźni wodnej, przesączyć, odparować 12 ml przesączu
i wysuszyć do stałej masy,
11) zapisać obserwacje i efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
suszarka,
−
zeszyty,
−
przybory do pisania
4.1.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) przeprowadzić reakcje charakterystyczne kationów i anionów?
¨
¨
2) przeprowadzić reakcje grupowe kationów i anionów?
¨
¨
3) identyfikować kationy i aniony?
¨
¨
4) identyfikować grupy funkcyjne?
¨
¨
5) oznaczyć gęstość piknometrem?
¨
¨
6) oznaczyć gęstość areometrem?
¨
¨
7) dokonać analizy mieszaniny kationów?
¨
¨
8) dokonać analizy mieszaniny anionów?
¨
¨
9) określić tożsamość środków leczniczych?
¨
¨
10) określić tożsamość substancji leczniczych?
¨
¨
11) określić czystość środków leczniczych?
¨
¨
12) określić czystość substancji leczniczych?
¨
¨

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
17
4.2. Analiza ilościowa
4.2.1. Materiał nauczania
Klasyczna (chemiczna) analiza ilościowa
Analiza wagowa (grawimetryczna)
Analiza wagowa polega na dokładnym określeniu masy oznaczanego składnika.
Oznaczany składnik przeprowadza się w trudno rozpuszczalny osad.
Do analizy wagowej należą metody, w których oznaczany składnik usuwa się z odważonej
próbki przez ogrzewanie i oblicza jego zawartość z ubytku masy.
Wytrącanie osadu
Osad w analizie wagowej musi być trudno rozpuszczalny, mieć ściśle określony skład
chemiczny, czysty i łatwy do sączenia. Strącenie osadu wykonuje się w zlewkach. Do
rozcieńczonej, podgrzanej analizy dodaje się rozcieńczony roztwór odczynnika z pipety
i miesza bagietką. Odczynnik strącający dodajemy w nadmiarze celem całkowitego wytrącenia
osadu.
Oddzielanie osadu
Osad należy oddzielić od roztworu macierzystego. Oddzielanie osadu od roztworu
macierzystego przeprowadza się poprzez sączenie na sączkach z bibuły lub lejki do sączenia
pod zmniejszonym ciśnieniem. Jeżeli osad wymaga prażenia, oddzielamy go na sączkach
z bibuły. Do sączenia, w zależności od tego czy osad jest grubo czy drobnokrystaliczny,
stosujemy sączki gęste, średnie i rzadkie. Wielkość sączka dobieramy do masy osadu. Sączenie
przeprowadza się przez lejek analityczny, w którym umieszcza się odpowiednio złożony
sączek z bibuły. Lejek z sączkiem umieszcza się w statywie. Na początku ostrożnie zlewamy
roztwór znad osadu po bagietce. Następnie przenosimy osad przy pomocy tryskawki. Aby
pozbawić osad zanieczyszczeń należy przemyć go wodą destylowaną. Najczęściej
przemywanie przeprowadza się na sączku. Osad zebrany na sączku przemywa się roztworem
przemywającym z tryskawki. Przemywanie przez dekantację stosujemy wtedy, gdy cały osad
opadł na dno. Przemywanie przeprowadza się w zlewce. Po przeniesieniu roztworu osad
spłukuje się ze ścianek zlewki i ponownie pozostawia do opadnięcia. Klarowną ciecz przenosi
się na sączek, a osad ponownie przemywa. Czynność powtarzamy 2-3 razy.
Sączenie pod zmniejszonym ciśnieniem przeprowadza się w tyglach szklanych z dnem
porowatym, gdy osad przed ważeniem nie wymaga suszenia.
Suszenie i prażenie osadu
Odsączony osad suszy się w suszarkach w odpowiedniej temperaturze. Suszy się
w suszarkach elektrycznych przez 1–2 godziny. Po wysuszeniu osad umieszcza się
w eksykatorze (naczynie, w którym znajdują się substancje pochłaniające wodę), gdzie
substancja stygnie, nie pochłania wilgoci i dwutlenku węgla.
Sączki z osadami wymagającymi prażenia umieszcza się w tyglu porcelanowym, spala
w palniku gazowym, a następnie praży osad w piecu elektrycznym do stałej masy.
Analiza objętościowa (miareczkowa)
Polega na tym, że do roztworu zawierającego oznaczaną substancję wprowadza się
niewielkimi porcjami – „miareczkami” – równoważną ilości odczynnika w postaci roztworu
mianowanego tj. roztworu o dokładnie znanym stężeniu. Miano roztworu ustala się na
substancję wzorcową. Zawartość oznaczanej substancji (w gramach) oblicza się na podstawie
zmierzonej objętości zużytego roztworu mianowanego - „titrantu”. Celem określenia
momentu, w którym została doprowadzona ilość odczynnika równoważna ilości składnika
oznaczanego (tzw. PR – punktu równoważności), stosujemy odpowiednie wskaźniki
(indykatory).

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
18
Wskaźnikiem jest najczęściej substancja zmieniająca barwę w chwili zakończenia reakcji
między roztworem mianowanym a roztworem miareczkowanym, rolę wskaźnika może również
pełnić odczynnik miareczkujący np. manganian (VII) potasu. Wskaźniki można podzielić na
dwubarwne (oranż metylowy) i jednobarwne (fenoloftaleina). Wskaźniki zmieniają barwy
odpowiednio do zmian zachodzących w roztworach: wskaźniki alkacymetryczne w zależności
od zmian pH, redoks w zależności od zmian potencjału redoks.
Zamiast wzrokowego określenia punktu równoważności można dokonać indykacji
punktu równoważności miareczkowania (PR) metodami instrumentalnymi (zmiany właściwości
elektrycznych lub optycznych).
Moment w którym wskaźnik zmienia barwę nazywa się punktem końcowym
miareczkowania (PK). Dążymy do tego aby punkt równoważności pokrywał się z punktem
końcowym miareczkowania.
Typy metod miareczkowych:
−
miareczkowanie bezpośrednie – polega na tym, że oznaczany składnik miareczkuje się
roztworem odczynnika,
−
odwrotne – dodajemy określoną ilość titrantu w nadmiarze, czekamy aby zaszła reakcja,
a następnie odmiareczkowujemy nie zużytą część titrantu innym odpowiednio dobranym
roztworem mianowanym,
−
podstawieniowe – polega na tym, że miareczkuje się nie oznaczany składnik lecz jego
podstawnik tzn. substancję, która jest produktem reakcji składnika oznaczanego
z jakimkolwiek odczynnikiem.
Klasyfikacja metod miareczkowych według typu reakcji zachodzącej podczas
miareczkowania.
Alkacymetria
Metody miareczkowe oparte na reakcji kwas – zasada, stosuje się je powszechnie do
oznaczania kwasów i zasad nieorganicznych i organicznych.
Kompleksometria
Podstawą kompleksometrii jest reakcja tworzenia trwałego, trudno dysocjującego,
rozpuszczalnego związku kompleksowego. Najczęściej stosuje się edetynian sodu,
powszechnie oznacza się go skrótem Na
2
EDTA. Metody kompleksometryczne stosuje się do
oznaczania metali. Służą między innymi do oznaczania bizmutu w preparatach
farmaceutycznych.
Redoksymetria
Dział analizy miareczkowej oparty na reakcjach utleniania i redukcji. Należą do niego
między innymi metody manganometryczne i jodometryczne.
Manganometria
Dział analizy miareczkowej w której roztworem mianowanym jest manganian (VII)
potasu, za pomocą którego oznaczamy reduktory. Przebieg redukcji manganianu (VII) potasu
zależy od odczynu środowiska. Funkcję wskaźnika pełni manganian (VII) potasu.
Jodometria
Dział redoksymetrii oparty na odwracalnej reakcji:
J
2
+ 2e ↔ 2J
-
(potencjał normalny E
o
=0,535 V)
Oznaczenia jodometryczne można podzielić na dwie grupy: substancje, których potencjały
utleniające są niższe od potencjału układu J
2
/2J
-
(reduktory) miareczkujemy bezpośrednio
mianowanym roztworem jodu. Substancje o wyższym potencjale utleniającym niż potencjału
układu J
2
/2J
-
(utleniacze) utleniają jony jodkowe do wolnego jodu, który odmiareczkowuje się
mianowanym roztworem tiosiarczanu sodu.
2S
2
O
3
2-
+ J
2
→ S
4
O
6
2-
+ 2J
-

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
19
Wskaźnikiem w oznaczeniach jodometrycznych jest skrobia, która tworzy z jodem
połączenie o szafirowym zabarwieniu.
Jod słabo rozpuszcza się w wodzie, dobrze rozpuszcza się w stężonym roztworze jodku
potasu. Roztwory jodu przechowuje się w butelkach z ciemnego szkła. Miano roztworu jodu
nastawia się na tlenek arsenu (III).
Miareczkowe metody wytrąceniowe.
Argentometria
Analiza wytrąceniowa polega na reakcji tworzenia się trudno rozpuszczalnego osadu
o ściśle określonym składzie chemicznym. W celu zauważenia końca miareczkowania stosuje
się odpowiednie wskaźniki, które są właściwe dla danego oznaczania.
Instrumentalne metody analizy
Metody instrumentalne to metody analizy chemicznej oparte na wykorzystaniu zjawisk
fizycznych lub fizykochemicznych. Do wykonania analiz konieczna jest odpowiednia aparatura
fizyczna lub fizykochemiczna. Metody instrumentalne charakteryzują się dużą czułością w
związku z tym dużą wykrywalnością i oznaczalnością. Ich stosowanie pozwala uzyskać
wyniki w krótkim czasie. Metody instrumentalne można podzielić na:
−
metody optyczne i metody spektroskopowe,
−
metody elektroanalityczne,
−
metody rozdzielcze,
−
metody radiometryczne.
Metody optyczne i spektroskopowe to metody analityczne wykorzystujące światło i jego
oddziaływanie na materię. Zalicza się do nich między innymi:
−
refraktometrię,
−
interferometrię,
−
polarymetrię,
−
nefelometrię,
−
turbidymetrię,
−
spektrofotometrię UV (zakres nadfioletu),
−
spektrofotometrię VIS (zakres światła widzialnego),
−
spektrofotometrię IR (zakres podczerwieni).
Metody elektroanalityczne. Wykorzystują zjawiska związane z przepływem prądu
elektrycznego przez roztwory elektrolitów lub związane z reakcjami zachodzącymi na
elektrodach, które są zanurzone w takich roztworach. Do metod elektroanalitycznych należą:
−
metody potencjometryczne,
−
metody elektrolityczne,
−
metody kulometryczne,
−
metody oparte na pomiarze przewodnictwa lub pojemności elektrycznej roztworu,
−
metody woltamperometryczne.
Metody rozdzielcze. Do najczęściej stosowanych instrumentalnych metod rozdzielczych
należą:
−
chromatografia,
−
ekstrakcja,
−
elektroforeza.
Metody radiometryczne. Polegają na pomiarze promieniowania jądrowego emitowanego
przez naturalne i sztuczne izotopy promieniotwórcze.
Spektrofotometria w świetle widzialnym tradycyjnie nazywana kolorymetrią, mimo, że
klasyczne oznaczenie kolorymetryczne polega na wizualnej ocenie intensywności barw
roztworów. W nowoczesnej kolorymetrii mierzy się absorbancję roztworu. Oznaczanie
kolorymetryczne składa się z dwóch etapów. Pierwszy – otrzymanie barwnego połączenia

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
20
(etap chemiczny), drugi – pomiar absorpcji światła przez roztwór (etap fizyczny). Pomiar
absorbancji możemy wykonać za pomocą różnych przyrządów: kolorymetru, fotokolorymetru
i spektofotometru.
Chromatografia
Chromatografia jest techniką rozdzielania mieszanin na poszczególne składniki pomiędzy
dwie nie mieszające się fazy z których jedna jest fazą nieruchomą (stacjonarną), druga jest fazą
ruchomą. Fazą nieruchomą może być ciało stałe lub ciecz, a fazą ruchomą gaz, ciecz.
Chromatografia gazowa (GC)
Chromatografia gazowa to wszystkie metody chromatograficzne, w których fazą ruchomą
jest gaz. Fazą nieruchomą może być ciało stałe lub ciecz. W związku z tym wyróżnia się
chromatografię w układzie gaz-ciecz (podziałowa) i gaz-ciało stałe (adsorpcyjna).
Chromatografia cieczowa to wszystkie metody chromatograficzne, w których fazą
ruchomą jest ciecz. Obecnie szerokie zastosowanie w analizie jakościowej i ilościowej znajduje
chromatografia kolumnowa z użyciem eluentu (faza ruchoma) pod wysokim ciśnieniem tzw.
HPLC chromatografia cieczowa wysokosprawna.
Chromatografia cienkowarstwowa (TLC)
Chromatografia cienkowarstwowa (TLC) obejmuje procesy chromatograficzne, w których
fazę nieruchomą stanowi cienka warstwa adsorbentu, a fazę ruchomą rozpuszczalnik.
Adsorbent o grubości ok. 0,2 mm umieszczony jest na płytkach szklanych, plastikowych, folii
aluminiowej. Na płytkę chromatograficzną nanosi się substancję badaną i substancję wzorcową
za pomocą mikropipety w odległości ok. 20 mm od krawędzi płytki. Po wyschnięciu płytki
umieszcza się ją w komorze chromatograficznej z faza ruchomą. Po przejściu fazy ruchomej
na wysokość ok. 15 cm wyjmujemy chromatogram i suszymy. W przypadku substancji
barwnych lub po wywołaniu substancji odczynnikiem chemicznym (odczynnik reaguje z
substancją tworząc z nią barwny produkt) ustalamy miejsce położenia plamki wizualnie w
świetle widzialnym i UV, w przypadku substancji bezbarwnych stosujemy odpowiedni
odczynnik wywołujący. Chromatografię cienkowarstwową używa się do badania tożsamości
środków farmaceutycznych oraz badania zanieczyszczeń w środkach farmaceutycznych.
Stosunek wielkości przesunięcia badanej substancji do wielkości przesunięcia się
rozpuszczalnika dla danego związku i dla danego rozpuszczalnika ma wartość stałą, zależną od
współczynnika podziału – oznaczamy go symbolem R
f
.
Znając wartość R
f
substancji można wykryć ich obecność na podstawie położenia plamek.
Ocena jakości produktów leczniczych
Analiza środków farmaceutycznych polega na identyfikacji (badaniu tożsamości), badaniu
czystości i określaniu zawartości związków stosowanych w terapii.
Oceny jakości środków leczniczych dokonuje się zgodnie z obowiązującymi normami.
Zbiór podstawowych wymagań (norm) odnoszących się do składu i jakości środków
farmaceutycznych oraz metod badania surowców farmaceutycznych i leków znajduje się
w Farmakopei Polskiej. Zawiera ona również podstawowe wymagania dla niektórych
artykułów sanitarnych i opakowań środków farmaceutycznych. Związek chemiczny przed
wprowadzeniem do lecznictwa musi odpowiadać, pod względem czystości, ściśle określonym
wymaganiom. Wskazówki dotyczące określania zanieczyszczeń zawarte są w odpowiednich
monografiach substancji. Oceny zanieczyszczeń dokonuje się w porównaniu z roztworami
wzorcowymi lub próbami kontrolnymi. Preparat uznaje się za czysty jeśli zawartość
zanieczyszczeń nie przekracza zawartości związków w próbie wzorcowej. Badanie czystości
środków farmaceutycznych można również przeprowadzić posługując się metodami TLC
i HPLC.
Badanie tożsamości polega na identyfikacji pierwiastków, kationów, grup funkcyjnych.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
21
Do badania zawartości wykorzystuje się metody analizy ilościowej, klasyczne
i instrumentalne. W badaniach ilościowych (zawartości) wykonujemy dwie równoległe próby a
wynik jest średnią arytmetyczną.
Ocena i interpretacja wyników analizy chemicznej
Czułość metody określa najmniejszą ilość substancji lub jej stężenie jakie można zmierzyć
daną metodą analityczną.
Dokładność metody oznacza stopień zgodności z wartością rzeczywistą.
Precyzja metody to zgodność uzyskanych wyników między sobą.
Szacowanie niepewności pomiarów i metody obliczania niepewności pomiarowych.
Niepewność pomiaru jest miarą rozrzutu wyników powtarzanych pomiarów badań.
Zapisując wynik analizy x należy napisać jednostkę podanej wartości i opatrzyć przedziałem
niepewności
∆
x:
x
±
∆
x
Z powodu występowania przypadkowych niepewności (błędów) pomiarowych
powtarzanie pomiaru daje różne wyniki. Otrzymane wyniki rozkładają się wokół wartości
rzeczywistej a ich rozrzut zależy od dokładności prowadzonych badań.
Występowanie niepewności przypadkowych podlega pewnym prawidłowościom. Małe
odchylenia od wartości rzeczywistej występują częściej niż odchylenia duże. Oszacowanie
wartości niepewności przypadkowych można wykonać korzystając z metod statystyki
matematycznej.
Oprócz statystycznie rozłożonych niepewności przypadkowych podczas badań mogą też
wystąpić błędy grube i błędy systematyczne.
Błędy grube powstają w wyniku nieprawidłowego odczytu lub pomyłki osoby
wykonującej analizę. Błędy grube powodują, że wyniki różnią się znacznie od pozostałych
wyników i można je łatwo zauważyć.
Błędy systematyczne występują stale podczas wykonywania oznaczenia daną metodą.
Ocena niepewności przypadkowych
Wielokrotne powtarzanie analizy pozwala otrzymać serię wyników x
1,
x
2,
x
3
...... x
n
gdzie n – liczba wykonanych pomiarów. Rzeczywista wartość analizy jest zbliżona do średniej
arytmetycznej otrzymanych wyników x
x
n
x
i
i
n
=
=
∑
1
1
Odchylenie standardowe jest miarą niepewności pojedynczego wyniku pomiaru.
(
)
σ =
−
−
=
∑
1
1
2
1
n
x
x
i
i
n
Miarą niepewności średniej arytmetycznej x jest:
(
)
σ
σ
x
i
i
n
n
n n
x
x
=
=
−
−
=
∑
1
1
2
1
(
)
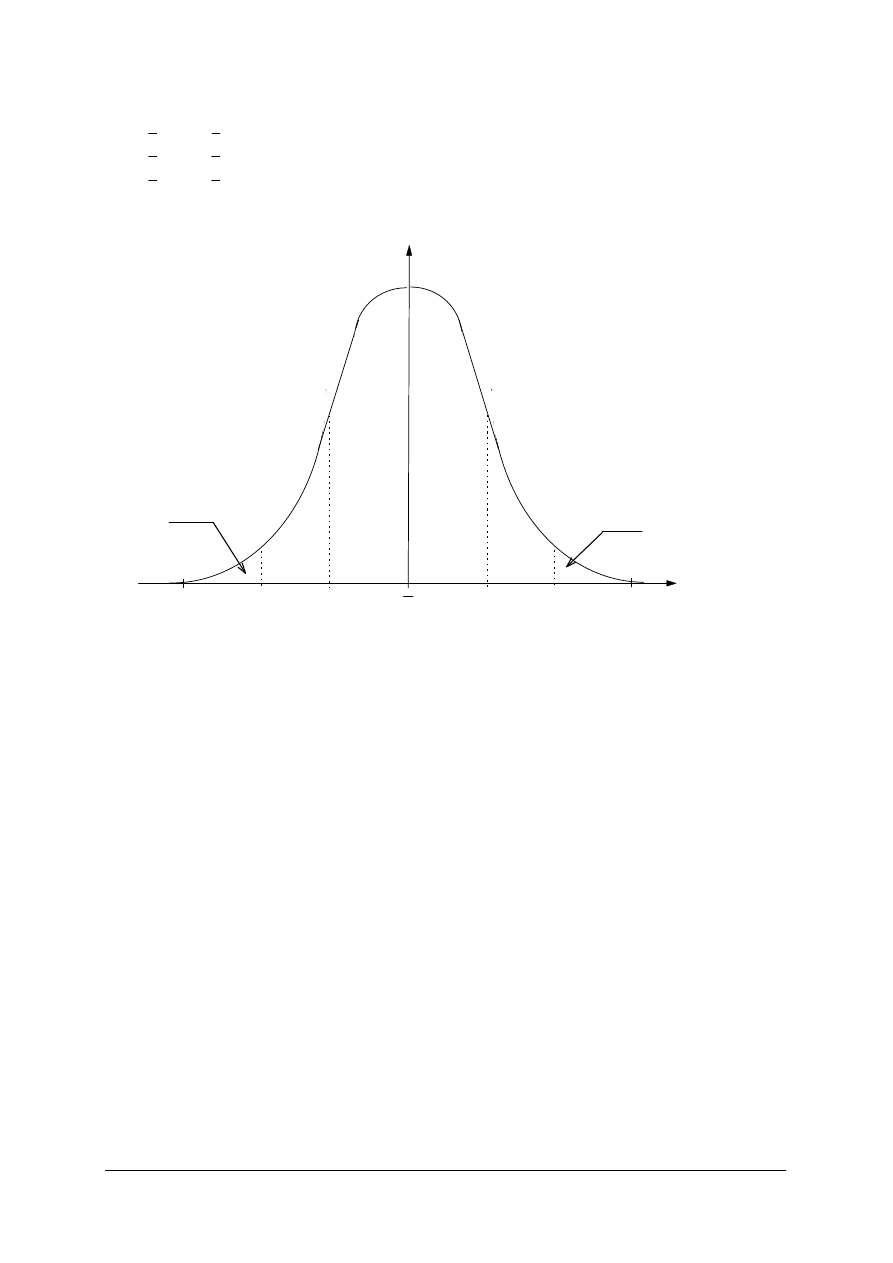
Rozkład prawdopodobieństwa Gaussa daje możliwość obliczenia prawdopodobieństwa, że
dowolny wynik pomiaru znajduje się w zadanym przedziale wartości x.
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
22
W przedziale:
(
x
-
σ
,
x
+
σ
) mieści się 68,27% wyników,
(
x
- 2
σ
,
x
+ 2
σ
) mieści się 95,45% wyników,
(
x
- 3
σ
,
x
+ 3
σ
) mieści się 99,73% wyników.
f(x)
x
x
σ
2
σ
3
σ
σ
2
σ
3
σ
2,14%
2,14%
13,60%
13,60%
34,13%
34,13%
Rys 2. Krzywa Gaussa rozkładu niepewności przypadkowych. Wartości liczbowe określają procentowe
prawdopodobieństwa pojawienia się wyniku pomiaru w wyznaczonych przedziałach.
4.2.2. Pytania sprawdzające
Odpowiadając na pytania, sprawdzisz, czy jesteś przygotowany do wykonania ćwiczeń.
1. Czy wiesz czym zajmuje się analiza ilościowa?
2. Czy znasz sposoby oddzielenia osadu ?
3. Czy potrafisz wyliczyć metody analizy miareczkowej?
4. Czy potrafisz scharakteryzować poszczególne sposoby przeprowadzania miareczkowania?
5. Czy wiesz jaki rodzaj reakcji zachodzi podczas miareczkowania kompleksometrycznego?
6. Czy wiesz na czym polegają metody manganometryczne?
7. Czy potrafisz scharakteryzować metody optyczne?
8. Na czym polega chromatografia cienkowarstwowa?
9. Co jest roztworem mianowanym w jodometrii?
10. Co to jest kolorymetria?

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
23
4.2.3. Ćwiczenia
Ćwiczenie 1
Oznacz alkacymetrycznie kwas cytrynowy.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej
2) uważnie przeczytać treść zadania,
3) odważyć na wadze analitycznej dokładnie ok. 0,50 g substancji,
4) rozpuścić substancję w 50 ml wody w kolbie stożkowej o pojemności 250 ml,
5) dodać 0,05 roztworu ml fenoloftaleiny,
6) miareczkować roztworem 0,5 mol/l wodorotlenku sodu,
7) obliczyć zawartość kwasu cytrynowego wykorzystując do obliczeń informację, że 1 ml
roztworu wodorotlenku sodu 0,5 mol/l odpowiada 32,02 mg bezwodnego kwasu
cytrynowego,
8) zapisać efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne: naczynko wagowe, zlewka, kolba stożkowa, biureta, pipety,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 2
Oznacz metodą jodometryczną kwas askorbowy.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) odważyć na wadze analitycznej dokładnie ok. 0,10 g substancji,
4) rozpuśćić substancję w 20 ml świeżo przegotowanej i ochłodzonej wody kolbie stożkowej
o pojemności 250 ml,
5) dodać do analizy 10 ml kwasu siarkowego o stężeniu 178 g/l,
6) dodać 1 ml skrobi (100g/l),
7) miareczkować roztworem jodu (0,05 mol/l) do pojawienia się trwałego niebieskiego
zabarwienia roztworu,
8) obliczyć zawartość kwasu askorbowego wiedząc, że 1 ml jodu 0,05 mol/l odpowiada
8,81 mg kwasu askorbowego,
9) zapisać wyniki pracy w zeszycie.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
24
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne: naczynko wagowe, zlewka, kolba stożkowa, pipety,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 3
Oznacz stratę masy po suszeniu w mieszance ziołowej.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) rozdrobnić i przesiać mieszankę ziołową przez sito o wielkości oczek 0,5 mm,
4) wysuszyć naczynko wagowe w suszarce przez 2 h w temperaturze 105
o
C,
5) ostudzić naczynko w eksykatorze,
6) zważyć naczynko z dokładnością do czwartego miejsca po przecinku,
7) powtórzyć czynności suszenia i ważenia do uzyskania stałej masy naczynka wagowego,
wyniki nie powinny się różnić między sobą o więcej niż 0,0002 g,
8) odważyć dokładnie ok. 2 g mieszanki ziołowej i suszyć w temperaturze 105
o
C przez 2 h,
ochłodzić w eksykatorze,
9) zważyć naczynko z mieszanką (i ponownie suszyć do stałej masy),
10) obliczyć zawartość wody,
11) zapisać efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia
−
literatura fachowa,
−
zestaw sit,
−
szkło laboratoryjne: naczynko wagowe,
−
waga analityczna,
−
eksykator,
−
suszarka,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 4
Oznacz glukozę w syropie przeciwkaszlowym.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
25
2) uważnie przeczytać treść zadania,
3) uzupełnić otrzymany roztwór do kreski, w kolbie miarowej o pojemności 100 ml, wodą
z dodatkiem 2-3 kropli wodorotlenku amonu (96 g/l),
4) napełnić roztworem rurkę polarymetryczną o długości 1 dm,
5) wykonać pomiar skręcalności optycznej w temperaturze 20
o
C w świetle sodowym,
6) obliczyć procentową zawartość glukozy posługując się wzorem:
α *100
c=
l*[ α]
D
20
c – stężenie roztworu
α – odczytany kąt skręcenia płaszczyzny polaryzacji (w stopniach)
α
D
20
– skręcalność dla linii D sodu (dcm)
l – długość rurki polarymetrycznej
7) zapisać obserwacje, wyniki i obliczenia w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
polarymetr,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje
−
waga analityczna,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 5
Oznacz żelazo metodą krzywej wzorcowej za pomocą kolorymetru.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) odmierzyć z biurety do kolb miarowych o pojemności 100 ml następujące objętości
roztworu wzorcowego żelaza (III) wg podanych objętości:
−
kolba 1 – 2 ml
−
kolba 2 – 4 ml
−
kolba 3 – 6 ml
−
kolba 4 – 8 ml
−
kolba 5 – x – badana analiza
4) dodać do każdej kolby 1 ml roztworu 6 mol/l kwasu azotowego,
5) następnie dodać do każdej z kolb po 5 ml roztworu 10% tiocyjanianu potasu,
6) uzupełnić roztwory w kolbach wodą do objętości 100 ml i dokładnie wymieszać,
7) zmierzyć po 5 minutach absorbancję stosując wodę jako odnośnik,
8) wyniki zestawić w tabeli,
9) wykreślić na podstawie uzyskanych wyników krzywą wzorcową A=f(C),
10) odczytać z krzywej wzorcowej stężenie roztworu badanego,
11) zapisać efekty pracy w zeszycie.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
26
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
szkło laboratoryjne: biureta, naczynko wagowe, zlewka, kolba miarowa, pipety,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
kolorymetr,
−
zeszyty,
−
przybory do pisania.
Ćwiczenie 6
Dokonaj
badania
czystości
hydroksybenzoesanu
etylu
metodą
chromatografii
cienkowarstwowej według Farmakopei Polskiej.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) sporządzić roztwór A substancji badanej w acetonie o stężeniu 50 mg/ml,
4) sporządzić roztwór B substancji badanej w acetonie o stężeniu 0,15 mg/ml,
5) nanieść po 5 μl roztworu A i B na płytkę chromatograficzną,
6) umieścić płytkę w komorze chromatograficznej zawierającej fazę ruchomą: chlorek
metylenu – octan etylu – kwas siarkowy (1,762 kg/l) w stosunku 90: 10 : 0,5,
7) rozwinąć chromatogram na wysokość 10 cm,
8) wyjąć płytkę i wysuszyć w temperaturze pokojowej,
9) obejrzeć chromatogram w świetle nadfioletowym (254 nm), jeżeli na chromatogramie
roztworu A wystąpią inne plamy poza plamą główną, nie mogą być większe ani
intensywniejsze niż plama główna na chromatogramie roztworu B,
10) zapisać obserwacje i efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
płytki chromatograficzne GF
254
,
−
lampa kwarcowa laboratoryjna,
−
komora chromatograficzna,
−
mikropipety,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
zeszyty,
−
przybory do pisania.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
27
Ćwiczenie 7
Oznacz chloramfenikol metodą spektrofotometryczną w zakresie światła nadfioletowego.
Sposób wykonania ćwiczenia
Aby wykonać ćwiczenie, powinieneś:
1) przeczytać materiał nauczania z poradnika dla ucznia i poszerzyć wiadomości z literatury
uzupełniającej,
2) uważnie przeczytać treść zadania,
3) odważyć dokładnie ok. 0,10 g substancji,
4) rozpuścić substancję ogrzewając w 80 ml wody w zlewce,
5) po ochłodzeniu roztwór przenieść ilościowo do kolby miarowej,
6) popłukać zlewkę, przenieść popłuczyny do kolby i uzupełnić do kreski,
7) odmierzyć 2,0 ml tego roztworu i rozcieńczyć do 100 ml wody,
8) zmierzyć absorbancję rozcieńczonego roztworu przy długości fali 278 nm,
9) obliczyć zawartość chloramfenikolu przyjmując absorpcję właściwą α
1%
1 cm
= 297
wykorzystując wzór:
A·b
c=
a
1cm
1%
·l·d
a
1cm
1%
– wartość absorpcji właściwej badanego związku
A – zmierzona absorbancja
b – współczynnik rozcieńczenia
l – grubość warstwy w cm
d – odważka w g
10) zapisać obserwacje i efekty pracy w zeszycie.
Wyposażenie stanowiska pracy:
−
poradnik dla ucznia,
−
literatura fachowa,
−
spektrofotometr,
−
szkło laboratoryjne,
−
odczynniki chemiczne, substancje,
−
waga analityczna,
−
zeszyty,
−
przybory do pisania.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
28
4.2.4. Sprawdzian postępów
Czy potrafisz:
Tak
Nie
1) określić co to jest Punkt Równoważnikowy?
¨
¨
2) jaka jest zasada wytrącania osadów w analizie wagowej?
¨
¨
3) oznaczyć kwas cytrynowy metodą alkacymetryczną?
¨
¨
4) oznaczyć stratę masy po suszeniu?
¨
¨
5) odmierzyć ciecz za pomocą naczyń miarowych?
¨
¨
6) sporządzić roztwór o określonym stężeniu?
¨
¨
7) przygotować roztwór z fiksanali?
¨
¨
8) wykonać obliczenia do ćwiczeń?
¨
¨
9) oznaczyć stężenie glukozy metodą polarymetryczną?
¨
¨
10) przeprowadzić identyfikację metodami chromatograficznymi?
¨
¨
11) wykonać krzywą wzorcową w oparciu o przeprowadzone pomiary
w kolorymetrii ?
¨
¨
12) oznaczyć spektrofotometrycznie substancje lecznicze?
¨
¨
13) nastawić miano roztworu?
¨
¨
14) oznaczyć kwas askorbowy metodą jodometryczną?
¨
¨

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
29
5. SPRAWDZIAN OSIĄGNIĘĆ
INSTRUKCJA DLA UCZNIA
1. Przeczytaj uważnie instrukcję.
2. Podpisz imieniem i nazwiskiem kartę odpowiedzi.
3. Zapoznaj się z zestawem zadań testowych.
4. Test zawiera 20 zadań dotyczących jednostki „Wykonywanie analizy jakościowej
i ilościowej produktów leczniczych”
5. Zadania są zamknięte wielokrotnego wyboru.
6. Tylko jedna odpowiedź jest prawidłowa.
7. Udzielaj odpowiedzi na załączonej karcie odpowiedzi.
8. Prawidłowe odpowiedzi zaznacz w odpowiedniej rubryce znakiem X.
9. Na rozwiązanie testu masz 45 minut.
10. Kolejność rozwiązań jest dowolna.
11. W przypadku pomyłki błędną odpowiedź zakreśl kółkiem, a następnie prawidłową
zaznacz X.
12. Uważnie czytaj zestaw zadań testowych.
13. Jeżeli będziesz miał problem z udzieleniem odpowiedzi na jakieś pytanie, zostaw je,
przejdź do następnych a do niego wrócisz na końcu.
14. Po zakończeniu rozwiązywania zadań, sprawdź w karcie odpowiedzi, czy dla wszystkich
zadań zaznaczyłeś odpowiedź.
15. Rozwiązuj zadania samodzielnie.
Powodzenia!

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
30
ZESTAW ZADAŃ TESTOWYCH
1. Próbka ogólna to
a) próbka jednostkowa pobrana z partii materiału.
b) próbka pierwotna pobrana z partii materiału.
c) połączone próbki pierwotne pobrane z partii materiału.
d) próbka przeznaczona do badań.
2. Pomiar skręcalności właściwej wykonujemy
a) refraktometrem.
b) polarymetrem.
c) spektofotometrem z przystawką TR.
d) spektofotometrem z przystawką TK.
3. Odczynnikiem grupowym kationów III grupy według podziału Bunsena jest
a) amid kwasu tiooctowego wobec HCl.
b) amid kwasu tiooctowego wobec NH
3
* H
2
O.
c) amid kwasu tiooctowego wobec NH
4
Cl.
d) amid kwasu tiooctowego wobec NH
3
*H
2
O,NH
4
Cl.
4. Aniony grupy II z roztworem AgNO
3
a) nie tworzą osadu.
b) wytrącają się w postaci białego osadu.
c) wytrącają się w postaci osadu rozpuszczalnego w HNO
3.
d) wytrącają się w postaci osadu nierozpuszczalnego w HNO
3.
5. W przypadku poparzenia ręki stężonym kwasem solnym w pierwszej kolejności należy
ranę
a) przemyć wodą utlenioną.
b) przemyć roztworem amoniaku.
c) przemyć dużą ilością wody.
d) przemyć 1 % roztworem NaHCO
3.
6. Miareczkowanie podstawieniowe polega na tym, że
a) miareczkujemy substancję będącą produktem reakcji składnika oznaczanego
z jakimkolwiek odczynnikiem.
b) miareczkujemy bezpośrednio mianowanym roztworem.
c) dodajemy
do
analizy
roztworu
mianowanego
w
nadmiarze,
nadmiar
odmiareczkowujemy.
d) miareczkujemy roztworem pomocniczym.
7. W metodach miareczkowych przy doborze wskaźnika wskazane jest aby
a) PK=PR.
b) PK>PR.
c) PK<PR.
d) PKP≠R.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
31
8. Do metod optycznych, które wykorzystują światło i jego oddziaływanie na materię nie
należy
a) spektrofotometria.
b) polarografia.
c) refraktometria.
d) polarymetria.
9. Zawartość kwasu askorbowego oznaczamy
a) metodą jodometryczną.
b) metodą manganometryczną.
c) metodą alkacymetryczna.
d) metodą argentometryczną.
10. Jodometria to dział analizy miareczkowej, w którym wskaźnikiem jest
a) roztwór jodu.
b) roztwór KMnO
4
.
c) skrobia.
d) wskaźniki redoks.
11. Stratę masy po suszeniu substancji sproszkowanych przeprowadzamy
a) w temperaturze 50-60
o
C.
b) w temperaturze 60-90
o
C.
c) w temperaturze 100-105
o
C.
d) w temperaturze dobranej do rodzaju substancji.
12. Roztwór mianowany to
a) roztwór odczynnika chemicznego.
b) roztwór do badania tożsamości.
c) roztwór do badania czystości.
d) roztwór o dokładnie znanym stężeniu.
13. Do wykonania miareczkowania potrzebny jest następujący zestaw szkła
a) biureta, kolba stożkowa, pipeta.
b) biureta, kolba miarowa, pipeta.
c) biureta, kolba miarowa, cylinder.
d) biureta, kolba miarowa, pipeta jednomiarowa.
14. Badanie tożsamości polega na
a) identyfikacji.
b) określeniu zawartości.
c) określeniu czystości.
d) wszystkie odpowiedzi są poprawne.
15. Sączenie osadów drobnoziarnistych przeznaczonych do prażenia przeprowadzamy na
a) sączkach gęstych.
b) sączkach średnich.
c) sączkach rzadkich.
d) saczkach z dnem porowatym.

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
32
16. Metodą rozdzielania i oczyszczania wykorzystującą prężność pary jest
a) ekstrakcja.
b) destylacja.
c) krystalizacja.
d) sublimacja.
17. W laboratorium najwłaściwszymi metodami badań są metody
a) precyzyjne i dokładne.
b) precyzyjne i mało dokładne.
c) mało precyzyjne i dokładne.
d) mało precyzyjne i mało dokładne.
18. Roztwór azotanu srebra używamy do określenia tożsamości:
a) jonów azotanowych (V) [NO
3
-
].
b) jonów chlorkowych [Cl
-
].
c) jonów wapnia [Ca
2+
].
d) jonów cynku [Zn
2+
].
19. Do badania tożsamości syropu zwykłego używamy
a) odczynnika Fehlinga.
b) roztworu Lugola.
c) odczynnika Nesslera.
d) odczynnika Schiffa.
20. Ile wodorotlenku sodu należy odważyć aby sporządzić 1 l roztworu o stężeniu
0,05 mol/l?
a) 0,2 g.
b) 2 g.
c) 0,5 g.
d) 5 g.
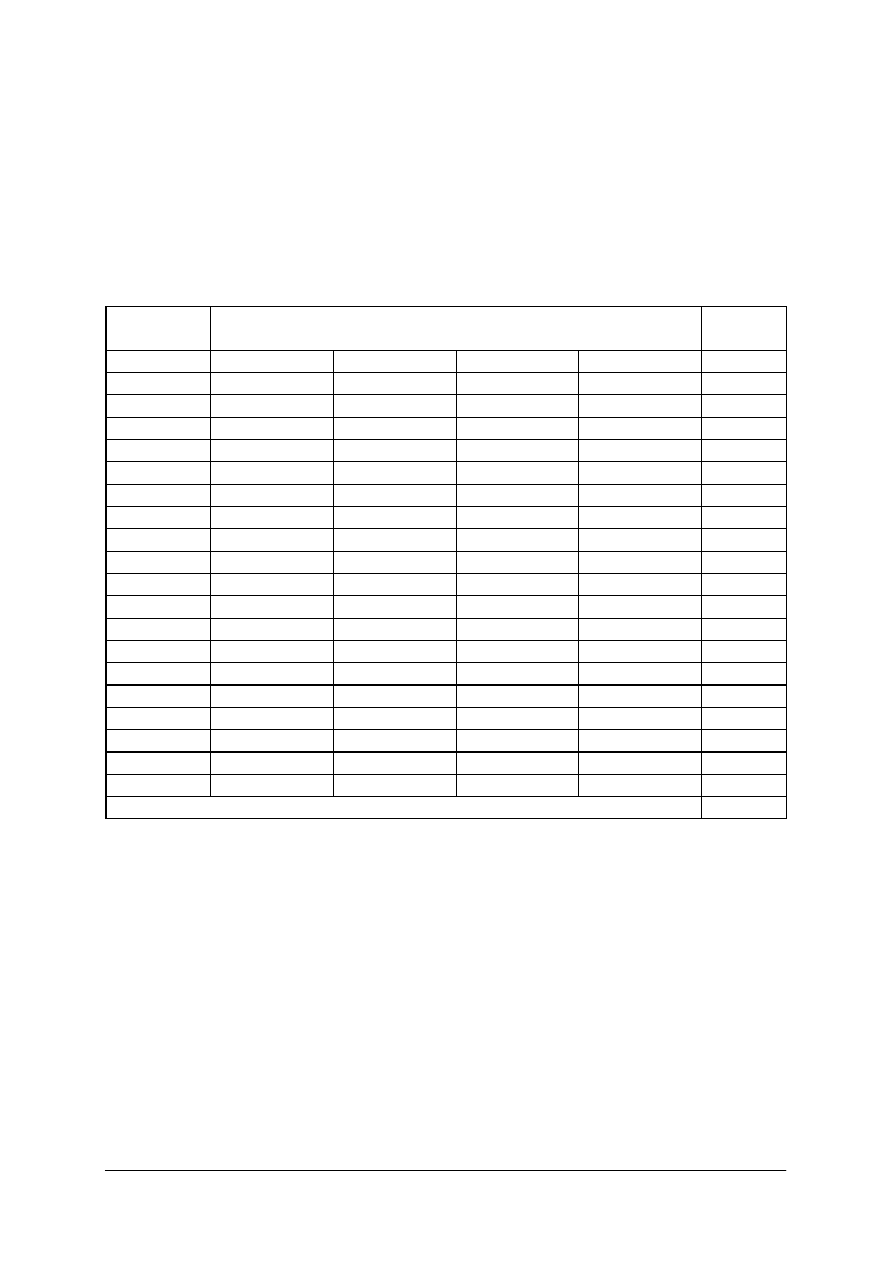
„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
33
KARTA ODPOWIEDZI
Imię i nazwisko..........................................................................................
Wykonywanie analizy jakościowej i ilościowej produktów leczniczych
Zakreśl poprawną odpowiedź
.
Nr
zadania
Odpowiedź
Punkty
1
a
b
c
d
2
a
b
c
d
3
a
b
c
d
4
a
b
c
d
5
a
b
c
d
6
a
b
c
d
7
a
b
c
d
8
a
b
c
d
9
a
b
c
d
10
a
b
c
d
11
a
b
c
d
12
a
b
c
d
13
a
b
c
d
14
a
b
c
d
15
a
b
c
d
16
a
b
c
d
17
a
b
c
d
18
a
b
c
d
19
a
b
c
d
20
a
b
c
d
Razem:

„Projekt współfinansowany ze środków Europejskiego Funduszu Społecznego”
34
6. LITERATURA
1. Cygański A.: Chemiczne metody analizy ilościowej. WNT, Warszawa 1999
2. Cygański A.: Metody spektroskopowe w analizie chemicznej. WNT, Warszawa 1997
3. Kocjan R. i wsp.: Chemia analityczna Tom I i II. PZWL, Warszawa 2001
4. Kohlmunzer S.: Farmakognozja. PZWL, Warszawa 2003
5. Lipiec T., Szmal Z.: Chemia analityczna z elementami analizy instrumentalnej. PZWL,
Warszawa 1996
6. Minczewski J., Marczenko Z.: Chemia analityczna Tom I i II. PWN, Warszawa 2001
7. Pawełczyk E., Płotkowiak Z., Zając M.: Chemiczna analiza leków. PZWL, Warszawa
1981
8. Sthal E.: Chromatograficzna i mikroskopowa analiza surowców roślinnych. PZWL,
Warszawa 1987
9. Strzelecka H., Kamińska J., Kowalski J., Walewska E.: Chemiczne metody badań
roślinnych surowców leczniczych. PZWL, Warszawa 1987
10. Szczepaniak H.: Metody instrumentalne w analizie chemicznej. PWN, Warszawa 2004
11. Zejc A., Gorczyca M.: Chemia leków. PZWL, Warszawa 2002
12. Farmakopea Polska IV PZWL, Warszawa 1970
13. Farmakopea Polska V PTFarm., Warszawa 1995-1999
14. Farmakopea Polska VI PTFarm, Warszawa 2002
15. Farmakopea Polska VII PTFarm, Warszawa 2006
Wyszukiwarka
Podobne podstrony:
05 Wykonywanie analizy jakościowej i ilościowej produktów
05 Wykonywanie analizy jakościowej i ilościowej produktów
wymagania jakościowe dla produktów leczniczych
Pomiar Dydaktyczny, 11 ocenianie, ANALIZA JAKOŚCIOWA I ILOŚCIOWA
02 Wykonywanie analiz jakosciow Nieznany (2)
Pomiar Dydaktyczny, 09 TYTUL, ANALIZA JAKOŚCIOWA I ILOŚCIOWA
02 Wykonywanie analiz jakościowych
02 Wykonywanie analiz jakościowych
04 Wykonywanie analiz ilosciowy Nieznany (2)
Chemiczne metody analizy ilościowe śr leczniczych Rajzer
05 Wykonywanie podstawowych analiz jakościowych
04 Wykonywanie analiz ilościowych
04 Wykonywanie analiz ilościowych
Wykonywanie podstawowych analiz jakościowych
Reklama produktów leczniczych
więcej podobnych podstron