
Formation of active inclusion bodies in the periplasm
of Escherichia coli
Jean-Philippe Arié,
†
Marika Miot,
†
Nathalie Sassoon
and Jean-Michel Betton*
Unité de Biochimie Structurale and CNRS URA2185,
25–28 rue du Docteur Roux, 75724 Paris Cedex 15,
France.
Summary
To examine the relationship between folding and
aggregation in the periplasm of Escherichia coli, we
have analysed the cellular fates of exported proteins
fused to either the wild-type maltose-binding protein
(MalE) or the aggregation-prone variant MalE31. The
propensity of fusion proteins to aggregate in the peri-
plasm was determined by the intrinsic folding char-
acteristics of the upstream protein. When
b-lactamase
or alkaline phosphatase was linked to the C-terminus
of MalE31, the resultant fusion proteins accumulated
in an insoluble form, but retained their catalytic
activity. In addition, these protein aggregates induced
an extracytoplasmic stress response, similar to
unfused MalE31. However, using a fluorescent sub-
strate, we found that alkaline phosphatase activity
was present inside periplasmic aggregates. These
results suggest that periplasmic inclusion body for-
mation may result in intermolecular interactions
between participating proteins without loss of func-
tion of the fused enzymes.
Introduction
Accumulation of aggregated proteins in Escherichia coli
occurs when cells are exposed to environmental stress,
such as elevated temperatures or hyperosmolarity, or
when they are overproduced by recombinant genes
acting as protein factories (Baneyx and Mujacic, 2004).
In these cases, the polypeptide chain fails to remain
correctly folded or to fold sufficiently rapidly, and con-
sequently forms aggregates if it escapes the cellular
quality-control systems (Wickner et al., 1999). Because
incompletely folded proteins expose some structures that
are buried in the native state, they are prone to associate
with other molecules in the crowded cellular environment.
Two different physiological consequences can be attrib-
uted to protein aggregation (Miot and Betton, 2004). The
first is a loss of protein function, which is often accompa-
nied by misplacement of the misfolded protein. Second,
the toxic properties of the aggregates can trigger a cellular
stress response and cause various metabolic changes.
Protein aggregates or inclusion bodies are generally
viewed as polymeric complexes of partially structured,
misfolded or unfolded proteins forced to associate by
non-specific
hydrophobic,
intermolecular
interactions
(Fink, 1998). However, recent studies suggest that these
protein aggregates might be formed from different confor-
mational states through a molecular mechanism that is
more specific than that commonly assumed (Ventura and
Villaverde, 2006).
Maltose-binding protein (MalE) is the soluble periplas-
mic receptor of the high-affinity transporter of maltodex-
trins in E. coli. In earlier work, we identified misfolding
associated amino acid substitutions in a structural turn
connecting the
a-helix I to b-strand B of the N-terminal
domain of MalE (Betton et al., 1996). Among these muta-
tions, the most defective folding variant, MalE31, corre-
sponded to the double substitution of Gly32-Ile33 to
Asp32-Pro33. In vitro studies provided evidence that
folding intermediates of MalE31 are kinetically trapped in
an off-pathway reaction leading to their aggregation
(Raffy et al., 1998). The crystal structure of MalE31 con-
firmed that the structural effect of the double substitution
is exerted at the level of folding intermediates, rather than
that of the native structure (Saul et al., 2003). In vivo, the
MalE31 precursor is correctly exported but the defective
folding of mature protein leads, at high levels of produc-
tion, to the formation of inclusion bodies in the periplasm
(Betton and Hofnung, 1996). At low levels of production,
misfolded MalE31 is rapidly degraded by DegP, a stress
protease involved in periplasmic quality control (Betton
et al., 1998). One physiological consequence for cells
producing MalE31 is the induction of an extracytoplasmic
stress response via the Cpx signalling pathway (Hunke
and Betton, 2003), but not that of the general
s
32
-
dependent stress response (Betton et al., 2002).
In this report, we have examined the cellular fates of
periplasmic proteins when fused either to the wild-type
MalE or to the aggregation-prone MalE31. By reversing
the configuration of fusion proteins, we have found
that the solubility of fusion proteins in the periplasm is
Accepted 18 August, 2006. *For correspondence. E-mail: jmbetton@
pasteur.fr; Tel. (
+33) 1 4568 8959; Fax (+33) 1 4568 8604.
†
These
authors contributed equally to this work.
Molecular Microbiology (2006) 62(2), 427–437
doi:10.1111/j.1365-2958.2006.05394.x
First published online 14 September 2006
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd
determined by the intrinsic folding characteristics of the
upstream protein. However, when soluble periplasmic
enzymes [
b-lactamase (Bla) and alkaline phosphatase
(PhoA)] were covalently linked to MalE31, the resultant
insoluble proteins retained their catalytic activity. Thus,
our findings indicate that the formation of periplasmic
inclusion bodies does not compromise the folding of other
proteins when fused to the C-terminus of MalE31.
Results
Configuration of MalE fusions determines their
cellular fates
It has been previously observed that the solubility of
aggregation-prone proteins is markedly improved when
fused to MalE (Kapust and Waugh, 1999), and that this
property is only effective when MalE is joined to the
N-terminal of these proteins (Sachdev and Chirgwin,
1998). To determine whether this solubility-enhancing
activity could also be observed in the periplasm, we
designed two fusion proteins connected to the defective
folding protein MalE31, which only differ by the configu-
ration of their mature sequence (Fig. 1A). Sodium
dodecyl-sulphate-polyacrylamide
gel
electrophoresis
(SDS-PAGE) analysis of whole cell lysates showed that
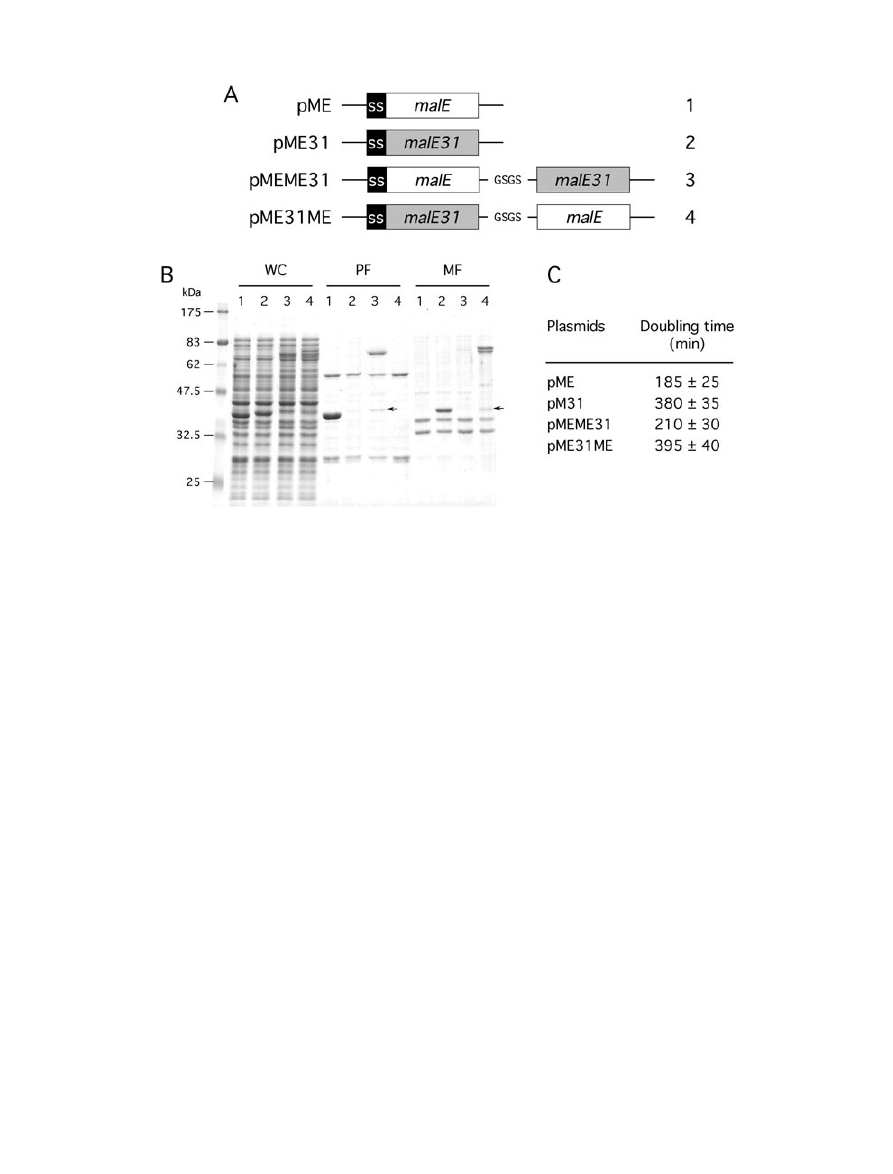
both full-length proteins were correctly produced, but at
lower levels than their unfused counterpart (Fig. 1B). The
formation of periplasmic inclusion bodies were biochemi-
cally determined by centrifugation from spheroplast
fractionation. As expected, MalE31 was present exclu-
sively in the insoluble membrane fraction (or final pellet),
whereas MalE was found in the soluble periplasmic frac-
tion (or first supernatant). When MalE was linked to the
N-terminus of MalE31, the corresponding protein was cor-
rectly exported and produced as soluble species in the
periplasm. In contrast, when it fused to the C-terminus of
MalE31, the resulting protein was mainly insoluble
(Fig. 1B). Subsequent analysis of the gel revealed the
presence of two prominent bands for MalE31-MalE,
visible from both whole cell and membrane fractions. As
the lower band migrated as the periplasmic MalE-MalE31,
these bands should correspond to the precursor and
mature species of MalE31-MalE. An additional faint MalE-
sized band (42 kDa) was also detected in the periplasmic
Fig. 1. Configuration of fusion proteins determines their cellular fates.
A. Schematic representation of genetic constructs used for the production of MalE fusion proteins under malE promoter control. The
N-terminal signal sequence of MalE (SS) and the four-residue peptide linker are indicated.
B. Cells carrying pME (1), pME31 (2), pMEME31 (3) or pME31ME (4) were grown at 30°C, then fractionated by spheroplast preparation.
Whole cell (WC), periplasmic (PF) and membrane (MF) fractions were analysed by SDS-PAGE and proteins stained by Coomassie blue. The
position of the 42 kDa breakdown product is indicated by an arrow.
C. Maltose phenotypes. Numbers are doubling times of cells producing the various fusions represented in (A) from cultures in liquid M63B1
maltose minimal medium at 30°C.
428
J.-P. Arié, M. Miot, N. Sassoon and J.-M. Betton
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437

and membrane fractions of both fusions, suggesting that
few proteins were cleaved in the linker region. However,
such a band was only detected by immunoblotting the
corresponding spheroplasted cells with anti-MalE anti-
body (data not shown). It was likely that limited proteolysis
yielding the MalE moiety occurred during the fractionation
procedure. As the primary structure of these fusion pro-
teins is very similar, the observed differences in fraction-
ation are only due to their folding behaviour. Thus, MalE
can function as a solubility enhancer in the periplasm as
well, but only when linked to the N-terminal of an
aggregation-prone protein.
Previously, we showed that MalE31 was unable to
complement the chromosomal deletion
DmalE444 for
maltose uptake, even when its periplasmic folding, facili-
tated by co-producing the FkpA chaperone, led to a
soluble protein (Saul et al., 2003). Therefore, the maltose
phenotype is a strict indicator of periplasmically active
MalE in the context of these fusion proteins. Cells produc-
ing MalE-MalE31 formed red colonies (Mal
+
) on maltose
MacConkey agar (Fig. S1A), and the maltose-binding
activity of MalE-MalE31 was confirmed by purifying this
full-length fusion protein on an amylose column (data not
shown). In contrast, cells producing MalE31-MalE formed
pale pink colonies (Mal
–
) on the maltose indicator agar
plate. Maltose phenotypes of these fusions were further
characterized by measuring their doubling time in liquid
M63B1-maltose minimal medium (Fig. 1C). Growth on
maltose indicated that the MalE moiety is functional within
the soluble MalE-MalE31 fusion, but is neither released
by proteolytic cleavage, nor functional within the insoluble
MalE31-MalE fusion. However, we cannot exclude that a
fraction of MalE might be actually natively folded within
this fusion protein, but embedded in inclusion bodies, and
therefore unable to functionally interact with the maltose
transporter.
MalE31 promotes insolubility of proteins to which
it is fused
In view of these results, two other fusion proteins were
designed to investigate the conformation of non-maltose
C-terminal proteins when linked to MalE31. Because
enzymatic activity is a very sensitive probe of the native
state, two different periplasmic enzymes, Bla and PhoA,
were fused to the C-terminus of MalE or MalE31 (Figs 2A
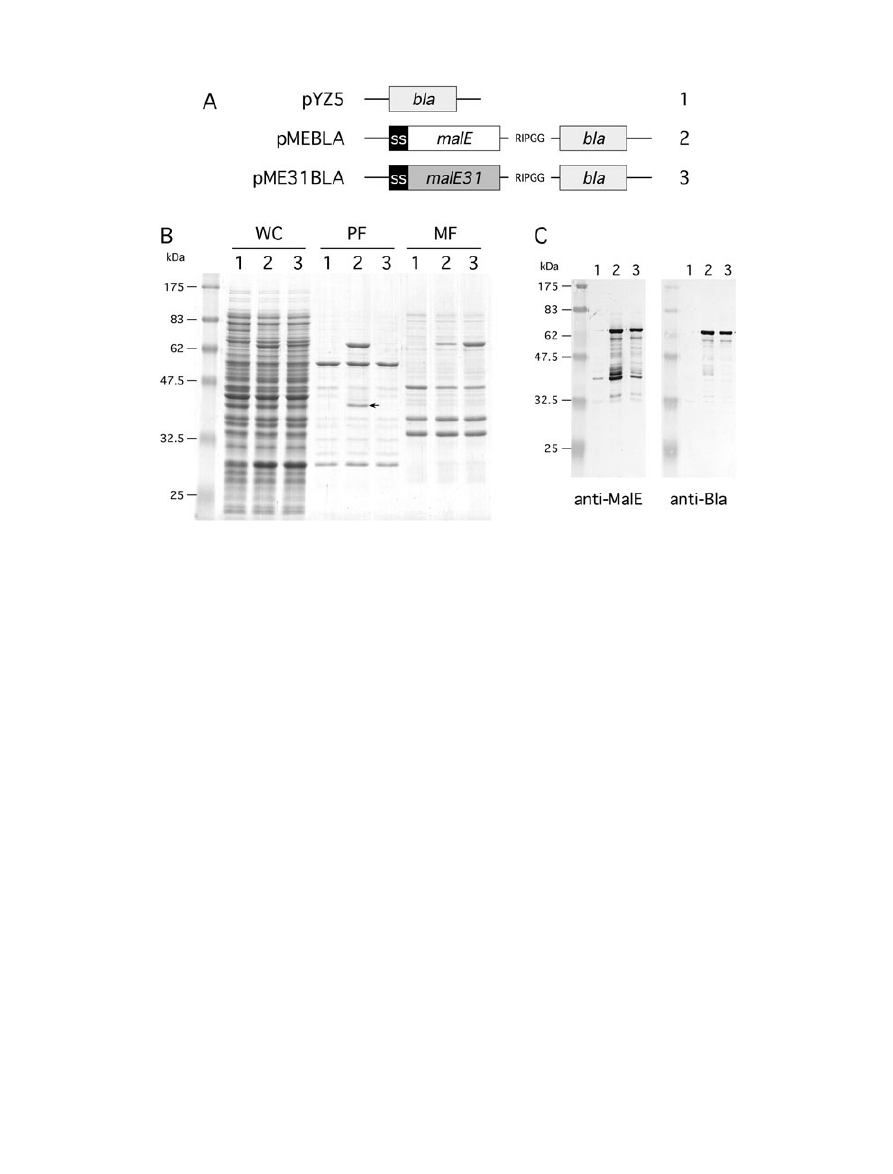
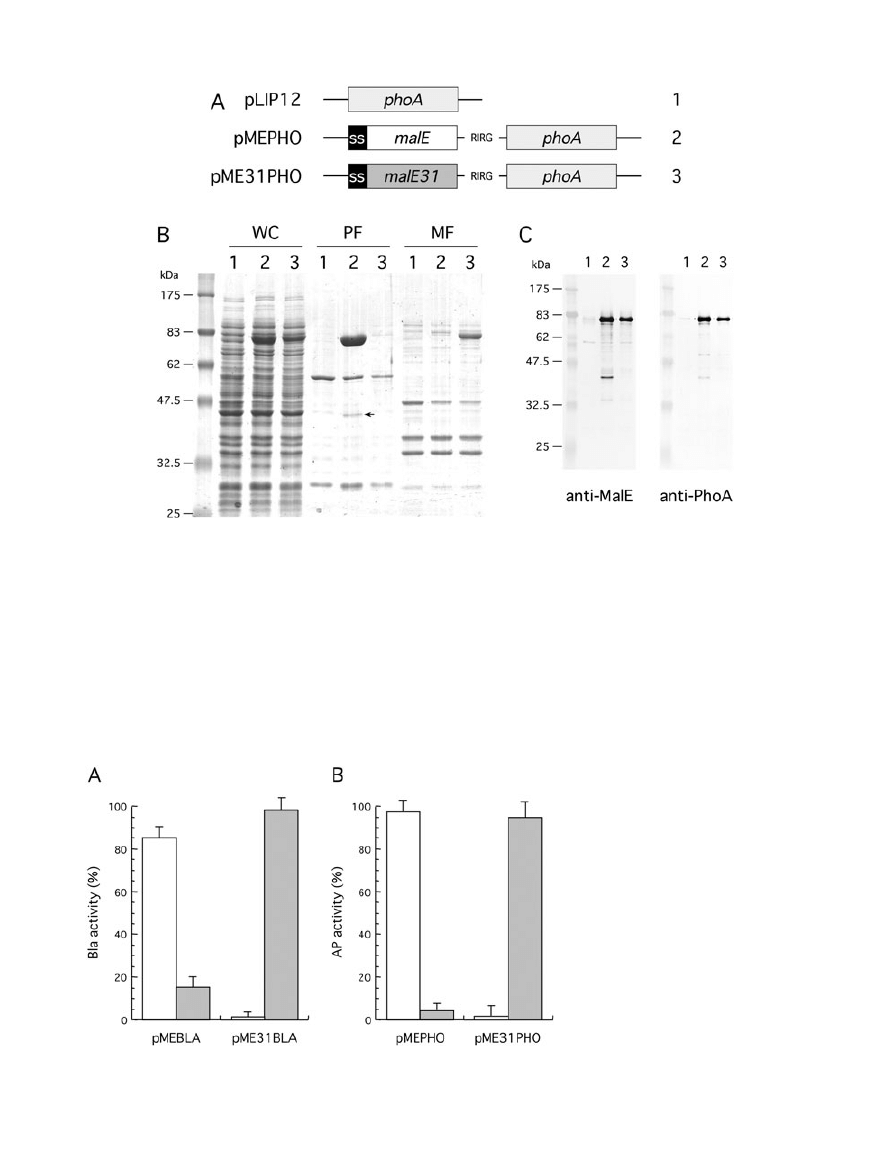
and 3A). MalE-Bla was produced at a lower level than
MalE-PhoA, and both full-length fusions were detected at
their expected size, respectively, 73 kDa and 87 kDa, in
the periplasm (Figs 2B and 3B). However, a significant
fraction (10–15%) of MalE-Bla was insoluble, while MalE-
PhoA was fully soluble. Previous studies have shown that
the overproduction of
b-lactamase resulted in the forma-
tion of inclusion bodies (Bowden and Georgiou, 1990),
probably from partially folded intermediates transiently
bound to the inner membrane (Minsky et al., 1986).
In agreement with the above results, MalE31-PhoA and
MalE31-Bla fractionated with the membrane fraction, con-
firming that MalE31 promotes the insolubility of these
linked periplasmic enzymes. Although a breakdown
product of approximately 42 kDa was visible from the
periplasmic fraction of MalE-Bla and MalE-PhoA, bands
larger than both full-length MalE31-Bla and MalE31-PhoA
were not detected in any fractions, contrary to that
observed above with MalE31-MalE. We hypothesized that
this polypeptide could be some MalE released by pro-
teolytic cleavage during fractionation.
We took advantage of Bla and PhoA phenotypes as
reliable indicators of functionally exported fusion proteins.
In contrast to maltose phenotypes that require productive
protein–protein interactions, Bla and PhoA phenotypes
only require that a diffusive small molecule be accessible
to the catalytic site of these enzymes. Because Bla
confers resistance to penicillins, when pop6499 cells were
plated onto agar containing ampicillin, those producing
active Bla species in the periplasm survived and formed
colonies (Fig. S1B). Surprisingly, cells producing MalE31-
Bla were also resistant to ampicillin as those producing
MalE-Bla, indicating that the Bla moiety was catalytically
active in both fusion proteins. Because PhoA has two
intrachain disulphide bonds required for its native struc-
ture, and therefore for its enzymatic activity, only gene
fusions encoding proteins, which displayed PhoA in the
periplasm, would produce an active enzyme. The corre-
sponding colonies can be readily detected on XP indicator
plates (Fig. S1C). Cells producing either MalE31-PhoA or
MalE-PhoA gave dark blue colonies (PhoA
+
) on these
agar plates at 30°C. The observed difference in colony
size indicated a growth defect for cells producing MalE31-
PhoA. Although MalE31-Bla and MalE31-PhoA accumu-
lated in an insoluble form like the unfused MalE31, the
positive phenotypes of these gene fusions suggested that
both encoded Bla and PhoA are active inside periplasmic
inclusion bodies.
To determine whether full-length fusion proteins or
enzymatically active fragments resulting from proteolytic
cleavages contributed to these phenotypes, whole cell
lysates were analysed by immunoblotting with anti-MalE
and either anti-Bla or anti-PhoA antibodies (Figs 2C and
3C). Under steady-state conditions, presumed degrada-
tion products were mainly detected in extracts of cells
producing MalE-Bla or MalE-PhoA, and were apparently
absent from extracts containing MalE31-Bla or MalE31-
PhoA. In both cases, the major fragment of approximately
42 kDa reacted only with the anti-MalE antibodies, sug-
gesting that this polypeptide lacks the Bla or PhoA moiety.
This result could indicate that the degradation by host
proteases occurred within these fused proteins. Thus,
Active periplasmic inclusion bodies
429
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437
soluble MalE fusions were partially degraded in the peri-
plasmic fraction, while insoluble MalE31 fusions remained
intact in the membrane fraction. It is conceivable that
insoluble fusion proteins which are sequestered in inclu-
sion
bodies
become
protected
against
proteolytic
degradation.
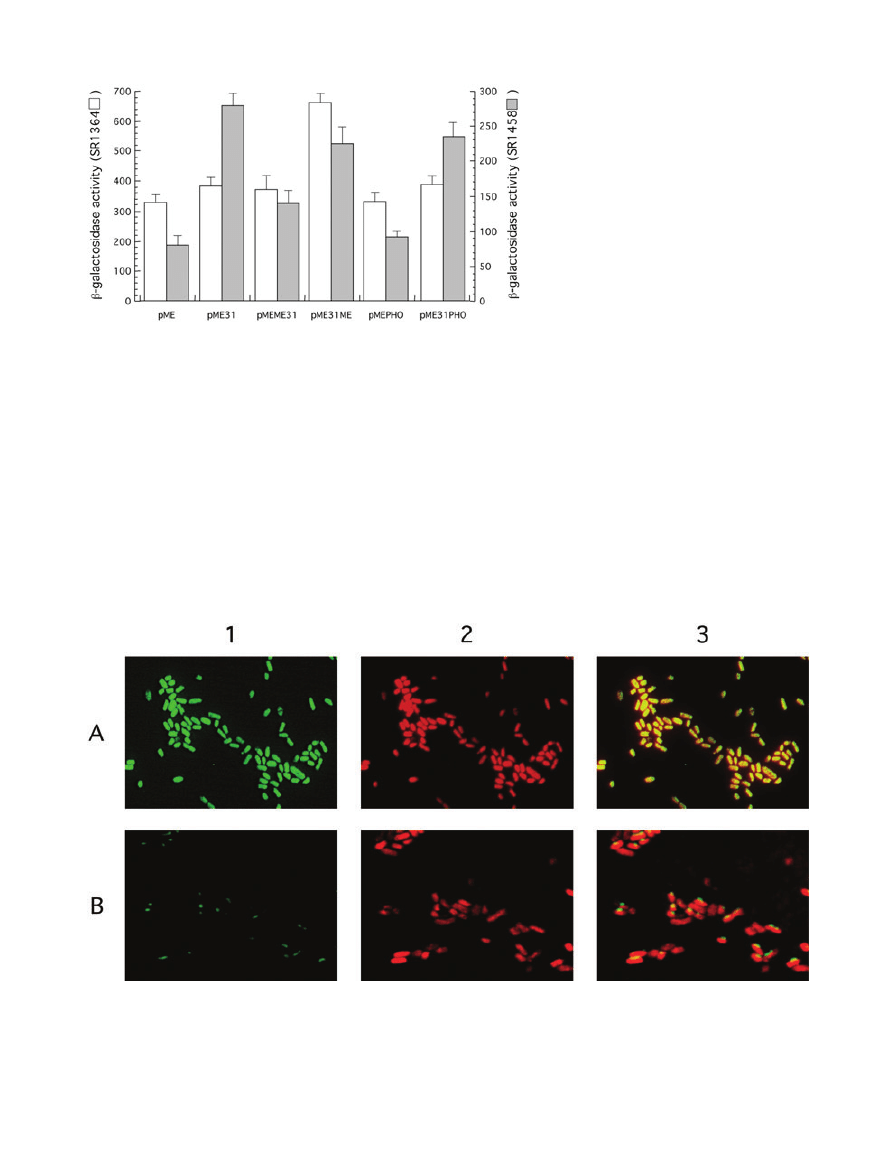
Enzymatic activity in the insoluble subcellular fraction
Evidence that insoluble MalE31-Bla and MalE31-PhoA
were enzymatically active came from the ability to
measure both Bla and alkaline phosphatase (AP) activi-
ties of subcellular fractions. When spheroplasts were cen-
trifuged, the Bla or AP activity was found mainly or
exclusively in the supernatant of MalE-Bla or MalE-PhoA,
but not in that of MalE31-Bla or MalE31-PhoA (Fig. 4A
and B). In contrast, the major fraction (
⬎ 95%) of Bla or
AP activity was present in the pellet of MalE31-Bla or
MalE31-PhoA, indicating that this particulate fraction con-
tained active Bla and PhoA enzymes, and that they are
either membrane associated or aggregated. The ampicil-
lin resistance of cells producing MalE31-Bla suggested
that this insoluble fusion protein is indeed exposed in
the periplasm. As disulphide bonds cannot be formed in
the cytoplasm (Derman et al., 1993), the AP activity in the
insoluble fraction established periplasmic folding of
MalE31-PhoA.
Overproduction of MalE31 fusions induces an
extracytoplasmic stress response
To directly address whether MalE31 fusion proteins
aggregated in the cytoplasm as precursors or in the peri-
plasm as mature proteins, stress responses were
assessed in two different strains, carrying either a single
copy of lon-lacZ (SR1364) or degP-lacZ (SR1458) tran-
scriptional gene fusions. E. coli stress responses induced
by the presence of misfolded proteins are compartmen-
talized into cytoplasmic responses, controlled by the
sigma factor
s
32
(Rist et al., 2003), and periplasmic
responses, controlled both by the
s
E
and Cpx signal trans-
duction pathways (Raivio and Silhavy, 1999). Thus, stress
promoter activity in both strains help to discern the cellular
compartment in which MalE31 aggregation occurred
Fig. 2. Production and fractionation of MalE-Bla fusions.
A. Schematic representation of genetic constructs used for the production of Bla fusion proteins under malE promoter control. The N-terminal
signal sequence of MalE (SS) and the five-residue peptide linker are indicated.
B. Cells carrying pYZ5 (1), pMEBLA (2) or pME31BLA (3) were grown at 30°C, then fractionated by spheroplast preparation. Whole cell (WC),
periplasmic (PF) and membrane (MF) fractions were analysed by SDS-PAGE and proteins stained by Coomassie blue. The position of the
42 kDa breakdown product that corresponds to near full-length MalE is indicated by an arrow.
C. Immunoblot of spheroplasted cells from cells carrying pYZ5 (1), pMEBLA (2) and pME31BLA (3) using anti-MalE and anti-Bla sera.
430
J.-P. Arié, M. Miot, N. Sassoon and J.-M. Betton
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437
(Betton et al., 2002). Consistent with the cellular fraction-
ation, the two protein fusions containing N-terminal MalE
did not increase lacZ expression from either stress gene
promoter (Fig. 5). All proteins that contain N-terminal
MalE31 increased lacZ expression from the degP
promoter. Although the extent of this stress response
varied, the production of all these proteins, except Mal31-
MalE, did not induce a cytoplasmic stress response. For
MalE31-MalE, the cytoplasmic accumulation of precur-
sors, previously suggested from the gel in Fig. 1B, can
Fig. 3. Production and fractionation of MalE-PhoA fusions.
A. Schematic representation of genetic constructs used for the production of PhoA fusion proteins under malE promoter control. The
N-terminal signal sequence of MalE (SS) and the four-residue peptide linker are indicated.
B. Cells carrying pLIP12 (1), pMEPHO (2) or pME31PHO (3) were grown at 30°C, then fractionated by spheroplast preparation. Whole cell
(WC), periplasmic (PF) and membrane fractions (MF) were analysed by SDS-PAGE and proteins stained by Coomassie blue. The position of
the 42 kDa breakdown product that corresponds to near full-length MalE is indicated by an arrow.
C. Immunoblot of spheroplasted cells from cells carrying pLIP12 (1), pMEPHO (2) and pME31PHO (3) using anti-MalE and anti-PhoA sera.
Fig. 4. Cellular fractionation of enzymatic
activities. The steady-state distribution of Bla
(A) or AP (B) activity in the soluble and
soluble fractions was determined from cells
producing the different fusion proteins
represented in Figs 2A and 3A respectively.
Spheroplasts were centrifuged, then Bla or AP
activity was determined on supernatants
(white bars) and pellets resuspended to the
same volume (grey bars). The enzymatic
activity in each fraction was calculated using
the average of four independent experiments
and normalized to the spheroplast activity
before centrifugation.
Active periplasmic inclusion bodies
431
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437
reflect the twofold increase in
b-galactosidase synthesis
directed from the
s
32
-dependent promoter. It seems likely
that a fraction of MalE31-MalE precursors aggregated in
the cytoplasm before export and processing. No evidence
for cytoplasmic misfolding of MalE31-PhoA was obtained
in these studies. This fusion protein induced solely an
extracytoplasmic stress response, in a manner similar to
that observed with the unfused MalE31.
Distribution of AP activity in live cells
To confirm that AP activity was physically associated with
periplasmic protein aggregates, we examined the in vivo
cellular localization of PhoA fusion proteins using the fluo-
rescent AP substrate, ELF-97 (Fig. 6). When cells produc-
ing MalE-PhoA were directly observed, the fluorescence
was uniformly distributed throughout the cells, consistent
with free diffusion of this protein in the periplasm. In
contrast, visualization of cells producing MalE31-PhoA
revealed a single fluorescent focus. Although a preferen-
tial cellular localization was hard to assess due to the
small size of the cells and the limited resolution of light
microscopy, the punctated fluorescence indicated the
presence of active PhoA within MalE31-PhoA aggregates.
Although slightly smaller than those formed from the
unfused MalE31, inclusion bodies were visible by trans-
mission electron micrography of cells producing MalE31-
PhoA (data not shown).
Fig. 5. Cellular stress responses induced by
the production of fusion proteins. The activity
of lon and degP promoters, both fused to the
lacZ gene, were assessed in E. coli strains
SR1364 or SR1458 respectively. Each strain
carrying the indicated plasmids was grown at
30°C in LB containing ampicillin (0.1 mg ml
-1
)
and maltose (0.2%). Miller units of lacZ
encoded
b-galactosidase were calculated
using the average of four independent
experiments.
Fig. 6. Intracellular localization of PhoA by fluorescence microscopy. Cells carrying pMEPHO or pME31PHO were grown at 30°C, then
incubated with the AP substrate, ELF-97, for 30 min. Fixed cells were stained by propidium iodide and samples observed by fluorescence
microscopy. Panels of representative micrographs show the desphosphorylated product in green (1), the stained nucleoids with propidium
iodide in red (2), and merged fluorescence (3) of cells producing MalE-PhoA (A) or MalE31-PhoA (B).
432
J.-P. Arié, M. Miot, N. Sassoon and J.-M. Betton
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437
Evolution of AP activity during cell growth at 30°C
As we have never detected the presence of soluble
MalE31-PhoA in the periplasm, the AP activity from cells
producing this fusion protein would monitor the periplas-
mic aggregation of this fusion protein. Previously, we have
reported that the accumulation of aggregated mature
MalE31 exerted a cell toxicity at 37°C, but not at 30°C
(Hunke and Betton, 2003). In the same genetic context,
the production of MalE31-PhoA induced a growth arrest
even at 30°C (Fig. 7A), and more deleterious effects
at 37°C. Therefore, the AP activity was followed from
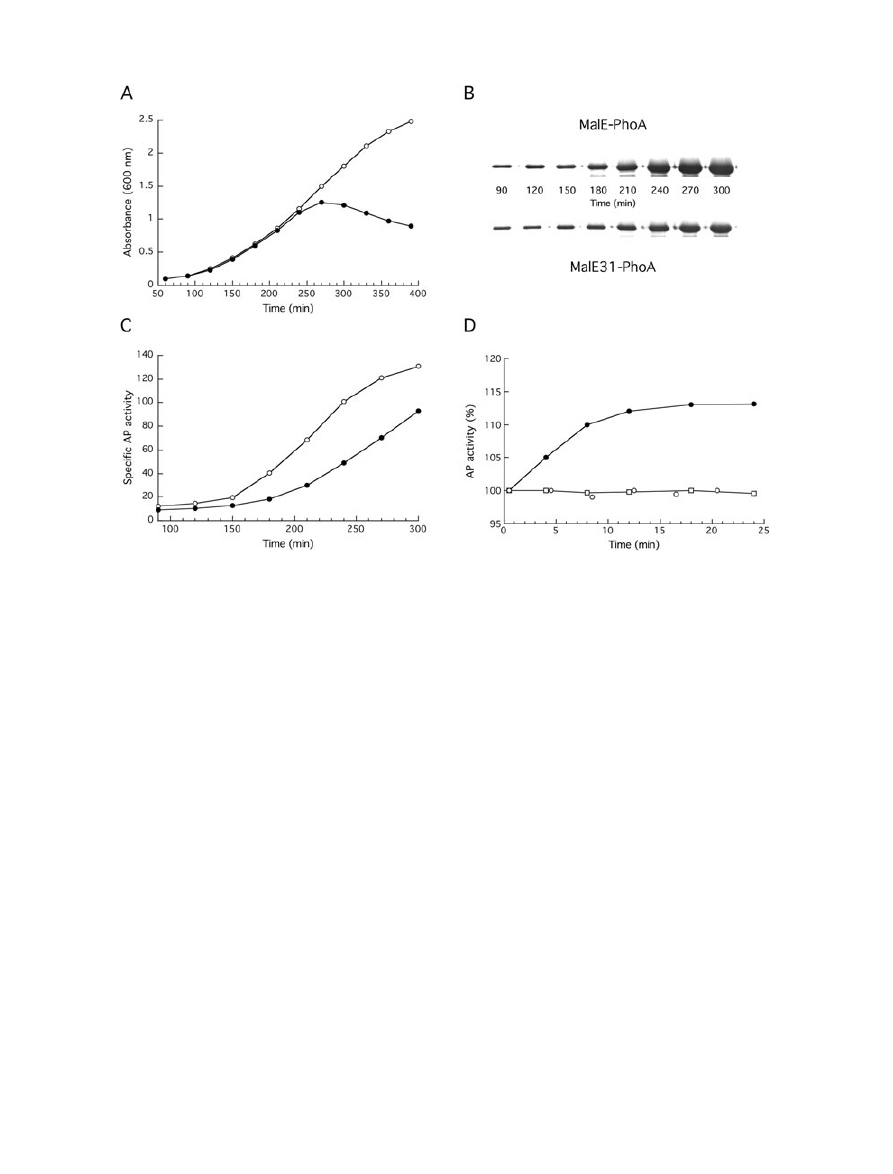
growing cells over 90–300 min at 30°C.
To take into account the difference in levels of fusion
protein present in the cells, the steady-state amounts of
MalE-PhoA and MalE31-PhoA were determined by den-
sitometry analysis of immunoblots using anti-PhoA anti-
bodies (Fig. 7B), and serial dilution of purified MalE-31
served as standard. When expressed as units per micro-
gram of protein, a steady increase in AP activity from
MalE-PhoA was found during the exponential growth, and
then a plateau was reached coinciding with the end of this
growth phase (Fig. 7C). The AP activity from MalE31-
PhoA followed approximately the same curve as for MalE-
PhoA, except for the presence of an important lag time.
This behaviour indicates that MalE31 imposed some con-
straints on the periplasmic folding of PhoA. Further, we
used the AP activity to monitor de novo MalE31-PhoA
folding when protein synthesis was inhibited by the addi-
tion of chloramphenicol to the growing cells (Fig. 7D).
While the AP activity of MalE-PhoA immediately stopped
increasing, the AP activity of MalE31-PhoA continued
to increase for about 10 min after the addition of
chloramphenicol. Although increments represented only
10–15% of the AP activity present at the time of adding
chloramphenicol, this increase in the absence of protein
synthesis indicates that the periplasmic folding of PhoA is
much slower when linked to MalE31 than to MalE. Finally,
Fig. 7. Evolution of AP activity during cell growth at 30°C.
A. Growth curves of cells carrying pMEPHO (open circle) or pME31PHO (filled circle) in LB medium containing ampicillin at 30°C.
B. At the indicated times, aliquots from both cultures were withdrawn, and intracellular levels of fusion proteins were analysed by
immunobloting with anti-PhoA antibodies.
C. AP activity of permeabilized cells producing MalE-PhoA (open circle) or MalE31-PhoA (filled circle) was measured from the same aliquots
and normalized to the relative amounts of fusion proteins.
D. After 210 min of growth at 30°C, chloramphenicol (150
mg ml
-1
) was added to the cultures (time 0). Samples were withdrawn at different
times and AP activity of permeabilized cells producing MalE-PhoA (open circles) or MalE31-PhoA (filled circles) was measured. AP activity of
MalE31-PhoA following translational arrest was determined after the addition of 5 mM iodoacetamide (open squares).
Active periplasmic inclusion bodies
433
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437

we verified that the increase in AP activity of MalE31-
PhoA was linked to the acquisition of disulphide bonds by
inhibiting their formation with iodoacetamide after transla-
tional arrest.
Discussion
Our observations provide evidence that both the wild-type
MalE and MalE31 variant have a strong influence on the
periplasmic fate of proteins to which they are fused. After
translocation across the inner membrane, newly exported
proteins encounter in the periplasm a kinetic partitioning
between folding, degradation and aggregation (Miot and
Betton, 2004). Our data indicate that the solubility of
fusion proteins is controlled by whether the protein emerg-
ing first from the SecYEG translocase folds into a soluble
or insoluble state in the periplasm of E. coli. Therefore,
MalE and MalE31 promote solubility and insolubility,
respectively, of periplasmic fusion proteins. Although
these opposing effects correlated well with the intrinsic
folding characteristics of MalE and MalE31, we show that
these two periplasmic fates do not interfere with the func-
tional folding of linked enzymes. For these fusion proteins,
the term insolubility denotes only a chemical characteris-
tic related to an operational definition (sedimentation), but
not to folding and the absence of function. Fusions with
MalE have been successfully used to enhance the cellular
folding of recombinant proteins (Kapust and Waugh,
1999). Although these MalE fusion proteins were gener-
ally produced in the cytoplasm, one study underlined that
MalE can efficiently assist the periplasmic oxidative
folding of a disulphide-rich protein from Plasmodium fal-
ciparum when fused to its C-terminus (Planson et al.,
2003). To explain this particular behaviour, it has been
proposed that MalE could function as an intramolecular
chaperone in the context of fusion proteins by binding to
folding intermediates and preventing their intermolecular
interactions (Fox et al., 2001). Such a chaperone-like
activity of MalE has also been reported in vitro (Richarme
and Caldas, 1997), but in these experiments MalE was
not covalently linked to aggregation-prone proteins, and
could not reveal how intramolecular interactions solubilize
the fusion protein.
In a similar vein, we have tested the role of MalE31 in
protein folding when fused to the periplasmic Bla or PhoA
proteins. Because MalE31 folds more slowly than MalE,
the degradation and aggregation of fusion proteins should
be favoured. However, aggregated MalE31 fusion pro-
teins displayed enzymatic activity. As the Bla or AP activity
did not originate from degradation products, it implies that
a significant fraction of both Bla and PhoA enzymes,
covalently linked to MalE31, were correctly folded within
insoluble and intact protein fusions. We did not know
whether aggregated fusion proteins represent true inclu-
sion bodies, like those formed from the unfused MalE31
which required high urea concentrations to be solubilized,
or membrane-bound proteins held by hydrophobic
domains of MalE31. Experiments to study the solubiliza-
tion of MalE31-PhoA by different molecules are underway
to shed light on the chemical nature of these intermolecu-
lar interactions. Nevertheless, it is important to consider
whether our observations are more related to character-
istics of the folding pathway of MalE31 rather than protein
folding in the periplasm. Although MalE31 influences the
folding rate of PhoA, the aggregation of MalE31-PhoA did
not compromise its AP activity, as generally suggested in
the context of fusion with an aggregation-prone protein
(see below). The most plausible explanation is that the
intermolecular interactions leading to fusion protein
aggregation occur only after the complete folding of PhoA.
However, even with the folding reaction preceding an
aggregation reaction, we never observed the presence of
soluble MalE31-PhoA in the periplasm. This observation
is consistent with the view that misfolded proteins are
either rapidly degraded or aggregated in the bacterial
periplasm (Betton et al., 1998). From real-time fluorescent
labelling of bacterial cells, it was observed that the cellular
aggregation of a retinoic acid-binding protein I (CRABP I)
variant displayed a concentration-dependent apparent lag
time (Ignatova and Gierasch, 2004). In that case, the slow
conversion of soluble misfolded to insoluble aggregated
protein was kinetically detectable, and the time-course of
cellular aggregation fitted well with the in vitro protein
aggregation reaction (Ignatova and Gierasch, 2005).
Although further work will be necessary to determine
kinetic parameters, our data suggest that the folding rate
of PhoA is significantly reduced by the presence of
MalE31, regardless of whether some of the newly trans-
located fusion proteins are degraded. Because active
PhoA requires a dimerization step, it would be conceiv-
able that PhoA first assembles, and then its active site
could be formed before being embedded within inclusion
bodies. Few other studies have reported that bacterial
inclusion bodies from overproduced recombinant proteins
can retain some biological activity (Worrall and Goss,
1989; Tokatlidis et al., 1991; Carrio et al., 2005; Garcia-
Fruitos et al., 2005). Therefore, the possibility that aggre-
gated proteins can also derive from native-like proteins
must be considered. It is also important to note that the
formation of inclusion bodies might even result in the
enrichment of active enzymes as recently suggested
(Garcia-Fruitos et al., 2005).
Aside from the phage display technique (Sieber et al.,
1998), several methods have been developed to monitor
protein folding in E. coli cells. Most of these methods rely
on genetic fusion between a target protein and a reporter
protein displaying a specific phenotype that is indepen-
dent of the target function (Waldo, 2003). Their common
434
J.-P. Arié, M. Miot, N. Sassoon and J.-M. Betton
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437

principle is based on the observation that when the target
protein folds into a soluble conformation, the fused
reporter will be functional. In contrast, when the target
protein aggregates, the fused reporter will be inactive,
resulting in an obvious null phenotype. Successful
reporter fusion proteins, including the green and blue
fluorescent proteins,
b-galactosidase, and chlorampheni-
col acetyltransferase, have been designed to monitor cel-
lular fluorescence (Waldo et al., 1999) or fluorescence
resonance energy transfer (Philipps et al., 2003), lactose
utilization (Wigley et al., 2001) and chloramphenicol resis-
tance (Maxwell et al., 1999) respectively. However, none
of these fusion reporters could be used to monitor protein
folding in the periplasm.
In order to develop such a genetic screen, we selected
several well-characterized exported enzymes. Among
these, the small nuclease A from Staphylococcus aureus
(NucA) displayed some potential advantages. However,
when MalE31 was fused to NucA in order to validate the
screening method, surprisingly we observed that periplas-
mic insolubility was not correlated to loss of nuclease
activity. At that time, we thought that because of its small
size NucA folds too rapidly into its active structure, and
therefore an useless reporter to monitor slow protein
aggregation. With the present results, we confirmed that
even linked to more complex proteins with slower folding
rates than NucA, the periplasmic aggregation of MalE31
does not interfere with the functional folding of a fused
reporter protein. Thus, the activity of a protein reporter in
folding screens is not a reliable indicator of protein solu-
bility, and great care must be taken to correctly interpret
the in vivo folding of fusion proteins. Nevertheless, split
protein folding reporters have been developed to over-
come these problems by rationalizing that structural
complementation between protein fragments relies only
on their accessibility to restore the reporter function
(Wigley et al., 2001; Cabantous et al., 2005). Finally, a
genetic selection based on folding quality control of the
twin-arginine translocation (Tat) pathway could also elimi-
nate the bias introduced by fusion protein folding (Fisher
et al., 2006).
Experimental procedures
Bacterial strains and growth conditions
The E. coli strain pop6499, a derivative of MC4100, was used
as host for plasmids encoding the various translational
fusions, as described previously. This strain carries a non-
polar deletion of malE (
DmalE444) and a malT
c
allele that
confers constitutive expression of the maltose operons.
Expression of Plon-lacZ and PdegP-lacZ transcriptional
fusions was monitored in SR1368 and SR1458 strains
respectively. Luria broth (LB) and M63B1 growth media and
MacConkey agar (Difco) were as described by Miller (1992).
Cells were generally grown in LB with appropriate antibiotics
at 30°C. The antibiotics ampicillin, kanamycin and chloram-
phenicol were used at 100, 30 and 150
mg ml
-1
respectively,
unless otherwise stated. The chromogenic indicator for PhoA,
5-Bromo
-4-chloro-3-indolyl phosphate (XP) was used in agar
plates at 0.1 mg ml
-1
.
Plasmid constructions
Plasmids pME and pME31 are pBR322 derivatives that carry
the wild-type malE and malE31 alleles (Betton et al., 1996),
respectively, under the control of their own MalT-dependent
promoter (PmalE). Plasmids pMEME31 and pME31ME were
constructed by subcloning a PCR-amplified DNA fragment of
the corresponding mature sequence of MalE and MalE31,
with primers containing a HindIII adaptator, into pME and
pME31 cut with the same enzyme respectively. Plasmid
pMEBLA carrying a malE-bla fusion, under control of the
PmalE promoter, was described previously (Betton et al.,
1997). Plasmid pMEPHO was constructed by subcloning a
2395 bp PstI/BamHI fragment from pPD140 (Duplay et al.,
1987) into pLIP12 (Dassa and Muir, 1993) cut with the same
enzymes. The substitution of malE by the malE31 allele was
performed by exchanging the 1406 bp ScaI/BglII fragment
from p31H into pMEPHO and pMEBLA respectively. Plasmid
pYZ5 (Zhang and Broome-Smith, 1990) is a pBR322 deriva-
tive that carries the bla gene without a promoter, and was
used as a negative control.
Cell fractionation
Cultures at an absorbance at 600 nm (A
600
) of 1.2 were
centrifuged, and cells were fractionated by spheroplast
preparation. The cell pellets, normalized to the same A
600
value, were resuspended in 10 mM Tris-HCl buffer (pH 7.5)
containing 0.5 M sucrose. Lysozyme (0.2 mg ml
-1
) and EDTA
(10 mM) were added, and suspensions were incubated for
20 min at 4°C. An aliquot of these suspensions was taken
and used as whole cell extracts. Then, the samples were
centrifuged for 5 min at 15 000 g, and supernatants contain-
ing the periplasmic fractions were withdrawn. Spheroplast
pellets were washed, freeze-thawed, and centrifuged at
25 000 g for 20 min. Supernatants were discarded and
pellets were washed with 10 mM Tris-HCl buffer (pH 7.5) and
resuspended into the same buffer to give the membrane
fractions. Proteins from whole cell extracts and the subcellu-
lar fractions were separated by SDS-PAGE. After electro-
phoresis, proteins were either stained with Coomassie blue
or electrotransferred onto nitrocellulose membranes, which
were incubated first with rabbit antiserum specific for MalE,
Bla or PhoA, and then with PhoA-coupled antiserum against
rabbit immunoglobulins. The immunoblots were developed
with nitroblue tetrazolium and XP. For quantitative analysis,
gels were scanned with an Image master VDS camera
(Amersham Biotech).
Enzymatic assays
b-Lactamase
activity
from
subcellular
fractions
was
determined spectrophotometrically at 490 nm using the
chromogenic substrate Nitrocefin, as described previously
Active periplasmic inclusion bodies
435
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437

(Betton et al., 1997). AP activity from subcellular fractions
was followed at 25°C in assay buffer (1 M Tris-HCl, pH 8,
containing 5 mM p-nitrophenyl phosphate), by monitoring the
release of p-nitrophenolate at 410 nm. AP assays of perme-
abilized cells were performed at 30°C and enzymatic units
were calculated as described previously (Manoil and Beck-
with, 1985). Cells were treated with 150
mg ml
-1
chloram-
phenicol and 5
mM iodoacetamide to stop both translation
and disulphide bond formation. The
b-galactosidase activity
in permeabilized cells were assayed as described previously
(Betton et al., 2002) from cultures induced by 0.2% maltose.
A minimum of three independent determinations was aver-
aged to obtain the indicated values.
Imaging of cellular MalE-PhoA and MalE31-PhoA
Cells were grown as described above to an A
600
of 0.6. Then,
ELF-97, a fluorogenic phosphatase substrate (Molecular
Probes Europe) was added at 80
mM to the cells, and incu-
bated at 37°C for 30 min. Cells were collected by centri-
fugation, and pellets were resuspended in 1 ml of sterile
phosphate buffer (0.1 M sodium phosphate pH 7.5). Samples
were filtered through black membrane filters (0.22
mm, Milli-
pore) and fixed with 2% (v/v) formaldehyde solution for
15 min. Cells were rinsed with the phosphate buffer and
stained 15 min with 60
mM propidium iodide (Live/Dead
BacLight Kit, Molecular Probes). After incubation, samples
were refiltered and quickly washed with the phosphate buffer.
Preparations were mounted with Live/Dead immersion oil on
glass microscope slides. Images were taken with a 100
¥ oil
immersion objective with a numerical aperture of 1.4 on an
Axiovert 200M microscope (Zeiss).
References
Baneyx, F., and Mujacic, M. (2004) Recombinant protein
folding and misfolding in Escherichia coli. Nat Biotechnol
22: 1399–1408.
Betton, J.M., and Hofnung, M. (1996) Folding of a mutant
maltose-binding protein of Escherichia coli which forms
inclusion bodies. J Biol Chem 271: 8046–8052.
Betton, J.M., Boscus, D., Missiakas, D., Raina, S., and
Hofnung, M. (1996) Probing the structural role of an alpha
beta loop of maltose-binding protein by mutagenesis: heat-
shock induction by loop variants of the maltose-binding
protein that form periplasmic inclusion bodies. J Mol Biol
262: 140–150.
Betton, J.M., Jacob, J.P., Hofnung, M., and Broome-Smith,
J.K. (1997) Creating a bifunctional protein by insertion of
beta-lactamase into the maltodextrin-binding protein. Nat
Biotechnol 15: 1276–1279.
Betton, J.M., Sassoon, N., Hofnung, M., and Laurent, M.
(1998) Degradation versus aggregation of misfolded
maltose-binding protein in the periplasm of Escherichia
coli. J Biol Chem 273: 8897–8902.
Betton, J.M., Phichith, D., and Hunke, S. (2002) Folding and
aggregation of export-defective mutants of the maltose-
binding protein. Res Microbiol 153: 399–404.
Bowden, G.A., and Georgiou, G. (1990) Folding and aggre-
gation of beta-lactamase in the periplasmic space of
Escherichia coli. J Biol Chem 265: 16760–16766.
Cabantous, S., Terwilliger, T.C., and Waldo, G.S. (2005)
Protein tagging and detection with engineered self-
assembling fragments of green fluorescent protein. Nat
Biotechnol 23: 102–107.
Carrio, M., Gonzalez-Montalban, N., Vera, A., Villaverde, A.,
and Ventura, S. (2005) Amyloid-like properties of bacterial
inclusion bodies. J Mol Biol 347: 1025–1037.
Dassa, E., and Muir, S. (1993) Membrane topology of MalG,
an inner membrane protein from the maltose transport
system of Escherichia coli. Mol Microbiol 7: 29–38.
Derman, A.I., Prinz, W.A., Belin, D., and Beckwith, J. (1993)
Mutations that allow disulfide bond formation in the cyto-
plasm of Escherichia coli. Science 262: 1744–1747.
Duplay, P., Szmelcman, S., Bedouelle, H., and Hofnung, M.
(1987) Silent and functional changes in the periplasmic
maltose-binding protein of Escherichia coli K12. I. Trans-
port of maltose. J Mol Biol 194: 663–673.
Fink, A.L. (1998) Protein aggregation: folding aggregates,
inclusion bodies and amyloid. Fold Des 3: R9–R23.
Fisher, A.C., Kim, W., and DeLisa, M.P. (2006) Genetic
selection for protein solubility enabled by the folding quality
control feature of the twin-arginine translocation pathway.
Protein Sci 15: 449–458.
Fox, J.D., Kapust, R.B., and Waugh, D.S. (2001) Single
amino acid substitutions on the surface of Escherichia coli
maltose-binding protein can have a profound impact on the
solubility of fusion proteins. Protein Sci 10: 622–630.
Garcia-Fruitos, E., Gonzalez-Montalban, N., Morell, M., Vera,
A., Ferraz, R.M., Aris, A., et al. (2005) Aggregation as
bacterial inclusion bodies does not imply inactivation of
enzymes and fluorescent proteins. Microb Cell Fact 4: 27.
Hunke, S., and Betton, J.M. (2003) Temperature effect on
inclusion body formation and stress response in the peri-
plasm of Escherichia coli. Mol Microbiol 50: 1579–1589.
Ignatova, Z., and Gierasch, L.M. (2004) Monitoring protein
stability and aggregation in vivo by real-time fluorescent
labeling. Proc Natl Acad Sci USA 101: 523–528.
Ignatova, Z., and Gierasch, L.M. (2005) Aggregation of a
slow-folding mutant of a beta-clam protein proceeds
through a monomeric nucleus. Biochemistry 44: 7266–
7274.
Kapust, R.B., and Waugh, D.S. (1999) Escherichia coli
maltose-binding protein is uncommonly effective at pro-
moting the solubility of polypeptides to which it is fused.
Protein Sci 8: 1668–1674.
Manoil, C., and Beckwith, J. (1985) TnphoA: a transposon
probe for protein export signals. Proc Natl Acad Sci USA
82: 8129–8133.
Maxwell, K.L., Mittermaier, A.K., Forman-Kay, J.D., and
Davidson, A.R. (1999) A simple in vivo assay for increased
protein solubility. Protein Sci 8: 1908–1911.
Miller, J. (1992) A Short Course in Bacterial Genetics. Cold
Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Minsky, A., Summers, R.G., and Knowles, J.R. (1986) Secre-
tion of beta-lactamase into the periplasm of Escherichia
coli: evidence for a distinct release step associated with a
conformational change. Proc Natl Acad Sci USA 83: 4180–
4184.
Miot, M., and Betton, J.M. (2004) Protein quality control in the
bacterial periplasm. Microb Cell Fact 3: 4.
Philipps, B., Hennecke, J., and Glockshuber, R. (2003)
436
J.-P. Arié, M. Miot, N. Sassoon and J.-M. Betton
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437

FRET-based in vivo screening for protein folding and
increased protein stability. J Mol Biol 327: 239–249.
Planson, A.G., Guijarro, J.I., Goldberg, M.E., and Chaffotte,
A.F. (2003) Assistance of maltose binding protein to the in
vivo folding of the disulfide-rich C-terminal fragment from
Plasmodium falciparum merozoite surface protein 1
expressed in Escherichia coli. Biochemistry 42: 13202–
13211.
Raffy, S., Sassoon, N., Hofnung, M., and Betton, J.M. (1998)
Tertiary structure-dependence of misfolding substitutions in
loops of the maltose-binding protein. Protein Sci 7: 2136–
2142.
Raivio, T.L., and Silhavy, T.J. (1999) The sigmaE and Cpx
regulatory pathways: overlapping but distinct envelope
stress responses. Curr Opin Microbiol 2: 159–165.
Richarme, G., and Caldas, T.D. (1997) Chaperone properties
of the bacterial periplasmic substrate-binding proteins.
J Biol Chem 272: 15607–15612.
Rist, W., Jorgensen, T.J., Roepstorff, P., Bukau, B., and
Mayer, M.P. (2003) Mapping temperature-induced confor-
mational changes in the Escherichia coli heat shock tran-
scription factor sigma 32 by amide hydrogen exchange.
J Biol Chem 278: 51415–51421.
Sachdev, D., and Chirgwin, J.M. (1998) Order of fusions
between bacterial and mammalian proteins can determine
solubility in Escherichia coli. Biochem Biophys Res
Commun 244: 933–937.
Saul, F.A., Mourez, M., Vulliez-Le Normand, B., Sassoon, N.,
Bentley, G.A., and Betton, J.M. (2003) Crystal structure of
a defective folding protein. Protein Sci 12: 577–585.
Sieber, V., Pluckthun, A., and Schmid, F.X. (1998) Selecting
proteins with improved stability by a phage-based method.
Nat Biotechnol 16: 955–960.
Tokatlidis, K., Dhurjati, P., Millet, J., Beguin, P., and Aubert,
J.P. (1991) High activity of inclusion bodies formed in
Escherichia coli overproducing Clostridium thermocellum
endoglucanase D. FEBS Lett 282: 205–208.
Ventura, S., and Villaverde, A. (2006) Protein quality in bac-
terial inclusion bodies. Trends Biotechnol 24: 179–185.
Waldo, G.S. (2003) Genetic screens and directed evolution
for protein solubility. Curr Opin Chem Biol 7: 33–38.
Waldo, G.S., Standish, B.M., Berendzen, J., and Terwilliger,
T.C. (1999) Rapid protein-folding assay using green fluo-
rescent protein. Nat Biotechnol 17: 691–695.
Wickner, S., Maurizi, M.R., and Gottesman, S. (1999) Post-
translational quality control: folding, refolding, and degrad-
ing proteins. Science 286: 1888–1893.
Wigley, W.C., Stidham, R.D., Smith, N.M., Hunt, J.F., and
Thomas, P.J. (2001) Protein solubility and folding moni-
tored in vivo by structural complementation of a genetic
marker protein. Nat Biotechnol 19: 131–136.
Worrall, D.M., and Goss, N.H. (1989) The formation of bio-
logically active
b-galactosidase inclusion bodies in Escheri-
chia coli. Aust J Biotechnol 3: 28–32.
Zhang, Y.B., and Broome-Smith, J.K. (1990) Correct inser-
tion of a simple eukaryotic plasma-membrane protein into
the cytoplasmic membrane of Escherichia coli. Gene 96:
51–57.
Supplementary material
The following supplementary material is available for this
article online:
Fig. S1. Phenotype of cells producing the different fusion
proteins.
A. Maltose MacConkey agar plate showing the phenotype of
cells carrying pME (1), pME31 (2), pMEME31 (3) or
pME31ME (4), after 24 h incubation at 30°C.
B. In vivo ampicillin sensitivity tests of cells carrying pYZ5 (1),
pMEBLA (2) or pME31BLA (3). Overnight cultures were
spotted after serial dilutions (as indicated) on a LB agar plate
supplemented with 0.5 mg ml
-1
ampicillin and incubated for
24 h at 30°C.
C. XP agar plate showing the phenotype of cells carrying
pLIP12 (1), pMEPHO (2) or pME31PHO (3) after 24 h incu-
bation at 30°C.
This material is available as part of the online article from
http://www.blackwell-synergy.com
Active periplasmic inclusion bodies
437
© 2006 The Authors
Journal compilation © 2006 Blackwell Publishing Ltd, Molecular Microbiology, 62, 427–437
Wyszukiwarka
Podobne podstrony:
Expression of correctly folded proteins in E coli
feminism and formation of ethnic identity in greek culture
Formation of heartwood substances in the stemwood of Robinia
Formation of a new chromosomes as a virulence mechanism in C glabrata
Andrew Garrett Convergence in the formation of Indo European subgroups
A Propagandist of Extermination, Johann von Leers and the Anti Semitic Formation of Children in Nazi
Far Infrared Energy Distributions of Active Galaxies in the Local Universe and Beyond From ISO to H
Rapid and efficient purification and refolding of a (His) tagged recombinant protein produced in E c
Role of the Structure of Heterogeneous Condensed Mixtures in the Formation of Agglomerates
Protein quality in bacterial inclusion bodies
Production of recombinant proteins in E coli
Inclusion bodies formation and utilisation
feminism and formation of ethnic identity in greek culture
Woziwoda, Beata; Kopeć, Dominik Changes in the silver fir forest vegetation 50 years after cessatio
Formation of heartwood substances in the stemwood of Robinia
Method for enhancing solubility of the expressed recombinant protein in E coli
1999 The past and the future fate of the universe and the formation of structure in it Rix
Secretory production of recombinant proteins in E coli
więcej podobnych podstron