
Inclusion Bodies: Formation and Utilisation
Beatrix Fahnert
1
· Hauke Lilie
2
· Peter Neubauer
3
1, 3
Biocenter Oulu, Department of Process and Environmental Engineering, Bioprocess
Engineering Laboratory, University of Oulu, P.O. Box 4300, 90014 Oulu, Finland
E-mail:
1
beatrix.fahnert@oulu.fi;
3
peter.neubauer@oulu.fi
2
Institute of Biotechnology, Martin-Luther-University Halle-Wittenberg, 06099 Halle
(Saale), Germany
E-mail: lilie@biochemtech.uni-halle.de
Abstract
The efficient in vivo folding of many heterologous proteins is a major bottleneck of
high level production in bacterial hosts and simple optimisation protocols have not been avail-
able yet. Therefore, inclusion body (IB) based processes play a major role as a potential strategy
for the production of complex recombinant proteins. These processes combine the advantages
of a high accumulation of the target protein in well-characterised bacteria such as Escherichia
coli, efficient strategies for IB isolation, purification and in vitro protein refolding without the
need of complicated coexpression systems. Recent advances in the molecular physiology of IB
formation and resolubilisation allow straight-forward optimisation of fermentation processes
to obtain a high-quality product. In addition, simple strategies have been developed to optimise
the purification and renaturation of disulfide bond containing proteins making a fast transfer
of such processes into the industrial production scale realistic.
Keywords
IBs · Aggregation · Proteolysis · Refolding · Stress response
1
Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
95
2
Protein Aggregation in Prokaryotes – The Formation of IBs . . .
96
2.1
Structural Characteristics of Proteins Favouring Aggregation . .
96
2.1.1
Disulfide Bonds . . . . . . . . . . . . . . . . . . . . . . . . . . .
97
2.1.2
Membrane Proteins . . . . . . . . . . . . . . . . . . . . . . . . .
98
2.1.3
Glycosylation . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
98
2.2
Composition and Structure of IBs and Kinetics of IB Formation
99
2.2.1
Architecture and Structure . . . . . . . . . . . . . . . . . . . . .
99
2.2.2
Composition of IBs . . . . . . . . . . . . . . . . . . . . . . . . . 100
2.2.3
Kinetics of In Vivo Aggregation . . . . . . . . . . . . . . . . . . . 101
2.2.4
Stability of IBs . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
2.3
The Physiology of IB Formation . . . . . . . . . . . . . . . . . . 103
2.3.1
The Metabolic Load of IB Synthesis . . . . . . . . . . . . . . . . . 103
2.3.2
The Response to Misfolded Protein . . . . . . . . . . . . . . . . . 107
2.3.2.1 Stress Responses . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
2.3.2.2 Chaperone Action . . . . . . . . . . . . . . . . . . . . . . . . . . 110
2.3.2.3 Periplasmic Response to Misfolded Protein . . . . . . . . . . . . 112
2.3.2.4 Response to Misfolded Proteins in Other Organisms . . . . . . . 112
2.3.3
Host Characteristics for High-Quality IBs . . . . . . . . . . . . . 113
2.4
IB Based Processes Versus Soluble Production . . . . . . . . . . . 114
2.4.1
Cultivation Conditions Promoting Aggregation . . . . . . . . . . 114
© Springer-Verlag Berlin Heidelberg 2004
Adv Biochem Engin/Biotechnol (2004) 89: 93– 142
DOI 10.1007/b93995

2.4.2
IBs as a Result of Failure in Formation of Correct Disulfide
Bonds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
2.4.3
How to Avoid IBs and to Favour Correctly Folded Proteins . . . . 115
2.4.3.1 Rate of Synthesis . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
2.4.3.2 Fusion Proteins . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
2.4.3.3 Coexpression of Chaperones and Foldases . . . . . . . . . . . . . 117
2.4.3.4 Cultivation Conditions and Addition of Folding Promoting
Agents . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
2.4.3.5 Cellular Redox Situation . . . . . . . . . . . . . . . . . . . . . . . 121
2.5
IBs in Prokaryotes Other than E. Coli . . . . . . . . . . . . . . . . 121
3
Production of IBs and Down-Stream Functionalisation . . . . . 122
3.1
Fermentation Process for IB Protein Production . . . . . . . . . . 122
3.2
Preparation of IBs . . . . . . . . . . . . . . . . . . . . . . . . . . 127
3.2.1
IB Isolation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
3.2.2
Purification of IBs . . . . . . . . . . . . . . . . . . . . . . . . . . 128
3.2.3
Solubilisation of IBs . . . . . . . . . . . . . . . . . . . . . . . . . 128
3.3
Refolding of Proteins from IBs . . . . . . . . . . . . . . . . . . . 129
3.3.1
Disulfide Bond Formation During Protein Renaturation . . . . . 131
3.3.2
Improving Renaturation . . . . . . . . . . . . . . . . . . . . . . . 132
3.4
Industrial Processes Based on Refolding of IB Proteins . . . . . . 134
3.4.1
Human Tissue-Type Plasminogen Activator (t-PA) . . . . . . . . 134
3.4.2
Antibody Fragments and Immunotoxins . . . . . . . . . . . . . . 135
3.5
The Future of IB Based Processes for Recombinant Proteins . . . 135
4
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136
List of Abbreviations
ATP
Adensine 5¢-triphosphate
DNA
Desoxyribonucleic acid
DOT
Dissolved oxygen tension [%]
DTE
Dithioeritrol
DTT
Dithiotreitol
E. coli
Escherichia coli
EDTA
Ethylene diamine tetra-acetic acid
F
Feed rate of concentrated carbon source solution [l h
–1
]
Fab
Antigen-binding fragment
Fv
Antibody fragment (variable regions)
FTIR
Fourier transform infrared spectroscopy
GdmCl
Guanidinium chloride
GSH/GSSG
Reduced/oxidised form of glutathione
Hsp
Heat shock protein
IBs
Inclusion bodies
IMAC
Immobilized metal affinity chromatography
94
B. Fahnert et al.

IPTG
Isopropyl-b-thiogalactopyranoside
LDAO
N,N-dimethyldodecylamine-N-oxide
m, m
max
(Maximum) specific growth rate [h
–1
]
mRNA
Messenger RNA
OD
500
Absorption (optical density) at 500 nm, measure for cell density
PDI
Protein disulfide isomerase
PEG
Polyethylene glycol
PTH
Parathyroid hormone
P
tac
, P
T7
, P
s32
Promoters designations
RNA
Ribonucleic acid
RNAP
RNA polymerase
RP-HPLC
Reversed phase high pressure liquid chromatography
S
0
Initial substrate concentration during the fermentation [g l
–1
]
SDS
Sodium dodecyl sulfate
S
i
Substrate concentration in the feed solution [g l
–1
]
t
D
Doubling time for cell growth [h
–1
]
t-PA
Tissue-type plasminogen activator
tRNA
Transfer RNA
V
Fstart
Fermenter volume [l] at feed start
VP1
Virus protein 1
X, X
Fstart
Cell dry weight [g l
–1
] (at feed start)
Y
X/S
Yield coefficient for cell growth on the carbon source [g g
–1
]
1
Introduction
The effective synthesis of proteins by recombinant hosts has become a widely ap-
plied strategy in research and industry. Although a number of different host sys-
tems are available today E. coli and other prokaryotes are still the first choice if
posttranslational modifications are not needed for protein function. Although
most of the proteins can be expressed at high concentration levels within a short
time in E. coli, in many cases the expressed product accumulates in a non-native
form intracellularly in dense particles called IBs.
We now know about many methods which can be tested to reduce aggregation
and to optimise the in vivo production of the soluble, active form of the protein,
e.g. cultivation at low temperature and limited induction, fusion of the protein to
solubilising partners, coexpression of chaperones and foldases, expression of the
protein in different cell compartments and expression in mutant strains [1–5].
However, the optimal process for a specific product is still unpredictable due to
the many factors influencing the synthesis and folding of proteins in the cellular
environment. The respective optimisation generally takes a lot of time and the fi-
nal yield of the active protein still is often unsatisfactory in comparison to the to-
tal product yield. Therefore, the purification and the refolding of IB proteins is
an attractive alternative, because the aggregates can be easily separated and
mostly contain the product in a high concentration. The in vitro refolding process
is the critical step, but the optimisation can be performed in a strategic way with
Inclusion Bodies: Formation and Utilisation
95

a step by step evaluation of the optimum conditions and additives contrarily to
the in vivo strategy. The major progress in the refolding of IBs was connected to
the development of industrial processes, including the first large-scale industrial
production process of a heterologous product in E. coli (human insulin).
This review aims to elucidate the internal and external factors leading to ag-
gregation. Thereby the paper not only concentrates on the strategies applied to
the production of IBs, but also summarises methods that can be used to avoid ag-
gregation to obtain a soluble (if desired) and active product. The biology of the
host cell is discussed in relation to the production of recombinant proteins and
strategies for the refolding of proteins from IBs are summarised allowing protein
production in a technical and industrial scale.
2
Protein Aggregation in Prokaryotes – The Formation of IBs
By the expression of proteins with cytoplasmic expression vectors product yields
of more than 50% of the total cellular protein can be obtained (see, e.g. [6]). In
case of a eukaryotic target protein the product is often enriched in IBs. These ag-
gregates usually contain the product in a non-native conformation, however in
a high concentration. The formation of IBs mainly depends on the kinetic com-
petition between protein-specific folding and aggregation rates connected to the
synthesis rate [4].Aggregation is a predominant feature in very strong expression
systems, but also increases with high inductor concentration, with the use of
complex growth media and at higher cultivation temperature [1, 7].According to
Rudolph [4] it can be concluded that the IB formation depends on the specific
folding behaviour rather than on the general characteristics of a protein such as
size, fusion partners, subunit structure and relative hydrophobicity. However,
folding-rate-limiting structural characteristics as disulfide bonds and certain
point mutations can significantly promote the formation of aggregates [8, 9].
2.1
Structural Characteristics of Proteins Favouring Aggregation
The primary structure of each protein provides any information necessary for
the functional conformation and activity (Anfinsen’s dogma) [10]. The number
of possible conformations is very high (10
30
for a protein of 100 amino acids with
each having two possible configurations, [11]). However, due to the finite time of
the folding procedure the protein cannot establish all of them (Levinthal’s para-
dox) [12]. Thus there are pathways guiding the folding to the native state being
thermodynamically stable and on a lower energetic level. The number of possi-
ble conformations of the polypeptide is decreased during the folding reaction by
non-covalent interactions (van der Waals contacts, salt bonds, polar interactions,
electrostatic interactions, hydrogen bonds). On the other hand different se-
quences can end in similar conformations (e.g. immunoglobulin G). This implies
an enzymatic folding assistance since thermodynamic processes are not suffi-
cient in every case [11, 13, 14]. Intermediates of the folding pathways can accu-
mulate due to aggregation caused by exposed hydrophobic regions [15]. That is
96
B. Fahnert et al.

why even small changes in the primary structure of a protein may affect its sol-
ubility [11, 16].
2.1.1
Disulfide Bonds
The formation of the correct disulfide bonds is usually the rate-limiting reaction
for the formation of the native structure of disulfide bond containing proteins
under otherwise optimum conditions. Due to the reducing redox conditions in
the cytosol of E. coli disulfide bond containing proteins are often accumulated as
IBs if they are produced in this compartment. Recent studies indicate that mu-
tations leading to a more oxidising intracellular milieu, such as the inactivation
of the genes for thioredoxin reductase (trxB) and glutathione reductase (gor),
promote the formation of correctly folded product and may decrease aggregation
of proteins containing disulfide bonds [17–19]. Although the gor and trxB gene
products are central in the known major reductive pathways in the E. coli cell the
mutations are not lethal and suppressor mutants with an oxidised cytosol can be
easily selected. They even grow well on complex and mineral salt media. The re-
sults from Bessette et al. [17] are very promising even in relation to proteins with
a high content of disulfide bonds, shown for a truncated form of t-PA with nine
disulfide bonds, if foldases such as DsbC or other proteins with a thioredoxin fold
are coexpressed. As a second interesting example recently also the production of
a correctly folded Fab antibody fragment in the cytosol of trxB mutants has been
published [20]. Despite the enthusiasm for the new systems the obtained prod-
uct yields are still low in comparison to processes based on in vitro refolding.
Other disulfide-bond-rich proteins containing more complex structures (e.g.
disulfide knots) have not been successfully produced in vivo in their correct fold
in high amounts yet. In vivo production of BMP2 has been thoroughly investi-
gated under different conditions and with various fusions. Although a soluble
product was obtained in vivo as a fusion to maltose binding protein (MBP) by
Fahnert [21] a high yield of active protein was gained only by in vitro renatura-
tion. In case of another disulfide knot-containing protein, human nerve growth
factor (hNGF), the natural prosequence was shown to support the folding of the
protein and an efficient in vitro refolding process from E. coli IBs was established
[22, 23].
Proteins consisting of different subunits, such as antibody Fab fragments –
containing both, intermolecular and intramolecular disulfide bonds – have been
traditionally produced as IBs. Both separate expression of the heavy and light
chains and coexpression systems have been used and in vitro refolding protocols
have been developed [24, 25]. During the last few years many Fab and single-
chain Fv antibody fragments have already been produced successfully in the E.
coli periplasm or displayed on the bacterial surface in correctly folded form
[26–32]. However, the optimisation studies are still time-consuming and the
yields are low in many cases. It is still not possible to predict the strategy for the
correct in vivo folding of a new Fab fragment even if similar molecules only dif-
fering in the sequence of the variable region have been expressed successfully be-
fore.
Inclusion Bodies: Formation and Utilisation
97

Small proteins with disulfide bonds such as human proinsulin still are mainly
produced as IBs as the preprotein, a fusion protein [33] or by separate expression
of the different chains. Although the formation of disulfide bonds is limited in
bacteria the limitation to produce proinsulin as a correctly folded product is not
due to disulfide bond formation, but to the lack of the formation of the correct
disulfide bonds and the stability of the product [34].
2.1.2
Membrane Proteins
Overexpression of membrane proteins has been a complicated challenge. Such
proteins have been considered as very toxic and difficult to express. Membrane
proteins are mostly enriched in small amounts, but nevertheless they are aggre-
gating certainly due to their surface-exposed hydrophobic regions.
Production of heterologous membrane proteins seems to be more problem-
atic for more and more E. coli membrane proteins such as OmpF, OmpC, PhoE
and LamB which have been successfully produced on very high levels (about 80%
of the total cellular membrane protein) [35]. Membrane proteins not only ag-
gregate but are also considered to be toxic to the E. coli cell.A breakthrough came
in 1996 when Miroux and Walker [36] succeeded in isolating specific E. coli mu-
tants allowing the production of membrane proteins. If membrane proteins are
overproduced in these mutants, new intracellular membranes proliferate con-
taining the recombinant product in the correct conformation [37]. The authors
reported the soluble production of the b subunit of F
1
F
0
ATP synthase, contain-
ing one transmembrane span, in the E. coli BL21(DE3) mutants C41 and C43.
Shanklin [38] also expressed an E. coli membrane protein, the acyl-acyl-carrier
protein synthase (80.6 kDa) efficiently in the C41 mutant. In this case a smaller
amount of this protein (1/3) was also correctly integrated in membranes of the
BL21(DE3) [39] strain. Promising results have also been reported by others for
the expression of active membrane bound cytochromes in these mutants [40, 41].
Saribas et al. [40] succeeded in producing as much as 200 mg l
–1
of cytochrome
P450 2B4 in shake flask cultures.
Alternatively, IB production with following renaturation of urea-dissolved IBs
in phosphate buffer containing n-dodecyl-N,N-dimethyl-1-ammonio-3-propane-
sulphonate (SB12) also seems to be promising. Jansen et al. [42] produced Neis-
seria meningitidis PorA using the E. coli BL21/pET system in high amounts as
very pure IBs at 37 °C and established an efficient in vitro refolding protocol of
this outer membrane protein.
2.1.3
Glycosylation
Many therapeutically interesting proteins from eukaryotes are glycosylated. How-
ever, glycosylation is often not a prerequisite for function but influences activity
and degradation characteristics, such as thermostability [43]. Therefore many
proteins can be produced in bacterial hosts lacking the eukaryotic glycosylation
system for therapeutic applications, but extensive tests are necessary to verify the
98
B. Fahnert et al.
function. Recombinant proteins produced in E. coli may even have beneficial
properties compared to the glycosylated forms. For example, a non-glycosylated
recombinant variant of human tissue plasminogen activator obtained by re-
folding from E. coli IBs showed a longer half-life and lower clearance rate in rats
[44, 45].
On the other hand, glycosylation may affect folding behaviour and solubility.
Therefore, glycosylated proteins may be prone to aggregation if produced in
E. coli.
2.2
Composition and Structure of IBs and Kinetics of IB Formation
Although the refolding of recombinant proteins from IBs has been applied as a
technology for many years, surprisingly only few recent studies have been di-
rected to the composition and structure of IBs, slowly improving our under-
standing. An excellent specific review on this subject was recently published by
Carrio and Villaverde [46].
2.2.1
Architecture and Structure
IBs are refractile intracellular protein aggregates. They can be seen as dense parti-
cles in electron microscopy, but are often even visible in light microscopy. Moreover
the production of IBs also affects the optical density measurement becoming ob-
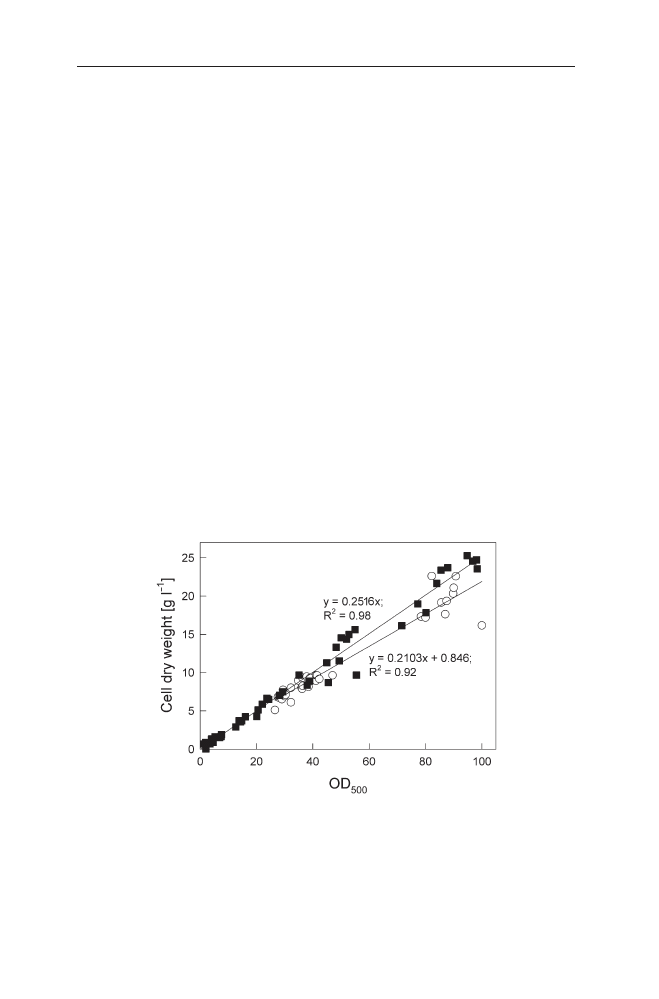
vious after plotting the optical density against the dry cell weight (see Fig. 1).
Inclusion Bodies: Formation and Utilisation
99
Fig. 1
Relation between the optical density of a culture (OD
500
) and the cell dry weight is in-
fluenced by the formation of IBs. Non-induced culture of E. coli RB791 pKK177glucC pUBS520
(filled squares) and culture induced with 1 mmol l
–1
IPTG (open circles). pKK177glucC contains
the gene for the S. cerevisiae a-glucosidase controlled by P
tac
. Data from fed-batch fermenta-
tions have been described in detail earlier [47, 48]. Induction was performed at approximately
OD
500
of 26. The data are from eight fermentations

Recently, interesting studies have been published by Villaverde and colleagues
concerning the fine-architecture of IBs. The authors show by proteolytic studies
that IBs of their model protein, a b-galactosidase fusion to the foot and mouth
disease virus VP1 capsid, are not homogenous structures, but consist of internal
sub-structures with a different accessibility to proteolysis [49]. The proteolytic
studies were supportingly illustrated by scanning electron micrographs showing
the sub-body structure emerging after protease treatment. The study also indi-
cates that the stability of IBs, measured as the digestion rate by trypsin, depends
on the fusion construct. The N-terminal fusion of VP1 to b-galactosidase
(LacVP1) was significantly more stable than the C-terminal fusion construct
(VP1Lac).
In most cases cytoplasmic IBs consist predominantly of the incorrectly folded
recombinant product. Stabilisation possibly occurs mainly via hydrophobic in-
teractions [4]. Some evidence exists that the product protein in IBs can be par-
tially structured [50–52]. Interestingly, decreases in a-helix- and increases in b-
sheet-contents have been detected in IBs in comparison to the native product
[51]. In b-lactamase IBs the b-sheet-content increases with increasing tempera-
ture independent of the location of the product in cytoplasmic or periplasmic ag-
gregates.
IBs can also be formed from native protein [53], but this seems to be the ex-
ception. By attenuated total reflection FTIR Oberg et al. found the secondary
structure in interleukin-1b IBs to be very similar to the native protein indicating
that these IBs contain a folding intermediate with a native-like secondary struc-
ture [52].
2.2.2
Composition of IBs
IBs consist predominantly of the recombinant product [46, 54, 55]. However,
IB preparations often contain a significant part of other cellular proteins
(membrane proteins, RNA polymerase, ribosomal subunit proteins), ribosomal
RNA and DNA [56–58]. These compounds are mainly integrated due to co-
precipitation of cell debris components during the process of IB preparation
[54, 57]. The co-purification of membrane components with the recombinant
product can be limited by the addition of sarkosyl to the lysis mixture [59].
However, cellular proteins may also be directly incorporated in IBs as shown
by Rinas and Bailey [55] for pre-b-lactamase and by Valax and Georgiou
[60] for IBs formed under different conditions. In the latter study contaminat-
ing polypeptides ranged from less than 5% to over 50% of the total protein
content. Here 0.5–13% of the IBs was phospholipids and nucleic acids repre-
sented only a minor impurity in both cytoplasmic and periplasmic IBs. Cyto-
plasmic IBs of mature b-lactamase had the lowest amount of impurities irre-
spective of the growth conditions, but large amounts of outer membrane
proteins and phospholipids were observed in periplasmic IBs from cells grown
at basic pH.
The protein charge can have a major influence on the composition of IBs as
concluded by Chaturvedi et al. [58]. The authors found high amounts of DNA in
100
B. Fahnert et al.

IBs of d-endotoxin, a crystalline protein from Bacillus thuringiensis with positive
electrostatic potential on most of its solvent accessible surface, after overexpres-
sion in E. coli.
In many cases the presence of the contaminating proteins in IBs is related to
incomplete purification after cell lysis. However, even after stringent washing of
IBs with detergents some E. coli proteins have still been found in a significant
concentration. To a major part these proteins belong to the groups of membrane
proteins such as OmpF, OmpC, OmpA [54] and OmpT [61]. These membrane
components could be a result of the cell lysis and washing procedure leading to
further aggregation of the IBs from different cells. However, additionally also
other proteins can be detected in IBs such as the molecular chaperones GroEL,
DnaK, the neomycin gene product and the small heat shock proteins IbpA and
IbpB [46, 61]. Both IbpA and IbpB seem to be generally connected to intracellu-
lar aggregates [62, 63]. Possibly they are among the first cellular detectors of mis-
folded proteins and act as aggregation controllers [64]. All the proteins men-
tioned above were also found in the soluble cytoplasmic fraction with exception
of IbpB. This protein was only detected in the insoluble fraction. It seems sur-
prising that translation-related proteins are not common in IBs, which should be
expected if aggregation is appearing in an uncontrolled way due to the close con-
nection between translation and folding/aggregation processes. There are only
few studies and at least Rinas and Bailey [54] found the ribosomal L7/L12 pro-
teins in b-lactamase IBs.
Although the recombinant product is mostly accumulated as the major com-
ponent in IBs it often does not appear as a unique spot in two dimensional SDS
gels, but as a smear or as multiple spots in the direction of the isoelectric fo-
cussing (e.g. [54, 57, 61]). This changed electrophoretic mobility of the full-length
protein points to chemical product modifications known from the downstream
processing, but may also originate from folding intermediates or from precipi-
tation during isoelectric focussing. Studies addressing this quality aspect during
the in vivo production of IBs have not been published to our knowledge. Fur-
thermore, IBs often contain truncated forms of the product either having the cor-
rect N- or the correct C-terminus. These fragments principally occur due to
pretermination of translation, internal translation start sites or proteolytic degra-
dation. Finally, other analyses also showed abnormally elongated polypeptides of
the product [65–67].
2.2.3
Kinetics of In Vivo Aggregation
In vivo protein aggregation mostly occurs by folding intermediates with surface-
exposed hydrophobic patches normally buried in the interior of a protein mol-
ecule. Such surface exposed patches are prone to aggregation themselves and this
may end up in a chain reaction if folding intermediates further accumulate or
proteins loose their correct three-dimensional structure, e.g. during heat stress.
To answer the question why preferably eukaryotic proteins aggregate in bac-
terial expression systems it is interesting to evaluate differences in the process of
protein synthesis in prokaryotes and eukaryotes.
Inclusion Bodies: Formation and Utilisation
101

Prokaryotic organisms are characterised by a fast translation process of up to
20 amino acids per second [68]. Therefore protein folding occurs in most cases
posttranslationally, although the translation rate can also be slowed down if the
mRNA contains a high number of rare codons.
In contrast the maturation of nascent proteins in eukaryotic cells often begins
cotranslationally in domains as a vectorial process and continues posttransla-
tionally after the release of the protein from the ribosome [69–71]. The rate of
translation in eukaryotes is slower than in E. coli but varies depending on the cell
type and protein. Proper glycosylation is connected to very slow translation rates,
for example [72]. Furthermore, it has been suggested that the slower rate of trans-
lation in eukaryotic cells has an important role in the proper folding by permit-
ting the sequential folding of individual domains during the translation process
[73]. This is especially relevant for proteins secreted into the endoplasmic retic-
ulum, because in its lumen many chaperones and foldases directly bind to the
protein during the translocation process and assist the folding [74].
Aside from the translation rate, aggregation of proteins is dependent on the
protein amount of unfolded peptides in a solution but is also influenced by the
total concentration of proteins [75]. Therefore in vitro refolding of proteins can
be performed by stepwise addition of denatured protein to the refolding solution
to keep the actual concentration of the unfolded protein low and by this the un-
wanted side reactions [76]. Similarly it is known that in vivo a high synthesis rate
of the target protein positively affects aggregation and that one way to decrease
the aggregation probability is to slow down the synthesis rate, e.g. by lowering the
cultivation temperature or the inducer concentration [77].
The kinetics of IB formation have been currently studied in detail for a b-
galactosidase fusion protein by Carrio et al. [78]. In this study the amount of
aggregated target protein was increasing with an approximately constant rate
over 5 h.
IBs are mainly accumulating at the proximal ends of a bacterium. Possibly
many primary micro aggregates fuse to a larger amorphous body if the concen-
tration of the unfolded protein is increasing. This suggestion is strongly sup-
ported by the electron microscopic visualisation of purified and protease treated
IBs by Carrio et al. [49]. Another interesting question is how many IBs accumu-
late per cell. Carrio et al. found mainly two distal IBs per cell shortly after in-
duction, but later the fraction of cells with two IBs decreased to only 20% [78].
The number of IBs and their size is changing with the genetic background of the
cell.A detailed study on VP1Lac by Carrio and Villaverde indicated a low amount
of IB protein, but a high number of aggregates (140) in a groE mutant [79]. This
recent study also strongly supports the hypothesis that the formation of IBs is not
a stochastic process, but that cellular components are involved in the construc-
tion of IBs. IBs were about two times larger and contained more recombinant
protein in a dnaK mutant in comparison to the WT. In contrast, IB formation was
strongly suppressed in a groEL mutant. Aside from being interesting for the de-
sign of new recombinant production strains, these results indicate GroE and
DnaK as the main antagonistic controllers of IB formation. DnaK prevents IB for-
mation by reducing aggregation. GroEL transits the protein between soluble and
insoluble fractions and positively participates in IB formation.
102
B. Fahnert et al.

2.2.4
Stability of IBs
As discussed above, IBs appear as a result of an imbalance between protein syn-
thesis, folding and aggregation reactions. Their formation is mainly supported by
tight hydrophobic interactions. In general IBs are highly stable and resistant to
proteases in vivo. Although IBs mostly contain incorrectly folded proteins, in
some cases enzymatic activities can be associated to enzyme-based IBs [53] and
native-like secondary structures have been detected in other IB proteins [52].
Such aggregation intermediates are prone to proteolysis and, correspondingly,
IBs disappear either due to proteolysis of the resolubilised polypeptide or due to
further protein functionalisation. The equilibrium between formation of aggre-
gates and their resolubilisation has currently obtained more interest, because
bacterial IBs could be seen as models for dynamic and structural analysis of pro-
tein aggregation as it also occurs in several degenerative diseases [79].
In vivo resolubilisation of IBs has been mainly observed in case of homolo-
gous host proteins and fusions to b-galactosidase, but IBs from heterologous pro-
teins often seem to be more resistant to degradation.An extensive study was pub-
lished by Carrio and Villaverde [80] investigating the in situ resolubilisation of
the P22 tailspike protein from IBs. This is interesting since the P22 tailspike pro-
tein is an example for a protein with a very slow and complex folding pathway.
The protein finally forms trimers to be integrated into functional bacteriophages.
Aside from refolding, IB proteins can also be degraded. It has been suggested that
this process is ATP-dependent, which is in agreement with recent molecular data,
showing that aggregated proteins can be solubilised in vivo by ClpB in connec-
tion with the Hsp70 chaperone system [81].
However, the exact network of the resolubilisation of IBs is still not fully un-
derstood.Also data from a larger group of proteins are necessary to evaluate with
respect to whether the in vivo solubilisation affects the quality of IBs in processes
aiming for the production of the target protein in an aggregated form.
2.3
The Physiology of IB Formation
2.3.1
The Metabolic Load of IB Synthesis
Metabolic consequences of the production of recombinant proteins are exclu-
sively reviewed in Chap. 2 by Hoffmann and Rinas. The following paragraph dis-
cusses the molecular physiological aspects in connection to conditions typical for
the production of IBs characterised by a very high synthesis rate from a differ-
ent point of view.
The accumulation of a specific protein population in a cell is connected with
a complex multifactorial synthetic pathway. Parameters include the general rates
of transcription, translation and protein folding, the specific rates of these
processes as well as the rate of formation of a single mRNA molecule, of a single
polypeptide or of a correctly folded protein molecule. Although the single
Inclusion Bodies: Formation and Utilisation
103

processes and their reactions have been thoroughly investigated in vitro and are
well understood, the complex cellular network is actually still too complex to be
thoroughly considered concerning the optimisation of a recombinant process.
Therefore all optimisation studies still have to be performed on a trial and error
basis.
The copy number of the target protein gene has a major impact on the rate of
product accumulation, because it is the basis of the signal amplification cascade
(see Fig. 2). The gene copy number is determined by the origin of replication of
the plasmid and is mostly in the range of 20–50 copies per cell in the case of
medium-copy plasmids or up to more than 100 copies for high-copy number
plasmids.
The copy number basically affects the sum of the cellular components engaged
in the synthesis process and therefore has a significant impact on the competing
reactions in the cell. At the start of a screening process for product optimisation
a low copy number of a stably maintained vector is strongly recommended, since
signal amplification is more easily and more accurately controlled at the level of
transcription and translation than at the level of copy number control. Further-
more, the gene copy number not only depends on the plasmid origin, but also
changes during a process depending on the strain, the growth medium, the
growth rate and the cultivation temperature. A proper control of the exact copy
number is rarely possible for vectors with a higher copy number.
Most optimisation strategies aim to control properly the transcription rate.
This includes the choice of an inducible promoter and the variation of the in-
ducer concentration based on a given expression system. It may be important to
consider that the inducer concentration influences the number of mRNA mole-
cules produced per time, but does not affect the synthesis rate of a single mRNA.
It is assumed that the E. coli RNA polymerase has a constant transcription rate
of about 40 to 50 nucleotides per second lowered by pausing sites leading to tran-
sient stoppage of the transcription especially under detrimental environmental
conditions and resulting in up to a 50% reduction of the total transcription rate
[82]. In case of E. coli RNA polymerase the transcription rate is in the order of the
translation rate of ribosomes varying dependent on the codon bias between ap-
proximately 15 nucleotides per second for rare codons and 62 nucleotides per
second for abundant codons [68]. These rates for transcription and translation
suggest that ribosomes stack closely to each other behind the E. coli RNA poly-
merase if the recombinant genetic construct contains a strong ribosome binding
104
B. Fahnert et al.
Fig. 2
Scheme of the signal amplification cascade from a recombinant gene to the final
product (a, b and c are factors indicating the degree of amplification in each step)

site (Fig. 3). In this case the synthesis time of a single protein molecule is limited
either by the transcription rate of the RNA polymerase or by the codon usage if
rare codons with low translation rates are contained at higher levels in the se-
quence of a recombinant gene.
T7 RNA polymerase in connection with a T7 promoter is generally applied as
an alternative strong expression system [83]. This polymerase synthesises the
mRNA with a four- to fivefold higher transcription rate (about 230 nucleotides
per second) than E. coli RNA polymerase [84–86]. As the initiation rate of both
RNA polymerases is similar (E. coli RNA polymerase: 1–3 per second; T7 RNA
polymerase: 1 per second) the higher accumulation of proteins in the T7 systems
is most probably due to the high elongation rate of the enzyme. Therefore in T7
RNA polymerase systems the synthesis rate of the single protein molecule is not
limited by the formation rate of the mRNA, but it is only limited by the codon us-
age of the gene. That is why many proteins show a higher accumulation rate than
in case of E. coli RNA polymerase based expression systems. However, the high
synthesis rate of mRNA may be also a disadvantage. The mRNA not covered by
ribosomes can be easily attacked by RNases or form stable secondary structures
(see Fig. 3B).
The higher rate of synthesis of proteins by the T7 RNA polymerase is
possibly also the reason why formation of IBs seems to be more significant with
this expression system than with E. coli RNA polymerase based expression
systems.
The described physiological data indicate that a high expression rate of the re-
combinant protein should be optimal for an IB production process. Therefore a
strong ribosome binding site is beneficial. In contrast a weaker ribosome bind-
ing site could improve the yield of correctly folded product by causing a higher
distance between the ribosomes which eventually should decrease the aggrega-
tion between different product molecules but may also decrease the mRNA sta-
bility.
The codon bias of a gene is a major factor for the synthesis rate of a target pro-
tein. The codon bias varies between different organisms and is well reflected by
the respective tRNA population [87]. If an mRNA from a heterologous target gene
is overexpressed in E. coli, differences in codon bias of the gene to the codon bias
of E. coli can impede translation. In practice the presence of a small number of
rare codons does not affect the target protein synthesis very much. However, the
accumulation of a recombinant gene product can be very low if the gene contains
clusters and/or numerous rare E. coli codons. The most severe effects have been
Inclusion Bodies: Formation and Utilisation
105
Fig. 3A, B
Schematic drawing of the hypothetical distribution of ribosomes on their cognate
mRNAs in relation to the RNA polymerase in recombinant systems with a strong ribosome
binding site and with: A E. coli RNA polymerase; B T7 RNA polymerase

observed in case of multiple consecutive rare codons situated near the N-termi-
nus of a coding gene sequence.
Analysis of the codon bias in all 4290 E. coli genes reveals a number of un-
derrepresented codons. In particular arginine codons AGG, AGA and CGA,
isoleucine codon AUA and leucine codon CUA represent less than 0.1% of the
codon bias of all genes and less than 8% of the population of the codons for the
specific amino acid [88]. On the assumption that the codon bias was evolution-
ary optimised to support fast growth of an organism, further rare codons can be
defined rarely contained in genes which are highly and continuously expressed
during fast growth [89]. If such abundantly expressed genes are analysed the
codons GGA for glycine, CGG for arginine and CCC for proline must also be con-
sidered as rare since they only represent less than 2% of the respective popula-
tions for a specific amino acid [90]. Problems with the codon bias of a recombi-
nant target gene can be solved either by redesigning the codons through
site-directed mutagenesis or by a higher expression of the rare tRNAs. The first
method can be very laborious and is only used in cases where multiple consec-
utive codons occur in a gene, but coexpression of tRNAs has become a valuable
tool and a number of vectors as well as E. coli strains for overexpression of dif-
ferent tRNAs are commercially available.
The problem of the codon bias seems to be very relevant for the production
of target proteins in IBs if a high synthesis rate of the protein is desired since the
codon bias directly influences the translation rate as described above. An exper-
imental indication that a higher translation rate stimulates aggregation was ob-
tained from overproduction of a recombinant yeast a-glucosidase rich in AGA
and AGG codons. Co-overexpression of the cognate argU tRNA increases the to-
tal a-glucosidase amount per cell from 4% of the total cell protein to about 16%.
The positive effect of argU on the translation speed is in accordance with the in-
vestigations of the translation rates for single codons by Soerensen and Peder-
sen [68] and with the analysis of the tRNA levels in E. coli by Kurland and
coworkers [87]. The higher speed of translation not only leads to a higher amount
of product, but also increases the relative amount of aggregated product. In con-
trast the expression of soluble a-glucosidase is decreased if argU was co-over-
expressed (Table 1).
The optimisation of the codon bias of a target gene is very important for the
production of high quality IBs because a non-optimised codon bias also increases
the rate of translation errors appearing due to codon hopping, pretermination
of translation, frame-shift mutations etc. However, the codon bias also directly
influences the maintenance of the cellular translation system. The preferential
withdrawal of rare tRNAs directly inhibits protein synthesis and cell viability [93].
A high synthesis rate of a protein with slow folding characteristics may exceed
the available resources of foldases, chaperones and proteases. In this context IBs
may represent reservoirs of protease-resistant protein steadily accumulating un-
til chaperones and proteases become available either for successful protein fold-
ing or for proteolysis. This model, recently suggested by Carrio and Villaverde
[46], is supported by data on higher IB accumulation in a dnaK mutant (see also
Fig. 5). In contrast, a groEL mutant had a lower content of IBs and a higher
amount of folded protein which might be explained by the early function of the
106
B. Fahnert et al.

Hsp70 chaperone system in the protein folding pathway and the GroEL system
as a “second stage” folding supporter (see “Molecular Components of Physio-
logical Stess Responses in E. coli” by L.M. Wick and T. Egli, this volume).
2.3.2
The Response to Misfolded Protein
2.3.2.1
Stress Responses
During their evolution all organisms have evolved mechanisms to respond to un-
favourable environmental conditions. In bacteria the corresponding adaptational
network ensuring a better survival of the cells is mainly controlled at the level of
transcription. An environmental stimulus such as heat induces a number of reg-
ulatory cascades. The different modules are grouped in dependence on their spe-
cific response regulators into regulons. The complexity of the bacterial response
network results from the possibility that one stimulus is inducing different reg-
ulons and that different genes can be multiple controlled.
Strong induction of recombinant protein expression is an artificial stress redi-
recting the metabolism of a bacterium to the production of a specific target pro-
tein.Although the metabolic network of a cell has some flexibility to increase cer-
tain pathways, the load of a high induction of a gene with a strong promoter and
a strong ribosome binding site is considerable with respect to necessary energy
resources [94] and imposes a stress situation. Compared to stress situations oc-
curring in natural evolution, recombinant protein synthesis is possibly most sim-
ilar to the induction of lysogenic bacteriophages or to bacteriophage infection in
general. In these situations the bacteriophage occupies the bacterial protein syn-
thesis system for its own purposes to produce daughter phages. In a similar way
Inclusion Bodies: Formation and Utilisation
107
Table 1
Influence of the co-overexpression of the rare argU tRNA on the concentration and
activity of a-glucosidase expressed in E. coli RB791
a-Glucosidase
a
–argU overexpression
+argU overexpression
–IPTG
+IPTG
–IPTG
+IPTG
Activity (U ml
–1
)
b
0.42
3.18
0.08
1.07
(U mg
–1
)
c
0.03
0.41
0.007
0.12
Total (mg ml
–1
)
b
0
111.44
0
446.13
(mg mg
–1
)
c
0
14.36
0
49.64
(% of TCP)
d
0
4
0
16
a
Data calculated at 3–4 h after induction.
b
mg a-glucosidase per ml cultivation broth.
c
Units or mg a-glucosidase per mg cell dry weight.
d
% of total cell protein.
Data according to Lin [91]. Experimental conditions: E. coli RB791 pUBS520 glucC containing
the glucC gene under P
tac
control. Coexpression of argU by pUBS520 [92]. Experiments were
performed under fed-batch conditions at 35 °C on glucose based mineral salt medium with
induction with 1 mmol l
–1
IPTG in the fed-batch phase as described earlier [47].

the induction of a recombinant gene channels the bacterial resources for the pro-
duction of the target protein. Although the strong synthesis of a recombinant
protein can principally induce a number of different stress signals connected to
cellular regulons such as the stringent response, the general stress response and
the SOS response it depends on the strength of the competition whether the cor-
responding gene expression pattern can be found, because all responses are con-
nected to the synthesis of new proteins. If the competition is strong, the protein
synthesis apparatus including transcription and translation, can be fully occu-
pied by the synthesis of the product and no cellular responses are observed, but
the cell will loose its ability to divide and slowly die [48]. However, if the synthe-
sis of the product allows the synthesis of cellular proteins in parallel, typical stress
responses can be observed, such as the SOS response [95]. Only a heat shock like
response is always found if misfolded forms of the target protein accumulate in-
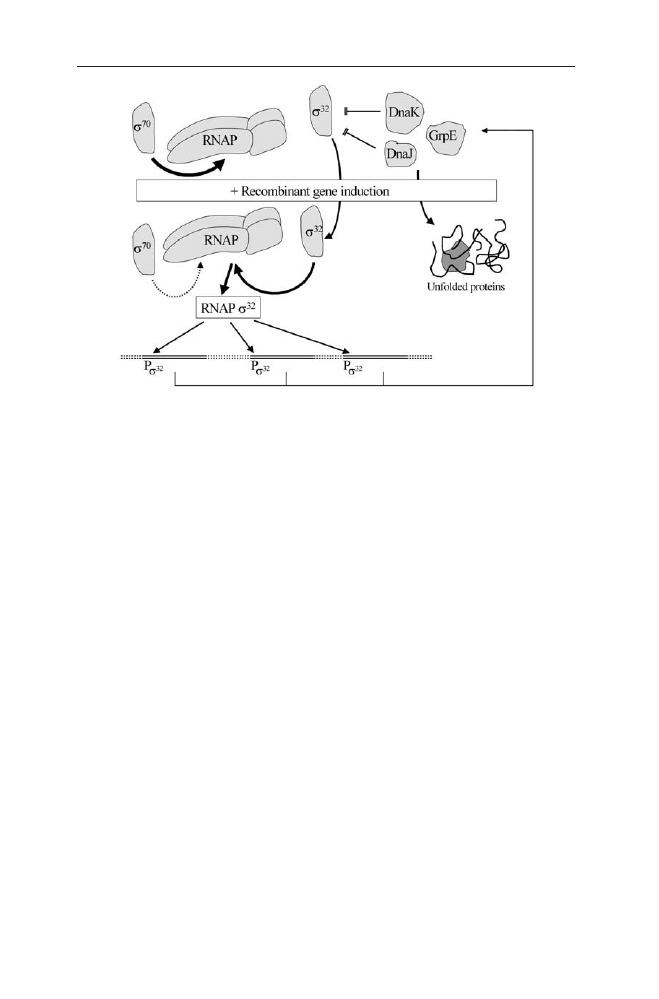
dependent of the expression system used (e.g. [96–99]). The high speed and com-
petitiveness of the heat shock response by s
32
can be explained from the regula-
tory mechanism of this response.
As discussed above, IBs result from aggregation of non-native polypetides. In
connection to bacterial stress responses the accumulation of proteins recognised
as non-functional or unfolded molecules is very critical and induces mechanisms
to counteract this process. Analogously, in nature unfolded proteins appear dur-
ing heat stress causing the unfolding and following aggregation of proteins. It has
been elucidated that the bacterial heat shock response is regulated by a sensitive
measure of the appearance of unfolded molecules. This regulation centrally in-
volves the heat-shock sigma factor s
32
and its binding to the DnaK and DnaJ
chaperones. Both chaperones contribute to downregulation of s
32
activity and
stability (see Fig. 4 and see “Molecular Components of Physiological Stess
Responses in E. coli” by L.M. Wick and T. Egli, this volume).
After a heat shock, while proteins are inactivated and aggregated proteins ap-
pear, s
32
is released from the chaperones leading to higher stability and activity
of s
32
. Consequently, s
32
edging out the house-keeping sigma factor s
70
of RNA
polymerase and the RNAPs
32
holoenzyme initiates transcription from promot-
ers belonging to the heat-shock regulon. Additionally the heat shock contributes
to transient inactivation of s
70
[100], which however is not relevant in recombi-
nant protein production without temperature shift.
Among others, the s
32
connected response leads to increased transcription of
dnaK, of other chaperone genes and of heat shock related protease genes such as
lon and clpP [101, 102]. Therefore the concentration of both the Hsp70 chaper-
one proteins (DnaK, DnaJ and GrpE) and the Hsp60 chaperonins (GroEL and
GroES) increases rapidly and transiently. This transient increase of the chaper-
ones furthermore leads to binding and degradation of s
32
causing the transient
character of the heat shock response [103, 104].
Unlike a heat shock primarily causing the unfolding of existing proteins, in-
duction of a recombinant protein causes the synthesis of misfolded proteins. Sim-
ilar to a heat shock this also induces a heat shock-like response and corre-
spondingly enhanced proteolytic activities [105–108] and increasing levels of
Hsp60 and Hsp70 chaperones [21, 97, 109–112]. Strong overexpression in con-
nection to protein aggregation also induces ibpA and ibpB gene expression and
108
B. Fahnert et al.
incorporation of these proteins in IBs [62, 110]. Although even the presence of
plasmids is known to increase the levels of heat shock proteins in comparison to
the plasmid-free host strain [113] a true induction of the heat shock like response
is found if a temperature sensitive promoter, such as the l P
L
and P
R
promoters
with the thermosensitive cI
857
repressor, is used for induction of the target pro-
tein [110]. However, a heat shock like response is also induced without a change
of the temperature by the accumulation of abnormally folded protein [108] and
by the accumulation of export-defective secretory protein precursors [114].
The regulation of the heat shock like response to misfolded recombinant pro-
teins is similar to a heat shock. The accumulation of newly synthesised misfolded
proteins also attracts the chaperones and leads to an activation and stabilisation
of s
32
[103, 115, 116]. However, this response is longer lasting [99, 117] as the syn-
thesis of the recombinant protein is continuing. Furthermore, as s
32
must com-
pete for the very effective synthesis of the recombinant protein, the accumulation
of new chaperones is lower than after a heat shock, inactivation of s
32
is retarded.
The detailed kinetics of this however remains to be evaluated.
Aside from a potential positive effect of the heat shock-like response on the
folding of a recombinant protein by increasing the level of chaperones, the in-
duction of the heat shock response also can negatively affect the production of
recombinant proteins because it is connected to induction of cytoplasmic (Lon,
Clp) and periplasmic (DegP, OmpT) proteases [118]. Therefore, methods to re-
duce the heat shock-like response and thus the proteolytic activities during over-
expression of target proteins can submit positive effects. Such positive effects
were obtained by lowering the cultivation temperature [108, 119], by using htpR
or lon mutants [119] and by antisense down-regulation of s
32
[120].
Inclusion Bodies: Formation and Utilisation
109
Fig. 4
Regulation of the s
32
connected unfolded protein response after induction of recombi-
nant genes

2.3.2.2
Chaperone Action
Although the correct folding of the three-dimensional structure of a protein is an
intrinsic characteristic, chaperones and foldases decrease the folding time and
protect the protein from aggregation. It has been calculated that in E. coli only
10–20% of the host’s proteins are folded with the help of the Hsp60 and Hsp70
chaperone systems during exponential growth and this increases to about 30%
under heat shock conditions [121–123]. In contrast, many of the interesting het-
erologous target proteins fold slowly and would therefore benefit from the avail-
ability of chaperones. Accordingly there are many examples where coexpression
of chaperones significantly increased the yield of correctly folded product.
The chaperones prevent aggregation of heat-inactivated proteins and other
misfolded proteins in an ATP-dependent manner. Supported by GrpE and ClpB,
Hsp40 (DnaJ) and Hsp70 (DnaK) prevent the aggregation of nascent protein mol-
ecules (in case of at least 20% of all cellular proteins sized 14–90 kDa) until their
synthesis is finished. DnaK binds to a short linear consensus motif of 4–5 hy-
drophobic amino acids (mainly leucine) flanked by basic ones [124–127]. Hsp60
substrates consist preferentially of partially structured, hydrophobic protein mol-
ecules [128]. In case of a misfolding (indicated by exposed hydrophobic areas) of
the now non-linear, compact protein it can enter the hydrophobic cavity of the
GroEL chaperonin. A special sequence is not needed [129], but the non-native
state is sufficiently attractive. This is the fate of about 30% of all cellular proteins
sized 10–55 kDa. An additional binding of trigger factor to GroEL even enhances
the interaction with the substrate. By assembling GroES (Hsp10) and consuming
ATP the conformation of GroEL changes. This causes the bound substrate pro-
tein to be released from the cavity surface of GroEL. So it is enabled to fold again
according to the principle of the Anfinsen cage (spontaneous self-assembly of
polypeptides [10]) in the hydrophilic milieu of the chaperon’s interior. The pro-
tein is released after the dissociation from the chaperone. If the correct confor-
mation were not achieved the protein could enter GroEL again [130, 131].
During the folding process proteins can be transferred from one chaperone
system to the other. Thereby the chaperones do not refold the protein actively to
the correct structure, but they lower the aggregation potential by repeated bind-
ing and releasing of the substrate and allow the protein successively to reach a
kinetically favourable state and finally the active conformation.
Irreversibly damaged proteins may be further presented to the Clp chaper-
ones. These proteins are structurally similar to GroEL in having an interior cham-
ber formed by one or two stacked rings of six or seven monomers. ClpB of E. coli
can apparently act exclusively as a molecular chaperone, whereas other Clp fam-
ily members such as ClpA and ClpX function both as chaperones and as com-
ponents of ATP-dependent proteases. Damaged proteins may be either directed
to further degradation by proteases such as ClpPX [132] or alternatively they can
be resolubilised by the ClpB chaperone in concert with the Hsp70 chaperone sys-
tem [81, 133, 134].
The mechanism behind this is that hydrophobic regions of polypeptides are
exposed either as the newly made proteins emerge from the ribosome or because
110
B. Fahnert et al.
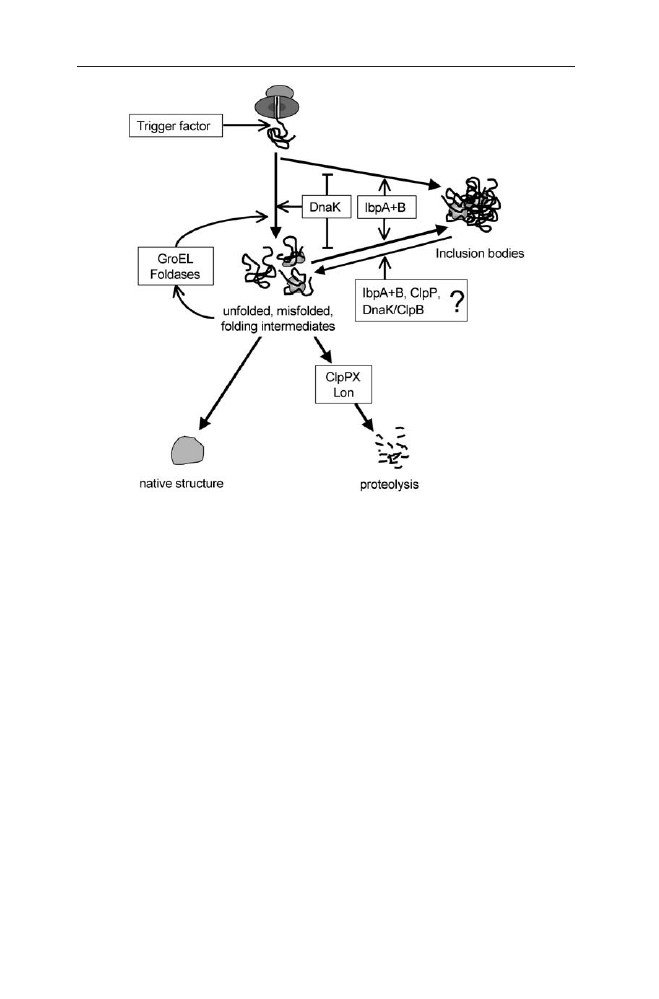
of subsequent misfolding or failure to assemble properly (see Fig. 5). These ex-
posed regions are subject to binding by any of the chaperones or by the ATP-de-
pendent proteases. Chaperone binding and release of folding intermediates may
allow proteins to reach their native conformation or may return them to the pool
of non-native proteins that can rebind chaperones or proteases. Protease bind-
ing followed by ATP-dependent unfolding and subsequent degradation removes
the protein from the pool of non-native proteins. At a high rate of synthesis of
polypeptides needing the chaperone and foldase systems, unfolded, misfolded
and partially folded proteins will eventually aggregate to IBs.
Although the chaperones act most generally to prevent aggregation, they are
also able to dissolve aggregates [133, 135, 136]. The resolubilisation of protein ag-
gregates seems also to be connected to the action of two low molecular weight
proteins, IbpA and IbpB [62]. The synthesis of these proteins is also induced by
heat shock through s
32
[137]. The function of these small proteins is still unclear
[138]; however there is indication that IbpB is a molecular chaperone that assists
the refolding of denatured proteins in the presence of other chaperones [64]. In
case of malate dehydrogenase and lactate dehydrogenase it has been shown that
IbpB-stabilised proteins alone do not refold spontaneously, but they are prefer-
entially delivered to the Hsp70 chaperone system where they refold in a strict
Inclusion Bodies: Formation and Utilisation
111
Fig. 5
Model of competitive pathways for aggregation, refolding and protein degradation for
the process of recombinant protein production (Æ positive effect,
negative effect)

ATPase-dependent manner. GroEL/GroES chaperones do not interact directly with
IbpB-released MDH, but mediate further processing of Hsp70 released folding in-
termediates. In this connection IbpB seems to bind and stabilise aggregation-
prone folding intermediates during stress and thus prevents the further irre-
versible aggregation of these proteins. On the other hand, as an integral part of
a cooperative multi-chaperone network IbpB may be involved in the active re-
folding of stress-denatured proteins by delivering them to the Hsp70 and Hsp60
chaperone systems [64].
2.3.2.3
Periplasmic Response to Misfolded Protein
In addition to the induction of the s
32
response by proteins overexpressed within
the bacterial cytoplasm, a s
24
(s
E
) dependent heat shock response is induced by
misfolded proteins in the periplasm. The response to protein misfolding in the
cell envelope is a finely tuned system regulated by a cascade of phosphorylation
and dephosphorylation reactions [139]. Interestingly, overexpression of periplas-
mic proteins not only induces the s
E
regulon, but also leads to a significant in-
crease of the expression of the cytoplasmic s
32
dependent chaperones GroEL,
GroES, DnaJ and DnaK as revealed by transcriptome analysis [21]. Similar to the
cytoplasm there exist chaperones and proteases in the periplasm contributing to
degradation or refolding of damaged proteins. One example is the protease DegP
consisting of a double ring to be entered by a protein before it is proteolytically
cleaved. Therefore the protein has to be partially unfolded or disaggregated, re-
spectively. Interestingly, DegP changes its tertiary structure and activity in a tem-
perature-dependent manner. It is only a protease at high temperatures but may
act as a chaperone in the lower temperature range [140, 141].
2.3.2.4
Response to Misfolded Proteins in Other Organisms
The response to unfolded proteins is not restricted to E. coli but common for
other microorganisms, too. In Bacillus subtilis overexpression of insoluble pro-
teins induces the so-called class I and class III heat shock genes (according to the
classification of Hecker et al. [142] and Derré et al. [143]) as detected by Mogk et
al. [144] and Jürgen et al. [145]. Mogk et al. [144] found non-native proteins to in-
duce the CIRCE regulon controlled by the HrcA repressor. The authors supposed
that high levels of non-native proteins to titrate the GroE chaperonins, which pre-
vents reactivation of the HrcA repressor and thus causes induction of class I heat
shock genes. Jürgen et al. [145] have confirmed these results for another recom-
binant protein and showed additionally induction of clpP, clpC and clpE, be-
longing to the class III heat shock proteins. Interestingly, the authors proved that
the Clp proteins ClpC, ClpP and ClpX are associated with the IBs, suggesting that
these proteins also in Bacillus subtilis contribute to resolubilisation of recombi-
nant aggregates. In contrast to E. coli, the authors found no significant induction
of the mRNAs of the proteases LonA and LonB, which they discuss in relation to
the major importance of the ClpP protease with its subunits ClpC or ClpX in
112
B. Fahnert et al.

Bacillus subtilis. Additional differences between the response of Bacillus subtilis
and E. coli have been detected by Jürgen et al. [145] in connection to the signif-
icant increase of the mRNA levels of genes encoding purine and pyrimidine syn-
thesis enzymes (purB, purC, purM, pyrA, pyrD) and ribosomal proteins (rpsA,
rpsB, rplJ) in Bacillus subtilis.
2.3.3
Host Characteristics for High-Quality IBs
In vivo protein aggregation is mainly connected to the competition of protein
synthesis, folding and misfolding of a certain recombinant protein. Parameters
in this process are the intrinsic characteristics of the protein, but this process in-
volves additionally external factors of the host system such as availability of
chaperones and foldases. Furthermore, aggregates principally can be resolu-
bilised by the bacterial chaperone and protease systems.
In conclusion the accumulation of IBs seems to be best at a high synthesis rate
of aggregation prone proteins in connection to a low content of chaperones (es-
pecially DnaK) and proteases. Therefore high induction at the transcription level
and strong ribosome binding sites are generally used, e.g. strong promoters such
as P
lacUV5
in connection to the P
10
promoter of the T7 phage or P
tac
. These pro-
moters are induced with a high concentration of IPTG (0.4 to 2 mmol l
–1
final
concentration).Also heat inducible systems, e.g. the phage l promoters P
L
and P
R
can be applied.
If the target protein is proteolytically sensitive or the IBs are very unstable,
chaperones and heat shock related proteases could be suppressed by the use of
rpoH mutants, protease mutants or by antisense titration of rpoH [120]. In this
context E. coli BL21 is considered to be a useful host strain for IB formation be-
cause of the lon mutation [146].
The small heat shock proteins IbpA and IbpB may be involved in the con-
struction and resolubilisation of IBs. Our own data on two different recombinant
proteins suggest that IBs produced in ibp mutants may be of higher purity (un-
published). However, in a recent study from Carrio and Vilaverde [46] an ibp mu-
tant also showed much lower production. More studies are needed to evaluate the
effects of such mutations with respect to different target proteins.
Changes in the concentration of the main chaperon systems may affect the
quality and concentration of IBs.A dnaK mutant contained a significantly higher
amount of aggregated VP1Lac in comparison with a WT and all product was ag-
gregated [79]. Additionally the IBs had a higher stability if protein synthesis was
inhibited suggesting that a dnaK mutant could be a good host strain for IB-based
recombinant processes.
As IB formation is normally connected with a short induction time (2–5 h)
and high product synthesis, the codon bias is a major factor determining the
quality of the aggregates. Overexpression of rare tRNAs can increase the total
yield but also the relative amount of product contained in IBs by increasing the
translation rate (see Table 1).
Inclusion Bodies: Formation and Utilisation
113

2.4
IB Based Processes Versus Soluble Production
2.4.1
Cultivation Conditions Promoting Aggregation
Aggregation is caused by protein-protein-interactions. Therefore the high con-
centration of unfolded proteins within the cell during recombinant protein pro-
duction is especially aggregation promoting. The so-called crowding effect [147]
is characterised by competing interchain and intrachain interactions leading to
wrong intermolecular contacts. Secreted proteins are less likely to aggregate be-
cause of the increased volume of the periplasm and the thereof resulting dilution
effect [16].
All the processes from transcription to folding affected by recombinant ex-
pression have to be considered on both sides (target protein and host) and in
their interaction in order to find the optimal cultivation conditions (temperature,
medium, cell density, host strain). The most common and still useful approach
is empirical testing since every target protein is different [16, 148, 149].
The following conditions have been shown to promote aggregation: high tem-
perature, high cell density, high concentrations of the inducer and a short induc-
tion time [77, 150, 151]. By varying the expression level and the input of thermal
energy, conditions leading to an optimised yield of IBs can be found. Complex me-
dia are favouring the formation of IBs as well [152]. The high temperatures help
to cross the thermodynamic threshold necessary for intermolecular reactions and
thus aggregation. All the expression conditions mainly influence the aggregation
kinetics by varying the protein concentration and synthesis rate.
2.4.2
IBs as a Result of Failure in Formation of Correct Disulfide Bonds
In prokaryotes, proteins mainly fold post-translationally due to the rapid trans-
lation (see above). However, a cotranslational folding of certain proteins might
also occur. In contrast, eukaryotic proteins are considered to fold cotranslation-
ally in domains. This is thought to be one reason for the often incorrect folding
of eukaryotic proteins in prokaryotic hosts [71, 73]. The tendency of IB forma-
tion is, among other parameters, dependent on the number of cysteines. Ac-
cording to this the cystine-rich mammalian proteins are reported to be especially
difficult to express in E. coli solubly [153].
Moreover, the natural folding environment is different in many other aspects.
In eukaryotes the disulfide bonds are formed within the endoplasmic reticulum.
It contains about 100 g l
–1
of protein. Permeases import ATP from the cytoplasm.
It is needed by some chaperones. As the endoplasmic reticulum is oxidising (ra-
tio of reduced to oxidised glutathione: 1–3:1 [154]) it provides the basis for disul-
fide bond formation. Different foldases are present. The PDIs (protein disulfide
isomerases, e.g. PDI, Ero1p) perform the disulfide shuffling and the PPIs (pep-
tidyl-prolyl-cis/trans-isomerases, e.g. cyclophilin B) the propeptidyl-bond iso-
merisation [155]. PDI has got two active site motifs (Cys-X-X-Cys, thioredoxin
114
B. Fahnert et al.

superfamily). One of them is oxidising and the other isomerising. The reoxida-
tion of PDI is probably performed by Ero1 [156]. There are also the heat shock
proteins 70 (e.g. BiP), 40 (e.g. Sec63p), 90 (Grp94) and lectins (calnexin, calreti-
culin, both of them being glycoprotein chaperones) [14]. Glycosylation improves
solubility and is often a prerequisite of correct folding. In the endoplasmic retic-
ulum ATP is consumed for forming disulfide bonds. In contrast the presence of
ATP is unlikely in the prokaryotic periplasm [157].
The catalysis of the formation of disulfide bonds is more complicated in the
periplasm than in the endoplasmic reticulum, because its oxidising milieu (redox
potential) is affected by the environment of the cell. Small molecules are con-
stantly diffusing through the cell envelope [158]. The foldases of the periplasm
are independent of ATP. Redox proteins catalyse the slow formation of disulfide
bonds. These disulfide bond forming proteins (Dsbs) also belong to the thiore-
doxin superfamily. DsbA oxidises the thiolgroups of proteins within the
periplasm and is reoxidised by DsbB. Spontaneous formation of disulfide bonds
and even the effective catalysis performed by DsbA is at random. DsbC, E and G
repair the non-native bonds and are reduced again by DsbD. It is not known so
far when the disulfide bonds are formed (co- or post-translationally) and
whether the involved cysteines are chosen specifically [17, 158–162].
Due to the known differences between disulfide bond formation in the natural
eukaryotic environment of the most target proteins and the recombinant
prokaryotic host there is no question about the likelihood of misfolding. More-
over the cytoplasm as the most important compartment for the production of IBs
is reducing. Thus the IBs formed there might be mainly due to the absence of the
correct disulfide bonds. Nevertheless IBs found in the periplasm are caused by
the factors (incorrect disulfide bond formation vs folding characteristics) dis-
cussed above.
2.4.3
How to Avoid IBs and to Favour Correctly Folded Proteins
The first problem to occur during heterologous protein expression in E. coli is the
high local concentration of molecules. Moreover the recombinant host is lacking
the specific foldases needed by the product, any compartments and post-trans-
lational modifications [163].
The results of the product expression depend on different factors such as
strain, expression plasmid, induction conditions, temperature and compartment.
The corresponding impact cannot be predicted [164]. So every problem of the
production (e.g. expression, solubility, functionality) has to be solved for every
target protein separately. Solubility, functionality and minimal proteolysis can be
achieved by combining different upstream strategies.
2.4.3.1
Rate of Synthesis
The expression rate and the correct folding of the product are among other pa-
rameters determined by the level of gene induction, by the gene codon bias and
Inclusion Bodies: Formation and Utilisation
115

by the mRNA stability [165]. In actual optimisation procedures for soluble,
correctly folded proteins, the rate of synthesis is mostly controlled at the in-
duction level. The best results are usually obtained by lowering the cultivation
temperature down to 18–25 °C and applying low inducer concentration
[77, 152]. Therefore processes for soluble proteins are mostly characterised by
a low specific synthesis rate and long cultivation time after induction (6–24 h)
[166, 167].
The rate of synthesis of a single polypeptide depends on the elongation rate
in the translation process dependent on the codon bias [68]. It is well known that
an accumulation of unfavourable codons within a gene (see above) causes read-
ing errors (e.g. frameshifts, hops), mRNA instability and degradation [168, 169],
misincorporation of other amino acids at rare codons [170] and even death of the
cells [93]. Therefore, extra copies of rare tRNAs, e.g. argU, ileY and leuW, are in-
troduced into the host cells resulting in up to a 100-fold increase of the product
yield [171]. Alternatively the sequence of the recombinant gene can be adapted.
The codon usage of the first 20 amino acids is crucial for the expression level of
the product [172–174]. However, it is not considered in most cases that an im-
provement of the codon bias to abundant codons increases the probability of ag-
gregation by a high specific synthesis rate. Moreover, although the rate of trans-
lation might have a profound effect on the in vivo folding reaction and the
accessibility of chaperones, it has not been possible yet to tune the translation
process.
2.4.3.2
Fusion Proteins
Aggregation problems and other limitations, such as inefficient translation ini-
tiation and an incomplete removal of the start methionine, can often be solved
by fusion of the target protein to other proteins. Both eukaryotic (glutathione-S-
transferase from Schistosoma japonicum) and prokaryotic (protein A from
Staphylococcus aureus; maltose binding protein, thioredoxin and DsbA from
Escherichia coli) proteins are used as partners [34, 175]. Additionally the folding
and solubility promoting effect of the prosequences of eukaryotic proteins dur-
ing their recombinant production have been shown [21, 176, 177]. Those prose-
quences (steric chaperones) are essentially supporting the correct folding due to
lowering the activation energy and covering hydrophobic regions causing a sta-
bilisation of folding intermediates within the folding pathway. Moreover,
dimerisation is supported [178].
Fusion proteins are applied both in basic research and biotechnology not only
because of their folding promoting effect, but also with respect to using them as
a purification and immobilisation tool [179].
The order of the fusion partners is often determining the solubility of the
product. An N-terminal fusion of maltose binding protein leads to a soluble ex-
pression in most cases, whereas IBs accumulate in case of C-terminal fusions.
Thus the preliminarily translated maltose binding protein prevents aggregation
of the partner protein during its translation. The yield of the renaturation of
those IBs is higher than that of the unfused target protein [180–182].
116
B. Fahnert et al.

A comparison of six different insoluble target proteins each fused to maltose
binding protein, glutathione-S-transferase or thioredoxin showed maltose bind-
ing protein to be the most effective one in promoting solubility independent of
the expression temperature. Thioredoxin was more effective than glutathione-S-
transferase [183]. So the success is not necessarily correlated with the relative size
of the fusion partners.
If the target protein is soluble but misfolded, aggregation could occur at a high
expression level in spite of the fusion. However, this is prevented in case of mal-
tose binding protein being the fusion partner [183]. Being a periplasmic protein
maltose binding protein directs by its native signal peptide the whole fusion to
the periplasm. The cytoplasmic variant of the fusion protein (without the signal
peptide) can also be analysed, because maltose binding protein is correctly folded
within the cytoplasm as well. The target protein can also be enabled by maltose
binding protein to form the functional oligomers [184].
The beneficial influence of maltose binding protein is caused by both its mol-
ecular characteristics and its interaction with the target protein. Comparable to
other periplasmic substrate binding proteins (hydrophilic with some hydropho-
bic amino acids, [185]) maltose binding protein has – with or without its ligand
bound – a chaperone-like effect [186]. Due to its interaction with membranes
maltose binding protein is able to contact the hydrophobic areas exposed by mis-
folded proteins [187]. Usually the expression of proteins is downregulated after
a heat shock, but the maltose binding protein level remains constant at about
1 mmol l
–1
in the periplasm (50-fold excess to the membrane partners) [188]. The
chaperone-like effect has already been seen at 0.01 mmol l
–1
although the affin-
ity to the actual substrate is 1 mmol l
–1
[187, 189]. Thus maltose binding protein
seems to be some kind of chaperone held in reserve by the cell [187].
Meanwhile the unique benefit of maltose binding protein has often been pub-
lished [150, 183, 190]. Although most of the maltose binding protein fusions are
soluble the target proteins are not always folded correctly [21, 181, 182, 191, 192].
There are different opinions about the reason for the nevertheless solubility
promoting effect. These are profoundly discussed by Fox and co-workers [193].
Four (may be even concurring) reasons are considered. Maltose binding protein
could form micelles comparable to detergents. Being a pace-setting folding an-
chor, maltose binding protein might reduce the number of possible conforma-
tions. It could also act as a chaperone magnet or via its own intrinsic chaperone
activity. Fox et al. [193] favour the folding rate of maltose binding protein to be
promoting the solubility of the target protein.
Thus it is not always known how fusions avoid IBs and achieve correctly folded
proteins, but the effect is undeniable.
2.4.3.3
Coexpression of Chaperones and Foldases
As discussed in Chap. 4 by Hoffmann and Rinas, chaperones are folding catalysts
via preventing aggregation of nascent chains and unfolding of misfolded pro-
teins. They do not feature any folding repair mechanism concerning the shuffling
of disulfide bonds. So the success of the catalysis depends on the folding envi-
Inclusion Bodies: Formation and Utilisation
117

ronment and even more on the target protein. This is also why chaperones are
sometimes only of little help once recombinant proteins aggregated due to other
reasons (e.g. average charge of the protein, amount of cysteines and prolines, hy-
drophobic or hydrophilic parts, size [153]. This is often the cause why simulta-
neous coexpression of a chaperone with the recombinant protein is not benefi-
cial. Thus the coexpression should start earlier than the one of the protein of
interest. The rate-determining steps in folding are oxidation of cysteines (catal-
ysed by Dsbs), the disulfide shuffling (in vivo catalysed by PDIs) and the peptidyl-
prolyl bond isomerisation (by PPIs) [155]. In E. coli disulfide bonds are formed
by the Dsbs which are located in the periplasm. However, also the cytoplasmic
thioredoxine and glutathione systems may be modulated to the formation
of a limited number of disulfide bonds. As IBs may be the result of incorrectly
formed disulfide bonds it is a useful approach to coexpress foldases and or
chaperones.
Again the effect is not predictable, but depends on the target protein and has
to be tested empirically [194]. Although the experimenter’s experience is bene-
ficial the conditions have to be individually optimised for every protein to be re-
combinantly produced [16]. Therefore the cellular processes of the host have to
be considered since expression conditions favouring the solubility of one target
protein may be detrimental to another one. Despite the well-known physiology
of E. coli the complex regulation is far from being understood.
For example the production of cystine-rich proteins demands especially high
amounts of DsbC and so DsbD as well. This has to be considered in the coex-
pression approach [162]. Moreover an overproduction of different combinations
of Dsbs has different effects. In case of nerve growth factor being the target pro-
tein, the expression could be especially increased by coproducing DsbABCD. In
contrast, a PDI coproduction did not have any advantageous effect at all [195]
whereas elsewhere different influences of PDI were reported [196]. Coproduction
of either DsbA or RotA did not always promote the correct folding [197]. Some-
times the yield is increased only, but IBs are not prevented [198]. A coproduction
of DnaK/J was seen to promote solubility independent of the cultivation tem-
perature, but GroEL/ES works only up to 30 °C [199]. Even cytoplasmic ATP-in-
dependent chaperones are used to support folding of proteins within the
periplasm [200]. Generally an increased solubility and yield of native product
might be achieved only in case of a product not being totally insoluble.
Coexpressed chaperones and foldases do not only prevent aggregation, but
they can remove misfolded proteins as well. DnaK selectively binds to misfolded
proteins after having detected areas of 4–5 hydrophobic amino acids on the sur-
face flanked by basic ones and guides the protein to the degradation pathway
[116, 201]. DnaK acts comparably to a detergent separating hydrophobic regions
and hence solubilising the denatured proteins. DnaK preferably attacks small ag-
gregates. Aggregation of proteins in the cytoplasm due to heat shock can be
avoided by co-overproduction of DnaK and ClpB [134, 202].The degradation of
non-native proteins by ClpP supported by GroEL/ES and trigger factor was also
reported [203]. ClpB shears larger aggregates by changing ATP-driven its own
conformation [204]. ClpB can substitute DnaK [205] and ClpA can be involved
as well [206].
118
B. Fahnert et al.

Although all these systems have an aggregation preventing or unfolding ac-
tivity they are not necessarily successful in the suppression of IB accumulation
per se. This is caused by the non-physiological character of recombinant protein
expression. The host cell is overcrowded with protein often remarkably different
to their own ones. Cellular systems are working at their limits. So the solubilis-
ing effect of the chaperones depends on the target protein and the expression
conditions. Thus sometimes an overexpression of certain folding catalysts works,
but this has to be found out empirically.
2.4.3.4
Cultivation Conditions and Addition of Folding Promoting Agents
The maintenance of intracellular conditions avoiding self-aggregation by
screening different cultivation conditions is often a more straightforward time-
saving alternative [16, 148, 149] than coexpressing homo-or heterologous chap-
erones and foldases which is a more or less successful experimental approach and
includes much time and effort. Even though many recombinant proteins are sol-
ubly expressed by choosing an appropriate combination of cultivation conditions
(e.g. low temperature, medium, strain, low inducer concentration at a long in-
duction duration [77, 150, 151]) they often lack their correct folding.
The addition of folding promoting agents then not only enhances solubility
but also has a beneficial effect on the folding of the target protein. However, the
influence is not predictable as always. Substances added to the medium affect the
periplasm more efficiently than the cytoplasm, because of the high permeability
of the outer membrane of E. coli for molecules smaller than 600 Da [207]. So the
prokaryotic periplasm is to be considered as a test-tube. Varying the redox po-
tential of the medium establishes different redox states within the periplasm. This
leads to modified conditions the disulfide bonds are formed under. Wunderlich
and Glockshuber [208] reported a fivefold increase in correctly folded target pro-
tein after adding reduced and oxidised glutathione to the medium. This effect was
even enhanced by coexpressing DsbA. This approach has been successfully con-
firmed by Bardwell [209] and many others.
Different other low molecular weight additives and their effect on recombinant
proteins are described.
Polyols and sucrose [210] are folding promoting due to their increasing the
viscosity of the solvent and the stability of protein solutions by excluded volume
effects. Many other additives cause protein protective stress responses (e.g. low
pH-value [152], ethanol [34, 149, 211], isopropanol [212]). Added cofactors such
as zinc (inhibiting proteases within the cell envelope, [213]), magnesium
[214] and calcium [195] or single amino acids can cope with limits or directly af-
fect the proteins. Glycine influences the folding of proteins prone to aggregation
[215]. l-Arginine increases the yield of native product [34, 200] due to its solu-
bilising effect [155, 216], but
D
-arginine has no effect (Neubauer and Ganjuurjav,
unpublished results). Growth on glycerol [152] or complex medium [34, 217] can
be advantageous for solubility and folding of the recombinant product. This ben-
efit of complex medium may be caused by an increased expression of foldases
[218].
Inclusion Bodies: Formation and Utilisation
119

Formamide, methyl formamide, acetamide, urea and urea derivatives are used
as well. They were among others found to increase the yield of both a truncated
tissue plasminogene activator (rPA) and a Fab fragment when periplasmatically
expressed in E. coli [200, 219]. In case of rPA the coupled coproduction of the ATP
independent chaperone DnaJ and the addition of low molecular weight com-
pounds at 25 °C increased the extractable in vivo activity considerably.
Nevertheless the effects are not always fully characterised. For example ethanol
increased the yield of cytoplasmically expressed preS2-S¢-beta-galactosidase.
This was considered to be an effect of the upregulation of heat shock protein syn-
thesis [211]. In contrast to this, in the same report the authors showed that
ethanol promotes the aggregation of human SPARC, a protein exhibiting chap-
erone dependence similar to that of preS2-S¢-beta-galactosidase.
Compatible solutes being osmoprotective towards the cells in the natural habi-
tat also have a protein stabilising effect when added to the medium. These sub-
stances (amino acids and their derivatives, sugars, polyols, quaternary amines
and their sulphur analogues, sulphate esters, N-acetylated diamino acids, pep-
tides) are water-soluble, uncharged at a neutral pH-value and can be accumulated
in high amounts. They are affecting the hydratation of macromolecules and thus
are used as chemical chaperones. For instance betaine is imported via ProU and
ProP. The expression of these transport proteins is increased by osmotic stress
[220, 221]. Betaine can reach an intracellular concentration of 50 mmol l
–1
and
prevents aggregation then. Its in vitro effect is comparable to DnaK [222].
Screening the influences of osmoprotectants (trimethylamine N-oxide, potas-
sium glutamate, betaine, sarcosine hydrochloride, glycerol, sucrose) on the fold-
ing of recombinant proteins helps to find the corresponding optimum. For this
purpose the formation of a complex of GroEL and the misfolded target protein
has been published. Aggregations are avoided by means of this. Thus the effects
of folding promoting agents can be conveniently analysed in vitro [223].
In an in vivo system the aggregation preventing and folding promoting influ-
ence of sorbitol and betaine has been analysed concerning the transcription of
cellular chaperones, proteases and stress response elements [21]. The cellular pro-
duction of periplasmic factors (FkpA, DegP, s
E
and its regulators) was not
changed after the addition. Also the amounts of the cytoplasmic factors GroEL
and GroES were not increased. Hence the addition of sorbitol and betaine may
stabilise newly synthesised recombinant protein during its folding in such a way
that the natural capacity of GroEL, ES within the cytoplasm and of the foldases
within the periplasm is sufficient. In contrast, the synthesis of some proteins of
the cytoplasm with unfolding abilities (DnaK, ClpA, ClpX) was increased. This
might be due to the fact that some recombinant protein already aggregated pre-
liminarily to the addition of sorbitol and betaine. The addition might also have
stabilised folding intermediates inducing the degradative pathways.
The benefit of folding promoting agents has been seen in many different cases
and is an approach worth to make with new target proteins in order to favour
their solubility and correct folding.
120
B. Fahnert et al.

2.4.3.5
Cellular Redox Situation
Due to its oxidising redox potential the periplasm is the compartment where
disulfide bonds are actually formed within the cell. By changing the redox state
of the cytoplasm from reducing to oxidising a new opportunity for producing re-
combinant disulfide-bonded proteins was created there.
The prokaryotic cytoplasm is oxidising in thioredoxin-reductase mutant
(trxB
–
) strains. In wildtype strains thioredoxin reductase reduces thioredoxin.
This then reduces proteins having accidentally formed disulfide bonds in the cy-
toplasm in order to re-establish their function [18]. In the mutants thioredoxin
is accumulated in an oxidised state. Hence it can act as a catalyst for the disulfide
bond formation [224]. Whether the correct folding of a soluble recombinant tar-
get protein can be achieved in the naturally oxidising periplasm or in the artifi-
cially oxidising cytoplasm cannot be predicted. As usual this has to be experi-
mentally tested. Nevertheless it is an alternative approach offering a higher
production capacity and a different folding environment (chaperones, proteases,
protein concentration, co-factors). The oxidising effect of the cytoplasm of those
mutant strains can even be enhanced by coexpression of thioredoxin or more ox-
idising mutants thereof [17].
2.5
IBs in Prokaryotes Other than E. Coli
IBs have been found so far in all microbial expression systems including lower
eukaryotic organisms, such as yeasts. A number of studies showed aggregation
of heterologous proteins produced in Bacillus subtilis (e.g. [145, 225–227]).An in-
teresting study by Jürgen et al. [145] has indicated that in Bacillus spec. a num-
ber of stress proteins are connected to recombinant IBs, such as GroEL, DnaK,
ClpC, ClpP, ClpX. The connection of the Clp proteins to IBs was revealed by off-
line analysis of the protein content of purified IBs and by immunogold labelling
and electron microscopy of IB containing cells. The latter method is especially
powerful and still has not been exploited very often. It could be applied to image
analysis to verify the molecular models of IB resolubilisation.
Natural protein crystals occur in Bacillus thuringiensis and Bacillus sphaeri-
cus. In difference to IBs discussed in this review, these aggregates contain the ac-
tive protein. If the expression pattern of the binary-component toxicity system
is changed, aggregation without crystallisation can be obtained, connected to a
loss of insect-larvae toxic activity [228, 229]. The crystallisation seems to be a
characteristic of the toxic proteins, because crystals can also be obtained if the
genetic system is transferred into other Bacillus strains [228].
Inclusion Bodies: Formation and Utilisation
121

3
Production of IBs and Down-Stream Functionalisation
3.1
Fermentation Process for IB Protein Production
The advantage of producing a protein in form of IBs is that high product amounts
are commonly obtained and that the aggregates can be easily separated and pu-
rified. Furthermore, the optimisation of the in vitro refolding conditions is
straightforward as the parameters are limited, in contrast to the complexity of
optimising soluble in vivo production.
For cytoplasmic IB production of a protein the corresponding coding gene is
inserted downstream of a strong inducible promoter. Therefore a number of
commercial systems can be applied using the promoters P
10
from T7 phage, the
tac or lacUV5 promoters, the araB promoter or the temperature inducible pro-
moters P
L
and P
R
from phage l.
The codon usage of the product gene is important as well. It should not limit
the expression in E. coli and avoid the formation of proteins with an incorrect
amino acid sequence or fragments.
In advance of fermentation a small-scale optimisation procedure is done in
shake flasks concerning the host strain and the medium. With regard to the host
strain, both E. coli K-12 strains, such as W3110 [230, 231], or B strains, such as
BL21 [39] can be used. However, other strains having specific characteristics, such
as RV308 [232] with low acetate production during growth on glucose-contain-
ing media, can also be used. Specific mutants, for instance in connection with the
stringent response, can also be included in the optimisation procedure, such as
K10. From the point of view of a fast process development of efficient high cell
density fermentations, auxotrophic mutants for amino acids or nucleotides, such
as C600, should be avoided to allow the use of a simple standard fed-batch pro-
tocol. This is important, because addition of amino acid containing complex ad-
ditives during fed-batch processes often causes problems, because the concen-
tration of the single amino acids cannot be easily controlled. Although complex
additives, such as yeast extract, may reduce proteolysis [7] and increased prod-
uct levels [233], they also can cause problems by high production of acetic acid
and variability of the substrate source if the process is scaled up. It is unlikely that
sufficient amino acids can be added to supply all the requirements at high cell
densities [7]. Furthermore, amino acids can be taken as alternative carbon source
under the carbon limiting conditions of fed-batch fermentation. As no fast on-
line methods exist for measuring the concentration of critical amino acids, toxic
effects due to accumulation of amino acids consumed at a low rate, such as
leucine [234], and negative effects on the protein due to depletion of critical
amino acids cannot easily be avoided.
Furthermore, from a physiological point of view, changes in the metabolic
flows after induction may cause problems for the cell and the product quality for
in strong induction systems the main energy flux is directed to the production
of the recombinant product. Thus the induction of new synthetic pathways for
the cellular production of amino acids or the synthesis of new transporters is
122
B. Fahnert et al.

problematic if an amino acid becomes exhausted after induction of the target
protein.Another problem may arise from the scale-up of a process to a larger fer-
menter. The metabolic rates and cellular responses may vary strongly on a pilot
scale or in production fermenters due to different fluid dynamics and mixing
characteristics.
Therefore we recommend the production on pure mineral salt medium with
glucose or glycerol as single carbon source. A corresponding recipe was suc-
cessfully used over years in our laboratory (shown in Table 2). By the use of this
medium we have seen a high reproducibility during the scale-up from shake flask
cultivations to a 100-l pilot scale. The basic medium has been used in high cell
density fermentations and up to more than 80 g l
–1
dry cell weight (320 g wet
weight) was obtained with the addition of only glucose, ammonia and magne-
sium sulphate during the process. The medium was also successfully used in E.
coli fed-batch cultivations in large-scale fermentations in 8 m
3
and 30 m
3
pro-
duction reactors.
The principle scheme of an inclusion body protein production process is de-
scribed in Fig. 6. The starting material can be either a frozen stock culture or
overnight plates of newly transformed cells. Generally two different precultures
are used, a first culture in nutrient broth (NBII or DYT), followed by a second cul-
ture in baffled shake flasks on the fermentation medium. Each preculture is used
at the exponential growth phase as inoculum for the next cultivation. Batch to
batch variations and plasmid instabilities may be avoided if the precultures do
not reach the stationary phase and all inocula are in the exponential phase of
growth. Selective conditions, e.g. by applying the corresponding antibiotics,
should be used to avoid plasmid loss in all precultures and the main fermenta-
tion.
In principle most fed-batch fermentations for IBs in the laboratory scale can
be carried out within one day (for example, see Fig. 7). Therefore the inoculation
is done in the morning and a batch phase at 35 °C is recommended.
Inclusion Bodies: Formation and Utilisation
123
Table 2
Composition of a high-cell-density fermentation medium based on mineral salts
(in g l
–1
)
Basic medium
Feeding solution
Na
2
SO
4
2.00 g
Na
2
SO
4
2.00 g
(NH
4
)
2
SO
4
2.47 g
(NH
4
)
2
SO
4
2.47 g
NH
4
Cl
0.50 g
K
2
HPO
4
14.60 g
K
2
HPO
4
14.60 g
NaH
2
PO
4
· 2H
2
O
4.00 g
NaH
2
PO
4
· 2H
2
O
4.00 g
(NH
4
)
2
-H-Citrate
1.00 g
(NH
4
)
2
-H-Citrate
1.00 g
Glucose
660.00 g
Glucose
20.00 g
Thiamine
0.10 g
Thiamine
0.10 g
Trace elements
2.00 ml
Trace elements
2.00 ml
MgSO
4
(1 mol l
–1
)
2.00 ml
Optional
Yeast extract, peptone,
20.00 g
or casamino acids
124
B. Fahnert et al.
Fig. 6
Principle flow chart of a fermentation process for the production of IBs
Fig. 7
Graph of a fed-batch fermentation of E. coli RB791 for production of IBs. The process
consists of three phases. An initial batch phase is followed by a constant feeding of a concen-
trated glucose solution (thick line in the lower graph) started at zero hours. Following the
growth of the culture proceeds quasi-linearly. Three hours after feed start IPTG is added to in-
duce the synthesis of the recombinant product (grey bars). The lower graph also shows the DOT
(thin black line) indicating by the increasing DOT that respiration is declining at about 2 h af-
ter induction

The basic fermentation is started with a batch with an initial substrate con-
centration of 20 g l
–1
. The pH is controlled so as not to drop below 7.0 by addi-
tion of 25% ammonia. Air flow (e.g. 0.02 to 2 vvm) and stirrer speed (e.g. 200 to
1500 rpm) are controlled in a cascade mode during the batch phase to keep the
dissolved oxygen tension (DOT) above 20%. The foam level is commonly con-
trolled by an antifoam electrode and addition of a chemical antifoam agent, such
as polypropyleneglycol 2000. The medium contains all other basic ions necessary
for growth to high cell densities of above 100 g l
–1
of cell dry weight with excep-
tion of MgSO
4
. A 1 mol l
–1
solution of MgSO
4
is either intermittently or continu-
ously added to the cultivations with a rate of 2 ml l
–1
for every 2.5 g of cell dry
mass additional growth.
The batch is followed until the initially added glucose is consumed. Most
strains grow on the described mineral salt medium at 35 °C at pH 7.0 with a spe-
cific growth rate m of about 0.69 h
–1
(corresponding to 1 h doubling time). Glu-
cose limitation is appearing at about 7 to 9.5 g l
–1
of dry cell mass, correspond-
ing to an optical density at 500 nm (OD
500
) of 28–38 in dependence on the host
strain. If strains are used which produce high growth inhibiting amounts of
acetate as overflow metabolite, the initial glucose concentration is lowered.
Feeding of a concentrated glucose solution (for recipe see Table 1) is started
after the batch glucose is consumed. Alternatively, if glycerol is used as a carbon
substrate, a high concentrated glycerol solution is used as feed solution. The ex-
haustion of glucose or glycerol gives a fast uprising signal of the dissolved oxy-
gen electrode. When a cascade controller for aeration and stirrer speed is used,
the decrease of these signals indicates glucose exhaustion. Care has to be taken
to start the glucose feed as fast as possible after the glucose exhaustion signal.
This is important to avoid established stress responses and to make the process
reproducible.
The feeding solution is added either simply in a constant way or by an expo-
nential feeding programme. Our experiences have shown that the addition by a
simple pump without fermenter control is well applicable. Two points are im-
portant to guarantee for a good yield: (a) the flow of the feed solution must be
low enough to allow glucose limitation in the fermenter; (b) the flow of the feed
solution must be regulated in a way that the specific growth rate does not de-
crease below 0.1 h
–1
until the point of induction. An appropriate initial feeding
rate F (in litres per hour) is calculated by the following equation:
1 X
Fstart
· ln(2) V
F =
3
·
002
·
4
[l h
–1
]
(1)
2 Y
X /S
· t
D
S
i
Thereby, X
Fstart
is the cell dry mass in g l
–1
at the end of the batch, t
D
is the doubling
time of the biomass in hours, V is the volume of the bioreactor in litres, S
i
is
the carbon substrate concentration in the feeding solution (in g l
–1
) and Y
X/S
is
the yield coefficient for biomass during the growth on the substrate. Y
X/S
can be
simply calculated by
X
Fstart
Y
X /S
=
01
.
(2)
S
0
Inclusion Bodies: Formation and Utilisation
125

After feeding starts the dissolved oxygen signal will stabilise to a value char-
acterised by the respiratory activity. The respiratory activity is controlled by the
available carbon source. The time until the DOT becomes constant depends on
the acetate concentration which has been accumulated during the batch and is
co-metabolised with the added feed-glucose at the start of the fed-batch. At the
time the dissolved oxygen signal has been stabilised the feed flow rate can be
slowly increased stepwise. Glucose limitation can be easily controlled by quickly
interrupting the glucose flow by stopping the pump for some seconds and check-
ing the DOT signal change, which should rise. By this procedure the feed flow rate
can be increased as high as a DOT level of 20 % can be kept.
In case an exponential feed protocol is used, this can be calculated according
to the following equation:
m
max
X
Fstart
· V
Fstart
F =
8
·
002
· e
(m
set
· t)
[l h
–1
]
(3)
Y
X /S
S
i
where m
set
is the specific growth rate during the fed-batch phase and should be
set in the range between 0.1 and 0.2 h
–1
and t is the time after feed start (t=0 at
feed start).
Also, in the case of exponential feed, one should ensure that glucose is limit-
ing the growth from time to time either by simply switching off the pump man-
ually for a short time and waiting for the rise of the DOT, which appears a mo-
ment after feed stop due to glucose exhaustion, or by measuring the glucose
concentration in samples from the fermentation broth.
The synthesis of the recombinant product is induced 3–5 h after the feed starts
in the same way as evaluated during the optimisation procedure in the shake
flasks. However, induction can be performed earlier if the time of the fermenta-
tion is decreased, or later if a higher cell density is wanted to increase the total
product yield (in g l
–1
). As discussed above, it is strongly recommended that the
specific growth rate at the point of induction is not below 0.1 h
–1
.
For most strong expression systems the fermentation could be stopped 3–5 h
after induction; however, this can vary depending on the expression system used,
the protein of interest and the product stability.
If the process is performed by the use of complex additives, such as yeast ex-
tract, casamino acids or peptone, the fermentation can be performed with addi-
tion of the complex substrate from the start. However, this sometimes leads to el-
evated acetic acid production.As a consequence of the pH regulation the balance
of ammonia can also be disturbed, leading to growth inhibition effects if am-
monia is used as the only nitrogen source. Therefore, ammonia should be mea-
sured during the optimisation of such processes. Furthermore, the higher growth
rate on complex media can cause oxygen insufficiency at lower cell density. The
oxygen signal due to glucose exhaustion can also be indistinct, since the cells can
start to use components of the extract as carbon source.
To overcome these problems, the batch phase may be performed on pure min-
eral salt medium without yeast extract or casamino acids. These are added only
about 1–2 h before induction for preconditioning of the cells to the changed en-
vironment. Therefore these additives are first added once at 10 g l
–1
. Afterwards
126
B. Fahnert et al.
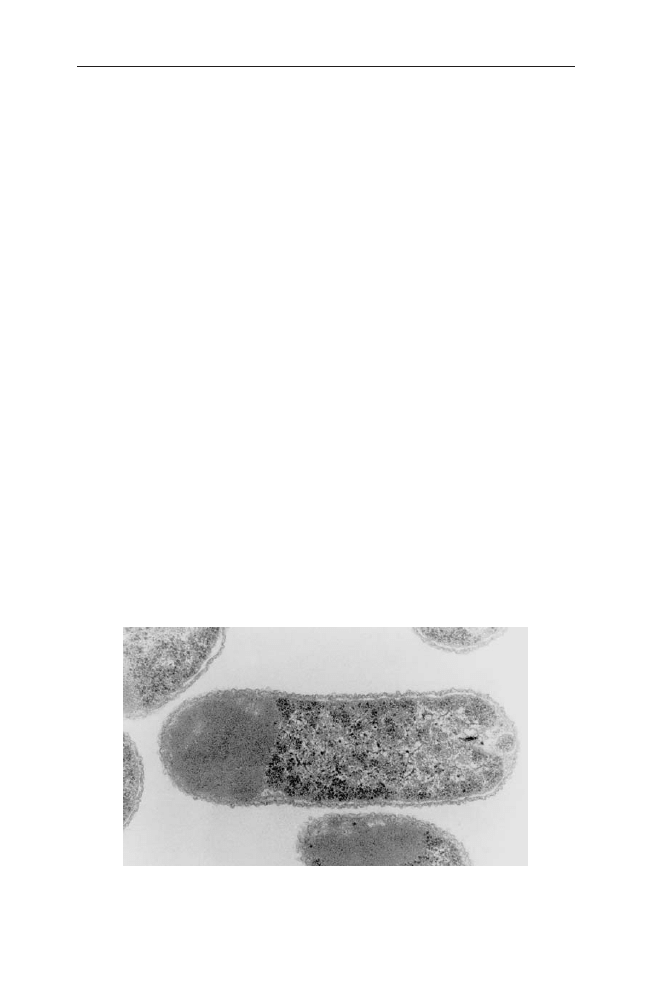
either a second addition is performed at the point of induction or a continuous
addition is started directly after the first pulse at a constant rate of about 3 g l
–1
h
–1
. Also, if complex additives are used, it should be always ensured that glucose
is the growth limiting nutrient.
Finally it should be stated that the fermentation protocol described above is
based on our experiences and corresponds to the basic method used in our lab-
oratories. Of course, other protocols and media can be obtained from different
sources. However, we believe that our protocol been used for production of some
ten proteins in IBs, is a very simple method which easily can be adapted to the
production of IBs at different production scales.
3.2
Preparation of IBs
3.2.1
IB Isolation
IBs obtained by cytosolic microbial overexpression of a recombinant protein are
large particles with an amorphous manifestation (Fig. 8). Since IBs are charac-
terised by a relatively high specific density of about 1.3 mg ml
–1
[235], they can
be harvested after cell lysis by centrifugation at moderate rotor speeds [236]. The
first step of this isolation procedure is maximal lysis of the E. coli cells by
lysozyme treatment, subsequent repetitive high pressure dispersion and, finally,
incubation with detergent, such as Triton X-100 and high salt. This combination
of cell disruption techniques guarantees the complete disintegration of all par-
ticulate matter (membrane fragments, cell wall debris, etc.) [237]. Provided that
the initial level of expression is sufficiently high, collection of the IBs by cen-
trifugation [212, 238] or filtration generally yields a rather homogeneous prepa-
ration. It may contain the recombinant protein with a purity of up to 90% [22].
However, certain host cell proteins such as elongation factor EF-Tu, outer mem-
Inclusion Bodies: Formation and Utilisation
127
Fig. 8
Electron micrograph of negatively stained E. coli cells displaying IBs

brane proteins or small heat-shock proteins may be enriched in IB isolates ([62,
67], also see above). These impurities may derive from co-precipitation upon
overexpression of the foreign gene product. Most of the impurities commonly
identified in IB isolates, however, may originate primarily from incomplete cell
lysis and removal of particulate host cell material.
3.2.2
Purification of IBs
IB isolates collected after maximum cell disruption are usually relatively homo-
geneous. In this case, the proteins can be renatured directly after solubilisation
without a further purification of the recombinant protein. However, proteina-
ceous and non-proteinaceous contaminations of IBs may interfere with subse-
quent renaturation of the recombinant protein [239]. Therefore, if the recombi-
nant expression and IB isolation result in recombinant material with a high
degree of impurities, these IBs can be further purified by additional washing
steps. These may utilise EDTA, low concentrations of denaturant such as urea or
GdmCl and detergents such as Triton X-100, deoxycholate, octylglycoside and
sarcosyl (Table 3). Furthermore, IBs in their particulate state can be purified by
gel filtration.
Alternatively, the recombinant protein can be purified from IBs after solubil-
isation. This is particularly attractive if the protein is expressed as fusion protein
with a purification tag such as a His-tag. In this case purification can be achieved
by IMAC under denaturing conditions [240, 241]. More generally, RP-HPLC may
be applied for chromatographic purification of solubilised IBs.
3.2.3
Solubilisation of IBs
Although IB proteins may contain a relatively high secondary structure content
[51, 52], they do not readily disintegrate under physiological solvent conditions.
A variety of methods can be used for solubilisation of IBs [248]. In most cases,
however, rather strong denaturants are employed. To this end chaotrophs such as
GdmCl or urea are used with GdmCl preferable to urea, because urea solutions
may contain isocyanate leading to carbamylation of free amino groups of the
128
B. Fahnert et al.
Table 3
Purification of IB material
Recombinant protein
Washing step
Reference
Human prourokinase
0.1% Triton X-100
[242]
Arginine deiminase
4% Triton X-100
[243]
Human t-PA
2% Triton X-100, 5 mol l
–1
urea
[244]
Horse radish peroxidase
2 mol l
–1
Urea
[245]
Human IGF-1
0.5% Sarcosyl
[246]
Bovine growth hormone
2% Deoxycholate
[247]
N-Terminal domain of PTH receptor
1.5% LDAO
[241]

polypeptide, especially upon long-term incubation at alkaline pH values [249].
The denaturants are usually employed at high concentrations (6–8 mol l
–1
) to en-
sure complete solubilisation and unfolding of the IB proteins. However, in a few
cases denaturants at low concentrations (1–2 mol l
–1
) proved to be more efficient
since other impurities in the particulate fraction were not solubilised under these
mildly denaturing conditions [250].
Another class of denaturants solubilising IBs are detergents. Cetyl trimethy-
lammonium salts (CTAC or CTAB) have been used for solubilisation of IBs of hu-
man growth hormone [251, 252]. Even though detergents might be advantageous
in some cases compared to GdmCl and urea, one has to keep in mind that they
may interfere with the following protein renaturation and subsequent purifica-
tion.
Besides denaturing agents, extremes of pH have been used to solubilise IBs.
Here 20% acetic acid was sufficient to solubilise a fusion protein of maltose bind-
ing protein with the acetylglucosaminidase F2 [253]. On the other hand alkaline
solutions (pH > 12) have been utilised in the preparation of growth hormones
[252] and proinsulin [254]. Although pH-induced solubilisation of IBs is a very
simple method, it is problematic because at extreme pH values certain residues
of the polypeptide chain of a protein may become chemically modified.
In the case of proteins containing cysteine, the isolated IBs usually contain a
certain amount of interchain disulfide bonds [255], which reduce the solubility
of the IBs in the absence of reducing agents. Addition of low-molecular weight
thiol reagents such as dithiothreitol, glutathione, cysteine or mercaptoethanol in
combination with chaotrophs allows reduction of the inter-chain disulfide bonds
by thiol-disulfide exchange [256]. Since the reactive species in thiol-disulfide ex-
change is the thiolate anion, IB solubilisation in the presence of reducing agent
is usually performed under mildly alkaline conditions.
3.3
Refolding of Proteins from IBs
Refolding of solubilised IB proteins requires removal of the denaturant. The clas-
sical procedure for changing the buffer is dialysis. This technique can also be ap-
plied for protein renaturation [241]. Dialysis of a denatured protein against re-
naturation buffer is characterised by a slow gradual removal of the denaturant.
Whereas for certain proteins this may be advantageous [257], in most cases pro-
teins tend to aggregate at intermediate denaturant concentrations, thus limiting
the yield of renaturation upon dialysis. Consequently, most protocols for rena-
turing proteins consist of dilution of the denatured protein in the renaturation
buffer. By this method the protein environment is switched to native conditions
immediately. Furthermore, dilution of the denatured protein not only results in
low denaturant concentrations but also in low protein concentration during re-
folding.
The most prominent unproductive side reaction of renaturation is aggrega-
tion. Since aggregation is a process of second order or higher order reaction ki-
netics [258] low protein concentrations (10–100 mg ml
–1
) are strictly required for
efficient renaturation. This, however, leads to large reaction volumes in order to
Inclusion Bodies: Formation and Utilisation
129
produce quantities of renatured IB proteins. To circumvent this technical prob-
lem, a stepwise renaturation procedure can be applied (Fig. 9). As completely
folded proteins are usually not prone to co-precipitation with folding proteins,
high yields of renatured protein per volume of refolding buffer can be obtained
by slow continuous or discontinuous addition of the denatured protein to the re-
folding buffer [25, 256, 259].
Size-exclusion chromatography (SEC) is an alternative buffer-exchange
method to remove high denaturant concentrations and promote renaturation.
Two different experimental strategies are described. First, application of the de-
natured protein on a gel filtration column, equilibrated in renaturation buffer.
Thus, chromatography separates the denaturant from the protein, thus facilitat-
ing renaturation [260, 261]. Second, the column may be equilibrated in denatur-
ing buffer. After penetration of the denatured protein in the gel matrix the chro-
matography buffer is changed to renaturation conditions [262].
Other kinds of chromatography have also been used for protein renaturation.
These techniques depend on the immobilisation of the denatured protein on a
solid matrix. After changing the buffer to native conditions the proteins refold,
still bound to the matrix, thus preventing the unfolded protein and folding in-
termediates from aggregation with other protein molecules. If the IB protein is
fused to a purification tag such as a His-tag or a polyionic fusion peptide, im-
mobilisation and subsequent renaturation can be achieved using IMAC [263, 264]
and ion exchange chromatography [265].
130
B. Fahnert et al.
Fig. 9
Stepwise renaturation of proinsulin. Denatured and reduced proinsulin was refolded at
15 °C by stepwise addition of the protein to the refolding buffer (10 mmol l
–1
Tris, 10 mmol l
–1
glycine, 1 mmol l
–1
EDTA, pH 10.5, 0.5 mmol l
–1
cysteine, 4.5 mmol l
–1
cystine). The final
concentration was 500 mg ml
–1
per pulse. After every 30 min samples were withdrawn, the
concentration of native proinsulin was analysed by RP-HPLC (filled circles) and new protein
was added to the renaturation buffer. Thirty pulses were performed (adapted from [259])

3.3.1
Disulfide Bond Formation During Protein Renaturation
Folding proteins with concomitant disulfide bond formation includes the for-
mation of both the native tertiary structure of the molecule stabilised by non-co-
valent interactions and the covalent disulfide bonds. For disulfide bond forma-
tion, the number of possible combinations increases dramatically with the
number of cysteine residues present in the polypeptide chain [237]. However,
pure statistics of disulfide bond formation are surpassed by the preponderance
of the correct folding pathway. Obviously, disulfide bond formation is directed to-
wards the correct pairing by the conformational energy gained upon formation
of the native conformation and vice versa.
The simplest method promoting disulfide bond formation is oxidation by
molecular oxygen, catalysed by metal ions such as Cu
2+
[266, 267]. As the low ef-
ficacy of disulfide bond formation by oxidation with molecular oxygen, thiol-
disulfide exchange reactions with low molecular weight thiols in reduced and
oxidised form are generally employed for protein disulfide bond formation
(Fig. 10).
The most commonly used oxido-shuffling reagents are reduced and oxidised
glutathione. However, other low molecular weight thiols such as cysteine/cystine,
cysteamine/cystamine, or di-hydroxyethyl disulfide/2-mercaptoethanol have
been utilised for disulfide bond formation of IB proteins (Table 4).As thiol-disul-
fide exchange reactions are rapidly reversible, oxido-shuffling reagents increase
both the rate and the yield of correct protein disulfide bond formation by rapid
reshuffling of improper disulfide bonds.
Another approach of oxidative refolding uses a two-step-mechanism. At first
all cysteines in the denatured protein are converted to mixed disulfides with glu-
Inclusion Bodies: Formation and Utilisation
131
Table 4
Oxidative protein renaturation
Protein
Thiol
Disulfide
Reference
Fab fragment
5 mmol l
–1
GSH
0.5 mmol l
–1
GSSG
[25]
N-Terminal domain
5 mmol l
–1
GSH
1 mmol l
–1
GSSG
[241]
of PTH receptor
Fab fragment
0.5 mmol l
–1
GSH
Mixed disulfides
[25]
Fab fragment
3 mmol l
–1
DTT
4 mmol l
–1
GSSG
[270]
Human t-PA
2 mmol l
–1
GSH
Mixed disulfides
[24]
Human t-PA
2 mmol l
–1
GSH
0.2 mmol l
–1
GSSG
[271]
Human IL2
10 mmol l
–1
GSH
1 mmol l
–1
GSSG
[272]
Proinsulin
0.5 mmol l
–1
cysteine
4.5 mmol l
–1
cystine
[259]
Fig. 10
Principle of “oxido-shuffling” during disulfide bond formation

tathione. In the subsequent renaturation step, formation of the correct disulfide
bonds is catalysed by adding catalytic amounts of the low molecular weight thiol
in its reduced form [24]. The advantage of this procedure compared to oxidative
refolding of the fully reduced protein is the introduction of charged residues
in the denatured state of the protein. These charges increase the solubility
of the denatured protein and early folding intermediates, thus decreasing the
propensity of aggregation during renaturation. Similarly, S-sulfonation is
frequently used to improve refolding of disulfide bond containing IB proteins
[268, 269].
3.3.2
Improving Renaturation
In vitro folding of small, single-domain proteins is often quantitative, i.e. all un-
folded polypeptide chains fold back to their native form. For larger, multi-domain
proteins the yield of in vitro folding is, however, often much lower, since unpro-
ductive side reactions (especially aggregation) compete with proper folding. In
this case, the yield can be improved by speeding up rate determining folding
steps, decelerating aggregate formation and/or destabilising off-pathway prod-
ucts. This can be achieved by optimising the folding conditions with respect to
buffer composition, ionic strength, pH, folding time, temperature, protein con-
centration, co-factors and, in the case of disulfide-bonded proteins, additives
which promote direct disulfide bond formation (see above).
Upon in vitro folding of disulfide-bonded as well as non-disulfide-bonded
proteins the yield of correct folding can be improved by supplementing the
refolding buffer with low molecular weight additives (Table 5). As an example,
renaturation with concomitant disulfide bond formation of reduced chymotryp-
sinogen A was only feasible in the presence of non-denaturing concentrations
of urea or GdmCl [273]. Analysis of oxidative folding vs aggregate formation
using lysozyme as a model protein showed that GdmCl decelerates aggregate
formation more strongly than folding [274]. Improving refolding by the addition
of non-denaturing concentrations of denaturants is, however, only possible if the
native state of the respective protein is sufficiently stable under these conditions.
Non-denaturing concentrations of these additives may, however, also have a
detrimental effect on folding in cases where intermediates are populated which
are prone to aggregation. As an alternative to GdmCl or urea, other chaotrophs
such as alkyl-urea or organic co-solvents like carbonic acid amides may be
employed to improve in vitro folding [24].
As shown for numerous proteins, the yield of correct folding can be improved
tremendously by adding the amino acid
L
-arginine to the refolding buffer in rel-
atively high molar concentrations [22, 25, 216, 241, 275]. Although containing a
guanidino group, arginine has only a minor effect on protein stability. As shown
for RnaseA, arginine only slightly destabilises the native protein conformation
[276]. On the other hand this additive strongly enhances the solubility of folding
intermediates [5, 277]. The increase in solubility of folding intermediates with-
out significant destabilisation of the final native structure results in an improved
refolding of many different proteins.
132
B. Fahnert et al.

In some cases additives, which are strongly stabilising native protein struc-
tures, are essential for successful folding. Human placental alkaline phosphatase,
for example, could only be refolded in vitro in the presence of stabilisers such as
sulphate or carbohydrates [278]. For bovine carbonic anhydrase B, stoichiomet-
ric amounts of polyethylene glycol (PEG) have been found to improve in vitro
structure formation [279]. In this case, PEG inhibits aggregation by complex for-
mation with a molten globule folding intermediate being otherwise prone to ag-
gregation.
Other low molecular weight additives such as detergents and mixed micelles
have been found to promote protein renaturation. These additives bind to fold-
ing intermediates, thus preventing aggregation. Proper folding requires the re-
lease of the detergent from the folding intermediate facilitated by the extraction
with cyclodextrin [280]. In this context one has to consider that cyclodextrin also
interacts with bulky hydrophobic amino acid side chains [281, 282]. Therefore,
cyclodextrin may also increase the solubility of folding intermediates.
As for protein purification, the optimum conditions for in vitro folding have
to be established on a case by case basis. The first round of the development of a
folding process comprises a crude variation of the folding conditions such as var-
ious additives, protein concentration, pH, temperature, time and ionic strength.
The second round of process development involves the fine-tuning of the solvent
Inclusion Bodies: Formation and Utilisation
133
Table 5
Renaturation procedure for IB proteins (see text for references)
Step
Conditions
IB solubilisation
2 h incubation at 20 °C in
6 mol l
–1
GdmCl
100 mmol l
–1
DTT (or DTE)
0.1 mol l
–1
tris-HCl, pH 8
c
p
≈5 mg ml
–1
Removal of reductant
Adjustment of pH to ca. pH 4.5,
dialysis against 6 mol l
–1
GdmCl, pH 4.5
Folding
1:200 dilution in tris-HCl buffer, pH 7.5
a
or pH 8.5
b
,
15 °C, 5 mmol l
–1
EDTA in the presence of additives such as:
– no additive
– 0.5 mol l
–1
L
-arginine
– 1 mol l
–1
tris-HCl
– 0.5 mol l
–1
Gdm/Cl
– 0.06 mg ml
–1
laurylmaltoside
– 33 mmol l
–1
chaps-mixed micelles consisting of Triton X-100
and phospholipids
– 20% glycerolmetal ions, ligands, etc.
c
a
For proteins containing cysteines but no disulfide bonds in the native state: add 2 mmol l
–1
DTE to folding buffer.
b
For proteins containing disulfide bonds in the native form, add 5 mmol l
–1
reduced and
1 mmol l
–1
oxidised glutathione to the folding buffer.
c
If the native protein contains metalions, EDTA should be omitted from the folding buffer.
For refolding add a fivefold molar excess of the respective metal ion to the folding buffer.
Similarly, add other ligands that bind to the authentic native protein.
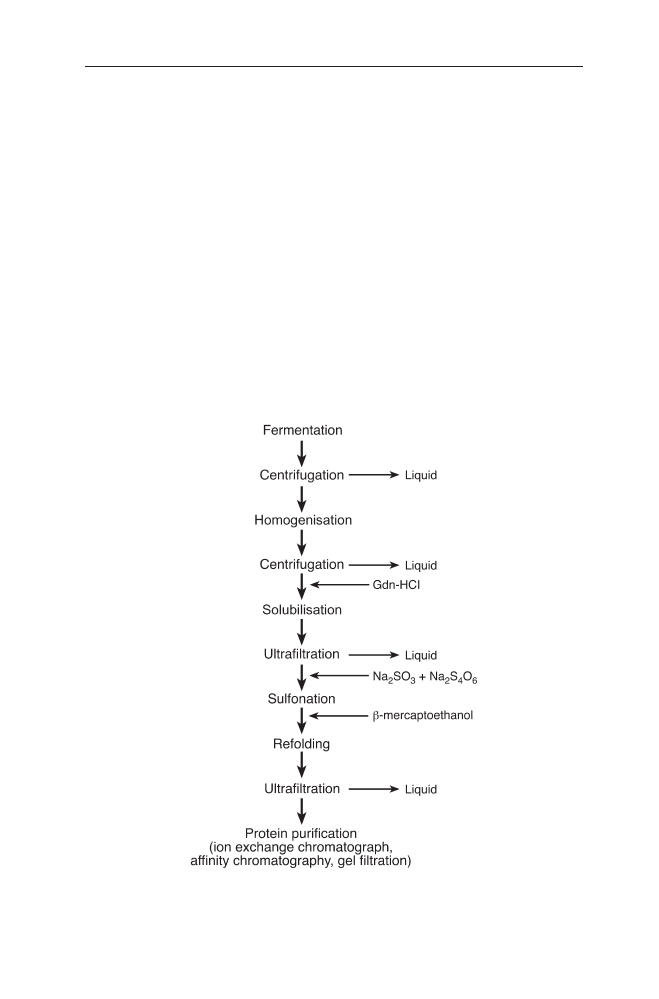
conditions. After careful optimisation, in vitro folding of any recombinant pro-
tein deposited in IBs will be likely to be successful.
3.4
Industrial Processes Based on Refolding of IB Proteins
3.4.1
Human Tissue-Type Plasminogen Activator (t-PA)
t-PA is a serine protease of the fibrinolytic pathway. It catalyses the proteolytic
conversion of plasminogen to the active protease plasmin which degrades fibrin
clots. t-PA is a glycosylated polypeptide of a size of 527 amino acids arranged in
five structural domains: a finger-domain, an EGF-like domain, two kringle do-
mains and the proteolytic active domain [283]. t-PA contains 35 cysteine residues
that form 17 disulfide bonds. Furthermore, t-PA has a very low solubility even in
the native state. Thus, this complex molecule exemplifies all the challenges for
protein renaturation. In Fig. 11 the overall process of production of recombinant
t-PA is summarised. This procedure is based on the in vitro renaturation of t-PA
134
B. Fahnert et al.
Fig. 11
Diagram of the production process of t-PA (adapted from Datar et al. [285])

according to the techniques described before. Essential features of the renatura-
tion comprises the formation of mixed disulfides of the denatured protein with
glutathione, the use of molar concentrations of
L
-arginine as low molecular
weight folding enhancer and a stepwise renaturation protocol [284]. These com-
bined techniques allowed high yield production of t-PA from IBs.
3.4.2
Antibody Fragments and Immunotoxins
Antibody fragments are used in large quantities for both diagnostic and thera-
peutic applications.As fusion proteins with toxins such as Pseudomonas exotoxin
they provide a strategy for cancer therapy. Antibody fragments and immuno-
toxins can be produced in large quantities in E. coli in the form of IBs. The re-
naturation of these multi-domain proteins, however is a complex process com-
prising domain folding, heterodimeric association and formation of intradomain
and interchain disulfide bonds. As in the case of t-PA similar methods of protein
renaturation allowed the refolding of a Fab fragment with a yield of about 40%
[25] and the production of immunotoxins on a technical scale [216].
3.5
The Future of IB Based Processes for Recombinant Proteins
The genomes from several organisms have been sequenced so far including
mouse and man. With this overwhelming sequence-based knowledge there is a
great demand on the elucidation of structural and functional properties of the
respective gene products. This can only be accomplished by recombinant pro-
duction of the concerning proteins. Although many organisms and expression
systems are now being used for recombinant protein production, E. coli as a host
is often preferred due to its simple genetic manipulation, established strategies
for recombinant protein expression, rapid growth and detailed physiological
knowledge. High-level expression in E. coli often leads to an accumulation of the
protein in IBs. This has been thought previously to be a problem; now it is ac-
knowledged as a chance happening. With careful optimisation of a protein re-
naturation procedure it is very likely that a certain target protein can be refolded
from the IB to its native state. Therefore, IB production guarantees large amounts
of recombinant protein, a simple but very efficient first purification step and to-
gether with an automated renaturation protocol an overall process of high re-
producibility and charge consistency.
Acknowledgements
Results of this work have been obtained by the financial support of the
European Community (BIO-CT95-0028, BIO-CT98-0167). BF has been supported by a Marie
Curie Fellowship of the European Community program “Quality of Life Individual Fellowships
of the Fifth Framework Programme” under contract number QLK3-CT-2001-51066.
Inclusion Bodies: Formation and Utilisation
135

4
References
1. Neubauer P, Winter J (2001) In: Merten O-W, Mattanovich D, LANG C, Larsson G,
Neubauer P, Porro D, Postma PW, Teixeira de Mattos J, Cole J (eds) Recombinant protein
production with prokaryotic and eukaryotic cells.A comparative view on host physiology,
Kluwer Academic Publishers, Dortrecht, The Netherlands, p 196
2. Baneyx F (1999) Curr Opin Biotechnol 10:411
3. Misawa S, Kumagai I (1999) Biopolymers 51:297
4. Rudolph R (1996) In: Cleland JL, Craik CS (eds) Principles and practice of protein fold-
ing. Wiley, New York
5. Rudolph R, Lilie H (1996) FASEB J 10:49
6. Viitanen MI, Vasala A, Neubauer P, Alatossava T (2003) Microb Cell Fact 2:1
7. Swartz JR (1996) In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB,
Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) Escherichia coli
and Salmonella: cellular and molecular biology. ASM Press, Washington, DC, p 1693
8. Rinas U, Tsai LB, Lyons D, Fox GM, Stearns G, Fieschko J, Fenton D, Bailey JE (1992)
Bio/Technology 10:435
9. Schulze AJ, Degryse E, Speck D, Huber R, Bischoff R (1994) J Biotechnol 32:231
10. Anfinsen CB, Haber E, Sela M, White FHJ (1961) Proc Natl Acad Sci USA 47:1309
11. Dobson CM, Karplus M (1999) Curr Opin Struct Biol 9:92
12. Levinthal C (1968) J Chim Physique 65:44
13. Karplus M (1997) Fold Des 2:S69
14. Stevens FJ, Argon Y (1999) Semin Cell Dev Biol 10:443
15. Ellis RJ (1997) Curr Biol 7:R531
16. Schein CH (1993) Curr Opin Biotechnol 4:456
17. Bessette PH, Aslund F, Beckwith J, Georgiou G (1999) Proc Natl Acad Sci USA 96:13703
18. Derman AI, Prinz WA, Belin D, Beckwith J (1993) Science 262:1744
19. Proba K, Ge L, Pluckthun A (1995) Gene 159:203
20. Levy R, Weiss R, Chen G, Iverson BL, Georgiou G (2001) Protein Expr Purif 23:338
21. Fahnert B (2001) PhD thesis, Friedrich-Schiller-University of Jena, Faculty of Biology and
Pharmaceutics
22. Rattenholl A, Lilie H, Grossmann A, Stern A, Schwarz E, Rudolph R (2001) Eur J Biochem
268:3296
23. Rattenholl A, Ruoppolo M, Flagiello A, Monti M, Vinci F, Marino G, Lilie H, Schwarz E,
Rudolph R (2001) J Mol Biol 305:523
24. De Bernadez Clark E, Schwarz E, Rudolph R (1999) Methods Enzymol 309:217
25. Buchner J, Rudolph R (1991) Bio/Technology 9:157
26. Plückthun A, Krebber A, Krebber C, Horn U, Knüpfer U, Wenderoth R, Nieba L, Proba K,
Riesenberg D (1996) In: Hoogenboom H, McCafferty J, Chiswell D (eds) Antibody engi-
neering: a practical approach. IRL Press, Oxford, p 203
27. Hayhurst A (2000) Protein Expr Purif 18:1
28. Horn U, Strittmatter W, Krebber A, Knüpfer U, Kujau M, Wenderoth R, Müller K, Matzku
S, Plückthun A, Riesenberg D (1996) Appl Microbiol Biotechnol 46:524
29. Humphreys DP, Weir N, Lawson A, Mountain A, Lund PA (1996) FEBS Lett 380:194
30. Kujau MJ, Riesenberg D (1999) Microbiol Res 154:27
31. Morbe JL, Riesenberg D (1997) Microbiol Res 152:385
32. Raffai R, Vukmirica J, Weisgraber KH, Rassart E, Innerarity TL, Milne R (1999) Protein
Expr Purif 16:84
33. Nilsson J, Jonasson P, Samuelsson E, Stahl S, Uhlén M (1996) J Biotechnol 48:241
34. Winter J, Neubauer P, Glockshuber R, Rudolph R (2000) J Biotechnol 84:175
35. Ghosh R, Steiert M, Hardmeyer A, Wang YF, Rosenbusch JP (1998) Gene Expr 7:149
36. Miroux B, Walker J-E (1996) J Mol Biol 260:289
37. Arechaga I, Miroux B, Karrasch S, Huijbregts R, de Kruijff B, Runswick MJ, Walker JE
(2000) FEBS Lett 482:215
136
B. Fahnert et al.

38. Shanklin J (2000) Protein Expr Purif 18:355
39. Studier FW, Moffatt BA (1986) J Mol Biol 189:113
40. Saribas AS, Gruenke L, Waskell L (2001) Protein Expr Purif 21:303
41. Mulrooney SB, Waskell L (2000) Protein Expr Purif 19:173
42. Jansen C, Wiese A, Reubsaet L, Dekker N, de Cock H, Seydel U, Tommassen J (2000)
Biochim Biophys Acta 1464:284
43. Solovicova A, Gasperik J, Hostinova E (1996) Biochem Biophys Res Commun 224:790
44. Martin U, Fischer S, Kohnert U, Opitz U, Rudolph R, Sponer G, Stern A, Strein K (1992)
Naunyn Schmiedebergs Arch Pharmacol 346:108
45. Mattes R (2001) Semin Thromb Hemost 27:325
46. Carrio MM, Villaverde A (2002) J Biotechnol 96:3
47. Teich A, Lin HY, Andersson L, Meyer S, Neubauer P (1998) J Biotechnol 64:197
48. Lin HY, Hanschke R, Nicklisch S, Nietsche T, Jarchow R, Schwahn C, Riemschneider S,
Meyer S, Gupta A, Hecker M, Neubauer P (2001) In: Merten O-W, Mattanovich D, Lang C,
Larsson G, Neubauer P, Porro D, Postma PW, Teixeira de Mattos J, Cole J (eds) Recombi-
nant protein production with prokaryotic and eukaryotic cells. A comparative view on
host physiology. Kluwer Academic Publishers, Dortrecht, The Netherlands, p 55
49. Carrio MM, Cubarsi R, Villaverde A (2000) FEBS Lett 471:7
50. Mitraki A, King J (1989) Bio/Technology 7:690
51. Przybycien TM, Dunn JP, Valax P, Georgiou G (1994) Protein Eng 7:131
52. Oberg K, Chrunyk BA, Wetzel R, Fink AL (1994) Biochemistry 33:2628
53. Tokatlidis K, Dhurjati D, Millet J, Beguin P, Aubert JP (1991) FEBS Lett 282:205
54. Rinas U, Bailey JE (1992) Appl Microbiol Biotechnol 37:609
55. Rinas U, Bailey JE (1993) Appl Environ Microbiol 59:561
56. Hartley DL, Kane JF (1988) Biochem Soc Trans 16:101
57. Rinas U, Boone TC, Bailey J-E (1993) J Biotechnol 28:313
58. Chaturvedi R, Bhakuni V, Tuli R (2000) Protein Expr Purif 20:21
59. Frankel S, Sohn R, Leinwand L (1991) Proc Natl Acad Sci USA 88:1192
60. Valax P, Georgiou G (1993) Biotechnol Prog 9:539
61. Jürgen B, Lin HY, Riemschneider S, Scharf C, Neubauer P, Schmid R, Hecker M, Schweder
T (2000) Biotechnol Bioeng 70:217
62. Allen SP, Polazzi JO, Gierse JK, Easton AM (1992) J Bacteriol 174:6938
63. Laskowska E, Wawrzynow A, Taylor A (1996) Biochimie 78:117
64. Veinger L, Diamant S, Buchner J, Goloubinoff P (1998) J Biol Chem 273:11032
65. Bowden GA, Georgiou G (1990) J Biol Chem 265:16760
66. Geli V, Baty D, Knibiehler M, Lloubes R, Pessegue B, Shire D, Lazdunski C (1989) Gene
80:129
67. Hart RA, Rinas U, Bailey JE (1990) J Biol Chem 265:12728
68. Sorensen MA, Pedersen S (1991) J Mol Biol 222:265
69. Frydman J, Erdjument-Bromage H, Tempst P, Hartl FU (1999) Nat Struct Biol 6:697
70. Chen W, Helenius J, Braakman I, Helenius A (1995) Proc Natl Acad Sci USA 92:6229
71. Ellis RJ, Hartl FU (1999) Curr Opin Struct Biol 9:102
72. Ujvari A, Aron R, Eisenhaure T, Cheng E, Parag HA, Smicun Y, Halaban R, Hebert DN
(2001) J Biol Chem 276:5924
73. Netzer WJ, Hartl FU (1997) Nature 388:343
74. Molinari M, Helenius A (2000) Science 288:331
75. Ellis RJ (2001) Trends Biochem Sci 26:597
76. Bazarsuren A, Grauschopf U, Wozny M, Reusch D, Hoffmann E, Schaefer W, Panzner S,
Rudolph R (2002) Biophys Chem 96:305
77. Schein CH, Noteborn MHM (1988) Bio/Technology 6:291
78. Carrio MM, Corchero JL, Villaverde A (1998) FEMS Microbiol Lett 169:9
79. Carrio MM, Villaverde A (2003) FEBS Lett 537:215
80. Carrio MM, Villaverde A (2001) FEBS Lett 489:29
81. Goloubinoff P, Mogk A, Zvi AP, Tomoyasu T, Bukau B (1999) Proc Natl Acad Sci USA
96:13732
Inclusion Bodies: Formation and Utilisation
137

82. Vogel U, Sorensen M, Pedersen S, Jensen K-F, Kilstrup M (1992) Mol Microbiol 2191
83. Studier FW, Rosenberg AH, Dunn JJ, Dubendorf JW (1990) Methods Enzymol 185:60
84. Iost I, Guillerez J, Dreyfus M (1992) J Bacteriol 174:619
85. Golomb M, Chamberlin M (1974) J Biol Chem 249:2858
86. Lyakhov DL, He B, Zhang X, Studier FW, Dunn JJ, McAllister WT (1998) J Mol Biol
280:201
87. Dong H, Nilsson L, Kurland CG (1996) J Mol Biol 260:649
88. Nakamura Y, Gojobori T, Ikemura T (2000) Nucleic Acids Res 28:292
89. Henaut A, Danchin A (1996) In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low
KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) Escherichia
coli and Salmonella typhimurium: cellular and molecular biology. ASM Press, Washing-
ton, DC, p 2047
90. Novy R, Drott D, Yaeger K, Mierendorf R (2001) inNovations 1
91. Lin HY (2000) PhD thesis, Martin-Luther-University Halle-Wittenberg, Department of
Biochemistry and Biotechnology, Halle, Germany
92. Brinkmann U, Mattes RE, Buckel P (1989) Gene 85:109
93. Zahn K (1996) J Bacteriol 178:2926
94. Da Silva NA, Bailey JE (1986) Biotechnol Bioeng 28:741
95. Aris A, Corchero JL, Benito A, Carbonell X,Viaplana E,Villaverde A (1999) Biotechnol Bio-
eng 64:127
96. Bahl H, Echols H, Straus DB, Court D, Crowl R, Georgopoulos CP (1987) Gene Dev 1:57
97. Dong H, Nilsson L, Kurland CG (1995) J Bacteriol 177:1497
98. Ito K, Akiyama Y, Yura T, Shiba K (1986) J Bacteriol 167:201
99. Parsell DA, Sauer RT (1989) Genes Dev 3:1226
100. Blaszczak A, Zylicz M, Georgopoulos C, Liberek K (1995) EMBO J 14:5085
101. Yura T, Nakahigashi K (1999) Curr Opin Microbiol 2:153
102. Bukau B (1993) Mol Microbiol 9:671
103. Straus DB, Walter WA, Gross CA (1987) Nature 329:348
104. Tilly K, Spence J, Georgopoulos C (1989) J Bacteriol 171:1585
105. Goff SA, Goldberg AL (1985) Cell 41:587
106. Kosinski MJ, Bailey JE (1991) J Biotechnol 18:55
107. Kosinski MJ, Bailey JE (1992) J Biotechnol 23:211
108. Kosinski MJ, Rinas U, Bailey JE (1992) Appl Microbiol Biotechnol 37:335
109. Snow A, Hipkiss AR (1987) Biochem Soc Trans 15:965
110. Rinas U (1996) Biotechnol Prog 12:196
111. Gill RT, DeLisa MP, Valdes JJ, Bentley WE (2001) Biotechnol Bioeng 72:85
112. Gill RT, Valdes JJ, Bentley WE (2000) Metab Eng 2:178
113. Birnbaum S, Bailey JE (1991) Biotechnol Bioeng 37:736
114. Wild J, Walter WA, Gross CA, Altman E (1993) J Bacteriol 175:3992
115. Craig EA, Gross CA (1991) Trends Biochem Sci 16:135
116. Bukau B, Horwich AL (1998) Cell 92:351
117. Kanemori M, Mori H, Yura T (1994) J Bacteriol 176:5648
118. Gill RT, DeLisa MP, Shiloach M, Holoman TR, Bentley WE (2000) J Mol Microbiol Biotech-
nol 2:283
119. Surek B, Wilhelm M, Hillen W (1991) Appl Microbiol Biotechnol 34:488
120. Srivastava R, Cha HJ, Peterson MS, Bentley WE (2000) Appl Environ Microbiol 66:4366
121. Ewalt KL, Hendrick JP, Houry WA, Hartl FU (1997) Cell 90:491
122. Teter SA, Houry WA, Ang D, Tradler T, Rockabrand D, Fischer G, Blum P, Georgopoulos C,
Hartl FU (1999) Cell 97:755
123. Deuerling E, Schulze-Specking A, Tomoyasu T, Mogk A, Bukau B (1999) Nature 400:693
124. Schmid D, Baici A, Gehring H, Christen P (1994) Science 263:971
125. Zhu Z, Zapata G, Shalaby R, Snedecor B, Chen H, Carter P (1996) Bio/Technology
14:192
126. Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B (1997) EMBO J 16:1501
127. Rudiger S, Buchberger A, Bukau B (1997) Nat Struct Biol 4:342
138
B. Fahnert et al.

128. Houry WA, Frishman D, Eckerskorn C, Lottspeich F, Hartl FU (1999) Nature 402:147
129. Fricke J, Neuhard J, Kelln R–A, Pedersen S (1995) J Bacteriol 517
130. Ellis RJ (1994) Curr Biol 4:633
131. Coates AR, Henderson B, Mascagni P (1999) Biotechnol Genet Eng Rev 16:405
132. Wickner S, Maurizi MR, Gottesman S (1999) Science 286:1888
133. Zolkiewski M (1999) J Biol Chem 274:28083
134. Mogk A, Tomoyasu T, Goloubinoff P, Rudiger S, Roder D, Langen H, Bukau B (1999) EMBO
J 18:6934
135. Glover JR, Lindquist S (1998) Cell 94:73
136. Motohashi K, Watanabe Y, Yohda M, Yoshida M (1999) Proc Natl Acad Sci USA 96:7184
137. Chuang SE, Burland V, Plunkett G III, Daniels DL, Blattner FR (1993) Gene 134:1
138. Thomas JG, Baneyx F (1998) J Bacteriol 180:5165
139. Missiakas D, Raina S (1997) Trends Biochem Sci 22:59
140. Kim KI, Park SC, Kang SH, Cheong GW, Chung CH (1999) J Mol Biol 294:1363
141. Pallen MJ, Wren BW (1997) Mol Microbiol 26:209
142. Hecker M, Schumann W, Völker U (1996) Mol Microbiol 19:417
143. Derre I, Rapoport G, Msadek T (1999) Mol Microbiol 31:117
144. Mogk A, Völker A, Engelmann S, Hecker M, Schumann W, Völker U (1998) J Bacteriol
180:2895
145. Jürgen B, Hanschke R, Sarvas M, Hecker M, Schweder T (2001) Appl Microbiol Biotech-
nol 55:326
146. Corchero JL, Viaplana E, Benito A, Villaverde A (1996) J Biotechnol 48:191
147. van den BB, Ellis RJ, Dobson CM (1999) EMBO J 18:6927
148. Murby M, Uhlén M, Stahl S (1996) Protein Expr Purif 7:129
149. Georgiou G, Valax P (1996) Curr Opin Biotechnol 7:190
150. Wang C, Castro AF, Wilkes DM, Altenberg GA (1999) Biochem J 338:77
151. Choi JH, Jeong KJ, Kim SC, Lee SY (2000) Appl Microbiol Biotechnol 53:640
152. Kopetzki E, Schumacher G, Buckel P (1989) Mol Gen Genet 216:149
153. Wilkinson DL, Harrison RG (1991) Bio/Technology 9:443
154. Hwang C, Sinskey AJ, Lodish HF (1992) Science 257:1496
155. Jaenicke R (1998) Biol Chem 379:237
156. Debarbieux L, Beckwith J (1999) Cell 99:117
157. Braakman I, Helenius J, Helenius A (1992) Nature 356:260
158. Missiakas D, Raina S (1997) J Bacteriol 179:2465
159. Raina S, Missiakas D (1997) Annu Rev Microbiol 51:179
160. Chung J, Chen T, Missiakas D (2000) Mol Microbiol 35:1099
161. Rietsch A, Bessette P, Georgiou G, Beckwith J (1997) J Bacteriol 179:6602
162. Rietsch A, Beckwith J (1998) Annu Rev Genet 32:163
163. Schein CH (1989) Bio/Technology 7:1148
164. Mburu DN, Roberts TK, Boettcher B (1999) Biochem Mol Biol Int 47:1009
165. Pines O, Inouye M (1999) Mol Biotechnol 12:25
166. Lavallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM (1993) Bio/Tech-
nology 11:187
167. Dracheva S, Palermo RE, Powers GD, Waugh DS (1995) Protein Expr Purif 6:737
168. Deana A, Ehrlich R, Reiss C (1996) J Bacteriol 178:2718
169. Coburn GA, Mackie GA (1999) Prog Nucleic Acid Res Mol Biol 62:55
170. Calderone TL, Stevens RD, Oas TG (1996) J Mol Biol 262:407
171. Kleber-Janke T, Becker WM (2000) Protein Expr Purif 19:419
172. Parker J (1989) Microbiol Rev 53:273
173. Spanjaard RA, Chen K, Walker JR, van Duin J (1990) Nucleic Acids Res 18:5031
174. Rosenberg AH, Goldman E, Dunn JJ, Studier FW, Zubay G (1993) J Bacteriol 175:716
175. Lavallie ER, McCoy JM (1995) Curr Opin Biotechnol 6:501
176. Hammonds RG, Schwall R, Dudley A, Berkemeier L, Lai C, Lee J, Cunningham N, Reddi
AH, Wood WI, Mason AJ (1991) Mol Endocrinol 5:149
177. Ellis RJ (1998) Trends Biochem Sci 23:43
Inclusion Bodies: Formation and Utilisation
139

178. Hidaka Y, Shimono C, Ohno M, Okumura N, Adermann K, Forssmann WG, Shimonishi Y
(2000) J Biol Chem 275:25155
179. Uhlen M, Forsberg G, Moks T, Hartmanis M, Nilsson B (1992) Curr Opin Biotechnol 3:363
180. Sachdev D, Chirgwin JM (1998) Biochem Biophys Res Commun 244:933
181. Sachdev D, Chirgwin JM (1998) Protein Expr Purif 12:122
182. Sachdev D, Chirgwin JM (2000) Methods Enzymol 326:312
183. Kapust RB, Waugh DS (1999) Protein Sci 8:1668
184. Blondel A, Bedouelle H (1990) Eur J Biochem 193:325
185. Boos W, Lucht JM (1996) In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB,
Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) Escherichia coli
and Salmonella typhimurium: cellular and molecular biology. ASM Press,Washington, DC,
p 1175
186. Richarme G, Caldas TD (1997) J Biol Chem 272:15607
187. Martineau P, Saurin W, Hofnung M, Spurlino JC, Quiocho FA (1990) Biochimie 72:397
188. Dietzel I, Kolb V, Boos W (1978) Arch Microbiol 118:207
189. Kellermann O, Szmelcman S (1974) Eur J Biochem 47:139
190. Furukawa H, Haga T (2000) J Biochem (Tokyo) 127:151
191. Louis JM, McDonald RA, Nashed NT,Wondrak EM, Jerina DM, Oroszlan S, Mora PT (1991)
Eur J Biochem 199:361
192. Saavedra-Alanis VM, Rysavy P, Rosenberg LE, Kalousek F (1994) J Biol Chem 269:9284
193. Fox JD, Kapust RB, Waugh DS (2001) Protein Sci 10:622
194. Nishihara K, Kanemori M, Yanagi H, Yura T (2000) Appl Environ Microbiol 66:884
195. Kurokawa Y, Yanagi H, Yura T (2000) Appl Environ Microbiol 66:3960
196. Zhan X, Schwaller M, Gilbert HF, Georgiou G (1999) Biotechnol Prog 15:1033
197. Knappik A, Krebber C, Pluckthun A (1993) Biotechnology NY 11:77
198. Joly JC, Leung WS, Swartz JR (1998) Proc Natl Acad Sci USA 95:2773
199. Thomas JG, Baneyx F (1996) J Biol Chem 271:11141
200. Schaffner J, Winter J, Rudolph R, Schwarz E (2001) Appl Environ Microbiol 67:3994
201. Huang H, Sherman MY, Kandror O, Goldberg AL (2001) J Biol Chem 276:3920
202. Gottesman ME, Hendrickson WA (2000) Curr Opin Microbiol 3:197
203. Kandror O, Sherman M, Goldberg A (1999) J Biol Chem 274:37743
204. Thomas JG, Baneyx F (2000) Mol Microbiol 36:1360
205. Diamant S, Ben-Zvi AP, Bukau B, Goloubinoff P (2000) J Biol Chem 275:21107
206. Wickner S, Gottesman S, Skowyra D, Hoskins J, McKenney K, Maurizi MR (1994) Proc Natl
Acad Sci USA 91:12218
207. Rosenbusch JP (1990) Experientia 46:167
208. Wunderlich M, Glockshuber R (1993) J Biol Chem 268:24547
209. Bardwell JC (1994) Mol Microbiol 14:199
210. Georgiou G, Valax P, Ostermeier M, Horowitz PM (1994) Protein Sci 3:1953
211. Thomas J–G, Baneyx F (1997) Protein Expr Purif 11:289
212. Kuivila R (2002) Diploma thesis, University of Oulu, Deptartment of Biochemistry, Fin-
land
213. Baneyx F, Ayling A, Palumbo T, Thomas D, Georgiou G (1991) Appl Microbiol Biotechnol
36:14
214. Beck R, Burtscher H (1994) Protein Expr Purif 5:192
215. Kaderbhai N, Karim A, Hankey W, Jenkins G,Venning J, Kaderbhai MA (1997) Biotechnol
Appl Biochem 25:53
216. Buchner J, Pastan I, Brinkmann U (1992) Anal Biochem 205:263
217. Moore JT, Uppal A, Maley F, Maley GF (1993) Protein Expr Purif 4:160
218. Wei Y, Lee JM, Richmond C, Blattner FR, Rafalski JA, LaRossa RA (2001) J Bacteriol 183:545
219. Ambrosius D, Rudolph R, Schäffner J, Schwarz E (1999)
220. Kempf B, Bremer E (1998) Arch Microbiol 170:319
221. Kempf B, Bremer E (1998) J Biosci 23:447
222. Caldas T, Demont-Caulet N, Ghazi A, Richarme G (1999) Microbiology 145:2543
223. Voziyan PA, Jadhav L, Fisher MT (2000) J Pharm Sci 89:1036
140
B. Fahnert et al.

224. Stewart EJ, Aslund F, Beckwith J (1998) EMBO J 17:5543
225. Dahan D, Srikumar R, Laprade R, Coulton JW (1996) FEBS Lett 392:304
226. Muttilainen S, Butcher SJ, Runeberg K, Nurminen M, Idanpaan-Heikkila I, Wahlstrom E,
Sarvas M (1995) Microb Pathog 18:365
227. Wang LF, Hum WT, Kalyan NK, Lee SG, Hung PP, Doi RH (1989) Gene 84:127
228. Charles JF, Hamon S, Baumann P (1993) Res Microbiol 144:411
229. Rang C, Lacey LA, Frutos R (2000) Curr Microbiol 40:200
230. Smith MW, Neidhardt FC (1983) J Bacteriol 154:336
231. Jensen KF (1997) J Bacteriol 175:3401
232. Morrisson TG, Malamy MH (1970) J Bacteriol 103:81
233. Tsai LB, Mann M, Morris F, Rotgers C, Fenton D (1987) J Ind Microbiol 2:181
234. Mizutani S, Mori H, Shimizu S, Sakaguchi K, Kobayashi T (1986) Biotechnol Bioeng 28:204
235. Mukhopadhyay A (1997) Adv Biochem Eng Biotechnol 56:61
236. Taylor G, Hoare M, Gray DR, Marston FAO (1986) Bio/Technology 4:553
237. Rudolph R, Böhm G, Lilie H, Jaenicke R (1997) In: Creighton TE (ed) Protein function. A
practical approach. IRL Press, Oxford, p 57
238. Middelberg AP, O’Neill BK (1991) Aust J Biotechnol 5:87
239. Maachupalli-Reddy J, Kelley BD, De Bernardez CE (1997) Biotechnol Prog 13:144
240. Hochuli E, Dobeli H, Schacher A (1987) J Chromatogr 411:177
241. Grauschopf U, Lilie H, Honold K, Wozny M, Reusch D, Esswein A, Schäfer W, Rücknagel
KP, Rudolph R (2000) Biochemistry 39:8878
242. Orsini G, Brandazza A, Sarmientos P, Molinari A, Lansen J, Cauet G (1991) Eur J Biochem
195:691
243. Misawa S, Aoshima M, Takaku H, Matsumoto M, Hayashi H (1994) J Biotechnol 36:145
244. Sarmientos P, Duchesne M, Denefle P, Boiziau J, Fromage N, Delporte N, Parker F, Lelievre
Y, Mayaux JF, Cartwright T (1989) Bio/Technology 7:495
245. Smith AT, Santama N, Dacey S, Edwards M, Bray RC, Thorneley RN, Burke JF (1990) J Biol
Chem 265:13335
246. Noguchi Y, Satoh S, Yamaguchi M, Watanabe K, Hayashi M, Yamada H, Saito Y, Kobayashi
M, Shimomura K (1996) J Ferment Bioeng 82:128
247. Langley KE, Berg TF, Strickland TW, Fenton DM, Boone TC, Wypych J (1987) Eur J
Biochem 163:313
248. De Bernadez Clark E (2001) Curr Opin Biotechnol 12:202
249. Hagel P, Gerding JJ, Fieggen W, Bloemendal H (1971) Biochim Biophys Acta 243:366
250. Li Y, Oelkuct M, Gentz R (1999) Patent No US5912327
251. Puri K, Crivelli E, Cardamone M, Fiddes R, Bertolini J, Ninham B, Brandon MR (1992)
Biochem J 285:871
252. Patra AK, Mukhopadhyay R, Mukhija R, Krishnan A, Garg LC, Panda AK (2000) Protein
Expr Purif 18:182
253. Reddy A, Grimwood BG, Plummer TH, Tarentino AL (1998) Glycobiology 8:633
254. Hartman JR, Mendelowitz S, Gorecki M (1999) Patent No US6001604
255. Schoemaker JM, Brasnett AH, Marston FA (1985) EMBO J 4:775
256. Fischer B, Perry B, Sumner I, Goodenough P (1992) Protein Eng 5:593
257. Maeda Y, Ueda T, Imoto T (1996) Protein Eng 9:95
258. Zettlmeissl G, Rudolph R, Jaenicke R (1979) Biochemistry 18:5567
259. Winter J, Lilie H, Rudolph R (2002) Anal Biochem 310:148
260. Fahey EM, Chaudhuri JB, Binding P (2000) Sep Sci Technol 35:1743
261. Fahey EM, Chaudhuri JB, Binding P (2000) J Chromatogr B Biomed Sci Appl 737:225
262. Muller C, Rinas U (1999) J Chromatogr A 855:203
263. Negro A, Onisto M, Grassato L, Caenazzo C, Garbisa S (1997) Protein Eng 10:593
264. Rogl H, Kosemund K, Kuehlbrandt W, Collinson I (1998) FEBS Lett 432:21
265. Stempfer G, Holl-Neugebauer B, Rudolph R (1996) Nat Biotechnol 14:329
266. Ahmed AK, Schaffer SW, Wetlaufer DB (1975) J Biol Chem 250:8477
267. Builder S, Hart R, Lester A, Reifsnyder D (1998) Patent No US5808006
268. Kuhelj R, Dolinar M, Pungercar J, Turk V (1995) Eur J Biochem 229:533
Inclusion Bodies: Formation and Utilisation
141

269. Mukhopadhyay A (2000) Vaccine 18:1802
270. Lilie H, McLaughlin S, Freedman R, Buchner J (1994) J Biol Chem 269:14290
271. Kohnert U, Rudolph R,Verheijen JH,Weening-Verhoeff EJ, Stern A, Opitz U, Martin U, Lill
H, Prinz H, Lechner M (1992) Protein Eng 5:93
272. Tsuji T, Nakagawa R, Sugimoto N, Fukuhara K (1987) Biochemistry 26:3129
273. Orsini G, Goldberg ME (1978) J Biol Chem 253:3453
274. Hevehan D, De Bernadez Clark E (1997) Biotechnol Bioeng 54:221
275. Lin WJ, Traugh JA (1993) Protein Expr Purif 4:256
276. Lin TY, Timasheff SN (1996) Protein Sci 5:372
277. Lilie H, Schwarz E, Rudolph R (1998) Curr Opin Biotechnol 9:497
278. Rudolph R, Lilie H, Schwarz E (1999) In: Rehm HJ, Reed G (eds) Biotechnology. Wiley
VCH, Weinheim, p 112
279. Cleland JL, Hedgepeth C, Wang DI (1992) J Biol Chem 267:13327
280. Rozena D, Gellma SH (1995) J Am Chem Soc 117:2373
281. Machida S, Ogawa S, Xiaohua S, Takaha T, Fujii K, Hayashi K (2000) FEBS Lett 486:131
282. Sharma L, Sharma A (2001) Eur J Biochem 268:2456
283. Ny T, Elgh F, Lund B (1984) Proc Natl Acad Sci USA 81:5355
284. Rudolph R, Buchner J, Lenz H (1994) Patent No EU0364926
285. Datar RV, Cartwright T, Rosen CG (1993) Bio/Technology 11:349
Received: August 2003
142
Inclusion Bodies: Formation and Utilisation
Wyszukiwarka
Podobne podstrony:
Formation of active inclusion bodies in E coli
2003 formatchain and network management gilda project
[12]Aging sensitizes towards ROS formation and lipid peroxidation in PS1M146L transgenic mice
Protein quality in bacterial inclusion bodies
Learning about protein solubility from bacterial inclusion bodies
C 2 Handout company formation and management
Formation and growth of calcium phosphate on the surface of
VP7 Data Format and Decoder Overview
Griffiths, Turnbullb And White Re Examining The Small Cap Myth Problems In Portfolio Formation And L
feminism and formation of ethnic identity in greek culture
E-Inclusion and the Hopes for Humanisation of e-Society, Media w edukacji, media w edukacji 2
Evidence for the formation of anhydrous zinc acetate and acetic
Atari 8 Bit Demopac 1 Strings and Formatting
Arnaiz P , Berruezo P P , de Haro R , Martínez R , Inclusive and Supportive Education Congress Inter
A Propagandist of Extermination, Johann von Leers and the Anti Semitic Formation of Children in Nazi
CAMBRIDGE ENGLISH SKILLS REAL LISTENING AND SPEAKING 2 WITHOUT ANSWERS Sally Logan Craig Thaine for
Jayne Raisborough Lifestyle Media and the Formation of the Self (2011)
feminism and formation of ethnic identity in greek culture
więcej podobnych podstron