SPEKTROFOTOMETRIA
Prawa absorpcji
I prawo absorpcji (prawo Bouguera-Lamberta). Wiązka światła monochromatycznego po przejściu przez jednorodny ośrodek absorbujący o grubości d ulega osłabieniu według równania:
gdzie: I0 - natężenie wiązki promieniowania monochromatycznego padającego na jednorodny ośrodek absorbujący; I - natężenie promieniowania po przejściu przez ośrodek absorbujący, k - współczynnik absorpcji, charakterystyczny dla danej substancji. Zależność tę wygodniej jest przedstawić w formie logarytmicznej:
A = ln I0/I= kd lub A = log I0/I= ad
gdzie: a = 0,4343k, A - zdolność pochłaniania, zwana absorbancją. Obok terminu absorbancja używane są także inne terminy, tj.: wartość absorpcji, ekstynkcja (E) lub gęstość optyczna (D). I prawo absorpcji można także sformułować w następujący sposób: absorbancja jest proporcjonalna do grubości warstwy absorbującej, jeżeli wiązka promieniowania monochromatycznego przechodzi przez jednorodny ośrodek absorbujący. Często przy określaniu ilości zaabsorbowanego promieniowania spotykamy się z wielkością nazywaną transmitancją (T), zdefiniowaną jako:
T = I0/I A zatem: A = log 1/T Transmitancja podawana jest najczęściej w procentach; w związku z tym:
%T = I / I0 100 = 100 T
II prawo absorpcji (prawo Beera). Prawo to dotyczy absorpcji promieniowania elektromagnetycznego przez roztwory i można je sformułować w następujący sposób: jeżeli współczynnik absorpcji rozpuszczalnika jest równy zeru, to wiązka promieniowania monochromatycznego, po przejściu przez jednorodny roztwór substancji absorbującej o stężeniu c, ulega osłabieniu według równania:
W formie logarytmicznej zależność ta może ma postać:
A = ln I0/I= kdc lub A = log I0/I= adc
Prawo to można sformułować w ten sposób: jeżeli współczynnik absorpcji rozpuszczalnika jest równy zeru, to absorbancja wiązki promieniowania monochromatycznego przechodzącego przez roztwór jednorodny jest wprost proporcjonalna do stężenia roztworu c i do grubości warstwy absorbującej d.
III prawo absorpcji (prawo addytywności absorpcji). Absorbancja roztworu wieloskładnikowego równa się sumie absorbancji poszczególnych składników:
A = A1 + A2 + ... + An
A1, A2, ... , An są to absorbancje poszczególnych składników. Prawo addytywności umożliwia analizę ilościową układów wieloskładnikowych. W równaniu na absorpcję: A = acd
wielkość a jest właściwym współczynnikiem absorpcji, gdy stężenie wyrażamy w g/cm3. natomiast, gdy stężenie wyrażamy w mol/dm3, to równanie to przybiera postać: A = εcd
Współczynnik ε nazywamy molowym współczynnikiem absorpcji. Współczynnik ten nie zależy od stężenia, a zatem na wykresie zależności ε = f (c) otrzymamy linię prostą, równoległą do osi odciętych. Współczynnik ten zależy natomiast od długości fali światła padającego, rodzaju substancji absorbującej światło i temperatury roztworu. Wartość A zależy od stężenia roztworu, a funkcja A = f (c) jest linią prostą, jeżeli roztwór spełnia prawo Beera. Absorbancja zależy także od długości fali λ światła padającego. Wykres zależności absorbancji od długości fali określa się mianem krzywej absorpcji lub elektronowym widmem absorpcyjnym.
Odchylenia od praw absorpcji
Odchylenia od praw absorpcji mogą być związane z podstawowymi ograniczeniami praw lub też z czynnikami chemicznymi bądź aparaturowymi. Prawa absorpcji spełnione są dla roztworów rozcieńczonych (c < 10-2 mol/dm3). W roztworach takich nie występuje zależność molowego współczynnika absorpcji od współczynnika załamania światła. W przypadku roztworów stężonych ε jest funkcją rzeczywistego molowego współczynnika absorpcji εrz i współczynnika załamania światła n zgodnie z równaniem:
Prawa absorpcji zakładają, że jedynym oddziaływaniem promieniowania elektromagnetycznego z substancją rozpuszczoną jest absorpcja promieniowania w myśl schematów:
X + hν
X*
Jednakże degradacja wzbudzonej cząsteczki (X*) do stanu podstawowego (X) jest możliwa również na drodze fluorescencji lub fosforescencji w myśl schematu:
X*
gdzie ν' - częstotliwość promieniowania na drodze fluorescencji lub fosforescencji. W takim przypadku pozorna transmitancja będzie podwyższona, a zatem pozorna absorbancja będzie niższa od rzeczywistej.
Czynniki chemiczne powodujące odchylenia od praw absorpcji są związane z reakcjami chemicznymi zachodzącymi w analizowanym roztworze. Przy zmianie pH roztworu i zmianie jego stężenia mogą zachodzić takie reakcje, jak: dysocjacja, asocjacja, różne reakcje kompleksowania, polimeryzacja i solwatacja. Zmiany właściwości optycznych analizowanych substancji, związane z tymi reakcjami, tłumaczą następujące przykłady:
Alkohol benzylowy w roztworze CCl4 w zależności od stężenia może występować w formie monomeru lub polimeru:
(λmax = 270 nm) (λmax = 300 nm)
Stąd, w zależności od długości fali, przy której będzie wykreślana krzywa wzorcowa, mogą nastąpić dodatnie lub ujemne odchylenia od prawa absorpcji.
W roztworach kwasów i zasad mogą, w zależności od pH, ustalać się różne równowagi kwasowo-zasadowe. Na przykład kwas benzoesowy w formie niezdysocjowanej i w formie zdysocjowanej charakteryzuje się różnymi wartościami λmax i ε:
C6H5COOH + H2O C6H5COO− + H3O+ (λmax = 273 nm, ε = 970) (λmax = 268 nm, ε = 560)
Dla takiego układu prawo absorpcji jest spełnione tylko w ściśle określonym pH roztworu i przy stałej sile jonowej.
W roztworze dwuchromianu potasowego, w miarę jego rozcieńczani, pomarańczowy K2Cr2O7 przechodzi w żółty K2CrO4 w myśl równania:
W zależności od długości fali, przy której mierzona jest absorbancja, może wystąpić dodatnie lub ujemne odchylenie od praw absorpcji. Równowaga tej reakcji może być kontrolowana poprzez buforowanie dwuchromianu silnym kwasem lub chromianu silną zasadą. W przypadku tworzenia kompleksów równowagi reakcji uzależnione są od stężenia ligandów. Wymagane jest stałe stężenie i to takie, które znacznie przekracza stężenie oznaczanego metalu. Na przykład układ Fe (III) - SCN− lub Cu (II) - NH3, w którym zachodzi stopniowe tworzenie się kompleksów, nie spełnia na ogół prawa absorpcji. Oddziaływanie rozpuszczalnika z badaną substancją powoduje często zmianę położenia λmax, zmienia kształt pasma, a także wpływa na wartość ε. Rozpuszczanie substancji powoduje najczęściej przesunięcie pasma absorpcyjnego w kierunku fal dłuższych w odniesieniu do pasma w fazie gazowej. Jest to tzw. przesunięcie czerwone lub efekt batochromowy. Efekt ten wzrasta ze wzrostem względnej przenikalności elektrycznej rozpuszczalnika. Wymienione wyżej czynniki mogą powodować dodatnie lub ujemne odchylenia od praw absorpcji.
Czynniki aparaturowe powodujące odchylenie od praw absorpcji są najczęściej związane z brakiem monochromatyczności promieniowania oraz występowaniem promieniowania rozproszonego. Brak monochromatyczności promieniowania stanowi główną przyczynę odchyleń od praw absorpcji. Polichromatyczność promieniowania jest bowiem pogwałceniem założeń praw absorpcji. Niedostateczna monochromatyczność występuje w przypadku pomiarów przy użyciu kolorymetrów i fotokolorymetrów, w których czynnikami monochromatyzującymi są filtry barwne i interferencyjne. Niekorzystnym zjawiskiem jest występowanie w przyrządach niższej klasy tzw. promieniowania rozproszonego. Jest to takie promieniowanie, które dociera do detektora, a nie przechodzi przez próbkę. Promieniowanie to powoduje wystąpienie ujemnego odchylenia od praw absorpcji.
Aparatura Dział metod analitycznych opartych na absorpcji promieniowania elektromagnetycznego w zakresie nadfioletu i części widzialnej widma, przyjęto nazywać spektrofotometrią absorpcyjną, a przyrządy absorpcjometrami. W zależności od budowy absorpcjometry można podzielić na 3 grupy: 1) absorpcjometry wizualne - kolorymetry, 2) absorpcjometry fotoelektryczne - fotokolorymetry, 3) spektrofotometry. Metody, stosujące odpowiednie przyrządy, przyjęto określać mianem kolorymetrii, fotokolorymetrii i spektrofotometrii. Poszczególne grupy przyrządów różnią się szczegółami budowy, jednak wszystkie mają takie same zasadnicze elementy, a schemat blokowy typowych absorpcjometrów składa się z 6 podstawowych układów:
Źródło promieniowania: w kolorymetrach - światła lub żarówka; w fotokolorymetrach - lampa wolframowa, lampa rtęciowa; w spektrofotometrach - lampa wolframowa na zakres widzialny i bliską podczerwień. Emituje ona promieniowanie ciągłe w zakresie od 380 do 2500 nm. W zakresie nadfioletu, od 180 do 380 nm, źródłem promieniowania ciągłego są lampy deuterowe lub wodorowe. Źródłem światłą dla zakresu widzialnego i nadfioletu jest wysokociśnieniowa łukowa lampa ksenonowa. Regulacja natężenia promieniowania: przesłona irysowa, szczelina regulowana, opór zmienny w obwodzie lampy. Szerokość szczeliny, w większości przyrządów, zmienia się od 0,01 do 1 mm. Od szerokości szczeliny zależy czułość i rozdzielczość przyrządów. Ogólnie biorąc, rozdzielczość jest tym większa, im węższa jest szczelina. Regulacja długości fali światła padającego: filtry barwne i interferencyjne w kolorymetrach oraz monochromatory (pryzmaty i siatki dyfrakcyjne) w spektrofotometrach. W zakresie widzialnym znajdują zastosowanie filtry wykonane ze szkła barwnego. W zakresie nadfioletu stosuje się filtry krystaliczne z odpowiednich soli lub roztworów soli w naczyniach kwarcowych.
Monochromatory mają zastosowanie w spektrofotometrach i są głównym czynnikiem odróżniającym tę klasę przyrządów od pozostałych absorpcjometrów. Są nimi siatki dyfrakcyjne i pryzmaty. Pryzmaty mają różną budowę. Spotyka się zarówno proste pryzmaty, jak i skomplikowane układy pryzmatyczne. Do najczęściej stosowanych należą: a) pryzmat Cornu: wykonany z dwóch sklejonych kryształów kwarcu lewo- i prawoskrętnego o sumarycznym kącie łamiącym 60o. Jest to pryzmat o średniej dyspersji, stosowany w przyrządach niższej klasy. b) Pryzmat typu Littrowa: wykonany z kwarcu o kącie łamiącym 30o; ścianka prostopadła do osi optycznej pokryta jest metalem dającym lustro, dzięki temu promień przechodząc przez pryzmat ulega dwukrotnie załamaniu. Pryzmat typu Littrowa stosowany jest do budowy monochromatorów o dużej dyspersji. c) Pryzmat typu Wadswortha: połączony ze zwierciadłem, najczęściej stosowany w spektrofotometrach na podczerwień. Siatki dyfrakcyjne stosowane są jako monochromatory, najczęściej w przypadkach, gdy wymagana jest większa dyspersja w zakresie fal długich. Dyspersja siatek dyfrakcyjnych nie zależy od długości fali promieniowania, a tylko od rodzaju siatki dyfrakcyjnej. Ujemną cechą siatek dyfrakcyjnych jest występowanie w widmie promieniowania rozproszonego i to jest główną przyczyną stosowania w spektrofotometrach monochromatorów pryzmatycznych.
Pomieszczenie dla substancji badanej: kiuweta, probówka. W zakresie widzialnym stosowane są kiuwety ze szkła lub mas plastycznych. W zakresie nadfioletu stosuje się kiuwety kwarcowe. Kiuwet kwarcowych można używać zarówno w zakresie widzialnym, jak i w nadfiolecie. Grubość kiuwety wynosi zazwyczaj 1 cm, ale można używać zarówno kiuwet o większej, jak i mniejszej grubości.
Detektor: w kolorymetrach - oko ludzkie, w fotokolorymetrach - fotoogniwa, fotokomórki, fotopowielacze; a) Fotoogniwa, zwane ogniwami zaporowymi, zbudowane są z trzech warstw. Na płytce z Fe lub Cu znajduje się warstwa półprzewodnika (Se lub CuO), a na niej cienka warstwa metalu: Ag, Pb lub Au. Wiązka światła, padając na Se, wytwarza fotoprąd, który można zmierzyć za pomocą galwanometru, włączonego szeregowo w obwód. Natężenie prądu płynącego w fotoogniwach selenowych jest stosunkowo duże i wynosi około 120 mA na lumen, stąd też fotoogniwa nie wymagają stosowania dodatkowych wzmacniaczy prądu. Fotoogniwo selenowe jest czułe na światło o długości fali 300 - 800 nm. b) Fotokomórki stosowane są najczęściej w spektrofotometrach w połączeniu z odpowiednim układem wzmacniającym. Rozróżnia się fotokomórki próżniowe i fotokomórki wypełnione gazem szlachetnym. W obu przypadkach są to bańki szklane, w których znajduje się katoda w formie blaszki, pokryta warstewką światłoczułą, i anoda w postaci obrączki lub spirali metalowej. Katoda połączona jest z biegunem ujemnym prądu, a anoda z dodatnim. W przypadku, gdy na katodę pada promieniowanie elektromagnetyczne, następuje emisja elektronów, które przyciągane są przez anodę na skutek różnicy potencjałów. Natężenie prądu fotoelektrycznego rejestruje włączony w obwód galwanometr. Natężenie powstającego fotoprądu zależy od natężenia promieniowania padającego na fotokomórkę, od długości fali λ światłą padającego, a także od różnicy potencjałów między elektrodami fotokomórki. Materiałem światłoczułym, pokrywającym katodę, są niektóre metale i tlenki metali. c) Fotopowielacz (fotomnożnik) jest wzmacniaczem bardzo słabych impulsów prądowych, wywołanych działaniem promieniowania elektromagnetycznego o niewielkim natężeniu. Jest to typ lampy elektrodowej zamkniętej z jednej strony fotokatodą, a z drugiej cokołem. Między fotokatodą a anodą znajduje się kilka lub kilkanaście elektrod, zwanych dynodami, do których przyłożone są zmniejszające się napięcia U1, U2, U3, ... . Działanie fotopowielacza polega na tym, że foton, padając na fotokatodę, wybija z niej elektrony, te zaś trafiają na dynodę, wywołując wtórną emisję elektronów, przy czym każdy z elektronów wybija z dynody kilka nowych. Proces ten powtarza się na kolejnych dynodach i w efekcie otrzymuje się wielokrotne wzmocnienie fotoprądu. Fotopowielacze mają zastosowanie w spektrofotometrach UV-VIS wyższej klasy, a także w innych przyrządach optycznych, np. w kwantomerach czy spektrofotometrach absorpcji atomowej. Wskaźnik lub rejestrator: galwanometr, samopis, potencjometr, oscyloskop.
Kolorymetry Kolorymetry, jako pierwsze absorpcjometry, odegrały w analizie instrumentalnej ważną rolę. Na szczególną uwagę zasługuje kolorymetr typu Duboscqa. Pomiar stężenia roztworu badanego, za pomocą tego przyrządu, przeprowadza się metodą zrównania barw. Osiąga się to na drodze zmian grubości warstwy roztworu badanego. Światło wysyłane przez żarówkę, ulega odbiciu od płytki ze szkła mlecznego i oświetla równomiernie dna naczyń pomiarowych. Jedno naczynie napełnia się roztworem badanym, a drugie roztworem tej samej substancji, o znanym stężeniu. Światło po przejściu przez naczynie pomiarowe i układ optyczny wpada do okularu, w którym pole widzenia, w postaci koła, podzielone jest linią na połówki. Oba naczynia pomiarowe są umieszczone na odpowiednich wspornikach, połączonych z mechanizmem umożliwiającym pionowe podnoszenie i opuszczanie naczyń. Grubość warstw roztworów reguluje się za pomocą szklanych walców. Pomiar polega na doprowadzeniu obu połówek pola widzenia do oświetlenia o identycznym natężeniu. Na podstawie otrzymanych wyników, grubości warstw roztworu badanego d1 i wzorcowego d2, można obliczyć stężenie badanego roztworu, korzystając z wzoru: c1d1 = c2d2.
Absorpcjometry fotoelektryczne Detektorami w absorpcjometrach fotoelektrycznych są fotoogniwa, fotokomórki lub fotopowielacze. Do grupy absorpcjometrów fotoelektrycznych zalicza się przyrządy o różnorodnych rozwiązaniach konstrukcyjnych. Można je podzielić na absorpcjometry jedno- i dwuwiązkowe, w zależności od tego, ile wiązek promieniowania biegnie od źródła do detektora. W absorpcjometrach dwuwiązkowych istnieje możliwość jednoczesnego porównywania absorbancji próbki i wzorca. Inny podział absorpcjometrów uwzględnia sposób wykonania pomiarów. Można tu rozróżnić absorpcjometry wychyleniowe i kompensacyjne. Do pierwszej grupy należą aparaty działające na zasadzie wychylenia wskazówki galwanometru wzdłuż podziałki, skalowanej w jednostkach absorbancji i procentach transmitancji. Do drugiej grupy należą przyrządy pracujące na zasadzie kompensacji potencjometrycznej. Są to aparaty typu zerowego, a ich wskazania są niezależne od wahań napięcia prądu, zasilającego źródło światła. Spośród dużej liczby produkowanych absorpcjometrów, stosunkowo najbardziej rozpowszechnionym w Polsce jest spektrokolorymetr „Spekol” produkowany przez Zakłady C. Zeiss w Jenie. Jest to absorpcjometr jednowiązkowy, wychyleniowy, o niewielkich rozmiarach i prostej obsłudze. Umożliwia on pomiary absorbancji w zakresie 365 - 750 nm. Zasadniczy przyrząd jest zaopatrzony w różne przystawki, pozwalające przeprowadzić miareczkowanie spektrofotometryczne, turbidymetryczne, fluorescencyjne oraz pomiar remisji, stopnia zmętnienia i natężenia fluorescencji. Światło, emitowane przez źródło, którym jest lampa wolframowa (6 V, 30 W) na zakres 420 - 750 nm (lub lampa rtęciowa HQE-40 na zakres 365 - 750 nm), przechodzi przez kondensor i po odbiciu od zwierciadła wchodzi przez szczelinę wejściową do kolimatora. Po przejściu przez układ achromatyczny pada na monochromator, którym jest siatka dyfrakcyjna. Za pomocą odpowiedniego układu dźwigni można obracać siatkę dyfrakcyjną i dzięki temu przepuszczać przez szczelinę wyjściową monochromatora wiązkę o żądanej długości fali. Szerokość spektralna przepuszczanych przez szczelinę wiązek monochromatycznych wynosi 12 nm. Wybrana długość widma, po przejściu przez soczewkę achromatyczną, trafia na szczelinę wyjściową i po przejściu przez kiuwetę z roztworem badanym i filtr barwny do pochłaniania promieniowania cieplnego pada na detektor. W standardowym przyrządzie detektorem jest fotoogniwo selenowe. Powstały w fotoogniwie fotoprąd ulega wzmocnieniu we wzmacniaczu tranzystorowym i dostaje się do urządzenia pomiarowego. Spektrofotometry Spektrofotometry UV-VIS należą do absorpcjometrów najwyższej klasy. Dzięki zastosowaniu pryzmatów i siatek dyfrakcyjnych otrzymuje się promieniowanie o wąskiej szerokości spektralnej, a przez wprowadzenie dodatkowych filtrów, praktycznie monochromatyczne. Detektorami w tych przyrządach są fotokomórki, z odpowiednimi układami wzmacniającymi fotoprąd, i fotopowielacze. Ze względu na sposób rejestracji, spektrofotometry UV-VIS można podzielić na dwie grupy: - spektrofotometry punktowe, w których absorbancję odczytuje się metodą wychyleniową, na skali galwanometru lub częściej metodą kompensacyjną, równoważąc układ pomiarowy za pomocą potencjometru, wyskalowanego w jednostkach absorbancji i procentach transmitancji, - spektrofotometry samopiszące, rejestrujące widma absorpcji w układzie %T = f (λ). Są to urządzenia o bardzo skomplikowanej budowie. Są to aparaty dwuwiązkowe, o szeroko rozbudowanym układzie optycznym, umożliwiającym dobrą monochromatyzację widma. Dzięki zastosowaniu, jako detektorów, fotopowielaczy charakteryzują się większą czułością.
Zastosowanie spektrofotometrii UV-VIS Spektrofotometria UV-VIS znalazła różnorodne zastosowanie w wielu dziedzinach chemii. Jest powszechnie stosowana w analizie ilościowej, a także do identyfikacji związków organicznych i do badania związków kompleksowych.
Analiza ilościowa Pomiar zawartości badanego składnika metodą spektrofotometryczną jest oparty na prawach absorpcji. Dokładność oznaczeń zależy od właściwego przygotowania roztworów wzorcowych, a także od dobrania optymalnej długości fali , przy której ma być wykonany pomiar spektrofotometryczny. Optymalną długość fali dobieramy za pomocą krzywej absorpcji, przedstawiającej zależność absorbancji od długości fali światła (A = f (λ)). Pomiary spektrofotometryczne wykonuje się najczęściej przy długości fali odpowiadającej λmax. Trudniejszy jest wybór analitycznej długości fali, gdy w celu oznaczenia kationu metalu M otrzymujemy barwny kompleks ML i gdy zarówno kompleks jak i odczynnik L absorbują promieniowanie. Należy wówczas wykreślić krzywą absorpcji odczynnika i badanego kompleksu. Analityczną długość fali ustalamy wówczas przy λopt, gdzie różnica absorbancji odczynnika i badanego kompleksu ΔA jest największa.Ilościowe oznaczanie badanego składnika przeprowadza się najczęściej metodą algebraiczną, metodą krzywej wzorcowej lub miareczkowania spektrofotometrycznego.
Metoda algebraiczna Stężenie cx oznaczanego składnika oblicza się bezpośrednio ze wzoru:
cx = A / εd
Współczynnik absorpcji ε można wyznaczyć mierząc, przy danej długości fali λmax, absorbancję kilku roztworów wzorcowych i z tych danych obliczyć wartość εśr. Inny sposób oznaczania stężenia cx analizowanej substancji polega na porównaniu absorbancji roztworu badanego i roztworu wzorcowego o znanym stężeniu substancji oznaczanej. Pomiar absorbancji roztworów przeprowadza się przy tej samej długości fali w naczyniach o takiej samej grubości warstwy. Absorbancję tych dwóch roztworów można wyrazić wzorami:
Ax = ελcxdx Awz = ελcwzdwz
Dzieląc równania stronami otrzymuje się:
gdy dx = dwz, a ελ = const, otrzymuje się:
Sposób porównywania absorbancji stosuje się często w przypadku pojedynczych oznaczeń. Modyfikacją tej metody, dającą pewniejsze wyniki, jest sposób z dwoma roztworami wzorcowymi c1 i c2 tak dobranymi, aby A1 < Ax < A2, wówczas nieznane stężenie roztworu można obliczyć z wzoru:
Metoda krzywej wzorcowej Polega ona na sporządzeniu serii (4 - 6) roztworów wzorcowych oznaczanej substancji, o różnych stężeniach, zmierzeniu absorbancji tych roztworów, przy wybranej długości fali i wykreśleniu krzywej wzorcowej A = f (c). Krzywa wzorcowa (krzywa analityczna) jest linią prostą, jeżeli roztwory spełniają prawa absorpcji. W celu określenia stężenia cx badanej substancji należy, w tych samych warunkach, oznaczyć jej absorbancję Ax i z krzywej wzorcowej odczytać szukane stężenie cx. Zarówno w metodzie algebraicznej jak i w metodzie krzywej wzorcowej jako roztwór odniesienia można stosować albo rozpuszczalnik, albo wzorcowy roztwór substancji badanej.
W metodzie fotometrii bezpośredniej roztworem odniesienia jest rozpuszczalnik lub roztwór, sporządzony z rozpuszczalnika i stosowanych w danym oznaczeniu odczynników. Przyrząd ustawia się na 0% transmitancji, gdy fotokomórka znajduje się w ciemności (zamknięta przesłona), i na 100% transmitancji, gdy promieniowanie przechodzi przez roztwór odniesienia (rozpuszczalnik). Po takim ustawieniu przyrządu można mierzyć dowolne natężenie promieniowania, przechodzącego przez roztwór badany, w zakresie 0 - 100% transmitancji. W tym konwencjonalnym sposobie postępowania popełnia się jednak duży błąd, szczególnie wtedy, gdy mierzone są roztwory bardzo stężone (T < 20%) lub bardzo rozcieńczone (T > 65%). W takich przypadkach stosuje się tzw. metodę spektrofotometrii różnicowej. W metodzie tej roztworem odniesienia jest roztwór badanej substancji o stężeniu c1. Zerową transmitancję ustawia się tu tak samo jak w metodzie fotometrii bezpośredniej, tzn. dla zaciemnionego detektora promieniowania. Natomiast transmitancję T = 100% nastawia się na najmniej stężony roztwór wzorcowy c1. takie postępowanie pozwala na rozszerzenie skali przyrządu, a tym samym na zwiększenie dokładności pomiarów. Można wykazać, że absorbancja różnicowa jest proporcjonalna do różnicy stężeń próbki badanej i wzorcowej: A = ε d (cx - c1)
Analogiczny tok postępowania można przyjąć w przypadku roztworów rozcieńczonych (T > 65%). Zerową transmitancję należy nastawić na najbardziej stężony roztwór wzorcowy, a T = 100% na czysty rozpuszczalnik. Jeżeli w metodzie konwencjonalnej transmitancja wzorca (c1) jest równa 90%, a transmitancja próbki badanej (cx) 93%, to po rozszerzeniu skali przyrządu dla c1 uzyskamy T = 0%, a dla cx otrzymamy T = 30%.
Metoda miareczkowania spektrofotometrycznego Miareczkowanie spektrofotometryczne stosuje się w celu ustalenia punktu końcowego miareczkowania wówczas, gdy zmiana barwy, towarzysząca temu punktowi, nie jest dostatecznie wyraźna, aby ją zaobserwować wizualnie, lub przebiega stopniowo, w miarę dodawania odczynnika miareczkującego. Pomiar polega na kolejnym mierzeniu absorbancji, zmieniającej się podczas miareczkowania roztworu badanego. Proces miareczkowania przedstawia się graficznie. Wykres sporządza się w układzie: A = f (V), gdzie V - objętość roztworu miareczkującego. Miareczkowanie przeprowadza się przy określonej długości fali w specjalnie do tego celu przystosowanych spektrofotometrach (np. Spekol z przystawką do miareczkowania Ti lub TiMi), w których roztwór zawarty w kiuwecie jest mieszany za pomocą mieszadełka elektromagnetycznego. W metodzie miareczkowania spektrofotometrycznego otrzymuje się różne typy krzywych miareczkowania. Miareczkowanie spektrofotometryczne można prowadzić zarówno bez indykatora, jak również wobec indykatora.
Miareczkowanie bez indykatora. Oto kilka przykładów takich miareczkowań: a) oznaczana substancja A i produkt miareczkowania AB nie absorbują światła, a odczynnik miareczkujący B absorbuje światło. Krzywa miareczkowania ma wówczas przebieg przedstawiony na rysunku. Przykładem jest miareczkowanie jonów Fe2+ za pomocą KMnO4 przy λ = 550 nm.
b) produkt miareczkowania AB absorbuje światło, a substancja oznaczana A i odczynnik miareczkujący B nie absorbują, np. miareczkowanie małych ilości Cu2+ za pomocą EDTA przy długości przy λ = 260 nm. Krzywa miareczkowania przyjmuje postać przedstawioną poniżej:
Podobny przebieg ma krzywa miareczkowania Fe3+ za pomocą NH4SCN.
c) oznaczania substancja A pochłania światło, a odczynnik miareczkujący B i produkt reakcji AB nie pochłaniają. Przykładem jest miareczkowanie dwuchromianu za pomocą jonów Fe2+ przy λ = 350 nm. Krzywa miareczkowania wygląda następująca:
d) oznaczana substancja A i odczynnik miareczkujący B pochłaniają światło, a produkt reakcji AB nie pochłania światła. Krzywa ma wówczas przebieg następujący:
Miareczkowanie wobec indykatora. Jeżeli żaden ze składników reakcji A, B, AB nie pochłania promieniowania, to często stosuje się indykator Y, który pozwala uchwycić punkt końcowy miareczkowania. Indykatorem takim może być substancja, która tworzy barwne połączenia typu AY, BY lub ABY; np. NH4SCN tworzy z jonami Fe3+ barwny kompleks, wykazujący maksimum absorbancji przy λ = 525 nm. Kompleks ten jest jednak mniej trwały od kompleksu Fe3+ - EDTA i dlatego w procesie miareczkowania barwnego rodanku żelazowego za pomocą EDTA następuje odbarwienie roztworu. W analogiczny sposób można miareczkować za pomocą EDTA: Bi3+ wobec tiomocznika przy λ = 400 nm, Cu2+ wobec NH4OH przy λ = 630 nm. Miareczkowanie spektrofotometryczne charakteryzuje się wieloma zaletami, a mianowicie:
Badanie składu związków kompleksowych Spektrofotometria jest ważną metodą pomiarową stosowaną do badania równowag w roztworach. Wykorzystuje się ją w szczególności do wyznaczania stałych dysocjacji kwasów i zasad oraz do ustalania składu i stałych trwałości związków kompleksowych. Związki kompleksowe powstają w wyniku współdziałania jonu centralnego M z ligandem L w myśl równania:
mM + nL MmLn
Badanie procesów kompleksowania sprowadza się do identyfikacji powstających kompleksów, ustalenia ich składu, ładunku oraz charakterystyki termodynamicznej, z którą związane jest obliczenie wartości stałej równowagi reakcji, określającej trwałość kompleksu. Ustalenie składu kompleksu MmLn sprowadza się do wyznaczenia wartości współczynników m i n. W przypadku, gdy m = 1, powstający kompleks nazywamy jednordzeniowym, gdy m > 1 - wielordzeniowym. Do najczęściej stosowanych metod wyznaczania składu kompleksu należą: metoda stosunków molowych i metoda zmian ciągłych (metoda serii izomolowych). W metodzie stosunków molowych mierzy się absorbancję serii roztworów, które zawierają zmienne ilości jednego składnika, a stałą ilość drugiego. Z kolei sporządza się wykres przedstawiający absorbancję jako funkcję stosunku molowego liganda i jonu centralnego A = f (cL/cM). Punkt przecięcia na krzywej odpowiada stosunkowi molowemu składników kompleksu. W metodzie serii izomolowych przygotowuje się serię roztworów o zmiennych składach molowych obydwu składników, ale przy ich stałym stężeniu sumarycznym. Otrzymana krzywa absorpcji wykazuje maksimum przy takim stosunku molowym, który odpowiada składowi kompleksu.
SPEKTROFOTOMETRIA W PODCZERWIENI
Promieniowanie elektromagnetyczne absorbowane przez cząsteczki powoduje nie tylko wzbudzenie elektronowe, ale wywołuje również zmiany energii oscylacyjnej i rotacyjnej. Absorpcja promieniowania IR w obszarze 10000-100 cm-1 wiąże się ze zmianami stanów oscylacyjnych w cząsteczce. Widmo oscylacyjne występuje w postaci pasm, ponieważ każdej zmianie energii oscylacji towarzyszy zmiana energii rotacji. Widma oscylacyjne są wynikiem dwóch rodzajów drgań cząsteczkowych: drgań walencyjnych (rozciągających) - zmiany zachodzą wzdłuż osi wiązania i zmianie ulegają długości wiązań i drgań deformacyjnych - polegają na zmianie kątów między wiązaniami lub na ruchu grupy atomów w stosunku do reszty cząsteczki, bez zmiany położenia atomów, względem siebie w grupie, należą do nich: drgania nożycowe, kołyszące skręcające i wahadłowe. Liczba stopni swobody cząsteczki jest równa sumie stopni swobody wszystkich atomów wchodzących w jego skład. Każdy z atomów ma trzy stopnie swobody, więc cząsteczka złożona z n atomów ma 3n stopni swobody. Cząsteczka nieliniowa - 3n-6 - oscylacyjne stopnie swobody (tzw. drgania podstawowe), cząsteczka liniowa - 3n-5 oscylacyjnych stopni swobody. W widmach Ir oprócz częstości odpowiadających drganiom podstawowym,
obserwuje się również pasma harmoniczne (nadtony) i pasma kombinacyjne, w postaci pasm o niskim natężeniu. Pasma harmoniczne odpowiadają częstością będącym wielokrotnością jednej z częstości podstawowych, a pasma kombinacyjne sumie lub różnicy dwóch różnych częstości podstawowych. Czasami w widmie IR występują pasma o dużym natężeniu, powstające w wyniku tzw. rezonansu Fermiego - ma miejsce, gdy któraś z częstości harmonicznych ma wartość bliską częstości drgania podstawowego. W podczerwieni aktywne są tylko te drgania, z którymi związana jest zmiana momentu dipolowego cząsteczki, natomiast w widmie ramanowskim czynne są tylko te drgania, z którymi związana jest znaczna polaryzowalność cząsteczki. Stąd zasada wzajemnego wykluczania: jeżeli cząsteczka ma środek symetrii, to w widmie IR czynne są drgania antysymetryczne względem środka symetrii i drgania te nie są czynne w widmie ramanowskim i na odwrót - w widmie ramanowskim czynne są drgania symetryczne względem środka symetrii i z kolei drgania te nie występują w widmie IR. Pewne grupy funkcyjne, niezależnie od zmian w pozostałych częściach cząsteczki, absorbują promieniowanie w wąskim zakresie częstości - grupowe częstości charakterystyczne. Przybliżone częstości poszczególnych drgań walencyjnych można wyznaczyć za pomocą prawa Hooke'a, które ustala zależność między częstością drgań, masą atomów i stałą siłową wiązania:
F - stała siłowa wiązania; równa jest 5 N/cm dla wiązania pojedynczego i odpowiednia dwa i trzy razy większa dla wiązania podwójnego i potrójnego. Źródła promieniowania podczerwonego. 1) włókno Nernsta - drucik o średnicy 1 mm i długości 1 cm, wykonany z mieszaniny tlenków ceru, cyrkonu, toru i itru. 2) włókno Globar - pręt silitowy z SiC. Monochromatory podczerwieni. Pryzmaty - muszą mieć wysoką przezroczystość w interesującym nas zakresie spektralnym i dużą dyspersję kątową. Najczęściej są wykonane z NaCl, ale stosuje się także inne substancje (np. SiO2, Al2O3, KCl itd.).Ujemne cechy tych materiałów to wrażliwość na wilgoć i porysowanie, a ponieważ większość jest dobrze rozpuszczalna w wodzie i łatwo matowieje, optykę aparatu umieszcza się w specjalnych komorach klimatyzowanych o temperaturze wyższej niż temperatura otoczenia. Siatki dyfrakcyjne - są typu odbiciowo - płaskiego, stanowią one powierzchnie szklane, pokryte warstwą glinu, z dużą liczbą równoległych rys w jednakowych odstępach. Siatka dyfrakcyjna powoduje rozszczepienie energii promienistej w widma różnych rzędów. Detektory w podczerwieni. Zadaniem detektora jest przemiana promieniowania podczerwonego w sygnał elektryczny. W spektroskopii IR zastosowanie znalazły detektory termiczne: 1) termopary - (termoogniwa) ich działanie opiera się na efekcie Peltiera - wytworzenie w obwodzie siły elektromotorycznej na skutek umieszczenia obu złącz w różnych temperaturach. 2)bolometr - termometr oporowy, w którym zmiana temperatury, spowodowana padającym promieniowaniem, powoduje zmianę jego oporności, a tym samym odpowiednią zmianę napięcia elektrycznego. 3)detektor pneumatyczny - to tzw. komórka Golaya - bardzo czuły termometr gazowy. Promieniowanie padające na element aktywy (błona z tworzywa sztucznego pokryta glinem) ogrzewa gaz w zamkniętej przestrzeni. Powstający w konsekwencji mały wzrost ciśnienia jest zamieniony na sygnał elektryczny.
Metody spektroskopii w podczerwieni. Metoda transmisyjna - umożliwia badanie gazów, cieczy i ciał stałych. Metody odbiciowe - (refleksyjne) metoda pojedynczego odbicie - efekt ten zachodzi, gdy promieniowanie pada od strony ośrodka o większej gęstości optycznej (pryzmat o współczynniku załamania n1) na próbkę (n2) pod kątem Φ większym od kąta granicznego Φgran. Fala elektromagnetyczna wnika (penetruje) podczas odbicia w ośrodek o najmniejszej gęstości optycznej (próbkę) na głębokość rzędu długości fali. Głębokość penetracji zależy od długości fali promieniowania, jego kąta padania oraz stosunków współczynników załamania światła n2/n1.
|
KONDUKTOMETRIA
Przewodnictwo elektryczne, właściwe, molowe.
Zdolność jonów roztworu elektrolitu do przewodzenia prądu możemy określać poprzez przewodność elektrolityczną, przewodność elektrolityczną właściwą lub przewodność elektrolityczną molową. Przewodność elektrolityczna G elektrolitu jest odwrotnością oporu elektrycznego (R) danego roztworu:
Przewodność elektrolityczna właściwa k roztworu elektrolitu definiuje się jako odwrotność oporu właściwego ρ (ρ=ρ(R · S) / l ) próbki elektrolitu o przekroju S i długości l za pomocą relacji:
Przewodność elektrolityczna właściwa jest związana z oporem poprzez stosunek k = l/S, który jest wielkością stałą zwaną stałą naczynka lub pojemnością oporową:
Pomiary przewodności i przewodności właściwej przeprowadza się w specjalnie skonstruowanych naczynkach z dwiema wbudowanymi elektrodami o powierzchni S, umieszczonymi w stałej odległości l. Wyznaczenie przewodności lub przewodności właściwej elektrolitów sprowadza sie wiec do pomiaru oporu danej próbki po uprzednim określeniu charakterystyki naczynka. Stała naczynka, równa stosunkowi odległości pomiędzy elektrodami do ich powierzchni, charakteryzuje wymiary słupa roztworu elektrolitu pomiędzy dwiema elektrodami. Wyznacza się ją mierząc opór naczynka napełnionego elektrolitem wzorcowym, którego przewodność właściwa została wyznaczona droga pomiarów bezpośrednich:
Przewodność elektrolityczną właściwą k dla badanego elektrolitu można następnie obliczyć z zależności:
wykorzystując wyznaczona wartość stałej naczynka k i zmierzone wartości oporu R. Osobnym typem naczynek są sondy konduktometryczne współpracujące z konduktometrami nowego typu. Maja ściśle określoną, podaną przez producenta, wartość pojemności oporowej. Pomiar z użyciem sondy przeprowadza sie zanurzając ją w roztworze elektrolitu. W przypadku elektrolitów do pomiarów oporu w zasadzie nie można stosować prądu stałego, gdyż powoduje to zmiany składu elektrolitu i zmiany powierzchni elektrod (polaryzacja elektrod). Przy zastosowaniu prądu zmiennego należy wyeliminować opory pozorne w obwodzie prądu. Przewodność elektrolityczna właściwa jest zależna zarówno od stężenia jonów jak i od prędkości ich poruszania sie w polu elektrycznym. Uwzględniając istnienie w roztworze elektrolitu zarówno jonów dodatnich jak i ujemnych możemy wyrazić przewodność właściwą za pomocą następującej relacji:
κ = F · (c+ · z+ · u+ + c- · |z-| · u-) = F · c (α+ · z+ · u+ + α- · |z-| · u-) gdzie: F - stała Faradaya, c - stężenie molowe elektrolitu, c i c - stężenia kationu i anionu, c+ = α+ · c, c- = α- · c, z+ i z- - wartościowości kationu i anionu, u+ i u- - ruchliwości kationu i anionu [m2/(V s)], α - stopień dysocjacji. Z warunku elektroobojętności: α+ · z+ = α- · |z-| κ = F · c · α+ · z+ · (u+ · u-) = F · c · α- · |z-| · (u+ · u-) Wyznaczenie przewodności właściwej elektrolitów sprowadza się do pomiaru oporu właściwego danej próbki przy zastosowaniu wykalibrowanych naczynek. Dla danego naczynka stosunek l/S jest wielkością stałą zwaną stałą naczynka lub pojemnością oporową. Wyznacza się ją mierząc opór naczynka napełnionego elektrolitem wzorcowym, którego przewodność właściwa została wyznaczona drogą pomiarów bezpośrednich. Krzywe zależności przewodności właściwej od stężenia elektrolitu mają charakterystyczny przebieg. W obszarze niskich stężeń obserwuje się wzrost przewodności właściwej związany z przyrostem liczby jonów w jednostce objętości roztworu wraz ze wzrostem stężenia. W obszarze wyższych stężeń wzrost oddziaływań miedzy jonami, zmniejszający ich ruchliwość, oraz zmniejszenie stopnia dysocjacji prowadzi do obniżenia wzrostu lub nawet do spadku przewodności właściwej.
dla słabych i mocnych elektrolitów Do analizy właściwości przewodników elektrolitycznych stosuje sie też pojęcie przewodności molowej:
którą definiuje się jako przewodność elektrolitu umieszczonego pomiędzy dwiema elektrodami odległymi o 1m o powierzchni przekroju S takiej, by zmieściła się pomiędzy nimi objętość roztworu zawierającego 1 mol elektrolitu. Przewodność molowa charakteryzuje, więc zdolność 1 mola elektrolitu do przewodzenia prądu elektrycznego. Przewodność molowa przy danym stężeniu jest proporcjonalna do stopnia dysocjacji i odpowiadającej temu stężeniu sumy ruchliwości jonowych: Λm = F · α- · |z-| · (u+ · u-) = F · α+ · z+ · (u+ · u-) Λm = α- · |z-| · λ- + α+ · z+ · λ+ Wielkości te zależą od stężenia roztworu i w pewnym stopniu od rodzaju drugiego jonu. Wielkości przewodności molowych jonów są na ogół zbliżone, co świadczy o podobieństwie ich ruchliwości, niezależnie od budowy i wartościowości. Znacznie większą przewodność wykazują jony H+ i OH-, co jest związane z innym mechanizmem ich poruszania sie w roztworze. Krzywe zależności przewodności molowej od stężenia wykazują odmienny przebieg dla elektrolitów mocnych i słabych. W obszarze niskich stężeń dla obu grup elektrolitów obserwuje sie monotoniczny spadek przewodności molowej ze stężeniem, jednak jest on znacznie szybszy dla elektrolitów słabych. W przypadku tych elektrolitów wartość przewodności spada do niskich wartości już w roztworach średnio stężonych. Tłumaczy sie to zmniejszaniem stopnia dysocjacji ze wzrostem stężenia. Wpływ wzrostu stężenia roztworów silnych elektrolitów na spadek wartości przewodności jest znacznie słabszy. W obszarze wysokich stężeń wartość przewodności dla tego typu elektrolitów pozostaje na znaczącym poziomie, ze względu na to, że mocne elektrolity są w roztworze praktycznie całkowicie zdysocjowane. Pomiary przewodnictwa roztworów elektrolitów są stosowane do wyznaczania różnych wielkości fizykochemicznych oraz do celów analitycznych.
Pojęcie stałej i stopnia dysocjacji, prawo rozcieńczeń Ostwalda.
Według Arrheniusa stopień dysocjacji elektrolitu zgodnie z prawem działania mas zmniejsza się wraz ze wzrostem stężenia roztworu. Zakładał on, że zależność przewodności od stężenia wywołana jest jedynie zmiana
stopnia dysocjacji i zaproponował metodę wyznaczania stopnia dysocjacji opartą o następującą zależność pomiędzy przewodnością molową w roztworze o danym stężeniu ( λm) i w roztworze nieskończenie rozcieńczonym ( λ0m) a stopniem dysocjacji (α):
Zależność tę oparto na założeniu, że ruchliwości jonów są niezależne od stężenia. Stosując prawo działania mas do reakcji dysocjacji słabego kwasu HA = H+ + A- Ostwald zdefiniował stała równowagi, która można zapisać następująco:
U podstaw prawa rozcieńczeń Ostwalda leży założenie o niezależności ruchliwości jonów od stężenia oraz założenie o słuszności opisu równowag jonowych za pomocą prawa działania mas.
Pomiar przewodnictwa metodą mostka.
Prąd zmienny ze źródła E biegnie do A, gdzie ulega rozdwojeniu. Część prądu płynie po chromonikielinowym drucie AB, a inna część przez drut ADB, w którym zawarty jest opór wzorcowy Rx, w którym jest naczynko z badanym roztworem. Oba prądy łączą się w punkcie B i wracają do E. Gdy natężenie prądu na odcinku CD osiąga minimum mostek zostaje zrównoważony (potencjał w punkcie D jest równy potencjałowi w punkcie C) i prąd płynie rozgałęzieniem CD.
mostek Wheatstone'a
Naczynka konduktometryczne i ich kalibrowanie.
Różne typy naczynek konduktometrycznych:
Ostwalda firmy Philips Arrheniusa
Naczynka pomiarowe mają różne kształty i są zaopatrzone w dwie elektrody umieszczone w stałej odległości od siebie. Naczynie pomiarowe wypełnione elektrolitem można scharakteryzować jego oporem. Opór naczynia zawierającego roztwór elektrolitu o przewodności właściwej κ oraz elektrody o powierzchni A umieszczone względem siebie w odległości l można obliczyć z równania:
Wartość k = Rκ
Prawo Kohlrauscha i jego interpretacja.
Prawo Kohlrauscha głosi, iż graniczne przewodnictwo molowe mocnych elektrolitów (Λ∞) jest wielkością addytywną. Oznacza to, że do obliczenia jego wartości wystarczy dodać wartości przewodnictw granicznych jonów wchodzących w jego skład: Λ∞ = Λ∞(kation) + Λ∞(anion) Prawo Kohlrauscha można też wykorzystać do obliczenia przewodnictwa granicznego elektrolitów słabych, które nie dysocjują całkowicie na swobodne jony, więc nie można w ten sam sposób obliczyć ich przewodnictwa jak w przypadku elektrolitów mocnych. W przypadku słabych kwasów (HA) w tym celu wykorzystuje się pomiar w układzie z mocnym kwasem (najczęściej HCl), rozpuszczalnej soli słabego kwasu (NaA) oraz soli mocnego kwasu: Λ∞HA = Λ∞HCl + Λ∞NaA - Λ∞NaCl W roztworach nieskończenie rozcieńczonych (rozpatrujemy przewodnictwo graniczne) kationy i aniony mogą poruszać się całkowicie swobodnie czyli niezależnie przenosić ładunek elektryczny.
Równanie Debye'a-Huckela-Onsagera.
Zakłada całkowitą dysocjację elektrolitów mocnych uwzględniając elektrostatyczne oddziaływania między jonami. Podstawowy postulat tej teorii stanowi, iż każdy jon (jon centralny) otoczony jest chmurą jonową o znaku przeciwnym. Sumaryczny ładunek chmury jonowej jest równy co do wielkości i przeciwny co do znaku, ładunkowi jonu centralnego. Gęstość ładunku jest największa w pobliżu jonu centralnego i maleje w miarę wzrostu odległości od niego. Równanie wyraża się wzorem:
k = A + BΛ0 gdzie A, B - stałe zależne od rodzaju rozpuszczalnika.
Zastosowanie konduktometrii do wyznaczania stałej i stopnia dysocjacji.
Posługując się prawem rozcieńczeń Ostwalda i postulatem Arrheniusa oraz przekształcając te równania otrzymujemy równanie opisujące stałą dysocjacji Ka słabego kwasu z wykorzystaniem przewodnictwa równoważnikowego:
Dalsze przekształcenie prowadzi do równania o następującej formie:
Równanie to jest równaniem prostej typu y = ax + b:
Po zmierzeniu przewodnictwa właściwego κ roztworu słabego kwasu dla różnych stężeń c można wyliczyć przewodnictwo równoważnikowe roztworu λ. Z wykresu funkcji 1/λ = f(cλ) w pierwszej kolejności wyznacza się λ∞ (z przecięcia prostej z osią y przy zerowej wartości na osi x), a następnie z nachylenia prostej stałą dysocjacji Ka.
Miareczkowanie konduktometryczne
POTENCJOMETRIA
Metody potencjometryczne wykorzystują zależność między stężeniem (aktywnością) oznaczanego jonu w roztworze i potencjałem elektrycznym odpowiedniej elektrody - wykonuje się pomiar siły elektromotorycznej (SEM) ogniwa, z którego jednym półogniwem jest elektroda wskaźnikowa, której potencjał jest zależny od stężenia oznaczanego jonu, a drugim - elektroda porównawcza, której potencjał ma wartość stałą. Obie elektrody są w kontakcie z badanym roztworem. Metody potencjometryczne polegają więc na pomiarze siły elektromotorycznej (SEM) ogniwa złożonego z dwu elektrod zanurzonych do badanego roztworu. Mierzona SEM zależy w określony sposób od stężenia w roztworze oznaczanego składnika. Za zmianę SEM odpowiedzialna jest jedna z elektrod, elektroda wskaźnikowa. Metody potencjometryczne dzielą się na dwie grupy: 1. Metody bezpośrednie polegające na wyznaczeniu stężenia oznaczanego składnika na podstawie wartości SEM ogniwa, którego kalibracji dokonano za pomocą próbek wzorcowych. Należą tu pomiary pH roztworów oraz oznaczenia za pomocą elektrod jonoselektywnych. 2. Metody pośrednie, czyli miareczkowe, stosowane do wyznaczania punktu końcowego miareczkowania - jest to tzw. miareczkowanie potencjometryczne. W miareczkowaniu tym wyznacza się zmiany SEM odpowiedniego ogniwa spowodowane dodawaniem mianowanego roztworu odczynnika miareczkującego.
Elektrody
1. Elektrody pierwszego rodzaju, odwracalne względem kationu i anionu, zbudowane z metalu będącego w równowadze z roztworem, zawierającym jony tego samego metalu. Potencjał takiej elektrody zależy od aktywności (stężenia) jonów metalu w roztworze. Reakcję elektrodową można przedstawić następująco: M0 * Mn+ + ne- Np. elektroda srebrowa (Ag0 * Ag+ + e-), chlorowa, wodorowa 2. Elektrody drugiego rodzaju, odwracalne względem wspólnego anionu, zbudowane są z metalu pokrytego warstwą jego trudno rozpuszczalnej soli. Potencjał takiej elektrody zależy od stężenia w roztworze anionu tej soli. Równanie reakcji elektrodowej ma postać: M0+A- * MA + e- np. elektroda kalomelowa, chlorosrebrowa (Ag0 + Cl- * AgCl + e-) 3. Elektrody trzeciego rodzaju, odwracalne względem wspólnego kationu - zbudowane są z metalu (M10) pokrytego cienką warstewką trudno rozpuszczalnej soli tego metalu (M1A) zawierającą jeszcze drugi kation, który ze wspólnym anionem tworzy sól łatwiej rozpuszczalną (M2A). Potencjał takiego półogniwa zależy od stężenia w roztworze drugiego kationu, zgodnie z równowagą opisaną równaniem: M10 + M2A * M1A + M2n+ + ne- Np. elektroda odwracalna względem jonów wapnia (Pb0 + Ca2C2O4 * PbC2O4 + Ca2+ + 2e-) 4. Elektrody utleniająco- redukujące (redoks) - elektrodę stanowi obojętny chemicznie metal (Pt,Au), spełniający funkcję przewodnika elektronów, zanurzony w roztworze zawierającym substancję w postaci utlenionej i zredukowanej (jony tego samego pierwiastka różniące się stopniem utlenienia). Potencjał takiej elektrody zależy od stosunku stężeń obu postaci. Elektrody redoks są najczęściej stosowane jako elektrody wskaźnikowe w miareczkowaniach redoksometrycznych. 5. Elektrody jonoselektywne - elektrody te reagują selektywnie na określony jon w obecności innych i z tego względu nazywane są elektrodami jonoselektywnymi. Często stosuje się też termin elektrody membranowe, gdyż istotnym elementem ich budowy jest zawsze membrana (stała lub ciekła), zawierająca substancję elektroaktywną. Elektrody te są elektrochemicznymi półogniwami, w których na granicy faz membrana/roztwór powstaje różnica potencjałów zależna od aktywności określonego jonu, który występuje w fazie roztworu i w fazie membrany, mogąc przechodzić łatwo z jednej fazy do drugiej. W przeciwieństwie do elektrod pierwszego, drugiego i trzeciego rodzaju, w procesach elektrodowych nie biorą udziału elektrony, a za powstawanie potencjału jest odpowiedzialna równowaga reakcji wymiany jonowej między roztworem a membraną, wyrażona równaniem: I r * I m gdzie: Ir - jon I w roztworze Im - jon I w fazie membrany 6. Elektrody wskaźnikowe - potencjał elektrod jest zależny od stężenia jonu (względem którego elektroda jest odwracalna) w badanym roztworze Elektrody wskaźnikowe decydują o możliwościach analitycznych potencjometrii, gdyż dany jon (substancję) można oznaczać potencjometrycznie tylko wówczas, gdy istnieje czuła na ten jon (lub czuła na jon wchodzący w reakcję analityczną z danym jonem) elektroda wskaźnikowa. Potencjometrycznie można oznaczać stężenia jonów wodorowych (pH) oraz wielu innych jonów (kationów i anionów).Przykładowe elektrody wskaźnikowe to elektroda srebrowa, elektroda platynowa, elektroda szklana, elektroda antymonowa, oraz elektrody jonoselektywne inne niż szklana. 7. Elektrody porównawcze - potencjał ich powinien być stały , niezależny od składu roztworu badanego Drugim elementem ogniwa pomiarowego (obok elektrody wskaźnikowej) jest elektroda porównawcza. Znajomość właściwości tej elektrody jest bardzo ważna, gdyż niewłaściwy dobór i przygotowanie elektrody porównawczej może być źródłem największych błędów przy pomiarach SEM. Elektrody porównawcze, zwane też elektrodami odniesienia, muszą charakteryzować się stałym, powtarzalnym potencjałem, praktycznie niezależnym od składu badanego roztworu. Spośród wielu możliwych układów odniesienia jedynie standardowa elektroda wodorowa, elektroda chlorosrebrowa i kalomelowa znalazły praktyczne zastosowanie.
Elektroda kalomelowa Półogniwo, elektroda, elektroda drugiego rodzaju, którą stanowi rtęć stykająca się z chlorkiem rtęci(I) (w celu zabezpieczenia elektrody przed obecnością Hg2+ do sporządzenia jej nie używa się czystego kalomelu, lecz pasty kalomelowej zawierającej niewielkie ilości rozdrobnionej rtęci) w roztworze chlorku potasu (KCl). Schematycznie tę elektrodę można przedstawić następująco: Hg | Hg2Cl2 | Cl- Reakcja elektrodowa: Hg2Cl2 + 2e- ⇌ 2Hg(c) + 2Cl-(r)
wskazuje, że potencjał takiego półogniwa zależy od stężenia jonów chlorkowych. Stosując nasycony roztwór jonów Cl- (np. KCl), uzyskuje się półogniwo o stałym potencjale, który w temperaturze 25°C wynosi E = + 0,2679 V.
Elektroda szklana
Jonoselektywna elektroda, czuła na jony wodorowe, jest to półogniwo, w którym membrana jest wykonana ze specjalnego gatunku szkła sodowego. Zwykle jest to wąska rurka szklana zakończona cienkościenną membraną w kształcie bańki. Wewnątrz znajduje się roztwór buforowy o dokładnie znanym pH, zawierającym chlorki. W roztworze tym jest zanurzona porównawcza (wyprowadzająca) elektroda wewnętrzna o znanym potencjale, zwykle chlorosrebrowa, która posiada wyprowadzenie na zewnątrz. Przypadku elektrody szklanej różnica potencjałów między szkłem a roztworem stykającym się z nim, zależy od pH tego roztworu.
Elektrody jonoselektywne
Elektrodami jonoselektywnymi nazywa się elektrody, których potencjał zależy liniowo od logarytmu aktywności danego jonu w roztworze (w określonym przedziale stężeń). Zależność prostoliniowa utrzymuje się na ogół w zakresie kilku rzędów stężenia (3-4). Wspólną ich cechą jest to, że na ich potencjał ma wpływ nie tylko stężenie oznaczanego jonu, lecz także stężenia innych jonów. Elektrody te są zaopatrzone w membranę jonowymienną, która oddziela odpowiednie półogniwo od roztworu badanego. Na potencjał elektrody membranowej składa się potencjał międzyfazowy na granicy faz membrana-roztwór, uwarunkowany wymianą jonową między roztworem i membraną oraz potencjał dyfuzyjny, wynikający z procesów zachodzących wewnątrz membrany, szczególnie w jej warstwie przylegającej do roztworu.
Potencjał elektrod membranowych jest opisany przez wzór Nikolskiego:
n - wartościowość jonu i, na który elektroda jest czuła z - wartościowość jonu przeszkadzającego j Kij - współczynnik selektywności (stała selektywności) elektrody czułej na jon i względem jonu j
Potencjał elektrody, równanie Nernsta
najprostsza elektroda wykonana z metalu i zanurzona w roztworze soli tego metalu. Ustala się równowaga między fazą stałą (elektroda) i fazą ciekłą (roztwór badany): M0 * Mn+ + e- Efektem tej równowagi jest potencjał powstający na granicy faz. Zależność potencjału elektrody od aktywności jonu metalu w roztworze, liczby elektronów biorących udział w tej reakcji i od temperatury opisuje równanie Nernsta:
gdzie: E - potencjał elektrody E0 - potencjał standardowy elektrody - wartość stała; charakterystyczna dla danej elektrody, odpowiadająca potencjałowi elektrody w roztworze o aktywności jonów Mn+ równej jedności; n - liczba elektronów biorących udział w reakcji elektrodowej R - stała gazowa T - temperatura w skali bezwzględnej F - liczba Faradaya a Mn+ - aktywność jonów Mn+ w roztworze aM0- aktywność metalu
Potencjometryczny pomiar pH
Metoda jest oparta na pomiarach SEM ogniwa złożonego z elektrody wskaźnikowej o potencjale zależnym od aktywności jonów wodorowych (wodorowa, szklana, chinhydrynowa) i elektrody odniesienia o stałym znanym i odtwarzalnym potencjale (najczęściej kalomelowa). SEM utworzonego ogniwa równa jest różnicy potencjałów obu elektrod.
gdzie: ∏0- normalny potencjał danej elektrody odniesiony do wartości potencjału normalnej elektrody wodorowej z - liczba elektronów biorących udział w reakcji elektronowej R - stała gazowa F - stała Faradaya T - temperatura bezwzględna Podstawiając pH=-logaH+ i przekształceniu powyższego równania względem pH, otrzymujemy związek między wartością pH badanego roztworu i wartością SEM ogniwa zbudowanego z tego roztworu i zanurzonych w nim dwóch elektrod: wskaźnikowej i odniesienia.
Miareczkowanie potencjometryczne
Polega na mierzeniu różnicy potencjałów między elektrodą wskaźnikową i porównawczą po dodaniu każdej porcji odczynnika miareczkującego. Jest ono możliwe do wykonania wówczas, gdy dobierze się elektrodę wskaźnikową, która będzie reagowała na zmiany stężenia składnika oznaczanego lub odczynnika miareczkującego, zachodzące podczas miareczkowania. Dodawanie odczynnika miareczkującego powoduje zmiany stężenia składnika oznaczanego. Początkowo względne zmiany stężenia oznaczanych jonów są niewielkie i zmiany potencjału są również niewielkie. Natomiast w pobliżu PR następuje skok potencjału. Krzywa miareczkowania, przedstawiająca zależność potencjału od objętości titranta wygląda następująco:
Metody wyznaczania PK w potencjometrii
KULOMETRIA
Jest metodą elektroanalityczną opartą na zjawisku elektrolizy. Polega ona na pomiarze ładunku elektrycznego przepływającego przez roztwór, koniecznego do przeprowadzenia reakcji elektroutleniania lub elektroredukcji. Ilościową zależność pomiędzy masą wydzielonej substancji a przepływającym ładunkiem podał Faraday w postaci dwóch praw. Pierwsze prawo stwierdza, że ilość substancji utlenionej lub zredukowanej na jednej z elektrod pod wpływem prądu elektrycznego jest proporcjonalna do ładunku, jaki przepłynął przez roztwór:
m - masa substancji wydzielonej na elektrodzie [g] i - natężenie prądu płynącego w obwodzie [A] t - czas elektrolizy [s] Q - ładunek, jaki przepłynął przez obwód [C] k - równoważnik elektrochemiczny [g/C], jest wielkością stała, charakterystyczną dla danej substancji ulegającej reakcji elektrodowej, i jest równy masie tej substancji, jaka wydzieli się na elektrodzie przy przepływie ładunku równemu 1C. Drugie prawo elektrolizy określa ilość elektryczności potrzebną do wydzielenia na elektrodzie masy substancji odpowiadającej przejściu jednego mola elektronów. Ta ilość elektryczności nosi nazwę stałej Faradaya. F = M / zk F - stałą Faradaya [C/mol] M - masa molowa substancji reagującej na elektrodzie [g/mol] z - ilość elektronów biorących udział w reakcji elektrodowej
Równanie to jest podstawą obliczeń w analizie kulometrycznej. W równaniu tym wszystkie wielkości są stałe, oprócz ładunku, który jest charakterystyczną dla kulometrii wielkością mierzoną. Ze względu na szybkość oznaczania substancji analizę kulometryczną dzielimy na: - bezpośrednią - oznaczana substancja bezpośrednio reaguje na jednej z elektrod (ulegając reakcji utleniania lub redukcji) W czasie elektroliza utrzymuje się stały, kontrolowany potencjał elektrody pracującej, natomiast w miarę zachodzenia reakcji natężenie prądu sukcesywnie maleje i jego spadek do zera wskazuje koniec reakcji. - pośrednią zwaną miareczkowaniem kulometrycznym - oznaczana substancja nie ulega reakcji elektrodowej, lecz wchodzi w reakcję z substancją wytwarzaną w trakcie elektrolizy na jednaj z elektrod. Zakończenie reakcji określone zostaje za pomocą wskaźników wizualnych lub metod instrumentalnych (amperometrycznie, potencjometrycznie, konduktometrycznie, spektrofotometrycznie) Ze względu na sposób wykonania analizy kulometrię dzielimy na: - potencjostatyczną - elektroliza prowadzona jest przy stałym potencjale elektrody pracującej (anody lub katody) - amperostatyczną - elektroliza prowadzona jest przy stałym natężeniu prądu płynącego w obwodzie
AMPEROMETRIA
Pod nazwą amperometria i miareczkowanie amperometryczne rozumiemy metody oparte na pomiarach zależności natężenia prądu od napięcia z zastosowaniem elektrod innych niż kroplowa elektroda rtęciowa. Największe zastosowanie w tych metodach znalazły mikroelektrody stałe przy określaniu zależności natężenia od napięcia, a wśród nich wirująca elektroda platynowa - wykonana z platynowego druciku o średnicy 0,5 mm i długości 1-4 mm, wystającego z wirującej rurki, prostopadle lub pod pewnym kątem do jej osi. Liczba obrotów rurki powinna być stała i wynosić 300-600 obrotów na minutę. Miareczkowanie amperometryczne z jedną elektrodą polaryzowaną. W metodzie tej elektrodą wskaźnikową jest wirująca elektroda platynowa. Miareczkowanie przeprowadza się przy stałym napięciu, odpowiadającym granicznemu prądowi dyfuzyjnemu danego depolaryzatora. Krzywe miareczkowania mają taki sam przebieg jak krzywe miareczkowania polarograficznego. Miareczkowanie amperometryczne z dwiema elektrodami polaryzowanymi (biamperometryczne). Polega na obserwacji zmian natężenia prądu stanowiącego funkcję przyłożonego napięcia i stężenia jonów uczestniczących w miareczkowaniu. Zestaw do miareczkowania składa się z dwóch elektrod platynowych, zanurzonych w roztworze badanym i połączonych poprzez galwanometr i opornicę ze źródłem prądu stałego. Do elektrod przykłada się nieduże stałe napięcie rzędu 10-150 mV. Tak mała wartość napięcia nie pozwala na przebieg elektrolizy w roztworze, tzn. w obwodzie pomiarowym nie płynie prąd. Jeśli jednak w roztworze badanym znajdą się dwie substancje, z których jedna może się utleniać, a druga redukować przy danym napięciu, to między elektrodami popłynie prąd. Taki przepływ prądu nastąpi wówczas, gdy w roztworze znajdą się związki tworzące odwracalny układ erdoks. W przypadku obecności w roztworze nieodwracalnego układu erdoks prąd może płynąc wtedy, gdy przyłożone do elektrod napięcie jest większe od sumy nadnapięcia anodowego i katodowego. Jeśli przyłożone napięcie jest mniejsze, prąd nie płynie. W punkcie końcowym miareczkowania następuje zmiana natężenia prądu bądź w postaci dużego wzrostu natężenia, bądź prawie całkowitego zaniku prądu. Promieniowanie padające na próbkę może ulec pochłonięciu, odbiciu lub rozproszeniu. Może ono także wzbudzić cząsteczkę lub atom i spowodować emisję nowego promieniowania. Jeżeli absorpcja promieniowania dotyczy cząsteczek, mamy do czynienia z metodą absorpcyjnej spektrofotometrii w nadfiolecie (UV), świetle widzialnym (VIS) i w podczerwieni (IR). Jeśli natomiast promieniowanie absorbowane jest przez atomy, mówimy o absorpcyjnej spektrofotometrii atomowej (UV-VIS) i absorpcji rentgenowskiej. Rozproszenie promieniowania przez cząstki ośrodka jest podstawą metod nefelometryczny. Oprócz metod spektroskopowych, opartych na zjawiskach absorpcji emisji i rozproszenia promieniowania, istnieją metody analityczne wykorzystujące inne zjawiska m.in. zjawisko załamana światła (refraktometria) lub zdolność skręcania płaszczyzny polaryzacji (polarymetria).
METODY OPARTE NA ZJAWISKU ELEKTROLIZY
W zakresie wymienionych metod wykorzystywane są zjawiska związane z reakcjami zachodzącymi na elektrodach, jak również przepływem prądu elektrycznego przez elektrolit. Metody elektroanalityczne można podzielić na dwie zasadnicze grupy: elektrograwimetryczne i elektrometryczne. Do metod elektrograwimetrycznych zaliczamy: Elektrolizę - czyli proces zachodzący w roztworze, zawierającym zdysocjowany związek chemiczny, pod wpływem przepływającego przez roztwór stałego prądu. W wyniku tego następuje przesuwanie się i wydzielanie kationów na katodzie oraz anionów na anodzie. Poszukiwany pierwiastek lub jego związek oznacza się przez wydzielenie go na elektrodzie, a następnie zważenie. W metodzie tej stosuje się prąd o natężeniu w przybliżeniu stałym, nie kontrolując potencjału elektrody, na której wydziela się oznaczoną substancję. Elektrolizę z kontrolowanym potencjałem katody - polegającą na kontrolowaniu potencjału tej elektrody, na której ma przebiegać całkowicie tylko jedna pożądana reakcja elektrodowa. Elektrolizę wewnętrzną - polegająca na wykorzystaniu siły elektromotorycznej (SEM) ogniwa elektrolitycznego do wydzielenia metalu na elektrodzie bez potrzeby przykładania napięcia z zewnętrznego źródła prądu. Do metod elektrometrycznych polegających na pomiarze jednej lub kilku wielkości elektrycznych, które są związane ze stężeniem bądź całkowitą ilością oznaczanego związku chemicznego, zaliczamy: Konduktometrię - polegającą na pomiarze zmian przewodnictwa elektrycznego roztworu, które zależą od stężenia analizowanej substancji w roztworze i zachodzących w tym roztworze reakcji chemicznych. Potencjometrię - w której dokonuje się pomiaru siły elektromotorycznej (SEM) ogniwa zbudowanego z elektrod zanurzonych w roztworze analizowanym. Potencjał jednej z elektrod zwanej wskaźnikową, zależny jest od stężenia jonu oznaczanego. Polarografię - polegającą na rejestracji zmian natężenia prądu płynącego przez elektrolit w zależności od napięcia przyłożonego do układu elektrod, z których jedna jest kroplową elektrodą rtęciową - do bezpośrednich badań jakościowych oraz ilościowych. Kulometrię - polegającą na oznaczaniu substancji w roztworze na podstawie pomiaru ładunku elektrycznego, który przepływał przez obwód w czasie elektrolizy, jeśli nie zaszły przy tym żadne reakcje uboczne. Te i inne metody elektroanalityczne znajdują szerokie zastosowanie w analizie chemicznej, farmaceutycznej, klinicznej, biochemicznej i innych.
|
ATOMOWA SPEKTROSKOPI ABSORPCYJNA
Atom pierwiastka w stanie podstawowym (o energii Ep) absorbuje foton promieniowania (energia hν ), co powoduje zmianę rozkładu elektronów w atomie, przeprowadzając go w stan o wyższej energii (Ep + hν), czyli stan wzbudzony. Zgodnie z zakazem Pauliego, opisy stanów energetycznych poszczególnych elektronów muszą się różnić wartością przynajmniej jednej liczby kwantowej. W stanie podstawowym atomu (w temp. ok. 20ºC) elektrony zapełniają kolejno poziomy energetyczne wg wzrastającej energii. Elektron walencyjny może zostać przeniesiony z poziomu podstawowego na poziom wzbudzony. Różnica energii między poziomem podstawowym a wzbudzonym (ΔE) równa się: ΔE = ( Ep + hν ) - Ep = hν = h c/ Ponieważ poziomy energetyczne mogą przyjmować tylko pewne ściśle określone wartości, więc i różnice energii między nimi nie są dowolne. Średni czas trwania atomu w stanie wzbudzonym jest bardzo krótki, rzędu 10-8 s. Po tym czasie elektron, wracając do stanu podstawowego, emituje energię dokładnie taką, jaka była potrzebna do przejścia w stan wzbudzony:
Porcje energii czyli promieniowanie o określonej częstotliwości lub określonej długości fali, które jest absorbowane przez dany atom jest emitowane podczas powrotu do stanu podstawowego. Oznacza to, że atom może absorbować promieniowanie elektromagnetyczne tylko o takiej długości fali, przy której może je emitować i jest ono charakterystyczne dla danego pierwiastka. Zjawisko to jest podstawą analizy jakościowej metodą atomowej spektrometrii absorpcyjnej. Przejściom elektronów pomiędzy różnymi poziomami energetycznymi odpowiadają różne częstotliwości promieniowania, których zbiór stanowi charakterystyczne dla danego pierwiastka widmo atomowe (widmo liniowe). Do celów analitycznych należy dokonać wyboru jednej z wielu różnych linii absorpcyjnych. W metodzie ASA wykorzystuje się zwykle linię związaną z przejściem elektronu walencyjnego ze stanu podstawowego na pierwszy (najniższy poziom wzbudzony) i nazywa się ją linią rezonansową, zaś najniższy stan wzbudzony - stanem rezonansowym. Miarą intensywności zjawiska absorpcji promieniowania elektromagnetycznego przez wolne atomy jest absorbancja (A) określana jako A = lg I0/I Zasada pomiarów metodą AAS polega na tym, że linia rezonansowa oznaczanego pierwiastka o natężeniu I0, emitowana ze źródła promieniowania przechodzi przez atomizer, w którym jest absorbowana przez obecne tam wolne atomy. Ta część promieniowania (linii rezonansowej), która nie została pochłonięta przez wolne atomy, dociera poprzez monochromator do detektora, który mierzy jej natężenie (I). Linie atomowe mają kształt krzywych Gaussa i charakteryzują się intensywnością oraz szerokością określaną przez szerokość połówkową mierzoną w połowie wysokości piku.
Naturalna szerokość linii w zakresie promieniowania UV-Vis stosowanych w AAS wynosi ok. 10-6 - 10-4 nm. W atomizerze szerokość linii absorpcyjnej będzie większa z powodu poszerzenia temperaturowego (tzw. poszerzenie dopplerowskie) oraz poszerzenia ciśnieniowego (zjawisko Lorentza). W obu przypadkach wartość tego poszerzenia wynosi ok. 10-3 nm i jest o dwa rzędy większa od szerokości naturalnej linii absorpcyjnej. Dlatego szerokość połówkowa linii emitowanej ze źródła promieniowania powinna być zdecydowanie mniejsza niż szerokość linii absorpcyjnej ze względu na czułość (im mniejsza szerokość linii emitowanej tym większy jej zakres będzie objęty absorpcją) i jak najmniejsza ze względu na specyficzność metody (możliwość nakładania się linii spektralnych innych pierwiastków). Uzyskuje się to przez zastosowanie wyższej temperatury w atomizerze od temperatury w lampie emitującej.
Zasada pomiarów metodą ASA
Aparatura do ASA
Aparaty AAS mogą być jedno- lub dwuwiązkowe. W spektrometrach dwuwiązkowych promieniowanie emitowane ze źródła jest dzielone na dwie wiązki, wiązkę przechodzącą przez atomizer i wiązkę odniesienia omijającą atomizer. Obie wiązki przechodzą przez ten sam monochromator a następnie są naprzemiennie rejestrowane przez ten sam detektor. Eliminuje się w ten sposób błąd pomiaru wynikający ze zmian intensywności promieniowania źródła w czasie trwania pomiaru lub zmian czułości detektora.
Źródła promieniowania
Źródła promieniowania stosowane w metodzie AAS muszą się charakteryzować dużą monochromatycznością promieniowania o częstotliwości zgodnej z częstotliwością rezonansową oznaczanego pierwiastka. Promieniowanie emitowane przez źródło powinno odznaczać się dużym natężeniem i stabilnością. W praktyce, w metodzie AAS stosuje się lampy z katodą wnękową lub wzbudzane wysoką częstotliwością (bądź mikrofalami) lampy bezelektrodowe. Lampy z katodą wnękową są rurkami szklanymi z okienkami kwarcowymi. Wewnątrz zamkniętej rurki znajduje się gaz szlachetny (Ne lub Ar) pod niskim ciśnieniem (2-8 hPa). Lampy te zawierają dwie elektrody. Anodą jest drut wolframowy, katodę stanowi wydrążony cylinder wykonany z metalu, który ma być oznaczany i którego linię rezonansową lampa ma emitować. Oś cylindra katody odpowiada osi optycznej przyrządu. Gdy między anodę i katodę zostanie przyłożone dostatecznie duże napięcie (rzędu kilkuset wolt), gaz wypełniający lampę zostanie zjonizowany. Dodatnie jony gazu,
bombardując katodę wybijają, z niej atomy metalu. Atomy metalu w stanie gazowym ulegają wzbudzeniu i emitują promieniowanie, które składa się z linii charakterystycznych dla atomów metalu, jonów metalu i gazu szlachetnego. Natężenie promieniowania można zmieniać regulując natężenie prądu płynącego w lampie. Odpowiednią linię można wyodrębnić z niezbyt skomplikowanego widma za pomocą prostych monochromatorów. Lampą z katodą wnękową można oznaczać tylko jeden pierwiastek, ten z którego została wykonana katoda. Produkuje się też lampy kilkupierwiastkowe, ale nie znajdują one szerszego zastosowania, głównie z powodu małego natężenia wycinanej wiązki promieniowania. Lampy bezelektrodowe ze wzbudzeniem wysoką częstotliwością są to wąskie, zamknięte rurki kwarcowe zawierające wewnątrz warstwę metalu, który ma być oznaczony lub/i warstwę soli tego pierwiastka (1 - 2 mg). Rurka wypełniona jest gazem szlachetnym (Ar, Ne) pod zmniejszonym ciśnieniem (0,2 - 0,8 hPa). Atomizację i wzbudzenie uzyskuje się przez działanie pola elektromagnetycznego o wysokiej częstotliwości. Lampy bezelektrodowe charakteryzują się dobrymi parametrami (natężenie linii i szerokość połówkowa) i są bardzo trwałe. Produkuje się je głównie dla pierwiastków, dla których nie można zbudować lamp HCL - Sb, As, Se, Te, P, Hg, Bi, Cs, Ge, K, Rb, Tl.
Atomizery
Zadaniem atomizerów jest otrzymywanie z dużą, powtarzalną wydajnością wolnych atomów z próbek analitycznych. Im większa wydajność wolnych atomów w stanie podstawowym, odniesiona do badanej próbki, tym większa czułość metody analitycznej. W procesie atomizacji musi występować prosta proporcjonalność między stężeniem oznaczanej substancji w próbce a stężeniem atomów w plazmie absorpcyjnej. Wytworzone atomy powinny w jak najmniejszym stopniu ulegać wzbudzeniu i jonizacji. Atomizer płomieniowy Atomizacja płomieniowa wymaga przeprowadzenia ciekłej próbki analitycznej w aerozol. Aerozol uzyskuje się najczęściej w nebulizerze pneumatycznym. W komorze nebulizera analizowany roztwór przeprowadza się w delikatną mgłę, następnie miesza aerozol z gazem palnym i wprowadza jednorodnie do palnika z zastosowaniem powierzchni rozpryskowych lub sit dla odrzucenia lub rozbicia większych kropel. Gazem zasysającym próbkę jest zawsze gaz utleniający. Mieszanina rozpuszczalnika, próbki, gazów utleniającego i palnego wprowadzana jest do palnika szczelinowego. Płomienie palnika muszą dostarczać energii wystarczającej do przeprowadzenia roztworu w wolne atomy. Sam płomień powinien absorbować tylko niewielką część promieniowania emitowanego przez źródło. Stosowane w metodzie AAS mieszaniny gazów to gaz miejski-powietrze (T = 1980 K), propan-butan-powietrze (T = 2200 K), acetylen-powietrze (T = 2600 K), acetylen-tlen (T = 3300 K), acetylen-tlenek azotu (I) (T 3220 K), wodór-powietrze (T= 2275 K) oraz wodór-tlen (T = 2825 K). Najczęściej stosuje się płomień acetylen-powietrze. Ma on wysoką temperaturę i dopiero poniżej 230 nm występuje wzrastająca absorpcja własna płomienia. Płomień acetylen-powietrze jest zalecany do oznaczania następujących pierwiastków: Mg, Ca, Sr, Cr, Mb, Mn, Tc, Fe, Ru, Os, Co, Rh, Ni, Pd, Pt, Cu, Ag, Au, Zn, Hg, Ga, In, Tl, Pb, Sb, Bi. Dla pierwiastków, które tworzą w płomieniu trwałe tlenki (np. Ba, Al., B, Be, Si) konieczne jest stosowanie płomienia redukującego z użyciem gazu utleniającego tlenku azotu (I). Analizowana próbka może być roztworem prostej soli MA (M+ i A-) lub roztworem zawierającym inne składniki. Po wprowadzeniu prostej soli do płomienia zachodzą w niej następujące przemiany fizykochemiczne i reakcje chemiczne: Odparowanie rozpuszczalnika M+ + A- (mgła) « MA (ciało stałe) Stopienie soli i przeprowadzenie jej w stan pary MA (ciało stałe) «MA (ciecz) « MA (gaz) Reakcja dysocjacji termicznej MA (gaz) « M(gaz) + A(gaz) Atomizery bezpłomieniowe. Atomizery bezpłomieniowe stosuje się dla ominięcia rozcieńczania próbek oraz uniknięcia wpływu matrycy. Najczęściej stosowanym sposobem atomizacji bezpłomieniowej jest atomizacja elektrotermiczna w kuwecie (rurce) grafitowej (piec Massmanna). Kuwety są to rurki grafitowe o dł. 20 - 50 mm i średnicy wewnętrznej 4 - 6 mm. Powierzchnia rurki pokryta jest warstwą grafitu pirolitycznego, co zapobiega dyfuzji atomów w głąb ścianek. Próbkę stałą lub ciekłą wprowadza się bezpośrednio do rurki grafitowej lub na specjalną płytkę grafitową (platforma Lwowa), która znajduje się w atmosferze bardzo czystego gazu obojętnego, najczęściej argonu. Ogrzewanie elektryczne, oporowe lub indukcyjne, odbywa się w sposób programowany, sterowany za pomocą komputera. Cykl pomiarowy składa się odparowania rozpuszczalnika, mineralizacji próbki (piroliza) i atomizacji, czyli przeprowadzenia oznaczanej substancji do plazmy termicznej w postaci wolnych atomów. Zaletami atomizacji elektrotermicznej są m.in. możliwość oddzielenia pierwiastka od składników matrycy, warunki sprzyjające atomizacji trwałych termicznie tlenków oraz całkowita, jednorazowa atomizacja wprowadzonej próbki z dużą wydajnością (w płomieniowej AAS - tylko kilka procent). Atomizery wodorkowe Zdolność tworzenia łatwo lotnych wodorków przez niektóre pierwiastki (Se, Te, As, Sb, Bi, Ge, Sn, Pb) wykorzystano do uwolnienia ich od matrycy. Wodorki tworzy się w reakcji z silnymi substancjami redukującymi, np. z borowodorkiem sodu w środowisku kwaśnym, następnie czyste wodorki wypłukuje się wodorem z mieszaniny reakcyjnej i wprowadza do kwarcowej kuwety pomiarowej, ogrzewanej płomieniem lub elektrycznie do temperatury ok. 1000ºC. W tej temperaturze wodorki ulegają rozpadowi na wolne atomy i gazowy wodór (atomizacja). Atomizery wykorzystujące zimne pary rtęci Stężenie par rtęci powietrzu w temperaturze 300 K może wynosić ok. 20 ng/cm3 i jest to wystarczające stężenie do oznaczenia rtęci metodą AAS. Rtęć w postaci jonów Hg2+ w roztworach można zredukować za pomocą Sn2+ i wolną rtęć wypłukać z mieszaniny reakcyjnej argonem. Rtęć w gazach można zatężyć na wacie złotej; z podgrzanego do 700 -800 K amalgamatu rtęć ulega desorpcji i może być przeniesiona do kuwety pomiarowej w strumieniu argonu. Kuweta pomiarowa (absorpcyjna) to ogrzewana rurka szklana z okienkami kwarcowymi znajdująca się w osi optycznej spektrometru absorpcji atomowej.
Monochromatory Zadaniem monochromatora jest eliminacja promieniowania własnego płomienia i wycięcie linii rezonansowej z promieniowania emitowanego przez lampę z katodą wnękową (źródła promieniowania liniowego). Monochromatory działają na zasadzie siatki dyfrakcyjnej naciętej na powierzchni zwierciadła, które jest umieszczone na obrotowym uchwycie, umożliwiającym kierowanie na szczelinę przepuszczającą do detektora różne długości fal (monochromatory typu Littrowa, Eberta i Czernego - Turnera). Spektrometry AA działają w zakresie od 193,7 do 852,1 nm. Detektory Detektorem w spektrometrze absorpcji atomowej jest fotopowielacz. Jest to układ składający się z fotokatody, szeregu dynod i anody. Zasada działania fotopowielacza polega na tym, że foton pada na katodę, wybija z niej elektrony, które trafiają na dynodę. Każdy elektron wybija kilka nowych elektronów z dynody. Proces ten jest powtarzany na kolejnych dynodach i w ten sposób otrzymuje się wielokrotne wzmocnienie prądu, który jest
proporcjonalny do liczby zaabsorbowanych fotonów. Prąd przekazywany jest do miernika lub innego urządzenia pomiarowego wyskalowanego w jednostkach absorbancji lub transmitancji. Jako rejestratory stosowane są komputery umożliwiające jednocześnie opracowanie statystyczne wyników.
Zakłócenia w metodzie ASA Metoda absorpcyjnej spektrometrii atomowej, podobnie jak inne metody instrumentalne, ograniczana jest zakłóceniami spowodowanymi obecnością w analizowanym roztworze substancji towarzyszących. Mogą one być przyczyną wielu błędów. Zakłócenia te (zwane interferencjami) można podzielić na trzy grupy: · zakłócenia wynikające z nakładania się linii emisyjnych i absorpcyjnych analizowanych pierwiastków, · zakłócenia wynikające z fizycznych właściwości roztworów i mające wpływ na wydajność nebulizacji, · zakłócenia chemiczne powodowane zakłóceniami chemicznymi zachodzącymi w atomizerze. W próbkach złożonych linia rezonansowa oznaczanego pierwiastka może nakładać się z liniami spektralnymi innych pierwiastków. Interferencje wywołane nakładaniem się linii można eliminować przez wykonanie pomiarów przy innej długości fali odpowiadającej innej linii spektralnej oznaczanego pierwiastka lub przez selektywne wyizolowanie pierwiastka oznaczanego lub zakłócającego. Absorpcja linii spektralnej oznaczanego pierwiastka może być pozornie zmniejszona przez emisję promieniowania przez wzbudzone w atomizerze atomy, cząsteczki czy cząstki ciał stałych. Duże cząstki plazmy termicznej mogą rozpraszać promieniowanie, przez co pozornie zwiększać absorpcję. Można temu zapobiec, zwiększając efektywność nebulizacji poprzez zmniejszenie rozmiaru kropel. Procesy emisji, absorpcji i rozpraszania promieniowania przez składniki plazmy nie będące agalitem, można wyeliminować aparaturowo poprzez tzw. korektę tła. Innym typem zakłóceń są interferencje chemiczne, przeważnie specyficzne dla poszczególnych pierwiastków. Nazywa się je często efektami matrycowymi, gdyż powodowane są składnikami matrycy. Składniki matrycy mogą powodować inną lepkość i napięcie powierzchniowe roztworu próbki niż roztworów wzorcowych a tym samym różną wydajność nebulizacji. Problemy takie występują zwłaszcza przy badaniu płynów fizjologicznych i olejów mineralnych. Lepkość i napięcie powierzchniowe można zmniejszyć przez dodatek substancji powierzchniowo-czynnych i rozpuszczalników organicznych. Innym problemem jest wpływ składu matrycy na tworzenie związków analizowanego pierwiastka różniących się lotnością i trwałością termiczną, wpływ na stopień dysocjacji termicznej lub możliwość jonizacji.
Zastosowanie ASA - Metodą AAS można oznaczać około 70 pierwiastków. Problematyczne jest oznaczanie niemetali - AAS jest typową metodą oznaczania pojedynczego pierwiastka. Zastosowanie spektrometrów wielokanałowych nie dało istotnego postępu w eliminacji - AAS jest metodą oznaczania pierwiastków śladowych i składników (bardzo rzadko stosuje się ją do oznaczania składników głównych). - Określany zakres stężeń odpowiada w przybliżeniu jednemu rzędowi wielkości. W przypadku możliwości pomiaru bardzo małych absorbancji zakres ten może objąć 2 - 3 - AAS jest metodą względną. Do wyznaczenia stężenia wykorzystuje się krzywe wzorcowe (wyniki dokładniejsze) lub metodę dodatków (metoda szybsza, ale mniej dokładna) - Metoda AAS jest podatna na wszelkiego rodzaju zakłócenia - stąd konieczność obsługi przez personel o wysokich kwalifikacjach. - AAS jest techniką stosowaną w rutynowych oznaczeniach w laboratoriach metalurgicznych, rolniczych, medycznych, biologicznych, geologicznych, ochrony środowiska i wszędzie tam, gdzie zachodzi konieczność oznaczeń śladowych ilości pierwiastków.
Spektralna analiza emisyjna
Powstawanie widma emisyjnego Jeżeli atomowi będącemu w stanie gazu lub pary dostarczymy energii to elektrony przechodzą z poziomu podstawowego Fp na wyższy poziom energetyczny Ek następnie po czasie ok.10-8 sek.. wracają na poziom, podstawowy emitując charakterystyczne promieniowanie dla danego pierwiastka równe różnicy dwóch jego poziomów wyrażonych wzorem: Ek - Ep = ΔE = hv Najniższy poziom wzbudzenia atomu odpowiadający przejściu elektronu na poziom n = 2 nazywa się rezonansowym. Największe prawdopodobieństwo przejścia P maja linie rezonansowe tj. linie ,wymagające najmniejszej energii wzbudzenia. Prawdopodobieństwo przejść kwantowych. Jest rożne a zatem różne natężenia będą mieć linie w badanym widmie. Natężenie linii widmowych I zależy od tego ile atomów emituje promieniowanie danego rodzaju i jest proporcjonalne do stężenia jego atomów w promieniującym źródle i jest słuszne dla małych zawartości pierwiastka c w próbce. Zależność ta wyrażona jest równaniem Łomakina-Scheibego I = a cb gdzie a i b są to stale dla danego zakresu stężeń
a = ao e-E/kT gdzie: E - energia wzbudzenia danej linii T — temp. źródła wzbudzenia w ok. k —stała Boltzmanna Dla stosunku natężeń dwóch linii pierwiastków wpływ zmian temperatury jest najmniejszy gdy energie wzbudzenia mają podobne wartości. Takie linie nazywa się parą linii homologicznych.. Parametr a zależy od szybkości parowania próbki, na którą dominujący wpływ mają pierwiastki towarzyszące obecne w próbce tj. łatwo, średnio i trudnolotne z uwagi na możliwość zachodzenia reakcji chemicznych w czasie wzbudzania. Dlatego przy wykonywaniu analiz ilościowych konieczne jest posługiwanie się wzorcami tj. substancjami o podobnym składzie chemicznym co badane próbki. Parametr b zależy od warunków absorpcji promieniowania w zewnętrznej warstwie płomienia łuku lub iskry a wiec od stężenia w tej warstwie. Wykrywalność pierwiastka zależy od energii potrzebnej do wzbudzenia oznaczanych pierwiastków.Pierwiastki można podzielić na 3 grupy: - o niskich potencjałach od 1,4 — 3 eV, pierwiastki łatwolotne - o średnich potencjałach od 3 — 10 ev, pierwiastki średniolotne - o wysokich potencjałach od 10 — 30 eV, pierwiastki trudnolotne
Źródła wzbudzenia O łatwości wzbudzenia decyduje temperatura wrzenia pierwiastka tub jego związku. Jako źródła wzbudzenia stosowane są najczęściej: płomień palnika, łuk różnego rodzaju, iskra, laser, palnik plazmowy. Dla pierwiastków łatwolotnych tj. pierwiastków. alkalicznych i ziem alkalicznych stosuje się płomień palnika gazowego o temp. 30000K. Wzbudza się wówczas próbki w postaci roztworów emisyjna metoda zwaną fotometria płomieniowa. Dla pierwiastków średniolotnych najczęściej stosowane są różnego rodzaju wzbudzenia łukowe o temp. od 4000 — 7000 0K jak np. łuk prądu stałego stosowany głównie do wzbudzenia próbek proszkowych - nie przewodzących przy wykrywaniu pierwiastków ( analiza jakościowa ). Do wykonywania analiz ilościowych stosuje się zwykle bardziej stabilne źródła
tj. łuk prądu zmiennego dla próbek metalicznych i stopów. Dla próbek zawierających pierwiastki trudnolotne stosuje się najbardziej stabilne źródło jak iskra skondensowana ewentualnie w kombinacji z laserem lub palnik plazmowy. Laser i palnik plazmowy są nowoczesnymi źródłami, bardzo kosztownymi i nie zawsze celowe jest ich zastosowanie. Natomiast szeroko stosowana jest automatyczna aparatura spektralna z wzbudzeniem iskrowym. Emitowane promieniowanie jest wzmacniane za pomocą fotopowielaczy a wyniki są drukowane z moż1iwością przekazywania na odległość. Urządzenia takie nazywane są kwantometrami, mają szerokie zastosowanie do wykonywania seryjnych analiz ilościowych bardzo szybkich w czasie wytopu stali, żeliwa i stopów o zaprogramowanym składzie chemicznym. który musi być szybko kontrolowany. Aparatura spektralna W skład jej wchodzą w/w źródła wzbudzenia, spektrograf, spektroprojektor i mikrofotrometr. Spektrografy Spektrografami nazywa się aparaty do rejestracji widma emitowanego w czasie wzbudzenia. Emitowane promieniowanie jest rozszczepiane za pomocą pryzmatów lub siatek dyfrakcyjnych. Średnia dyspersja liniowa jest podstawą podziału spektrografów dla obszaru widma ultrafioletowego i widzialnego na: małe, średnie i duże. Zdolność rozdzielcza układu optycznego zależy również od właściwości przyrządu rejestrującego tj. kliszy spektralnej. Dobór spektrografu zależy od rodzaju i liczby oznaczanych pierwiastków a w pewnym stopniu i ich stężeń. Dobór wielkości dyspersji spektrografu zależy od rodzaju głównych składników a zwłaszcza gdy występują one w ilościach dużych. Sposób rejestracji i pomiar natężenia są zależne od zakresu widma oraz od żądanej precyzji. Dobór zakresu widma W zakresie widma 2000 — 2550 Å widma pierwiastków wykazują niewielką liczbę linii i wystarczy zastosować spektrografy o średniej dyspersji ( gdzie różnica dwóch sąsiadujących linii dλ jest nie mniejsza niż 0,3 wtedy linie te nie koincydują ze sobą. Natomiast w obszarze poniżej 2400 Å jest mała czułość klisz spektralnych ) maleje współczynnik kontrastowości co ma wpływ na obniżenie precyzji ilościowych wyników analiz. Do analiz wykorzystuje się zakres widma od 2550 - 3360 Å gdyż czułość i wsp. kontrastowości zmieniają się nieznacznie, w tym obszarze występuje większość linii charakterystycznych pierwiastków, na podstawie których można wykrywać i oznaczać śladowe i większe stężenia pierwiastków. Powyżej 3500 Å dyspersja kwarcu, z którego wykonany jest pryzmat w spektrografie jest mała jak również występuje duża zmienność charakterystyki klisz. Dlatego należy stosować aparaty o dużej dyspersji (różnica dλ poniżej 0,15 Å ponadto w przedziale 3000 - 4600 Å występuje w widmach większość linii pierwiastków ziem rzadkich, metali alkalicznych i ziem alkalicznych. W zakresie 4000 - 7000 Å racjonalne jest posługiwanie się spektrografami z optyką szklaną. Z uwagi na to, że dyspersja kwarcu maleje z długością fali celowe jest stosowanie do rejestracji widma z siatką dyfrakcyjną. Identyfikacji linii widmowych (określenie długości fali) poszczególnych pierwiastków dokonuje się na spektroprojektorze SP-2 f-my Zeissa o powiększeniu 20x, atlasu Kalinina ( tablic ) o tym samym powiększeniu. Pomiar zaczernień linii S dokonuje się na mikrofotometrze Zelssa. Światło przechodzące przez analizowaną linię przechodzi do fotokomórki. Fotoprąd, który daje fotoelement mierzy się czułym galwanometrem (jego wychylenie na skali oznacza ilość światła I przechodzącą przez obraz linii). Przyjmując, że wychylenie galwanometru jest proporcjonalne do ilości światła padającego na fotoelement dla wartości zaczernienia otrzymuje się:
WOLTAMPEROMETRIA
Technika analityczna, której podstawą jest pomiar zależności natężenie prądu - potencjał elektryczny w układzie elektrody pracującej i odniesienia zanurzonych w roztworze badanym zawierającym oznaczaną substancję i elektrolit podstawowy. Elektroda porównawcza (odniesienia) jest niepolaryzowana (np. elektroda kalomelowa), natomiast elektroda pracująca jest polaryzowaną obojętną, i może to być: - kroplowa elektroda rtęciowa (ker) - inna elektroda rtęciowa - mikroelektroda stała Pomiary wykonywane z zastosowaniem ker są nazywane polarografią zgodnie z terminologią IUPAC, choć często pojęcie to jest utożsamiane z woltamperometrią. Metody woltamperomatryczne Ta grupa różnorodnych metod elektroanalitycznyh polega na rejestracji krzywych będących obrazem zmian wielkości prądu płynącego przez układ w zależności od jego składu (zawartości substancji oznaczanych) i napięcia przyłożonego do elektrod. Istnieje wiele odmian metod woltamperometrycznych a poszczególne techniki różnią się kształtem przebiegu napięcia przykładanego do elektrod (stałe, liniowo zmienne, impulsy prostokątne, przebiegi sinusoidalne) i rodzajem stosowanych elektrod. Do metod woltamperometrycznych należą: - polarografia z odmianami: stałoprądowa, oscylopolarografia, polarografia prądu maksymalnego, pulsowa, zmiennoprądowa - woltamperomatria i jej odmiany: chronowoltamperometria inwersyjna, różnicowa, cykliczna, różniczkowa - amperometria
POLAROGRAFIA
Elektrochemiczna metoda analityczna polegająca na przyłożeniu liniowo wzrastającego potencjału elektrycznego do kroplowej elektrody rtęciowej będącej elektrodą pracującą z cyklicznie zmieniającą się w trakcie pomiaru powierzchnią i rejestracji natężenia prądu płynącego przez nią. Wartość natężenia prądu jest proporcjonalna do stężenia obecnej w roztworze substancji ulegającej utlenieniu lub redukcji. Krzywa zależności natężenia prądu od liniowo rosnącego potencjału, rejestrowana za pomocą aparatu zwanego polarografem, w postaci tzw. krzywej polarograficznej pozwala zidentyfikować substancję badaną i określić jej stężenie.
AB - prąd szczątkowy BC - prąd dyfuzyjny CD - strefa depolaryzatora Analiza jakościowa Wielkością charakterystyczną dla substancji ulegającej utlenieniu lub redukcji w warunkach polarograficznych jest potencjał półfali.
Analiza ilościowa Polarograficzna metoda analizy ilościowej opiera się na proporcjonalnej zależności wysokości fali od stężenia. Metody: porównania ze wzorcem (metoda ta polega na przygotowaniu w identycznych warunkach roztworów badanej próbki o stężeniu oznaczonym. Wykonujemy analizę polarograficzną i sporządzamy wykres zależności stężenia badanej substancji od wysokości piku polarograficznego), dodatku wzorca (metoda wielokrotnego dodatku wzorca. W tym wariacie dodawano wzorca o znanym stężeniu do próbki badanej).
CHROMATOGRAFIA
To technika analityczna lub preparatywna służąca do rozdzielania lub badania składu mieszanin związków chemicznych. W każdej technice chromatograficznej najpierw rozdziela się badaną mieszaninę, a następnie przeprowadza się detekcję poszczególnych składników. Rozdział substancji następuje w wyniku przepuszczenia roztworu badanej mieszaniny przez specjalnie spreparowaną fazę rozdzielczą (złoże), zwaną też fazą stacjonarną. Fazą rozdzielczą są substancje wykazujące zdolności sorpcyjne lub zdolne do innych oddziaływań na substancje przepływające. Podczas przepływu eluenta (fazy ruchomej) przez fazę rozdzielczą następuje proces wymywania zaadsorbowanych (lub związanych) substancji. Intensywność tego procesu jest różna dla poszczególnych składników mieszaniny. Jedne składniki są więc zatrzymywane w fazie dłużej, a inne krócej, dzięki czemu może następować ich separacja. Czas przebywania danego składnika w kolumnie określany jest mianem czasu retencji (wielkość równa ilości czasu potrzebnego do przejścia przez całą długość rozdzielczej fazy rozdzielczej określonego składnika analizowanej mieszaniny).
Rodzaje chromatografii W zależności od rodzaju eluentu czyli substancji w której rozpuszcza się badaną mieszaninę rozróżnia się następujące techniki chromatograficzne: - chromatografia cieczowa - w której eluentem jest ciekły rozpuszczalnik lub mieszanina rozpuszczalników - chromatografia gazowa - w której eluentem jest gaz (zwykle hel, argon lub wodór, czasem azot). - chromatografia nadkrytyczna - w której eluentem jest gaz w stanie nadkrytycznym. W zależności od rodzaju i sposobu przygotowania fazy rozdzielczej: - TLC - chromatografia cienkowarstwowa - w której fazę rozdzielczą stanowi cienka warstwa fazy stałej naniesiona na sztywną płytkę. Na tak spreparowaną płytkę nanosi się próbkę roztworu, po czym na skutek działania sił kapilarnych, grawitacji lub pola elektrycznego następuje przepływ i rozdział mieszaniny; - chromatografia bibułowa - w której fazę rozdzielczą stanowi pasek lub arkusz bibuły filtracyjnej lub specjalnego typu bibuły chromatograficznej; - chromatografia kolumnowa - w której faza rozdzielcza jest umieszczona w specjalnej kolumnie, przez którą przepuszcza się następnie roztwór badanej mieszaniny. Przepływ roztworu przez kolumnę można wymuszać grawitacyjnie lub stosując różnicę ciśnień na wlocie i wylocie kolumny; - chromatografia powinowactwa - w której odpowiednio spreparowana faza rozdzielcza jest zdolna do oddziaływań chemicznych o zmiennym powinowactwie wobec rozdzielanych substancji; - chromatografia jonowymienna - w której substancje oddziaływają ze złożem za pomocą oddziaływań jonowych. W zależności od parametrów procesu: HPLC - wysokosprawna/ciśnieniowa chromatografia cieczowa - odmiana cieczowej chromatografii kolumnowej z użyciem eluentu pod wysokim ciśnieniem; FPLC - szybka, białkowa/szybkosprawna chromatografia cieczowa - odmiana HPLC działająca na niższych ciśnieniach, stosująca prócz złóż sorpcyjnych, także zwykłe złoża typu sit molekularnych, służąca głównie do rozdziału białek i polipeptydów. UPLC -, ultrasprawna chromatografia cieczowa - odmiana cieczowej chromatografii kolumnowej. Działa na wyższych ciśnieniach i mniejszych przepływach, a kolumny mają mniejsze ziarno (1,7 - 1,8 μm). Pozwala uzyskiwać krótsze czasy retencji i wyższe rozdzielczości. GPC - chromatografia żelowa - odmiana kolumnowej chromatografii cieczowej. Polega na rozdziale składników mieszaniny na sitach molekularnych ze względu na ich wielkość (masę). Stosowana m.in. do określania średnich mas cząsteczkowych polimerów Chromatografia cieczowa Rodzaj chemicznej techniki analitycznej - chromatografia, w której eluentem jest ciecz, zwykle jakiś rozpuszczalnik. Istotą każdej chromatografii cieczowej jest rozdział analizowanej mieszaniny na poszczególne związki chemiczne poprzez przepuszczanie roztworu tej mieszaniny przez stałe lub żelowe złoża, co w pewnym sensie przypomina filtrację. Na skutek oddziaływań międzycząsteczkowych między związkami tworzącymi mieszaninę a złożem jedne z nich przechodzą przez złoże szybciej a inne wolniej. Istnieje bardzo wiele rodzajów chromatografii cieczowej, które można podzielić na trzy ogólne rodzaje: - chromatografia cienkowarstwowa - służąca do identyfikacji oraz oczyszczania mieszanin związków chemicznych. Faza rozdzielcza, złoże, czyli faza stacjonarna o właściwościach sorpcyjnych (silikażel, tlenek glinu, rzadziej celuloza lub ziemia okrzemkowa) jest umieszczona jako cienka (do 0.01-2 mm) warstwa na płytce szklanej, aluminiowej lub wykonanej z tworzywa sztucznego. Substancje rozdzielane nanosi się punktowo przy dolnej krawędzi płytki, po czym płytkę umieszcza się w komorze chromatograficznej zanurzonej na kilka milimetrów w eluencie, tak by naniesione substancje nie zostały zanurzone. Eluent stopniowo wspina się po płytce dzięki zjawisku kapilarnemu. Powoduje to przemieszczanie się rozdzielanych substancji ku górze. Prędkość ruchu poszczególnych składników rozdzielanej mieszaniny jest zależna od oddziaływań międzycząsteczkowych między związkami chemicznymi obecnymi w analizowanej próbce, a fazą rozdzielczą i eluentem. Gdy czoło eluenta dotrze do górnej krawędzi płytki rozdział jest zakończony. Zależnie od swojej natury fizykochemicznej - składniki rozdzielanej mieszaniny docierają w tym czasie na różną wysokość płytki (współczynnik Rf), a równomierność rozmieszczenia plam substancji można charakteryzować ilościowo tzw. funkcjami. W przypadku, gdy substancje nie są naturalnie barwne, ich obecność można stwierdzić używając, zależnie od ich właściwości, światła UV, roztworów wywołujących (w których płytka jest zanurzana lub nimi spryskiwana - reagują one specyficznie i barwnie z rozdzielanymi substancjami) lub innych metod (np. wywoływanie w parach jodu, piroliza). - chromatografia kolumnowa - Polega na rozdzieleniu mieszanin poprzez wprowadzenie jej na stałą fazę stacjonarną umieszczoną w cylindrycznej kolumnie i rozdzieleniu jej na składniki przy użyciu ciekłej fazy ruchomej, wprowadzanej z odpowiedniego zbiornika lub dolewanej bezpośrednio na kolumnę. Przepływ fazy ruchomej może być grawitacyjny lub wymuszony przez niewielkie nadciśnienie wprowadzanego eluentu (uzyskiwane np. za pomocą pompy perystaltycznej lub sprężonego gazu), lub przez niewielkie podciśnienie u wylotu kolumny (uzyskiwane np. za pomocą pompki wodnej). W zależności od wielkości kolumn i ilości rozdzielanego materiału chromatografia kolumnowa może być wykonywana w skali analitycznej, laboratoryjnej (półpreparatywnej, preparatywnej) i przemysłowej. - elektroforeza - która w zasadzie jest formą chromatografii cienkowarstwowej, w której jednak najpierw nasącza się złoże mieszaniną, a rozdział następuje na skutek działania pola elektrycznego
Chromatografia gazowa analityczna technika chromatograficzna, w której fazą nośną jest gaz (najczęściej hel, argon, coraz rzadziej wodór). Technika ta umożliwia procentowe ustalenie składu mieszanin związków chemicznych, w których występuje ich nawet kilkaset. Stosując klasyczną detekcję umożliwia przybliżoną identyfikację składników mieszaniny, pełną identyfikację z detektorem masowym. Chromatografia gazowa jest najczęściej stosowaną metodą do szybkiej analizy złożonych mieszanin związków chemicznych oraz oceny czystości tych związków, zarówno w przemyśle jak i w rozmaitych laboratoriach. Chromatografię gazową stosuje się m.in. w: - przemyśle petrochemicznym - np. do oceny składu chemicznego produkowanej benzyny; ochronie środowiska - do oceny stopnia zanieczyszczenia, gleby, powietrza i wody; - kryminalistyce - np. do analizy źródła pochodzenia narkotyków na podstawie składu zawartych w nich zanieczyszczeń; - kontroli antydopingowej - gdzie aparaty GC-MS stanowią podstawową metodę wykrywania niedozwolonych substancji w krwi, pocie, moczu i ekstrakcie z włosów sportowców. - w przemyśle spożywczym - do badania składu surowców i produktów żywnościowych oraz do wykrywania zafałszowań żywności Zaletą chromatografii gazowej jest możliwość użycia bardzo niewielkiej próbki analizowanej substancji - od nawet 0,01 µl do maksymalnie 100 µl. Chromatografia bibułowa to rodzaj chemicznej techniki analitycznej, podtyp chromatografii cieczowej, w której fazę rozdzielczą stanowi specjalna bibuła o wysokiej czystości i określonych parametrach. Technika ta bywa też stosowana do oczyszczania i identyfikacji mieszanin związków chemicznych. Prędkość ruchu poszczególnych składników rozdzielanej mieszaniny jest uzależniona od oddziaływań międzycząsteczkowych między związkami chemicznymi tworzącymi analizowaną próbkę, a fazą rozdzielczą i rozpuszczalnikiem zwanym w tym przypadku eluentem. Substancje rozdzielane nakrapia się punktowo przy dolnej krawędzi bibuły, po czym umieszcza się ją w komorze chromatograficznej zanurzoną na kilka milimetrów w eluencie, tak by nakropione substancje nie zostały zanurzone. Dzięki zjawisku występowania sił kapilarnych eluent stopniowo wznosi się po bibule ciągnąc za sobą z różną prędkością związki chemiczne tworzące analizowaną mieszaninę. Gdy czoło eluenta dotrze do górnej krawędzi bibuły rozdział jest zakończony. Ten etap analizy nazywany jest rozwijaniem chromatogramu. Dzięki różnicy prędkości wędrówki rozdzielanych związków chemicznych w momencie zakończenia analizy znajdują się one na bibule na różnych wysokościach względem czoła eluentu. Ze względu na kierunek przepływu eluentu przez bibułę chromatografię bibułową dzieli się: - wstępującą - prowadzoną w sposób opisany powyżej, tzn. z eluentem wędrującym po bibule z dołu do góry - zstępującą - w której bibuła jest nasączana od góry a eluent wędruje z góry do dołu; tego rodzaju chromatografia wymaga specjalnej konstrukcji komory chromatograficznej. Po zakończonym etapie rozwijania, gdy przebiegał on prawidłowo, rozdzielone związki tworzą na bibule obszary o kształtach zbliżonych do łez. Gdy rozdzielanie związki są barwne obszary te można bezpośrednio obserwować w świetle widzialnym lub przy innej długości fali, którą analizowane związki absorbują. W przypadku aromatycznych związków organicznych jest to najczęściej światło UV. Gdy związki nie są barwne działa się na chromatogram odpowiednimi odczynnikami, które reagują z analizowanymi związkami nadając im przy tym barwę. Najczęściej stosowanym odczynnikiem wywołującym są pary jodu. Chromatografia jonowymienna Rodzaj cieczowej chromatografii kolumnowej. Jest to metoda preparatywna używana do wydzielenia z mieszaniny żądanej substancji. W tej metodzie chromatografii faza stacjonarna, złoże, jest obdarzona ładunkiem. Stanowi je zazwyczaj żywica jonowymienna, zawierająca obdarzone ładunkiem grupy funkcyjne, oddziałujące z przeciwnie naładowanymi grupami związków, które mają zostać zatrzymane przez nośnik: - pozytywnie naładowany jonowymieniacz (anionit) wiąże aniony - negatywie naładowany jonowymieniacz (kationit) wiąże kationy. Związki związane z jonowymieniaczem mogą być wymyte z kolumny przez stopniową elucję, a także poprzez zmianę stężenia soli lub pH. Tego rodzaju chromatografii używa się do oddzielania takich związków jak aminokwasy, peptydy i białka. Chromatografia jonowymienna jest powszechnie stosowana do oczyszczania białek w systemie FPLC. Odmianą chromatografii jonowymiennej jest chromatografia jonowa, w której stosuje się wysokosprawne kolumny rozdzielające wypełnione jednorodnymi żywicami o małych cząsteczkach i najczęściej z automatyczną detekcją konduktometryczną.
CHROMATOGRAFICZNE METODY ANALIZY
Chromatografia obejmuje metody rozdziału mieszanin jednorodnych różnych substancji oparte na podziale składników mieszaniny fazę nieruchomą (stacjonarną) i ruchomą układu chromatograficznego. Klasyfikacja metod chromatograficznych. Klasyfikacja wynikająca z natury zjawisk będących podstawą rozdziału chromatograficznego. Biorąc pod uwagę mechanizm oraz naturę zjawisk rozdziału chromatograficznego, chromatografię można podzielić na następujące grupy: 1) adsorpcyjną - oparty jest na zjawisku adsorpcji składników mieszaniny na odpowiednio dobranej powierzchni fazy nieruchomej, zwanej adsorbentem. Głównym warunkiem rozdziału są różne wartości współczynników adsorpcji poszczególnych składników mieszaniny. 2) podziałową - oparte jest na procesie ekstrakcji, tzn. na podziale składników mieszaniny między dwie niemieszające się fazy, z których jedna- faza stacjonarna (ciecz) jest naniesiona na specjalny nośnik, a druga - faza ruchoma (ciecz lub gaz) przepływa nad fazą stacjonarną. Głównym warunkiem rozdziału są różne wartości współczynników rozpuszczalności poszczególnych składników mieszaniny w ciekłej fazie stacjonarnej. 3) jonowymienną - opiera się na reakcji wymiany jonowej między rozdzielanymi jonami, zawartymi w roztworze, a jonami związanymi z makrocząsteczką wymieniacza jonowego. 4) osadową - opiera się na wytrącaniu osadów w wyniku reakcji chemicznych, zachodzących między osadzonym na nośniku odczynnikiem a znajdującymi się w roztworze rozdzielanymi jonami. Głównym warunkiem są różne wartości rozpuszczalności wytrącanych osadów. 5) sitową - opiera się na efekcie sitowym. Głównym warunkiem rozdziału są różne wartości masy cząsteczkowej rozdzielanych związków. Klasyfikacja uwzględniająca stan skupienia fazy nieruchomej i ruchomej. Fazą stacjonarną w procesie chromatograficznym może być ciało stałe mające właściwości adsorpcyjne (chrom. Adsorpcyjna) lub ciecz naniesiona na nośnik w celu wytworzenia granicy faz większej powierzchni wymiany (chrom. Podziałowa). Fazą ruchomą może być ciecz lub gaz (para substancji lotnej). Chromatografia gazowa. Jest skuteczną i szybką metodą rozdzielania mieszanin związków lotnych. Substancja rozdzielana jest przenoszona za pomocą gazu nośnego (faza ruchoma) przez kolumnę wypełnioną fazą nieruchomą. W zależności od typu stosowanej fazy nieruchomej rozróżnia się: a) chromatografię gazową podziałową (fazą nieruchomą jest ciecz naniesiona na stały nośnik) b) chromatografię gazową adsorpcyjną (fazą nieruchomą jest adsorbent) c) chromatografię gazową kapilarną (fazą nieruchomą jest ciecz naniesiona bezpośrednio na ścianki kolumny).
Chromatografia gazowa znalazła również zastosowanie do identyfikacji i oznaczeń ilościowych małych ilości mieszanin gazowych. Rozdział mieszanin metodą chromatografii gazowej prowadzi się najczęściej techniką wymywania (elucji). Na szczyt kolumny chromatograficznej nanosi się próbkę analizowanej mieszaniny, a następnie przepuszcza się przez kolumnę gaz nośny, który pełni rolę czynnika wymywającego. Wielkościami które w określonych warunkach rozdziału charakteryzują daną substancję pod względem jakościowym są: 1) czas retencji : a) całkowity czas retencji - danego składnika tr - czas liczony od momentu wprowadzenia próbki do momentu pojawienia się na wyjściu kolumny maksymalnego stężenia wymywanego związku. b) martwy czas retencji - tM - czas retencji gazu nie ulegającego adsorpcji c) zredukowany czas retencji - tR' - czas retencji danego składnika liczony od martwego czasu retencji: tR'=tR - tM
d) skorygowany czas retencji tRO : tRo = jtR , j - to współczynnik korekcyjny, uwzględniający spadek ciśnienia w kolumnie: pi - ciśnienie na wejściu kolumny, po - ciśnienie na wyjściu kolumny
e) względny czas retencji : 2) objętość retencji: a) całkowita objętość retencji danego składnika VR - objętość gazu nośnego potrzebna do wymycia tego składnika: VR = FotR , Fo - objętościowa prędkość wypływy gazu nośnego z kolumny. b) martwa (zerowa) objętość retencji VM: VM = FotM c) zredukowana objętość retencji VR' d) skorygowana objętość retencji VRo e) absolutna (rzeczywista lub całkowicie poprawiona) objętość retencji VN, zależy od parametrów kolumny, temperatury kolumny, ilości fazy stacjonarnej i rodzaju badanej substancji. f) właściwa objętość retencji Vg - absolutna objętość retencji przeliczona na 1g fazy ciekłej (lub 1g adsorbentu) i temp. 0oC:
Gdzie: Tc - temp. kolumny w K, wL - masa fazy stacjonarnej w kolumnie w g. g) względna objętość retencji:
Właściwa i względna objętość retencji zależy od temperatury kolumny i współczynnika podziału, a nie zależy od parametrów) kolumny. Parametry chromatograficzne.
1) współczynnik podziału K:
2) stosunek podziału k' zwany również liczbą podziału lub współczynnikiem pojemności kolumny: Stosunek podziału decyduje o tym, jaki ułamek masy związku chromatografowanego znajdzie się w fazie stacjonarnej, a jaki w fazie ruchomej. Istnieje następująca zależność między absolutną objętością retencji VN a współczynnikiem podziału i stosunkiem podziału: VN = KVL = k'VG. Za pomocą parametrów retencji można wyznaczyć takie parametry chromatograficzne, jak indeks retencji i współczynnik selektywności (stopień rozdziału) 3) indeks retencji Ix substancji x:
gdzie tRx' - zredukowany czas retencji związku x, eluowanego między węglowodorami o z i z+1 atomach węgla. 4) współczynnik selektywności αj,i zwany również stopniem rozdziału, określa w sposób ilościowy możliwość rozdzielenia dwóch substancji j i i:
Selektywność rozdziału zależy nie tylko od rodzaju analizowanych składników, ale również od rodzaju fazy ciekłej, gdy faza ciekła zostanie źle dobrana, tzn, gdy wartości współczynników podziału substancji j i i będą zbliżone, wówczas mimo sprawnej kolumny nie uda się rozdzielić od siebie obu składników. W przypadku gdy substancja i nie ulega podziałowi między dwie fazy, współczynnik selektywności oblicza się za pomocą następującego wyrażenia :
Analiza jakościowa. Analiza jakościowa chromatografii gazowej opiera się na danych retencyjnych. Przeprowadza się ją najczęściej porównując dane retencyjne związków badanych, zdanymi retencyjnymi związków wzorcowych. Duże znaczenie mają zwłaszcza względne parametry retencyjne, które zmierzono już dla znacznej liczby związków i umieszczono w opracowaniach monograficznych z chromatografii gazowej. Najprostszy problem jakościowy polega na potwierdzeniu lub wykluczeniu obecności określonego związku w analizowanej mieszaninie. W tym celu wykonuje się w identycznych warunkach chromatograf substancji wzorcowej i badanej. Jeżeli na chromatografie substancji badanej nie występuje pik o czasie retencji równym czasowi retencji substancji wzorcowej, dowodzi to nieobecność związku wzorcowego w badanej próbie. Jeżeli na chromatografie substancji badanej występuje taki pik, to obecność związku wzorcowego nie jest przesądzona w sposób definitywny, gdyż wiele związków należących do różnych klas chemicznych może mieć identyczny lub prawie identyczny czas retencji. Identyfikację należy potwierdzić przez powtórzenie analizy na innej kolumnie. Identyczność czasów retencji substancji nieznanej i związku wzorcowego na różnych kolumnach uważa się za dowód identyczności tych dwóch substancji. Analiza ilościowa. Analiza ilościowa jest oparta na liniowej zależności między sygnałem detektora (powierzchnią piku) a stężeniem substancji badanej w gazie nośnym. Powierzchnia piku jest proporcjonalna do ilości oznaczanej substancji. Można ją wyznaczyć geometrycznie, planimetrycznie lub przez całkowanie, za pomocą integratorów. Czasami stosuje się również metodę ważenia wyciętych pików. Zastosowanie chromatografii gazowej. W chemii analitycznej chromatografia gazowa jest stosowana głównie do identyfikacji i ilościowego oznaczania składników mieszaniny. |
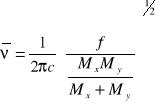
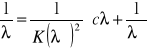
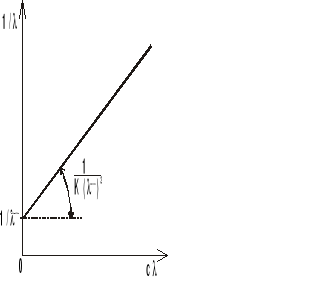
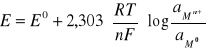
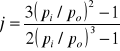
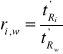
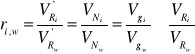
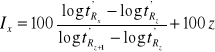
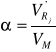
Wyszukiwarka
Podobne podstrony:
Strzałka ugięcia, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
Piotr Milejski Projekt przenośnika taśmowego, AGH, 6 semestr, Maszyny i Urządzenia transportowe
pomiar, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
Untitled 1, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
Druty, Druty - MiU, Maszyny i urządzenia transportowe
MiUT test ćw 5 Bembenek, WIMiR AGH, Semestr VI, Maszyny i Urządzenia Technologiczne (MiUT), Kolokwia
liny, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
Untitled 20, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
matryca do wyrobu sciag, MASZYNY I URZĄDZENIA TRANSPORTOWE
Olszyna, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
Dane do projektu przenośnika zgrzebłowego, Uczelnia, Maszyny i urządzenia transportowe
Sprawko liny, AGH - IMIR - IMIM, III ROK, Maszyny i urządzenia transportowe
labendziorek, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
mut teroia 2, MASZYNY I URZĄDZENIA TRANSPORTOWE
Sprawozdanie Maszyny i Urządzenia Transportowe
Nadwyżka, IMiR - st. inż, sem.6 od sołtysa, Maszyny i urządzenia transportowe
F4C Maszynista urzadzen transportu pneumatycznego
więcej podobnych podstron