Wytwarzanie i wzrost nanostruktur
Teoria zarodkowania
Wytwarzanie cienkich warstw na stałym podłożu o charakterze nieorganicznym jest możliwe przy wykorzystaniu metody naparowywania w próżni.
W pierwszym etapie formowania się warstwy tworzą się tzw. zarodki krystalizacji. Zarodki powiększają swoje rozmiary w miarę zwiększania ilości atomów zaadsorbowanych na powierzchni. Następnie tworzą się większe klastery, które ulegają procesowi koalescencji, aż do utworzenia warstwy ciągłej.
W kolejnych etapach następuje dalszy wzrost prostopadły do podłoża oraz dalsza rekrystalizacja w miarę zwiększania się grubości warstwy.
Mechanizm tworzenia się zarodków krystalizacji i przebieg kolejnych etapów wzrostu jest uzależniony od wielu czynników.
Do najważniejszych z nich należą temperatura podłoża, jego typ i orientacja krystalograficzna, jak również obecność w układzie gazów resztkowych i ich rodzaj oraz geometria układu źródło - podłoże.
Przy naparowywaniu w bardzo niskich temperaturach dyfuzja powierzchniowa zachodzi bardzo powoli, uniemożliwiając rekrystalizację krystalitów i ich koalescencję. Powoduje to wzrost kryształów w kształcie kolumn, pomiędzy którymi występują puste miejsca.
Zazwyczaj, podczas naparowywania metali w próżni, źródło ma temperaturę o wiele wyższą od podłoża. Tworzy to stan przesycenia par metalu, w którym szybkość adsorpcji atomów na podłożu jest większa od szybkości ich desorpcji.
Proces tworzenia się zarodków fazy stałej występuje zawsze podczas tworzenia się warstwy, ponieważ na każdym etapie tworzenia warstwy zachodzi zjawisko dyfuzji powierzchniowej.
Na skutek zderzeń atomy łączą się w większe skupiska, a następnie są pułapkowane w charakterystycznych miejscach na powierzchni, tworząc zarodki fazy krystalicznej.
Dynamikę tego procesu opisuje tzw. teoria zarodkowania, w której szybkość tworzenia się zarodków określa się badając przebieg pewnej funkcji nazywanej energią swobodną (G - energia swobodna Gibbsa).
Termodynamiczna teoria zarodkowania zakłada, że przyrost energii swobodnej przy tworzeniu się kulistego zespołu, złożonego z pojedynczych atomów, opisuje wyrażenie:
![]()
(1.1.1)
gdzie zmiana energii swobodnej dla jednostki objętości wynosi:
![]()
(1.1.2)
Wielkości fizyczne w powyższych wyrażeniach oznaczają:
r - promień kulistego zespołu
γkg - napięcie powierzchniowe na granicy kondensat - pary metalu
kB - stała Boltzmana
T - temperatura bezwzględna
V - objętość atomowa
s - stopień przesycenia, tj. stosunek aktualnej prężności par metalu w temperaturze podłoża T do prężności równowagowej (p/p0).
p - prężność par metalu w temperaturze T
p0 - równowagowa prężność par.
Rys. 1.2 - Zależność zmiany całkowitej energii swobodnej skupiska atomów ΔG0 od jego promienia r. Wartość r*k odpowiada promieniowi zarodka krytycznego. W miarę zwiększania przesycenia par gazu w układzie s > s' > s'', promień krytyczny zmniejsza się.
Krytyczną wartość przyrostu energii swobodnej ΔG*k i wartość krytyczną promienia kulistego zarodka otrzymać można z warunku na ekstremum funkcji ΔG0: (∂G0/∂r = 0). Wyrażając zmianę energii swobodnej ΔG0 przy użyciu (1.1.3) otrzymujemy:
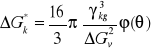
(1.1.4)
oraz
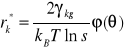
, ![]()
(1.1.5)
Możliwość tworzenia się zarodków krystalizacji powstaje w warunkach silnego przesycenia par metalu. Jak wynika z równania (1.1.5) i rysunku (Rys. 1.2) wartość promienia krytycznego maleje ze wzrostem przesycenia. Teoria ta daje wartości r*k rzędu promieni atomowych.
Na przykład dla srebra osadzanego z prędkością 0,1 [nm/s] na podłoże w temperaturze pokojowej otrzymujemy r*k równe około 0,22 [nm], co w przybliżeniu odpowiada rozmiarom jednego atomu srebra. W takiej sytuacji jednoatomowy zarodek nie może ulec rozpadnięciu i możliwy jest tylko jego dalszy wzrost.
Epitaksjalny wzrost warstw
W dalszym etapie powstawania warstwy zarodki krytyczne krystalizacji przyłączają kolejne atomy tworząc coraz większe krystality. Stopień ich orientacji może być różny i zależy od rodzaju użytych materiałów i warunków panujących podczas naparowywania. Jeżeli tworząca się warstwa jest monokryształem o orientacji określonej orientacją monokrystalicznego podłoża, to tego rodzaju proces nazywamy wzrostem epitaksjalnym.
W przypadku, gdy warstwa epitaksjalna powstaje z tego samego materiału, co podłoże, ma taką samą strukturę i orientację krystalograficzną, to proces ten nazywamy homoepitaksją. Heteroepitaksja zachodzi wówczas, gdy mamy do czynienia ze wzrostem monokryształu o innym, niż podłoże składzie chemicznym, strukturze i orientacji.
Otrzymywanie warstw homoepitaksjalnych jest znacznie łatwiejsze, niż heteroepitaksjalnych ze względu na problem niedopasowania stałych sieciowych i powstawanie dyslokacji.
Metoda wytwarzania |
Rok odkrycia |
Cechy charakterystyczne |
Epitaksja z fazy gazowej |
1958 |
Do transportu materiału wykorzystuje się halogenki bądź wodorki metali; metoda łatwa i bezpieczna; duża prędkość parowania; służy do otrzymywania grubych warstw; |
Epitaksja z fazy ciekłej |
1963 |
Wzrost następuje poprzez umieszczenie przesyconej cieczy na podłożu; metoda łatwa i tania; mało precyzyjna kontrola grubości warstwy przy otrzymywaniu ultra-cienkich warstw. |
Epitaksja z wiązki molekularnej MBE (Molecular Beam Epitaxy) |
1967 |
Osadzanie w warunkach ultra-wysokiej próżni; bardzo dokładna kontrola ilości odparowanego materiału; utrudniony wzrost materiałów o dużej prężności par |
Depozycja chemicznych par metalotlenkowych |
1968 |
Transport materiału na powierzchnię zachodzi poprzez hydrodynamiczny napływ organo-metalicznych molekuł. Rolę gazu transportującego pełni H2; materiałów w przypadku niektórych materiałów wydzielają się toksyczne składniki. |
Tab. 1.1 - Metody otrzymywania warstw epitaksjalnych.
Rys. 1.4 - Fizyczny model epitaksji z uwzględnieniem charakterystycznych procesów zachodzących na powierzchni podczas wzrostu warstwy. Efektywność procesu jest proporcjonalna do wartości ![]()
exp(Ei/kBT), gdzie Ei - oznacza odpowiednio energię: adsorpcji Eads, desorpcji Edes, energię tworzenia zarodków Enukl oraz energię dyfuzji powierzchniowej Edyf. Przy czym w procesie dyfuzji zależność jest odwrotnie proporcjonalna.
Pierwszej klasyfikacji rodzajów wzrostu warstw epitaksjalnych dokonał w 1958 roku E. Bauer. Wykorzystując termodynamiczne zależności pomiędzy swobodnymi energiami powierzchniowymi podłoża γp, kondensatu γk oraz ich interfejsu γi wyszczególnił trzy rodzaje wzrostu (Rys. 1.5):
Wzrost Franka - van der Merwe (FM) (wzrost warstwa po warstwie): następuje, gdy atomy adsorbatu oddziałują silniej z atomami podłoża, niż ze sobą nawzajem; przed rozpoczęciem tworzenia kolejnej warstwy poprzednia jest całkowicie uformowana.
Wzrost Volmera - Webera (VW) (wzrost wyspowy): ma miejsce, gdy atomy adsorbatu silniej oddziałują ze sobą, niż z atomami podłoża; następuje wzrost krystalitów (typu 3D) bezpośrednio na podłożu aż do utworzenia ciągłej warstwy na skutek koalescencji, przy odpowiedniej ilości osadzonego materiału.
Wzrost Stranskiego - Krastanova (SK) (wzrost mieszany): energia interfejsu rośnie wraz ze wzrostem grubości warstwy; początkowo następuje wzrost warstw po warstwie, aby po osiągnięciu pewnej krytycznej grubości przejść do fazy wzrostu wyspowego.
Rys. 1.5 - Podstawowe rodzaje wzrostu warstw epitaksjalnych. θ określa ilość osadzonego materiału wyrażonego liczbą monowarstw ML.
r [Å]
ΔG*k
r*k
ΔG0
Energia
powierzchniowa
Energia
objętości
s > s' > s''
Desorpcja
Adsorpcja
chemiczna
na defektach
Nukleacja
Interdyfuzja
Dyfuzja
powierzchniowa
Edes
Edyf
Enukl
Eads
Adsorpcja
fizyczna
Narastająca
warstwa
Strumień atomów ze źródła
warstwowy: (F-M)
warstwowo - wyspowy: (S-K)
wyspowy: (V-W)
θ < 1ML 1ML < θ < 2ML θ > 2ML
Wyszukiwarka
Podobne podstrony:
El en i środowisko 13 14 1, Prywatne, EN-DI semestr 4, Elektroenergetyka, wykład + ćwiczenia
mini mikro, ~WSB GDYNIA WSB GDAŃSK, 2 semestr, Mikroekonomia (wykłady) dr Katarzyna Gregorkiewicz
fiza, BUDOWNICTWO PŁ, Semestr I, fizyka wykład
Narazenia od pól elektromagnetycznych 13 14 1, Prywatne, EN-DI semestr 4, Elektroenergetyka, wykład
TEST fila, SEMESTR 1, Standardy, Wykłady, EGZAMIN
Grupa, Studia Pwr, Semestr 1, Psychologia (wykład)
Narazenia od pól elektromagnetycznych 13 14 2, Prywatne, EN-DI semestr 4, Elektroenergetyka, wykład
nanotechnologia wykłady Wykład 1 (22 03 2012)
STATECZNOŚĆ IV SEMESTR EGZAMIN WYKŁADÓW SZOZDA
Moc bierna 13 14 1, Prywatne, EN-DI semestr 4, Elektroenergetyka, wykład + ćwiczenia
PSYCHOPATOLOGIA - materiały, WSFiZ - Psychologia, VI semestr, Psychopatologia - wykłady
chemia wyklady wskrzynka(1), BUDOWNICTWO PŁ, Semestr I, chemia wykład
Wyklad 1 z enzymologii, Studia, Przetwórstwo mięsa - Semestr 1, Enzymologia, Wykłady
Zoologia wykł. 1. (2), Semestr 1, zoologia, wykłady
pytania oczyszczanie wody egz (3), Politechnika Wrocławska, Ochrona Środowiska W7, Semestr V, SOW- w
Biofizyka pytania z kola, Biotechnologia PWR, Semestr 5, Biofizyka - Wykład, Biofizyka - materiały
DD - Opór powietrza, Transport Polsl Katowice, 5 semestr, ŚT, Wyklady, Srodki transportu
więcej podobnych podstron