Andrzej Michalski 17.04.2008 r.
Biotechnologia; Wydział: AEiI; Grupa Dziekańska: AU2; Sekcja 2
SPRAWOZDANIE
Ćwiczenie 4
Składające się z 3 części:
I. Trawienie DNA faga λ za pomocą enzymu restrykcyjnego EcoRI
II. Analiza elektroforetyczna produktów reakcji trawienia.
III. Oznaczanie DNA wyizolowanego z komórek na ćwiczeniu poprzednim.
I. Trawienie DNA faga λ za pomocą enzymu restrykcyjnego EcoRI
Wstęp teoretyczny
Nasze pierwsze ćwiczenie było związane z enzymem restrykcyjnym EcoRI. Jest to endonukleaza wyizolowana po raz pierwszy ze szczepu RY13 E.coli i jest częścią bakteryjnego systemu modyfikacji restrykcyjnych. Enzymy z grupy endonukleaz przecinają nić DNA w miejscu wyznaczanym przez specyficzną sekwencję DNA. Rozpoznawana sekwencja z reguły ma charakter symetryczny o długości od 4 do 8 par zasad. I tak jest właśnie w enzymie EcoRI. Przy cięciu DNA tworzy on lepkie końce z przedłużonymi końcami 5'. Sekwencją rozpoznawaną i ciętą przez enzym jest palindromiczna sekwencja 5'-G▼AATTC-3'.
Z powodu swojej selektywności i specyficznego miejsca cięcia, które następnie może ulec ligacji, enzym ten jest używany w biologii molekularnej, szczególnie w technice klonowania, screeningów DNA, czy mutagenezy ukierunkowanej.
Opis wykonywanego ćwiczenia
Obliczanie ilości odczynników potrzebnych do trawienia faga λ
Aby można było skutecznie strawić DNA faga λ należało dokładnie określić ilość potrzebnych odczynników
Mieliśmy do dyspozycji takie odczynniki jak:
DNA faga λ o stężeniu 0.25 μg/μl
Bufor do trawienia 5x stężony
Enzym restrykcyjny EcoRI 2.5 U*/μl
Wodę dejonizowaną
U* - jednostka enzymatyczna - jest to ilość enzymu przekształcającego 1 jednostkę masy (g, mg, μg) substratu w jednostce czasu (godzina, minuta) w temperaturze i warunkach optymalnych dla działania enzymu. W przypadku enzymów restrykcyjnych jedna jednostka enzymatyczna jest to ilość enzymu przecinającego 1 μg DNA w czasie 1 godziny w temperaturze 370C (dla nielicznych enzymów restrykcyjnych optymalna temperatura inkubacji jest inna) .
Cała mieszanina reakcyjna powinna mieć objętość 10 µl i być w odpowiednich stężeniach:
0,5 μg DNA faga λ
5 U enzymu EcoRI
Wszystko powinno być w 1x stężonym buforze do trawienia o składzie :
* 50 mM NaCl
* 100 mM Tris:HCl pH 7.9
* 5 mM MgCl2
* 0.025% Triton X-100
W tym celu należało przeprowadzić obliczenia, aby móc określić ilość poszczególnych substancji:
* DNA faga λ:
0.25 μg - 1 μl
0.5 μg - x μl
x = 2 μl
* Enzym restrykcyjny EcoRI:
2.5 U - 1 μl
5 U - x μl
x = 2 μl
* Bufor do trawienia:
Posiadamy bufor 5x stężony a chcemy mieć 10 μl roztworu, w którym bufor ten jest 1x stężony.
Dlatego na 1 jednostkę buforu należy przeznaczyć 4 jednostki innych odczynników. Na 10 μl
całej mieszaniny reakcyjnej, 1 jednostka zajmuje 2 μl (10 μl / 5 jednostek), dlatego potrzebujemy:
1 * 2μl = 2μl buforu do trawienia
4 * 2μl = 8μl reszty odczynników
* Woda dejonizowana:
Wodą dejonizowaną należy dopełnić mieszaninę reakcyjną do 10μl. Posiadamy już:
2μl+2μl+2μl=6μl innych odczynników, dlatego należy dodać 4μl H2O dejonizowanej.
Mieszanie odczynników oraz trawienie
Materiał genetyczny jest bardzo często materiałem labilnym, dlatego ważna jest kolejność dodawania odczynników. Zapewni to właściwe proporcje substratów i ułatwi aktywność enzymatyczną.
W tym przypadku do eppendorfki odczynniki dodawaliśmy w takiej kolejności:
4μl H2O
2μl Buforu do trawienia
2μl DNA faga λ
2μl enzymu EcoRI
Następnie wytrząsaliśmy przez kilka sekund w wortex'ie, zwirowaliśmy programem „short” (przycisk przytrzymany około 10 sekund) i po tych czynnościach wstawiliśmy probówkę do inkubacji w temperaturze 370C przez 1 godzinę.
II. Analiza elektroforetyczna produktów reakcji trawienia.
Wstęp teoretyczny
Elektroforeza jest powszechnie stosowana w laboratoriach do rozdzielania aminokwasów, białek, fragmentów DNA i RNA. Jest to zjawisko poruszania się naładowanych cząstek koloidalnych - pod działaniem pola elektrycznego - w stosunku do nieruchomego ośrodka rozpraszającego. DNA i RNA mają ładunek ujemny i zgodnie z tym ładunkiem , nadawanym przez grupy fosforanowe , migrują w polu elektrycznym w kierunku anody. Szybkość wędrowania cząsteczek zależy przede wszystkim od ich wielkości, posiadanego ładunku i masy cząsteczkowej, co zezwala na rozdział układów o różnej wielkości i budowie cząsteczek. W naszym ćwiczeniu za podłoże do elektroforetycznego rozdzielenia kwasów nukleinowych stosowaliśmy żel agarozowy.
Do elektroforezy potrzebowaliśmy jeszcze barwnik , który informował nas jak daleko zaszły najszybsze cząsteczki (DNA w żelu nie widać) oraz związek fluoryzujący, który pozwoli nam zlokalizowanie prążków w świetle UV.
Opis wykonywanego ćwiczenia
Przygotowanie saneczek z grzebieniem oraz żelu agarozowego
Na samym początku przygotowaliśmy specjalne saneczki z grzebieniem, do których później była wlewana płynna agaroza. Saneczki należało dobrze uszczelnić poprzez dokładne ich dokręcenie, aby płynna agaroza z nich nie wyciekała. Następnie w dobrze dokręconych saneczkach trzeba było umieścić specjalny grzebień, dzięki któremu w stężonej agarozie zostały utworzone małe zagłębienia, zwane studzienkami. Został on umieszczony blisko jednego z brzegów saneczek.
Kolejnym etapem było przygotowanie żelu agarozowego.
Osoba prowadząca ćwiczenia już wcześniej przygotowała dla nas żel agarozowy z dodatkiem bromku etydyny. Dodanie bromku etydyny umożliwiało nam w późniejszym etapie ćwiczeń obserwacje rozdzielonego DNA dzięki promieniom UV.
Przygotowana wcześniej agaroza była już stężona, dlatego musieliśmy ją rozpuścić. Jeszcze przed rozpuszczeniem na prośbę prowadzącego ćwiczenia musieliśmy dodać trochę sproszkowanego agaru, aby agaroza była sztywniejsza. Teraz mogliśmy przystąpić do rozpuszczania. W tym celu umieściliśmy kolbę ze stężoną agarozą w mikrofalówce. Mikrofalówka była włączana na kilka-kilkanaście sekund, po czym kolbę wyciągaliśmy, mieszaliśmy zawartą w niej agarozę i z powrotem wkładaliśmy do mikrofalówki. W trakcie rozpuszczania dosypaliśmy jeszcze raz trochę sproszkowanego agaru. Robiliśmy to tak długo, aż cała agaroza się uległa roztopieniu.
Następnie rozpuszczoną agarozę zostawiliśmy do ostygnięcia, do temperatury około 50oC, czyli do momentu, aż kolba przestała parzyć w dłonie podczas trzymania jej prze około 1 minutę. Wyższa temperatura mogła uszkodzić saneczki.
Po ostygnięciu wylaliśmy całą agarozę na saneczki z umieszczonym w nich grzebieniem i zostawiliśmy do stężenia.
Przygotowanie prób DNA faga λ
Po godzinnej inkubacji prób DNA faga λ wyciągnęliśmy probówkę z inkubatora i dodaliśmy buforu obciążającego, który składał się z:
0.25% błękit bromofenolowy;
0.25% cjanol ksylenu;
30% glicerol
Bufor obciążający powodował zwiększenie gęstości naszej próbki, co uniemożliwiało jej wypłynięcia podczas elektroforezy. Obecność błękitu bromofenolowego była bardzo ważna, ponieważ umożliwiała nam śledzenie jak daleko zaszły najszybsze cząsteczki, ponieważ samego DNA w żelu nie widać. Następnie probówkę wytrząsaliśmy przez kilka sekund w wytrząsarce i włożyliśmy do wirówki na około 10 sekund (program short).
Dodatkowo jedna z sekcji przygotowała jedną próbę nietrawionego faga λ w składzie:
- 1 μl DNA faga λ
- 5 μl H2O
- 1 μl buforu obciążającego.
Natomiast nasza sekcja zajęła się przygotowaniem 1x stężonego buforu TAE (na TAE składają się takie substancje jak: Tris:octan oraz EDTA) w ilości: 700ml, który ma zdolność przewodzenia prądu. Mieliśmy do dyspozycji flakonik z 50x stężonym buforem TAE dlatego musieliśmy zastosować odpowiednie rozcieńczenie. Aby je uzyskać, musieliśmy do 1 jednostki buforu TAE dodać 49 jednostek H2O dejonizowanej. Jedna jednostka wyniosła 700/50 = 14ml, dlatego do dużego cylindra wlaliśmy 14ml 50x stężonego buforu TAE a następnie 686ml H2O dejonizowanej.
Elektroforeza
Po stężeniu agarozy, ostrożnie wyjęliśmy z niej grzebień, dzięki któremu powstały w niej małe studzienki. Wyjęliśmy saneczki z aparatu uszczelniającego i włożyliśmy je do aparatu do elektroforezy. Wszystko zalaliśmy 700ml wcześniej przygotowanego buforu TAE tak, aby bufor ten zakrywał naszą stężoną agarozę (kilka milimetrów ponad powierzchnią żelu agarozowego). Następnie każda sekcja umieszczała swoje próby DNA faga λ (12μl) za pomocą pipety automatycznej w studzienkach żelu agarozowego. W studzience szóstej umieściliśmy marker, a w siódmej - próbę niestrawionego faga λ. Na marker składają się cząsteczki DNA pocięte enzymami restrykcyjnymi na fragmenty o określonej i znanej masie cząsteczkowej. Ułatwia on ocenę wielkości innych porównywanych cząsteczek, na podstawie porównania szybkości wędrówki podczas elektroforezy. Natomiast obecność niestrawionego DNA faga λ pokazywała nam, że trawienie jest konieczne, aby cząsteczki mogły nam się lepiej rozdzielić, ponieważ cząsteczki niestrawionego DNA są zbyt duże.
Po wypełnieniu wszystkich studzienek próbami DNA przykryliśmy przykrywką urządzenie do elektroforezy, w którym wzdłuż przeciwległych krawędzi agarozy biegną elektrody do których przykłada się napięcie elektryczne. Następnie podłączyliśmy przewody doprowadzające prąd w taki sposób, aby cząsteczki DNA przemieszczały się w stronę dalszego brzegu saneczek. Jak wiadomo DNA ma ładunek ujemny, dlatego migruje on w stronę anody. Podczas przepływu prądu widoczne były liczne pęcherzyki powietrza wydzielające się na elektrodach zanurzonych w buforze TAE.. Napięcie, które przyłożyliśmy wynosiło 110 V. Elektroforezę przeprowadzaliśmy około 1 godziny, po czym wyłączyliśmy urządzenie i przenieśliśmy saneczki z agarozą do urządzenia emitującego promienie UV. Dzięki obecności bromku etydyny mogliśmy obserwować rozdzielone DNA. Jednakże 1 godzina nie wystarczyła na wystarczające rozdzielenie się DNA, dlatego włożyliśmy saneczki z powrotem do urządzenia do elektroforezy na dodatkowe 30 min, po czym ponownie przenieśliśmy je nad światło UV, pod którym widzieliśmy, że DNA było już lepiej rozdzielone.
Wnioski
Na zdjęciu poniżej są wyniki naszej elektroforezy. Prążki ze studzienki nr 2 są rozdzielonymi fragmentami DNA przygotowanymi przez moją sekcję, tzn. sekcję 2. W studzience 6 znajduje się marker, natomiast w studzience 7 niestrawione DNA faga λ.
Niestety, zakres naszej wiedzy na temat odczytywania informacji z prążków powstałych po elektroforezie, jest niewystarczający. Dlatego jesteśmy w stanie wyciągnąć tylko kilka wniosków:
DNA niestrawione znajdujące się w studzience 7 uległo tylko nieznacznemu rozdzieleniu. Powodem tego jest fakt, że w elektroforeza polega na migracji fragmentów DNA - im mniejszy fragment tym szybciej się przesuwa. Niestrawione DNA jest bardzo duże, dlatego bardzo wolno przesuwa się agarozie.
W studzience 6 znajduje się dosyć dobrze ( w porównaniu do reszty) rozdzielony marker. Jak widać, żadnej sekcji DNA nie rozdzieliło się tak dobrze, aby można było je porównywać z markerem.
Niektórym sekcjom ( np. 1, 3, 4 …) DNA nie uległo odpowiedniemu strawieniu lub było go za dużo, ponieważ nie uległo zbyt mocnemu różnicowaniu w żelu.
Niektórym sekcjom ( np. 11, 12 …) DNA zostało zbyt mocno strawione, bądź było go za mało, ponieważ jest słabo widoczne w żelu.
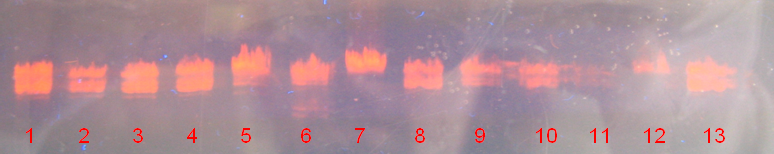
Zdjęcie przedstawiające żel agarozowy z DNA po elektroforezie umieszczony w promieniowaniu UV.
III. Oznaczanie DNA wyizolowanego z komórek na ćwiczeniu poprzednim.
Wstęp teoretyczny
Podczas tego ćwiczenia badaliśmy DNA za pomocą biofotometru firmy eppendorf. Biofotometr jest to przyrząd pomiarowy do szybkiego i pewnego oznaczania ilościowego dsDNA, ssDNA, RNA, oligonukleotydów, protein i bakteryjnej gęstości komórek (pomiary zmętnienia).
My zastosowaliśmy go do sprawdzenia czystości wyizolowanego DNA.
Działanie biofotometru polega na wysyłaniu wiązki światła o określonej długości fali przez próbkę badanej substancji. Każda substancja potrafi absorbować światło o różnej długości fali. Podczas naszego ćwiczenia pomiar absorpcji światła odbywał się dla takich długości fal:
* 230 nm - zanieczyszczenia białkowe - wiązania peptydowe, węglowodory;
* 260 nm - szczyt absorpcji dla DNA;
* 280 nm - szczyt absorpcji dla białek - aminokwasy aromatyczne, fenole;
* 320 nm - wytrącenia w roztworze, brud.
Opis wykonywanego ćwiczenia
Przygotowanie próbki do badania w biofotometrze
Na naszych poprzednich ćwiczeniach wyizolowaliśmy DNA z ludzkich komórek. Wyizolowane DNA zostało dla nas przechowane w zamrażarce. Teraz należało je odpowiednio przygotować, aby można je było zbadać w biofotometrze. Na samym początku rozmroziliśmy ją w rękach, zmierzyliśmy jej objętość która wyniosła 49.5μl, zwortexowaliśmy przez kilka sekund oraz wirowaliśmy 10 sekund programem short. Następnie należało uzyskać rozcieńczenie 50x, czyli do 98μl dejonizowanej H2O należało dodać 2μl naszego DNA, po tym etapie próbkę również zmieszaliśmy w wytrząsarce i wirowaliśmy kilka sekund.. Oprócz tego, należało jeszcze przygotować próbę kontrolną czyli 100μl wody dejonizowanej.
Badanie próbki DNA w biofotometrze
Na samym początku na dno sterylnej kuwetki wprowadziliśmy 100μl H2O dejonizowanej w celu wykalibrowania urządzenia. Kuwetkę chwytaliśmy tylko i wyłącznie za matowe ścianki, żeby swoimi odciskami palców nie zakłócić przepływu światła co spowodowało by błędne wyniki. Wartość absorpcji dla czystej wody dejonizowanej wyszła nam 0.00, dlatego mogliśmy przejść do badania naszej próbki DNA. Za pomocą pipety automatycznej wylaliśmy wodę z kuwetki i wprowadziliśmy naszą próbkę DNA. Kuwetkę włożyliśmy do urządzenia i zmierzyliśmy absorpcję światła dla naszego DNA. Następnie przepisaliśmy wyświetlone przez biofotometr informacje do zeszytu. Kuwetkę wyjęliśmy, wylaliśmy próbkę DNA i przepłukaliśmy kuwetkę wodą dejonizowaną. Do ćwiczenia mogła przystąpić kolejna grupa.
Wyniki badania oraz obliczenia
W badaniu biofotometrem uzyskaliśmy takie oto wyniki:
Długość światła |
Wynik badania |
|
|
A 230 |
0,174 |
A 260 |
0,198 |
A 280 |
0,135 |
A 320 |
0,061 |
Otrzymaliśmy również takie informacje, jak stosunek A260/A280 oraz A260/A230
A 260 / A 280 |
1,47 |
A 260 / A 230 |
1,14 |
Następnie za pomocą odpowiedniego wzoru obliczyliśmy stężenie DNA w naszej próbce:
C = (A260 -A320) * rozcieńczenie * 0.05 [μg/μl]
C = ( 0,198 - 0,061 ) * 50 * 0,05
C = 0,34 μg/μl
Zmierzyliśmy, że objętość naszej próbki DNA wynosiła 49,5μl. Dlatego całkowita ilość DNA w próbce z wyizolowanym przez nas na poprzednim ćwiczeniu DNA wynosi:
X = C * 49,5
X= 16,95 μg
Wnioski
Miarą czystości DNA po jego izolacji jest stosunek A260/A280, który powinien wynosić 1.5-1. --> 9[Author:S] , oraz stosunek A260/A230, który powinien być wyższy niż 2. Stosunek A260/A280 informuje nas o zanieczyszczeniach białek i RNA, jeśli wynosi on poniżej 1,5 mamy do czynienia z zanieczyszczeniem białkami, a jeśli powyżej 1,9 - zanieczyszczenie RNA. Nasz wynik dla A260/A280 wynosi 1.47 co oznacza, iż nasza próbka jest zanieczyszczona białkami. Stosunek A260/A230 wyniósł u nas 1.14 i jest on dużo niższy niż 2, co potwierdza, że nasze DNA jest zanieczyszczone.
Powodem tego mogło być złe oddzielenie warstwy wodnej od warstw białek i fenolu podczas naszych ostatnich ćwiczeń.
Ile wynosi maksimum dla DNA?
Wyszukiwarka
Podobne podstrony:
Plan ćwiczeń II semestr I rok 2008, Semestr II, biologia MO, Laboratorium 1
Laboratorium 6 - Instrukcja - PCR, Semestr II, biologia MO, Laboratorium 6
Kolokwium - zagadnienia, Semestr II, biologia MO, Laboratorium 5
Laboratorium 2 - Instrukcja - Obserwacja komorek zwierzecych w mikroskopie swietlnym, Semestr II, bi
Laboratorium 4 - Instrukcja - Trawienie DNA faga za pomoca enzymu restrykcyjnego EcoRI, Semestr II,
Laboratorium 3 - Instrukcja - Izolacja DNA z hodowanych komórek eukariotycznych, Semestr II, biologi
Wzór sprawozdania - chemog dzienne lab, Inżynieria środowiska, inż, Semestr II, Chemia ogólna, labor
sprawozdanie chemia michał, Budownictwo UZ semestr I , II, Chemia budowlana, Sprawozdania od Seweryn
Ćwiczenie nr 35, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwicz
Ćwiczenie nr 44, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwicz
Ćwiczenie nr 50a, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwic
Ćwiczenie nr 33a, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwi
ZASADY ZALICZANIA ĆWICZEŃ AUD. z ekologii 2012-13, Semestr II, Ekologia, Ćwiczenia audytoryjne
Cwiczenie nr 83, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwicz
Ćwiczenie nr 72c, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwic
Ćwiczenie nr 13, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwicz
Ćwiczenie nr 34a, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwic
Ćwiczenie nr 34b, studia, Budownctwo, Semestr II, fizyka, Fizyka laborki, Fizyka - Labolatoria, Ćwic
więcej podobnych podstron