Spis treści
Ćwiczenie |
Temat |
Strona |
1 |
Organizacja zajęć. Przepisy BHP w pracowni chemicznej. |
2 |
2 |
Podstawowy sprzęt laboratoryjny. Wagi i ważenie. |
2 |
3 |
Stężenia procentowe, molowe; przeliczanie stężeń - zadania. |
4 |
4 |
Przygotowywanie roztworów o określonym stężeniu procentowym i molowym. |
4 |
5 |
Określenie i pomiar odczynu roztworu o nieznanym pH. |
7 |
6 |
Wyznaczanie pojemności buforowej. |
13 |
7 |
Nastawianie miana roztworu NaOH na 0,1 M roztwór HCl. |
17 |
8 |
Nastawianie miana roztworu KMnO4 na Na2C2O4. |
20 |
9 |
Oznaczanie jonów tiocyjanianowych metodą strąceniową |
24 |
10 |
Wykrywanie jonów. |
27 |
11 |
Oddzielanie kationu od anionu za pomocą kationitu. |
31 |
12 |
Oznaczanie żelaza metodą fotokolorymetryczną. |
33 |
13 |
Spektrofotometryczne oznaczanie stężeń barwników w mieszaninie. |
33 |
14 |
Chromatografia bibułowa. |
40 |
15 |
Uzupełnienie i zaliczenie ćwiczeń. |
|
ĆWICZENIE 1
Organizacja zajęć. Przepisy BHP w pracowni chemicznej.
Regulamin porządkowy obowiązujący w laboratorium.
Zajęcia w laboratorium powinny odbywać się jedynie pod nadzorem pracownika uczelni.
W czasie wykonywania ćwiczeń studenci powinni ściśle podporządkować się poleceniom prowadzącego ćwiczenia.
Prowadzący zajęcia w laboratorium ma obowiązek zapoznania studentów z zasadami bezpieczeństwa i higieny pracy oraz ochrony przeciwpożarowej.
Student może przystąpić do wykonywania ćwiczeń jeśli zaliczy wiadomości z zakresu BHP.
Podczas pracy w laboratorium studenci powinni być ubrani w fartuchy laboratoryjne.
Studenci mają obowiązek uporządkowania stanowiska pracy po zakończeniu ćwiczeń.
W przypadku uszkodzenia sprzętu i aparatury studenci powinni powiadomić o tym prowadzącego ćwiczenia.
Palenie tytoniu oraz spożywanie posiłków w laboratorium jest bezwzględnie zabronione.
W razie zaistnienia wypadku prowadzący zajęcia ma obowiązek udzielić pierwszej pomocy, zabezpieczyć miejsce wypadku oraz powiadomić kierownika zakładu i służby BHP.
W razie potrzeby wezwać pogotowie ratunkowe.
ĆWICZENIE 2.
Podstawowy sprzęt laboratoryjny. Wagi i ważenie.
Zakres teoretyczny:
Sprzęt metalowy, drewniany, porcelanowy, gumowy i z tworzywa sztucznego.
Sprzęt szklany.
Naczynia miarowe i ich użytkowanie.
Rodzaje wag i ich charakterystyka.
Zasady poprawnego ważenia na wadze technicznej i analitycznej.
Ze sprzętem labolatoryjnym (wyglądem, budową i przeznaczeniem) studenci zapoznają się na zajęciach.
Wagi i ważenie.
Ważenie polega na określaniu masy określonej substancji. Do tego celu używa się różnego rodzaju wag. Wielkościami charakteryzującymi wagę są: nośność wagi oraz jej czułość bezwzględna i względna.
Nośność wagi jest to największe dopuszczalne jej obciążenie, które nie spowoduje odkształcenia belki i zniszczenia tego urządzenia.
Czułość bezwzględna jest to najmniejsza masa, która umieszczona na szalce spowoduje wychylenie się wskazówki z położenia zero.
Czułość względna jest to stosunek czułości bezwzględnej do całkowitego obciążenia wagi w danym momencie.
Podział wag w zależności od czułości
Zwyczajne 5-10kg - czułość 1g.
Techniczne (laboratoryjne) 100g-1kg - czułość 10mg.
Analityczne do 200g - czułość 0,1mg.
Mikroanalityczne 1-3g czułość 0,001mg.
Zasady prawidłowego ważenia.
Waga powinna znajdować się w miejscu, które zapewnia jej ochronę przed niszczącym działaniem atmosfery laboratoryjnej. W celu zabezpieczania wagi przed wstrząsami ustawia się ją na stabilnym, dobrze wypoziomowanym stole.
Nie wolno wagi obciążać powyżej wskazanej nośności wagi.
Przed użyciem wagi należy koniecznie ją „wyzerować” - ustalić punkt zerowy.
Podczas odczytywania wahań wagi drzwiczki powinny być całkowicie zamknięte w celu uniknięcia zakłóceń spowodowanych prądami powietrza.
Nie wolno kłaść niczego na szalkach dopóki waga nie jest wyłączona (zaaretowana). Zarówno przedmiot ważony jak i odważniki należy umieszczać na środku szalek. Na szalkach nie wolno umieszczać przedmiotów gorących, zbyt zimnych i mokrych.
Substancji ważonych nie wolno umieszczać bezpośrednio na szalkach. Do tego celu należy używać naczyń wagowych, tygielków lub szkiełek zegarkowych.
Zaaretowywanie i odaretowywanie wagi należy robić ostrożnie, płynnym ruchem.
Nakładanie i zdejmowanie z szalek przedmiotów ważonych i odważników należy robić przy zaaretowanej wadze. Odważniki należy nakładać i zdejmować z szalek za pomocą szczypczyków.
Po zakończeniu ważenia należy sprawdzić czy waga jest unieruchomiona, odważniki zdjęte z szalek i drzwiczki zamknięte.
ĆWICZENIE 3. i 4.
Stężenia procentowe, molowe; przeliczanie stężeń - zadania.
Zakres teoretyczny:
Pojęcie mola, masy molowej, gęstości.
Definicja pojęcia stężenia procentowego i molowego.
Przygotowywanie roztworów o określonym stężeniu procentowym i molowym.
Zakres teoretyczny:
Technika przygotowywania roztworów o stężeniu procentowym i molowym.
Pomiar gęstości roztworu przy pomocy aerometru.
Mol
Jest jednostką liczebności substancji (liczba cząstek) równa liczbie atomów zawartych w masie 0,012 kg węgla-12.
Masa molowa
Wielkość fizyczna wyrażona w gramach; jest równa liczbowo masie atomowej lub cząsteczkowej.
Gęstość
Gęstość roztworu to stosunek masy tego roztworu do jego objętości - ilość gramów danej substancji w 1 ml roztworu
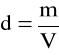
[g/ml]
Stężenie roztworu określa zawartość substancji w jednostce masy lub jednostce objętości. Najważniejsze są dwa rodzaje stężeń: stężenie procentowe i stężenie molowe.
Stężenia procentowe.
Najczęściej określa się stężenie w procentach wagowych (% m/m), jest to liczba gramów danej substancji zawarta w 100 gramach roztworu.
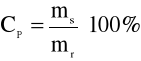
CP - stężenie procentowe [%]
ms - masa substancji [g]
mr - masa roztworu [g]
100% - mnożnik
Masa roztworu (mr) jest sumą masy substancji (ms) i masy rozpuszczalnika (mrozp):
![]()
![]()
Czasami wyraża się również stężenie w procentach objętościowych (% V/V), który mówi o liczbie części objętościowych substancji w 10 częściach objętościowych roztworu. Stężenie można również określić w procentach wagowo-objętościowych (% m/V), które określają liczbę gramów substancji rozpuszczonych w 100 ml roztworu.
Stężenie molowe
Stężenie molowe (Cm) jest to stosunek liczby moli (n) substancji rozpuszczonej do objętości roztworu (Vr). Roztworem 1 molowym nazywamy roztwór zawierający 1 mol danej substancji w 1 litrze roztworu [mol/l].
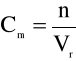
[mol/l]
Cm - stężenie molowe [mol/l]
n - liczba moli [mol]
Vr - objętość roztworu [l].
Zadanie1.
Oblicz stężenie molowe 96% H2SO4 o gęstości d = 1,84 g/ml.
Zadanie2.
Oblicz stężenie procentowe 0,5 - molowego roztworu siarczku sodu (Na2S). Gęstość roztworu d = 1,04 g/ml.
Ćwiczenie: Przygotowanie roztworu o określonym stężeniu procentowym
Sporządzić 350 g 2% roztworu KCl.
W tym celu należy odważyć w naczyniu szklanym na wadze analitycznej obliczoną ilość substancji. W cylindrze miarowym odmierzyć odpowiednią ilość wody destylowanej (gęstość wody przyjąć równą 1 g/cm3).
Naważkę przenieść ilościowo do zlewki o pojemności 500 ml i rozpuścić w przygotowanej wodzie mieszając roztwór bagietką. Następnie zmierzyć gęstość otrzymanego roztworu za pomocą areometru w cylindrze o pojemności 250 ml. Obliczyć stężenie molowe otrzymanego roztworu.
Ćwiczenie: Przygotowanie roztworu o określonym stężeniu molowym
Sporządzić 250 ml 0,1 mol/l roztworu zasady sodowej (NaOH) metodą rozcieńczenia roztworu o wyższym stężeniu.
W tym celu należy za pomocą pipety odmierzyć odpowiednią (obliczoną wcześniej) objętość 1 mol/l NaOH, przenieść do kolby miarowej o pojemności 250 ml, a następnie rozcieńczyć wodą destylowaną do żądanej objętości 250 ml (do kreski). Obliczyć stężenie procentowe otrzymanego roztworu. d = 1,003 g/cm3.
ĆWICZENIE 5.
Określenie i pomiar odczynu roztworu o nieznanym pH.
Zakres teoretyczny:
Zjawisko dysocjacji; stała i stopień dysocjacji.
Autodysocjacja wody; iloczyn jonowy wody; wskaźnik pH.
Indykatory odczynu roztworu.
pH-metr: budowa i zasada działania.
Obliczenia.
Stała i stopień dysocjacji.
Substancje chemiczne można podzielić na elektrolity i nieelektrolity. Elektrolity to substancje, które po rozpuszczeniu lub roztopieniu ulegają dysocjacji elektrolitycznej (rozpadowi na jony). Elektrolitami są kwasy, zasady i sole, natomiast do nieelektrolitów należy większość substancji organicznych (alkohole, etery, ketony). Cechą charakterystyczną roztworów elektrolitów (w przeciwieństwie do nieelektrolitów) jest przewodzenie prądu elektrycznego.
Zjawisko rozpadu wodnych roztworów elektrolitów na jony zaobserwował po raz pierwszy Svante Arrhenius. Podał on w latach 1885-87 tzw. teorię dysocjacji elektrolitycznej, czyli rozpadu elektrolitów na jony.
Jony są to atomy lub grupy atomów obdarzone ładunkiem elektrycznym, kationy - ładunkiem dodatnim, aniony - ujemnym. Dysocjacja elektrolityczna, jest procesem odwracalnym, przebiegającym całkowicie dopiero w dużym rozcieńczeniu.
Stopniem dysocjacji (oznaczany literą α) nazywamy stosunek liczby cząsteczek zdysocjowanych do ogólnej liczby cząsteczek rozpuszczonych danej substancji (przed dysocjacją).
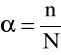
lub w procentach 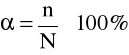
Stopień dysocjacji jest liczbą ułamkową (α<1), która określa, jaka część ogólnej liczby rozpuszczonych cząsteczek elektrolitu uległa rozpadowi na jony. Im silniejsza dysocjacja tym wartość stopnia dysocjacji jest bliższy jedności. Stopień dysocjacji elektrolitu zależy od:
- natury substancji rozpuszczonej,
- jej stężenia,
- natury rozpuszczalnika
- temperatury
Cząsteczki elektrolitu słabego (przykładowo słabego kwasu) ulegają w roztworze wodnym częściowej dysocjacji na jony według równania:
HA ![]()
H+ + A-
Reakcja dysocjacji jest odwracalna - obok dysocjacji zachodzi proces odwrotny zwany rekombinacją lub molaryzacją. W określonej temperaturze między cząsteczkami a odpowiadającymi im jonami ustala się natychmiast stan równowagi, tzn. liczba cząsteczek rozpadających się w jednostce czasu jest równa liczbie cząsteczek powstających w wyniku łączenia się jonów.
Zgodnie z prawem działania mas, stała równowagi tej reakcji, zwana stałą dysocjacji słabego kwasu, wyraża się wzorem:
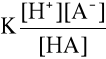
gdzie nawiasy kwadratowe oznaczają stężenie poszczególnych rodzajów cząsteczek (lub jonów) wyrażone w molach na litr roztworu.
Stała dysocjacji (inaczej stała jonizacji) jest to więc stosunek iloczynu stężeń jonów, na które rozpadł się elektrolit, do stężenia cząstek niezdysocjowanych. Wielkość ta określa moc kwasu lub zasady. Ponieważ stała dysocjacji nie zależy od stężenia, lecz wyłącznie od temperatury i rodzaju rozpuszczalnika, charakteryzuje elektrolit znacznie lepiej niż stopień dysocjacji,
W przypadku elektrolitów mocnych zamiast rzeczywistych stężeń molowych poszczególnych jonów należy używać tzw. aktywności (inaczej stężeń pozornych, aktywnych stężeń) jonów, które stanowią wypadkową różnych wpływów, jakim ulegają jony w roztworze (przyciąganie się jonów różnoimiennych, wpływ niepełnej dysocjacji cząsteczek, wpływ hydratacji i inne).
Zależność między stężeniem a aktywnością wyraża wzór:
![]()
a - aktywność
C - stężenie
f - współczynnik aktywności określający jaka część molowego stężenia stanowi stężenie aktywne.
Zatem dla elektrolitów mocnych (w których nie możemy pominąć oddziaływań między jonami) wzór na stałą dysocjacji (1) przyjmuje postać:
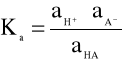
a wartość Ka nazywana jest termodynamiczną stałą dysocjacji.
Aktywności powinny być stosowane we wszystkich dokładnych obliczeniach we wzorach opartych na prawie działania mas. Jednakże dla elektrolitów słabych i roztworów silnie rozcieńczonych można z powodzeniem stosować wzory oparte na stężeniach.
Iloczyn jonowy wody. Pojęcie pH.
Proces dysocjacji wody (autodysocjacji) polega na tym, że dipolowa cząsteczka wody, oddziałując swym ujemnym, tlenowym biegunem na jedno z jąder wodorowych innej cząsteczki wody może spowodować jej rozpad. Woda jest bardzo słabym elektrolitem, który tylko w nieznacznym stopniu ulega rozpadowi jony:
H2O ![]()
H+ + OH-
Dla uproszczenia pomijamy powstawanie jonów hydroniowych H3O+, dlatego w dalszych rozważaniach, dla uproszczenia, będziemy się posługiwali symbolem H+.
Stałą dysocjacji wody zgodnie z prawem działania mas, jak dla każdej reakcji odwracalnej, można wyrazić wzorem:
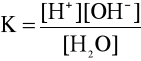
Wielkość ta w temperaturze 22°C wynosi 1,8 ⋅ 10-16.
Ze względu na znikomo mały stopień dysocjacji cząsteczek wody, stężenie wody [H2O], może być uważane za wielkość stałą. Mnożąc obie strony równania przez [H2O], otrzymujemy:
Kw = [H+] [OH-]
Wielkość Kw, czyli iloczyn stężeń jonów H+ i OH- nosi nazwę iloczynu jonowego wody i w danej temperaturze wielkością stałą. Określa ona zależność pomiędzy stężeniami tych jonów niezależnie skąd te jony pochodzą (z samej wody, czy też z kwasu lub zasady do niej dodanej). Ponieważ Kw jest wielkością stałą, to przy zwiększaniu stężenia jonów H+ tyle samo razy musi zmaleć stężenie jonów OH-.
Ponieważ stężenie wody wynosi 55,5 mol/l (1000g/Mw = 1000g/18g/mol), więc:
Kw = K[H2O] = [H+][OH-] = 1,8 ⋅ 10-16 ⋅ 55,5 = 1 ⋅ 10-14
Wzrost temperatury zwiększa wartość Kw ponieważ wraz z temperaturą zwiększa się stopień dysocjacji wody.
W czystej wodzie stężenia jonów wodorowych i wodorotlenowych są sobie równe:
[H+] = [OH-] czyli [H+]2 = 1 ⋅ 10-14
Stężenie jonów wodorowych i wodorotlenowych w czystej wodzie, w temperaturze pokojowej wynosi zatem:
[H+] = [OH-] = ![]()
= 10-7 mol/l
Taki roztwór, w którym [H+] = [OH-] ma odczyn obojętny.
Odczyn środowiska oznaczamy zwykle w sposób wprowadzony przez Sőrensena (1909). Stężenie jonów zastępujemy wykładnikiem stężenia jonów wodorowych - pH, czyli ujemnym logarytmem dziesiętnym stężenia jonów wodorowych.
[H+] = 10-pH
po zlogarytmowaniu:
pH = -lg[H+]
W temperaturze 22°C pH odczynu obojętnego wynosi 7, roztwory kwasowe mają
0 <pH < 7, a roztwory alkaliczne 7 < pH < 14. Możliwe jest uzyskanie roztworów stężonych kwasów, których pH < 0 jak i roztworów alkalicznych o pH > 14 lecz do celów analitycznych nie mają one praktycznego znaczenia.
Metody badania wartości odczynu roztworu.
Wskaźniki pH.
Są to substancje, które wykazują właściwość zmiany barwy w zależności od pH roztworu. Możemy ich używać do określenia odczynu roztworu, a także w alkacymetrii (dziale analizy miareczkowej) do uchwycenia punktu nasycenia równoważnikowego miareczkowania (punktu zobojętnienia roztworu). Wskaźniki pH to grupa związków organicznych, o charakterze słabych kwasów lub słabych zasad, których jony są inaczej zabarwione niż cząsteczki niezdysocjowane. Barwa roztworu zależy od stosunku stężeń obu form wskaźnika. Każdy wskaźnik ma charakterystyczną wartość pH, przy której następuje zmiana jego zabarwienia. Jest to wykładnik wskaźnika (pKInH). W praktyce zmiana barwy zachodzi w granicach ok. 2 jednostek pH ( ten zakres zmiany barwy nazywamy zakresem wskaźnikowym danego wskaźnika).
Rozróżnia się wskaźniki jedno-, dwu- i wielobarwne w zależności od ilości form wykazujących zabarwienie. Wskaźniki jedno- i dwubarwne stosuje się same bądź w mieszaninach z barwnikiem obojętnym, na którego tle zmiana zabarwienia właściwego barwnika jest wyraźniejsza. Duże znaczenie praktyczne mają wskaźniki uniwersalne - mieszaniny różnych wskaźników pH, których barwa zmienia się w szerokim zakresie pH, przechodząc kolejno przez różne zabarwienia.
Wskaźniki mogą występować w postaci roztworów wodnych bądź alkoholowych dodawanych do roztworu badanego, lub w postaci tzw. papierków wskaźnikowych (pasków bibuły nasączonych roztworem wskaźnika i wysuszonych), na które nakrapla się badany roztwór i obserwuje zmianę barwy papierka.
Pomiar pH za pomocą pH-metru.
Pomiaru pH można również dokonać wykorzystując zależność potencjału elektrody (wodorowej, chinhydronowej, antymonowej lub szklanej) od stężenia jonów wodorowych w roztworze w jakim się znajduje. Ze wszystkich stosowanych obecnie metod badania pH, metody potencjometryczne są najdokładniejsze.
W urządzeniach zwanych pH-metrami stosowana jest elektroda szklana (rurka szklana zakończona cienkościenną banieczką ze szkła elektrodowego). We wnętrzu rurki i banieczki znajduje się elektrolit o stałym stężeniu (najczęściej 0,1 mol/l HCl) oraz elektroda wyprowadzająca (najczęściej chlorosrebrowa Ag/AgCl), która zachowuje w środowisku o niezmiennym stężeniu jonów Cl- stały potencjał.
Na granicy zetknięcia się membrany szklanej z roztworem zawierającym jony wodorowe następuje wymiana tych jonów z niektórymi jonami sieci szkła jak i też włączanie się tych jonów w puste miejsca w tej sieci. Po obu stronach membrany powstaje wtedy potencjał zależny od stężenia jonów H+ w roztworze, a między roztworami: z wnętrza elektrody i badanym, tworzy się różnica potencjałów, która ze względu na stałość pH roztworu elektrodowego jest funkcją jedynie stężenia jonów wodorowych roztworu badanego. Pomiar pH polega więc na porównaniu potencjału elektrody szklanej w roztworze badanym z potencjałem tej elektrody we wzorcowym roztworze buforowym o znanej wartości pH.
Należy jeszcze zwrócić uwagę, że wartość pH mierzona potencjometrycznie odzwierciedla aktywność jonów wodorowych w roztworze a nie ich stężenie (właściwie dla oznaczenia pomiaru za pomocą pH-metru powinno się używać symbolu paH zamiast pH).
Ćwiczenie: Określenie i pomiar odczynu roztworów o nieznanym pH
Przy pomocy dostępnych wskaźników określić odczyn badanych roztworów.
I. W badanym roztworze zanurzyć szklaną bagietkę lub koniec pipety i przenieść 1 kroplę na papierek wskaźnikowy. Po ok. 15 sek z załączonej skali barw odczytać pH roztworu.
II. Do dwóch szeregów probówek (po 5) przenieść po ok. 5 ml badanych roztworów i dodać 1-2 krople odpowiedniego indykatora. Określić pH roztworu na podstawie podanych zakresów zmian barwy wskaźników.
III. Dokonać pomiaru pH za pomocą pH-metru.
Wskaźniki pH i zakresy zmian ich barwy.
1. Zieleń bromokrezolowa pH < 3,8 barwa żółta
3,8 < pH < 5,4 barwa zielona
pH > 5,4 barwa niebieska
2. Oranż metylowy pH < 3,1 barwa czerwona
3,1 < pH < 4,4 barwa pomarańczowa
pH > 4,4 barwa żółta
3. Fenoloftaleina pH < 8,2 bezbarwna
8,2 < pH < 10,0 barwa różowa
pH > 10,0 barwa malinowa (purpurowo-czerwona)
4. Czerwień metylowa pH < 4,2 barwa czerwona
4,2 < pH < 6,2 barwa pomarańczowa
pH > 6,2 barwa żółta
5. Błękit tymolowy pH < 1,2 barwa czerwona
1,2 < pH < 2,8 barwa pomarańczowa
2.8 < pH < 8,0 barwa żółta
8,0 < pH < 9,6 barwa zielona
pH > 9,6 barwa niebieska
Obserwacje zapisz w tabeli:
wskaźniki |
roztwór I |
roztwór II |
||
|
obserwacje |
wnioski |
obserwacje |
wnioski |
|
|
|
|
|
ĆWICZENIE 6.
Wyznaczanie pojemności buforowej.
Zakres teoretyczny:
Zjawisko hydrolizy; stała i stopień hydrolizy.
Roztwory buforowe; pH buforu, rodzaje buforów i sposób ich przygotowania oraz zastosowanie.
Obliczenia.
Roztwory buforowe.
Roztwory buforowe są to roztwory, które wykazują dużą stałość wartości wykładnika wodorowego pH pomimo rozcieńczenia wodą, jak również pod wpływem wprowadzenia do nich niewielkich ilości mocnych kwasów lub zasad, a więc utrzymują pH roztworu na określonym, stałym poziomie.
Najprostsze roztwory buforowe są to mieszaniny roztworów słabego kwasu i jego soli utworzonej z mocnej zasady (np. CH3COOH i CH3COONa) lub odwrotnie, słabej zasady i jej soli utworzonej z mocnego kwasu (np. NH3 . H2O i NH4Cl).
Dla słabego kwasu i jego soli (kwas jest słabo dysocjowany):
HA + H2O ![]()
H3O+ + A- (1)
i jego stała wyraża się wzorem:
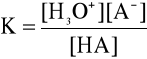
Obecność dobrze zdysocjowanej soli tego kwasu powoduje zmniejszenie jego dysocjacji ze względu na zwiększenie stężenia anionu. Ponieważ kwas jest zdysocjowany w bardzo małym stopniu, w przybliżeniu możemy przyjąć, że stężenie niezdysocjowanych cząsteczek kwasu [HA] będzie się praktycznie równało całkowitemu stężeniu kwasu Ck, a stężenie jonów słabego kwasu [A-] stężeniu jego soli Cs
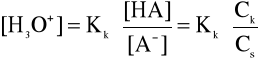
(2)
Jest to równanie Hendersona - Hasselbacha
Po zlogarytmowaniu równania otrzymujemy wyrażenie:
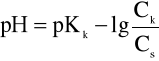
![]()
(3)
Jeżeli do omawianego roztworu dodamy mocnego kwasu, to wprowadzone jony H3O+ będą się łączyły z anionem A- w nie dysocjowane cząsteczki kwasu HA (odwrócenie reakcji (1)). Dlatego nieznacznie tylko zwiększy się stosunek stężeń [HA] : [A-] i początkowe pH roztworu praktycznie się nie zmieni. W przypadku wprowadzania do roztworu mocnej zasady, jony OH- przereagują z nie dysocjowanymi cząsteczkami kwasu.
Jeśli bufor jest mieszaniną słabej zasady i jej soli z mocnym kwasem, stężenie jonów wodorowych oblicza się podobnie:
B + H2O ![]()
BH+ + OH-
stała dysocjacji słabej zasady B wyraża się wzorem:
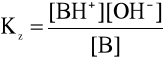
po podstawieniu [OH] wyrażeniem wynikającym z iloczynu jonowego wody otrzymujemy:
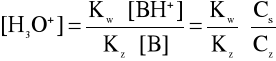
(4)
Po przeprowadzeniu przekształceń otrzymujemy ostateczne wzór na pH takiego buforu:
pH = pKw - pKz -lgCs + lgCz (5)
Ze wzorów (2) i (4) wynika, że stężenie jonów wodorowych (czyli również i pH) roztworu buforowego składającego się ze słabego kwasu i jego soli zależy od stałej dysocjacji kwasu HA oraz od stosunku stężeń kwasu HA i sprzężonej z nim zasady A-, natomiast dla buforu złożonego ze słabej zasady i jej soli z mocnym kwasem pH jest funkcją stosunku stężeń słabej zasady (np. NH3) i sprzężonego z nią kwasu (np. NH4).
Zgodnie z teorią Brőnsteda do obliczania pH roztworów buforowych obydwu omówionych typów wystarcza jeden wspólny wzór:
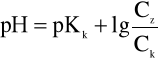
ponieważ w obu przypadkach mamy do czynienia ze sprzężonymi układami kwasu i zasady.
Otrzymane równania umożliwiają obliczenie pH buforów o dowolnym składzie, posługujemy się nimi gdy chcemy przygotować roztwory o określonym stężeniu jonów wodorowych.
Rozcieńczenie roztworu buforowego nie wpływa na wielkość pH. Jeżeli chodzi o temperaturę to pH roztworu buforowego zawierającego słaby kwas i jego sól nie zależy od temperatury, natomiast w roztworach mieszanin słabych zasad i ich soli pH zmienia się wraz z temperaturą, ponieważ temperatura wpływa na wartość Kw.
Roztwory buforowe odgrywają szczególnie ważną rolę w przemyśle farmaceutycznym i w biochemii, gdzie często przeprowadzane procesy wymagają stałości pH środowiska. Często używa się ich również w procesach technologicznych oraz w chemii analitycznej (przy rozdzielaniu kationów i anionów).
Najczęściej używane bufory:
- bufor fosforanowy (NaH2PO4 i Na2HPO4)
- bufor amoniakalny (NH4+ i NH3)
- bufor octanowy (CH3COOH i CH3COONa)
- bufor węglanowy (H2CO3 i NaHCO3).
Pojemność buforowa.
Pojemność buforowa (oznaczana symbolem β) to zdolność buforowania roztworu. Jest to liczba moli mocnej zasady lub kwasu, która musi być dodana do 1 litra roztworu, aby spowodować zmianę jego pH o jednostkę.
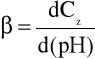
lub w uproszczeniu 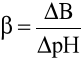
- dCz - nieskończenie mały przyrost stężenia zasady,
- ΔB - dodana ilość mocnej zasady (w molach na litr),
- d(pH) i ΔpH - odpowiednio nieskończenie mały lub skończony przyrost pH
Jeśli do roztworu wprowadzano mocny kwas, we wzorze należy umieścić znak „-” (minus).
W miarę zwiększania ilości dodawanego kwasu lub zasady pojemność buforowa zmniejsza się i staje się równa zeru, gdy cała zawarta w buforze sól zamieni się w słaby kwas, lub też cały kwas zostanie przeprowadzony w sól.
Największą pojemność buforową mają roztwory, w których stosunek stężeń soli i kwasu (ewentualnie zasady) jest równy jedności. Dla takich roztworów pH = pKk lub pH =pKz.
Praktyczny zakres stosowania buforu jest ograniczony w przybliżeniu do wartości pH w granicach pK ± 1. Pojemność buforowa zależy również od stężenia roztworów buforowych, ich rozcieńczanie powoduje zmniejszenie pojemności buforowej, choć nie zmienia ich pH. Dla danego stężenia C roztworu maksymalna wartość pojemności buforowej (przy pH = pKk) wyraża się uproszczonym wzorem
β = 0.58 × C
Jeśli chcemy buforować roztwór, którego pH znacznie odbiega od żądanej wartości to należy doprowadzić go do przybliżonej wartości pH, dodając kwasu lub zasady i dopiero potem dodać buforu.
Ćwiczenie: Wyznaczanie pojemności buforowej
Do zlewki o pojemności 100 ml odmierzyć za pomocą cylindra miarowego 50 ml buforu. Taką samą ilość wody destylowanej odmierzyć do drugiej zlewki o pojemności 100 ml. Zmierzyć pH buforu i wody destylowanej pH-metrem.
Następnie do obu zlewek dodać po 2,5 ml. 0,1 mol/l roztworu HCl. Roztwory wymieszać i ponownie zmierzyć ich pH.
Na podstawie wyników pomiarów i innych obserwacji:
Obliczyć pojemność buforową badanego buforu i wody destylowanej wg. wzoru:
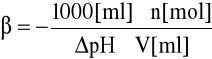
gdzie:
n - ilość moli mocnego kwasu dodanego do roztworu
V - objętość buforu lub wody (ml)
ΔpH - obserwowana zmiana wartości pH roztworu (wartość ujemna)
1000 - przelicznik objętości roztworu na litry
Określić charakter badanego buforu i podać jego nazwę.
ĆWICZENIE 7.
Nastawianie miana roztworu NaOH na 0,1 M roztwór HCl.
Zakres teoretyczny:
Podstawy analizy miareczkowej:
klasyfikacja metod miareczkowych,
PR i PK miareczkowania, wskaźniki PK,
roztwory mianowane.
Technika analizy miareczkowej.
Alkacymetria.
Analiza miareczkowa
Analiza miareczkowa polega na tym, że do roztworu oznaczanego wprowadza się porcjami (miareczkami) równoważną chemicznie ilość roztworu mianowanego (o znanym stężeniu). Stężenie (lub miano) oznaczanej substancji oblicza się na podstawie zmierzonej dokładnie objętości zużytego roztworu o znanym stężeniu (lub mianie).
Do rozpoznania momentu, w którym ilość wprowadzonego mianowanego roztworu zrównoważyła ilość składnika oznaczanego służy punkt równoważnikowy lub punkt nasycenia równoważnikowego (PR). Żeby móc wzrokowo zaobserwować punkt równoważnikowy do roztworu miareczkowanego wprowadza się wskaźnik (indykator), który zmienia barwę w chwili zakończenia reakcji między roztworem mianowanym, a roztworem miareczkowanym. Moment, w którym indykator zmienia swoje zabarwienie nazywa się punktem końcowym miareczkowania (PK).
Zasadniczo PK i PR powinny się pokrywać. W praktyce jednak punkt końcowy miareczkowania następuje tuż przed lub tuż po punkcie równoważnikowym. Różnica pomiędzy tymi punktami nazywa się błędem miareczkowania.
Zmiany zachodzące podczas miareczkowania można przedstawić graficznie. Na osi odciętych oznacza się ilość zużytego roztworu mianowanego (w mililitrach bądź w procentach całkowitej ilości potrzebnej do nasycenia równoważnikowego), na osi rzędnych wartości jakiegoś parametru związanego ze stężeniem oznaczanego składnika. Takim parametrem może być pH, potencjał red-oks, przewodność elektrolityczna itp. Otrzymamy wykres, który nosi nazwę krzywej miareczkowania z wyraźnie zaznaczonym skokiem miareczkowania. Pod nazwą „skok miareczkowania” rozumie się różnicę pH (potencjału red-oks, przewodności elektrolitycznej itp.) zmiareczkowanego roztworu w punktach odpowiadających doprowadzeniu 99,9% oraz 100,1% teoretycznej ilości roztworu mianowanego.
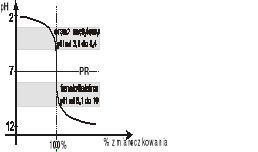
Krzywa miareczkowania mocnego kwasu mocną zasadą
Reakcje w analizie miareczkowej muszą spełniać kilka warunków:
Reakcja między roztworem miareczkowanym, a mianowanym powinna zachodzić szybko.
Reakcja między obydwoma odczynnikami musi zachodzić stechiometrycznie, zgodnie z równaniem.
Wprowadzany odczynnik nie może reagować z innymi substancjami obecnymi w roztworze.
Potrzebny jest odpowiedni indykator pozwalający dokładnie określić PK.
Klasyfikacja metod miareczkowych według typu reakcji zachodzącej podczas miareczkowania.
Alkacymetria - opiera się na reakcjach kwas - zasada.
Kompleksometria - metody oparte na tworzeniu trwałych, łatwo rozpuszczalnych związków kompleksowych.
Precypitometria - wykorzystuje reakcje, w których powstają związki trudno rozpuszczalne - metoda wytrąceniowa.
Redoksymetria - wykorzystuje reakcje utleniania i redukcji.
Roztwory mianowane
Roztwory mianowane są to roztwory o dokładnie znanym stężeniu, które wyznaczono poprzez nastawienie jego miana. Stężenie roztworu mianowanego wyraża się w [g/ml]. Często stężenie roztworów mianowanych wyraża się ich molowością [mol/l]. W alkacymetrii jako roztworów mianowanych najczęściej używa się roztworów mocnych kwasów i zasad. Z kwasów są to najczęściej; kwas solny lub siarkowy. Jako zasady najczęściej używa się roztworu wodorotlenku sodu.
Alkacymetria
Alkacymetria skupia metody miareczkowe oparte na reakcjach kwas-zasada. Metody te są również nazywane metodami zobojętniania. Oprócz kwasów i zasad można nimi również oznaczać sole słabych kwasów i mocnych zasad, bądź też odwrotnie. Alkacymetrię można podzielić na:
alkalimetrię - metoda oznaczania kwasów
acydymetrię - metoda oznaczania zasad
W większości oznaczeń alkacymetrycznych roztwór miareczkowany nie ma w punkcie równoważnikowym odczynu dokładnie obojętnego.
Obliczanie molowości roztworu
Molowość roztworu (C) (mol/l) najprościej można obliczyć z zależności;
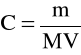
m - masa substancji rozpuszczonej (g)
M - masa molowa tej substancji (g/mol)
V - objętość otrzymanego roztworu (l)
Przy znanym mianie (T) roztworu (g/ml) bardzo łatwo można obliczyć jego stężenie molowe;
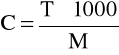
Podczas miareczkowania molowość roztworu można określić przy pomocy drugiego roztworu o znanym mianie. Do tego celu podstawą jest równanie reakcji chemicznej.
![]()
Z równania tego wynika, że a moli substancji A reaguje z b molami substancji B.
![]()
![]()
VA i VB - objętości roztworów A i B (ml lub l)
CA i CB - stężenia molowe roztworów A i B (mol/l)
Jeżeli znane są trzy z w/w wielości to można wówczas obliczyć wielkość czwartą.
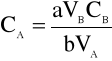
Ćwiczenie: Oznaczanie miana roztworu CH3COOH za pomocą 0,2 mol/l NaOH
Do kolby stożkowej o pojemności 250 ml odmierzyć pipetą 25 ml analizowanego roztworu kwasu octowego. Następnie dodać do kolby 50 ml wody destylowanej, 2 krople fenoloftaleiny i miareczkować 0,2 mol/l roztworem NaOH do pojawienia się trwałego różowego zabarwienia analizowanego roztworu kwasu.
W celu ustalenia dokładnego wyniku należy zmiareczkować co najmniej 3 próbki kwasu octowego. Obliczyć stężenie molowe analizowanego roztworu i jego miano w g/ml.
Ćwiczenie: Oznaczanie miana roztworu NaOH przez miareczkowanie go 0,05 mol/l roztworem H2SO4.
Do kolby stożkowej o pojemności 250 ml odmierzyć pipetą 25 ml analizowanego roztworu wodorotlenku sodu. Następnie dodać do kolby 50 ml wody destylowanej, 2-3 krople oranżu metylowego i miareczkować 0,05 mol/l roztworem H2SO4 do pierwszej zmiany barwy.
W celu ustalenia dokładnego wyniku należy zmiareczkować co najmniej 3 próbki NaOH. Obliczyć stężenie molowe analizowanego roztworu i jego miano w g/ml.
ĆWICZENIE 8.
Nastawianie miana roztworu KMnO4 na Na2C2O4.
Zakres teoretyczny:
Reakcje utleniania i redukcji, równania połówkowe, stopień utlenienia.
Redoksometria:
podział metod redoksometrycznych,
wskaźniki w redoksometrii
Redoksymetria.
Reakcje utleniania i redukcji polegają na wymianie elektronów pomiędzy reagentami.
Redukcja polega na przyłączeniu elektronów, a przez to na zwiększeniu ładunku ujemnego (zmniejszenie dodatniego) i obniżeniu stopnia utlenienia. Utlenianie zaś polega na oddawaniu elektronów, co powoduje zwiększenie ładunku dodatniego (zmniejszenie ujemnego) i podwyższenie stopnia utlenienia.
Przebieg procesu redukcji jest możliwy tylko wtedy gdy jednocześnie zachodzi proces utleniania. Oba te procesy stanowią wspólnie reakcję utleniania i redukcji czyli reakcję red-oks.
Utleniacze są to atomy, jony lub cząsteczki, które mają zdolność przyłączania elektronów - elektronobiorcy.
Reduktory są to atomy, jony lub cząsteczki mające zdolność odłączania elektronów - elektronodawcy.
Wszystkie reakcje red-oks można przedstawić w postaci układu sprzężonego, składającego się z dwóch reakcji połówkowych. Ogólnie reakcję red-oks możemy wyrazić w następujący sposób:
Oks1 + ne ![]()
Red1
Red2 ![]()
Oks2 + ne
Oks1 + Red2 + ne ![]()
Red1 + Oks2 + ne
Oks - utleniacz, Red - reduktor, n - liczba elektronów
Przykładem może być reakcja spalania magnezu w tlenie
2 Mg0 ![]()
Mg2+ + 2e
1 O20 + 4e ![]()
2O-2
2Mg0 + O20 ![]()
2Mg2+ + 2O-2
Stopniem utlenienia pierwiastka wchodzącego w skład określonej substancji nazywamy liczbę dodatnich lub ujemnych ładunków elementarnych jakie przypisalibyśmy atomom tego pierwiastka gdyby cząsteczki tej substancji miały budowę jonową.
Zasady oznaczenia stopnia utlenienia:
Stopień utlenienia pierwiastka w stanie wolnym równy jest zero.
Stopień utlenienia pierwiastka w postaci jonu prostego równa się jego elektronowartościowości (wartościowości jonu).
Suma stopnia utlenienia wszystkich atomów wchodzących w skład jonu złożonego równa jest ładunkowi jonu.
Suma stopnia utleniania wszystkich atomów wchodzących w skład cząsteczki obojętnej wynosi zero.
Fluor we wszystkich swych połączeniach występuje na -1 stopniu utlenienia.
Tlen w połączeniach ma stopień utlenienia -2. Wyjątki stanowią: fluorek tlenu OF2, w którym tlen ma stopień utlenienia +2.
Redoksymetria stanowi dział analiz miereczkowych skupiający metody oparte na reakcjach utleniania i redukcji. Oksydometria skupia metody oparte na miareczkowaniu mianowanym roztworem utleniacza, z kolei reduktometria skupia metody w których miareczkowanie przeprowadza się mianowanym roztworem reduktora.
Do typowych metod oksydometrycznych należą:
nadmanganianometria - utleniaczem jest KmnO4
cerometria - utleniaczem jest Ce(SO4)2
chromianometria - utleniaczem jest K2Cr2O7 lub K2CrO4
bromianometria - utleniaczem jest KBrO3
Typową metodą reduktometryczną jest tytanometria wykorzystująca zdolności redukujące tytanu. W metodach tych stosuje się również związki dwuwartościowe chromu, a także kwas askorbinowy.
Bardzo ważnym działem analizy miareczkowej jest jodometria znajdująca się na pograniczu oksydometrii i reduktometrii. Stosuje ona wolny jod jako utleniacz lub KIO3 jako reduktor.
W miarę przebiegu reakcji red-oks różnica potencjałów między reagentami maleje, gdyż z jednej strony obniża się potencjał utleniacza na skutek zwiększenia stężenia jego formy zredukowanej, a z drugiej strony wzrasta potencjał reduktora z powodu zwiększenia się stężenia jego formy utlenionej. W końcu następuje moment zrównoważenia się potencjałów utleniających obu układów i od tej chwili ustala się w roztworze stan równowagi dynamicznej pomiędzy składnikami obu układów.
Ered1 = Eutl2 = EPR
W punkcie równoważnikowym stosunek stężeń postaci utlenionej do zredukowanej reduktora musi być równy stosunkowi stężeń postaci zredukowanej do utlenionej utleniacza.
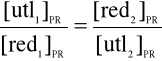
Reakcje utleniania i redukcji przebiegają stosunkowo powoli. Z reguły też reakcje te nie przebiegają bezpośrednio lecz etapami, ponieważ reduktor zwykle oddaje swe elektrony stopniowo, a utleniacz również może przyłączać je w kilku fazach, np.:
Mn7+ + 3e → Mn4+ + e → Mn3+ + e → Mn2+
Nadmanganianometria
Nadmanganianometria jest działem analizy miareczkowej obejmującej oznaczenia reduktorów za pomocą miareczkowania mianowanym roztworem nadmanganianu potasu (KMnO4). Związek ten ma bardzo wysoki potencjał utleniający. Ma on również silne zabarwienie dzięki czemu odpada konieczność stosowania wskaźników. Przebieg redukcji nadmanganianu zależy od pH środowiska;
W roztworze kwaśnym MnO4- + 8H+ + 5e ![]()
Mn2+ + 4H2O
W roztworze obojętnym MnO4- + 2H2O + 3e ![]()
MnO2 + 4OH-
W roztworze zasadowym MnO4- + e ![]()
MnO42-
Szybkość rozkładu nadmanganianu potasu zwiększa się wraz ze zwiększeniem stężenia jonów wodorowych. Dlatego kwaśne jego roztwory łatwiej ulegają rozkładowi niż obojętne. Proces ten przebiega znacznie szybciej w podwyższonych temperaturach, a zwłaszcza w temperaturze wrzenia.
Ćwiczenie: Nastawianie miana nadmanganianu potasu.
Najlepszą substancją do nastawiania miana nadmanganianu potasu jest szczawian sodu.
2KMnO4 + 5Na2C2O4 + 8H2SO4 → 2MnSO4 + 10CO2 + K2SO4 + 5Na2SO4 + 8H2O
2MnO4- + 5C2O42- + 16H+ → 2Mn2+ + 10CO2 + 8 H2O
Z powyższych równań wynika, że 2 mole KMno4 reagują z 5 molami Na2C2O4.
Do kolby stożkowej o pojemności 250 ml odmierzyć pipetą 10 ml 0,05 mol/l roztworu szczawianu sodu. Następnie dodać 50 ml wody destylowanej oraz 10 ml roztworu H2SO4 (1+3) (odmierzonego pipetą przy pomocy gruszki).
Otrzymany roztwór ogrzewać w łaźni wodnej do temperatury 70-80°C (tj. do momentu gdy nad roztworem zacznie się pojawiać mgiełka pary wodnej), a następnie miareczkować ostrożnie na gorąco roztworem, którego miano należy wyznaczyć (KMnO4).
Początkowo należy wprowadzać roztwór KMnO4 bardzo powoli, po kropli, czekając każdorazowo na zupełne odbarwienie się roztworu miareczkowanego. Gdy reakcja zachodzi już szybciej, można przyspieszyć proces, nie zapominając o mieszaniu w celu wyrównywania stężeń. Zbliżający się koniec miareczkowania poznać po tym, że zmniejsza się szybkość zaniku barwy nadmanganianu potasu. Miareczkowanie jest zakończone, gdy po dodaniu 1 kropli KMnO4 w nadmiarze, roztwór przybiera barwę różową, utrzymującą się w ciągu 1 minuty.
W celu ustalenia dokładnego wyniku należy zmiareczkować co najmniej 3 próbki.
Zanotować wyniki pomiarów, obliczyć stężenie (mol/l) i miano (g/ml) roztworu KMnO4 oraz napisać równanie zachodzącej reakcji (wraz z równaniami połówkowymi).
ĆWICZENIE 9.
Oznaczanie jonów tiocyjanianowych metodą strąceniową
Zakres teoretyczny:
Precypitometria - zasada metody strąceniowej
Iloczyn rozpuszczalności
Miareczkowanie strąceniowe.
Miareczkowa analiza strąceniowa (wytrąceniowa) polega na wydzieleniu oznaczanej substancji w postaci trudno rozpuszczalnego osadu przy użyciu mianowanego roztworu odpowiedniej substancji. Te reakcje wykorzystuje się w analizie chemicznej do oznaczania jonów. Ilość (a co za tym idzie stężenie) oznaczanego składnika oblicza się na podstawie objętości dodanego roztworu mianowanego. W celu określenia końca miareczkowania (PK miareczkowania) obserwuje się, czy dodana kropla odczynnika wytrąca jeszcze nową porcję osadu, lub stosuje się odpowiedni wskaźnik, właściwy dla danego oznaczenia.
Podstawą oznaczeń wytrąceniowych jest tworzenie się związków trudno rozpuszczalnych (praktycznie nierozpuszczalnych) w środowisku wodnym.
Jeżeli jon M+ reaguje z jonem A- z utworzeniem trudno rozpuszczalnego związku MA, to równowagę reakcji odwracalnej
M+ + A- ![]()
MA (↓) (1)
można opisać stałymi równowagi
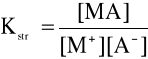
(2)
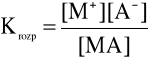
![]()
(3)
W przypadku osadów trudno rozpuszczalnych szczególnie duże znaczenie praktyczne ma stała reakcji rozpuszczania tych osadów. We wzorze (3) [MA] jest wielkością stałą (faza stała), wzór ten można zatem uprościć i przedstawić go w postaci
![]()
(4)
lub
![]()
(5)
Wielkość Kso zwana jest iloczynem rozpuszczalności trudno rozpuszczalnej substancji MA. Określa się go jako iloczyn stężeń jonów tej substancji w roztworze pozostającym w równowadze z osadem. Iloczyny rozpuszczalności są wielkościami stałymi dla danego rozpuszczalnika i w danej temperaturze.
Dla reakcji ogólnej typu
mMn+ + nAm- ![]()
MmAn (↓) (6)
iloczyn rozpuszczalności trudno rozpuszczalnego związku MmAn wyrazi się wzorem:
Kso = [Mn+]m[Am-]n (7)
W rzeczywistości należałoby rozpatrywać termodynamiczny iloczyn rozpuszczalności, w którym zamiast stężeń podstawia się współczynniki aktywności. W praktyce podczas obliczania iloczynów rozpuszczalności ma się do czynienia z roztworami trudno rozpuszczalnych substancji o małej sile jonowej, można więc korzystać ze wzoru (7), gdyż współczynniki aktywności są wtedy bowiem bliskie jedności.
Rozpuszczalność tworzącego się osadu odgrywa decydującą rolę w analizie wagowej i w oznaczeniach precipitometrycznych. Jednak gdy w analizie wagowej można zmniejszyć ilość oznaczanej substancji pozostającej w roztworze (zmniejszyć rozpuszczalność osadu) przez dodanie nadmiaru odczynnika wytrącającego, w miareczkowej analizie wytrąceniowej ilość dodanego odczynnika musi być ściśle równoważna chemiczne ilości składnika oznaczanego. Dlatego w oznaczeniach wytrąceniowych można stosować tylko takie reakcje, w których tworzą się osady praktycznie nierozpuszczalne.
Źródłem błędów w miareczkowej analizie wytrąceniowej może być również adsorbcja. na powierzchni osadu jonów, z których osad został utworzony. Tym sposobem część oznaczonej substancji może zostać uwięziona wewnątrz cząstek osadu, powodując zmniejszenie wyników miareczkowania. Aby zmniejszyć błędy spowodowane adsorpcją należy przeprowadzać miareczkowanie powoli, energicznie mieszając. Innym źródłem błędów jest często tworzenie się roztworów koloidowych. Taki osad odznacza się większą reaktywnością, wobec czego łatwiej przebiegają reakcje uboczne, wpływając niekorzystnie na wyniki oznaczenia.
Krzywe miareczkowania w oznaczeniach wytrąceniowych mają wygląd zupełnie podobny, jak w przypadku oznaczeń alkacymetrycznych. W miarę dodawania odczynnika wytrącającego stężenie oznaczanego składnika początkowo maleje bardzo powoli, później zmniejszanie stężenia jest coraz szybsze i wreszcie w pobliżu punktu równoważnikowego stężenie zmniejsza się skokowo (na osi rzędnych zaznacza się ujemny logarytm dziesiętny molowego stężenia oznaczanego składnika). Zgodnie z prawem iloczynu rozpuszczalności stężenie oznaczanego jonu nigdy nie może zmniejszyć się do zera, może natomiast przyjąć wartości tak małe, że dla celów praktycznych nie maja one znaczenia.
Najczęściej stosowana metoda miareczkowania stąceniowego jest argentometria, w której roztworem mianowanym jest azotan (V) srebra. Ponadto wykorzystuje się metody, w których roztworami mianowanymi są: azotan (V) rtęci (I) - metoda merkurometryczna, heksacyjanożelazian (II) potasu lub jony ołowiu.
Wskaźniki w miareczkowaniu stąceniowym.
W miareczkowych metodach strąceniowych nie ma wskaźników uniwersalnych. Prawie dla każdej metody istnieje specjalny, właściwy dla tej metody sposób określania PK miareczkowania.
W analizie strąceniowej stosuje się przede wszystkim dwie grupy wskaźników:
specyficzne, tworzące z nadmiarem odczynnika strącającego barwne osady lub barwne związki kompleksowe,
wskaźniki adsorbcyjne Fajansa
wskaźniki zewnętrzne
Najczęściej używanym wskaźnikiem specyficznym jest chromian (VI) potasu, stosowany do oznaczania chlorków, bromków i jodków. W punkcie równoważnikowym pierwsza kropla nadmiaru jonów Ag+ reaguje z jonami chromianowymi (VI), tworząc brunatnoczerwony osad Ag2CrO4. Do oznaczania halogenków metodą Volharda stosuje się w charakterze wskaźnika sole Fe (III). Po całkowitym strąceniu Ag+ w postaci AgSCN, pierwsza kropla nadmiaru jonów SCN- reaguje z Fe3+, tworząc czerwony kompleks [Fe(SCN)]2+.
Wskaźniki adsorbcyjne to barwne związki organiczne - słabe kwasy (np. eozyna lub fluoresceina) o ogólnym wzorze ROH lub słabe zasady (np. pochodne rodaminy) o ogólnym wzorze RH. Istotę ich działania można wytłumaczyć tym, że wskaźniki zaadsorbowane na powierzchni osadu tworzą z nim związek o innej barwie niż wolna forma wskaźnika w roztworze.
W miareczkowaniu strąceniowym niekiedy stosuje się barwniki zewnętrzne. Reakcję pomiędzy takim wskaźnikiem i roztworem miareczkowanym przeprowadza się na zewnątrz roztworu np. na bibule nasyconej wskaźnikiem lub sposobem kroplowym na płytce porcelanowej.
Ćwiczenie: Oznaczanie jonów tiocyjanianowych metodą strąceniową
Do kolby stożkowej o pojemności 250 ml odmierzyć pipetą 20 ml 0,1 mol/l AgNO3, rozcieńczyć wodą do ok. 100 ml. Następnie roztwór zakwasić 5 ml HNO3 (1:1) oraz dodać
2 ml wskaźnika (10% roztwór siarczanu (VI) amonu i żelaza (III)). Tak przygotowany roztwór miareczkować próbą KSCN o nieznanym stężeniu do pojawienia się pomarańczowego zabarwienia utrzymującego się przez 1 min.
Do oznaczenia przygotować dwie próbki.
Zanotować wyniki miareczkowania, obliczyć stężenie (mol/l) i miano (g/ml) KSCN.
ĆWICZENIE 10.
Wykrywanie jonów.
Zakres teoretyczny:
Podstawy analizy jakościowej.
Podział anionów i kationów na grupy analityczne.
Odczynniki grupowe, reakcje charakterystyczne i maskowania.
Analiza jakościowa.
Jakościowa analiza chemiczna ma na celu określenie jakościowego składu badanego ciała, tzn. ustalenie, jakie rodzaje materii - pierwiastki, atomy, jony, rodniki, związki chemiczne itp. - w tym ciele się znajdują. Można ją przeprowadzić rozmaitymi metodami, takimi jak badanie widma emisyjnego, barwienie płomienia palnika gazowego, zabarwienie pereł fosforanowych lub boraksowych, spektrografia, spektrometria fluorescencji rentgenowskiej, polarografia czy chromatografia. W przypadku niektórych substancji ich skład jakościowy można określać na podstawie lotności, zdolności odparowywania (sublimacji) lub rozkładu podczas ogrzewania (wszystkie te metody opierają się na badaniu substancji na sucho bez przeprowadzania jej do roztworu).
W chemicznej analizie jakościowej do identyfikacji poszczególnych jonów wykorzystuje się reakcje chemiczne, w których wydzielają się osady (białe lub barwne) trudno rozpuszczalnych związków, powstają barwne rozpuszczalne kompleksy, wydzielają się gazy bezwonne (np. dwutlenek węgla, wodór) lub o specyficznym zapachu (np. siarkowodór). Bliższe informacje o otrzymanych osadach uzyskuje się na podstawie badań ich rozpuszczalności w roztworach kwasów, zasad i środków kompleksujących lub w rozpuszczalnikach organicznych.
Badana substancja lub mieszanina, jak i stosowany odczynnik znajdują się najczęściej w roztworach wodnych jako elektrolity w stanie zdysocjowanym, to znaczy, że przeprowadzane reakcje zachodzą między jonami. W toku analizy najpierw rozpuszcza się badaną substancję, a następnie na otrzymany roztwór działa się odpowiednimi odczynnikami. Zwykle roztwór poddaje się systematycznej analizie, która składa się z badań wstępnych i kolejnych operacji zgodnie z przyjętym schematem postępowania. Analizę chemiczną w roztworach nazywa się analizą na mokro.
W analizie jakościowej możemy wyróżnić dwa etapy postępowania: oddzielanie i rozdzielanie jonów, a następnie ich identyfikację.
Odczynniki w analizie chemicznej.
specyficzne - w określonych warunkach dają reakcję tylko z danym jonem, to znaczy pozwalają na wykrycie danego jonu w obecności innych,
selektywne - dają podobną reakcję z pewną ograniczoną liczba jonów,
grupowe - wykazują zdolność wytrącania pewnej określonej kategorii jonów z roztworu i pozwalają na rozdzielenie jonów znajdujących się w badanym roztworze na poszczególne grupy analityczne
charakterystyczne - pozwalające na rozdział jonów w danej grupie analitycznej,
maskujące - łączą się z danym jonem ubocznym, wiążą go w dostatecznie trwałe kompleksy i tym samym wyłączają go z udziału w roztworze lub zmniejszają znacznie jego stężenie.
Analityczny podział jonów.
Podział kationów.
W stąceniowych metodach oddzielania i rozdzielania kationów wykorzystuje się różnice w rozpuszczalności różnych związków w wodzie. Na metodach strąceniowych opiera się klasyczny schemat rozdzielania kationów. Podzielono w nim kationy na pięć grup (na podstawie strącania z różnymi odczynnikami grupowymi).
Podział kationów na grupy analityczne (wg Lipiec i Szmal)
Grupa |
Odczynnik grupowy |
Wykrywane jony |
Skład osadu |
Uwagi |
I |
3 mol/l HCl |
Ag+, Pb2+, Hg22+ |
AgCl, PbCl2, Hg2Cl2 |
chlorki nierozpuszczalne w wodzie i w rozc. HCl |
II |
H2SO4 |
Ba2+, Sr2+, Ca2+, (Pb2+) |
BaSO4, SrSO4, CaSO4, (PbSO4) |
Siarczany nirozpuszczalne w wodzie i w rozc. H2SO4 |
III |
H2S lub CH3CSNH2 w rozc. H2SO4 |
Hg2+, Bi2+, Cu2+, Cd2+, Sn2+, Sn4+, As3+, As5+, Sb3+, Sb5+ |
HgS, Bi2S2, CuS, CdS, SnS, SnS2, As2S3, As2S5, Sb2S3, Sb3S5 |
siarczki nierozp. w rozc. HCl i H2SO4 |
|
(NH4)2S lub CH3CSNH2 wobec NH3 . H2O i NH4Cl |
Al3+, Cr3+, Fe3+, Fe2+, Ni2+, Co2+, Mn2+, Zn2+ |
Al.(OH)3, Cr(OH)3, FeS, Fe2S3, NiS, CoS, MnS, ZnS |
Siarczki nierozp. W H2O , rozpuszczalne w rozc. kwasach |
V |
brak |
Mg2+, Na+, K+, NH4+ |
|
Nie wytrącają się poprzednimi odczynnikami |
Podział anionów.
Wyróżnia się 7 grup analitycznych , przy czym odczynnikami grupowymi są azotan srebra i chlorek baru.
Podział anionów na grupy (Lipiec i Szmal)
I grupa |
Jony Ag+ wytrącają osad nierozpuszczalny w rozcieńczonym HNO3. Jony Ba2+ nie wytrącają osadu. |
Cl-, Br-, I-, CN-, NCS-, Fe(CN)64-, Fe(CN)63-, ClO- |
II grupa |
Jony Ag+ wytrącają osad rozpuszczalny w rozcieńczonym HNO3. Jony Ba2+ nie wytrącają osadu. |
S2-, CH3COO-, NO2- |
III grupa |
Jony Ag+ wytrącają biały osad rozpuszczalny w HNO3. Jony Ba2+ wytrącają biały osad rozpuszczalny w HNO3. |
SO32-, CO32-, C2O42-, C4H4O62-, BO2- |
IV grupa |
Jony Ag+ wytrącają barwne osady rozpuszczalne w HNO3. Jony Ba2+ wytrącają osad rozpuszczalny w HNO3. |
S2O32-, CrO42-, Cr2O72-, AsO33-, AsO43-, PO43- |
V grupa |
Jony Ag+ nie wytrącają osadu. Jony Ba2+ nie wytrącają osadu. |
NO3-, ClO3-, ClO4-, MnO4- |
VI grupa |
Jony Ag+ nie wytrącają osadu. Jony Ba2+ wytrącają osad. |
SO42-, F-, SiF62- |
VII grupa |
Jony Ag+ wytrącają żółty osad rozpuszczalny w HNO3. Jony Ba2+ wytrącają biały osad rozpuszczalny w HNO3. Przy odparowaniu ze stęż. HCl wytrąca się nierozpuszczalny osad. |
SiO32- |
Ćwiczenie: Wykrywanie jonów.
Wykrywanie kationów
W pierwszej kolejności ćwiczenia należy opisać sześć probówek (1, 1a, 2, 2a, 3, 3a). Otrzymujemy trzy nieznane roztwory, w których należy wykryć obecność określonych kationów. Pierwszy otrzymany roztwór należy rozpipetować do probówek 1 i 1a po 3 ml. Analogiczną czynność wykonać dla drugiego i trzeciego roztworu odmierzając odpowiednio po 3 ml do probówek 2 i 2a oraz 3 i 3a.
Do probówek oznaczonych 1, 2, i 3 dodać po 0,5 ml zasady sodowej i obserwować reakcje.
Do probówek 1a, 2a, 3a dodać po 0,5 ml fosforanu dwusodowego i obserwować reakcje.
Wykrywany jon |
Odczynnik |
obserwacje |
Fe2+ |
NaOH Na2HPO4 |
osad zielonkawy - brunatniejący osad biały - sinoniebieski |
Ag+ |
NaOH Na2HPO4 |
osad brunatny osad żółty |
Cu2+ |
NaOH Na2HPO4 |
osad niebieski osad zielonkawo niebieski |
Wykrywanie anionów
W pierwszej kolejności ćwiczenia należy opisać dziewięć probówek (1, 1a, 1b 2, 2a, 2b, 3, 3a, 3b).
Otrzymujemy trzy nieznane roztwory, w których należy wykryć obecność określonych anionów. Pierwszy otrzymany roztwór należy rozpipetować do probówek 1, 1a i 1b po 3 ml. Analogiczną czynność wykonać dla drugiego i trzeciego roztworu odmierzając odpowiednio po 3 ml do probówek 2, 2a i 2b oraz 3, 3a i 3b.
Do probówek oznaczonych 1, 2, i 3 dodać po 0,5 ml chlorku baru i obserwować reakcje.
Do probówek 1a, 2a, 3a dodać po 0,5 ml azotanu srebra i obserwować reakcje.
Do probówek 1b, 2b, 3b dodać po 0,5 ml kwasu siarkowego w celu zakwaszenia a następnie po 0,5 ml nadmanganianu potasu i obserwować reakcje.
Wykrywany jon |
Odczynnik |
obserwacje |
I- |
BaCl2 AgNO3 KMnO4 + H2SO4 |
brak osadu osad żółty odbarwienie (żółty wolny jod) |
SCN- |
BaCl2 AgNO3 KMnO4 + H2SO4 |
brak osadu osad biały odbarwienie |
CO32- |
BaCl2 AgNO3 KMnO4 + H2SO4 |
osad biały osad biały ciemniejący brak reakcji |
Zanotować obserwacje, podać nazwy wykrytych jonów i napisać jonowo przebieg obserwowanych reakcji.
ĆWICZENIE 11.
Oddzielanie kationu od anionu za pomocą kationitu.
Zakres teoretyczny:
Wymiana jonowa:
jonity - budowa i zasada działania
wykorzystanie wymiany jonowej do oddzielania i zagęszczania.
Wymiana jonowa.
Jonity (wymieniacze jonowe - kationity i anionity) to nierozpuszczalne, syntetyczne, wielkocząsteczkowe polimery organiczne o charakterze kwasów, zasad albo soli, zawierające czynne chemicznie ugrupowania jonowe, które mają właściwości wymiany jonów wchodzących w ich skład na jony z roztworu. Kationity, czyli wymieniacze kationowe zawierają grupy kwasowe (sulfonowe, karboksylowe, fenolowe), w których jony wodorowe ulegają wymianie na inne kationy. Anionity zawierają w swej strukturze czynne grupy zasadowe: czwartorzędowe amoniowe oraz trzecio-, drugo- i pierwszorzędowe aminowe.
Wymiana jonowa jest odwracalnym procesem wymiany jonów między jonitem a roztworem. Ustalający się stan równowagi między jonitem a otaczającym go roztworem podlega prawu działania mas i zależny jest od powinowactwa określonych jonów do danego jonowymieniacza.
Do wymiany jonowej stosuje się metodę kolumnową. Kolumnę (najczęściej szklaną) napełnia się namoczonymi ziarnami odpowiedniej żywicy, od której ilości i pojemności jonowymiennej zależy zdolność jonowymienna kolumny. Pojemność jonowymienna jonitu określana jest jako liczba milimoli jonów pierwiastka zatrzymanych przez jednostkę masy wymieniacza.
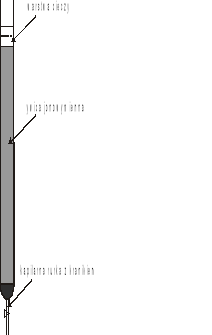
Kolumna jonowymienna
Po wysyceniu kolumny zatrzymanymi kationami lub anionami należy przeprowadzić jej regenerację tzn. przepuścić przez kolumnę kwas solny (w przypadku kationitu) lub roztwór amoniaku, bądź wodorotlenku sodu (dla anionitu).
Wymianę jonową stosuje się między innymi do rozdzielania pierwiastków i oddzielania mikroskładników od makroskładników, demineralizacji wody (wodę demineralizowaną wykorzystuje się często zamiast wody destylowanej), zagęszczania jonów z roztworów bardzo rozcieńczonych przed procesem analizy.
Ćwiczenie: Oznaczanie stężenia soli przez oddzielenie kationu od anionu za pomocą kationitu.
Przygotowanie kolumny z kationitem w postaci wodnej; regeneracja
Jonit umieszczony w kolumnie przemyć 50 ml wody destylowanej. Wodę przepuszczać dość szybko, a pod koniec sprawdzić odczyn eluatu przy pomocy papierka wskaźnikowego.
Odmierzyć cylindrem miarowym 20 ml 1 mol/l kwasu solnego i ostrożnie wlać go do kolumny. Następnie powoli spuszczać roztwór z kolumny, czyli tak aby po otworzeniu zacisku umieszczonego na wężu gumowym ciecz z kolumny wypływała kropla po kropli. Wprowadzić na kolumnę 30 ml wody destylowanej i tuż przed ukończeniem przepuszczania jej przez kolumnę sprawdzić odczyn kapiącego eluatu.
Następnie przepuścić już szybciej przez kolumnę 170 ml wody destylowanej, aż do usunięcia nadmiaru kwasu niezwiązanego z jonitem, czyli osiągnięcia obojętnego odczynu eluatu.
Oddzielenie kationu od anionu
Na przygotowany jonit wlać 20 ml badanego roztworu NaCl. Pod ujście węża podstawić czystą kolbkę o pojemności 300 ml, w której będzie prowadzone miareczkowanie. Kroplami spuszczać do niej roztwór z kolumny i po osiągnięciu ok. 1 cm warstwy cieczy nad jonitem sprawdzić odczyn kapiącego eluatu. Następnie wprowadzić na kolumnę 100 ml wody destylowanej i spuszczać ją po woli do tej samej podstawionej wcześniej kolbki. Sprawdzić odczyn eluatu.
Do zebranego w kolbce roztworu dodać 2 krople oranżu metylowego i miareczkować go 0,1 mol/l roztworem NaOH aż do zmiany zabarwienia roztworu z różowego na żółte.
Obliczyć stężenie soli w badanym, wprowadzonym na kolumnę roztworze, które przy założeniu 100%-owej sprawdzalności kolumny, będzie równe stężeniu kwasu czyli jonów wodorowych znajdujących się w miareczkowanym roztworze. Jony te zostały usunięte ze złoża i zastąpione przez kationy pochodzące z soli.
Zapisać obserwacje (odczyn eluatu) i wnioski dotyczące każdego etapu ćwiczenia.
Uwaga! Podczas spuszczania roztworu z kolumny należy pamiętać o tym, aby zawsze nad jonitem znajdowała się kilku mililitrowa warstwa cieczy.
ĆWICZENIE 12 i 13.
Oznaczanie żelaza metodą fotokolorymetryczną.
Zakres teoretyczny:
Podział i ogólna charakterystyka metod instrumentalnych.
Podstawy absorpcjometrii:
prawo Lamberta i Beera,
odstępstwa od praw absorpcji,
widmo absorpcji i krzywa wzorcowa.
Zasada działania spektrofotometru.
Oznaczanie stężenia żelaza metodą rodankową.
Podział promieniowania elektromagnetycznego; zakresy długości fal.
Budowa cząsteczki a absorpcja.
Zależność między absorpcją promieniowania i zabarwieniem.
Metody optyczne do których zaliczamy wszystkie metody analityczne wykorzystujące światło i jego oddziaływania z materią. Pod pojęciem światła rozumiemy wszystkie rodzaje promieniowania elektromagnetycznego.
Metody elektrochemiczne polegające na analizie zjawisk zachodzących w elektrodach pod wpływem prądu płynącego przez dana substancję lub jej roztwór. Metody te wykorzystują elektrody jonoselektywne.
Metody chromatograficzne służące do rozdzielania i analizy mieszanin. Metody te wykorzystują różnice w zachowaniu się poszczególnych składników mieszanin w układzie dwóch faz; ruchomej i stacjonarnej.
Liczba pasm absorpcji (maksimów)
Długość fali (lub częstotliwość) odpowiadająca maksimum
Natężenie i kształt pasma absorpcji.
Pojęcie chromatografii.
Rodzaje chromatografii i ich charakterystyka.
Chromatografia bibułowa:
Chromatografię adsorbcyjną - fazę stacjonarną stanowi ciało stałe (adsorbent), fazę ruchomą stanowi ciecz lub gaz. Rozdział substancji opiera się na różnicy współczynników adsorbcji jakie wykazuje adsorbent w stosunku do poszczególnych składników mieszaniny.
Chromatografia podziałowa (rozdzielcza) - układ faz stanowią dwie ciecze lub ciecz i gaz. Mieszanina zostaje rozdzielona na składniki w toku ciągłego procesu ekstrakcji przebiegającego między tymi fazami, przy czym wykorzystuje się różnice współczynników podziału.
Chromatografia jonowymienna - fazę nieruchomą stanowi jonowymieniacz, fazę ruchomą stanowi woda lub roztwór elektrolitu. W tej technice wykorzystuje różnice w sile wiązania poszczególnych składników (jonów) przez wymieniacz jonowy.
Chromatografia żelowa - fazę nieruchomą stanowi spęczniały żel, fazę ruchomą stanowi woda lub inny rozpuszczalnik. Chromatografia ta wykorzystuje różnice w zdolnościach poszczególnych składników rozdzielanej mieszaniny do dyfundowania do cząstek żelu (różnice w wielkościach cząstek).
Chromatografia osadowa - fazę nieruchomą stanowi substancja tworząca ze składnikami mieszaniny osady, fazę ruchomą stanowi rozpuszczalnik rozpuszczający osady. Technika ta umożliwia rozdzielenie składników mieszaniny na podstawie różnic w szybkości powstawania osadów oraz ich trwałości.
Zastosowanie odczynników dających charakterystyczne zabarwienie z substancjami poddanymi rozdziałowi.
Zastosowanie odczynników dających połączenia wykazujące fluorescencję w świetle nadfioletowym.
Zanurzenie bibuły w rozpuszczalniku znajdującym się na dnie komory - metoda wstępująca (rozpuszczalnik przemieszcza się z dołu do góry).
Zanurzenie bibuły w rozpuszczalniku znajdującym się w korytku ponad bibułą - metoda zstępująca (rozpuszczalnik przesuwa się z góry w dół).
Doprowadzenie rozpuszczalnika do punktu na środku arkusza bibuły - metoda pierścieniowa (rozpuszczalnik przesuwa się odśrodkowo po bibule).
Spektrofotometryczne oznaczanie stężeń barwników w mieszaninie.
Zakres teoretyczny:
Metody instrumentalne w chemii
Metody instrumentalne w chemii dzieli się na kilka bardzo szerokich działów.
Spektrofotometria należy do metod optycznych wykorzystujących absorpcję światła w zakresie widzialnym (VIS), nadfiolecie (UV) i podczerwieni (IR). Opiera się ona na selektywnej absorpcji promieniowania elektromagnetycznego przez roztwór badanej substancji.
Długości fal promieniowania elektromagnetycznego |
|
Promieniowanie gamma Promieniowanie rentgenowskie Promieniowanie nadfioletowe Promieniowanie widzialne Promieniowanie podczerwone Fale radiowe: - Mikrofale - Fale radiowe ultrakrótkie - Fale radiowe krótkie - Fale radiowe średnie - Fale radiowe długie - Fale radiowe bardzo długie |
10-14 - 5 x 1011 m 10-12 - 10-8 m 3,6 x 10-7 - 10-10 m 3,6 x 10-7 - 7,8 x 10-7 m 7,8 x 10-7 - 10-3 m
10-5 - 1 m 1 - 10 m 10 - 100 m 100 - 1000 m 103 - 104 m 104 - 105 m |
Światło widzialne składa się z fal elektromagnetycznych w zakresie 380-780 nm. Barwa ciała świadczy o tym, że przepuszcza ono lub absorbuje promieniowanie w w/w zakresu długości fal. Zabarwienie jest dopełnieniem barwy promieniowania absorbowanego.
Zależność między absorpcją promieniowania i zabarwieniem. |
||
absorbowane promieniowanie |
zabarwienie obserwowane |
|
długość fali (nm) |
barwa |
|
380-420 420-440 440-470 470-500 500-520 520-550 550-580 580-620 620-680 680-780 |
fioletowa fioletowoniebieska niebieska niebieskozielona zielona żółtozielona żółta pomarańczowa czerwona purpurowa |
zielonożółte żółte pomarańczowe czerwone purpurowe fioletowe fioletowoniebieskie niebieskie niebieskozielone zielone |
Podstawy absorpcjometrii.
Natężenie promieniowania elektromagnetycznego monochromatycznego (I0) przy przejściu przez ośrodek absorbujący częściowo ulega odbiciu (IR), częściowo ulega absorpcji (IA), a reszta zostaje przepuszczona prze ten ośrodek (IT).
I0 = IR + IA + IT
W przypadku roztworów odbicie światła jest znikome więc można je pominąć.
I0 = IA + IT
Bouguer i Lambert wykazali zależność między natężeniem światła przepuszczonego (IT), a grubością warstwy roztworu (l)
IT = I0 × 10-kl
Wielkość (k) jest współczynnikiem ekstynkcji i jest on odwrotnie proporcjonalny do grubości warstwy roztworu (w cm), powodującej 10-krotne zmniejszenie natężenia przechodzącego przez nią światła.
Beer stwierdził, że współczynnik ekstynkcji (absorpcji) roztworu jest proporcjonalny do stężenia substancji absorbującej światło (C).
k = k1 × C
Poprzez połączenie obu tych praw otrzymujemy prawo Bouguer - Lambert i Beera, które mówi, że absorbancja jest proporcjonalna do stężenia substancji oraz do grubości warstwy roztworu.
![]()
![]()
lg I0/ IT = k1 × l × C = A
A - oznacza absorbcję (ekstynkcję), wielkość mierzona na spektrofotometrze równa logarytmowi stosunku promieniowania padającego I0 do promieniowania przepuszczonego IT.
Współczynnik k1 jest wielkością stałą zależną od długości fali światła padającego, natury substancji rozpuszczonej i od temperatury. Jeżeli stężenie wyrażone jest w mol/l, k1 nosi nazwę molowego współczynnika absorpcji i oznaczany jest symbolem (ε).
A = ε c l
c - stężenie substancji (mol/l)
l - grubość warstwy (cm)
ε - molowy współczynnik absorpcji (l × mol-1 × cm-1)
Krzywa wzorcowa
Jeżeli barwny roztwór spełnia prawo Beera to przy użyciu roztworów wzorcowych o znanych stężeniach można ustalić zależność absorbancji od stężenia roztworu i przedstawić ją w układzie współrzędnych. Zależność ta jest linią prostą. W identycznych warunkach można odczytać absorbancję roztworu o nieznanym stężeniu i z równania prostej (krzywej wzorcowej) wyliczyć stężenie próby badanej.
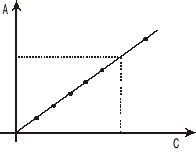
Krzywa zależności absorbancji od stężenia
Odstępstwa od prawa Bougera-Lamberta i Beera.
Liniowa zależność absorbancji od stężenia zgodna z prawem Beera zachowana jest jedynie dla niskich stężeń roztworów c<10-2 mol/l. Przy wyższych stężeniach może dojść do odchyleń od tego prawa wywołanych oddziaływaniami między cząsteczkami. Najlepiej jest tak dobierać stężenia badanych roztworów aby wartość absorbancji nie przekraczała 1. W nowoczesnych spektrofotometrach absorbancja może dochodzić nawet do 2,5.
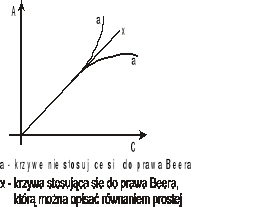
Przyczyną innych odchyleń od tego prawa mogą być również niedoskonałości aparaturowe (zbyt duża szerokość spektralna wiązki światła, zbyt duży poziom światła rozproszonego, niska czułość detektora itp.).
Widmo absorpcji.
Wybór długości fali przy której mierzy się absorbancję roztworu zależy od kształtu widma absorpcji. Długość fali (max) odpowiadająca maksymalnej absorbancji (Amax) służy zazwyczaj do wyznaczania krzywej wzorcowej tego roztworu oraz do oznaczania jego stężeń nieznanych. Widmo absorpcji promieniowania danego rodzaju cząsteczek określa się w funkcji długości fali (λ). Zazwyczaj pasmo absorpcyjne ma kształt zbliżony do krzywej dzwonowatej opisanej funkcją Gaussa.
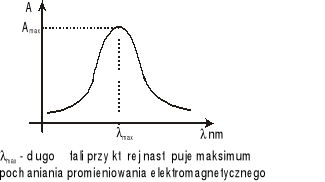
Kształt widma absorpcji dla danego rodzaju cząsteczek zależy od wielu czynników: stanu skupienia, temperatury, rozpuszczalnika, pH, obecności zanieczyszczeń.
Charakterystykę widma absorpcji stanowią;
Budowa cząsteczki a absorpcja promieniowania.
Spektrofotometria jest grupą optycznych metod analitycznych, w których badanym układem jest cząsteczka. W początkowym okresie rozwoju tej metody zaobserwowano, że barwne związki mają wiązania nienasycone, a ich barwa zależy od grupy atomów powiązanych z tymi wiązaniami. Takie grupy atomów nazwano chromoforami. Każdy chromofor ma charakterystyczne dla siebie pasmo absorpcyjne niezależnie od tego w jakim związku występuje. Przykłady najprostszych chromoforów pokazano poniżej;
>C=C< >C=O −N=N− −C≡C− −C≡N
W cząsteczce chromofor znajduje się w otoczeniu innych atomów lub chromoforów co powoduje, że charakterystyczne dla niego pasmo absorpcji ulega zmianie. Zmiany takie mogą prowadzić do przesunięcia pasma absorpcji w stronę fal dłuższych, a nazywane jest to przesunięciem batochromowym. Przesunięcie pasma absorpcji w stronę fal krótszych zwane jest z kolei przesunięciem hipsochromowym. Natężenie pasma absorpcji noże również ulec podwyższeniu lub obniżeniu - nazywa się to odpowiednio efektem hiperchromowym i hipochromowym. Grupy atomów, wpływające na tego typu zmiany widma chromoforu, które nie absorbują promieniowania, ale ich obecność powoduje zmiany charakterystyk spektralnych nazywa się auksochromami. Do auksochromów należą;
−NH2 −NR2 −SH −OH −OR
Budowa spektrofotometru.
Początkowo barwny roztwór badany porównywano z roztworami wzorcowymi w szklanych cylindrach umieszczonych na białym tle. W latach pięćdziesiątych pojawiły się spektrofotometry pozwalające dokładnie wyznaczyć natężenie światła zaabsorbowanego przez układ.
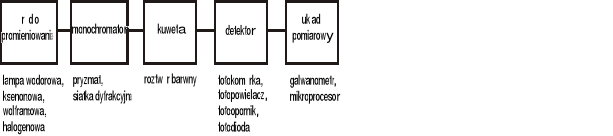
schemat blokowy spektrofotometru
Źródłem promieniowania w spektrofotometrach są różnego typu lampy emitujące promieniowanie z zakresu światła widzialnego. Wiązka promieniowania przechodzi następnie przez siatkę dyfrakcyjną lub pryzmat w celu monochromatyzacji promieniowania o wąskim, ściśle określonym zakresie. Roztwór badany umieszcza się w kuwecie szklanej (dla zakresu widzialnego) najczęściej o grubości 1 cm. Kuwetę umieszcza się w komorach zapewniając takie ustawienie aby roztwór znajdował się na drodze zmonochromatyzowanego promieniowania. Promieniowanie po przejściu przez kuwetę trafia do detektora, który przetwarza promieniowanie elektromagnetyczne na energię elektryczną. Sygnał elektryczny proporcjonalny jest do odbieranego sygnału optycznego.
Ćwiczenie: Oznaczanie żelaza ogólnego metodą rodankową.
Jony rodankowe NCS- reagują z jonami żelaza Fe3+ tworząc Fe(SCN)2+, Fe(SCN)2+ itd., aż do Fe(SCN)63- o barwie czerwonej. Stężenie reagentów i pH środowiska mają wpływ na to, które z kompleksów przeważają w roztworze. W oznaczeniach spektrofotometrycznych, w których stosuje się duże rozcieńczenia przeważają kompleksy pierwsze z szeregu. Ponieważ kompleksy o większym stosunku SCN : Fe mają intensywniejsze zabarwienie.
Przygotować 6 kolb stożkowych i opisać je kolejno cyframi 1-4 oraz próba badana i próba ślepa. Do kolby stożkowej „próba ślepa” wlać 100 ml wody destylowanej.
Do kolby stożkowej „próba badana” odmierzyć za pomocą kolby miarowej 100 ml roztworu żelaza o nieznanym stężeniu. Wszystkie próby przygotować według tabeli:
próba |
Fe(III) 0,1 mg/ml (ml) |
Woda dest (ml) |
HCl (1:1) (ml) |
H2O2 (ml) |
KSCN (ml) |
st. Fe(III) (mg/100ml) |
pr. ślepa |
0 |
100 |
10 |
0,5 |
5 |
0 |
1 |
0,3 |
do 100 |
10 |
0,5 |
5 |
0,03 |
2 |
0,7 |
do 100 |
10 |
0,5 |
5 |
0,07 |
3 |
1,2 |
do 100 |
10 |
0,5 |
5 |
0,12 |
4 |
2,0 |
do 100 |
10 |
0,5 |
5 |
0,20 |
pr. badana |
0 |
0 |
10 |
0,5 |
5 |
x |
Do kolby miarowej o pojemności 100 ml odmierzyć odpowiednią objętość roztworu wzorcowego żelaza Fe(III) o stężeniu 0,1 mg/ml, dopełnić do 100 ml wodą destylowaną i przelać do kolby stożkowej z odpowiednim numerem.
W kolbach stożkowych od 1 do 4 znajduje się odpowiednio 0,03; 0,07; 0,12; 0,20 mg Fe/100ml. Następnie do każdej kolby odmierzyć 10 ml roztworu HCl (1:1), 0,5 ml H2O2 (3%) i zamieszać. Po upływie 5 minut dodać 5 ml rodanku potasowego (KSCN) i ponownie roztwór zamieszać. Następnie zmierzyć dwukrotnie absorbancję na spektrofotometrze przy długości fali 480 nm. Jako odnośnik użyć próbę ślepą.
Zanotować wyniki. Narysować krzywą kalibracyjną (należy posłużyć się średnią wartością zmierzonej absorbancji). Z krzywej odczytać wartość stężenia żelaza w próbie badanej (mg/100ml), a następnie obliczyć stężenie Fe (III) w mg/l.
Ćwiczenie: Spektrofotometryczne oznaczanie stężenia barwników w mieszaninie.
Do kuwety spektrofotometrycznej wlać ok. 1,5 ml roztworu oranżu metylowego o stężeniu 50 μmol/l. Dokonać pomiarów absorbancji roztworu w zakresie od 360 nm do 600 nm co 10 nm, oraz od 600 nm do 750 nm co 50 nm, pamiętając o zmianie filtra przy długości fali 620 nm. Jako odnośnika użyć wody destylowanej, zerując spektrofotometr przy każdej zmianie długości fali.
Takich samych pomiarów dokonać dla błękitu metylenowego o stężeniu 10 mol/l w zakresie od 360 nm do 550 nm co 50 nm, oraz od 550 nm do 750 nm co 10 nm. Takie samo widmo wyznaczyć dla mieszaniny barwników o nieznanych stężeniach, w zakresie długości fali od 360 nm do 750 nm co 10 nm.
Wykreślić widma oranżu metylowego, błękitu metylenowego oraz mieszaniny tych barwników na papierze milimetrowym. Podać maksima absorpcji dla poszczególnych barwników, obliczyć ich współczynniki absorpcji molowej, a na ich podstawie stężenia poszczególnych barwników w mieszaninie.
Uwaga: pamiętać każdorazowo o zmianie filtra przy długości fali 620 nm.
ĆWICZENIE 14.
Chromatografia bibułowa.
Zakres teoretyczny:
- współczynnik podziału,
- rodzaje chromatografii bibułowej,
- technika chromatografii bibułowej.
Analiza chromatograficzna.
Chromatografię można określić jako technikę rozdzielania mieszanin substancji na poszczególne składniki lub ich grupy.
Technika ta wykorzystuje różnice zachowania się poszczególnych składników w układzie dwóch faz, z których jedna jest nieruchoma (stacjonarna), a druga ruchoma poruszająca się w określonym kierunku względem fazy nieruchomej.
Układ chromatograficzny zawiera trzy elementy: fazę nieruchomą, fazę ruchomą i rozdzielaną mieszaninę.
Metody chromatograficzne dzielimy na:
Chromatografia bibułowa
Jest rodzajem chromatografii podziałowej. Nośnikiem fazy stacjonarnej (wody) jest bibuła. Fazę ruchomą stanowią rozpuszczalniki organiczne lub ich mieszaniny.
Zasada chromatografii bibułowej polega na nanoszeniu niewielkich ilości badanych mieszanin w postaci roztworu na bibułę w określonym wcześniej punkcie startowym. Po wyschnięciu bibuły doprowadza się do niej rozpuszczalnik tak aby mógł swobodnie przepływać po bibule. Następuje wówczas rozwijanie chromatogramu polegające na podziale substancji między dwie fazy ciekłe - wodę i rozpuszczalnik. Dany związek zostaje wówczas w mniejszym lub większym stopniu przesunięty względem punktu startowego. Każdy składnik mieszaniny skupia się na bibule w postaci oddzielnej „plamki”.
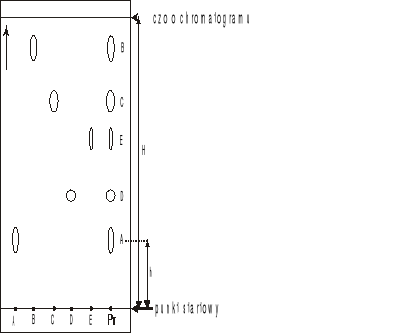
Chromatogram bibułowy: (A, B, C, D, E) - substancje wzorcowe, Pr-próba (mieszanina)
Stosunek wielkości przesunięcia danej substancji do przesunięcia rozpuszczalnika (czoła chromatogramu) nazywa się współczynnikiem podziału (Rf). W celu identyfikacji poszczególnych składników mieszaniny na chromatogramie rozwija się również roztwory substancji wzorcowych. Tak więc można wyznaczyć współczynnik Rf dla poszczególnych składników mieszaniny oraz roztworów wzorcowych i na tej podstawie określić skład badanej mieszaniny.
![]()
Jeżeli rozdzielane składniki mieszanin są barwne wówczas nie ma problemu z ich identyfikacją na chromatografie. Jeżeli jednak mamy doczynienia z substancjami bezbarwnymi wówczas należy odpowiednio wywołać chromatogram poprzez:
Bibuła chromatograficzna musi odznaczać się dużym stopniem czystości. Najczęściej stosuje się bibułę Whatmana. Nośnik ten nie może zawierać zanieczyszczeń żelaza. Przed użyciem bibułę nasyca się parami rozpuszczalnika, który będzie później stosowany jako faza ruchoma.
Komorę chromatograficzną mogą stanowić różne naczynia laboratoryjne (zlewki, cylindry lub komory fabryczne). Komory chromatograficzne powinny być szczelne aby umożliwić równomierne rozwinięcie chromatogramu.
Przepływ rozpuszczalnika osiąga się przez:
Rozpuszczalniki stosowane do rozdziału chromatograficznego muszą być odpowiednio dobrane, tak aby zachowywały się obojętnie w stosunku do bibuły i badanych substancji oraz powinny wykazywać różne współczynniki podziału dla badanych substancji.
Ćwiczenie; Chromatografia bibułowa.
Przygotować eluent do rozdziału chromatograficznego - 20 ml (50% metanol). Wlać eluent do zlewki o pojemności 300 ml i przykryć szalką Petriego.
Przygotować bibułę chromatograficzną o wymiarach 10 na 6 cm z zaznaczonym kierunkiem rozwijania chromatogramu.
Narysować prostą przez całą szerokość bibuły w odległości 1 cm od doły paska i zaznaczyć na niej 6 punktów w równych odległościach
Na zaznaczone punkty za pomocą wykałaczki nanieść kolejno po 2 krople rozdzielanych barwników (5 barwników + mieszanina). Po naniesieniu 1 kropli odczekać chwilę do jej wyschnięcia, a następnie nanieść 2 kroplę.
Tak przygotowana bibułę umieścić w pozycji pionowej w zlewce z eluentem, tak aby poziom eluentu był nieco poniżej linii startowej zaznaczonej ołówkiem. Zlewkę ponownie przykryć szalką Pertiego i pozostawić do rozwinięcia się chromatogramu.
Rozwijanie chromatogramu zakończyć gdy czoło rozpuszczalnika osiągnie przeciwległy brzeg bibuły (ok. 1 cm przez końcem bibuły).
Bibułę delikatnie wyjąć, zaznaczyć czoło rozpuszczalnika i wysuszyć. Następnie ołówkiem obrysować powstałe plamy barwników.
Policzyć Rf dla każdego barwnika, a w przypadku mieszaniny podać jej skład.
(rozdzielane substancje; barwnik 1, barwnik 2, Czerwień Kongo, Rodamina, Oranż Metylowy, mieszanina barwników)
1
Wyszukiwarka
Podobne podstrony:
LABORATORIUMCHEMANAL, Biotechnologia PWR, Semestr 4, Podstawy chemii analitycznej - Laboratorium, śc
Zn, Biotechnologia PWR, Semestr 4, Podstawy chemii analitycznej - Laboratorium, ściągi
reakcje, Biotechnologia PWR, Semestr 4, Podstawy chemii analitycznej - Laboratorium, ściągi
2 kolo z analitycznej laborki sciaga, Biotechnologia PWR, Semestr 4, Podstawy chemii analitycznej -
reakcje z analitycznej, Biotechnologia PWR, Semestr 4, Podstawy chemii analitycznej - Laboratorium,
cos, Technologia INZ PWR, Semestr 4, Podstawy Chemii Analitycznej, Podstawy chemii analitycznej - La
Synteza octanu n-butylu, Biotechnologia PWR, Semestr 3, Podstawy chemii organicznej - Laboratorium (
Zajecia 4, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Zajecia 3, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organicznej
Instrukcja NMR, Technologia INZ PWR, Semestr 3, Podstawy Chemii Organicznej, Podstawy chemii organic
Bufory sprawko, Biotechnologia PWR, Semestr 2, Podstawy chemii nieorganicznej Laboratorium, Instrukc
zestaw51 04, WIiTCH, semestr I, podstawy chemii, zestawy obliczenia chemiczne
Podstawy chemii analitycznej La Nieznany
Odp 21 54, WIiTCH, semestr I, podstawy chemii, zestawy obliczenia chemiczne
Pohl, podstawy chemii analitycznej, pytania egzamin
więcej podobnych podstron