Cząsteczka - budowa cząsteczki
Pod pojęciem cząsteczki rozumiemy trwały układ złożony z co najmniej z dwóch atomów (tego samego pierwiastka lub różnych pierwiastków) powiązanych wiązaniem chemicznym dowolnego typu. Cząsteczka ma zdefiniowany skład i budowę. Połączenia atomów, które nie spełniają powyższych warunków nie są cząsteczkami, nawet jeśli w zapisach reakcji chemicznych zapisujemy je tak jak inne cząsteczki. Dobrym przykładem jest tu sól kuchenna - chlorek sodu - NaCl, który mimo zapisu sugerującego istnienie cząsteczki dwuatomowej złożonej z atomu sodu i atomu chloru nigdy w takiej postaci nie występuje. Jako ciało stałe jest tzw. kryształem jonowym, czyli makrocząsteczką o wzorze NaxClx, gdzie x jest liczbą zmienną i nieokreśloną (zależy od wielkości kryształu); w roztworze występuje w postaci jonów Na+ i Cl- nie tworzących zdefiniowanej co do składu i budowy cząsteczki. Także w postaci ciekłej (stopiony kryształ) tworzy mieszaninę równej ilości jonów sodowych i chlorkowych. Należy zatem być ostrożnym w używaniu pojęcia cząsteczka, bowiem w wielu przypadkach jest to, z chemicznego punktu widzenia, określenie nieprawidłowe. Cząsteczek nie tworzą prawie wszystkie silne elektrolity, które tak jak NaCl w różnych układach są mniej lub bardziej uporządkowaną mieszaniną jonów. Nie można również, z formalnego punktu widzenia, mówić o cząsteczce polimeru, tak naturalnego (np. celuloza, białko, skrobia) jak i sztucznego (np. polistyren, polichlorek winylu, polidimetylosiloksan), bowiem substancje te nie są jednoznacznie określone co do składu i budowy lecz stanowią mieszaniny cząsteczek bardzo podobnych, ale jednak nie identycznych. Z przyczyn praktycznych dość często nie zwracamy większej uwagi na rzeczywistą postać związku chemicznego (cząsteczki, pary jonowe, asocjaty itp.), są jednak momenty gdy trzeba dokładnie przyjrzeć się formie występowania danego związku, by zrozumieć pewne jego właściwości (np. wysoką temperaturę wrzenia wody czy wartość ciśnienia osmotycznego roztworu danej substancji).
Cząsteczkę chemiczną charakteryzują przede wszystkim dwa czynniki - skład pierwiastkowy i struktura. Pierwszy określa ile i jakich atomów (których pierwiastków) tworzy cząsteczkę, drugi zaś - jak te atomy są połączone między sobą. Na przykład cząsteczkę glukozy opisuje wzór sumaryczny C6H12O6, który mówi, że cząsteczkę tworzą atomy węgla (6 atomów) wodoru (12 atomów) i atomy tlenu (6 atomów). Wzór ten jednak nic nie mówi na temat, jak te atomy łączą się między sobą, a co gorsza istnieje wiele substancji mających taki sam wzór sumaryczny a zupełnie inne właściwości chemiczne i fizyczne. Opis struktury cząsteczki, czyli określenie który atom tworzy wiązania z którym, identyfikuje konkretny związek o wiele bardziej precyzyjne, nie jest jednak opisem całkowicie wystarczającym, bowiem niekiedy właściwości chemiczne i fizyczne związku zależą także od tego, jak poszczególne atomy i grupy atomów (zwane grupami funkcyjnymi) są rozmieszczone względem siebie w przestrzeni (szczególnie istotne jest to w przypadku reakcji w organizmie żywym - patrz biochemia).
W praktyce najczęściej wystarczają nam schematyczne wzory strukturalne, zapisywane w sposób mniej lub bardziej uproszczony - nie wolno nam jednak zapominać, że o właściwościach chemicznych decyduje prawdziwa struktura, o wiele bardziej skomplikowana niż to co zapisujemy na papierze jako wzór chemiczny. Powinniśmy pamiętać, że między rzeczywistą strukturą a zapisywanymi przez nas wzorami jest relacja podobna do tej, jaka panuje między brzmieniem muzyki a jej zapisem nutowym.
Najprostszym zapisem symbolicznym cząsteczki jest wzór elementarny. Mówi on, z atomów jakich pierwiastków składa się cząsteczka związku i w jakich proporcjach ilościowych występują one w cząsteczce. Nie mówi jednak nic o rzeczywistej ilości atomów budujących cząsteczkę. Tę ostatnia wiadomość przekazuje nam wzór sumaryczny. Wzór sumaryczny można łatwo wywieść z wzoru elementarnego, jeżeli znamy masę cząsteczkowa substancji. Masa cząsteczkowa to suma mas atomowych wszystkich atomów tworzących dana cząsteczkę. Pewne podstawowe wiadomości na temat struktury cząsteczki dają nam wzory strukturalne - od uproszczonych, schematycznych po przestrzenne modele. Ze względu na trudności w przedstawieniu przestrzennej struktury na płaszczyźnie, dość często stosuje się umowne symbole, pomagające zrozumieć rzeczywistą budowę przestrzenną związku. Dwa związki zapisane poniżej (wzorami pod kreską) są identyczne jeśli chodzi o wzór sumaryczny i wszelkie uproszczone zapisy wzorów strukturalnych na płaszczyźnie. Dopiero umowa o zaznaczaniu w różny sposób wiązań skierowanych nad płaszczyznę papieru i pod płaszczyznę, pozwala wyraźnie rozróżnić te dwa związki. Różnica polega wyłącznie na innym usytuowani wiązania zaznaczonej na czerwono grupy hydroksylowej. Poniżej (nad kreską) mamy cztery różne zapisy tego samego związku (ester etylowy kwasu fenylooctowego).
Trzeba również pamiętać, że wzory zapisywane na papierze najczęściej nie uwzględniają również kątów między wiązaniami, wielkości (ani stosunków rozmiarów) poszczególnych atomów oraz długości wiązań. Ponadto pamiętać też trzeba, że wszystkie elementy cząsteczki są w ciągłym ruch - oscylacje, rotacje, deformacje kątów między wiązaniami - tak że żaden wzór nie jest w stanie oddać pełnej rzeczywistości.
W bardzo dużym uproszczeniu można powiedzieć, że cząsteczka to atomy i wiązania. O ile atomy w różnych konfiguracjach nie ulegają jakimś zasadniczym zmianom, o tyle wiązania łączące atomy w cząsteczkę są bardzo różne i mogą ulegać zmianom pod wpływem innych elementów cząsteczki. Wiązanie chemiczne jest realizowane przez elektrony ostatniej powłoki elektronowej danego pierwiastka, która nosi nazwę powłoki walencyjnej. Każdy atom, poprzez tworzenie wiązań stara się uzyskać konfiguracje oktetu (osiem elektronów) na swojej powłoce walencyjnej. "Klasyczne" wiązanie chemiczne polega na takim zbliżeniu dwóch atomów, że odpowiednie ich orbitale powłok walencyjnych nakładają się na siebie (część przestrzeni między atomami jest wspólna dla obydwu orbitali tworzących wiązanie) a znajdujące się na orbitalach elektrony (po jednym na każdym orbitalu) tworzą parę o przeciwnych spinach i znajdują się w przestrzeni wspólnej dla obu atomów. Takie wiązanie nazywamy wiązaniem atomowym, kowalencyjnym. W praktyce spotykamy je dość rzadko - takie wiązanie występuje między atomami tego samego pierwiastka, a więc najczęściej w przypadku pierwiastków gazowych N2, O2, Cl2 lub Br2 czy I2.
Drugim typem wiązań, najczęściej chyba spotykanym, (szczególnie w chemii organicznej) jest wiązanie atomowe spolaryzowane. Różni się ono od opisanego powyżej jedynie tym, ze ponieważ w wiązaniu biorą udział dwa atomy różnych pierwiastków, o różnych elektroujemnościach, "środek ciężkości" ładunku elektronów wiązania jest przesunięty w kierunku atomu o większej elektroujemności, przez co zatraca się równowaga elektryczna i w obrębie wiązania powstaje układ biegunowy (dipol). Jeden z atomów przyjmuje ładunek δ- a drugi δ+, i choć cząsteczka jako całość nadal jest elektrycznie obojętna, to przesunięcia ładunku w jej obrębie powodują powstanie miejsc bardziej podatnych na reakcje chemiczne. Ponieważ powstałe ładunki nie są równe elementarnemu ładunkowi zaznaczamy je symbolem δ
W dużym uproszczeniu można powiedzieć, że aktywność chemiczna cząsteczki, jej podatność na przemiany chemiczne zależy głównie od wielkości polaryzacji wiązań, choć nie wolno nam zapominać, że znaczna rolę odgrywa tu także przestrzenne usytuowanie poszczególnych atomów.
Jeżeli różnica elektroujemności dwóch atomów tworzących wiązanie jest większa niż 1,4 dochodzi do powstania wiązania jonowego. Wiązanie jonowe tym się różni od atomowego spolaryzowanego, że siłą wiążącą dwa atomy nie jest już wspólnota pary elektronowej lecz siły przyciągania elektrostatycznego dodatniego ładunku kationu (który oddał swój elektron, lub dokładniej, któremu elektron został zabrany) i ujemnego ładunku anionu (który z kolei powstał przez zabranie całego elektronu z kationu). Ponieważ następuje tu przesunięcie całego ładunku elementarnego, kationy i aniony zaznaczamy znakiem plus lub minus, już bez symbolu delty. Zupełnie inna jakość wiązania jonowego powoduje, że reakcje między związkami o charakterze jonowym przebiegają wyraźnie inaczej niż reakcje między związkami o wiązaniach atomowych. Ponieważ wiązania atomowe spolaryzowane możemy uznać za stan pośredni, między wiązaniem atomowym (brak polaryzacji wiązania) a wiązaniem jonowym ("pełna", maksymalna polaryzacja) czasem w różnych opracowaniach możemy spotkać się z określaniem procentowego udziału wiązania jonowego w wiązaniu atomowym, dla bardziej precyzyjnego określenia charakteru wiązania spolaryzowanego.
Specyficznym rodzajem wiązania chemicznego jest wiązanie koordynacyjne, lub inaczej donorowo-akceptorowe. Ta druga nazwa dokładnie oddaje charakter wiązania. Jest to wiązanie pozornie takie jak atomowa - wspólna para elektronów łączy dwa atomy - lecz różni się tym, że oba elektrony pochodzą od jednego atomu (donora) i są wykorzystywane także przez drugi atom (akceptor). Tego typu wiązania spotykamy w związkach, gdzie występują atomy na stopniu utlenienia większym od +4.
Ponieważ generalnie elektronów walencyjnych wokół każdego atomu może być tylko osiem (w przypadku wodoru dwa), to np. dla kwasu siarkowego(VI), trzymając się formalnych wartościowości, uzyskamy wzór (a). Wokół atomu siarki znajduje się wówczas aż 12 elektronów walencyjnych (sześć wiązań po 2 elektrony). Jest to niezgodne z podanym wcześniej warunkiem, więc musimy przyjąć, że dwa wiązania siarki z atomami tlenu są wiązaniami donorowo (siarka daje) akceptorowymi (tlen przyjmuje), co oznaczamy strzałkami od donora do akceptora. W takim układzie -wzór (b) - wokół wszystkich atomów jest odpowiednia ilość elektronów walencyjnych (po osiem, przy wodorach po dwa). Związki o takiej strukturze wiązań są najczęściej silnymi utleniaczami (kwas azotowy(V), nadmanganiany, kwas chlorowy(VII) itp.)
Zdarzają się odstępstwa od tej reguły, ale o nich powiemy w odpowiednim czasie i miejscu.
Ostatnim typem wiązania jest tzw. wiązanie wodorowe. Jest to nazwa nieco myląca, bowiem wiązanie wodorowe nie jest wiązaniem stricte chemicznym, nie wiąże atomów w cząsteczce, jest raczej pewnym typem oddziaływania między atomem wodoru w cząsteczce a elektroujemnym atomem innego pierwiastka, lecz ze względu na charakter tego oddziaływania otrzymało ono nazwę wiązania. Jest to dość słabe oddziaływanie występujące pomiędzy atomem wodoru połączonego z atomem silnie elektroujemnego pierwiastka (np. tlenu lub azotu) a elektroujemnym atomem, posiadającym tzw. wolne pary elektronowe, tzn. elektrony z powłoki walencyjnej, nie biorące udziału w wiązaniu. "Siłą napędową" powstania takiego wiązania jest częściowy ładunek dodatni na atomie wodoru, który powstaje wskutek polaryzacji wiązania wodór-atom elektroujemny (najczęściej tlen, np. w grupie -O-H) i związany z tym cząstkowy ładunek ujemny na atomie elektroujemnego pierwiastka. Jednak charakter sił wiążący atom wodoru z atomem elektroujemnego pierwiastka nie ma charakteru oddziaływań elektrostatycznych, tak jak ma to miejsce między dipolami, lecz posiada dość wyraźnie zaznaczony charakter wiązania chemicznego, aczkolwiek niezmiernie słabego. Udział w nim biorą wolne pary elektronowe atomu silnie elektroujemnego pierwiastka. Mimo niewielkiej siły wiązanie wodorowe jest przyczyna wielu ważnych zjawisk fizykochemicznych. Jemu zawdzięczamy istnienie naszego życia, bowiem gdyby ni wiązania wodorowe, woda (podstawa życia na Ziemi) nie byłaby cieczą o znanych nam właściwościach, lecz podobnie jak metan byłaby gazem, na co wskazuje jej formalna masa cząsteczkowa (metan 16, woda 18, amoniak 17, azot 28, tlen 32). W rzeczywistości wiązania wodorowe między cząsteczkami wody powodują, że zachowuje się ona jak substancja o masie o wiele większej (100-150). Silnie elektroujemny atom tlenu powoduje polaryzację wiązań H-O, umożliwiając powstanie oddziaływań o charakterze wiązań wodorowych, tak jak podano na przykładzie obok.
Zjawisko hybrydyzacji orbitali prowadzi między innymi do ustalenia kątów między poszczególnymi wiązaniami, a więc pośrednio również jest odpowiedzialne za właściwości chemiczne cząsteczki, a czasem także w pewnym stopniu za jej właściwości fizyczne. Dla łatwiejszego zapamiętania wzajemnych relacji między orbitalami w różnych typach hybrydyzacji przyjmijmy hipotezę, że przyroda stara się o jak największa harmonię i wobec tego orbitale względem siebie będą starały się zachować maksimum symetrii. Założenie to nie jest zresztą tak bardzo odległe od prawdy, choć pojęcie harmonii w przyrodzie może czasem nieco odbiegać od powszechnie przyjętego wśród ludzi.
Kierując się powyższą regułą zachowania harmonii, możemy przypuszczać, że wiązania w cząsteczce fluorku boru BF3 ulegną hybrydyzacji sp2 (bor ma trzy elektrony walencyjne, a więc układ - 1s2 2s2p1), bowiem nie może obsadzić więcej niż trzy orbitale, czyli utworzy trzy wiązania. Ponieważ trzy punkty wyznaczają płaszczyznę, najbardziej "harmonicznym" układem będą trzy wiązania leżące na jednej płaszczyźnie, a kąty między nimi będą wynosić 120°. Stąd ważny dla chemizmu tej substancji wniosek, że jej cząsteczki są płaskie.
Na podstawie powyższego przykładu można by mniemać, że określenie kształtu cząsteczki i kątów między wiązaniami to sprawa prosta, nieomal jak rebus w świątecznej gazecie. Należy jednak zachować czujność i ostrożność, na przykład bowiem z pozoru bardzo podobna sytuacja w amoniaku NH3 nie daje wcale płaskiej cząsteczki lecz piramidę o podstawie trójkąta i kąty między wiązaniami około 110°. Dzieje się tak dlatego, że chociaż azot występuje tu jako pierwiastek trójwartościowy (analogia do boru) to elektronów walencyjnych ma 5 (to zasadnicza różnica względem boru) i może obsadzić cztery wiązania, ulega zatem hybrydyzacji sp3. Trzy z czterech hybrydyzowanych wiązań wiążą trzy atomy wodoru, zaś czwarty orbital obsadzony jest przez pozostałe dwa elektrony tworząc niewiążącą, tzw. wolna parę elektronową. Para ta w środowisku kwaśnym tworzy wiązanie koordynacyjne z jonem wodorowym, dając jon amoniowy NH4+. Zatem przewidując kształt cząsteczki i kąty między wiązaniami należy brać pod uwagę ilość możliwych orbitali walencyjnych w danym atomie (to zależy od okresu w którym znajduje się dany pierwiastek), ilość elektronów walencyjnych (to z kolei jest zależne od grupy układu okresowego, do której należy pierwiastek) i wynikającą stąd możliwość utworzenia odpowiedniego typu hybrydyzacji, od której zależą kąty między wiązaniami, a co za tym idzie, również kształt cząsteczki. Kształt poszczególnych typów orbitali i przestrzenne ich ułożenie po hybrydyzacji podano w haśle budowa atomu.
Mówiąc i myśląc o kształcie konkretnej cząsteczki należy pamiętać, że wszystkie jej elementy są w ciągłym ruchu - oscylacyjnym, rotacyjnym, deformacyjnym itp. Należy zatem mówić raczej o pewnym zarysie kształtu, lub o kształcie przestrzeni zajmowanej przez cząsteczkę a nie o konkretnym kształcie cząsteczki, bo taki nie istnieje. Należy też mieć na względzie rozmiary poszczególnych atomów, które mogą znacznie różnić się między sobą. Na przykład atom węgla jest około dwa razy większy od atomu wodoru, a atom bromu aż cztery razy. Także długości wiązań, w zależności od typu wiązania i atomów te wiązania tworzących, różnią się czasem dość znacznie między sobą. Stąd wzory "strukturalne" na płaszczyźnie zazwyczaj nie odzwierciedlają proporcji i wzajemnych konfiguracji atomów w cząsteczce - a zatem i kształtu cząsteczki.
Kształt cząsteczki często może mieć olbrzymi wpływ na jej reaktywność. Jeśli uzmysłowimy sobie (o czym szczegółowiej będzie w rozdziale o reakcjach chemicznych), że aby mogła zajść reakcja, konkretne elementy dwóch cząsteczek (tzw. grupy funkcyjne) muszą zbliżyć się do siebie na odległość równą mniej więcej długości wiązania chemicznego, to zrozumiałym staje się, że "obfite kształty" cząsteczki mogą takie zbliżenie uniemożliwić. Mówimy wówczas o istnieniu przeszkody sterycznej utrudniającej, bądź wręcz uniemożliwiającej reakcję chemiczną między cząsteczkami, mimo że inne cząsteczki z tej samej grupy związków, o takich samych grupach funkcyjnych, ulegają danej reakcji bardzo łatwo.
Szczególnym, i to bardzo ważnym, przypadkiem wpływu kształtu cząsteczki na jej reaktywność jest zjawisko izomerii optycznej, lub dokładniej konfiguracji D i L, gdzie dwa, z pozoru identyczne związki, różniące się tylko tym, że ich cząsteczki mają się do siebie tak jak przedmiot i jego lustrzane odbicie, maja różną reaktywność. Zjawisko to pełni bardzo ważna rolę w przemianach biochemicznych, gdzie często jedna konfiguracja bierze czynny udział w przemianach ustrojowych w żywym organizmie, natomiast jej odmiana "lustrzana" jest całkowicie nie aktywna. Więcej i dokładniej na ten temat będzie mowa w rozdziale o izomerii.
Izomeria
Pod pojęciem izomerii (iso-mer - taki sam skład) rozumiemy zjawisko występowania różnych związków, o różnych właściwościach chemicznych i fizycznych a o takim samym składzie pierwiastkowym i masie cząsteczkowej. Podziału izomerii na typy i rodzaje dokonujemy w oparciu o rodzaj różnic między izomerami. Bez względu jednak na zastosowany podział izomerami nazwać możemy jedynie związki o takim samym składzie pierwiastkowym (jakościowym i ilościowym - identyczny wzór sumaryczny) i o różnych właściwościach chemicznych lub fizycznych.
Izomery strukturalne różnią się wzajemnymi powiązaniami między atomami wchodzącymi w skład cząsteczki, izomery przestrzenne mają identyczna budowę jeśli chodzi o powiązania między atomami - jednak różne jest usytuowanie przestrzenne tych atomów względem siebie. Podział na poszczególne grupy ma jak zwykle charakter umowny. Podany poniżej podział jest najczęściej spotykanym, najpopularniejszym, lecz można wyobrazić sobie także inne usystematyzowanie tego zjawiska. Jak zwykle chodzi jedynie o pewna systematyczność ułatwiającą zapamiętanie różnic i ułatwiająca komunikacje między poszczególnymi osobami.
Typ izomerii |
Grupa |
Uwagi |
strukturalna |
związki o różnym charakterze chemicznym, należące do różnych grup (np. alkohol CH3CH2OH i eter CH3OCH3); także związki nienasycone o różnym położeniu wiązań nienasyconych (np. penten-1 i penten-2) |
|
|
izomery różnią się kształtem łańcucha, np. n -pentan i izo -pentan |
|
|
podstawienia (położenia) |
izomery różniące się miejscem podstawienia atomu innego niż węgiel lub innej grupy funkcyjnej |
|
zjawisko izomerii, polegające na istnieniu w równowadze dwóch form danego związku, powstających przez przemieszczenie się pojedynczego atomu w obrębie cząsteczki; np. keton-enol |
|
przestrzenna |
geometryczna (cis-trans ) |
występuje w przypadku związków nienasyconych, etylenowych, gdy po przeciwnych stronach wiązania ma minimum dwa różne podstawniki; występuje również w związkach pierścieniowych, wówczas podstawniki mogą być po tej samej lub po różnych stronach płaszczyzny pierścienia. |
|
syn -anti |
analogicznie jak cis -trans , tylko w odniesieniu do podwójnego wiązania C=N- lub -N=N- |
|
Metameria:
Rozumie się tu przede wszystkim istnienie poważnych różnic chemicznych między izomerami - przynależność każdego z izomerów do innej grupy związków. Teoretycznie można by zaliczyć tu każdy rodzaj izomerii strukturalnej nie należący do innej grupy.
Łańcuchowa:
Szczególny rodzaj izomerii podstawienia (położenia), dotyczy związków z łańcuchem węglowym o różnej strukturze. Np. wszystkie poniższe związki mają wzór sumaryczny C6H14:
Podstawienia:
Izomery o różnym miejscu podstawienia. Należy bardzo uważać aby nie popełnić pomyłki. Na przykład związki A i B mają identyczna strukturę, tylko narysowane są nieco inaczej. (z innego punktu widzenia). Oba to 3-bromoheksan. Związek B wystarczy obrócić o 180° wokół osi pionowej i poziomej by otrzymać dokładnie taki sam obraz jak związku A.
Izomery podstawienia spotykamy równie często w grupie związków alifatycznych jak i aromatycznych. Szczególną uwagę należy zwrócić na związki cykliczne, gdzie izomeria ta występuje dopiero przy dwóch podstawnikach, lub w przypadku niehomogennego pierścienia (pierścień zbudowany z różnych pierwiastków - hetrocykle). Generalnie izomeria podstawienia występuje tam gdzie możliwe jest jednoznaczne określenie miejsca podstawienia.
|
|
Przykład związków A (benzen i cykloheksan) to przykład związków na tyle symetrycznych, że każda pozycja podstawienia (każdy atom węgla pierścienia) jest równocenna. Nie mogą zatem istnieć tu izomery jednopodstawione. |
|
|
Związki B są też symetryczne, lecz podstawnik tę symetrie nieco zakłócił. Następny podstawnik może zająć pozycje 1,2 lub 3. Mogą zatem istnieć trzy dwupodstawione izomery. |
|
|
W przypadku C (heterocykle) jest podobnie. Zakłócenie idealnej symetrii cząsteczki z przypadku A powoduje powstanie trzech różniących się miejsc podstawienia w przypadku dwupodstawionych izomerów. |
|
|
Istnienie dwóch, niesymetrycznych podstawników (atom hetero i podstawnik) na tyle psuje symetrię cząsteczki, że każdy nowy podstawnik może zostać przyłączony do jednego z czterech różnych miejsc. Paradoksalnie - przy zmniejszonej liczbie fizycznych miejsc do przyłączenia nowego podstawnika (z pięciu węgli do czterech) zwiększyła się liczba możliwych izomerów (z trzech do czterech). |
Tautomeria:
W reakcji przyłączenia wody do acetylenu otrzymać powinniśmy alkohol winylowy (enol - alkohol z grupą OH przy węglu z podwójnym wiązaniem):
otrzymujemy natomiast aldehyd octowy. Dzieje się tak za sprawą zjawiska tautomerii. Tautomeria ta polega na przemieszczeniu się atomu wodoru od grupy hydroksylowej do podwójnego wiązania. W wyniku tej migracji z nienasyconego alkoholu winylowego otrzymujemy aldehyd octowy:
Ogólny zapis tego zjawiska wygląda następująco:
Ponieważ najczęściej R, otrzymany związek należy do grupy ketonów, stąd nazwa tej tautomerii - enolowo-ketonowa.
Izomeria cis-trans
Izomeria cząsteczek, w których można określić jakąś płaszczyznę, w stosunku do której podstawniki tej cząsteczki leżą po dwóch stronach, a brak symetrii uniemożliwia nałożenie obu form na siebie poprzez proste przemieszczenia w przestrzeni. Taką płaszczyzną może być płaszczyzna podwójnego wiązania (płaszczyzna orbitali p tworzących wiązanie ) lub pierścień.
Izomeria optyczna
Izomeria optyczna jest szczególnym przypadkiem izomerii, gdzie o właściwościach izomerów decyduje kolejność rozmieszczenia czterech różnych podstawników przy atomie węgla. Taki atom węgla w cząsteczce, z którym łączą się cztery różne podstawniki nazywamy węglem asymetrycznym. Brak symetrii ugrupowania powoduje, że nie da się poprzez obroty w przestrzeni ustawić tak drugiej cząsteczki tego związku, by tworzyły one parę lustrzanych odbić. Cząsteczka stanowiąca "lustrzane odbicie" ma inną konfigurację podstawników przy asymetrycznym atomie węgla i oczywiście nie da się poprzez obroty w przestrzeni "nałożyć" na tę pierwszą. Zatem dla związków zawierających asymetryczny atom węgla istnieją dwa rodzaje cząsteczek o tym samym składzie a o różnej konfiguracji podstawników przy asymetrycznym atomie węgla w cząsteczce. Nazywamy je izomerami o konfiguracji D lub L. Jeżeli patrząc na przestrzenny wzór cząsteczki aldehydu glicerynowego (który jest "wzorcem" dla określania konfiguracji innych substancji) od strony grupy aldehydowej będziemy podążać od atomu wodoru H poprzez grupę OH do grupy CH2OH, to w zależności od konfiguracji zatoczymy łuk zgodnie z ruchem wskazówek zegara lub w kierunku przeciwnym.
|
|
|
|
|
|
|
|
|
|
|
|
aldehyd D - (+) - glicerynowy |
|
aldehyd L - (-) - glicerynowy |
Takie dwie cząsteczki stanowią swe lustrzane odbicie, ale nie jest możliwe otrzymanie jednej z drugiej poprzez jakikolwiek obrót w przestrzeni. Ustawiając je tak, żeby zgodne położenie przyjęły dwie grupy, pozostałe dwie grupy zajmują zawsze stanowiska przeciwne:
ustawienie lustrzane (zielony za płaszczyzną i czerwony - zgodne)
zgodne ustawienie w przestrzeni grup czerwonej i żółtej (niebieski i zielony - lustrzane odbicie)
Ponieważ w przypadku wielu związków, szczególnie tych o bardziej złożonej strukturze, wyprowadzenie ich konfiguracji L lub D od konfiguracji aldehydu glicerynowego jest trudne i często niejednoznaczne, wprowadzono określanie konfiguracji bezwzględnej, określanej literami R i S. Określanie konfiguracji R, S związku nie bazuje na podobieństwie do konfiguracji wzorca, jak to ma miejsce w przypadku konfiguracji D i L. W najkrótszym ujęciu ustalenie konfiguracji bezwzględnej polega na wykonaniu kolejnych czynności:
- ustalamy, według dość jasnych reguł, tzw. reguł pierwszeństwa, kolejność „ważności” każdego z czterech podstawników w centrum chiralności;
- ustawiamy cząsteczkę tak, żeby podstawnik najmniej znaczący przy atomie chiralnym (np. atom wodoru) znalazł się jak najdalej od nas. (Na rysunku poniżej czerwony) Jeżeli teraz, idąc od grupy o najwyższej „ważności” (zielona, np. chlor) do grupy o kolejno niższych „ważnościach” (żółta np. siarka i dalej niebieska, np. tlen) posuwamy się zgodnie z ruchem wskazówek zegara, to konfiguracje określamy jako R. W przeciwnym razie występuje konfiguracja S.
konfiguracja absolutna S |
|
konfiguracja absolutna R |
Obecność ugrupowania asymetrycznego w cząsteczce powoduje, że cząsteczka skręca płaszczyznę światła spolaryzowanego. Jeżeli w cząsteczce występuje jeden węgiel asymetryczny, możliwe są dwa izomery różniące się kierunkiem skręcenia płaszczyzny polaryzacji (jak wyżej). W przypadku wystąpienia w jednej cząsteczce większej ilości węgli asymetrycznych ilość możliwych izomerów wynosi 2n; gdzie n - ilość węgli asymetrycznych w cząsteczce.
Fale elektromagnetyczne w wiązce światła drgają we wszystkich kierunkach, prostopadłych do kierunku rozchodzenia się tej wiązki. Po przejściu światła przez ośrodek o odpowiedniej budowie (np. kryształ dwójłomny - kryształ szpatu islandzkiego - CaCO3) drgania te zostają uporządkowane w jednym kierunku, wyznaczając tzw. płaszczyznę polaryzacji
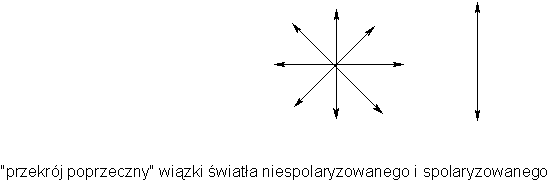
Światło spolaryzowane przebiegając koło atomu cząsteczki, oddziałuje z polem elektrycznym i magnetycznym tego atomu (jest przecież falą elektromagnetyczną), co powoduje skręcenie jego płaszczyzny polaryzacji. Każdy element cząsteczki, a zatem i każda cząsteczka, powoduje takie zjawisko. Jednak w roztworach substancji nie zawierających w swojej strukturze centrów asymetrii (węgli asymetrycznych), światło na swojej drodze spotka praktycznie taką samą ilość elementów skręcających płaszczyznę polaryzacji w prawo co i skręcających ją w lewo. Jest to wynik ustawienia się cząsteczek na drodze światła w taki sposób, że tworzą one pary "lustrzanych odbić". W sumie światło po przejściu przez roztwór takiej substancji będzie drgać w tej samej płaszczyźnie.
W roztworze izomerycznej odmiany o określonej konfiguracji, światło na swojej drodze nie może spotkać par będących lustrzanym odbiciem, a spotyka tylko jeden typ układu przestrzennego (biorąc pod uwagę tylko węgiel asymetryczny) powodujący skręcenie płaszczyzny w jedną, konkretną stronę.
Izomery optyczne będące swoimi odbiciami lustrzanymi nazywamy enencjomerami. Samo zjawisko zwie się z greckiego hiralnością (cząsteczki enancjomerów są jak prawa i lewa ręka). Roztwory enancjomerów, o takim samym stężeniu, skręcają płaszczyznę polaryzacji dokładnie o taki sam kąt, różniący się tylko znakiem.
Jeżeli w cząsteczce występuje więcej niż jedno centrum asymetrii powstaje więcej izomerów (2n), które tworzą kilka par enancjomerów. Cząsteczki, które należą do danej grupy izomerów optycznych a nie są względem siebie enancjomerami, nazywamy diastereoizomerami. Na przykład, przy trzech węglach asymetrycznych, cząsteczki izomeryczne mają konfigurację:
L,L,L; L,L,D; L,D,D; D,D,D; D,D,L; D,L,L; L,D,L; D,L,D;
Pary: L,L,L i D,D,D L,D,L i D,L,D L,L,D i D,D,L oraz L,D,D i D,L,L to pary enencjomerów (lustrzane odbicia), natomiast wszelkie inne zestawienia należą do diastereoizomerów - czyli izomerów o różnej wartości skręcalności, nie będących jednocześnie enancjomerami.
Wspomnieć należy także o jeszcze jednej formie mogącej wystąpić wśród izomerów konfiguracyjnych o parzystej ilości węgli asymetrycznych. Jest to forma mezo. Jest to forma optycznie nieaktywna (nie skręca płaszczyzny polaryzacji), w której symetria cząsteczki i identyczność układów pary (lub kilku par) węgli asymetrycznych powoduje, że skręcalność w jedna stronę przez jeden węgiel asymetryczny jest kompensowana skręcalnością w drugą stronę przez drugi węgiel asymetryczny. Przykładem może być tu kwas winowy:
pierwsza konfiguracja skręca płaszczyznę w lewo, trzecia w prawo, a środkowa jest optycznie nieaktywna, na skutek tego, że lewa skręcalność górnego węgla jest kompensowana przez identyczną, ale odwrotną co do znaku, skręcalność dolnego węgla. Ta identyczność wielkości obu skręcalności wynika z symetrii cząsteczki.
Kształt a właściwości chemiczne i fizyczne
Mówiąc o kształcie cząsteczki, nie możemy ani na chwilę zapominać, że używamy tu pewnego skrótu myślowego - bowiem poszczególne elementy cząsteczki, będąc w wiecznym ruchu (skracanie i wydłużanie wiązań, zmiana kątów miedzy wiązaniami, dyslokacja elektronów itp.) tworzą nie jeden, ściśle zdefiniowany kształt, lecz pulsującą strukturę. Mówiąc więc o kształcie cząsteczki mamy na myśli raczej pewne generalne zależności i proporcje między poszczególnymi elementami niż jakiś konkretny, zdefiniowany kształt. Mimo, że pojęcie - kształt cząsteczki - nie definiuje ostro rzeczywistości, jest wystarczającym przybliżeniem dla rozpatrzenia wpływu kształtu cząsteczki na jej niektóre właściwości chemiczne i fizyczne.
SYMETRIA
Zdolność związków chemicznych do uleganiu konkretnym przemianom i reakcjom chemicznym zależy w przeważającej mierze od polaryzacji wiązań i grup chemicznych w obrębie cząsteczki. Ze względu na różnice w elektroujemności pierwiastków zdecydowana większość substancji chemicznych wykazuje mniejszy lub większy moment dipolowy, z którym związana jest reaktywność cząsteczki, jej rozpuszczalność, zdolność tworzenia struktur krystalicznych itp. Jeżeli jednak cząsteczkę charakteryzuje wysoki poziom symetrii, poszczególne momenty dipolowe wiązań znoszą się nawzajem dając wypadkowy moment dipolowy bliski zero, co oczywiście rzutuje na właściwości chemiczne jak i fizyczne cząsteczki. Gwałtownemu osłabieniu ulegają siły międzycząsteczkowe, zdolność tworzenia kryształów, rozpuszczalność w polarnych rozpuszczalnikach lub zdolność rozpuszczania polarnych substancji.
Tetrachlorek węgla CCl4 jest dość lotną, praktycznie niepolarną cieczą, choć różnica w elektroujemności atomów tworzących jego wiązania jest znaczna (około 1,3). Spowodowane jest to symetria cząsteczki - wszystkie kąty między czterema wiązaniami C-Cl są równe i ich momenty dipolowe znoszą się nawzajem, dając cząsteczkę pozbawiona wypadkowego momentu dipolowego. Ten brak momentu dipolowego determinuje większość właściwości tego związku.
Podobnie symetria cząsteczki CO2 czy acetylenu C2H2 (cząsteczki liniowe) powoduje, że są to związki niepolarne, pomimo silnego spolaryzowania poszczególnych wiązań w obrębie cząsteczki.
ZAWADA STERYCZNA
W sytuacji, gdy jest to tylko możliwe (czyli w przeważającej większości cząsteczek) dwie części cząsteczki połączone wiązaniem pojedynczym σ wirują względem siebie. Ten ruch jest często przyczyną "uśredniania" sytuacji pewnego elementu cząsteczki powodując, że każdy atom takiego ugrupowania ma takie same właściwości. Wyobraźmy sobie, że w pewnej cząsteczce grupa metylowa jest nieruchoma względem pozostałej części cząsteczki. Każdy z wodorów grupy metylowej jest zatem w konkretnym i stałym otoczeniu innych atomów cząsteczki, które to otoczenie determinuje jego zachowanie. Mielibyśmy zatem trzy wodory w trzech różnych sytuacjach - nierównocenne chemicznie. Każdy z nich brałby udział w reakcji chemicznej w charakterystyczny dla siebie sposób. Jeżeli jednak wrócimy do koncepcji wirowania grupy metylowej względem pozostałej części cząsteczki, każdy z atomów wodoru tej grupy jest w identycznej sytuacji - wszystkie trzy wodory są równocenne chemicznie, tak samo reagują na bodźce chemiczne, ulegają identycznym reakcjom chemicznym (następuje uśrednienie sytuacji każdego z nich w ten sam sposób).
Jeżeli w cząsteczce występuje element hamujący lub wręcz uniemożliwiający rotację poszczególnych elementów może dojść do zróżnicowania właściwości chemicznych elementów, które zazwyczaj charakteryzują się równocennościa chemiczną. Najczęściej spotykanym przypadkiem hamowania swobodnej rotacji grup jest występowanie wiązania p między nimi. Są to jednak przypadki, które dają się przewidzieć i każdy chemik bierze je pod uwagę. Trudniejsze do przewidzenia są przypadki (i o nich tu mówimy) kiedy rotacje uniemożliwia przeszkoda przestrzenna (steryczna) w postaci dużego objętościowo podstawnika, tak zorientowanego w przestrzeni, że nie pozwala on fizycznie na swobodną rotacje grupy, która w innych warunkach rotuje bez przeszkód. Takimi dużymi, "przeszkadzającymi" podstawnikami mogą być np. pierścienie, grupy CBr3, grupy tert-butylowe (trimetylometylowe) itp.
Takie przeszkody steryczne mogą nie tylko uniemożliwiać rotacje jakiejś grupy i tym sposobem zmieniać jej właściwości, ale także mogą uniemożliwiać dostęp do miejsca reakcji innym cząsteczkom i w ten sposób czynić niereaktywną cząsteczkę posiadającą reaktywne grupy funkcyjne. Siła działania zawady sterycznej jest czasem tak duża, że np. umożliwia trwanie wolnych rodników przez długi czas, choć w zwykłych warunkach czas ich życia to ułamki sekund.
Szczególnym, bardzo ważnym - szczególnie w biochemii - jest przypadek szczególnej asymetrii cząsteczek spowodowanej obecnością tzw. węgla asymetrycznego, czyli atomu węgla połączonego z czterema rożnymi podstawnikami. Substancje o takiej asymetrii mogą występować w dwóch odmianach izomerycznych oznaczanych D i L i najczęściej charakteryzuje je zdolność skręcania płaszczyzny światła spolaryzowanego. Najistotniejszą cechą takich związków jest to, że najczęściej tylko jedna odmiana (L) jest biochemicznie reaktywna (patrz izomeria optyczna).
Jeśli więc spotkamy substancję, która zachowuje się inaczej, niż inni przedstawiciele danej grupy czy klasy związków, która ma inne właściwości niż podobne z pozoru inne związki, zastanówmy się, czy przypadkiem powodem tej "inności", wyjątkowości, nie jest fizyczna cecha cząsteczki, jakiś szczególny układ w jej fizycznej strukturze.
Energię wewnętrzną cząsteczki można podzielić na:
energię zawartą w jądrach atomowych (tym zajmują się raczej fizycy),
energię związaną z ruchami całej cząsteczki (energia translacji, termicznych ruchów cząsteczki jako całości),
energię związaną z zmianami długości wiązań i kątów między nimi (energia oscylacji),
energię związaną z ruchami obrotowymi cząsteczki (energia rotacji),
energię związaną z elektronami walencyjnymi (energia elektronowa).
W procesie ustalania struktury cząsteczki związku chemicznego znaczenia maja przede wszystkim trzy rodzaje energii - oscylacyjna, rotacyjna i elektronowa. Śledząc ich wartość, oraz zmiany pod wpływem bodźców zewnętrznych możemy wyciągać daleko idące wnioski dotyczące struktury.
Zmiany energii oscylacji i rotacji są tego rzędu, co kwanty energii niesione przez promieniowanie elektromagnetyczne z zakresu podczerwieni (IR). Zmiany energii oscylacji dotyczą zakresu około 4 000 - 600 cm-1, zaś energia rotacji odpowiada dłuższym falom (niższym energiom) z zakresu poniżej 1 000 cm-1. Zmiany energii elektronów walencyjnych, związane ze zmianą ich lokalizacji w cząsteczce (przejście z orbitalu wiążącego na antywiążący) wymaga dostarczenia energii związanej z zakresem promieniowania ultrafioletowego i widzialnego (100-350-700 nm). O energii jądra atomowego dyskutują raczej fizycy, natomiast dla badań strukturalnych bardzo interesująca jest energia związana ze zmianą orientacji wektora magnetycznego jądra atomowego (NMR, częstotliwość promieniowania rzędu kilkuset kHz).
Przy okazji warto zauważyć jak różnymi jednostkami posługujemy się przy określaniu długości fali. W przypadku ultrafioletu i światła widzialnego najczęściej używamy nanometrów (10-9 m) do określenia długości fali. Omawiając widma w podczerwieni prawie wyłącznie używamy do określenia długości fali tzw. liczb falowych (cm-1), rzadziej mikrometrów (mikronów, µm) a w NMR operujemy przede wszystkim częstotliwością (Hz). Wszystkie te jednostki są ze sobą powiązane, łatwo przeliczyć jedne na drugie, natomiast ta różnorodność wynika ze względów praktycznych.
długość fali () - odległość między dwoma ekstremami fali (1 nm = 10-9 m; 1 µm = 10-6 m)
liczba falowa (
) - ilość fal, która występuje na drodze 1 cm (cm-1), czasem zwana częstością (nie mylić z częstotliwością)częstotliwość () - ilość zmian w jednostce czasu (1 Hz = 1zmiana w czasie 1 sekundy)
Tak więc 3 000 cm-1 to jednocześnie:
Wiemy już, że wszelkie zmiany energetyczne w cząsteczce są kwantowane, wymagają odpowiedniego co do wielkości kwantu energii. Wiemy też, że wszystkie elementy cząsteczki są w ciągłym ruchu, czyli ciągle zmienia się ich energia (też w sposób kwantowy). Ponieważ wszystkie te elementy tworzą jedną całość - cząsteczkę, zmiany aktualnej energii jednych nie mogą pozostać bez wpływu na pozostałe. Tak więc pochłonięcie przez cząsteczkę kwantu energii niesionego przez falę o długości 250 nm spowoduje przejście elektronu walencyjnego z orbitalu wiążącego na antywiążący, nie pozostanie to jednak bez wpływu również na energię oscylacyjną tego wiązania (a także i innych wiązań). I choć kwant energii związany z długością fali 250 nm "nie pasuje" do przejść oscylacyjnych (rząd wielkości długości fali dla przejść oscylacyjnych to 4 000 nm) to jednak jest w stanie te oscylacje zmienić w sposób pośredni. Także samorzutne zmiany energii oscylacji poszczególnych wiązań będą miały wpływ na wielkość kwantu pochłoniętego przez cząsteczkę. Świadomość tych powiązań między odległymi od siebie przejściami energetycznymi jest konieczna do zrozumienia pojęć naturalnej szerokości połówkowej pasma, wpływu oddziaływań między- i wewnątrzcząsteczkowych na szerokość i położenia pasma oraz pojęcia pasm kombinacyjnych.
Ważnym dla zrozumienia zjawisk występujących w praktycznej spektroskopii jest też pamiętanie, że objaśnianie poszczególnych zjawisk, ich teoretycznej natury, odbywa się zazwyczaj na przykładach jednostkowych (jedna cząsteczka czy nawet tylko jakiś jej element) zaś w praktyce spektroskopowej mamy wielkie zbiory cząsteczek i otrzymane informacje (widma) dotyczą tych zbiorów a nie pojedynczej cząsteczki. Tak więc jedna cząsteczka w danej chwili może pochłonąć tylko falę o konkretnej długości (kwancie energii) ale olbrzymi zbiór cząsteczek zawarty w analizowanym roztworze może pochłaniać różne długości fal z pewnego wąskiego zakresu, bowiem poszczególne cząsteczki tego zbioru są w tej samej chwili w różnych stanach energetycznych, co ma wpływ na konkretną wartość pochłanianego przez konkretną cząsteczkę kwantu energii. W obrazie widmowym daje to efekt poszerzenia pasma. Pasmo nigdy nie jest, jak by to wynikało z teoretycznych rozważań o kwantowaniu energii, pojedynczą linią określoną jedną długością fali a zawsze pasmem o pewnej szerokości.
Reaktywność chemiczna
Podstawę do zrozumienia reaktywności danej grupy związków (lub nawet konkretnego związku) stanowi niewątpliwie zrozumienie istoty elementarnego procesu reakcji, omówionego także w rozdziale Wstęp działu dotyczącego reakcji chemicznych. Ograniczmy się do najczęściej spotykanego przypadku reakcji między dwiema cząsteczkami różnych związków: A i B. Na co dzień mówimy krótko, że cząsteczka A reaguje z cząsteczką B dając w wyniku reakcji cząsteczkę C. Należy jednak zdawać sobie sprawę, że ten tak krótko ujęty proces jest w rzeczywistości o wiele bardziej złożony i nawet w największym uproszczeniu trzeba opisać jego przebieg w kilku punktach:
w pewnym miejscu w przestrzeni spotkały się dwie, mogące ze sobą reagować cząsteczki A i B (np. cząsteczka kwasu organicznego i cząsteczka alkoholu); ich energie translacji (ruchu w przestrzeni) muszą być tak dobrane, by cząsteczki "zatrzymały się na chwilę" a nie odbiły się od siebie jak kule bilardowe. Jednym słowem reakcja wymaga, by cząsteczki przebywały obok siebie przez pewien okres czasu (bardzo krótki, ale jednak określony);
samo spotkanie, to za mało. Reagują przecież nie cząsteczki jako całość, a jedynie ich elementy, zwane grupami funkcyjnymi. W naszym przykładzie grupa karboksylowa -COOH i alkoholowa -OH. Zatem cząsteczki, aby móc przereagować musza być tak usytuowane wzajemnie w przestrzeni, aby ich grupy funkcyjne mogły się spotkać;
jeśli nawet dojdzie do spotkania grup funkcyjnych, to aby reakcja mogła się rozpocząć cząsteczki muszą mieć odpowiednio wysokie energie (energię wewnętrzną), pozwalające na przebieg reakcji;
wzajemne oddziaływanie elektronów walencyjnych atomów poszczególnych grup funkcyjnych, a pośrednio całych atomów, powoduje rozluźnienie jednych wiązań i zapoczątkowuje powstawanie nowych. Powstaje twór zwany kompleksem aktywnym (AB*), który już nie jest tylko dwiema cząsteczkami A i B, ale nie jest też jeszcze nową cząsteczką C (czyli już nie kwas+alkohol ale jeszcze nie ester);
powstały kompleks aktywny może, ale nie musi, przekształcić się w produkt. Jeśli tak się stanie - proces reakcji się zakończy. Może jednak dojść do rozpadu kompleksu aktywnego z powrotem na cząsteczki A i B i w ujęciu makroskopowym - mimo, że reakcja była już w bardzo zaawansowanym stanie - powiemy, że reakcja nie nastąpiła.
Podane powyżej elementarne etapy procesu reakcji to tylko jego główne elementy. Każdy z nich w dużej mierze jest zjawiskiem losowymi, czyli trywializując, zależy głównie od przypadku. Jednak prawdopodobieństwo zaistnienia tego przypadkowego zdarzenia zależy też w pewnym stopniu od czynników zewnętrznych - struktury reagujących cząsteczek i parametrów procesu (przede wszystkim stężenia lub ciśnienia reagentów oraz temperatury).
Trzeba też zdać sobie sprawę z tego, że to co potocznie nazywamy reakcją chemiczną zapisaną schematycznie jako A + B = C jest najczęściej wieloetapowym procesem, przebiegającym przez stadia pośrednie: A + B = X; X + D = Y; ..... = C.
Pojęcie ogólnej reaktywności substancji jest pojęciem dość nieostrym i pozwala na rozróżnienia między substancjami znacznie różniącymi się między sobą, natomiast jest zbyt mało precyzyjny by różnicować substancje o podobnych właściwościach.. Można spokojnie stwierdzić, że chlorek kwasu octowego jest o wiele bardziej reaktywny niż cykloheksan, natomiast porównywanie reaktywności np. alkoholi i kwasów organicznych nie ma większego sensu - brak ostrych kryteriów na podstawie których jednoznacznie można by przypisać jedną z tych grup do większej reaktywności. Z tego powodu często nie mówimy o reaktywności jakiejś substancji "w ogóle" lecz tylko w stosunku do konkretnej reakcji lub grupy reakcji. W stosunku np. do wodorotlenków kwasy organiczne są bardziej reaktywne niż alkohole, natomiast alkohole cechuje wyższa, niż w przypadku kwasów, reaktywność w rekcjach utleniania.
Reaktywność w praktyce oznacza zazwyczaj ilość reakcji w jakie wchodzi dana substancja czy grupa substancji, oraz wydajność tych reakcji w warunkach niezbyt odbiegających od normalnych. Jeżeli jakaś reakcja dla przeprowadzenia z wydajnością liczoną w dziesiątkach procent wymaga temperatury rzędu kilkuset stopni i ciśnienia kilkuset atmosfer, nie bierzemy jej pod uwagę w trakcie rozpatrywania reaktywności danej substancji. Natomiast wszelkie reakcje przebiegające z dość dużą wydajnością w temperaturach do paruset stopni i kilku atmosfer uznajemy za "właściwość" chemiczna danego związku. Z pobieżnej nawet analizy przebiegu reakcji chemicznej (patrz wyżej) jasno wynika, że cecha zwana reaktywnością jest wypadkową kilku innych właściwości cząsteczki reagującej. Prześledźmy te cechy i ich wpływ na reaktywność.
Spotkanie cząsteczek.
Dla cząsteczek niepolarnych czynnikiem decydującym o spotkaniu jest wyłącznie przypadek. Gdy reagentami są cząsteczki polarne dochodzi do tego aktywne oddziaływania ładunków dipoli (przyciąganie ładunków różnoimiennych), zwiększające ilość spotkań a zatem i szybkość reakcji. Ciśnienie bądź temperatura oraz wzrost stężenia "biernie" zwiększają prawdopodobieństwo spotkania się reagujących cząsteczek.
Tak więc pierwszy etap reakcji w danych warunkach zachodzi szybciej (łatwiej) w przypadku substancji polarnych.
Także "unieruchomienie" na jakiś czas cząsteczek reagujących, poprzez siły elektrostatyczne zwiększa szanse zajścia reakcji.Usytuowanie przestrzenne.
Ponieważ reakcje (przegrupowanie atomów i wiązań między reagującymi cząsteczkami) najczęściej zachodzą między elementami cząsteczki (grupami funkcyjnymi) obdarzonymi cząstkowym ładunkiem ujemnym (reakcje elektrofilowe) lub cząstkowym ładunkiem dodatnim (reakcje nukleofilowe) takie spotkanie i "unieruchomienie" cząsteczek reagujących w pozycji (+) (-) ułatwia przegrupowanie atomów i ładunków między reagującymi grupami.Aspekty energetyczne.
Tu, w tym krótkim opracowaniu, wspomnijmy tylko, że odpowiedni poziom energii wewnętrznej obu reagujących cząsteczek i różnica energii między substratami i produktami oraz poziom energii aktywacji decydują o wyniku reakcji. Szczegółowsze dane na temat czynnika energetycznego w przebiegu reakcji znajdziesz w rozdziale o energii w reakcjach i energii cząsteczek.Kształt cząsteczek substratów.
Kształt cząsteczki w powiązaniu z polarnością odgrywa dużą, czasem wręcz decydującą rolę w procesie przemian chemicznych. Przeszkody steryczne (fizyczne, geometryczne) mogą uniemożliwić spotkanie się grup reaktywnych. Przeszkodą w zbliżeniu się grup reagujących na odległość umożliwiającą zajście reakcji może być także silne pole elektryczne podstawników (szczególnie silnie elektroujemnych i "objętościowych" - brom, jod, grupa nitrowa, pierścień fenylowy). Jednocześnie w wielu przypadkach silnie elektroujemne podstawniki na drodze indukcji zwiększają ładunek dodatni dipola cząsteczki ułatwiając reakcje nukleofilowe. Tak więc nie można tu sformułować jednej reguły opisującej wpływ podstawnika na reaktywność cząsteczek.
Podsumowując: najistotniejszym i "najpewniejszym" czynnikiem wpływającym na reaktywność jest polarność - tak całej cząsteczki, jak i wiązań w obrębie centrum reakcji. Wpływ innych czynników mogących mieć znaczenie dla reaktywności danej grupy związków trzeba rozpatrywać kompleksowo, w powiązaniu z daną reakcją i warunkami jej przeprowadzania. Trzeba pamiętać, że także rozpuszczalnik i jego relacje z reagującymi w nim substancjami (solwatacja) mogą w sposób znaczący wpływać na reaktywność.
|
|
alkohol metylowy reaguje z kwasem octowym; nic nie stoi na przeszkodzie do powstania estru metylowego kwasu octowego (octanu metylu) |
|
|
|
|
|
ta sama reakcja między kwasem trichlorooctowym i trichlorometanolem napotyka na duże przeszkody związane z oddziaływaniem (odpychaniem) ładunków ujemnych na elektroujemnych atomach chloru; jednocześnie atomy chloru indukcyjnie zwiększają polarność wiązań w obrębie centrum reakcji (-OH i -COOH) |
|
|
|
|
|
wzór na papierze, jakimi najczęściej operujemy, nie oddaje prawidłowo rzeczywistości; wydaje się, że nic nie broni dostępu do grupy alkoholowej; wzór po lewej wyraźnie pokazuje, że dostęp do tej grupy (czerwono-niebieska) możliwy jest tylko "od dołu", dostępu z innych kierunków "bronią" pierścienie fenylowe, grupy nitrowe (granatowo-czerwone) i atom bromu (zielony) |
Ustalanie struktury cząsteczki
Znajomość struktury cząsteczki jest niezmiernie ważnym elementem - tak w projektowaniu procedur analitycznych prowadzących do wykrywania i oznaczania tych substancji, jak i w planowaniu przemian chemicznych (reakcji) prowadzących do modyfikacji struktury, mającej na celu otrzymanie związków o założonych właściwościach fizycznych i chemicznych. W niektórych przypadkach wystarczy nam znajomość struktury na poziomie pozwalającym napisać uproszczony wzór strukturalny, innym razem konieczna jest głęboka znajomość wszelkich relacji między składowymi cząsteczki (długości wiązań, kątów między nimi, budowa sieci krystalicznej itp.). Badania struktury cząsteczki należą do jednych z bardziej złożonych i skomplikowanych i wymagają głębokiej wiedzy chemicznej, fizycznej i ... matematycznej, oraz "naukowego zacięcia" - bowiem są to badania nie tylko dość złożone, ale na dodatek pośrednie. O strukturze wnioskujemy (obliczamy) na podstawie obserwacji zachowań cząsteczki w różnych warunkach. Trochę to przypomina próby opisania legendarnego yeti, którego nikt nie widział, lecz podejmowano próby jego opisania posługując się wyłącznie znajomością śladów, które pozostawiał (o ile to były jego ślady !).
Poniżej znajdziesz króciutkie omówienie poszczególnych metod i technik przydatnych przy określaniu budowy cząsteczki. Obok większości z nich znajdziesz linki do nieco mniej ogólnych opisów tych metod, lecz nie miej złudzeń - nawet dość obszerny opis to tylko encyklopedyczny skrót. Na temat każdej z tych metod napisano opasłe podręczniki zawierające, tak teoretyczne rozważania jak i techniczne opisy zastosowań. Jeśli zainteresują Cię te techniki, to materiału do studiowania starczy Ci na długo - może nawet na całe życie - bo ich praktyczne zastosowania wciąż ulęgają zmianom, doskonali się technika, pojawiają się nowe zastosowania itp.
Chciałbym tu od razu wyjaśnić, że wymieniając tu akurat te, a nie inne metody, kieruję się ich popularnością i znaczeniem w procesie ustalania struktury (a także w procesie poznawania zasad rządzących przyrodą) - nie są to jednak jedyne i nawet czasem nie najważniejsze metody. Bez obawy popełnienie błędu można bowiem uznać, że wnioski dotyczące struktury cząsteczki można wysnuwać praktycznie z każdych badań o charakterze chemicznym czy fizykochemicznym, że wspomnę tylko o refraktometrii, polarymetrii czy konduktometrii.
Najpopularniejszymi metodami analitycznymi, pozwalającymi w stosunkowo prosty sposób uzyskać wiele informacji na temat struktury cząsteczki są metody spektroskopowe. Spektroskopia jest to dział analityki zajmujący się oddziaływaniem niesprężystym promieniowania elektromagnetycznego i materii. Promieniowanie elektromagnetyczne to rozchodzenie się w przestrzeni kolejno pola elektrycznego i indukowanego przez niego pola magnetycznego, które z kolei indukuje pole elektryczne. Z długością fali promieniowania elektromagnetycznego związana jest ściśle wielkość kwantów energii niesionej przez to promieniowanie:
E = h·
gdzie: E - wielkość energii kwantu niesionego przez promieniowanie, - częstotliwość promieniowania, h - stała Plancka
Ponieważ częstotliwość jest odwrotnie proporcjonalna do długości fali ( = c, gdzie c - prędkość światła w próżni a długość fali) promieniowanie o dłuższych falach niesie mniejsze kwanty energii. Dlatego promieniowanie γ o bardzo dużych energiach kwantów (długość fali poniżej 0,1 nm) jest zdolne do destrukcji cząsteczek i jest dla istot żywych bardzo niebezpieczne, zaś dobrze nam znane promieniowanie z zakresu światła widzialnego (długości fal 350 - 900 nm) zmienia właściwości materii w bardzo ograniczonym zakresie (reakcje fotochemiczne, np. naświetlenie AgBr w fotografii) i jest do życia większości istot wręcz konieczne.
[±²] Główna cechą, różniącą zachowania w mikroświecie cząsteczek, atomów i cząstek elementarnych (mechanika kwantowa) od zachowań w świecie makro (mechanika niutonowska) jest zasada kwantowania energii. Polega ona w największym skrócie na tym, że jeżeli np. elektron na orbitalu s charakteryzuje się energią E1, a na orbitalu p - E2, to aby przenieść elektron z s na p należy mu dostarczyć energię równą E = E2 - E1 w postaci pojedynczego kwantu (porcji). Można wywołać to przejście działając na niego np. promieniowaniem elektromagnetycznym o długości fali niosącej takie właśnie kwanty energii (patrz wzór powyżej). Mówimy wtedy, że dzięki zgodności niesionego przez promieniowanie o długości fali kwantu energii z różnicą poziomów energetycznych elektronu na orbitalach s i p nastąpiło pochłonięcie energii i zjawisko przejścia elektronu na wyższy poziom energetyczny. Jeżeli na ten elektron podziałamy falą elektromagnetyczną o innej długości, zjawisko przejścia elektronu z orbitalu s na orbital p nie nastąpi, bowiem dostępny kwant energii będzie albo "za duży" (krótsze fale) albo "za mały" (dłuższe fale).
Pochłonięty kwant energii, jeżeli nie spowoduje nieodwracalnych zmian w cząsteczce, np. rozerwania wiązania, zostaje po jakimś czasie z cząsteczki wyemitowany, często też w postaci energii promieniowania elektromagnetycznego. Te dwa zjawiska - pochłaniania przez cząsteczkę określonych kwantów energii i ich emitowania (fluorescencja, fosforescencja, promieniowanie rentgenowskie, emisja wzbudzonych atomów) leżą u podstaw spektroskopii. Analizując długości fal pochłanianych i emitowanych przez cząsteczkę i porównując je z energiami przejść w obrębie poszczególnych elementów cząsteczki (znanych bądź z doświadczenia bądź z wyliczeń mechaniki kwantowej) można wnioskować o obecności tych elementów w badanej strukturze. Należy tu pamiętać, że nieznaczne zmiany wielkości pochłanianych kwantów energii będą występować w grupie przemian podobnego typu (np. przejścia elektronów walencyjnych - UV/Vis czy zmiany energii o obrębie wiązań - IR) zaś o rzędy wielkości różnić się będą kwanty powodujące przemiany w różnych elementach struktury (UV - elektrony walencyjne, IR - energie wiązań, NMR - orientacja jąder w polu magnetycznym). Należy też być świadomym tego, że o strukturze cząsteczki będziemy mogli mówić dopiero wtedy, gdy połączymy informacje cząstkowe, uzyskane poszczególnymi metodami. Nie ma (i chyba nigdy nie będzie) takiej metody, która dostarczała by informacji o wszystkich elementach składowych struktury cząsteczki.
UV
Promieniowanie elektromagnetyczne z zakresu ultrafioletu (~100-350 nm) i światła widzialnego (~350-900 nm) niesie kwanty energii zgodne, co do wielkości, z różnicami poziomów energetycznych elektronów walencyjnych cząsteczki. Wykorzystywany do badań tzw. ultrafiolet kwarcowy (200-350 nm) jest w stanie powodować przejścia elektronów wiązań typu (wiązania wielokrotne), a światło z zakresu widzialnego, niosąc jeszcze mniejsze kwanty energii, powoduje przejścia o jeszcze mniejszej różnicy między stanem podstawowym a wzbudzonym (sprzężone wiązania wielokrotne, przeniesienie elektronu), stąd zdecydowana większość związków chemicznych jest bezbarwna, tzn. nie pochłania fal z zakresu widzialnego. Tak więc na podstawie wartości pochłanianej długości fali (max) i natężenia tego zjawiska (max - molowy współczynnik absorpcji) możemy wnioskować o istnieniu wiązań wielokrotnych, ich sprzęganiu (wzajemne położenie) oraz istnieniu w cząsteczce układów aromatycznych. Spektroskopia w ultrafiolecie i świetle widzialnym jest dość prosta w wykonaniu i interpretacji, niesie jednak stosunkowo niewiele informacji o budowie cząsteczki i to na dość niskim poziomie szczegółowości.
IR
Spektroskopia w podczerwieni dotyczy zmian energetycznych zachodzących w wiązaniach jako całości, a wiec zmian spowodowanych oscylacją wiązań (wydłużanie i skracanie się wiązań) jak również ich deformacja (zmianami kątów między sąsiednimi wiązaniami). Daleka podczerwień (fale dłuższe niż 20 000 nm; 20 m) powoduje zmiany energii rotacji całej cząsteczki, ale ponieważ dotyczy szczególnych przypadków, nie będziemy się nią tu zajmować. Podobnie rzecz się ma z bliską podczerwienią (~1500 nm; 1,5 m). Zakres podczerwieni analitycznej najczęściej wykorzystywany i niosący najwięcej szczegółowych informacji to zakres 4 000 cm-1 do 400 cm-1 (2,5 - 25 m; 2 500 - 25 000 nm).
Wszystkie wiązania w cząsteczce ulegają dwóm podstawowym typom zmian - oscylacji (skracanie i wydłużanie wiązania z określoną amplitudą i częstotliwością) oraz deformacji (zmiany kąta między danym wiązaniem a określonym elementem cząsteczki - innym wiązaniem czy płaszczyzną pierścienia lub wiązania podwójnego). Każda z tych zmian wiąże się z pochłonięciem odpowiedniego kwantu energii, czyli zdolnością absorpcji promieniowania podczerwonego o ściśle określonej długości fali. W widmie IR manifestuje się to podstawowymi pasmami walencyjnymi (oscylacje) i deformacyjnymi (zmiany kątów). Ponieważ jednak cząsteczka to nie prosta suma wiązań, ale pewna całość, występują w niej również złożone ruchy - np. wiązanie oscylujące jednocześnie wychyla się poza płaszczyznę pierścienia. Taki złożony ruch to nowa jakość, pochłonięcie innego kwantu niż tylko związanego z oscylacja i deformacją. W widmie pojawiają się pasma kombinacyjne.
Analiza ilości, położenia i natężenia poszczególnych pasm walencyjnych, deformacyjnych i kombinacyjnych, zwana interpretacja widma, przynosi dużą ilość wiadomości na temat poszczególnych elementów struktury, które należy złożyć w spójną całość. Interpretacja widm IR nie należy do rzeczy łatwych, ale poprawnie przeprowadzona dostarcza bardzo dużo i bardzo szczegółowych wiadomości na temat budowy cząsteczki.
NMR
NMR (Nuclear Magnetic Resonance) jest techniką bazującą na zjawisku rezonansu jądrowego, tzn. pochłanianiu przez cząsteczkę (dokładniej - jądra atomowe budujących ją atomów) promieniowania elektromagnetycznego o częstotliwości charakterystycznej dla danego pierwiastka i danych okoliczności. Odkrywcy tego zjawiska otrzymali Nagrody Nobla w latach pięćdziesiątych dwudziestego wieku, zaś za twórcze jego zastosowanie w medycynie (tomografia komputerowa NMR) przypadł Nobel roku 2003.
Jądra atomowe, jako elementy ruchome (wirowanie wokół osi) i obdarzone ładunkiem elektrycznym (dodatnim) wytwarzają wokół siebie pole magnetyczne. Są więc paramagnetykami, które w zewnętrznym polu magnetycznym ustawiają się zgodnie z liniami sił tego pola (ustawienie równoległe). Ze względu jednak na działanie sił zewnętrznych, głównie słabych pól magnetycznych, pochodzących od sąsiednich jąder w cząsteczce, wektor magnetyczny jądra nie ustawia się idealnie równolegle do linii sił pola zewnętrznego, tylko wiruje pod niewielkim kątem do nich, tak że jego wektor zatacza okręg precesji.
Częstotliwość ruchu precesyjnego zależy między innymi od charakterystyki jądra (rodzaju pierwiastka) zaś od częstotliwości precesji i otoczenia w jakim znajduje się badane jądro (budowa cząsteczki) zależy częstotliwość absorbowanego promieniowania elektromagnetycznego. Tak więc znajdując częstotliwość pochłanianego promieniowania możemy ustalić rodzaj pierwiastka i scharakteryzować jego chemiczne otoczenie - a więc w sposób pośredni określić strukturę cząsteczki. Ponieważ zjawisko rezonansu magnetycznego w cząsteczce jest bardzo czułe na wpływy środowiska (struktury cząsteczki) wartości absorbowanej częstotliwości promieniowania elektromagnetycznego bardzo precyzyjnie opisują badana strukturę. Podobnie jak w spektroskopii w podczerwieni, widmo NMR dostarcza bardzo wielu i bardzo precyzyjnych wiadomości o strukturze - wymaga jednak dużej wiedzy i doświadczenia, bowiem inaczej odkodowanie tych wiadomości jest niemożliwe.
Spektrometria mas
Jest to metoda dostarczająca informacji o masie cząsteczkowej badanego związku i o jego "słabych punktach", czyli wiązaniach ulegających najszybciej rozerwaniu. Cząsteczki badanego związku w źródle jonów są przeprowadzane w jony dodatnie (tzw. macierzyste) poprzez wybicie z cząsteczki jednego elektronu walencyjnego. Jony macierzyste w większości przypadków zyskuję w czasie jonizacji taki nadmiar energii, że ulegają szybko dalszemu rozpadowi na jony potomne o różnym składzie, obojętne cząsteczki i rodniki. Pod wpływem pola elektrycznego, a potem i magnetycznego, dodatnio naładowane jony macierzyste i potomne są przyspieszane, "segregowane" według masy i wychwytywane przez detektor. Wynikiem analizy masowej jest zestawienie zawierające udziały poszczególnych jonów potomnych i jonu macierzystego w całkowitym prądzie jonowym - tzw. widmo masowe. Na podstawie widma masowego określa się masę cząsteczkową badanej substancji (masa jonu macierzystego) i masę oraz skład poszczególnych jonów. Pozwala to na ustalenie prawdopodobnej struktury związku. Podobnie jak w innych metodach o strukturze dowiadujemy się nie wprost a jedynie poprzez logiczne powiązanie składu i struktury powstałych jonów ze strukturą badanej cząsteczki.
Spektrometria mas może z powodzeniem zastąpić analizę elementarną i termograwimetryczną w badaniach strukturalnych i badaniach czystości substancji. Niestety jedną (jedyną?) z jej wad jest stosunkowo wysoka cena koniecznej aparatury.
Atomowa spektroskopia absorpcyjna i emisyjna dość luźno są związane z ustalaniem struktury, należą jednak ściśle do tematu spektroskopia i dlatego też zostaną tu pokrótce omówione.
Jak sama nazwa wskazuje, badaniom zostają poddane atomy poszczególnych pierwiastków - prawie wyłącznie metali. Atomizacji dokonuje się różnymi technikami, do najpopularniejszych należy rozpylenie roztworu badanej substancji w płomieniu (najczęściej acetylen-powietrze). Zatomizowany pierwiastek może pochłonąć ściśle określony kwant energii (a więc i ściśle określoną długość fali promieniowania elektromagnetycznego z zakresu UV-Vis). W takim przypadku mówimy o spektroskopii atomowej absorpcyjnej. Pewna część atomów pierwiastka w wysokiej temperaturze płomienia może "samoistnie" przejść w stan wzbudzony i następnie wyemitować nadmiar energii w postaci fali elektromagnetycznej, także o ściśle określonej, charakterystycznej dla danego pierwiastka długości. Wówczas mówimy o atomowej spektroskopii emisyjnej. To ostanie zjawisko wykorzystujemy także do prostej, wizualnej analizy jakościowej pierwiastków - świecenia płomienia na różne kolory w zależności od pierwiastka.
Mierząc długość fali absorbowanej bądź emitowanej przez badane atomy, oraz natężenie tego promieniowania, możemy oznaczyć jakościowo i ilościowo dany pierwiastek. Obecnie metoda AAS najpopularniejsze zastosowanie ma w badaniu różnych substancji na zawartość metali ciężkich - toksycznych dla człowieka.
Analiza elementarna powinna być właściwie omawiana na początku, jako że jest metodą stosowana zazwyczaj jako pierwsza w całym ciągu kolejnych badań strukturalnych. Nie daje nam ona bezpośrednich danych o strukturze, a jedynie określa skład pierwiastkowy - jakościowy i ilościowy. Jednak bez znajomości tego składu interpretacja wyników dalszych badań strukturalnych byłaby znacznie utrudniona.
Analiza elementarna polega na mineralizacji związku organicznego, tzn. utlenienia go aż do uzyskania prostych związków nieorganicznych (CO2; H2O; N2; SO3 itp.). Utlenienie to odbywa się w specjalnym piecu, w wysokiej temperaturze, często z użyciem katalizatorów zapewniających całkowite utlenienie całej badanej próbki. Powstałe w trakcie utlenienia gazy są wraz z gazem nośnym (powietrze lub czysty tlen) przepuszczane przez specjalne absorbery, zawierające substancję pochłaniającą wybiórczo każdy z wymienionych gazów. Na podstawie przyrostu masy każdego z absorberów określa się ilość danego związku nieorganicznego, a na tej podstawie ilość pierwiastka zawartego w analizowanej próbce. Znajomość procentowych zawartości danego pierwiastka pozwala na wyznaczenie wzoru elementarnego, z którego można obliczyć rzeczywisty wzór sumaryczny wykorzystując znajomość masy cząsteczkowej (otrzymaną np. podczas badań MS).
Termograwimetria (TG) i termiczna analiza różnicowa (TDA) choć dość luźno łączą się z tematem badania struktury, podobnie jak analiza elementarna, mogą w sposób szybki prosty i stosunkowo tani dostarczyć wiadomości bardzo pomocnych przy ustalaniu struktury. Zasada metody polega na tym, że próbka kilku-kilkudziesięciu miligramów badanej substancji jest ogrzewana w platynowym tygielku do wysokiej temperatury (nawet 1500° C), i w czasie tego ogrzewania rejestrowana jest masa próbki (TG) i temperatura. Jednocześnie rejestrowana jest różnica temperatur między próbka a atmosferą pieca ogrzewającego próbkę (TDA). W momentach, gdy w próbce następuje reakcja rozkładu bądź spalania, a ogólniej proces egzotermiczny lub endotermiczny, temperatura próbki gwałtownie rośnie lub maleje w stosunku do temperatury otoczenia. Te energetyczne efekty procesów zachodzących w badanej próbce w powiązaniu z ubytkami masy próbki i temperaturą przemiany pozwalają na wyciąganie pewnych wniosków dotyczących struktury, w sposób podobny jak dzieje się to przy interpretacji widm masowych.
Związek chromatografii z badaniem struktury substancji jest dość luźny, niemniej czasami ustalenie struktury związku, szczególnie naturalnego, bez wyizolowania go metodą chromatografii z jego naturalnego otoczenia (tzw. matrycy) byłoby niemożliwe.
Pojęciem chromatografii obejmujemy techniki analityczne wykorzystujące zjawisko różnej szybkości migracji składników analizowanej mieszaniny. Różnica w szybkości migracji poszczególnych składników mieszaniny spowodowana może być przez różne czynniki - rozpuszczalność, współczynnik podziału, adsorpcje, reakcje chemiczne itp. - stąd podstawowy podział technik chromatograficznych na adsorpcyjne, podziałowe i jonowymienne. Nazwa chromatografia ("pisanie barwami") wzięła się od pierwszych doświadczeń w tej dziedzinie rosyjskiego botanika Cwieta, który na początku XX wieku przeprowadzał tą metodą rozdział barwnych związków naturalnych.
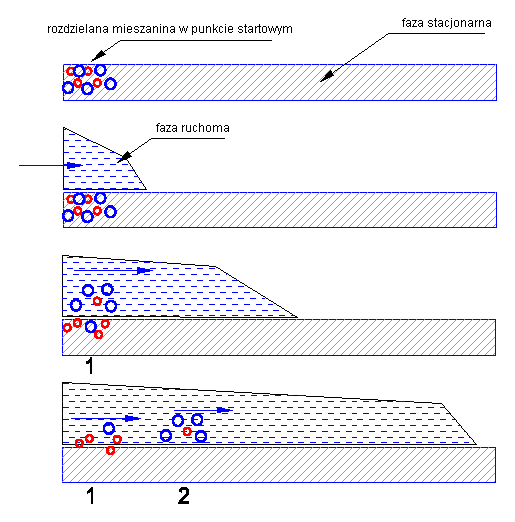
Klasyczny układ chromatograficzny to nieruchoma faza stacjonarna, w stosunku do której przesuwa się faza ruchoma. Składniki rozdzielanej mieszaniny wykazują powinowactwo zarówno do fazy stacjonarnej jak i do fazy ruchomej, jednak wielkość tego powinowactwa jest różna dla różnych składników. W efekcie tych różnic konkretny składnik mieszaniny przesuwa się wzdłuż drogi rozwijania chromatogramu tylko wtedy, kiedy znajduje się w fazie ruchomej. Składniki mieszaniny o większym powinowactwie do fazy ruchomej poruszają się zatem szybciej ("częściej" przebywają w fazie ruchomej), składniki o większym powinowactwie do fazy stacjonarnej - wolniej ("częściej" tkwią nieruchomo na fazie stacjonarnej). Ponieważ szybkość migracji każdego składnika mieszaniny jest wypadkową działania dwóch sił (powinowactw do dwóch faz chromatograficznych), dla większości mieszanin dość łatwo dobrać układ faz (układ chromatograficzny), który zapewni takie różnice w szybkości migracji, by możliwe było fizyczne odseparowanie cząsteczek poszczególnych substancji od siebie.
|
Mieszanina rozdzielanych substancji (czerwone i niebieskie cząsteczki) zostaje naniesiona na punkt startowy na fazie nieruchomej (stacjonarnej)
|
|
Rozpoczyna się ruch fazy ruchomej. Na punkt startowy napływa czysta faza ruchoma. |
|
Lepiej rozpuszczalna substancja niebieska w większości przechodzi do fazy ruchomej i wraz z nią wędruje wzdłuż drogi rozwijania chromatogramu. Substancja czerwona o większym powinowactwie do fazy stacjonarnej i słabej rozpuszczalności w fazie ruchomej w większości pozostaje w punkcie startowym na fazie stacjonarnej |
|
Ruch fazy powoduje, że nad punkt 1 napływa czysty rozpuszczalnik, co przyspiesza rozpuszczanie się w nim cząsteczek czerwonych, zaś cząsteczki które wcześniej poruszały się z faza ruchoma znalazły się nad punktem 2, co spowodowało ich częściowe przejście do fazy stacjonarnej, aż do uzyskania stanu równowagi |
|
Kolejne etapy rozwijania chromatogramu to przechodzenie cząsteczek z fazy ruchomej do stacjonarnej w miejscach, gdzie roztwór w fazie ruchomej styka się z "nie zajętą" fazą stacjonarną, oraz przechodzenie cząsteczek z fazy stacjonarnej do fazy ruchomej tam, gdzie "czysta" faza ruchoma styka się z faza stacjonarną obsadzoną cząsteczkami mieszaniny. |
|
Elementarne procesy przechodzenia z jednej fazy układu chromatograficznego do drugiej odbywają się pod wpływem dwóch czynników - powinowactwa do fazy ruchomej (zdolności rozpuszczania w fazie ruchomej) i powinowactwa do fazy stacjonarnej. |
|
Wypadkowa wartość tych dwóch przeciwstawnych czynników określa średni czas przebywania cząsteczek każdej substancji rozdzielanej mieszaniny w fazie ruchomej. Czas przebywania w fazie ruchomej określa natomiast średnią szybkość migracji wzdłuż drogi rozwijania chromatogramu, bowiem cząsteczki migrują tylko wraz z fazą ruchomą. |
|
Odpowiednio dobrany układ chromatograficzny powoduje uzyskanie takich różnic w szybkości migracji poszczególnych składników rozdzielanej mieszaniny, że odległości między skupiskami poszczególnych cząsteczek po zakończeniu procesu, są wystarczająco duże dla fizycznego odseparowania ich od siebie - dokonania rozdziału chromatograficznego |
W najprostszym, schematycznym ujęciu można to przedstawić następująco - cząsteczki czerwone mające silniejsze powinowactwo do fazy stacjonarnej poruszają się "krótszymi krokami" (linia czerwona) niż cząsteczki niebieskie, które szybciej przechodzą do fazy ruchomej i trudniej są z niej wychwytywane przez fazę stacjonarna (linia niebieska)
W chwili obecnej proces chromatograficznego rozdzielania możemy przeprowadzać na wiele sposobów, wieloma technikami. Podziału technik chromatograficznych można dokonywać na wiele sposobów, biorąc pod uwagę różne czynniki procesu. Wszystkie podziały są dość umowne i mają jedynie służyć łatwiejszemu porozumiewaniu się "chromatografistów", a nowym adeptom sztuki chromatograficznej maja ułatwić poruszanie się w gąszczu technik i ich odmian.
Ze względu na podstawowe zjawisko fizykochemiczne prowadzące do uzyskania rozdziału składników chromatografowanej mieszaniny wyróżniamy chromatografię adsorpcyjną, podziałową i jonowymienną. Ze względu na rodzaje stosowanych faz dzielimy chromatografię na gazową (faza ruchoma jest gazem), cieczową grawitacyjną (fazą ruchoma jest ciecz przepływająca przez złoże fazy stacjonarnej pod wpływem siły grawitacji) i cieczową ciśnieniową (przepływ fazy wymuszony jest wysokim ciśnieniem wytwarzanym przez specjalne pompy). Inny podział wyróżnia techniki planarne (np. cienkowarstwowa lub bibułowa) i kolumnowe (faza stacjonarna wypełnia szklana lub metalową rurkę - kolumnę). W ostatnich latach coraz częściej stykamy się z pojęciem chromatografii z użyciem odwróconych faz. Ponieważ do tej pory najczęstszym układem chromatograficznym był układ, w którym faza stacjonarna była bardziej polarna (bądź miała większe powinowactwo do substancji polarnych) niż faza ruchoma - układ, w którym faza ruchoma jest bardziej polarna niż stacjonarna uzyskał miano układu faz odwróconych (RP - reverse phase). Konkretną technikę można zazwyczaj zaliczyć do kilku grup jednocześnie, jako że kryteria poszczególnych podziałów najczęściej nie wykluczają się nawzajem.
O chromatografii adsorpcyjnej mówimy wówczas, gdy głównym zjawiskiem determinującym przebieg procesu chromatograficznego jest zjawisko adsorpcji. Zjawisko to polega z grubsza na tym, że cząsteczka substancji łączy się w sposób nietrwały (głównie siłami o charakterze wiązania wodorowego) z powierzchnia adsorbenta (np. żel krzemionkowy, tlenek glinu). W chwili gdy zaadsorbowana cząsteczka styka się z "czystą" fazą ruchomą następuje zjawisko desorpcji i cząsteczka przechodzi ponownie do fazy ruchomej.
W chromatografii podziałowej zjawiskiem decydującym o przebiegu procesu jest zjawisko podziału substancji między dwie ciekłe, nie mieszające się fazy. Ciekłą fazę stacjonarną uzyskujemy przez osadzenie na ziarnach stałego nośnika cienkiej warstewki odpowiednio dobranej cieczy (również pod względem parametrów fizycznych, głównie lepkości i związanej z tym siły przylegania - adhezji). Zgodnie z prawem podziału Nernsta, stosunek stężeń w obu fazach w stanie równowagi jest wielkością stałą i charakterystyczną w danych warunkach dla danej substancji. Tak więc, gdy stężenie składnika w fazie ruchomej jest większe, niż to wynika z prawa podziału, substancja przechodzi do fazy ciekłej stacjonarnej. Jeśli po chwili napłynie faza ruchoma o mniejszym stężeniu tej substancji nastąpi częściowe przejście z fazy stacjonarnej do fazy ruchomej (aż do ustalenia odpowiedniego stosunku stężeń w obu fazach).
Jest to typ chromatografii dość znacznie odbiegający od innych. Tu decydującą rolę pełni reakcja chemiczna, i to reakcja z zakresu oddziaływań elektrolitów. Prawie zawsze jest to technika kolumnowa, gdzie wypełnienie kolumny stanowi najczęściej granulowano drobno żywica jonowymienna. Jest to substancja z grupy polimerów, posiadająca na swej powierzchni grupy aktywne chemicznie - najczęściej o charakterze kwaśnym (kwasy sulfonowe) lub zasadowym. Te aktywne grupy chemiczne wychwytują z przepływającego przez kolumnę roztworu kationy lub aniony (w zależności od rodzaju wypełnienie), które później można wymyć z kolumny odpowiednim eluentem. Stosując odpowiednie wypełnienia i eluenty lub łącząc szeregowo kolumny o różnym wypełnieniu można doprowadzić do rozdzielenia mieszaniny kationów lub anionów.
Większe znaczenie żywice jonowymienne maja w przypadku stosowania ich do demineralizacji wody. Woda przepuszczana przed złoże kationitu a później anionitu, zostaje pozbawiona elektrolitów, co w niektórych przypadkach jest konieczne dla sprawnego działania urządzeń (kotły parowe, ogrzewanie wodne itp.). Jest to dość prosta i stosunkowo tania metoda uzdatniania wody. Zużyte kationity i anionity można następnie regenerować, przemywając je kwasem lub zasadą.
Chromatografia gazowa (Gas Chromatography - GC) ma jedno ważne ograniczenie - można ją stosować tylko dla związków lotnych w temperaturze chromatografowania (do ~350°C) i trwałych w tej temperaturze. W praktyce oznacza to najczęściej związki o masie cząsteczkowej poniżej 400 D.
Chromatografowanie przeprowadza się w kolumnie wypełnionej odpowiednim adsorbentem lub ciekłą fazą stacjonarną osadzoną na nośniku. Kolumna w czasie procesu chromatograficznego ogrzana jest do odpowiedniej temperatury. Nastrzyknięta na czoło kolumny mieszanina pod wpływem wysokiej temperatury przechodzi w postać gazową i wraz z gazem nośnym (wodór, azot, hel, itp.), stanowiącym w tej technice fazę ruchomą, przepływa przez kolumnę. Faza stacjonarna wychwytuje cząsteczki z fazy ruchomej poprzez adsorpcje lub rozpuszczanie w fazie ciekłej, a następnie oddaje do fazy ruchomej, zgodnie z zasadami zjawiska adsorpcji-desorpcji lub rozpuszczania gazów. Różna lotność składników analizowanej mieszaniny w połączeniu z różnym powinowactwem do fazy stacjonarnej daje w efekcie rozdzielenie składników.
W chromatografii gazowej dużą role odgrywają detektory, przy pomocy których wykrywamy i oznaczamy opuszczające kolumnę w gazie nośnym składniki mieszaniny. Ze względu na dużą różnorodność chemiczną i fizyczną analizowanych substancji wykorzystuje się wiele detektorów o różnych zasadach działania i bardzo różnej konstrukcji.
Wysokosprawna chromatografia cieczowa (HPLC - High Performance Liquid Chromatography; High Pressure Liquid Chromatography) jest dziś najpopularniejszą i najbardziej uniwersalną techniką chromatograficzną. Jest to technika kolumnowa, ciśnieniowa, w której faza ruchoma przepływa przez kolumnę wypełnioną drobnoziarnistą fazą stacjonarną pod dużym ciśnieniem (najczęściej 200-400 atm). Dzięki temu faza stacjonarna może być drobnoziarnista i gęsto upakowana (duża powierzchnia styku z fazą ruchomą, ale i duże opory dla przepływu fazy ruchomej), a faza ruchoma przepływać z prędkością kilku- kilkunastu mililitrów na minutę (w standardowych kolumnach) co pozwala skrócić czas analizy do sensownych rozmiarów (od kilkunastu minut do paru godzin). Stałość przepływu i wysokie ciśnienie zapewnia specjalna pompa, a jako detektor najczęściej stosuje się spektrofotometr UV, który sprzężony z rejestratorem lub komputerowym urządzeniem rejestrującym wykreśla chromatogram, na podstawie którego można jakościowo (na podstawie czasów retencji, czyli czasu między rozpoczęciem analizy a opuszczeniem przez składnik kolumny chromatograficznej) i ilościowo (na podstawie wielkości pola pod analizowanym pikiem chromatogramu) określić konkretny składnik mieszaniny. Kolumny w HPLC to grubościenne rurki stalowe o średnicy wewnętrznej 2- mm i długości 10-30 cm. Przewody doprowadzające fazę ruchomą pod ciśnieniem też są wykonane z metalu i są grubościennymi kapilarami o średnicy wewnętrznej rzędu 0,1 mm
HPLC jest stosowana jako chromatografia podziałowa, adsorpcyjna, w układzie faz odwróconych, zarówno izokratycznie (taki sam skład fazy ruchomej przez cały proces) jak i gradientowo. Ten ostatni sposób polega na tym, że w czasie procesu faza ruchoma zmienia swój skład (w sposób kontrolowany), tak aby wszystkie składniki uległy rozdzieleniu a czas analizy nie był zbyt długi.
Chromatografia cienkowarstwowa (TLC), mimo wielu mankamentów, ze względu na taniość i prostotę jest wciąż bardzo popularna techniką jakościowej analizy mieszanin. Często wykorzystuje się ją do szybkiej analizy na obecność niepożądanych zanieczyszczeń. Przy jej zastosowaniu możliwa jest również analiza ilościowa, lecz tu znacznie przewyższa ją technika HPLC.
Rozwijanie chromatogramu odbywa się na płytkach szklanych lub z folii aluminiowej pokrytych cienką warstewką adsorbentu (stąd nazwa). Na suchą płytkę nanosimy na linię startową niewielką ilość rozdzielanej mieszaniny, nakrapiając kilka miniaturowych kropel roztworu mieszaniny i odparowując rozpuszczalnik. Następnie płytkę wstawiamy do naczynia z fazą ruchomą, tak by poziom fazy był około 1 cm poniżej linii startowej. Całość wstawiamy do komory chromatograficznej, naczynia szczelnie zamkniętego i nasyconego parami fazy ruchomej, aby w czasie procesu chromatograficznego, jako skutek parowania poszczególnych składników, nie zmieniał się skład fazy ruchomej.
Faza ruchoma, dzięki siłom kapilarnym, samorzutnie pełznie po płytce do góry, co nazywamy rozwijaniem chromatogramu. Zakończenie procesu następuje zazwyczaj po osiągnięciu przez czoło rozpuszczalnika wysokości 10 - 15 cm od linii startowej (wartość parametru d). Mierząc na płytce wartości a i b obliczamy dla poszczególnych substancji wartości tzw. współczynnika retencji Rf = droga substancji / droga czoła fazy ( Rf =a/d dla substancji niebieskiej i Rf = b/d dla substancji fioletowej - patrz rysunek poniżej). Współczynnik ten jest charakterystyczny dla danej substancji w danych warunkach procesu chromatograficznego i może służyć do wstępnej identyfikacji substancji.
Jeżeli rozdzielane substancje nie są barwne, plamy możemy obserwować w świetle ultrafioletowym (wykorzystujemy tu zjawisko fluorescencji) lub spryskujemy płytkę odpowiednim odczynnikiem, z którym składniki rozdzielanej mieszaniny dają barwne produkty reakcji. Prostym i dość skutecznym sposobem uwidaczniania bezbarwnych plam jest wstawienie płytki z rozwiniętym chromatogramem na parę minut do komory zawierającej pary jodu. W celu otrzymania par jodu wystarczy nasypać na dno komory parę kryształków pierwiastkowego jodu i poczekać kilka minut (sublimacja jodu).
Wyszukiwarka
Podobne podstrony:
Cząsteczka, CHEMIA, semestr 1, chemia ogólna
ATOM I CZĄSTECZKA CHEMIA
Cząsteczka (VB), CHEMIA, semestr 1, chemia ogólna, wykłady
Masa atomowa i cząsteczkowa, NAUKA, chemia, lab
chemia, poprawa I kolo, Adsorpcja apolarna- zachodzi ona wówczas gdy czasteczka związku jest adsobow
Chemia labolatorium, Grupy funkcyjne, Grupa funkcyjna jest to charakterystyczne ugrupowanie atomów w
Chemia Fizyczna masa czasteczkowa id 112216
kartk. - masa czasteczkowa i prawo stałości składu, chemia, kartkówki
Cząsteczka (MO), CHEMIA, semestr 1, chemia ogólna, wykłady
Budowa atomów i cząsteczek, LICEUM różne, CHEMIA
atom i czasteczka wzory reakcje chemia gimnazjum
Chemia Atom, cząsteczka, wiązanie chemiczne
Cząsteczki elementarne, Chemia
oznaczanie średniej wiskozymetrycznej masy cząsteczkowej polimerów, Chemia fizyczna, laboratorium, C
Biłyk,chemia wody, Budowa cząsteczki wody
Sprawdzian kl IG ATOM I CZĄSTECZKA, GIM CHEMIA
więcej podobnych podstron