![]()
TERMOCHEMIA
Termochemia jest małym działem termodynamiki chemicznej, będącym praktycznym zastosowaniem pierwszej zasady termodynamiki do ciepeł reakcji chemicznych i przemian fizykochemicznych jak przemiany fazowe czy rozpuszczanie substancji. Każdej reakcji towarzyszy jakiś efekt cieplny, który można zmierzyć. Reakcje zachodzące z wydzieleniem ciepła nazywamy egzotermicznymi, z pochłanianiem ciepła z otoczenia, endotermicznymi.
Do pomiaru ciepła reakcji chemicznych służą urządzenia zwane kalorymetrami. Wyróżniamy kalorymetry :
Adiabatyczne, w których nie ma wymiany ciepła pomiędzy wnętrzem kalorymetru a otoczeniem. Tego typu kalorymetry są urządzeniami o złożonej konstrukcji, wymagającymi w obsłudze. Stosowane są zwykle w precyzyjnych pomiarach, przy ustalaniu ciepeł reakcji wykorzystywanych jako wzorcowe w pomiarach rutynowych. Zwykle jest to kalorymetr z dużym płaszczem wodnym i bardzo dobrą izolacją cieplną. W kalorymetrze takim istnieje specjalny system sterujący temperaturą płaszcza (grzałką i chłodnicą w płaszczu), tak aby była ona ciągłe taka sama jak w naczyniu, w którym prowadzona jest reakcja.
Diatermiczne, w których istnieje ograniczona wymiana ciepła z otoczeniem. Jest to zwykle naczynie o odpowiednio zaizolowanych termicznie ściankach (np. termos czyli naczynie Dewara), w którym przeprowadza się badaną reakcję (zwykle w środowisku wodnym) i śledzi się przebieg temperatury w czasie. Aby prawidłowo zmierzyć efekt cieplny reakcji, należy ocenić wymianę ciepła pomiędzy kalorymetrem a otoczeniem. W tym celu należy obserwować zmiany temperatury w kalorymetrze przed i po reakcji przez pewien przeciąg czasu. Kalorymetry diatermiczne są najczęściej stosowanymi w praktyce.
Izotermiczne, w których utrzymywana jest stała temperatura w trakcie pomiaru. W poprzednich dwu typach kalorymetrów ciepło reakcji jest znajdowane poprzez zmiany temperatury kalorymetru. W kalorymetrze izotermicznym, temperatura jest niezmienna, więc powstaje pytanie, jak mierzone jest ciepło. Typowym przykładem takiego kalorymetru jest kalorymetr lodowo-wodny, mierzący ciepła reakcji w temperaturze 0ºC. Płaszcz tego kalorymetru wypełniony jest mieszanina wody i lodu i dobrze zaizolowany termicznie. Przemianie pomiędzy wodą a lodem towarzyszy istotna zmiana objętości : wzrost przy krzepnięciu, spadek przy topnieniu. Mierząc zmiany objętości mieszaniny wody i lodu można dokładnie określić, jaka ilość wody skrzepła bądź stopiła się, a znając ciepło topnienia/krzepnięcia obliczyć ilość ciepła pochłoniętą bądź oddaną przez płaszcz.
Do pomiarów ciepeł spalania substancji organicznych stosuje się specjalne urządzenie zwane bombą kalorymetryczną, które wstawia się do odpowiedniego kalorymetru. Bomba kalorymetryczna to naczynie za stali kwasoodpornej, zamykane szczelnie, które może wytrzymać wysokie ciśnienie. W bombie takiej umieszcza się tabletkę ze sprasowanej substancji, nabija się ją tlenem pod wysokim ciśnieniem. Zapłon tabletki wywoływany jest przez przepływ prądu przez drucik znajdujący się w tabletce. Ponieważ bomba jest naczyniem zamkniętym, czyli reakcja spalania zachodzi w tym wypadku w stałej objętości, to mierzone ciepło jest zmianą energii wewnętrznej. Należy dodać, że w większości wypadków pomiary kalorymetryczne prowadzone są jako pomiary porównawcze. Ciepło badanej reakcji jest porównywane z ciepłem reakcji wzorcowej. Najpierw w kalorymetrze przeprowadza się reakcję wzorcową, której ciepło jest dokładnie znane i na tej podstawie ustala się pojemność cieplną kalorymetru (ilość ciepła potrzebną do ogrzania kalorymetru o jeden stopień) czyli tak zwaną stałą kalorymetru, a następnie przeprowadza się reakcję badaną i oblicza się poszukiwane ciepło. W obu wypadkach do obliczeń służy wzór kalorymetryczny.
![]()
gdzie : K - stała kalorymetru Q - ciepło reakcji, która zaszła w kalorymetrze T - zmiana temperatury kalorymetru spowodowana zajściem reakcji
Szczegóły pomiarów kalorymetrycznych, opracowanie wyników, a szczególnie znajdowania T, obliczania ciepeł reakcji poznajecie na zajęciach laboratoryjnych i nie będziemy tu ich omawiać.
Z pierwszej zasady termodynamiki wynika, że w układzie zamkniętym, przy braku pracy nieobjętościowej, ciepło w procesie izochorycznym jest równe zmianie energii wewnętrznej, a w procesie izobarycznym zmianie entalpii.
Ponieważ energia wewnętrzna i entalpia są funkcjami stanu i ich zmiana nie zależy od drogi przemiany, to w określonych uprzednio warunkach ciepło nie zależy od drogi procesu. W odniesieniu do reakcji chemicznych ten wniosek z pierwszej zasady termodynamiki formułuje się jako prawo Hessa.
Prawo Hessa
Prawo Hessa pozwala na obliczanie efektów cieplnych reakcji w oparciu o ciepła innych reakcji. Jeśli możemy stworzyć równanie reakcji nas interesującej przez kombinację liniową innych reakcji, to analogiczne działania wykonujemy na ich efektach cieplnych i otrzymujemy poszukiwane ciepło reakcji. Używa się go do znajdowania efektów cieplnych reakcji, których nie można przeprowadzić w praktyce z rozmaitych względów (zachodzą zbyt wolno lub w sposób niestechiometryczny). Wykorzystywane jest też do rozmaitych obliczeń z wykorzystaniem danych tablicowych.
Czasem prawo Hessa bywa formułowane następująco (w odniesieniu do reakcji przebiegających pod stałym ciśnieniem). Jeśli reakcja może zachodzić przez etapy pośrednie, to całkowita zmiana entalpii reakcji (ciepło pod stałym ciśnieniem) jest równa sumie zmian entalpii etapów pośrednich.
Rysunek 1. Ilustracja prawa Hessa
W przypadku przedstawionym na rysunku 1, entalpia całej reakcji wyrazi się jako :
rHcałkowita = rH1 + rH2
Uwagi o zapisie reakcji i symbolach
W termochemii, a właściwie w całej chemii fizycznej reakcje chemiczne zapisujemy w pewien specyficzny sposób, podając jako indeksy dolne stan reagentów. Jest to istotne, gdyż na przykład ciepło reakcji tworzenia wody będzie inne, gdy powstaje ona jako ciecz czy jako gaz (para wodna).
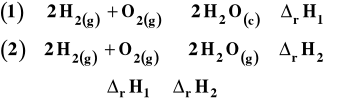
Czasem dodatkowo dodajemy informacje o stężeniach substancji w roztworze bądź ciśnieniach reagentów gazowych. W przypadku stałych reagentów czasem niewystarczające jest podanie informacji o stanie skupienia, lecz trzeba dodać, o jaką postać alotropową nam chodzi. Na przykład w odniesieniu do węgla nie wystarczy podać, że jest stały, ale należy dopisać czy jest to grafit czy diament.
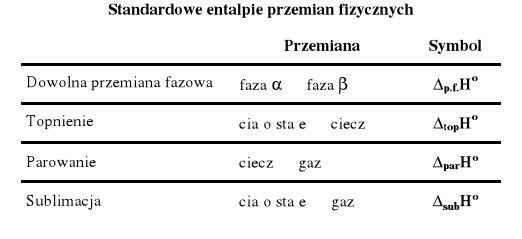
Wszystkie zmiany funkcji stanu np. standardowe entalpie tworzenia, standardowe entropie reakcji, standardowe entalpie swobodne tworzenia itp. zapisujemy w określony sposób, który jest wyjaśniony na rysunku 2 poniżej. Należy przestrzegać tej formy zapisu !
Dodajmy, że dla każdej reakcji chemicznej czy przemiany fizykochemicznej mamy do czynienia ze zmianą funkcji termodynamicznej. Podkreślamy to dodając zawsze przed symbolem funkcji znak oznaczający zmianę. Formalnie rzecz biorąc mamy więc zmianę energii wewnętrznej reakcji czy zmianę entalpii reakcji, ale w uproszczeniu mówi się o energii wewnętrznej reakcji czy entalpii.
Rysunek 2. Sposób zapisu symboli zmian funkcji termodynamicznych
Związek pomiędzy energią wewnętrzną i entalpią reakcji (ciepłem reakcji w stałej objętości i pod stałym ciśnieniem)
Zwykle mierzymy ciepła reakcji pod stałym ciśnieniem. Również większość danych w tablicach fizykochemicznych dotyczy entalpii reakcji. Jednak czasem wiemy, że dana reakcja w praktyce będzie prowadzona w zamkniętym reaktorze czyli w stałej objętości i chcemy ocenić jej efekt cieplny. I na odwrót, ciepła spalania mierzymy w warunkach izochorycznych (w zamkniętej bombie kalorymetrycznej), a chcemy następnie takie ciepło będące zmianą energii wewnętrznej przeliczyć na zmianę entalpii. Skorzystamy z różniczki entalpii.
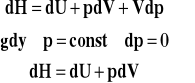
Ostatecznie :
![]()
Symbol rV oznacza zmianę objętości w reakcji chemicznej czyli różnicę objętości produktów i substratów. Zmianę tę można wyrazić jako różnicę objętości molowych produktów pomnożonych przez odpowiednie współczynniki stechiometryczne minus to samo dla substratów.
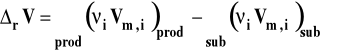
gdzie : i - współczynnik stechiometryczny danego reagenta Vm,i - objętość molowa reagenta
W przypadku reagentów w fazach skondensowanych (stałych, ciekłych, w roztworach) zmiany objętości w reakcji są znikomo małe (rzędu cm3 czyli 10-6 m3) i można je zaniedbać. W tym wypadku ciepło reakcji w stałej objętości i pod stałym ciśnieniem są praktycznie sobie równe.
![]()
Objętość molową reagenta gazowego można w przybliżeniu określić z równania stanu gazu doskonałego.
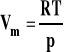
Wobec tego zmianę objętości można wyrazić jako :
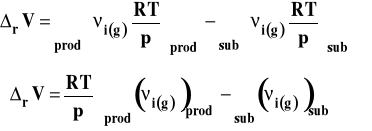
Ostatecznie ciepło reakcji pod stałym ciśnieniem i w stałej objętości można przeliczać nawzajem na siebie korzystając ze z wzoru :
![]()
gdzie 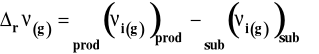
Pamiętajmy, że w obliczeniach uwzględniamy tylko reagenty gazowe ! Na przykład :
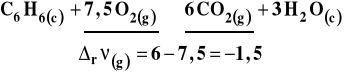
Na tym przykładzie widać, jak ważne jest notowanie w reakcjach stanu reagentów.
Pojęcie stanu standardowego i standardowych entalpii
Aby móc porównywać entalpie reakcji, prowadzić obliczenia z ich udziałem, musimy mieć wartości odnoszące się do tych samych warunków. W tym celu definiujemy stan standardowy i wielkości standardowe.
Stan standardowy
Zauważmy, że w definicji stanu standardowego nie ma określonej temperatury, a tylko ciśnienie standardowe. W myśl obecnych ustaleń Unii Chemii Czystej i Stosowanej (IUPAC) nie istnieje pojęcie temperatury standardowej.
Standardowa entalpia reakcji chemicznej
Standardową entalpię reakcji oznaczamy ![]()
.
Stan standardowy dla roztworu
Ponieważ wiele reakcji chemicznych przebiega w roztworach, musimy zdefiniować, co rozumiemy przez stan standardowy roztworu.
W związku z tą definicją definiujemy standardową entalpię rozpuszczania ![]()
.
Określamy entalpie przemian fazowych jako zmianę entalpii podczas przemiany jednego mola substancji z jednej fazy do drugiej. W zależności od typu przemiany możemy wyróżnić standardową entalpię topnienia, parowania, sublimacji, przemiany alotropowej (np. siarki rombowej w jednoskośną). Standardowe entalpie przemian fazowych są podawane zwykle dla temperatury, w której dana przemiana zachodzi pod ciśnieniem standardowym.
Standardowa entalpia tworzenia związku chemicznego
Jak należy rozumieć tę definicję ? Reakcja, w której powstaje dany związek z pierwiastków, musi być tak zapisana, aby powstawał jego jeden mol. Na przykład reakcję powstawania Na2CO3 stałego zapiszemy następująco :
![]()
Zmiana entalpii w tej reakcji będzie w warunkach standardowych standardową entalpią tworzenia. Jak wiadomo, rozmaite substancje mogą występować w rozmaitych postaciach. Na przykład tlen występuje w postaci cząsteczek O2 i O3 (ozon). Standardową postacią jest tlen o cząsteczkach dwuatomowych. Węgiel w stanie stałym występuje w kilku różnych postaciach alotropowych : grafit, diament i rozmaite fullereny. Jako postać standardową uważa się grafit. Formalnie reakcje tworzenia grafitu i diamentu można zapisać jako :
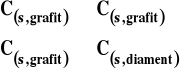
Ponieważ entalpia tworzenia to zmiana entalpii (różnica pomiędzy stanem końcowym i początkowym), to w pierwszej reakcji jest ona równa zero, a w drugiej nie. Wobec tego standardowa entalpia tworzenia grafitu wynosi zero, a diamentu jest różna od zera. Dla stałej siarki odmianą standardową jest siarka rombowa. Ponieważ entalpię tworzenia definiujemy jako zmianę, to w definicji musi być zawarte określenie tego, co przyjmujemy jako poziom zerowy. Bez określenia tego poziomu odniesienia definicja nie jest pełna. Może to wyjaśni lepiej analogia z geografii.
Rysunek 3. Kto ma rację w określeniu wysokości pagórka w myśl zasad geograficznych ?
Bezwzględną wysokość w geografii określamy w odniesieniu do umownego poziomu morza przyjętego jako zero. Czyli rację ma tylko ten, kto stoi na poziomie morza (h3). W definicji standardowej entalpii tworzenia pierwiastki w najtrwalszej odmianie to taki „poziom morza”. A dlaczego mówimy o czystych i rozdzielonych pierwiastkach. Jak dowiemy się później, tworzeniu mieszanin towarzyszy pewna zmiana entalpii i to musi być wyeliminowane.
Standardowa entalpia tworzenia jonu
Na przykład reakcję tworzenia jonów Cl- (w roztworze wodnym) zapiszemy następująco :
![]()
gdzie symbol „aq” oznacza nadmiar wody, a reakcje tworzenia jonu wodorowego :
![]()
przy czym entalpia tej reakcji w myśl definicji wynosi zero. Standardowe entalpie tworzenia jonów wprowadzono, aby móc łatwo obliczać efekty cieplne reakcji jonowych zachodzących w roztworach.
Powstaje pytanie, w jaki sposób można wyznaczyć standardowa entalpie tworzenia jonu, skoro nie można przeprowadzić w żaden sposób reakcji, w której powstawałyby tylko dane jony. Zsumujmy przedstawione reakcje tworzenia jonów H+ i Cl-.
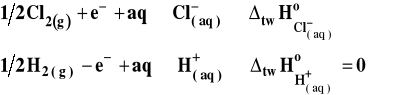
![]()
Można na podstawie pomiarów doświadczalnych wyznaczyć standardowe entalpie tworzenia gazowego HCl i rozpuszczania gazowego HCl w wodzie, które można zapisać następująco :
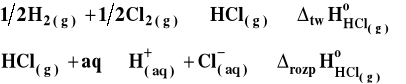
![]()
Po zsumowaniu, jak widać, otrzymujemy taką samą reakcję jak poprzednio. Wobec tego :
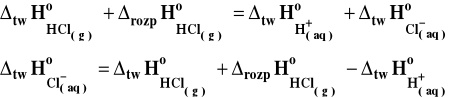
Na mocy definicji standardowa entalpia tworzenia jonu wodorowego wynosi zero, co pozwala na obliczenie standardowej entalpii tworzenia jonu chlorkowego. Mając tą wartość, można następnie napisać podobny cykl termodynamiczny dla na przykład NaCl i znaleźć standardową entalpię tworzenia jonu sodowego, i tak dalej.
Standardowe entalpie tworzenia związków chemicznych i jonów są znane i stabelaryzowane w zbiorach danych fizykochemicznych dla ogromnej liczby związków. Podawane są zwykle dla umownie wybranej temperatury 25°C (298 K). Temperatura ta dawniej zwana była temperaturą standardową i w starszych podręcznikach możecie spotkać takie pojęcie. W myśl obecnej definicji stanu standardowego, nie ma pojęcia temperatury standardowej ! Mogą one być wykorzystane do obliczenia entalpii dowolnej reakcji (o ile są tylko odpowiednie dane).
Rysunek 4. Schemat wyznaczania entalpii reakcji na podstawie standardowych entalpii tworzenia związków.
Z prawa Hessa wynika, że :
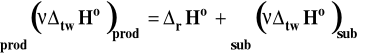
i standardową zmianę entalpii reakcji obliczamy ze wzoru :
Standardowa entalpia spalania
W zasadzie standardowe entalpie spalania są określane tylko dla związków organicznych. Standardowe entalpie spalania są stabelaryzowane zwykle dla umownej temperatury 25ºC. Wykorzystuje się do obliczania efektów cieplnych reakcji organicznych.
Rysunek 5. Schemat wyznaczania entalpii reakcji w oparciu o standardowe entalpie spalania związków.
Zauważmy, że w tym wypadku poziomem odniesienia „zerowym” są produkty spalania. Wobec tego standardowa entalpia spalania wody czy dwutlenku węgla wynosi zero. Z prawa Hessa wynika, że :
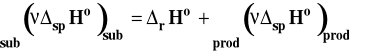
i standardową entalpię reakcji obliczamy ze wzoru :
Energia wiązania
Energie wiązań przydatne są do obliczania energii całych cząsteczek i dyskusji efektów energetycznych w nich jak energia napięcia pierścienia w małych węglowodorach alifatycznych czy energia rezonansu w węglowodorach aromatycznych. Jak należy rozumieć energię i wiązania i jak się ją znajduje można pokazać na poniższym przykładzie.
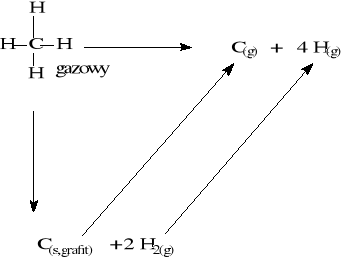
Aby określić energię wiązania C-H, bierzemy pod uwagę entalpię rozpadu metanu na atomy w stanie gazowym. Entalpia tej reakcji będzie równa czterem energiom wiązania C-H, przy czym zakładamy, że energia każdego z nich jest jednakowa czyli są one równocenne energetycznie. Aby to osiągnąć, tworzymy alternatywną drogę reakcji, poprzez rozmaite etapy pośrednie, przy czym muszą to być takie reakcje, których entalpie są znane. Na mocy prawa Hessa zmiana entalpii dla obu dróg przejścia od metanu do atomów jest taka sama. Wobec tego :
![]()
a energię wiązania C-H obliczymy ostatecznie jako :
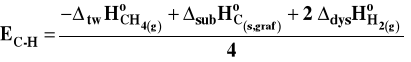
Wszystkie dane potrzebne do obliczenia energii wiązania można wyznaczyć w sposób mniej lub bardziej bezpośredni na podstawie wyników eksperymentalnych.
Zależność ciepeł reakcji od temperatury
Ciepło każdej reakcji zależy od temperatury. Wobec tego musimy umieć przewidzieć, jak zmiana temperatury wpłynie na ciepło reakcji i przeliczyć efekt cieplny z jednej temperatury na drugą. Służy temu prawo Kirchoffa.
Weźmy pod uwagę reakcję prowadzoną w układzie zamkniętym pod stałym ciśnieniem.
![]()
Formalnie możemy napisać, że entalpia tej reakcji to różnica molowych entalpii produktów i substratów w stanie standardowym, pomnożonych przez odpowiednie współczynniki stechiometryczne.
![]()
Zróżniczkujmy obie strony tego równania po temperaturze przy stałym ciśnieniu.
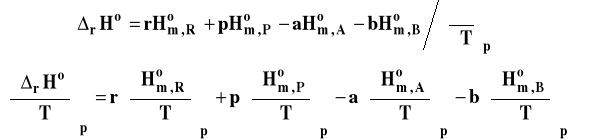
Wiemy, że pochodna entalpii molowej po temperaturze przy stałym ciśnieniu, to ciepło molowe izobaryczne.
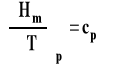
Wobec tego :
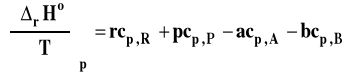
Prawo Kirchoffa
Prawo Kirchoffa można zapisać wzorem w postaci różniczkowej.
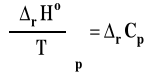
gdzie 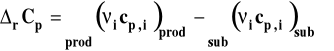
Prawo (wzór) Kirchoffa w postaci różniczkowej pozwala przewidzieć jakościowo wpływ temperatury na entalpię reakcji. Zauważmy, że po lewej stronie mamy pierwszą pochodną entalpii reakcji po temperaturze. Znak pierwszej pochodnej określa, czy funkcja jest malejąca czy rosnąca. Wobec tego wpływ temperatury na entalpię reakcji zależy od znaku zmiany pojemności cieplnej po prawej stronie wzoru Kirchoffa. Jeśli rCp < 0 to, gdy temperatura rośnie to entalpia reakcji maleje. Natomiast jeśli rCp > 0, to wraz ze wzrostem temperatury entalpia reakcji też rośnie.
Jednakże w większości przypadków prawo Kirchoffa w postaci różniczkowej jest niewystarczające, gdyż najczęściej chcemy obliczyć wartość entalpii reakcji w jakiejś temperaturze znając jej wartość w innej. Na przykład dysponujemy wartościami tablicowymi podawanymi dla temperatury 25ºC, a chcemy obliczyć entalpię reakcji w 80ºC, bo w takiej rzeczywiście będziemy ją prowadzić. Bądź też mierzymy ciepło reakcji w temperaturze na przykład 0ºC, a chcemy ją przeliczyć na 25ºC. Do tego celu trzeba równanie Kirchoffa przekształcić do postaci scałkowanej. W tym celu rozdzielamy zmienne, a następnie całkujemy w granicach od T1 do T2.
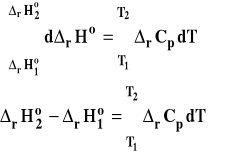
Ostatecznie otrzymujemy :
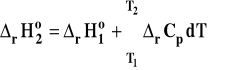
Aby móc skorzystać z tego wzoru w obliczeniach, musimy oczywiście znać wszystkie ciepła molowe izobaryczne reagentów biorących udział w reakcji. Sposób całkowania zależy od tego, czy wartości ciepeł molowych są stałe czy też zależą od temperatury, ale to już w zasadzie czysta matematyka.
W analogiczny sposób można zapisać wzory Kirchoffa dla reakcji przebiegających w stałej objętości, gdzie miarą ciepła reakcji jest energia wewnętrzna.
postać różniczkowa 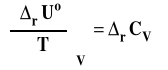
postać scałkowana 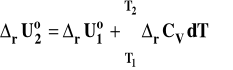
gdzie 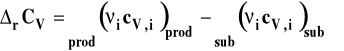
Sposób korzystania z obu tych zależności jest analogiczny jak w poprzednim przypadku, tylko używamy do obliczeń ciepeł molowych izochorycznych.
Ciepło reakcji chemicznej prowadzonej w układzie zamkniętym w stałej temperaturze i pod stałym ciśnieniem lub w stałej objętości, przy braku pracy nieobjętościowej, nie zależy od drogi reakcji, a jedynie od stanu początkowego i końcowego (substratów i produktów).
rH1
rH2
rHcałkowita
stan końcowy (produkty)
stan początkowy (substraty)
produkty pośrednie
symbol przyrostu
dolny indeks - dodatkowe informacje np. jakiej substancji dotyczy, czasem informacje o temperaturze, itp..
skrót rodzaju reakcji lub przemiany (jako dolny indeks) np.
r -reakcja
tw-tworzenia
p.f.-przemiana fazowa
itp.
symbol funkcji
standardowy
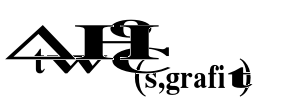
Stan standardowy substancji to jej stan w czystej postaci pod ciśnieniem 105 Pa w dowolnej ustalonej temperaturze.
ciśnienie standardowe po= 105 Pa
Standardowa entalpia reakcji (standardowa zmiana entalpii reakcji) bądź przemiany fizycznej to różnica entalpii czystych, rozdzielonych produktów i czystych, rozdzielonych substratów w stanie standardowym w tej samej, określonej temperaturze.
Stanem standardowym dla roztworu jest stan roztworu nieskończenie rozcieńczonego w warunkach standardowych, przy czym elektrolity są całkowicie zdysocjowane na jony, a cząsteczki bądź jony są solwatowane.
Standardowa entalpia rozpuszczania jest to zmiana entalpii towarzysząca rozpuszczeniu 1 mola czystego związku w warunkach standardowych, w nieskończenie dużej liczbie moli rozpuszczalnika z utworzeniem roztworu nieskończenie rozcieńczonego, w którym jony bądź cząsteczki są solwatowane.
Standardowa entalpia tworzenia związku chemicznego to zmiana entalpii towarzysząca utworzeniu 1 mola związku z czystych, rozdzielonych pierwiastków w warunkach standardowych, przy czym powstający związek jak i pierwiastki muszą być w najtrwalszej w tych warunkach odmianie termodynamicznej. Standardowa entalpia tworzenia pierwiastków w ich najtrwalszej odmianie termodynamicznej wynosi zero.
Standardowa entalpia tworzenia jonu to zmiana entalpii towarzysząca powstaniu 1 mola solwatowanych jonów w rozcieńczeniu nieskończenie wielkim z czystych, rozdzielonych pierwiastków w warunkach standardowych. Standardowa entalpia tworzenia jonu wodorowego w dowolnym rozpuszczalniku i temperaturze wynosi zero.
morze
h2
h3
h1
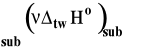
![]()
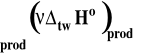
pierwiastki
produkty
substraty
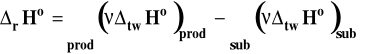
Standardowa entalpia spalania to zmiana entalpii towarzysząca pełnemu utlenieniu 1 mola związku w czystym tlenie, przy czym jako produkty reakcji powstają : CO2(g), H2O(c), N2(g), SO2(g), w warunkach standardowych.
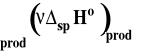
CO2(g) , H2O(c) , N2(g) , SO2(g)
![]()
![]()
substraty
produkty
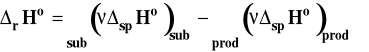
Pierwsza pochodna entalpii reakcji po temperaturze przy stałym ciśnieniu jest równa zmianie pojemności cieplnej reagentów w reakcji czyli różnicy ciepeł molowych produktów i substratów pomnożonych przez odpowiednie współczynniki stechiometryczne.
Jeśli rCp < 0 to, gdy T Ⴍ to rHo Ⴏ.
Jeśli rCp > 0 to, gdy T Ⴍ to rHo Ⴍ.
Energia wiązania jest to pewna umowna wielkość przypisywana wiązaniu w cząsteczce w ten sposób, że suma energii wiązań jest równa entalpii rozpadu 1 mola związku w stanie gazowym na atomy w stanie gazowym w temperaturze 0 K, przy czym analogiczne wiązania traktuje się jako równocenne energetycznie.
subHo
2 dysHo
4 EC-H
- twHo