DSC03655 (2)

Klasyczne ekstrakcje ciecz-ciecz prowadzone w rozdzielaczach, wymagaj!) stosowania od 10 do 1000 mL każdej z faz. W jednorazowej ekstrakcji, w celu ilościowego odzysku oznaczanej subastancji (>99%). wartość Kn musi być wysoka f>10). natomiast przy niskim K(> analitu i V=l. odzysk w wyżej wymienionym r zakresie gwarantuje trzykrotna ekstrakcja próbki świeżym rozpuszczalnikiem. Wyjaśnia to fakt. że w porównaniu do ekstrakcji jednorazowej. wielokrotna ekstrakcja jest procesem wydajniejszym. gwarantującym większe stężenie substancji w ekstrakcie (6.22).
Ekstrakcja ciecz-ciecz powszechnie nazywana ekstrakcją rozpuszczalnikiem jest jednym z bardziej popularnych sposobów ekstrakcji próbek ciekłych. Jest to metoda rozdziału składników próbki pomiędzy dwie nie mieszające się fazy (6.22,37). Jedną z nich stanowi roztwór wodny (woda lub wodne roztwory kwasów, soli. związków kompleksujących i chelatują-cych), a drugą rozpuszczalnik organiczny (eter dietylowy, chlorek metylenu, chloroform, octan etylu, toluen, ksylen, alifatyczne węglowodory i alkohole) (22,37). Podstawowymi kryteriami przy wyborze odpowiedniego rozpuszczalnika są: nie mieszanie się z fazą wodną, mała rozpuszczalność w wodzie (poniżej 10%). polar-ność. lotność (22). W celu zapewnienia kontaktu między fazami konieczne jest wytrząsanie. Zbyt silne wytrząsanie dla próbek zawierających środki powierzchniowo czynne lub tłuszcze. prowadzi do utworzenia emulsji. Jej złamanie odbywa się poprzez odwirowanie, wprowadzenie roztworu soli do fazy wodnej lub dodanie małych ilości odpowiednich rozpuszczalników organicznych (22,37).
W ekstrakcji próbek ciekłych, w celu zwiększenia wydajności procesu, dąży się do zwiększenia powierzchni kontaktu nie mieszających się faz m.in. przez: rozpylenie cieczy, energiczne mieszanie czy wprowadzenie w przeciwprądzie.
W przypadku trudności w rozdzieleniu faz stosuje się odwirowanie, a w niektórych procesach podwyższone lub obniżone ciśnienie (22).
Klasyczną ekstrakcję ciecz-ciecz można prowadzić jako proces ciągły lub jednorazowy.
W ekstrakcji ciągłej rozpuszczalnik o ciężarze właściwym większym od wody jest oddestylo-wywany ze zbiornika kondensacyjnego zawierającego ekstrakt, a skroplone pary w formie kropli przechodząc przez ciekłą matrycę ekstrahują substancję. Odpowiednio skonstruowa
ne ckstraktory umożliwiają także użycie jako rozpuszczalników cieczy o ciężarze właściwym mniejszym od wody. W tym przypadku oddestylowane krople rozpuszczalnika ekstrahują oznaczane związki przechodząc przez porowatą koócówkę lejka o wydłużonej nóżce, zanurzonego w ciekłej matrycy (rycina 4) (6.22,37). Zastosowanie hydrofitowych błon dodatkowo usuwa obecną w otrzymywanym ekstrakcie wodę. eliminując dodatek chemicznych środków suszących. Ciągła ekstrakcja ciecz-ciecz umożliwia przygotowanie próbek analitycznych o niskiej stałej K0 (22). Negatywną stroną metody są straty składników lotnych podczas destylacji roztworu ekstrakcyjnego oraz możliwość rozkładu termolnbilnych składników próbek (6.22,37).
Formą ekstrakcji ciecz-ciecz o małych ob-jętościach fazy organicznej jest mikroekstrak-cja. W metodzie tej wartość parametru V (2) waha się w granicach 0,001-0.01. Mikrockst-rakcję charakteryzuje obok niskiego zużycia rozpuszczalników, wysoka koncentracja ekstrahowanych substancji w fazie organicznej przy jednocześnie ich niskim odzysku z matrycy. Ekstrakcję tego typu przeprowadza się w kolb-kach miarowych, z wąską szyjką umożliwiającą łatwe oddzielenie malej objętości ekstraktu (I mL) od ciekłej próbki (22).
W preparatywnej skali rozdziału próbek o bardzo niskich wartościach K0 (I) lub stanowiących złożone mieszaniny związków o zbliżonych właściwościach najczęściej stosuje się dystrybucję w przeciwprądzie (countercurrent extraction) (6,22,37). Oznaczana substancja lub substancje są pozyskiwane na bazie współczynnika podziału między dwie nie mieszające się warstwy cieczy - górną i dolną, w wyniku tysiąca lub nawet kilku tysięcy procesów podziałowych zachodzących w zautomatyzowanym systemie zbudowanym z szeregu separacyjnych kapilar, stanowiących naczynka ekstrakcyjne. Gdy obie fazy. górna i dolna, po ustaleniu stanu równowagi rozdzielą się, każdy z dwóch rozpuszczalników osiąga kolejne stany równowagi z nowymi porcjami drugiego rozpuszczalnika w ciągłym procesie przeciwprądo-wym (6,22). Dystrybucja w przeciwprądzie byłą podstawą do opracowania, między innymi, metod wirówkowej ekstrakcji podziałowej - Cenirifugal Parlilion Exiraciion i wirówkowej chromatografii podziałowej - Cenirifugal Parlilion Chroinatography, w których ciekła faza stacjonarna jest utrzymywana przez silę
416
IFamlupjp 1'oM.j. Tum SN. nr V. 2002
odśrodkową, a ruchoma pompowana w przeciwprądzie w formie mikrokropli (18). Technika (a jest obecnie wykorzystywana do oczyszczania wielu grup związków naturalnych, m.in. alkaloidów (4.14.27.32,38.39).
Odśrodkowa ekstrakcja cieczowa (Cenirifugal Parlilion Exiraclinn) z zastosowaniem probówek VectaSep CLE (Whatman, Wielka Brytania) (rycina 5) polega na kontrolowanej dyspersji próbki przez mikroporowatą błonę do warstwy selektywnego rozpuszczalnika. Tą drogą mogą być ekstrahowane próbki o objętości 1,5 mL w czasie 30-00 min (44).
W ciągu ostatnich 15 lat w przygotowaniu próbek do analizy systematycznie wzrasta znaczenie i popularność metod ekstrakcji do fazy
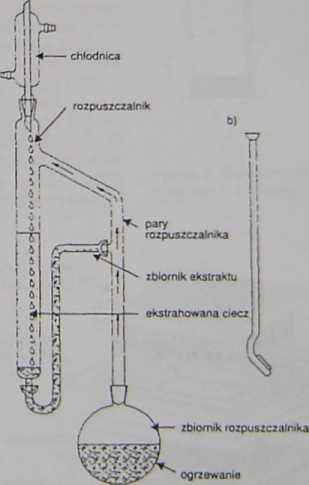
Rycina 4. Aparat do ekstrakcji ciągłej ciecz-ciecz: a) dla rozpuszczalników o ciężarze właściwym większym od wody, b) dla rozpuszczalników o ciężarze właściwym mniejszym od wody z użyciem lejka z wydłużoną szyjką zaopatrzoną w porowaty, szklany krążek umożliwiający przepływ kropli kondensatu przez próbkę - nadmiar ekstraktu przelewa się przez boczne ramię ekstraktora do zbiornika.
stałej - Solid Plunę Estraclion (SPE). stanowiącej różnego typu adsorbenty (26). Po raz pierwszy zastosowano tę metodę pod koniec lat 70. W SPE próbka migruje przez fazę stacjonarną. a ekstrahowane substancje są rozdzielane zgodnie ze stopniem, w jakim każda z nich ulega podziałowi iub jest adsorbowana przez fazę stacjonarną (19.22.26.29). Pomimo, że podstawy izolacji związków SPE przejęła od nowoczesnej chromatografii cieczowej, to jednak wyraźnie się od niej różni. W chromatografii ważny jest kształt piku i krótki czas retencji oznaczanego związku, natomiast w SPE nazywanej ..chromatografią cyfrową" zasadą jest otrzymanie czystej substancji z odpowiednim odzyskiem. Ekstrakcyjne procedury oparte na elementach chromatografii w SPE obejmują zatrzymanie - retencję analizowanej substancji na fazie stacjonarnej z następującą w kolejnym etapie izolacją i oczyszczaniem oraz szybką elucję w możliwie malej objętości fazy ruchomej. poprzedzającą analizę. Innymi słowy współczynnik podziału k'. którego wartość w chromatografii cieczowej dla rozdzielanych związków powinna wahać się w zakresie 1-10. w SPE wynosi odpowiednio >1000 dla retencji i <0.001 dla elucji (29).
Metoda SPE umożliwia zatężanie. oczyszczanie i izolację substancji analizowanej w rezultacie trzech selektywnych procesów zależnych od rodzaju adsorbentu i desorpcyjnego solwentu. Są to mianowicie:
- retencja - przez odpowiedni dobór fazy stacjonarnej i ruchomej zachodzi retencja substancji oznaczanej, zanieczyszczenia nie są zatrzymywane:
- wymywanie zanieczyszczeń - w wyniku zastosowania fazy ruchomej, wystarczająco silnej do usunięcia z matrycy zanieczyszczeń, ale zbyt słabej do usunięcia zaadsorbowanej substancji analizowanej:
- elucja - wymywanie analizowanych związków za pomocą eluentu, który pozostawia w złożu (fazie stałej) silnie zaadsorbowane zanieczyszczenia (6.10,29).
Ważnym elementem w wyborze sorbentu jest uwzględnienie charakteru oddziaływań fizykochemicznych z analizowaną substancją, ponieważ może być on zmieniany przez jej silniejsze wiązanie z matrycą (29).
Obecnie SPE obejmuje różne techniki w zależności od formy i postaci „fazy stałej", są to tradycyjne kolumienki SPE z selektywnymi
417
Farmacja IHil.ka. T«nn 58. nr 1. 21)02
Wyszukiwarka
Podobne podstrony:
DSC03616 (4) r 1 B m?CZU Wdzianego w ciągu doby przez zdrowego człowieka wynosi od 600 do 2000 ml i
PwTiR067 132 Rozdział 5 być stosowane wyłącznie w odniesieniu do obiektów hotelarskich w rozumieniu
SKMBT?5007122709470�26 Rozdział 8Społeczeństwo Zacznijmy od faktów Do dziś nie potrafię wymazać ze s
IMGi01 (4) 94 Rozdział 2 hierarchię przetwarzania - od prostych do bardziej złożonych. W miarę przec
u Historia sztuki dla bystrzaków Rozdział 25: Fotografia — od nauki do
DSC03617 (4) Diureza Ilość moczu wydalanego w ciągu doby przez zdrowego człowieka wynosi od 600 do 2
wymagane przez te narządy znajdują się w zakresach od -10 do 5°C. Są to urządzenia uniwersalne, posi
Nowy 10 OA Niech taneczny, lekki krok, będzie z Tobą cały rok, Niech prowadzi Cię bez stresu, od suk
DifiraSPIS TREŚCI Prowadzenie działalności gospodarczej w Internecie. Od e-commerce do e-businessu
DSCN0360 [Rozdzielczość Pulpitu] Waga psa w kilogramach od 10 do 15 kq od 15 do 20 od 20 d
Rozdział 21 4.A.I. Ekstrakcja ciecz-ciecz Ponieważ ekstrakcja ciecz-ciecz wymaga zwykle bardzo czyst
więcej podobnych podstron