WĘGLOWODANY
1. Wykrywanie węglowodanów (reakcja Molischa)
Zasada:
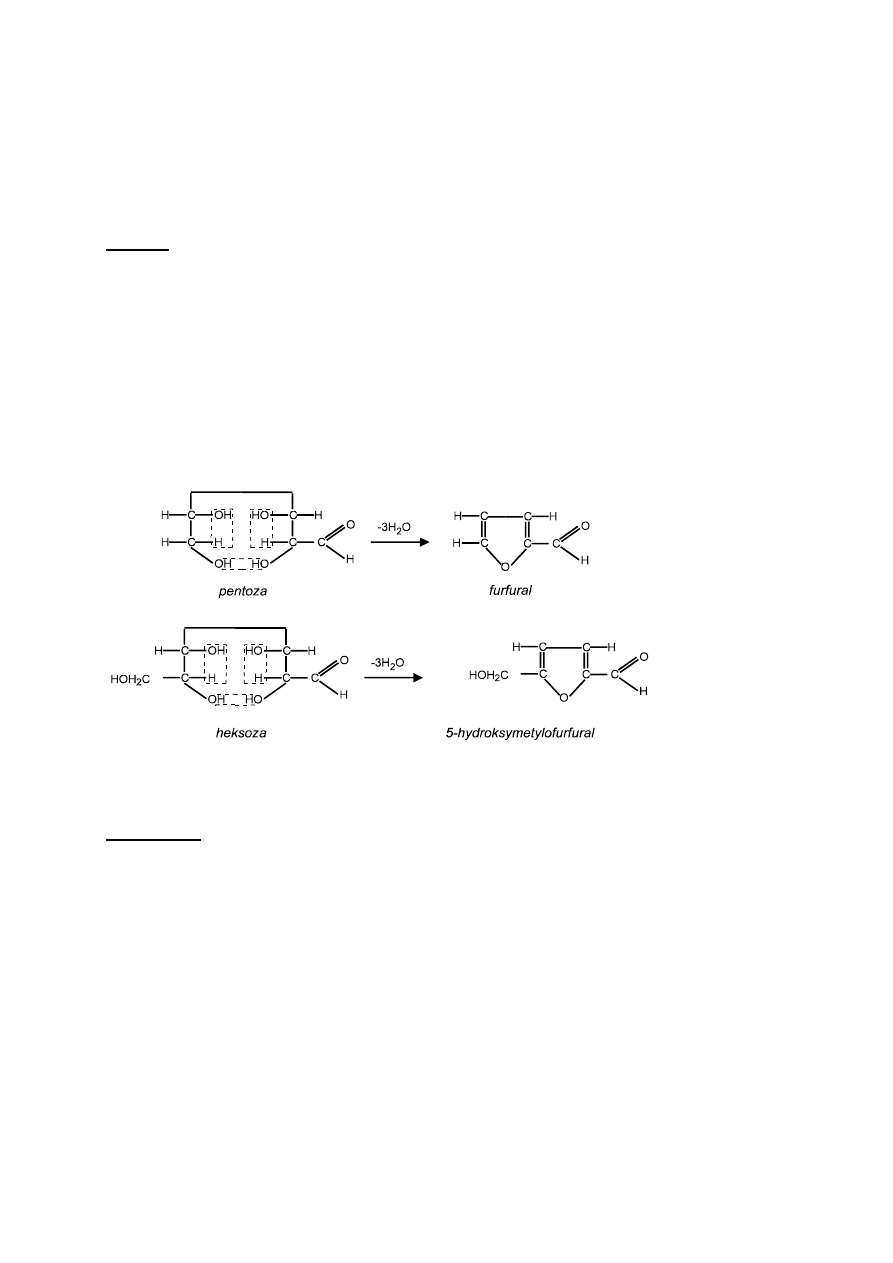
Jest to najbardziej ogólna reakcja charakterystyczna, którą wykazują wszystkie cukrowce. Pod
wpływem stężonego kwasu siarkowego pierścieniowe formy cukrów ulegają odwodnieniu
tworząc furfural (pentozy) lub 5-hydroksymetylofurfural (heksozy) (Rys. 1). Związki te reagują
z α-naftolem lub benzydyną, dając barwne połączenia.
Rys. 1. Reakcja odwodnienia pentoz i heksoz
Wykonanie:
Do probówek zawierających 1 cm
3
różnych cukrów prostych i złożonych (glukoza, fruktoza,
ryboza, sacharoza, skrobia) dodać 1–2 krople odczynnika Molischa, czyli świeżo
przygotowanego 10-procentowego alkoholowego roztworu α-naftolu (benzydyny). Zawartość
probówki wymieszać, po czym lekko przechylić i po ściance nalać 1 cm
3
stężonego kwasu
siarkowego (OSTROŻNIE!), tak by obydwie ciecze nie zmieszały się. Na granicy faz pojawia
się czerwony lub czerwonofioletowy pierścień.
2. Wykrywanie ketoz (Reakcja Seliwanowa)
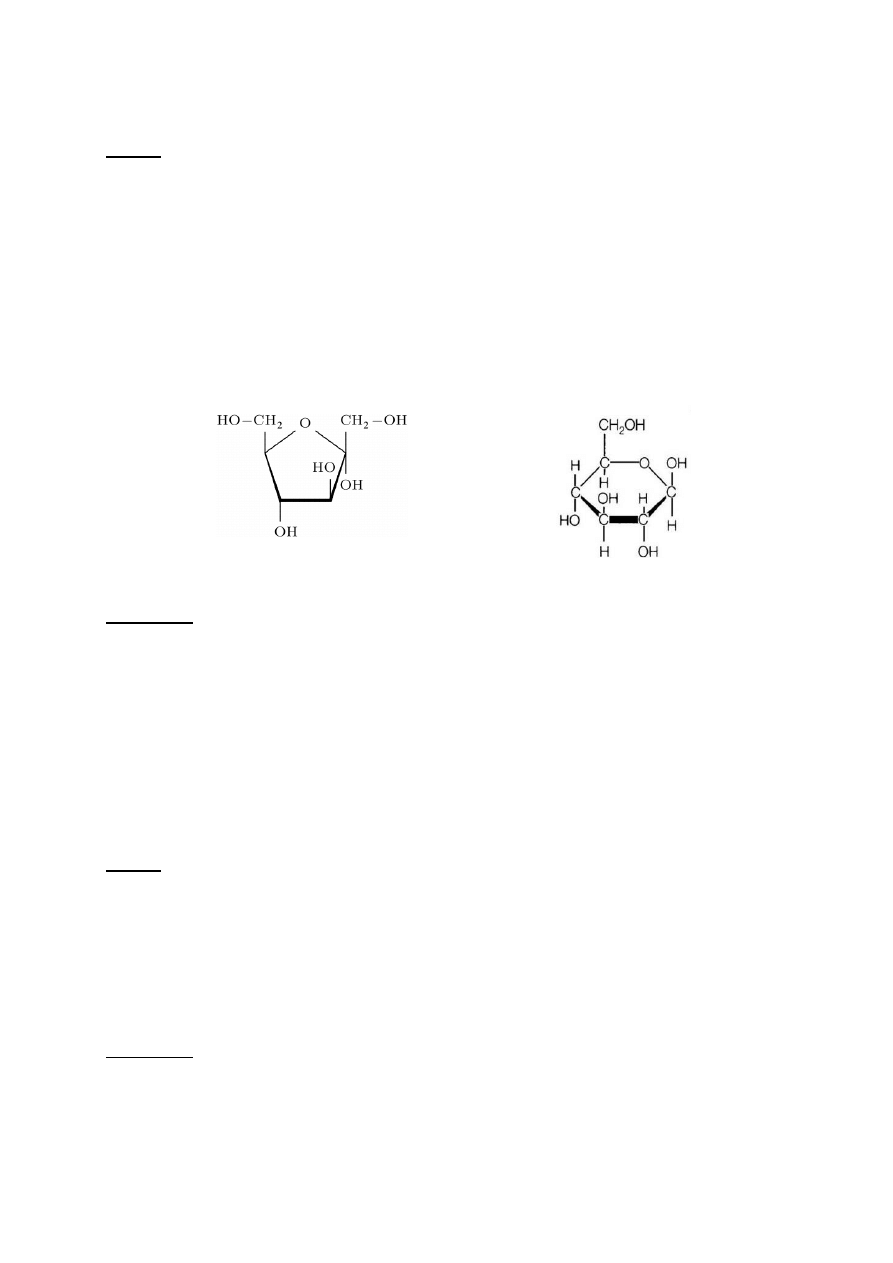
Zasada:
Pierścieniowe formy cukrów ulegają odwodnieniu pod wpływem mocnych kwasów, tworząc
furfural (pentozy) lub 5-hydroksymetylofurfural (heksozy). Hydroksymetylofurfural tworzy
następnie barwne połączenie z rezorcyną. Jednakże pod wpływem rozcieńczonego kwasu solnego
odwadnianie ketoz następuje znacznie szybciej aniżeli aldoz, co jest podstawą ich odróżniania.
Rys. 2. Wzór strukturalny A. α-D-fruktozy (ketozy) i B. α-D-glukozy (aldozy)
A. B.
Wykonanie:
Do probówek zawierających oddzielnie po 1 cm
3
roztworów A, B, C, które zawierają cukier dodać
po 1 cm
3
odczynnika Seliwanowa (skład odczynnika: 100 cm
3
stężonego kwasu solnego, 300 cm
3
wody, 150 mg rezorcyny) i ogrzewać na wrzącej łaźni wodnej. W przypadku ketozy po ogrzewaniu
przez ok. 0,5-1 min roztwór zabarwia się na kolor czerwony. Aldoza może również tworzyć
podobnie zabarwione połączenie z rezorcyną, jednakże po znacznie dłuższym ogrzewaniu.
3. Reakcja Biala wykrywająca pentozy (odróżnianie pentoz od heksoz)
Zasada:
Pierścieniowe formy cukrów ulegają odwodnieniu pod wpływem mocnych kwasów, tworząc
furfural (pentozy) lub 5-hydroksymetylofurfural (heksozy). Furfural daje z orcyną w obecności
jonów żelazowych zielone lub zielono-niebieskie zabarwienie, podczas gdy 5-
hydroksymetylofurfural w tych warunkach daje roztwór zabarwiony na żółto-brązowy kolor.
Wykonanie:
Do 3 probówek nalać po 2 cm
3
odczynnika Biala (skład odczynnika: 500 cm
3
stężonego HCl, 500
cm
3
H
2
O, 1 g orcyny, 20 kropli 10% wodnego roztworu FeCl
3
) i ogrzewać na łaźni wodnej w
temperaturze 90°C przez ok. 3 min, a następnie dodać do każdej probówki oddzielnie po 1 cm
3
analizowanych roztworów A, B, C, zawierających glukozę, fruktozę lub rybozę). Zamieszać i
wstawić do łaźni wodnej. Po kilku minutach zanotować powstające zabarwienie.
Na podstawie wyników reakcji 2 i 3 zidentyfikować cukry A, B, C spośród wymienionych
powyżej.
4. Odróżnianie cukrów redukujących od nieredukujących (reakcja Benedicta)
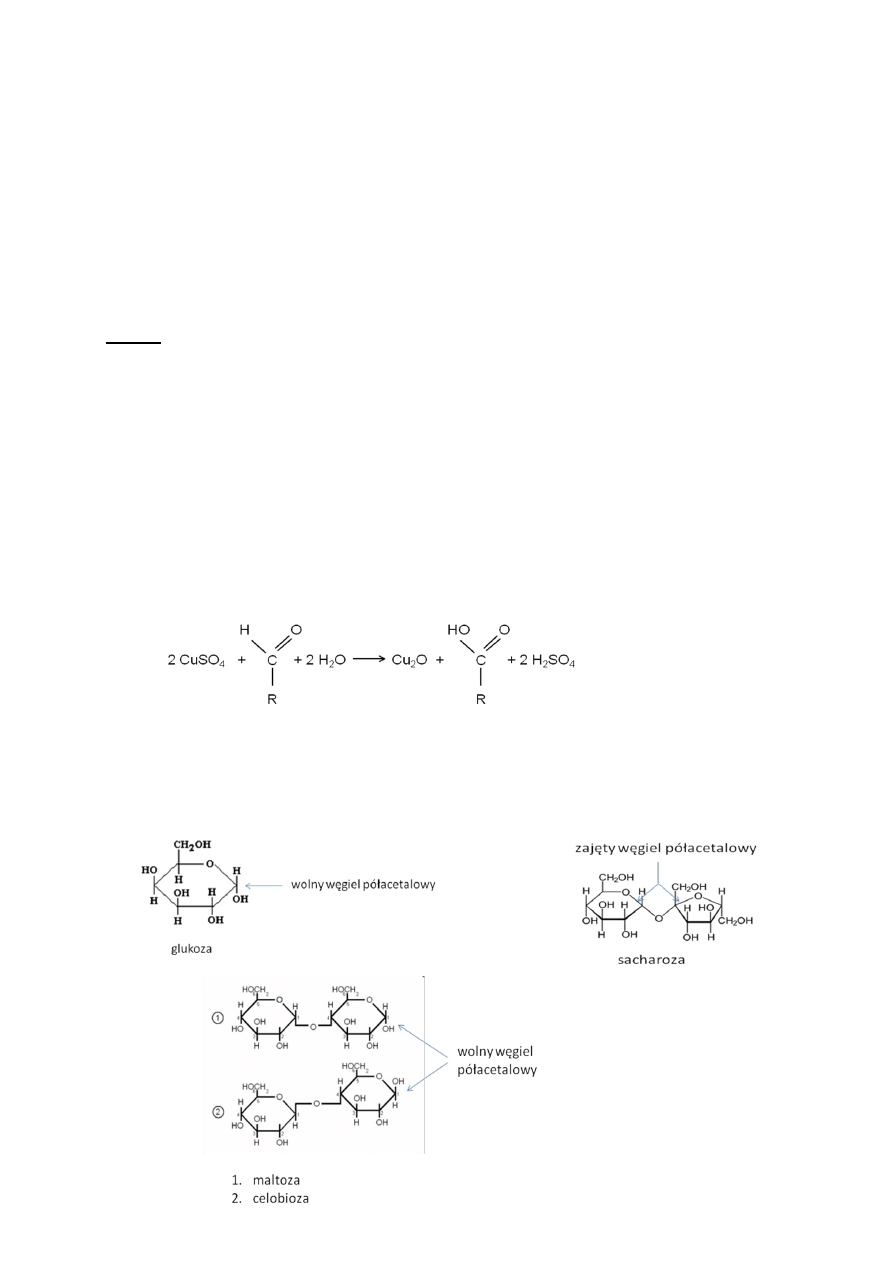
Zasada:
Cukry o właściwościach redukujących mają zdolność redukowania jonów miedzi Cu
2+
do jonów
Cu
+
o charakterystycznej, czerwonopomarańczowej barwie tlenku Cu
2
O (Rys. 3). Właściwości
redukcyjne charakteryzują tylko te cukry, które posiadają wolny węgiel z grupą aldehydową lub
ketonową, a więc wszystkie monosacharydy i te spośród oligosacharydów, które mają wolny
przynajmniej jeden węgiel półacetalowy (czyli węgiel karbonylowy, uczestniczący w tworzeniu
formy pierścieniowej cząsteczki) (Rys. 4).
Rys. 3. Reakcja redukcji jonów miedzi pod wpływem dowolnego cukru prostego
Rys. 4. Wzory strukturalne redukujących cukrów: glukozy, maltozy i celobiozy z zaznaczeniem
wolnego węgla półacetalowego, oraz sacharozy z zaznaczeniem zablokowanego węgla
półacetalowego w wiązaniu glikozydowym.
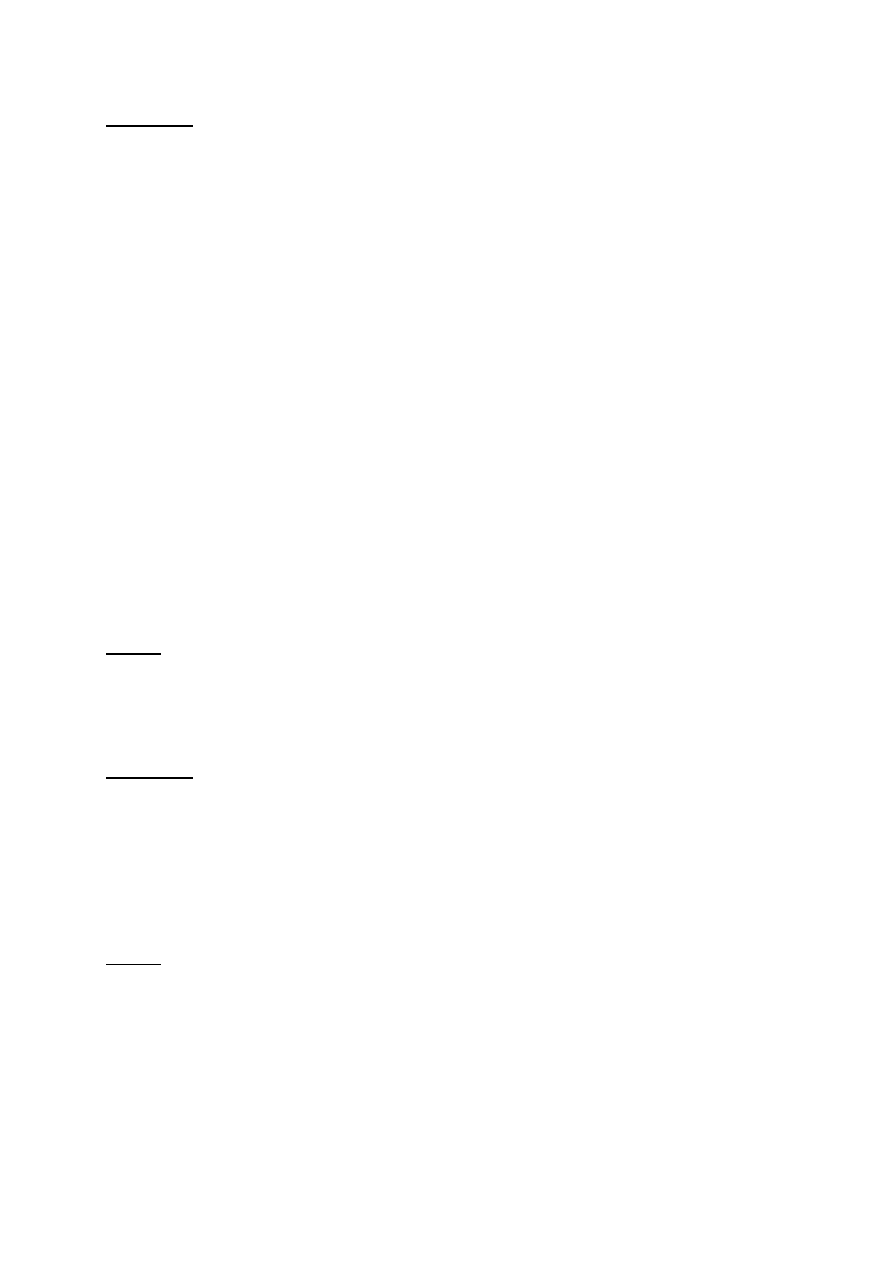
Wykonanie:
Do dwóch probówek odmierzyć po 1 cm
3
cukrów D i E (sacharozy lub maltozy), a następnie dodać
po 2,5 cm
3
odczynnika Benedicta (skład odczynnika: 173 g cytrynianu sodowego, 100 g
bezwodnego węglanu sodowego, 800 cm
3
wody + roztwór 17,3 g CuSO
4
w 100 cm
3
wody
uzupełnić wodą do 1 dm
3
), i ogrzewać na łaźni wodnej w temperaturze 60°C przez kilka minut. W
obecności cukru redukującego wytwarza się ceglastoczerwony osad. Przy mniejszej zawartości
cukru może powstać jedynie zielone zabarwienie albo osad o barwie od żółtej do pomarańczowej.
Na podstawie wyników reakcji 4 rozróżnić cukry D i E spośród wymienionych
Sprawozdanie:
Podać jakie związki były oznaczone literami A, B, C, D, E i na jakiej podstawie poszczególne
cukry zostały rozróżnione.
5. Hydroliza skrobi
a. Otrzymywanie skrobi z ziemniaka
Zasada:
W metodzie tej wykorzystuje się znaczny ciężar właściwy ziaren skrobiowych, które uwolnione z
komórek łatwo sedymentują.
Wykonanie:
Kilka ziemniaków umyć, obrać, zetrzeć na miazgę i rozcieńczyć równą objętością wody. Tkankę
odcisnąć przez kilka warstw gazy, a roztwór odwirować przy ok. 1000 obr./min przez 5 min. Zlać
ciecz znad osadu, a skrobię przepłukać etanolem i wysuszyć na bibule.
b. Hydroliza skrobi i próba jodowa
Zasada:
Cząsteczki glukozy tworzą w cząsteczce skrobi rozgałęzione helisy. Do wnętrza tych helis może
wnikać jod, rozmieszczając się regularnie po 1 atomie na 1 skręt helisy. Powoduje to powstanie
zabarwionego kompleksu jodowo-skrobiowego o barwie od fioletowo-czerwonej (amylopektyna)
do niebieskiej (amyloza). Po ogrzaniu zabarwienie znika z powodu rozkręcania się helis i rozpadu
kompleksu. Procesowi postępującej hydrolizy skrobi towarzyszy stopniowa zmiana barwy z nie-
bieskiej w fioletową, czerwoną i ostatecznie jej zanik.
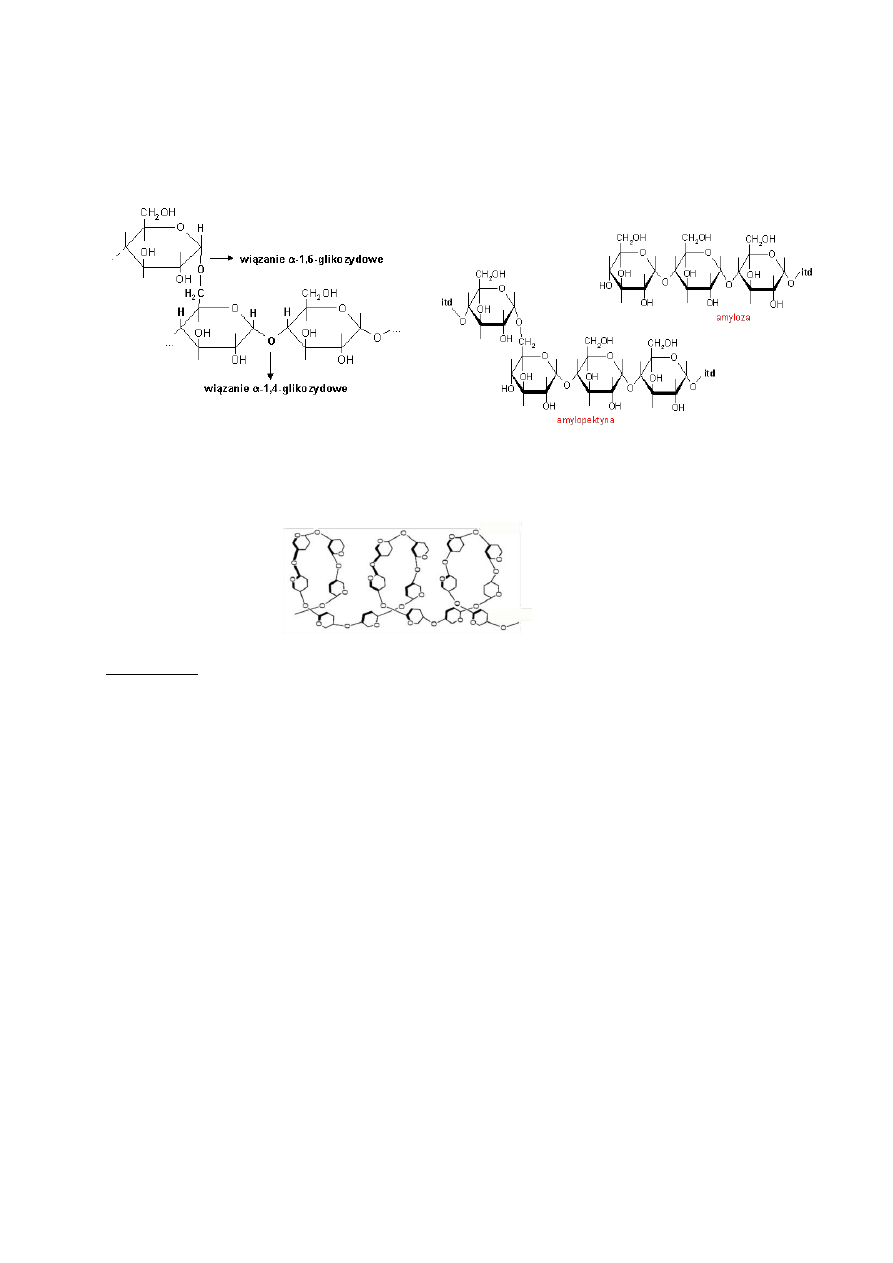
Rys. 5. Wiązania glikozydowe występujące w amylozie i amylopektynie
Rys. 6. Zwinięty przestrzennie łańcuch amylozy, tworzący pętle
Wykonanie:
Przygotowanie kleiku skrobiowego: 3 g skrobi rozmieszać w 50 cm
3
zimnej wody destylowanej.
150 cm
3
wody zagotować. Do gotującej wody powoli wlewać rozmieszana skrobię ciągle
mieszając. Kleik gotować przez 1 minutę. Ostudzić.
Przygotować 10 probówek zawierających po 1 cm
3
0,002% roztworu Lugola (jod w jodku potasu).
Do ok. 10 cm
3
kleiku skrobiowego otrzymanego ze skrobi wyizolowanej w części a) dodać 1 cm
3
stężonego kwasu solnego i ogrzewać na łaźni wodnej. Co ok. 1 min pobierać kilka kropel kleiku i
wlewać do kolejnej probówki. Obserwować postępującą hydrolizę skrobi i wywołaną tym zmianę
zabarwienia w kolejnych probówkach.
6. Oznaczanie zawartości cukrów rozpuszczalnych w tkankach roślinnych metodą
antronową
Sporządzenie krzywej kalibracyjnej
Przygotować wzorcowy roztwór glukozy o stężeniu 1 mg/cm
3
(Przykładowo 10 mg glukozy
rozpuścić w 10 cm
3
). Przygotować 5 probówek dla wzorcowych roztworów glukozy o stężeniu
0,2; 0,1; 0,05; 0,025 i 0,0125 mg glukozy w 1 cm
3
. Do pierwszej probówki pobrać 2 cm
3
wzorca
o stężeniu 1 mg/cm
3
i uzupełnić 8 cm
3
wody, uzyskując w ten sposób stężenie glukozy 0,2
mg/cm
3
. Zamieszać. Z probówki tej pobrać 3 cm
3
roztworu glukozy do następnej probówki i
dodać 3 cm
3
wody, uzyskując rozcieńczenie wynoszące 0,1 mg/cm
3
. Powtarzając postępowanie
uzyskujemy kolejne rozcieńczenia. Następnie pobrać do 5 nowych probówek po 1 cm
3
każdego
z rozcieńczeń. Do każdej z nich dodać 2 cm
3
0,2% antronu w stężonym H
2
SO
4
, zmieszać
roztwór na wstrząsarce laboratoryjnej i ogrzewać przez 3 minuty na łaźni wodnej o temperaturze
90
C. Po ostygnięciu próbek do temperatury pokojowej zmierzyć w szklanych kuwetkach
spektrofotometrycznych absorbancję (ABS) przy
= 620 nm względem ślepej próby (zamiast
roztworu cukru z roztworem antronu zmieszać 1 cm
3
wody).
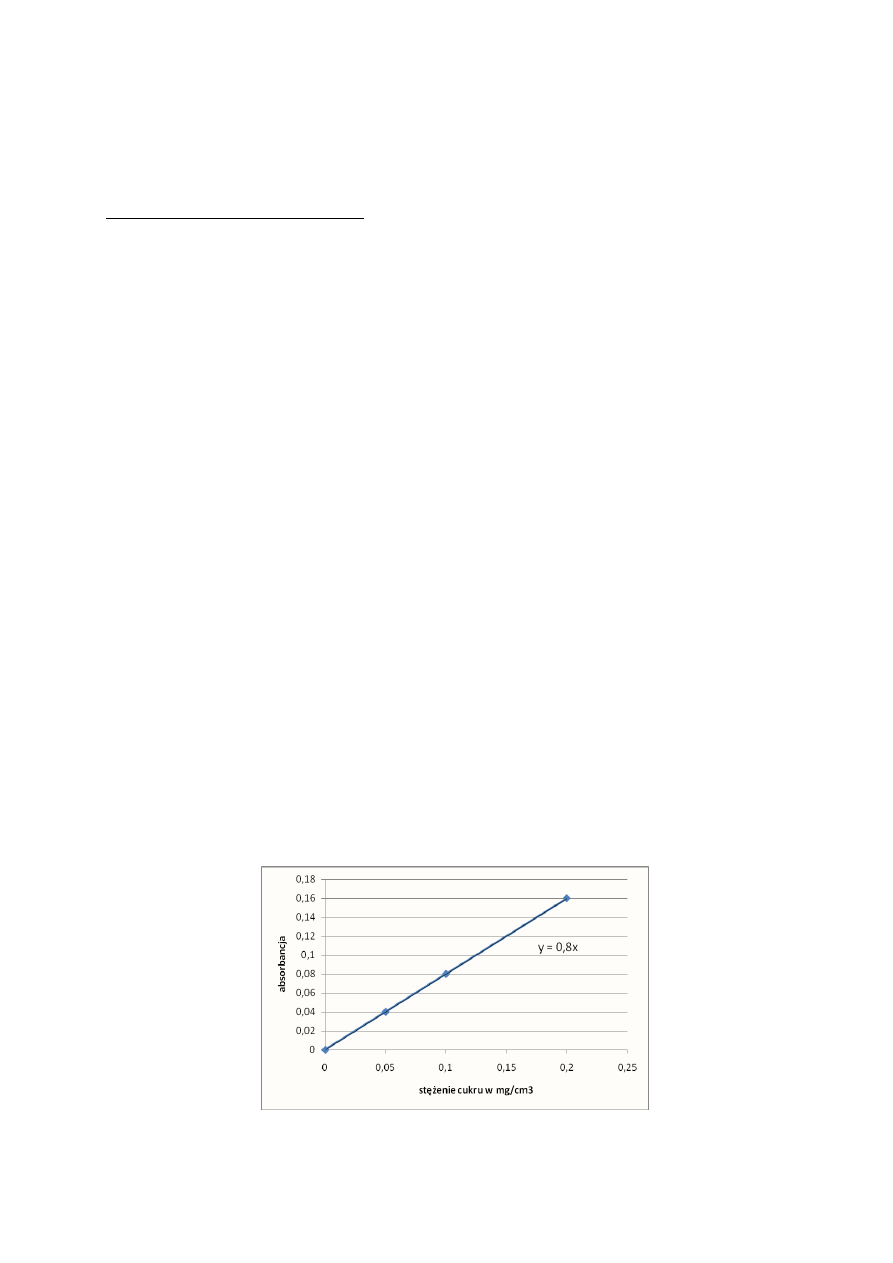
Krzywą wzorcową uzyskać w arkuszu Excela. Na podstawie danych dotyczących absorbancji
dla kolejnych stężeń glukozy sporządzić wykres punktowy, wstawić linię trendu i odczytać
równanie y = ax + b, gdzie y oznacza ABS, a – współczynnik (tangens kąta nachylenia krzywej
w stosunku do osi OX), x – stężenie cukru w mg/cm
3
, b – punkt przecięcia linii trendu z osią
OY. Jeżeli założymy, że zerowe stężenie cukru daje ABS=0, współczynnik b będzie też równał
się zero. Na rys. 7. przedstawiono przykładowa krzywą wzorcową.
Rys. 7. Przykładowy wykres krzywej wzorcowej

Oznaczenie zawartości cukrów rozpuszczalnych w materiale roślinnym
Ekstrakcja cukrów rozpuszczalnych
Pobrać dwukrotnie po 0,5 g świeżej masy liścia i każdą z prób utrzeć oddzielnie w moździerzu w
ciekłym azocie. Po odparowaniu azotu, podczas ucierania dodawać stopniowo 5 cm
3
wody.
Homogenat przenieść do szklanej probówki, a moździerz przepłukać jeszcze 5 cm
3
wody i zlać
je również do homogenatu w probówce. Próbki gotować przez 15 minut na wrzącej łaźni
wodnej, a następnie zawartość przenieść dokładnie do probówek wirówkowych. Tkankę
odwirować przez 10 minut przy 15 000 obr./min. Nadsącz zlać do nowej probówki i uzupełnić
wodą do 10 cm
3
.
Oznaczanie zawartości cukrów rozpuszczalnych
Pobrać 0,5 cm
3
nadsączu i rozcieńczyć go dodając 9,5 cm
3
wody. Stopień rozcieńczenia należy
dobrać tak, by odczytywane wartości absorbancji mieściły się w zakresie krzywej kalibracyjnej.
Rozcieńczony ekstrakt wymieszać na wstrząsarce laboratoryjnej i pobrać z niego dwukrotnie po
1 cm
3
. Do każdej z probówek dodać po 2 cm
3
0,2% antronu w stężonym H
2
SO
4
(POCh),
wstrząsnąć probówkę na wstrząsarce i ogrzewać przez 3 minuty na łaźni wodnej o temperaturze
90
C. Po ostygnięciu próbki do temperatury pokojowej zmierzyć absorbancję przy
= 620 nm
względem ślepej próby (zamiast roztworu cukru z roztworem antronu zmieszać 1 cm
3
wody).
Wartość zmierzonej absorbancji próbek wstawić do wzoru uzyskanego w punkcie a. Wyliczyć
wartość stężenia cukru (x) w mg/cm
3
roztworu. Chcąc wyznaczyć zawartość cukru w liściu czyli
w mg/g świeżej masy należy wartość wyliczonego stężenia x pomnożyć przez objętość
wyjściowego roztworu (tj. 10 cm
3
). Otrzymujemy w ten sposób zawartość cukru w całej
objętości tego roztworu. Roztwór ten powstał z homogenizacji 0,5 g liścia, chcąc zatem
otrzymać zawartość cukru w liściu w przeliczeniu na 1 g świeżej masy, należy uzyskany wynik
podzielić przez masę próbki liścia tj,. 0,5 g.
Sprawozdanie
a) Opisać zabarwienie w próbówkach na poszczególnych etapach hydrolizy skrobi.
b) Za pomocą arkusza kalkulacyjnego MS Excel wykreślić krzywą kalibracyjną i wyznaczyć
równanie regresji liniowej zależności absorbancji od stężenia glukozy (wzorca) w próbce.
c). Obliczyć stężenie cukru z pomocą równania regresji liniowej otrzymanego w podpunkcie a).
Następnie stężenie cukru w próbce przeliczyć na jego zawartość w świeżej masie liścia.

7. Oznaczanie zawartości ketoz
Wykonanie:
Odważoną tkankę roślinną homogenizować dodając stopniowo 5 cm
3
wody destylowanej.
Ekstrakt odwirować przez 20 min przy 4000 obr./min, po czym zlać supernatant. W celu
strącenia białek do 2,5 cm
3
supernatantu dodać 0,5 cm
3
bezwodnego octanu cynku (230g · dm
-3
)
oraz 0,5 cm
3
żelazocjanku potasu (150g · dm
3
), zmieszać na wstrząsarce laboratoryjnej oraz
odwirować.
Do supernatantu odpowiednio rozcieńczonego wodą do objętości 1 cm
3
dodać 1,5 cm
3
5%
rezorcyny w etanolu i 1,5 cm
3
(NH
4
)
2
Fe(SO
4
)
2
w HCl. Po wymieszaniu na wstrząsarce
laboratoryjnej próbki wstawić do łaźni wodnej o temp. 80°C na 40 minut. Po schłodzeniu próbek
w lodzie, zmierzyć absorbancję przy długości fali λ = 480 nm. Stężenie cukru obliczyć na
podstawie krzywej kalibracyjnej przygotowanej dla roztworu fruktozy.
8. Oznaczanie zawartości skrobi
Ekstrakcja i hydroliza enzymatyczna
Osad pozostały z wirowania w podpunkcie 6b rozpuścić w 5 cm
3
0,2 mol/dm
3
KOH i przenieść
do szklanej probówki. Gotować 30 minut na wrzącej łaźni wodnej utrzymując poprzez
dolewanie wody stałą objętość próbki (poziom cieczy należy zaznaczyć). Po ochłodzeniu ustalić
pH w probówce na 5,5 przez dodanie 1 cm
3
kwasu octowego o stężeniu 1 mol/dm
3
. Następnie
dodać 5 cm
3
dializowanej amyloglukozydazy (z Aspergillus oryzae, firmy SIGMA) (enzym
rozpuścić w proporcjach 35 U/cm
3
50 mM buforu octanowego o pH 4,5, wlać do woreczka
dializacyjnego, i umieścić go w zlewce z wodą destylowaną na całą noc) i inkubować probówki
30 minut w 55°C. Następnie próbkę zagotować (by unieczynnić enzym), przelać do probówki
wirówkowej i odwirować przez 10 minut przy 15 000 obr/min. Supernatant zlać i uzupełnić
wodą do 10 cm
3
.
Oznaczanie zawartości skrobi w równoważnikach glukozy
Postępować analogicznie jak w punkcie 6b, przy oznaczaniu zawartości cukrów
rozpuszczalnych.
Piśmiennictwo:
Ashwell G., 1975. Colorimetric analysis of sugars. Methods Enzymol., 3: 467-471.
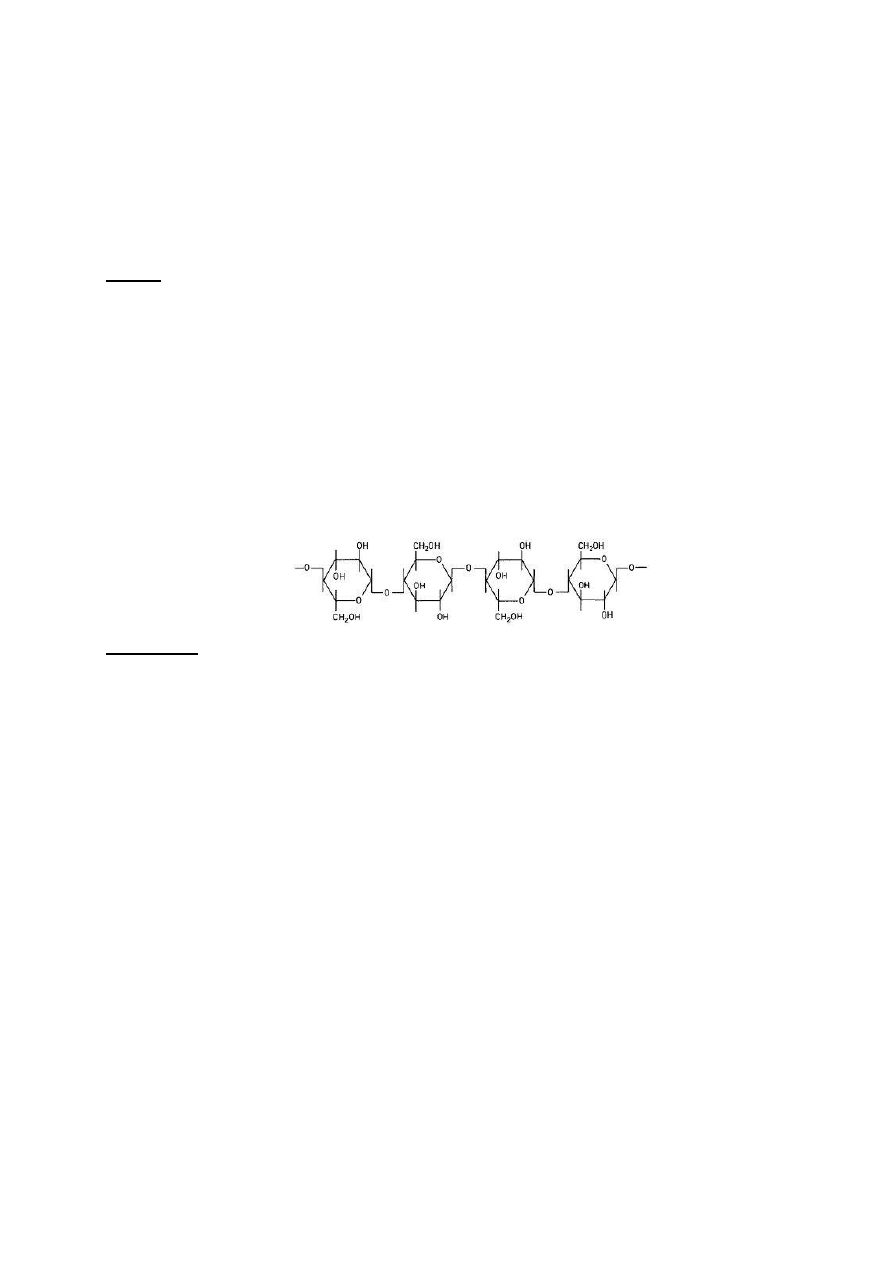
Rufty W.T. jr. and Huber S.C. 1983. Changes in starch formation and activities of sucrose
phosphate synthase and cytoplasmic fructose-1,6-biphosphatase in response to source-sink
alterations. Plant Physiol., 72, 474-480.
9. Rozpuszczanie celulozy w odczynniku Schweitzera
Zasada:
Celuloza różni się od skrobi przede wszystkim tym, że cząsteczki glukozy połączone są wiązaniami
β-glikozydowymi zamiast α (Rys. 8). Konsekwencją tego faktu jest bardzo duża odporność celulozy
na działanie czynników hydrolizujących, rozpuszczających oraz na rozkład enzymatyczny. Dopiero
stężone kwasy oraz takie specyficzne rozpuszczalniki jak odczynnik Schweitzera rozpuszczają
celulozę.
Rys. 8. Wzór strukturalny fragmentu łańcucha ligniny
Wykonanie:
Kawałek bibuły, strzępek waty lub skrawek płótna zalać pod wyciągiem 50 cm
3
odczynnika
Schweitzera (do 50 cm
3
0,3 M roztworu CuSO
4
dolać mieszając 50 cm
3
0,6 M NaOH, wytrącony
osad rozpuścić w 75 cm
3
28% roztworu amoniaku) po czym mieszać do rozpuszczenia. Po wlaniu
tego roztworu do rozcieńczonego kwasu solnego (10 cm
3
stężonego kwasu rozcieńczyć 90 cm
3
wody) celuloza ponownie się wytrąca. Jeśli roztwór celulozy wytłaczać przez bardzo wąskie dysze,
wówczas może w ten sposób powstać sztuczne włókno celulozowe.
UWAGA! Stężony roztwór amoniaku drażni drogi oddechowe, stąd eksperyment należy
wykonywać koniecznie pod sprawnym wyciągiem.

LIPIDY
Najczęściej stosowane metody chromatograficzne w analizie ilościowej i jakościowej
lipidów
Termin chromatografia oznacza efekt rozdziału (separacji) mieszaniny substancji na jej
składniki, obserwowany podczas przepływu fazy ruchomej wzdłuż powierzchni fazy
nieruchomej. Cząsteczki składników mieszaniny, które słabo oddziałują z fazą nieruchomą, są
szybciej unoszone przez płynącą fazę ruchomą, zaś cząsteczki przyciągane mocniej poruszają się
wolniej. Fazą ruchomą może być gaz lub ciecz, zaś fazą nieruchomą - ciecz, żel lub porowate
ciało stałe. Zastosowanie chromatografii w analityce chemicznej polega na separacji mieszaniny
substancji (związków chemicznych) na proste składniki, a następnie na pomiarze ich ilości.
Wykorzystywanie różnych faz ruchomych i nieruchomych, oraz odmiennych co do
fizykochemicznej natury oddziaływań powodujących rozdział chromatograficzny, doprowadziło
do powstania wielu różnych technik chromatograficznych.
Chromatografia gazowa (GC - gas chromatography)
Jest to metoda analityczna wykorzystująca efekt rozdziału chromatograficznego z użyciem gazu
(np. He) jako fazy ruchomej, oraz porowatego ciała stałego lub filmu polimeru organicznego
jako fazy nieruchomej. Stosowana do analiz składu złożonych mieszanin związków
chemicznych, zwłaszcza lotnych związków organicznych i nieorganicznych.
Najważniejsze elementy chromatografu gazowego to dozownik, służący do wprowadzenia
próbki do strumienia gazu nośnego, kolumna analityczna zawierająca fazę stacjonarną oraz
detektor. Kolumna chromatograficzna ma postać długiej i cienkiej rurki (stalowej, kwarcowej
lub szklanej), zwiniętej w zwój. W kolumnie kapilarnej faza stacjonarna tworzy cienką warstwę
na wewnętrznej powierzchni ścianek. Kolumna jest umieszczona w termostatowanym piecu,
wyposażonym w programator temperatury. W chromatografii gazowej najczęściej
wykorzystywane są detektory przewodnictwa cieplnego (TCD, HWD), detektory płomieniowo-
jonizacyjne (FID), detektory wychwytu elektronów (ECD) oraz detektory masowe (MSD).

Wysokosprawna chromatografia cieczowa (HPLC - high performance liquid chromatography)
Wysokosprawna chromatografia cieczowa jest uniwersalną metodą analityczną, stosowaną
głównie do analiz złożonych próbek, zwłaszcza zawierających nielotne, wielkocząsteczkowe
związki chemiczne, w szczególności substancje biologicznie czynne. Metoda HPLC
wykorzystuje efekt rozdziału chromatograficznego z użyciem cieczy jako fazy ruchomej. Skład
fazy ciekłej i rodzaj fazy stacjonarnej jest uzależniony od składu badanych próbek oraz typu
oddziaływań
wykorzystywanych
do
osiągnięcia
separacji
ich
składników.
Typowy zestaw do HPLC składa się ze zbiornika fazy ciekłej (eluentu), pompy, dozownika,
kolumny analitycznej, detektora i zbiornika na zużyty eluent. Zbiornik eluentu jest często
wyposażony w urządzenia do odpowietrzania i filtrowania cieczy. Najczęściej stosowane
mechaniczne pompy tłokowe umożliwiają osiąganie bardzo wysokich ciśnień i stabilnych
szybkości przepływu. Jako dozowniki zwykle wykorzystywane są zawory z pętlą dozującą o
określonej objętości. Kolumny analityczne to rurki stalowe wypełnione cząstkami fazy
stacjonarnej. Do detekcji składników opuszczających kolumnę najczęściej stosuje się detektory
absorpcji promieniowania UV-VIS, detektory fluorescencyjne, detektory refraktometryczne oraz
detektory
elektrochemiczne
(polarograficzne,
woltamperometryczne,
kulometryczne,
konduktometryczne).
Chromatografia cienkowarstwowa (TLC - thin layer chromatography)
Chromatograficzna metoda analityczna, wykorzystująca jako fazę stacjonarną cienką warstwę
porowatego sorbentu, oraz ciecz jako fazę ruchomą (eluent). Rozdział chromatograficzny
zachodzi w wyniku przepływu eluentu przez warstwę sorbentu, wciąganego przez sorbent na
skutek działania sił kapilarnych. Metoda TLC, szeroko stosowana jako podstawowa metoda
rozdziału i identyfikacji związków organicznych, jest ceniona ze względu na prostotę i szybkość
oraz niskie koszty.
W chromatografii cienkowarstwowej stosowane są na ogół te same sorbenty, co w kolumnowej
chromatografii cieczowej (najczęściej modyfikowany żel krzemionkowy). Rozdział
chromatograficzny prowadzi się w specjalnej, zamkniętej komorze, zawierającej eluent i
atmosferę nasycona jego parami. Do wizualizacji efektów rozdziału wykorzystywane są
specjalne reagenty, tworzące ze związkami organicznymi barwne związki. Można też posłużyć
się światłem ultrafioletowym, wywołującym fluorescencję sorbentu, maskowaną przez
rozdzielone składniki próbki.
Chromatografia fluidalna / Chromatografia płynem nadkrytycznym (SFC - supercritical fluid
chromatography)
Jest to nowa metoda chromatograficzna, łącząca cechy chromatografii gazowej i cieczowej. Jako
fazę ruchomą wykorzystuje gaz w warunkach nadkrytycznych (np. dwutlenek węgla w temp.
powyżej 31°C i 7 MPa), w których zanikają różnice pomiędzy fazą ciekłą i gazową.
Chromatografia fluidalna jest stosowana m.in. do rozdziału substancji trudno rozpuszczalnych,
gdyż fazy nadkrytyczne wykazują zdolność do rozpuszczania substancji złożonych z dużych,
niepolarnych cząsteczek, np. wyższych alkanów lub węglowodorów poliaromatycznych.
Aparatura SFC przypomina układ do HPLC i zawiera pompę dozującą eluent, zawór służący do
wprowadzania próbek, kolumnę (pakowaną lub kapilarną), regulator ciśnienia oraz detektor
(zwykle znany z chromatografii gazowej detektor płomieniowo-jonizacyjny). Układ może być
wyposażony w programator ciśnienia eluentu, umożliwiający osiągnięcie optymalnych
warunków rozdziału.
Ćwiczenia
1. Reakcje charakterystyczne tłuszczów prostych
a. Wykazanie obecności kwasów nienasyconych w tłuszczach
Zasada:
Pod wpływem KMnO
4
następuje utlenienie nienasyconych kwasów tłuszczowych. W miejscu
podwójnego wiązania zostaje przyłączony tlen i kwas tłuszczowy rozpada się na dwa fragmenty.
Analiza ich pozwala ustalić miejsce położenia nienasyconego wiązania w łańcuchu kwasu
tłuszczowego.
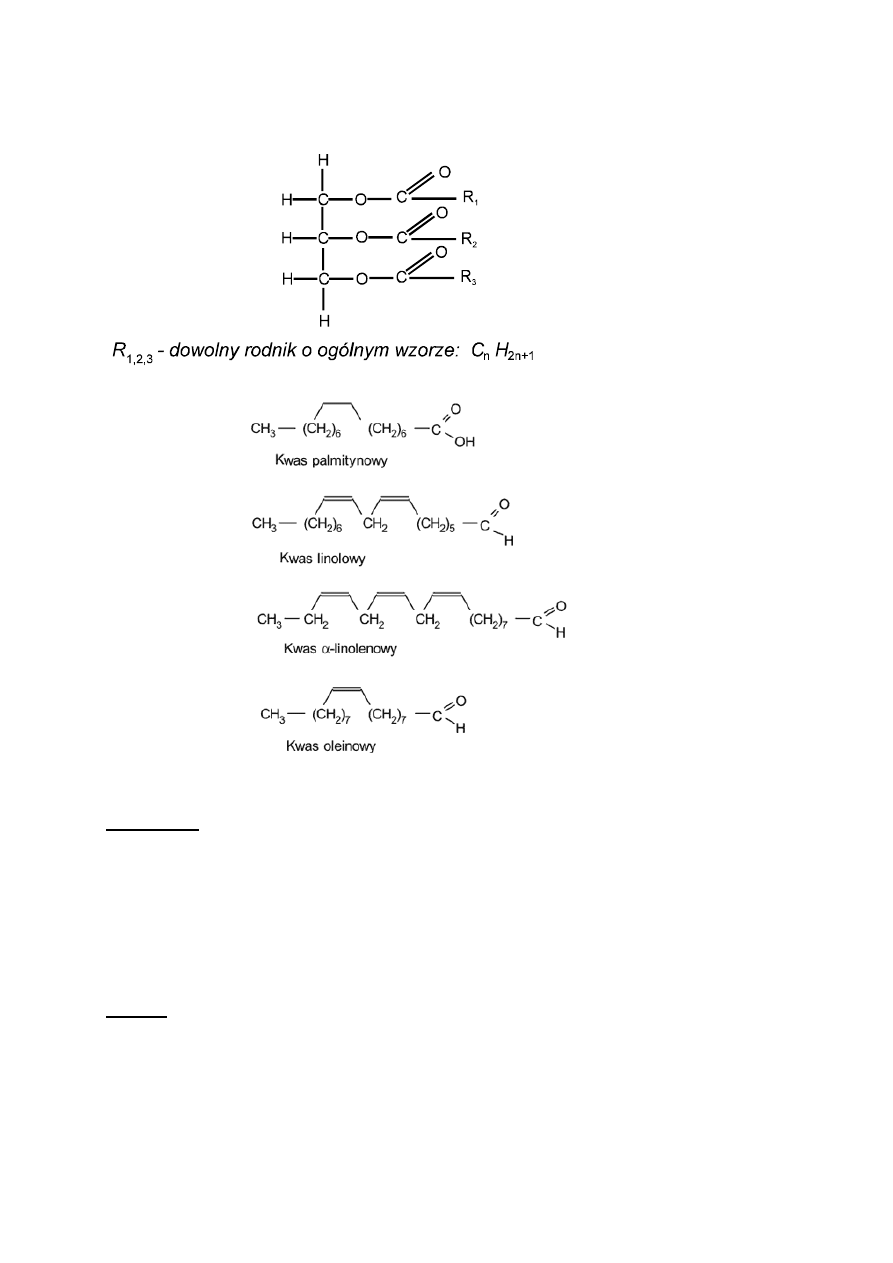
Rys. 9. Wzór strukturalny lipidu właściwego oraz przykłady kwasów tłuszczowych
Wykonanie:
Do probówki zawierającej dwie krople oliwy dodać 5 cm
3
Na
2
CO
3
o stężeniu 1 mol·dm
-3
.
Zawartość probówki lekko ogrzać i wstrząsając, dodawać po kropli KMnO
4
o stężeniu 0,01
mol·dm
-3
aż do wystąpienia trwającego ok. 2 minuty różowego zabarwienia.
b. Wykrywanie glicerolu
Zasada:
Jedną z reakcji służących do wykrywania glicerolu jest zdolność tworzenia w środowisku
zasadowym z jonami miedziowymi kompleksu o zielononiebieskiej barwie (reakcja Wagenaara).
Kompleks chelatowy tworzą dwie cząsteczki glicerolu i jeden atom miedzi. Inny sposób
wykrywania glicerolu polega na odwodnieniu glicerolu. Pod wpływem ogrzewania z KHSO
4

przekształca się on w akroleinę – nienasycony aldehyd o charakterystycznym drażniącym zapachu
(Rys. 10).
Rys. 10. Reakcja odwodnienia glicerolu
Wykonanie:
Do wykrycia glicerolu można wykorzystać następujące reakcje:
a). Umieścić w probówce kroplę glicerolu, dodać 3 cm
3
5% etanolowego roztworu NaOH i 0,5
cm
3
nasyconego roztworu CuSO
4
. W obecności glicerolu powstaje zielononiebieski osad.
b). Do dwóch kropli glicerolu umieszczonych w suchej probówce dodać około 100 mg KHSO
4
.
Zawartość probówki ogrzewać ostrożnie (!) nad płomieniem palnika aż do wystąpienia
charakterystycznego ostrego zapachu.
2. Ekstrakcja i frakcjonowanie lipidów z żółtka jaja kurzego
Wykonanie:
1 łyżeczkę żółtka jaja kurzego zalać 18 cm
3
mieszaniny składającej się z chloroformu i metanolu
zmieszanych w stosunku 1:2 i mieszać starannie przez kilka minut. Następnie mieszaninę wlać do
plastikowych próbówek wirówkowych i odwirować (5 min., 15.000 obr/min). Odessać (pipetą)
ciecz znad osadu, dodać do niej 5 cm
3
wody, dokładnie wymieszać, a następnie odessać górną
warstwę wodną. Chloroformowy ekstrakt lipidowy wlać do cylindra miarowego i uzupełnić
chloroformem do 10 cm
3
i pozostawić do dalszych doświadczeń (punkt a i d)
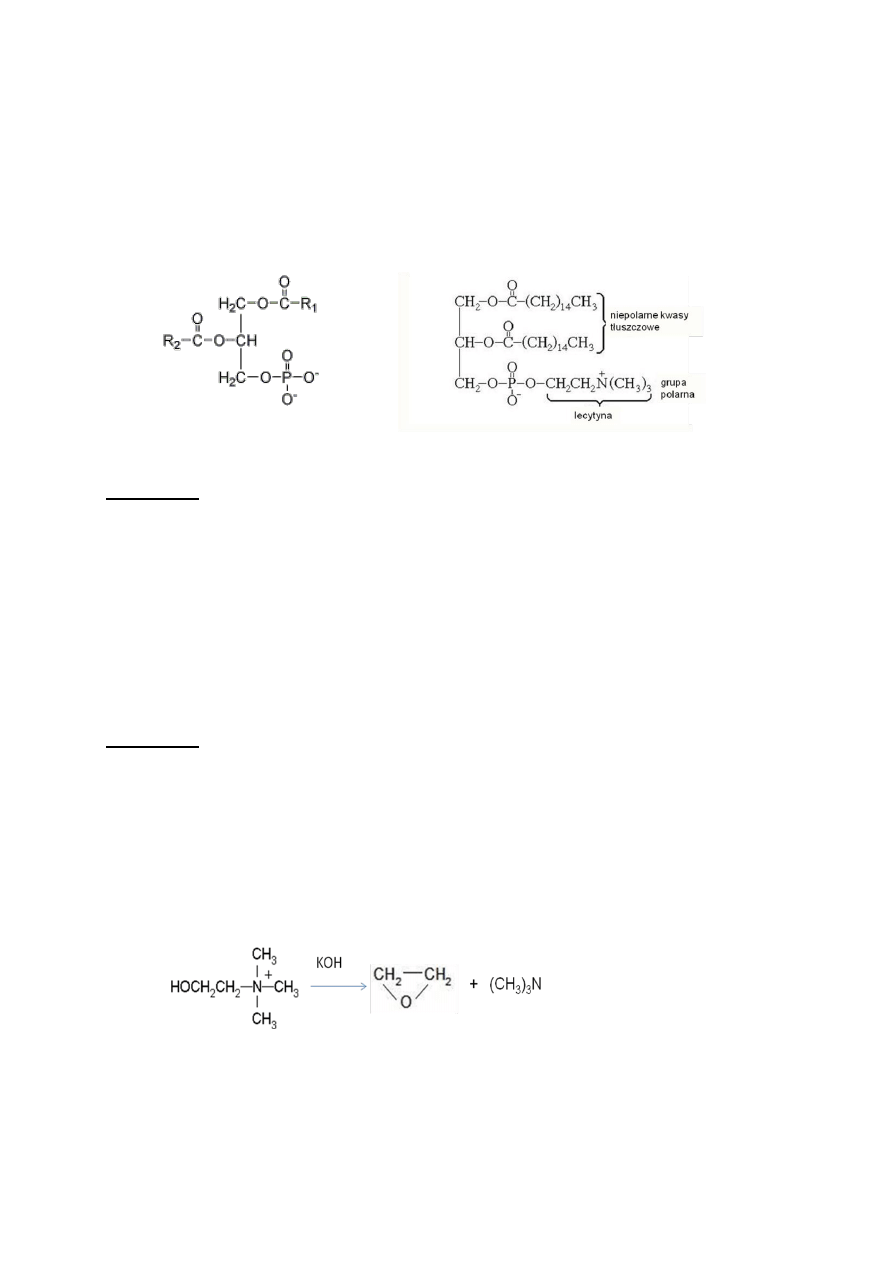
a. Wytrącanie pochodnych kwasu fosfatydowego
Rys. 11. Wzór strukturalny A. kwasu fosfatydowego i B. lecytyny
A. B.
Wykonanie:
7,5 cm
3
roztworu otrzymanego powyżej przelać powoli do cylindra z 15 cm
3
acetonu. Wytrąca się
osad glicerofosfolipidów, głównie lecytyn. Przelać zawartość do 2 plastikowych próbówek
wirówkowych (po równo) i odwirować (jak wyżej), a następnie przenieść delikatnie do 3 cm
3
wody. Wykryć obecność choliny według procedury opisanej w podpunkcie b i jonów
ortofosforanowych według procedury opisanej w podpunkcie c.
b. Wykrywanie choliny
Wykonanie:
Odmierzyć do probówki 1 cm
3
roztworu otrzymanego w podpunkcie a, dodać 1 cm
3
10% roztworu
KOH i ostrożnie (!) ogrzewać nad małym płomieniem palnika aż do pojawienia się
charakterystycznego zapachu. Zachodząca reakcja została przedstawiona poniżej:
Rys. 12. Schemat reakcji wykrywania choliny
cholina
tlenek etylenu + trójmetyloamina

c. Wykrywanie jonów ortofosforanowych
Zasada:
Obecność fosforanu można wykryć stosując kwaśny roztwór molibdenianu amonowego. Tworzy
się w tych warunkach kompleks soli amonowej heteropolikwasu czterotrójmolibdeniano-
fosforowego o żółtej barwie.
(NH
4
)
2
MoO
4
PO
4
3-
(NH
4
)
3
[P(Mo
3
O
10
)
4
]
Wykonanie:
Odmierzyć do probówki 1 cm
3
roztworu otrzymanego w punkcie a i dodać 0,5 cm
3
20% roztworu
NaOH. Zawartość probówki dobrze wymieszać, wrzucić "porcelankę" i gotować nad palnikiem
przez 2 minuty. Następnie dodać 2 cm
3
odczynnika molibdenianiowego (2% (NH
4
)
2
MoO
4
).
Roztwór barwi się na kolor żółty, co świadczy o obecności fosforanów.
UWAGA: poza żółknięciem roztworu można obserwować także wydzielanie się warstwy kwasów
tłuszczowych na powierzchni cieczy
3. Oznaczanie zawartości tłuszczu w mleku
Wykonanie:
3,5 cm
3
mleka umieścić w plastikowej probówce wirówkowej, a następnie bardzo powoli,
wstrząsając, dodać po kropli 3 cm
3
stężonego kwasu siarkowego. Unikać zagrzania probówki !
Dodać 0,3 cm
3
alkoholu amylowego, zmieszać i umieścić probówkę w wirówce. Wirować przez 5
minut przy 10000 obr./min. Po odwirowaniu zaobserwować wysokość słupa tłuszczu w górnej
części probówki.
4. Chromatografia cienkowarstwowa (TLC) fosfolipidów
Wykonanie:
Skład fosfolipidowy próbki określić metodą chromatografii cienkowarstwowej na płytkach
Kieselgel 60 (0,25 mm) firmy Merck. Przed nałożeniem próbek płytki należy aktywować w 110°C
przez 30 minut.
Komorę chromatograficzną (Rys. 13) napełnić 60 cm
3
mieszaniny rozwijającej (chloroform :
metanol : kwas octowy zmieszane w stosunku 65:25:8; v/v/v) i pozostawić zamkniętą przez 30
minut dla zrównoważenia (osiągnięcia stanu równowagi pomiędzy parami a mieszaniną).

Rys. 13. Komora chromatograficzna
Otrzymywanie ekstraktu lipidowego:
Jeden gram zlofilizowanego materiału roślinnego zmieszać z 10 cm
3
mieszaniny chloroformu i
metanolu 1:2 v:v. Odwirować 10 000 obr./min. Do analizy pobrać nasącz (ekstrakt).
Za pomocą mikropipety automatycznej nanieść na płytkę po 5
l chloroformowego ekstraktu
lipidów oraz standardy lipidowe firmy Sigma. Poszczególne roztwory winny być nakładane w
formie 5 mm paska (odstęp pomiędzy próbkami 1,5 cm; odstęp od dolnej krawędzi 1,5 cm; odstęp
od krawędzi bocznych 1,7 cm), przed zmianą nakładanego roztworu należy bezwzględnie
zmieniać końcówki pipety. Płytkę umieścić w komorze chromatograficznej zanurzając płytkę na
głębokość 5 cm w mieszaninie rozwijającej. Po rozwinięciu chromatogramu (po około 40-50
minut) płytki wysuszyć na powietrzu, wysuszone płytki umieścić w komorze wypełnionej parami
jodu i po uwidocznieniu się plamek obrysować je ołówkiem.
5. Wyznaczanie liczby kwasowej
Zasada:
Liczba kwasowa charakteryzuje ilość wolnych (niezobojętnionych) reszt kwasów organicznych
zawartych w tłuszczu. Określa się ją liczbą mg KOH potrzebnych do zobojętnienia wolnych reszt
kwasowych zawartych w 1 g tłuszczu. Podwyższona wartość liczby kwasowej może świadczyć o
procesach hydrolizy (jełczenia) tłuszczu, np. przy zbyt długim przechowywaniu.
Wykonanie:
Do kolbek odważyć po 5 g świeżego i starego tłuszczu. Każdą z próbek rozpuścić dokładnie w 50
cm
3
rozpuszczalnika benzynowego (ewentualnie OSTROŻNIE!!! podgrzewając). Po ostudzeniu
zmiareczkować próbkę 0,1 M etanolowym roztworem KOH w obecności fenoloftaleiny do
uzyskania trwałej, różowej barwy. Powtórzyć miareczkowanie w czystym rozpuszczalniku. (Ilość
KOH, którą zużyto do miareczkowania czystego rozpuszczalnika należy odjąć od ilości zasady
zużytej do miareczkowania tłuszczu). Obliczyć ilość KOH zużytą na zobojętnienie samego
tłuszczu, następnie wyznaczyć liczbę mg KOH potrzebną do zobojętnienia 1 g tłuszczu. W
obliczeniach przyjąć masę cząsteczkową KOH równą 56,11.
Przykładowe obliczenia:
Jeżeli na zobojętnienie 5g tłuszczu zużyto 20 cm
3
0,1 M (0,1 mola w 1000 cm
3
) KOH, to
wyliczamy, że w 20 cm
3
znajdowało się 50 razy mniej moli KOH czyli 2 milimole. 1 mol KOH
waży 56,11 g, a 2 milimole 112 mg. Na zobojętnienie 5 g tłuszczu zużyto zatem 112 mg KOH,
czyli na 1 g tłuszczu potrzeba 2,24 mg tej zasady.
6. Wyznaczanie liczby zmydlania
Zasada:
Liczba zmydlania charakteryzuje całkowitą ilość zestryfikowanych oraz wolnych
(niezobojętnionych) reszt kwasów organicznych zawartych w tłuszczu. Określa się ją liczbą mg
KOH potrzebnych do zobojętnienia wszystkich reszt kwasowych zawartych w 1 g tłuszczu. Liczba
ta pozwala wyznaczyć średnią masę cząsteczkową kwasów tłuszczowych zawartych w tłuszczu
(długość łańcucha węglowodorowego).
Wykonanie:
Do kolbek odważyć po 1 g tłuszczu stałego i tłuszczu płynnego, a następnie dodać 10 cm
3
0,5 M
roztworu KOH i 50 cm
3
etanolu, po czym ogrzewać przez 20 min na wrzącej łaźni wodnej i pod
chłodnicą zwrotną, albo nakrywając wylot kolby (lub lepiej zlewki) okrągłodenną kolbą, napełnioną
w połowie zimną wodą w celu skraplania oparów etanolu. Następuje wówczas hydroliza tłuszczu.
Po ostudzeniu zmiareczkować nadmiar niezobojętnionego KOH 0,5 M roztworem HCl wobec
fenoloftaleiny do zaniknięcia różowej barwy. Równolegle wykonać próbę kontrolną bez tłuszczu
(powtórzyć pomiar w czystym rozpuszczalniku).

7. Zmydlanie tłuszczów, wytwarzanie mydła, wytrącanie mydła
Zasada:
Tłuszcze jako estry kwasów tłuszczowych i glicerolu są podatne na hydrolizę. Jeśli hydrolizę
prowadzi się za pomocą wodorotlenku sodowego wówczas wydziela się wolny glicerol i sodowa
sól kwasu tłuszczowego, czyli mydło. Mydła sodowe i potasowe są rozpuszczalne, natomiast mydła
magnezowe, wapniowe i innych metali są nierozpuszczalne w wodzie.
Z roztworu wodnego można wydzielić mydło przez wysalanie w nasyconym roztworze NaCl albo
przez dodanie roztworu dowolnej soli, która wytwarza nierozpuszczalne mydło.
Rys. 14. Reakcja zmydlania tłuszczu
Wykonanie:
a) Do zlewki odważyć ok. 5 g tłuszczu, a następnie dodać 8 cm
3
30% NaOH i 5 cm
3
etanolu.
Podgrzewać łagodnie przez kilka minut, stale mieszając, aż utworzy się jednolita masa. Wówczas
dodać ok. 200 cm
3
gorącej destylowanej wody i dalej podgrzewać aż do rozpuszczenia mydła.
b) Do 20 cm
3
otrzymanego powyżej roztworu mydła dodać stopniowo stały chlorek sodowy aż do
uzyskania stanu wysycenia. Z roztworu wytrąca się wówczas osad mydła sodowego
nierozpuszczalnego w tym roztworze, które można wydzielić przez dekantację, wirowanie albo
sączenie.
c) Do 4 probówek wlać po 2 cm
3
roztworu mydła otrzymanego w podpunkcie a), dodać po 1 cm
3
rozcieńczonych roztworów Ca(NO
3
)
2
lub CaCl
2
, BaCl
2
, CuSO
4
i Pb(CH
3
COO)
2
. W każdym
przypadku wytrącają się osady mydeł nierozpuszczalnych pomimo dodawania większej objętości
wody.

8. Wyznaczanie liczby jodowej
Zasada:
Liczba jodowa charakteryzuje ilość podwójnych wiązań w kwasach tłuszczowych (stopień ich
nienasycenia). Określa się ją liczbą gramów jodu przyłączonych przez nienasycone kwasy
tłuszczowe zawarte w 100 g tłuszczu. Tłuszcz rozpuszcza się w rozpuszczalniku i dodaje bromek
jodu JBr, który przyłącza się do podwójnego wiązania w tłuszczu.
Rys. 15. Wzory przykładowych kwasów tłuszczowych nienasyconych oraz przyłączanie się jodu i
bromu do podwójnego wiązania
Nadmiar JBr reaguje z jodkiem potasu tworząc bromek potasu i uwalniając jod cząsteczkowy (J
2
).
JBr + KJ = J
2
+ KBr
Ostatecznie wydzielony jod miareczkuje się tiosiarczanem sodu Na
2
S
2
O
3
w obecności skrobi jako
wskaźnika:
J
2
+ 2Na
2
S
2
O
3
= Na
2
S
4
O
6
+ 2NaJ
Płynne tłuszcze zawierające nienasycone kwasy tłuszczowe charakteryzują się wysokimi
wartościami liczby jodowej.

Wykonanie:
Do dwóch kolbek ze szlifem o pojemności 200-300 cm
3
odważyć po 0,5 g tłuszczu płynnego i
stałego, a następnie rozpuścić w 10 cm
3
chloroformu. Następnie dodać 15 cm
3
odczynnika Hanusa
(1g JBr rozpuszczony w 50 cm
3
lodowatego kwasu octowego), wymieszać, zamknąć starannie i
odstawić na 30 min w ciemne miejsce. Po tym czasie dodać 50 cm
3
3% wodnego roztworu KJ,
spłukując starannie szyjkę i korek. Wymieszać i miareczkować 0,1 M roztworem Na
2
S
2
O
3
w
obecności skrobi jako wskaźnika (dodać kroplę kleiku skrobiowego) do zaniku niebieskiej (czasem
czarnej) barwy. Równolegle wykonać analizę kontrolną w ślepej próbie nie zawierającej tłuszczu.
Różnica objętości tiosiarczanu zużytego w ślepej próbie i próbie pomiarowej pozwala wyliczyć
ilość jodu wydzieloną w reakcji badanej próbki tłuszczu. Porównać wartości liczby jodowej dla
obydwu badanych tłuszczów, zinterpretować wyliczoną różnicę.
Jak wynika z powyższego równania 1 mol cząsteczkowego jodu reaguje z 2 molami tiosiarczanu
sodu. 1 cm
3
roztworu 0,1 M tiosiarczanu sodu zużytego w miareczkowaniu zawiera 0,1 mM
tiosiarczanu, który proporcjonalnie reaguje z 0,05 mM jodu, czyli z 22 mg J
2
. Należy zatem
przeliczyć, ile milimoli tiosiarczanu zużyto na zmiareczkowanie wolnego jodu i z proporcji
obliczyć liczbę miligramów jodu. Im mniej jest wiązań podwójnych w badanym tłuszczu, tym
mniej bromku jodku jest wiązane, a więc więcej wolnego jodu wydziela się w reakcji z jodkiem
potasu. A zatem więcej tiosiarczanu sodu zużywa się podczas miareczkowania. Duża liczba jodowa
świadczy o wysokim stopniu nasycenia kwasów w badanym tłuszczu (czyli o małej liczbie wiązań
podwójnych).
Wyszukiwarka
Podobne podstrony:
Biochemia skrypt AGH
BIOCHEMIA SKRYPT
BIOCHEMIA skrypt 2010 id 86508 Nieznany
chemia zywności wykłady, Zachomikowane, Naukowe, Medycyna, Biochemia, Skrypty
Podział cukrów biochemia ćwiczenia cukry reakcje?rwne
Cukry i lipidy
biochemia skrypt aminokwasy i bia id 86612
Podział cukrów biochemia ćwiczenia cukry reakcje?rwne
Biochemia 1 kolo cukry, Zootechnika UP Lublin, biochemia
biochemia sprawko cukry, BIOCHEMIA
BIOCHEMIA Sprawozdanie cukry id Nieznany (2)
Biochemia skrypt, Fizjoterapia OSW zaoczne, BIOCHEMIA
Cwiczenie 11 cukry i lipidy
biochemia druk cukry i polisacharydy
Biochemia skrypt AGH
BIOCHEMIA SKRYPT
BIOCHEMIA skrypt 2010 id 86508 Nieznany
więcej podobnych podstron