
Filogenetyka
Katedra Genetyki, Hodowli i Biotechnologii
Roślin, SGGW
Dr inż. Magdalena Święcicka, dr hab. Marcin Filipecki

Filogenetyka
Cel
– rekonstrukcja historii ewolucji
wszystkich organizmów
Klasyczne podejście:
historia ewolucji jest odtwarzana na
podstawie porównań cech
morfologicznych i fizjologicznych
badanych organizmów.

Filogenetyka
Molekularne podejście:
zadaniem filogenetyki molekularnej jest
zrekonstruowanie związków filogenetycznych między
badanymi sekwencjami
Podstawowe założenie w filogenetyce molekularnej:
sekwencje przodka mutują w sekwencje potomków
podobne gatunki są genetycznie blisko spokrewnione

Mechanizmy ewolucji
Mutacje w genach
Mutacje są rozprzestrzeniane w
populacji poprzez dryf genetyczny i/lub
selekcję naturalną
Duplikacja i rekombinacja genów

tempo mutacji zależy od regionu w genomie, genie, rodzaju genu;
częściej obserwuje się podstawienia w III pozycji kodonów;
CCG (prolina) zmiana G na jakikolwiek nt nie powoduje zmiany
aminokwasu
CTG (leucyna) zmiana C-T nie powoduje zmian
zmiana SYNONIMICZNA
zmiana NIESYNONIMICZNA
częściej obserwuje się podstawienia typu tranzycji (puryna-
puryna, pirymidyna-pirymidyna) niż transwersji;
częściej obserwowane są podstawienia między aminokwasami
podobnymi do siebie, ze względu na swoje właściwości
biochemiczne, biofizyczne, np.:
izoleucyna – lecyna
walina – izoleucyna
Kwas asparaginowy – kwas glutaminowy

OBOWIĄZUJĄCE SYMBOLE AMINOKWASÓW
Symbol
3-literowy
znaczenie
kodony
A
Ala
Alanina
GCT, GCC, GCA, GCG
B
Asp, Asn
Asparagina, Asparaginian
GAT, GAC, AAT, AAC
C
Cys
Cysteina
TGT, TGC
D
Asp
Asparaginian
GAT, GAC
E
Glu
Glutaminian
GAA, GAG
F
Phe
Fenyloalanina
TTT, TTC
G
Gly
Glicyna
GGT, GGC, GGA, GGG
H
His
Histydyna
CAT, CAC
I
Ile
Izoleucyna
ATT, ATC, ATA
K
Lys
Lizyna
AAA, AAG
L
Leu
Leucyna
TTG, TTA, CTT, CTC, CTA, CTG
M
Met
Metionina
ATG
N
Asn
Asparagina
AAT, AAC
P
Pro
Prolina
CCT, CCC, CCA, CCG
Q
Gln
Glutamina
CAA, CAG
R
Arg
Arginina
CGT, CGC, CGA, CGG, AGA, AGG
S
Ser
Seryna
TCT, TCC, TCA, TCG, AGT, AGC
T
Thr
Treonina
ACT, ACC, ACA, ACG
V
Val
Walina
GTT, GTC, GTA, GTG
W
Trp
Tryptofan
TGG
X
Xxx
Nieznany
Y
Tyr
Tyrozyna
TAT, TAC
Z
Glu, Gln
Glutaminian, Glutamina
GAA, GAG, CAA, CAG
*
End
Terminator
TAA, TAG, TGA

rzadko obserwuje się podstawienia między
aminokwasami bardzo różniącymi się swoimi
właściwościami:
tryptofan – izoleucyna
rzadko obserwuje się podstawienia między
aminokwasami pełniącymi ważne role w białkach:
tryptofan (
T
G
G
) na kodon stop (
T
A
G
)
mutacje
missens
– jeden aminokwas zastępowany
innym
mutacje
nonsens
– terminacja translacji
zmiana ramki odczytu
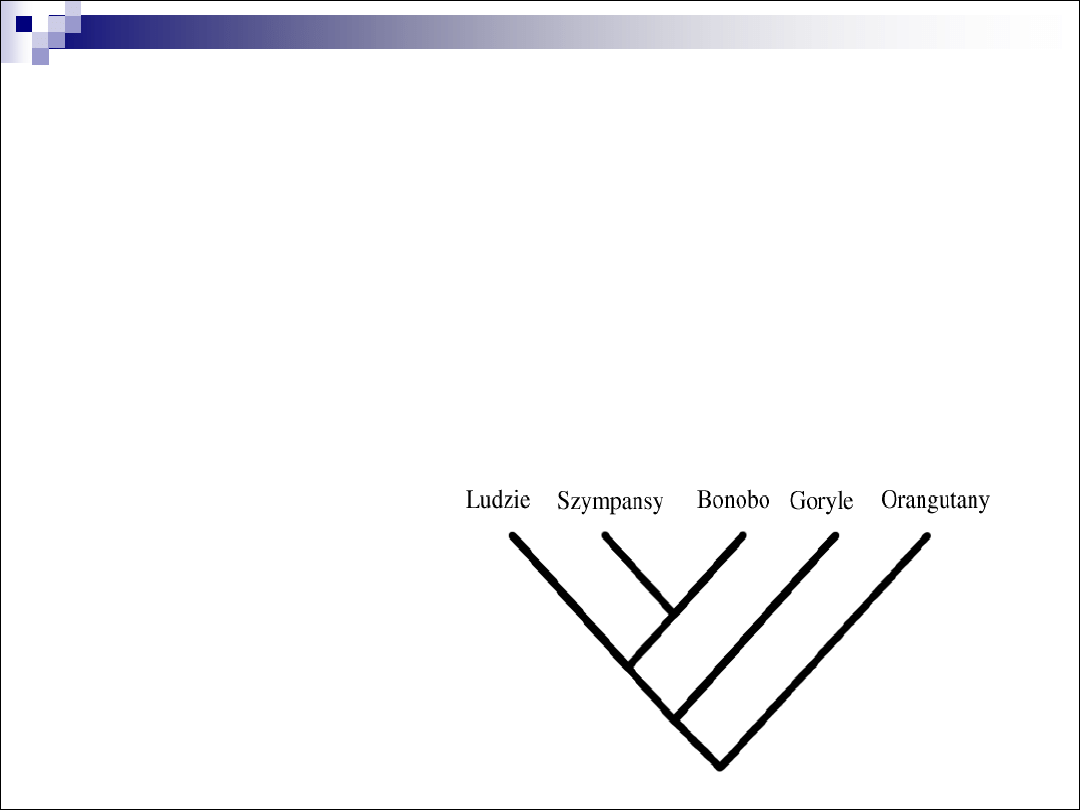
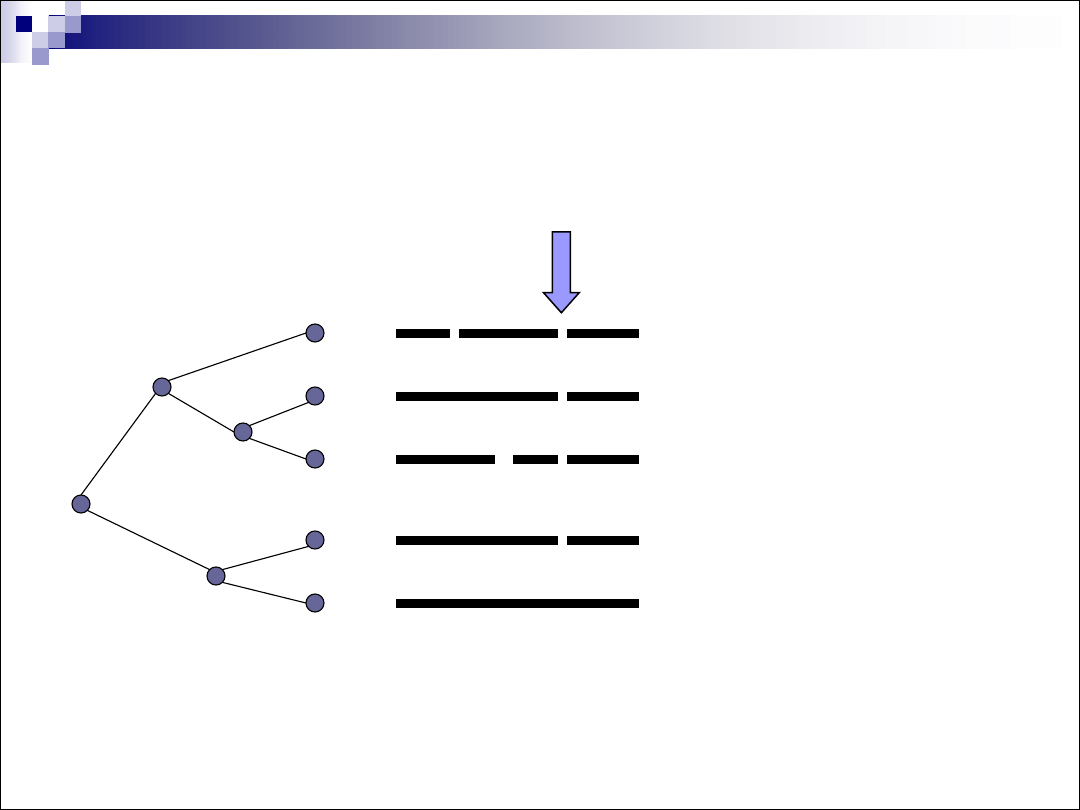
Wyrazem analiz filogenetycznych są
drzewa
filogenetyczne
między cząsteczkami –
drzewo genów
lub organizmami –
drzewo gatunków

Korzeń
– wspólny przodek dla wszystkich taksonów
Gałąź
– obrazuje związki ewolucyjne między
porównywanymi jednostkami taksonomicznymi
Długość gałęzi
– zazwyczaj reprezentuje liczbę zmian,
które się zdarzyły w danej linii ewolucyjnej
Węzeł
– reprezentuje miejsce rozgałęzień jednostek
taksonimicznych (populacji, organizmu, genu).
Liść
– reprezentuje aktualnie analizowaną jednostkę
taksonomiczną

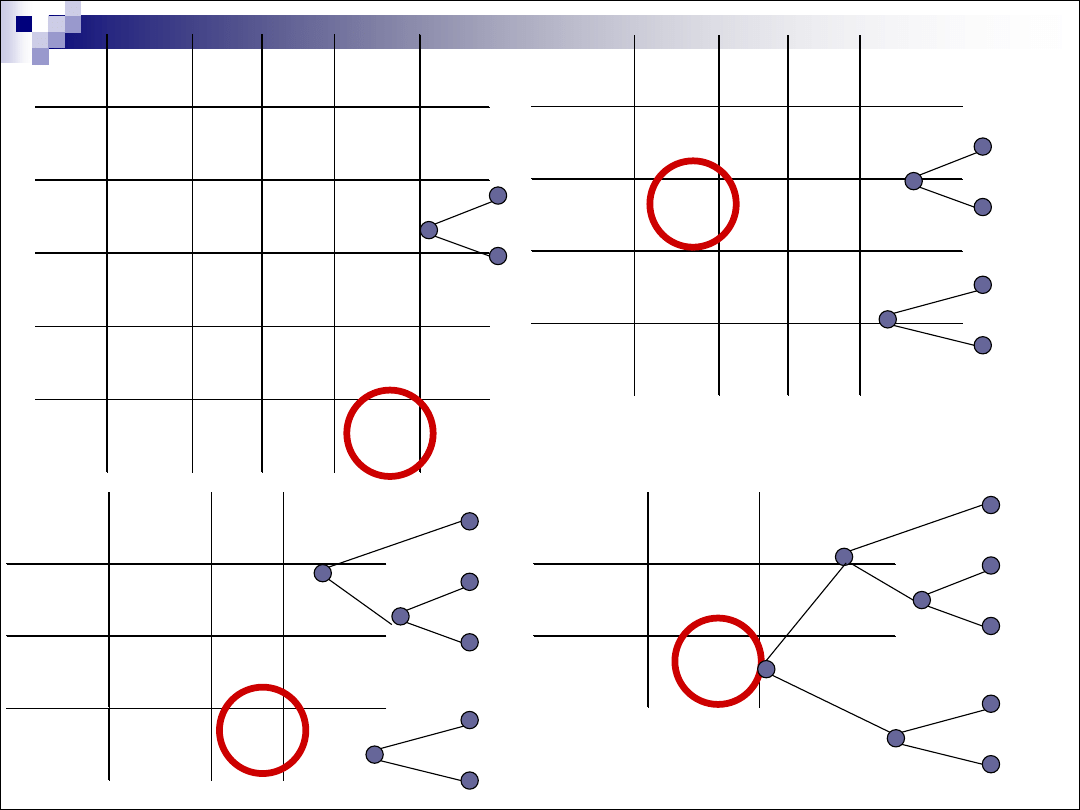
Drzewa
ukorzenione
i
nieukorzenione
znany wspólny przodek lub istnieje hipoteza na temat
wspólnego przodka / nieznany wspólny przodek
Topologia drzewa
Długość gałęzi (czas ewolucji, ilość zmian)
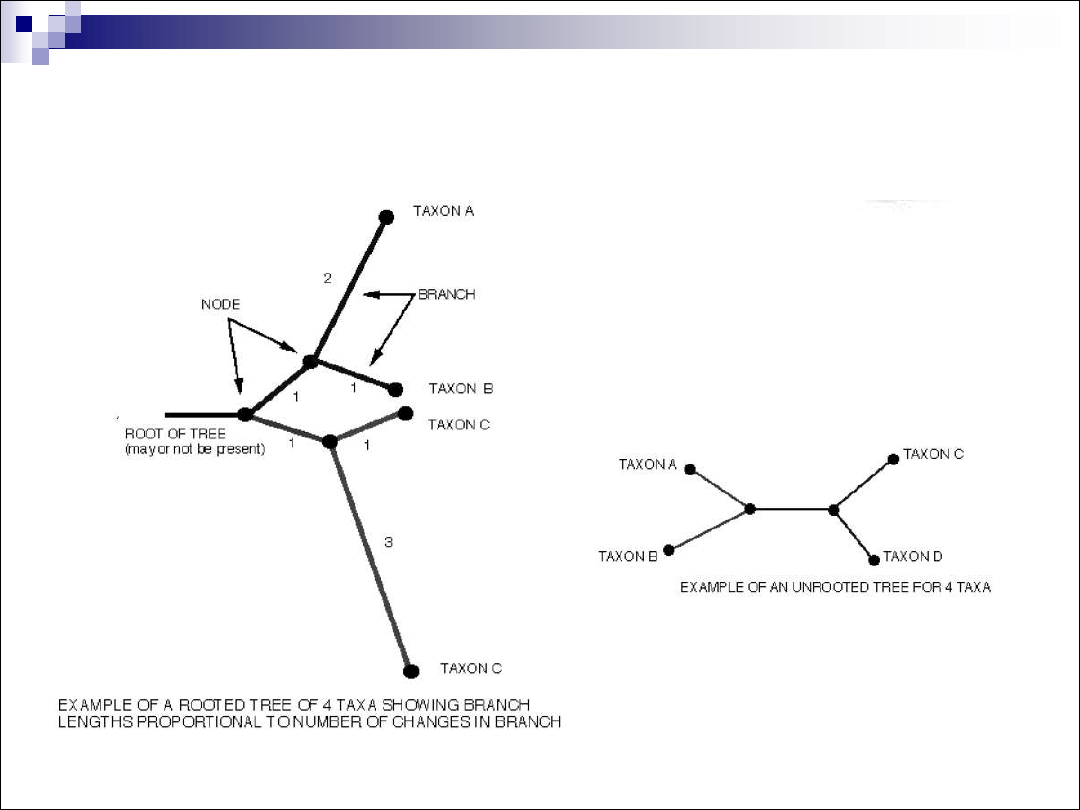
Przykładowe drzewa filogenetyczne

Po co konstruuje się drzewa filogenetyczne?
•Poznanie i zrozumienie historii ewolucyjnej
•Mapowanie różnicowania szczepów patogennych do
opracowania szczepionek
•Wsparcie dla epidemiologów
– Choroby infekcyjne
– Defekty genetyczne
• Narzędzie do przewidywania funkcji nowo odkrytych
genów
• Badania różnicowania układów biologicznych
• Poznanie ekologii mikroorganizmów

Filogenetyka zwana jest czasem
kladystyką
Klad
– zbiór potomków pochodzących od pojedynczego przodka
Podstawowe założenia kladystyki
:
1. każda grupa
organizmów
jest spokrewniona przez
pochodzenie od wspólnego przodka
2. kladogeneza ma charakter
bifurkacyjny
(rozwidlający się)
3. zmiany
w cechach pojawiają się w liniach
filogenetycznych z upływem czasu
Drzewo genów: bifurkacja – mutacja
Drzewo gatunków: bifurkacja – specjacja
Mutacja –
warunek niezbędny, ale nie zawsze wystarczający do
specjacji
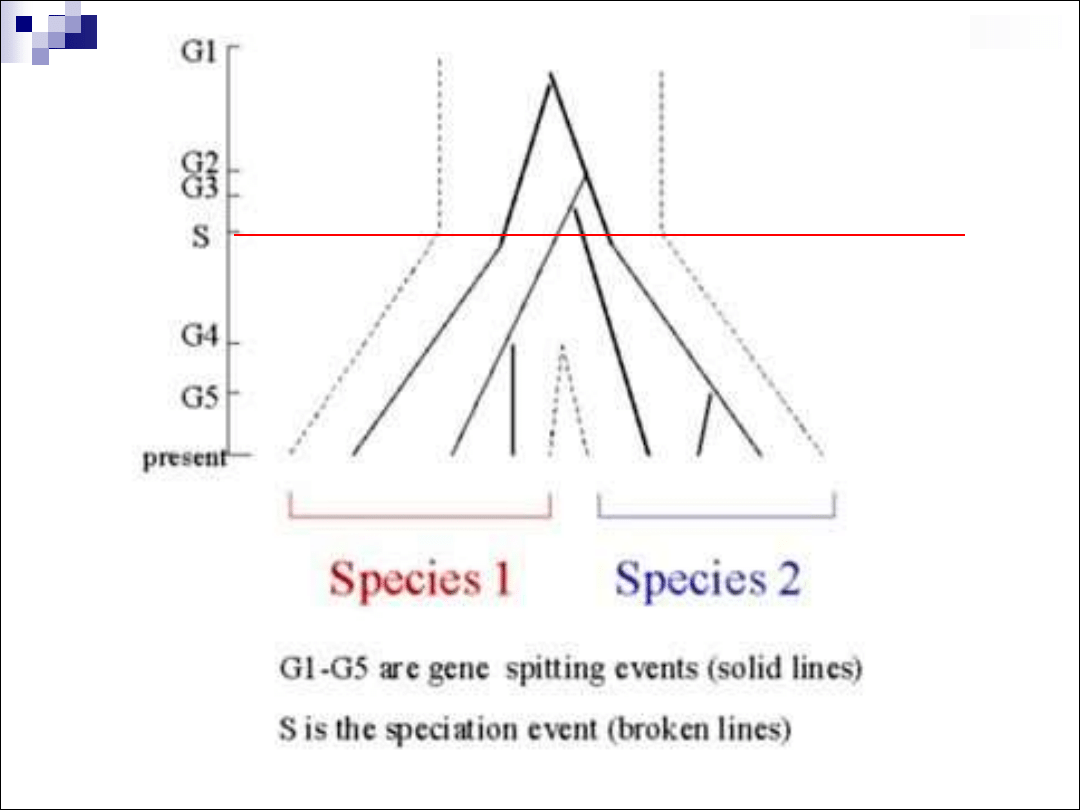

Często zapominamy o:
I Domniemany znak równości między podobieństwem
zestawu cech (np. nukleotydów), a pochodzeniem
II Mutacje somatyczne ≠ mutacje genetyczne
Mutacja – DNA lub białka wydziela się z tkanek
somatycznych, dla filogenezy istotne są tylko mutacje
w gametach
III Cechy używane do budowy drzewa gatunków mają
się nijak do cech używanych do budowy drzewa
genów


Cechy, które mogą być użyte do budowy drzewa rzędów owadów:
Poruszanie się
Okrycie stwardniałym oskórkiem lub kokonem,
Widoczność niezupełnie rozwiniętych narządów
Widoczność niecałkowicie wykształconych i nie funkcjonujących
odnóży,
Widoczność zawiązków skrzydeł
Widoczność aparatu gębowego
Zdolność do aktywnego poruszania się
Pełne wykształcenie narządów lokomotorycznych
Pełne wykształcenie zmysłów
Obecność członowanych odnóży krocznych
Liczba członowanych odnóży krocznych
Obecność pseudopodiów
Liczba pseudopodiów
Geny, które bierze się najczęściej do budowy drzew genów:
Cytochrom B
NADH dehydrogenase subunit I (ND1)
18S RNA
28S RNA
Horyzontalny transfer genów

Niektóre domyślne założenia kladystyki:
• sekwencje są poprawne
• sekwencje są
homologiczne
Podobieństwo
– to wielkość obserwowalna, którą można
określić np. jako % identycznych aminokwasów.
Homologia
– określa
wspólne pochodzenie
porównywanych
genów (to może być wniosek wyciągnięty z analizy
podobieństwa)
Termin
homologiczne
oznacza
odziedziczone po
wspólnym przodku

Niektóre domyślne założenia kladystyki (cd):
• każda pozycja
w sekwencjach dopasowanych (alignment)
jest homologiczna z każdą odpowiednią pozycją w tym
dopasowaniu
• różnorodność sekwencji w danym zbiorze jest na tyle
duża, że zawiera filogenetyczne sygnały, odpowiednie do
rozwiązania postawionego problemu

Jakich sekwencji użyć ?
•DNA (mt, rDNA, powoli czy szybko ewoluujące)
– Bardzo szczegółowe, niejednolite tempo mutacji
•cDNA/RNA
– Użyteczne dla bardziej odległych sekwencji homologicznych
•Sekwencje białkowe
– Użyteczne do badania większości odległych sekwencji
homologicznych, możliwość konstrukcji bardzo rozległych
ewolucyjnie drzew, bardziej jednolite tempo zmienności
mutacyjnej, więcej elementów zmienności

Jacek Leluk
Interdyscyplinarne Centrum Modelowania Matematycznego i Komputerowego, Uniwersytet Warszawski
Sekwencje rybosomowego 16S RNA
•Występują we wszystkich organizmach
•Są wysoce konserwatywne
•Nadają się do konstruowania bardzo rozległych
ewolucyjnie drzew
•Znane dla kilkudziesięciu tysięcy organizmów, głównie
prokariotycznych

Co jest obliczane?
Topologia drzewa
–porządek (kolejność) odgałęzień i korzeń
Długość odgałęzień (czas ewolucji)
Sekwencje przodków
Wartości pokrewieństwa (np.
prawdopodobieństwo poszczególnych przemian)
Wiarygodność drzewa

Jacek Leluk
Interdyscyplinarne Centrum Modelowania Matematycznego i Komputerowego, Uniwersytet Warszawski
Dopasowywanie sekwencji
(Multiple Sequence Alignment)
• Dopasowanie spokrewnionych sekwencji w taki
sposób, żeby odpowiadające sobie pozycje
znajdowały się w tej samej kolumnie
• Wypełnienie brakujących miejsca kreskami
(delecje, insercje)
• Każda kolumna znaków staje się pojedynczym
elementem do dalszych obliczeń filogenetycznych
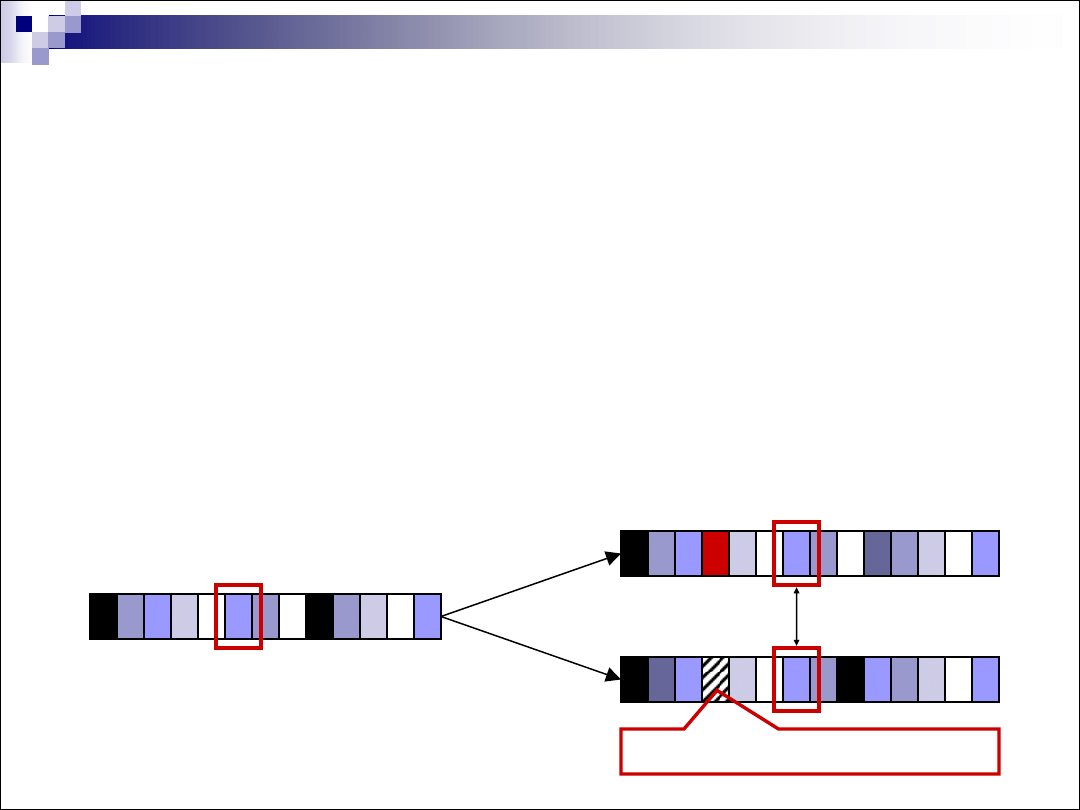
Dopasowanie i porównanie wielu sekwencji
Celem porównania wielu sekwencji jest ułożenie w
kolumnach aminokwasów (nukleotydów) pochodzących
od jednego aminokwasu (nukleotydu) w białku (genie)
wspólnego przodka we wszystkich sekwencjach od
niego pochodzących.
Wstawienie przerwy

Porównanie parami
Porównanie parami wszystkich sekwencji
- seq_1 & seq_ 2
0.91
seq_ 1 & seq_ 3
0.23
…
seq_ 8 & seq_ 9
0.87

Porównanie wielu sekwencji
W oparciu o
dendrogram przewodni
zaczyna
się porównywanie grup sekwencji.
Drzewo przewodnie
wskazuje, które
sekwencje są najbliższe – a więc najpierw
porównuje się te „łatwe”, a trudniejsze
zostawia się na potem.

Sekwencje nieułożone
a mthislgslyshktaktingsdeaskmewhf
b mthvslgsmyshktgrtingsdqaskkmewhy
c mshisitmyshktartidgseqaskmewhy
d mthipigsmyshktaravngseqasklqwhy
e mthipigsmystartincseqasklewhy
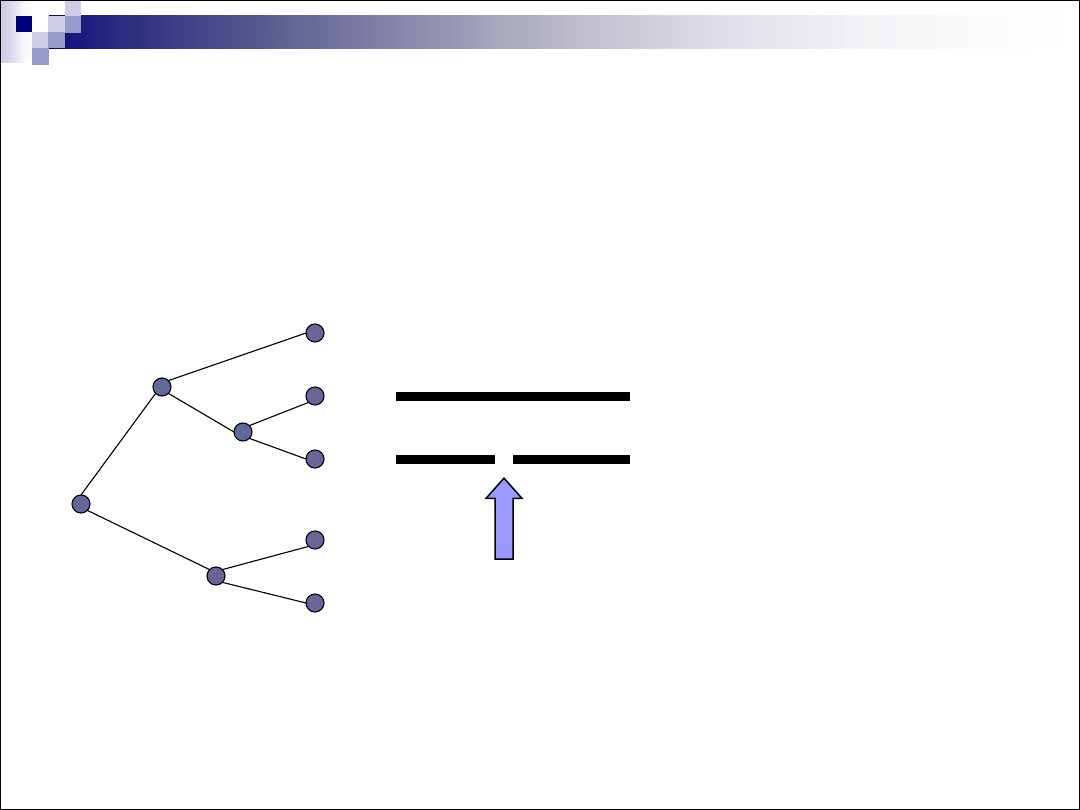
Porównanie wielu sekwencji
D
E
C
A
B
mthipigsmyshktaravngseqasklqwhy
mthipigsmys--tartincseqasklewhy
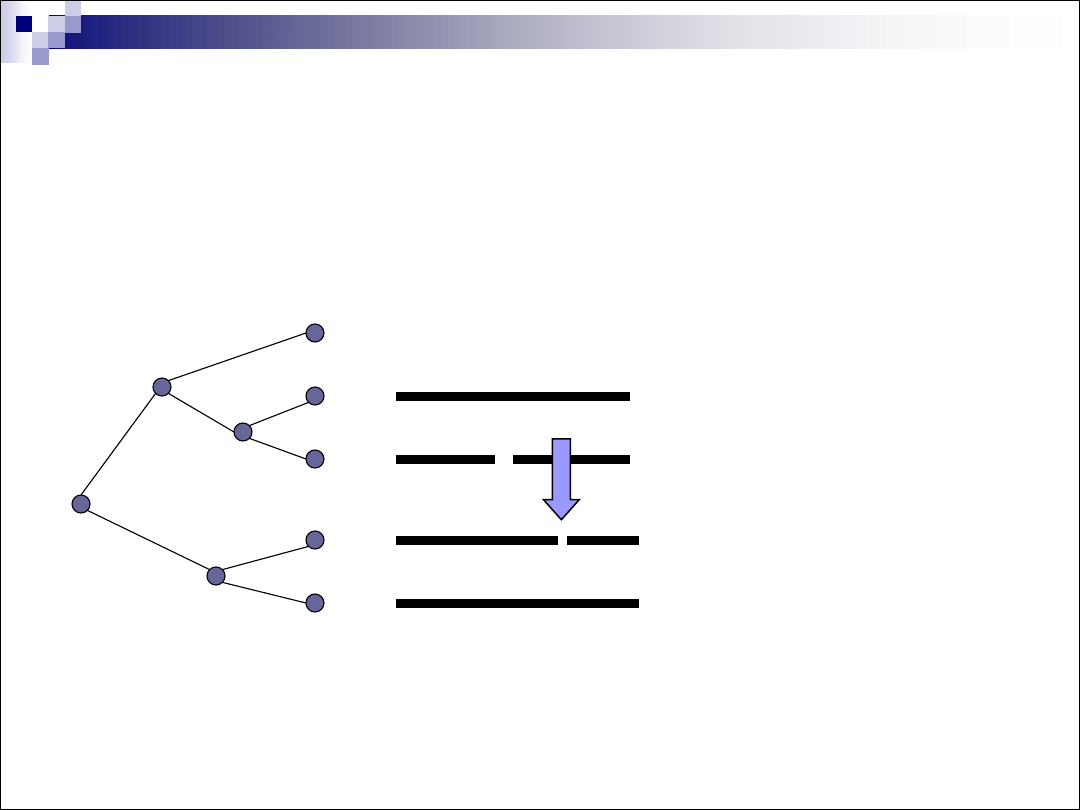
Porównanie wielu sekwencji
D
E
C
A
B
mthipigsmyshktaravngseqasklqwhy
mthipigsmys--tartincseqasklewhy
mthislgslyshktaktingsdeas-kmewhf
mthvslgsmyshktgrtingsdqaskkmewhy
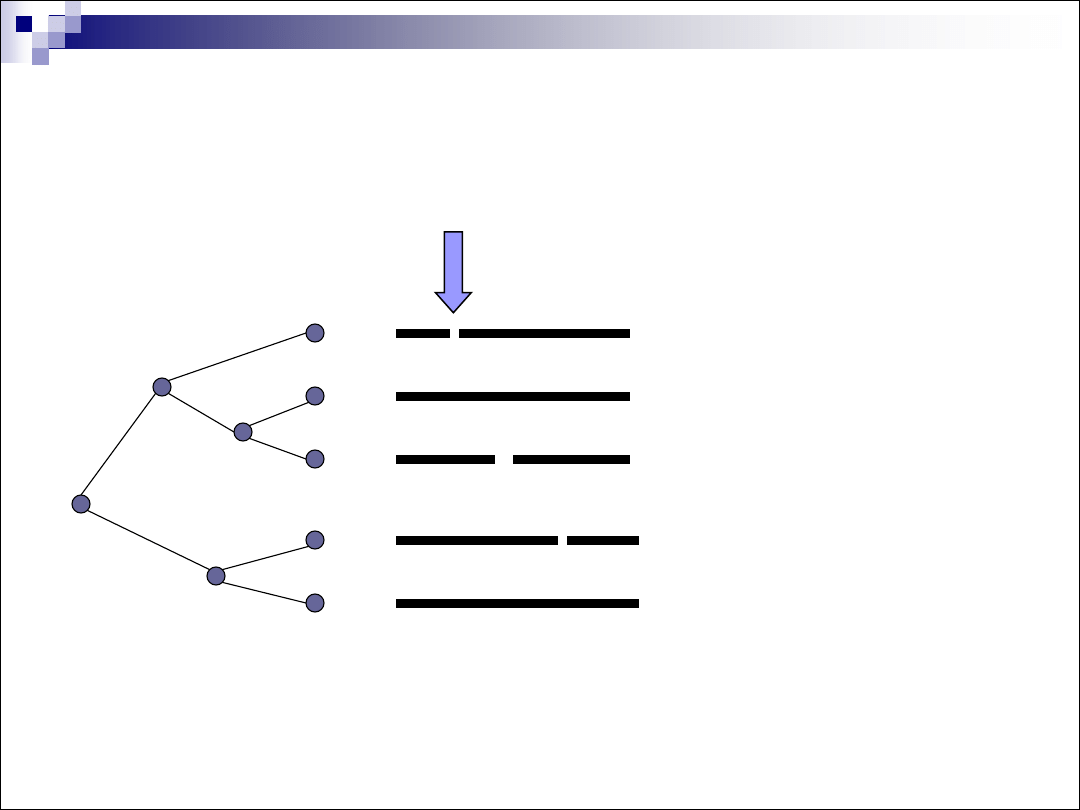
Porównanie wielu sekwencji
D
E
C
A
B
mshisi-tmyshktartidgseqaskmewhy
mthipigsmyshktaravngseqasklqwhy
mthipigsmys--tartincseqasklewhy
mthislgslyshktaktingsdeas-kmewhf
mthvslgsmyshktgrtingsdqaskkmewhy
Porównanie wielu sekwencji
D
E
C
A
B
mshisi-tmyshktartidgseqas-kmewhy
mthipigsmyshktaravngseqas-klqwhy
mthipigsmys--tartincseqas-klewhy
mthislgslyshktaktingsdeas-kmewhf
mthvslgsmyshktgrtingsdqaskkmewhy

Sekwencje ułożone
a mthislgslyshktaktingsdeas-kmewhf
b mthvslgsmyshktgrtingsdqaskkmewhy
c mshisi-tmyshktartidgseqas-kmewhy
d mthipigsmyshktaravngseqas-klqwhy
e mthipigsmys--tartincseqas-klewhy

Metody obliczeniowe konstruowania drzew
filogenetycznych
•
Metody analizy odległościowe (distance methods)
– met. średnich połączeń – (UPGMA; unweighted pair group method
with arithmetic mean,
- met. przyłączania sąsiadów (NJ; neighbor joining)
- met. Fitch-Margoliash (FM)
- met. minimalnych odległości (ME)
•
Metody oparte na cechach (character based methods)
- met. największej oszczędności (MP; Maximum Parsimony)
- met. największej wiarygodności (ML; Maximum Likelihood)
•
Łączenie drzew - drzewa konsensusowe, superdrzewa

Budowa dendrogramu przewodniego
Skonstruowanie dendrogramu przewodniego w
oparciu o porównania parami
Metoda średnich połączeń
- UPGMA
– unweighted pair
group method with arithmetic mean (PileUp & Clustal
V)
Metoda przyłączania sąsiada
- Neighbor-Joining (NJ)
(Clustal W, Clustal X)

Metody odległościowe
Odległość wyrażana jest w ułamkach
miejsc, którymi różnią się między sobą 2
sekwencje w wielokrotnym przyrównaniu
Para sekwencji różniąca się w 10% miejsc
jest bliżej spokrewniona niż para różniąca
się w 30%.

Metody odległościowe
przodek linia potomna
liczba zmian
A
C – A
0
A
C – G
1
A
C
0
C

Metoda nieważona grupowania parami ze średnią
arytmetyczną UPGMA
program znajduje najpierw
parę taksonów
, którą
dzieli
najmniejsza różnica
i ustala
punkt
rozejścia
między nimi, czyli węzeł,
w
połowie
odległości
.
łączy je
w klaster i wpisuje do nowej macierzy
odległości dzielące ten klaster od pozostałych
powtarzanie
tych etapów, aż macierz zostanie
zredukowana do 1 obiektu
A B C D E
A
0 6 9 11 9
B
6 0 7
9 7
C
9 7 0
8 6
D 11 9 8
0 4
E
9 7 6
4 0
A B C DE
A
0
6
9
10
B
6
0
7
8
C
9
7
0
7
DE 10
8
7
0
AB C DE
AB
0
8
9
C
8
0
7
DE
9
7
0
AB CDE
AB
0
8.5
CDE
8.5
0
D
E
D
E
A
B
D
E
C
A
B
D
E
C
A
B
1.
2.
3.
4.

UPGMA
Hipoteza zegara molekularnego
– ewolucja
różnych gatunków zachodzi w takim samym
tempie (FAŁSZ)
Rzadko używana metoda przez filogenetyków,
nadal popularna w epidemiologii
drobnoustrojów

Metody odległościowe –
przyłączanie
sąsiadów (NJ)
umożliwia konstruowanie
nieukorzenionych
drzew
drzewa addytywne
– odległość pomiędzy
gatunkami reprezentowanymi przez liście drzewa
są równe sumie długości łączących je gałęzi
(odległości od obu taksonów do węzła nie muszą
być identyczne)
i
j
n

Metody oparte na cechach
metoda
największej oszczędności
(MP)
metoda
największej wiarygodności
(ML)

Metoda największej oszczędności (MP)
Metoda
parsymonii
(oszczędności) –
najodpowiedniejsze jest takie drzewo, w
którym potrzebujemy najmniejszej liczby
zmian do wyjaśnienia danych występujących
jako przyrównanie sekwencji.
Kryterium parsymonii
A B
C D
+
A
D C
B
+
*
A
C D
B
+ +
Które drzewo jest najprostszym wytłumaczeniem
obserwowanego zróżnicowania cechy między gatunkami?
+ wykształcenie się cechy
* utracenie cechy

Metoda największej wiarygodności
Poszukiwanie drzewa, które zgodnie z określonym
modelem ewolucji maksymalnie uwiarygodnia dane.
Wiarygodność obliczamy dla:
topologii drzewa
długości gałęzi
wartości wskaźników tempa podstawień (częstość występowania
zasady, liczba tranzycji / liczby transwersji)
Wyznaczenie wartości ML może posłużyć do utworzenia
rankingu alternatywnych drzew.

Metoda bootstrap
Pozwala oszacować wiarygodność rozgałęzień w
drzewach
Porównuje topologię drzewa dla losowo
wygenerowanych dopasowań sekwencji (100 –
1000 dopasowań)
Drzewo z
wartościami bootstrap
(odsetek
wygenerowanych drzew, w których obserwowano
dokładnie takie samo rozgałęzienie linii
ewolucyjnych)

Dobór właściwego algorytmu
•Niedyskretny charakter zmiennych jednostek, duża ilość
danych, niewielkie zasoby obliczeniowe ==> Metoda
najbliższego sąsiedztwa (Neighbor joining)
•Dyskretny charakter zmiennych, niewielka liczba
mutacji/homoplazja ==> Maximum Parsimony
•Dyskretny charakter zmiennych, ograniczona długość
sekwencji, występowanie zjawiska homoplazji
==>Maximum Likelihood
• Dyskretny charakter zmiennych, wiele gatunków
==>Superdrzewo
•Kompletne genomy ==>Filogeneza całych genomów
Wyszukiwarka
Podobne podstrony:
FILOGENETYKA
26 Spalik, Piwczynski, Rekonstrukcja filogenezy i wnioskowanie filogenetyczne w badaniach ewolucyjny
filogeneza gatunków
Filogeneza układu pokarmowego
Filogeneza i systematyka organizmów żywych, Podręczniki , Biologia, Biologia -- TESTY MATURALNE !!!
Analiza filogenetyczna
filogeneza
16 Filogeneza i podział tkanek, FARMACJA, ROK 1, BOTANIKA
pytania z analizy filogenetycznej (1)egzamin
FILOGENEZA2
=1 Filogeneza roślin
Budowanie drzewa filogenetycznego
Filogeneza układu nerwowego i ewolucja układu nerwowego, psychologia uś, rok I
antropologia fizyczna filogeneza człowieka part2
6 Proces nabywania kultury w procesie filogenetycznym
FILOGENETYKA
więcej podobnych podstron