
Cechy uwarunkowane genetycznie
Cechy monogenowe uwarunkowane genetycznie mogą być:
–
autosomalne
–
dominujące
–
recesywne
–
sprzężone
–
autosomalnie
–
powiązanie
–
odpychanie
–
z płcią
–
z chromosomem X (gonosomalne)
–
dominujące
–
recesywne
–
z chromosomem Y (holandryczne)
Cechy autosomalne dominujące
Otoskleoroza
1/ 330
Hipercholesterolemia rodzinna
1 / 500 (heterozygoty) 1/1 mln (homozygoty)
–
heterozygoty
1 / 500
–
homozygoty
1/1 mln
Choroba Huntingtona
4-7 / 100 000
Skrzywienie palca (kamptodaktylia)
1 / 1500
Nerwiakowłókniakowatość
1 / 3500
Naczyniakowatość twarzy i mózgu
1 / 5000
Achondroplazja
1 / 15 000 – 40 0000
Siatkówczak oka (retinoblastoma)
1 / 20 000
Wielopalczastość (polidaktylia)
1 / 25 000
Zespół błękitnych białkówek
1 / 25 000
Cechy autosomalne recesywne
Mukowiscydoza
1 / 2000 – 4000
–
Czarnoskórzy
1 / 12 000 – 17 000
–
Chińczycy
1 / 90 000
Niedobór alfa-1-antytrypsyny
1 / 4000
Albinizm
–
OCA1
1 / 35 000
–
OCA 2
1 / 38 000
–
OA
1 / 54 000*
* z „Genetyki medycznej” Drewy
Fenyloketonuria
1 / 10 000
Galaktozemia
1 / 23 000 – 44 000
Zespół nadnerczowo płciowy
1/ 490 – 67 000
Alkaptonuria
1 / 200 000
Choroba Tay-Sachsa
1 / 200 000
Ketoacyduria
1 / 250 000
Homochromatoza
1 / 200 – 400
Mukopolisacharydoza
1 / 24 000
Mukopolisacharydoza (zespół Sanfilippo) – defekt glukozoaminidazy. Występują zniekształcenia
twarzy, upośledzenie umysłowe. Śmierć następuje w drugiej dekadzie życia (do 20 lat).
Ketoacyduria – choroba syropu klonowego. Brak dekarboksylazy leucyny odpowiedzialnej
za dekarboksylację leucyny, izoleucyny i waliny. Jedyne dostępne leczenie to brak tych
aminokwasów w diecie; zapobiega ono zmianom w mózgu i wczesnej śmierci.
Dziedziczenie pośrednie (Zea*)
Anemia
NN
Ciężkie uszkodzenie kośćca
Sierpowatość
Nn
Brachydaktylia
Zdrowy
nn
Zdrowy
*zea to po łacinie kukurydza
Anemia
Homozygota dominująca ma maksymalne natężenie cechy, np. anemia sierpowata – zmniejszenie
liczby krwinek.
Homozygota recesywna jest całkiem zdrowa.
Heterozygoty wykazują cechę pośrednią – sierpowatość krwinek, czyli zmianę kształtu przy
zmniejszonym ciśnieniu parcjalnym tlenu.
Brachydaktylia
Homozygota dominująca wykazuje ciężkie uszkodzenie kośćca.
Homozygota recesywna jest zdrowa.
Heterozygota wykazuje brachydaktylię.
Zasada: w zadaniach genetycznych przeliczamy udział procentowy na tych potomków, którzy
przeżyli.
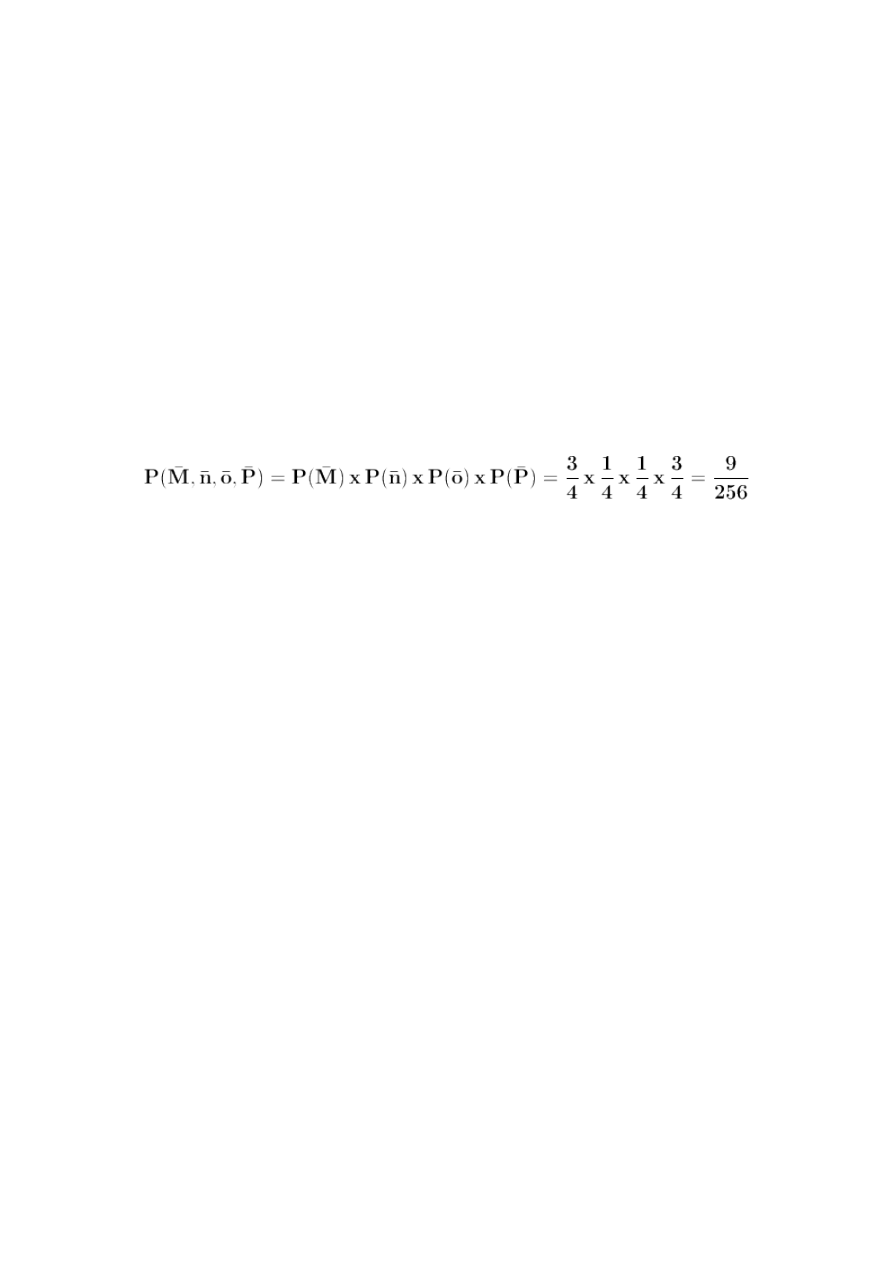
Wzory
Liczba gamet
2
n
Liczba potomstwa (=2
n
x 2
n
)
4
n
Liczba różnych fenotypów
2
n
Liczba różnych genotypów
3
n
Częstość cechy dominującej (D)
¾
Częstość cechy recesywnej (d)
¼
Cecha dla jednej cechy wynosi 0? → zadanie nierozwiązane, trzeba udowodnić, że z powodu danej
cechy fenotyp nie wystąpi.
Prawo prawdopodobieństw
Sprzężenia związane z płcią
Z chromosomem X: gonosomalne
Z chromosomem Y: holandryczne
Przypadkowej segregacji podlegają nie pojedyncze allele, lecz chromosomy zawierające
ułożone liniowo geny (geny sprzężone).
Cechy dominujące sprzężone z chromosomem X
Zespół Blocha-Sulzbergera
1 / 40 000 – 50 000
Krzywica oporna na działanie witaminy D
1 / 20 000
Zespół Retta
1 / 10 000 – 15 000
Zespół łamliwego chromosomu X (FraX), inaczej z. Martina-Bella
1 / 1250 – 2000
Właściwy zapis
X
D
X
D
X
D
Y
X
D
X
d
X
d
Y
X
d
X
d
Na przykład matka ma krzywicę oporną na witaminę D:
X
D
X
d
cc
, a ojciec ma włosy ciemne
(w domyśle: matka jasne, a ojciec zdrowy pod względem krzywicy):
X
d
YCC.
Gamety matki:
X
D
c, X
D
c, X
d
c, X
d
c
Gamety ojca:
X
d
C, YC, X
d
C, YC

Cechy recesywne sprzężone z chromosomem X
Deficyt dehydrogenazy glukozo-6-fosforanowej (DG6P)
1 / 3 – 1 / 100
Dystrofia mięśniowa Duchenna
1 / 3600
Hemofilie
–
hemofilia A
1 / 7 000 – 19 000
–
hemofilia B (Christmasa)
1 / 30 000
Zespół jąder feminizujących
1 / 62 400
Daltonizm
80-150 / 1000
Hyperamonemia typu II
1 / 14 000 – 80 000
Deficyt dehydrogenazy glukozo-6-fosforanowej (całkowity brak DG6P jest letalny), inaczej
fawizm – anemia hemolityczna, która spowodowana jest hemolizą erytrocytów. Pojawia się po
zadziałaniu czynnika zewnętrznego. Ma charakter wieloczynnikowy, działa po zadziałaniu
czynników środowiskowych; wywołuje ją np.: zjedzenie rośliny strączkowej, niektóre antybiotyki
czy sulfonamidy. Objawy to bóle brzucha, dreszcze, żółtaczka. Choroba ta chroni przed malarią.
Daltonizm – u chłopców najczęstsza jest deuteranomalia.
Hyperamonemia typ II – może doprowadzić do śpiączki wywołanej nadmiarem amoniaku.
X
R
X
R
X
R
Y
X
R
X
r
(nosicielka)
X
r
Y
X
r
X
r
Pewna nosicielka to kobieta, która ma co najmniej dwóch chorych synów lub chorego syna
i siostrzeńca, albo chorego syna i brata. Również pewnymi nosicielkami są córki chorego
mężczyzny.
Cechy sprzężone z chromosomem Y
Y*. Owłosiony płatek ucha czy rybia łuska chyba są sprężone z Y; istnieją kontrowersje.
Cecha występuje tylko u osobników płci męskiej i jest przekazywana synom.
Dziedziczenie mitochondrialne
Choroby przekazywane wyłącznie od matki. Otrzymywanie mitochondriów zachodzi od matki,
stanowią bowiem składnik komórki jajowej. Plemnik daje tylko materiał genetyczny, po trzecim
podziale mitochondria plemnika zanikają.
Cecha charakterystyczna to specyficzność tkankowa, a także nasilanie się objawów wraz
z wiekiem.

W komórce jajowej znajduje się około 100 000 cząsteczek mitochondrialnego DNA, w plemniku
ilość 100 cząsteczek mitochondrialnego DNA.
Objawy pojawiają się wtedy, gdy u potomka 85% cząsteczek mitochondrialnego DNA zawiera
mutację.
Zespół Lebera (LHOH) – wrodzona neuropatia nerwu wzrokowego.
MELAS – charakteryzuje się encefalopatią z kwasicą mleczanową i napadami drgawek
udaropodobnymi.
CPEO – porażenie mięśni odwodzących oka.
MERFF – padaczka, cukrzyca i głuchota.
Objawy chorób mitochondrialnych ujawniają się głównie u dorosłych, zaś u dzieci rzadko.
Może wystąpić głuchota po antybiotykach aminoglikozydowych i średni czas wystąpienia
głuchoty to 5 lat od podania antybiotyku. Jeżeli antybiotyki nie są podawane, a występuje choroba
dziedziczona mitochondrialnie, to choroba się pojawi, ale średni czas wystąpienia objawów to 20
lat, czyli znacznie później.
Objawy w jednej rodzinie mogą się różnić, a mogą też być zupełnie odmienne, zależy to od:
–
ilości zmutowanego DNA,
–
miejsca, do którego trafią cząsteczki zmutowane, czyli tkanki, w której wylądują.
Objawy kliniczne mogą w zasadzie dotyczyć wszystkich narządów i układów.
Wykazanie, że choroba dziedziczy się w linii matczynej nie jest proste, ponieważ rodzina może być
niezbyt liczna lub mogą wystąpić kłopoty z ustaleniem rodowodu tej rodziny.
Uważa się, że musi być zmutowane co najmniej 95% cząsteczek, jeśli zmiana dotyczy tRNA, gdzie
występują mutacje punktowe. Jeśli są to delecje, wystarczy 60% zmutowanego DNA.
W rodzinie wszyscy mogą być mutantami, a tylko niektórzy mieć objawy... Efekty mutacji zależą
od tego, w której tkance jest najwięcej cząsteczek zmutowanego DNA, bo w czasie podziału
komórkowego mitochondria rozdzielają się losowo, brak mutantów w jednej tkance nie świadczy
o nieobecności w innej.
Mitochondrialne DNA ulega eliminacji w tkankach dzielących się, np. we krwi, natomiast
w tkankach postmitotycznych jak mięśnie czy mózg – nie. Znane są przypadki, że zmutowane DNA
mitochondrialne jest znajdywane w jednej tkance, np. tylko w mięśniowej.
Diagnostyka opiera się na analizie DNA i na analizie mutacji genowych.
Choroby genetyczne charakterystyczne dla określonej grupy etnicznej
Różnice genetyczne obejmują cechy warunkujące przynależność do grupy etnicznej.
Afrykanie:
–
hemoglobinopatie HbS i HbC
–
talasemie alfa i beta
–
niedobór dehydrogenazy glukozo-6-fosforanowej

Chińczycy:
–
talasemie alfa (beta znacznie rzadziej)
–
niedobór dehydrogenazy glukozo-6-fosforanowej
Eskimosi:
–
wrodzony przerost nadnerczy
Mieszkańcy basenu Morza Śródziemnego:
–
talasemie beta
–
hemoglobinopatie HbS
Indianie Hopi:
–
albinizm
Finowie:
–
nerczyca wrodzona
–
wrodzona biegunka chlorkowa
–
ceroidolipofuscynoza
Europejczycy:
–
mukowiscydoza
–
fenyloketonuria
Meksykanie:
–
cukrzyca,
–
rozszczep warg i podniebienia
Apacze:
–
ciężki niedobór odporności
Japończycy:
–
rozszczep warg i podniebienia
Irlandczycy:
–
bezmózgowie
Rdzenni Amerykanie:
–
rozszczep warg i podniebienia
–
cukrzyca
Cechy różniące to najczęściej dziedziczące się recesywnie i wieloczynnikowo.
Selekcja naturalna jest wynikiem pewnych różnic między populacjami, np. niedokrwistość
sierpowata, niedobór dehydrogenazy glukozo-6-fosforanowej czy talasemia stały się częste w
niektórych populacjach, ponieważ geny tych chorób chronią przed zarażeniem malarią.
Pas równikowy: od 20-40% nosicieli niedokrwistości sierpowatej, hemoglobiny S; są tam gdzie
najczęściej występuje malaria.

Gen unikatowy w populacji
Mózgowo-kostna nerkowa dysplazja występująca w populacji bliźniąt Hutterite. Populacja
bliźniąt Hutterite jest to populacja nieliczna, zatem konieczne są związki osób blisko
spokrewnionych.
Społeczne skutki chorób genetycznych
Są duże – rozpoczynają się w dzieciństwie i trwają całe życie.
Są też przyczyną 30-50% przyjęć do szpitali pediatrycznych i 10% przyjęć do szpitali dla
dorosłych.
Jest to też główny powód śmiertelności noworodków.
Poradnictwo genetyczne
Forma specjalistycznej pomocy medycznej, której celem jest udzielanie informacji i porady
osobom konsultowanym i członkom ich rodzin, u których występuje podwyższone ryzyko
wystąpienia choroby uwarunkowanej genetycznie.
Zadania lekarza:
–
wyjaśnienie, czy dana choroba jest uwarunkowana genetycznie,
–
następnie oszacowanie ryzyka wystąpienia choroby u innych członków tejże rodziny,
–
ocena, czy istnieje zwiększone ryzyko urodzenia dziecka z ryzykiem choroby genetycznej,
bądź też czy istnieje sposób na zmniejszenie ryzyka.
Zadania w przypadku nowotwór – 5-10% nowotworów ma podłoże genetyczne i są dziedziczne.
Rak sutka, jajnika, jelita grubego, rak rdzeniasty tarczycy – następuje dziedziczenie się w sposób
dominujący. Poradnictwo genetyczne polega na badaniu osób z rodzin o predyspozycji do
występowania.
Etapy postępowania lekarza
1. Wywiad
2. Konstrukcja i analiza rodowodu
3. Badanie fizykalne
4. Badanie diagnostyczne
5. Rozpoznanie
6. Udzielenie porady

Konstruowanie diagramów
–
Płeć żeńska po prawej, męska po lewej stronie diagramu.
–
Cyfry rzymskie – pokolenia od najwcześniejszego.
–
Rysowanie rozpoczyna się od pokolenia najmłodszego.
–
Członkowie tego samego pokolenia są w tej samej linii poziomej.
–
Do oznaczania poszczególnych osób w każdym pokoleniu stosuje się liczby arabskie.
–
Potomstwo zaznacza się w kol urodzeń, umieszczając i numerując od lewej do prawej.
–
Trzeba uwzględnić w rodzinie pierwszego członka, u którego wykryto chorobę (probanta)
i wśród rodzeństwa osoby chore, zdrowe, zmarłe oraz adopcje.
–
Należy ustawić wszystkie urodzenia w porządku chronologicznym.
–
Należy ustalić, czy występują poronienia samoistne, martwe urodzenia, urodzenia dzieci
z wrodzonymi wadami rozwojowymi
–
Wywiad powinien obejmować co najmniej 3 pokolenia i wszystkich krewnych.
Rozpoznanie choroby genetycznej
Musi nastąpić zebranie wywiadu i badanie fizykalne.
Podstawowe badanie genetyczne to oznaczanie kariotypu. Badanie i analizę wykonuje się
w okresie postnatalnym i prenatalnym.
Ocena kariotypu jest przeprowadzana, gdy w rodzinie szansa na chorobę jest większa niż
w populacji:
–
wystąpiła aneuploidia w poprzedniej ciąży,
–
obecna jest aberracja strukturalna chromosomowa u rodziców,
–
matka jest starsza niż 35 lat.
Document Outline
- Cechy uwarunkowane genetycznie
- Cechy autosomalne dominujące
- Cechy autosomalne recesywne
- Dziedziczenie pośrednie (Zea*)
- Wzory
- Prawo prawdopodobieństw
- Sprzężenia związane z płcią
- Cechy dominujące sprzężone z chromosomem X
- Cechy recesywne sprzężone z chromosomem X
- Cechy sprzężone z chromosomem Y
- Dziedziczenie mitochondrialne
- Choroby genetyczne charakterystyczne dla określonej grupy etnicznej
- Gen unikatowy w populacji
- Społeczne skutki chorób genetycznych
- Poradnictwo genetyczne
- Etapy postępowania lekarza
- Konstruowanie diagramów
- Rozpoznanie choroby genetycznej
Wyszukiwarka
Podobne podstrony:
Genetyka 3 (19 12 2012)
Jak powstało radio 05.12.2012, BACHAMAS, Kronika 2012 2013
grama 05 12 2012
9 podstawy nauki o materiałach 05 12 2012
Wykład 9 (05 12 2012)
Genetyka 2 (12 12 2012)
Genetyka 3.12.2012, Biologia, ściągi
05.12.2009GENETYCZNA STRUKT. POPULACJI, dietetyka II rok, genetyka
IS 2011 12 wyklad 12 05 01 2012 MDW
Genetyka 2 (12 12 2012)
Ściskanie sprawko 05 12 2014
05 12 2011
05 09 2012 INTERNA
Mikołajki 06.12.2012, BACHAMAS, Kronika 2012 2013
więcej podobnych podstron