- 1 -
FX ChemStruct
Wersja 1.203.2
Portable
http://efofex.com/fxchemstruct.php
Korzystanie z programu
FX Stat ChemStruct
Przekład
Robert Wi
ś
niewski
http://chomikuj.pl/bobwis
Struktury chemiczne s
ą
niezwykle trudne do narysowania przy u
ż
yciu standardowych narz
ę
dzi
edycyjnych i nawet wyspecjalizowane narz
ę
dzia mog
ą
by
ć
powolne i uci
ąż
liwe. Nauczyciele
potrzebuj
ą
szybkiego sposobu rysowania struktur chemicznych - FX ChemStruct to ułatwia. Po prostu
wpisujesz to zechcesz, wybierasz opcj
ę
wy
ś
wietlania i kilka chwil pó
ź
niej b
ę
dziesz miał diagramy
o jako
ś
ci publikacyjnej

- 2 -
SPIS TRE
Ś
CI
1. Mo
ż
liwo
ś
ci programu
1.1. Podstawowe koncepcje
2. Korzystanie z programu FX ChemStruct
2.1. Dost
ę
p do FX ChemStruct
2.2. Jak wprowadza
ć
struktury
2.2.1. Struktury podstawowe
2.2.2. Struktury rozgał
ę
zione
2.2.3. Pier
ś
cienie benzenowe
2.2.4. Nieformatowane teksty / komentarze
2.2.5. Klikanie na zwi
ą
zkach I
2.2.6. Klikanie na zwi
ą
zkach II
2.2.7. Obracanie i odbijanie
2.2.8. Anulowanie obracania i odbijania
2.2.9. Izomery
2.2.10. Strzałki
2.2.11. Spacje
2.2.12. Rodniki R i R’
2.2.13. Jon nitroniowy
2.2.14. Wprowadzanie informacji fazowej
2.2.15. Ładunki
2.2.1.6. Zwi
ą
zki jonowe
2.2.17. Jony wieloatomowe
2.2.18. Struktury pier
ś
cieniowe
5.2.19. Zapobieganie tworzeniu wi
ą
za
ń
2.2.20. Stopnie
2.3. Opcje wy
ś
wietlania
2.3.1. Ukrywanie wi
ą
za
ń
?-H
2.3.2. Wy
ś
wietlanie ładunków
2.3.3. Zwi
ą
zki bez wi
ą
za
ń
2.3.4. Zezwolenie na pary elektronowe
2.3.5. Wy
ś
wietlanie wszystkich elektronów
2.4. Przykłady
2.4.1. Struktury rezonansowe
2.4.2. W
ę
glowodany
2.4.3. EDTA
2.5. Edycja struktur
2.6. Zmiana Co(NO3)2 na CO(NO3)2
2.7 Zmiana struktur
2.7.1. Narz
ę
dzia / Czcionki
2.7.2. Narz
ę
dzia / Opcje / Równania
2.7.3. Narz
ę
dzia / Opcje / Struktury
2.8. Moje struktury nie s
ą
poprawnie formatowane !
- 3 -
1. Mo
ż
liwo
ś
ci programu
1.1. Podstawowe koncepcje
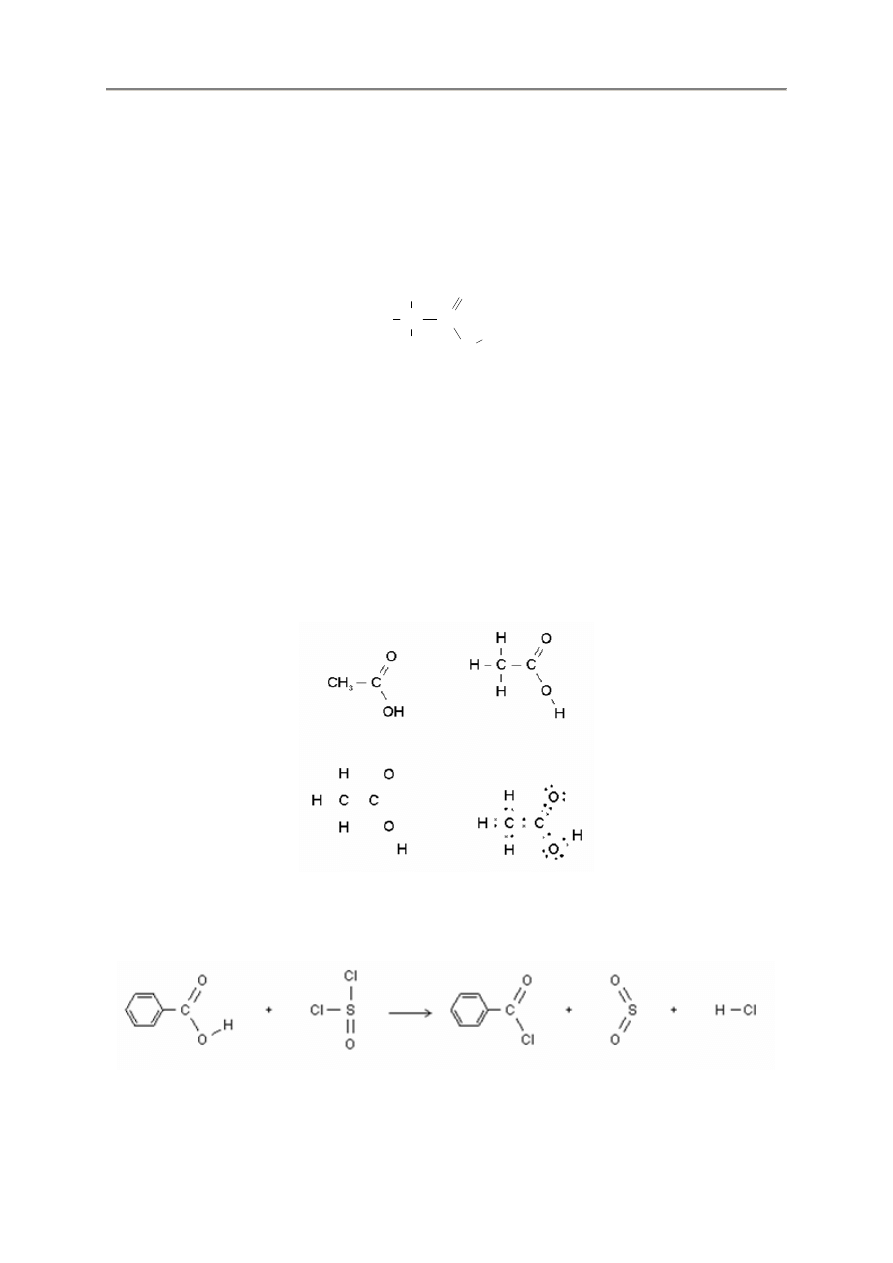
Program FX ChemStruct pozwala na łatwe rysowanie struktur chemicznych, Narysowanie wzoru
strukturalnego tak prostego jak np. kwas octowy jest czasem pracochłonnym i trudnym zadaniem, ale
FX ChemStruct pozwala na łatwe jego wykonanie.
C
H
H
H
C
O
O
H
•
Załadowa
ć
FX ChemStruct
•
Wpisa
ć
w polu edycji ch3cooh
FX ChemStruct pobiera t
ą
informacj
ę
i formatuje j
ą
do powy
ż
szej struktury. Nie trzeba nic wskazywa
ć
jak to wykona
ć
– wystarczy wpisa
ć
tylko sam
ą
formuł
ę
. U
ż
ytkownicy FX ChemStruct o oceniaj
ą
ten
program jako bardzo przyjazny, tworz
ą
cy na podobnych zasadach ró
ż
ne wyniki.
FX ChemStruct NIE mo
ż
e rysowa
ć
wszystkich struktur chemicznych, ale mo
ż
e rysowa
ć
szybko i łatwo
wi
ę
kszo
ść
najwa
ż
niejszych strukturalnych wzorów chemicznych.
FX ChemStruct mo
ż
e rysowa
ć
struktury chemiczne w ró
ż
nych stylach, nawet struktury bez wi
ą
za
ń
chemicznych,
Poni
ż
ej pokazano mały przykład mo
ż
liwo
ś
ci FX ChemStruct. Równanie to mo
ż
na uzyska
ć
wpisuj
ą
c:
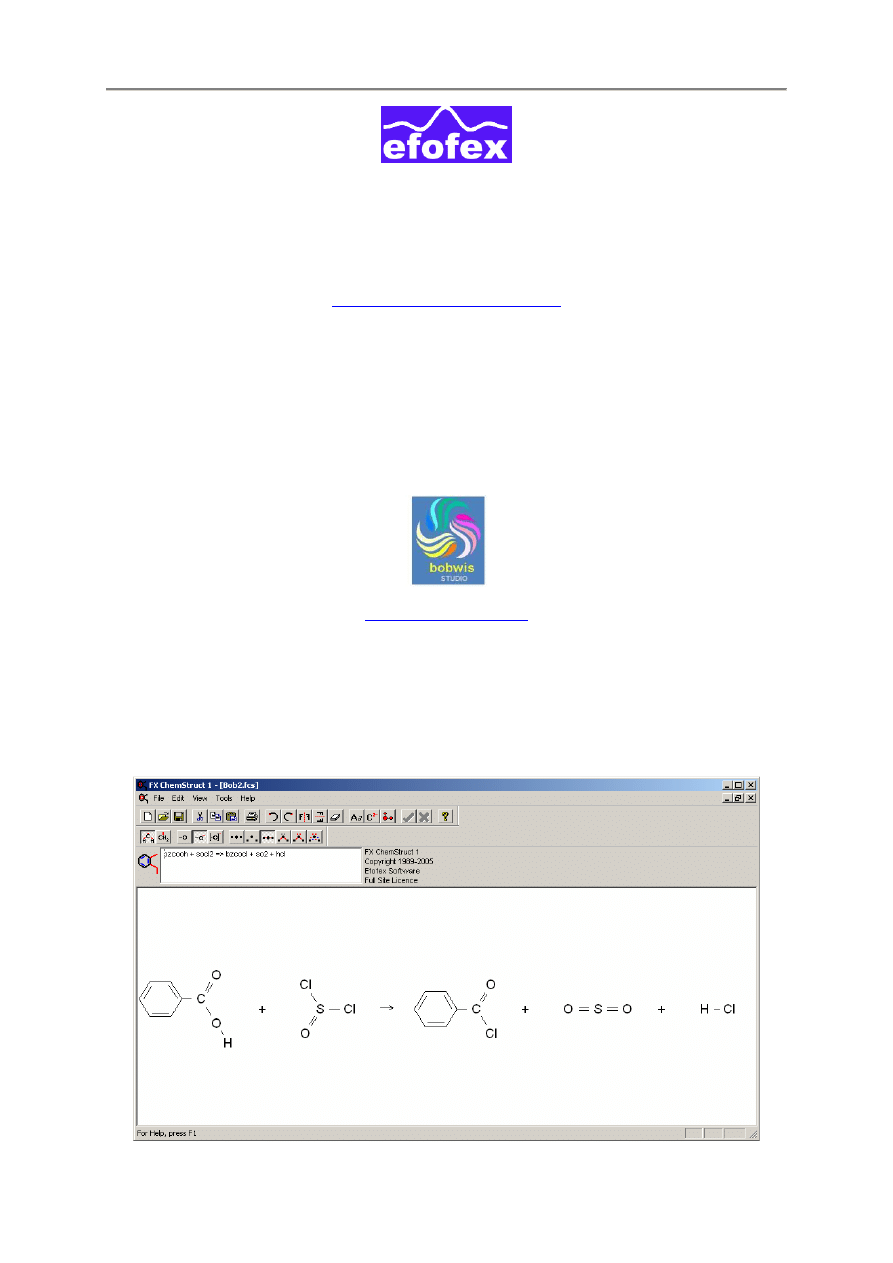
bzcooh + socl2 => bzcocl + so2 + hcl
FX ChemStruct został pierwotnie zaprojektowany do rysowania cz
ą
steczek organicznych, ale
dodano do niego mo
ż
liwo
ść
rysowania wielu cz
ą
steczek nieorganicznych i jonów wieloatomowych.

- 4 -
2. Korzystanie z programu FX ChemStruct
2.1. Dost
ę
p do FX ChemStruct
Istnieje kilka sposobów korzystania z programu FX ChemStruct - jako samodzielnego programu lub
jako obiektu w dokumencie procesora tekstu.
Tryb samodzielny
Gdy akceptujemy standardow
ą
instalacj
ę
programu FX ChemStruct, mo
ż
emy go uruchamia
ć
jako
program samodzielny znajduj
ą
c go w systemowym menu Start. Chocia
ż
jest to mo
ż
liwie, nie jest to
najlepszy sposób korzystania z FX ChemStruct. Wygodnej z niego korzysta
ć
z poziomu procesora
tekstu, np. Word.
Tryb obiektu w dokumencie
1
FX ChemStruct jest zaprojektowany do wstawiania jako obiekt w dokumencie. Mo
ż
na wstawia
ć
dowolny obiekt FX ChemStruct do dokumentu przez klikanie jego przycisku w pasku narz
ę
dzi lub
otworzy
ć
ulubiony procesor tekstu (np. Word) i wybra
ć
w nim polecenie Wstaw | Obiekt. Otworzy si
ę
lista dost
ę
pnych obiektów na której mo
ż
na wybra
ć
opcj
ę
FX ChemStruct aby uruchomi
ć
ten program.
Korzystamy wówczas z niego w taki sam sposób jak w trybie samodzielnym. Jedyna ró
ż
nica polega
na powrocie do Worda.
Zielony przycisk znacznika
powoduje powrót do Worda z obrazem ze wszystkimi wprowadzonymi
zmianami.
Czerwony przycisk krzy
ż
yka
powoduje powrót do Worda bez prowadzania zmian.
Po sko
ń
czeniu rysowania obrazu, klikamy znacznik
i nasz diagram zostaje wstawiony do Worda.
2.2. Jak wprowadza
ć
struktury
Wystarczy wpisa
ć
w polu edycji wymagan
ą
informacj
ę
. Nie trzeba wprowadza
ć
du
ż
ych liter symboli
chemicznych (np. wystarczy wpisa
ć
sn dla symbolu cyny Sn). Program FX ChemStruct rozpozna to
samodzielnie. Istnieje kilka trików do nauki, ale wystarczy na tylko jedna minuta.
By
ć
mo
ż
e najwi
ę
kszym trikiem przy wprowadzaniu struktur jest to,
ż
e trzeba poda
ć
do programu
pewne informacje takie jak przy r
ę
cznym wpisywaniu struktur. Przykładowo, gdy chcemy narysowa
ć
struktur
ę
propanu, nie trzeba wpisywa
ć
c3h8. Program FX ChemStruct skorzysta z tej informacji
i b
ę
dzie próbował budowa
ć
poni
ż
sz
ą
(tutaj poprawn
ą
) struktur
ę
, co jednak czasem mo
ż
e by
ć
niejednoznaczne.
1
Wariant ten jest dostępny tylko w wersji instalacyjne programu FX ChemStruct.

- 5 -
Zamiast tego lepiej wprowadzi
ć
ch3ch2ch3. Mówi to programowi gdzie trzeba wstawia
ć
poszczególne
wodory.
Wi
ę
cej informacji na ten temat b
ę
dzie podane dalej.
2.2.1. Struktury podstawowe
Wi
ę
kszo
ść
prostych struktur wprowadzamy tak jak przy ich r
ę
cznym pisaniu. Podkre
ś
la si
ę
,
ż
e nie
musimy korzysta
ć
z w du
ż
ych liter. FX ChemStruct zwykle rozpoznaje o co nam chodzi.
Alkany
ch3ch2ch2ch3
ch3(ch2)4ch3
Alkeny
ch2ch2
Alkiny
chch
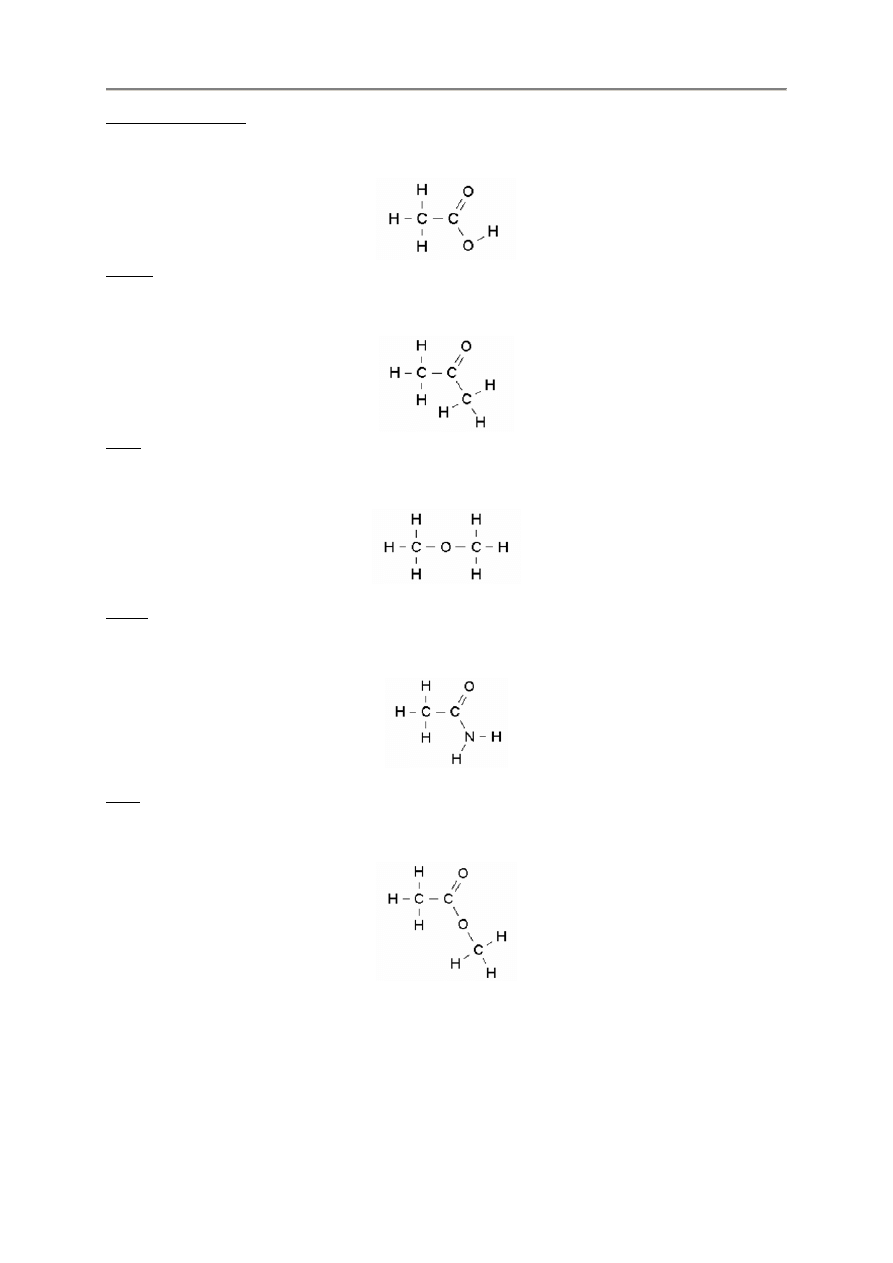
Aldehydy
ch3ch2coh
- 6 -
Kwasy karboksylowe
ch3cooh
Ketony
ch3coch3
Etery
ch3och3
Amidy
ch3conh2
Estry
ch3cooch3
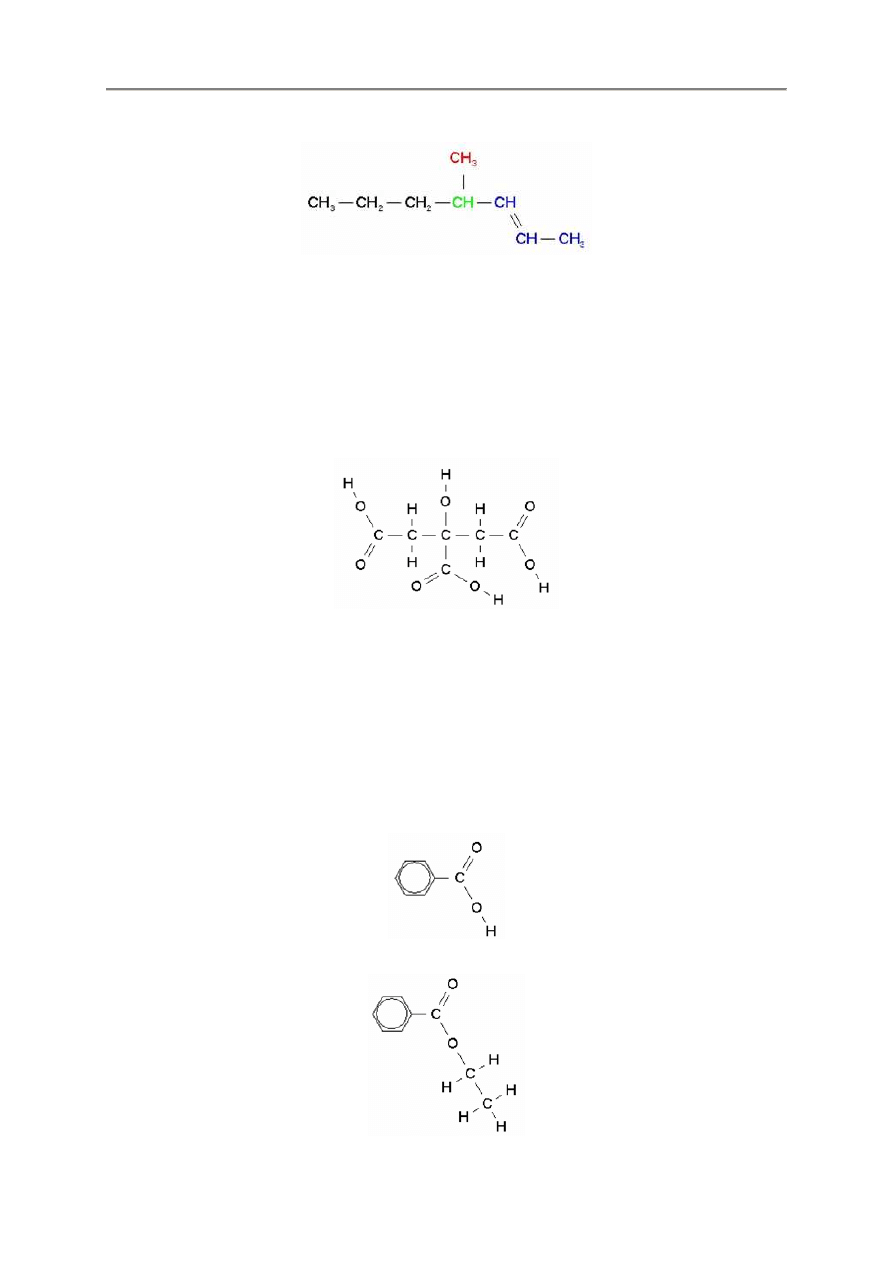
2.2.2. Struktury rozgał
ę
zione
Struktury rozgał
ę
zione s
ą
nieco trudniejsze do wprowadzania, ale niezbyt trudne do zapami
ę
tania
podstawowej zasady.
Wprowadzamy kompletnie ka
ż
d
ą
gał
ąź
przed rozpocz
ę
ciem nast
ę
pnej gał
ę
zi.
- 7 -
Przykładowo, w celu wprowadzenia 4-methyl-2-heptene:
wprowadzamy struktur
ę
do zielonych pierwiastków, nast
ę
pnie wprowadzamy jedn
ą
z gał
ę
zi (tutaj
czerwon
ą
), po czy wprowadzamy gał
ąź
niebiesk
ą
ch3ch2ch2
ch
ch3
chchch3
Mo
ż
na w ten sposób wprowadza
ć
bardzo skomplikowane struktury, np. kwasu cytrynowego.
coohch2cohcoohch2cooh
2.2.3. Pier
ś
cienie benzenowe
Pier
ś
cienie benzenowe mo
ż
na wprowadza
ć
korzystaj
ą
c z kodu bz. Je
ś
li w menu Tools | Options |
Structures mamy wybran
ą
opcj
ę
Use Ph fo Phenol, wówczas mo
ż
emy równie
ż
stosowa
ć
kod ph
do wskazywania pier
ś
cienia benzenowego
Przykłady:
bzcooh
phcooch2ch3

- 8 -
bzbz
bzbzbz
lub
2.2.4. Nieformatowane teksty / komentarze
Program FX ChemStruct zawsze próbuje nada
ć
„sens chemiczny” temu co wprowadzamy. Mo
ż
e to
jednak prowadzi
ć
do problemów. Przykładowo, gdy wpiszemy słowo katalizator Catalyst do struktury,
wówczas program FX ChemStruct b
ę
dzie próbował interpretowa
ć
go jako pierwiastki i sformatuje
wynik w poni
ż
szy sposób:
Wyst
ę
puj
ą
tu wap
ń
(Ca), tantal (Ta) oraz „l” (który nie mo
ż
e by
ć
przekształcony na pierwiastek),
itr (Y). siarka (S) oraz renegat „t”. Zapewne tego nie oczekiwali
ś
my ? Mo
ż
emy zmusi
ć
FX ChemStruct
aby pozostawił ten tekst bez zmiany przez otovczenie go cudzysłowami „Catalyst”. Uzyskamy wtedy
to czego oczekiwali
ś
my.
2.2.5. Klikanie na zwi
ą
zkach I
Formuła chemiczna jest dwuznaczna, a wi
ę
c gdy wprowadzamy struktur
ę
, istnieje cz
ę
sto wi
ę
cej ni
ż
jeden poprawny sposób jej wy
ś
wietlania. Jak przekaza
ć
do programu która wersja nas interesuje ?
W programie FX ChemStruct mo
ż
na zmienia
ć
„struktur
ę
” przez klikanie jej pierwiastków.
Przykładowo, gdy wpiszemy:
ch3chch3ch2chch3ch3
wówczas zobaczymy:
Ale chcemy uzyska
ć
:
Mamy pewien sposób aby program to zmienił. W celu uzyskania nowej wersji, wystarczy klikn
ąć
na
grupie CH aby otrzyma
ć
jej kolorow
ą
wersj
ę
po klikni
ę
ciu pierwiastka. FX ChemStruct b
ę
dzie
kolorował j
ą
na zielono, a wi
ę
c mo
ż
emy
ś
ledzi
ć
który pierwiastek został klikni
ę
ty.
Klikni
ę
cie pierwiastka przesuwa jego wi
ą
zania.

- 9 -
2.2.6. Klikanie na zwi
ą
zkach II
Klikanie pierwiastka przesuwa jego wi
ą
zania, ale gdy liczba wi
ą
za
ń
wzrasta, wówczas istnieje kilka
wariantów ich rozmieszczania.
Najcz
ęś
ciej chcemy skorzysta
ć
tylko z głównego rozmieszczenia atomów i FX ChemStruct zmienia je
cyklicznie te główne rozmieszczenia w miar
ę
klikania pierwiastka.
Czasem chcemy mie
ć
dost
ę
p do mniej popularnych rozmieszcze
ń
. Gdy przytrzymamy klawisz Shift
przy klikni
ę
ciu, wówczas FX ChemStruct b
ę
dzie cyklicznie przechodził przez wszystkie mo
ż
liwe
rozmieszczenia.
Przykłady:
ch3chclchclch3
Gdy klikniemy na drugiej grupie CH, b
ę
dziemy mogli zmienia
ć
dwie wersje tej cz
ą
steczki.
Gdy b
ę
dziemy klikali z klawiszem Shift, wówczas mo
ż
emy uzyska
ć
poni
ż
sze wersje:
2.2.7. Obracanie i odbijanie
Wszystkie struktury mo
ż
na obraca
ć
i odbija
ć
.
Te cztery przyciski s
ą
aktywne tylko dla ostatnio klikni
ę
tego zwi
ą
zku. Gdy nic nie jest klikni
ę
te, działaj
ą
tylko na pierwszy zwi
ą
zek.
Pierwsze dwa przyciski obracaj
ą
struktur
ę
z przyrostem co 15
°
w wybranym kierunku.
Ostatnie dwa przyciski tworz
ą
odbicie lustrzane struktury w pionie lub w poziomie.
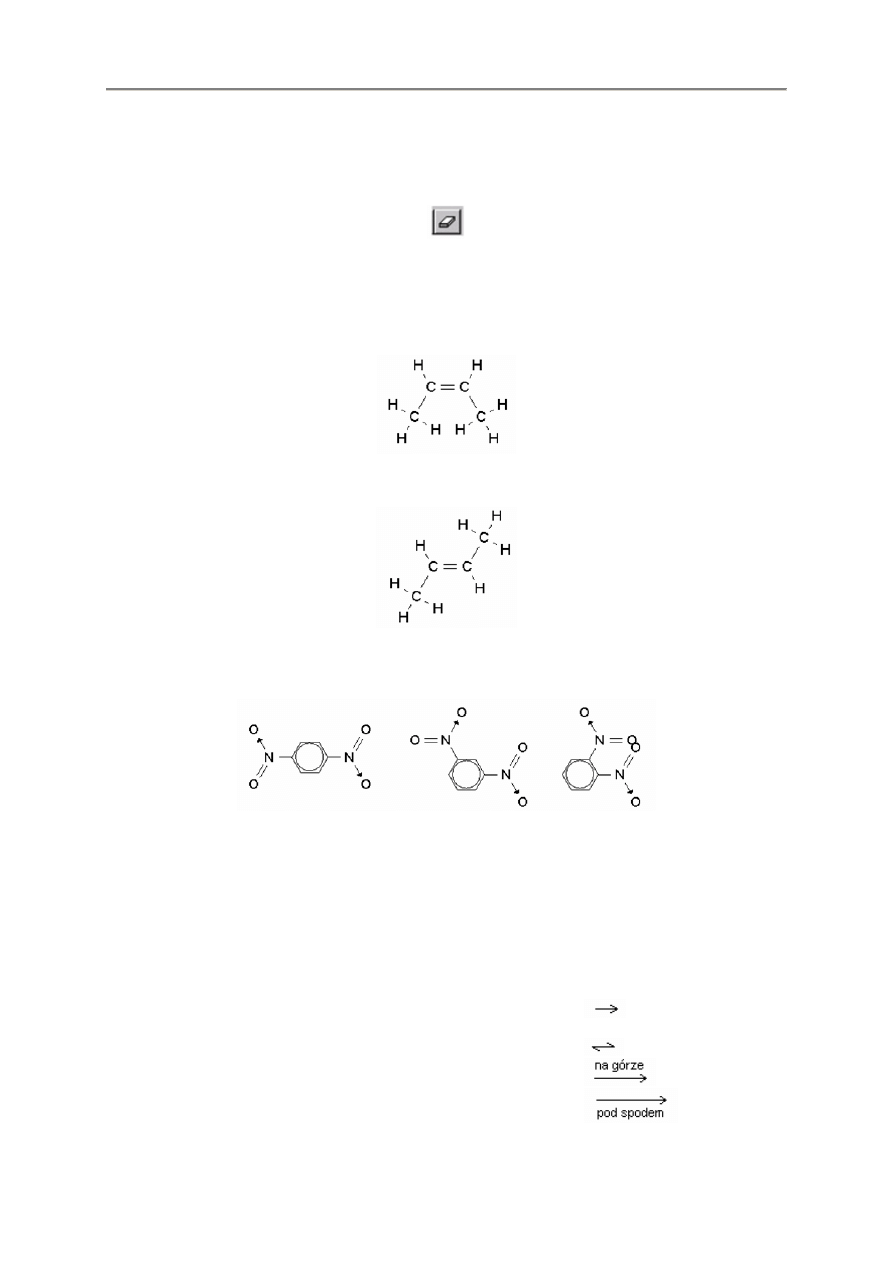
- 10 -
2.2.8. Anulowanie obracania i odbijania
Czasem klikniemy, obracamy lub odbijamy diagram i chcemy rozpocz
ąć
od nowa. W tym celu
wystarczy klikn
ąć
przycisk gumki aby cofn
ąć
wszystkie modyfikacje.
2.2.9. Izomery
Klikni
ę
cie atomu w strukturze tworzy ró
ż
ne izomery, np.
ch3chchch3
Uzyskujemy cis-2-buten. Jedno dodatkowe klikni
ę
cie tworzy trans-2 buten
To samo odnosi si
ę
do zwi
ą
zków benzenowych.
bzno2no2
Para, meta i orto dwunitrobenzen
2.2.10. Strzałki
Strzałki reakcji chemicznych pojawiaj
ą
si
ę
w wielu równaniach i trzeba pami
ę
ta
ć
kody niezb
ę
dne do
ich tworzenia.
Program FX ChemStruct korzysta z takich samych kodów i metod jak FX Chem.
=>
Tworzy pojedyncz
ą
strzałk
ę
<>
Tworzy podwójn
ą
strzałk
ę
=”na górze”>
Dodaje komunikat nad strzałk
ą
=,”pod spodem”>
Przecinek dodaje komunikat pod strzałk
ą
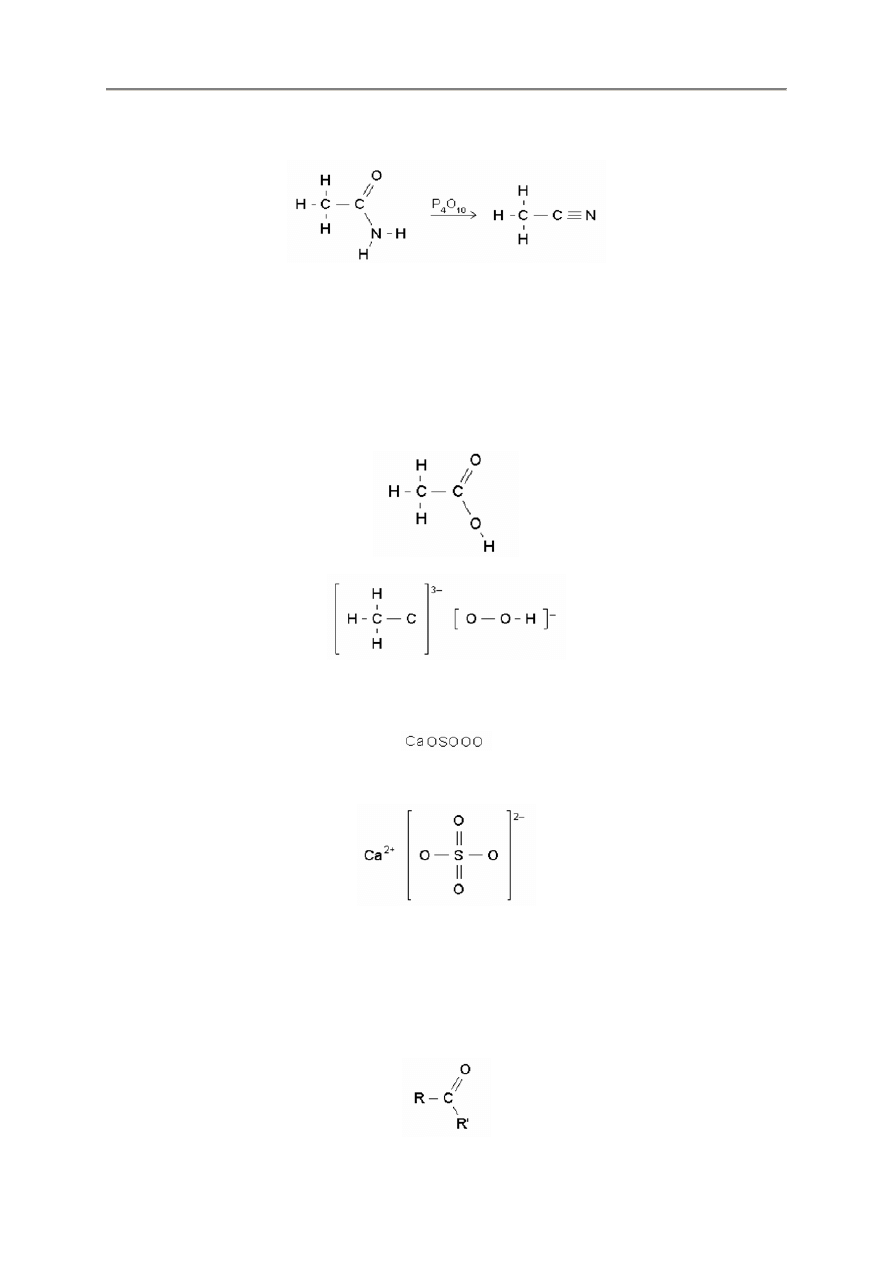
- 11 -
Gdy nie doł
ą
czymy cudzysłowów, program FX ChemStruct b
ę
dzie interpretował tekst nad lub pod
strzałkami jako równanie chemiczne, a nie jak struktur
ę
chemiczn
ą
.
2.2.11. Spacje
Spacje mo
ż
na stosowa
ć
mi
ę
dzy zwi
ą
zkami do tworzenia odst
ę
pów je
ś
li jest to konieczne. Spacja
w zwi
ą
zku rozrywa go na dwie osobne cz
ęś
ci. FX ChemStruct nie próbuje doł
ą
cza
ć
drugiej cz
ęś
ci do
pierwszej. Cz
ę
sto oznacza to,
ż
e to co wpisali
ś
my nie ma
ż
adnego sensu.
ch3cooh
ch3c ooh
Spacje s
ą
cz
ę
sto konieczne w zwi
ą
zkach jonowych.
caso4
ca so4
2.2.12. Rodniki R i R’
FX ChemStruct obsługuje korzystanie z rodników R i R’ w strukturach chemicznych, np.
rcor'
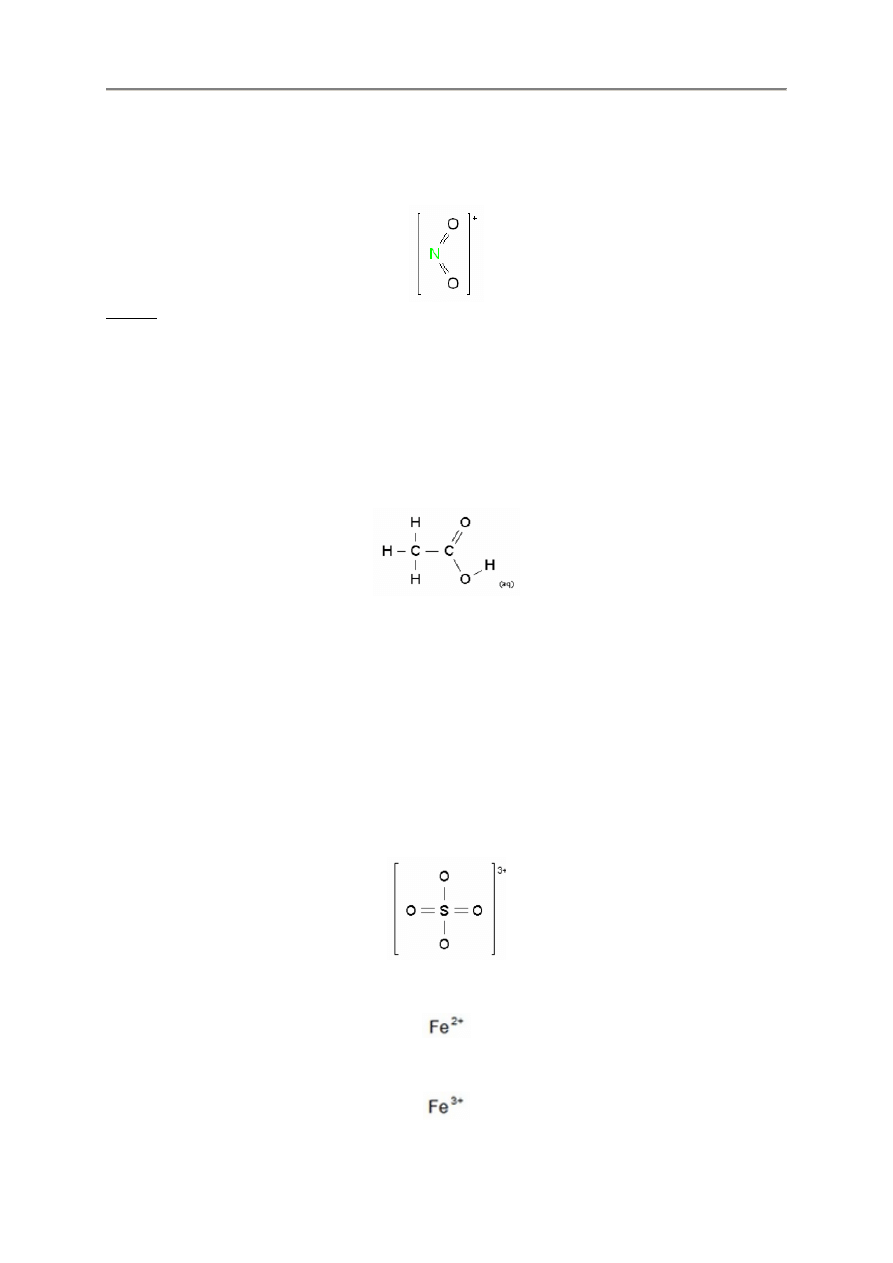
- 12 -
2.2.13. Jon nitroniowy
Mo
ż
na wprowadza
ć
jon nitroniowy (NO2+) wpisuj
ą
c:
ono
Uwaga:
Jest to tymczasowe rozwi
ą
zanie problemu.
2.2.14. Wprowadzanie informacji fazowej
Program FX ChemStruct akceptuje informacje fazowe w taki sam sposób jak FX Chem, np.
ch3cooh(aq)
2.2.15. Ładunki
Program FX ChemStruct automatycznie oblicza ładunki pierwiastków i jonów.
Gdy chcemy nadpisa
ć
ładunek swoj
ą
własn
ą
warto
ś
ci
ą
, wystarczy wpisa
ć
ładunek za jonem.
Główne tego zastosowanie ma miejsce w jonach metalicznych maj
ą
ce ró
ż
ne poziomy utlenienia.
Mo
ż
liwo
ść
ta pozwala na tworzenie naładowanych struktur, które mog
ą
by
ć
niepoprawne, np.
so43+
fe
fe3+
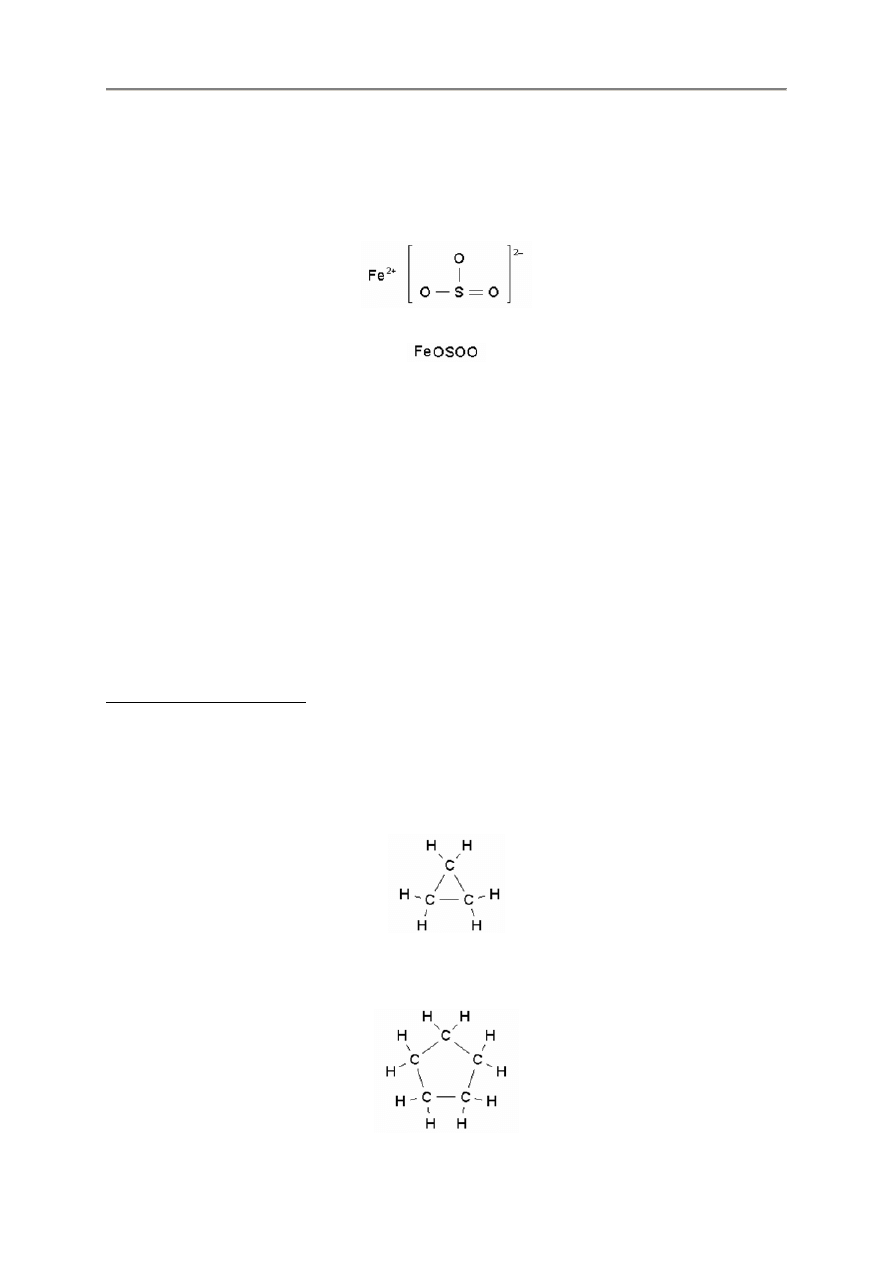
- 13 -
2.2.1.6. Zwi
ą
zki jonowe
W celu utworzenia zwi
ą
zków jonowych, trzeba doda
ć
spacj
ę
mi
ę
dzy kationem i anionem. Zapobiega
to przed prób
ą
wi
ą
zania tych dwóch cz
ęś
ci ze sob
ą
i pokazuje jawnie ładunki ka
ż
dego jonu, np.
fe so3
feso3
2.2.17. Jony wieloatomowe
Wi
ę
kszo
ść
jonów wieloatomowych jest rozpoznawana przez FX ChemStruct, ale trzeba otoczy
ć
jon
spacjami aby program nie próbował ł
ą
czy
ć
jonów z inn
ą
struktur
ą
,
2.2.18. Struktury pier
ś
cieniowe
W FX ChemStruct Istnieje ograniczone wspomaganie struktur pier
ś
cieniowych. Program mo
ż
e
obsługiwa
ć
automatycznie struktury pier
ś
cieniowe, ale ma to do
ść
ograniczon
ą
funkcjonalno
ść
, bo
u
ż
ytkownik musi z góry wskaza
ć
pier
ś
cie
ń
.
Mo
ż
na wł
ą
czy
ć
lub wył
ą
czy
ć
automatyczne rozpoznawanie struktur pier
ś
cieniowych za pomoc
ą
okienka dialogowego Tools | Options | Structures zaznaczaj
ą
c opcj
ę
Automatically Detect Rings.
Automatyczne rozpoznawanie
Gdy opcja ta jest wł
ą
czona, program FX ChemStruct szuka wprowadzonych pierwiastków, które mog
ą
tworzy
ć
pier
ś
cie
ń
. Gdy jest to mo
ż
liwe, wówczas program ł
ą
czy je w pier
ś
cie
ń
. Automatyczne
rozpoznawanie jest bardzo ograniczone i najlepiej działa na glownych pier
ś
cieniach benzenowych, np.
ch2ch2ch2
ch2ch2ch2ch2ch2
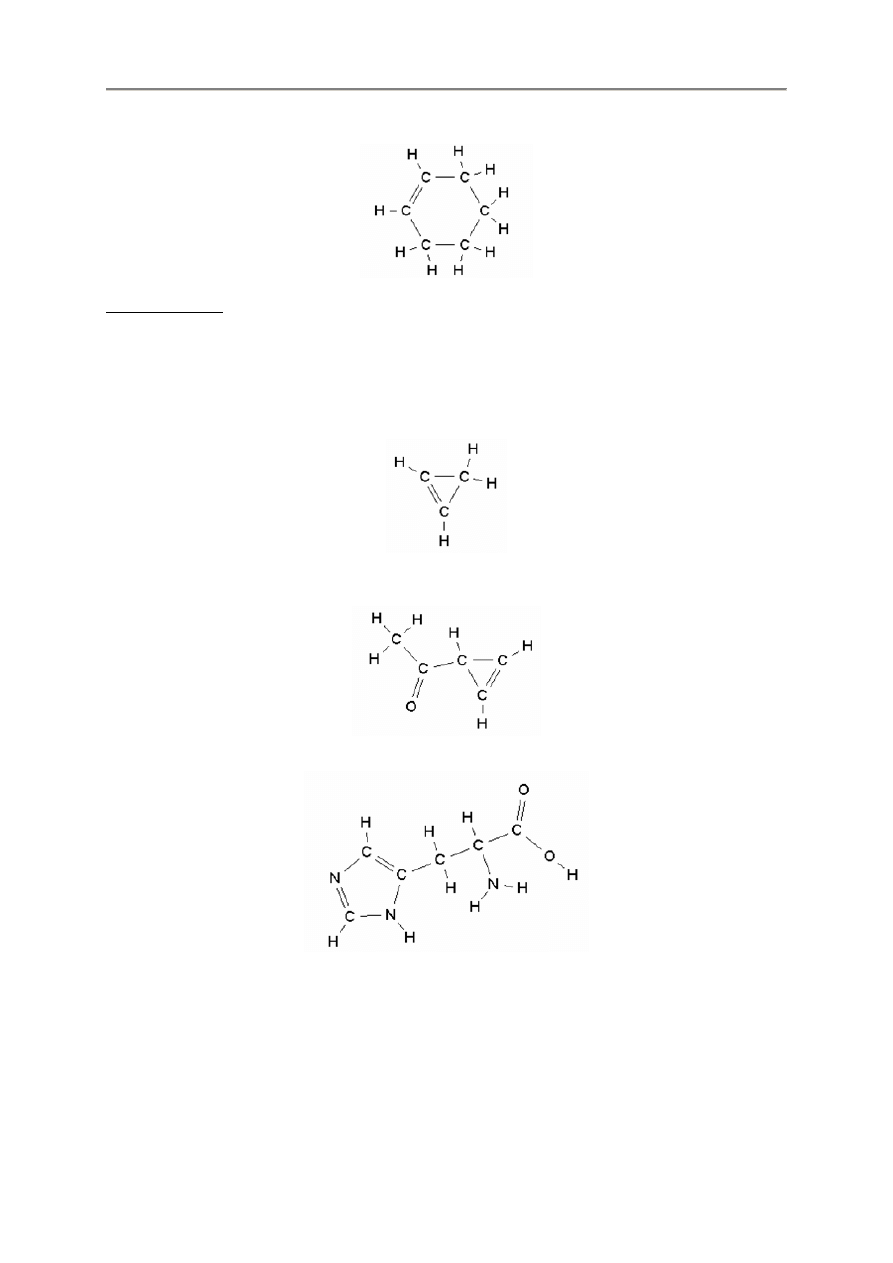
- 14 -
ch2ch2ch2chchch2
Wskazanie jawne
Gdy automatyczne rozpoznawanie nie jest wł
ą
czone, wówczas trzeba z góry wskaza
ć
struktury
pier
ś
cieniowe. W tym celu nale
ż
y otoczy
ć
pierwiastki struktury pier
ś
cieniowej nawiasami klamrowymi,
a program spróbuje je uformowa
ć
w pier
ś
cie
ń
, np.
{chchch2}
{chchch}coch3
{cchnchnh}ch2chnh2cooh
Jawne wskazanie pier
ś
cieni pozwala na rysowanie wi
ę
kszej liczby struktur, ale trzeba podkre
ś
li
ć
,
ż
e nie wszystkie struktury mo
ż
na wprowadza
ć
w ten sposób.
2.2.19. Zapobieganie tworzeniu wi
ą
za
ń
Program FX ChemStruct korzysta ze zło
ż
onego algorytmu do sprawdzania gdzie nast
ę
pny pierwiastek
ma by
ć
doł
ą
czany do istniej
ą
cej struktury. Czasem to mo
ż
e stwarza
ć
problemy gdy chcemy zapobiec
tworzeniu wi
ą
za
ń
, np.
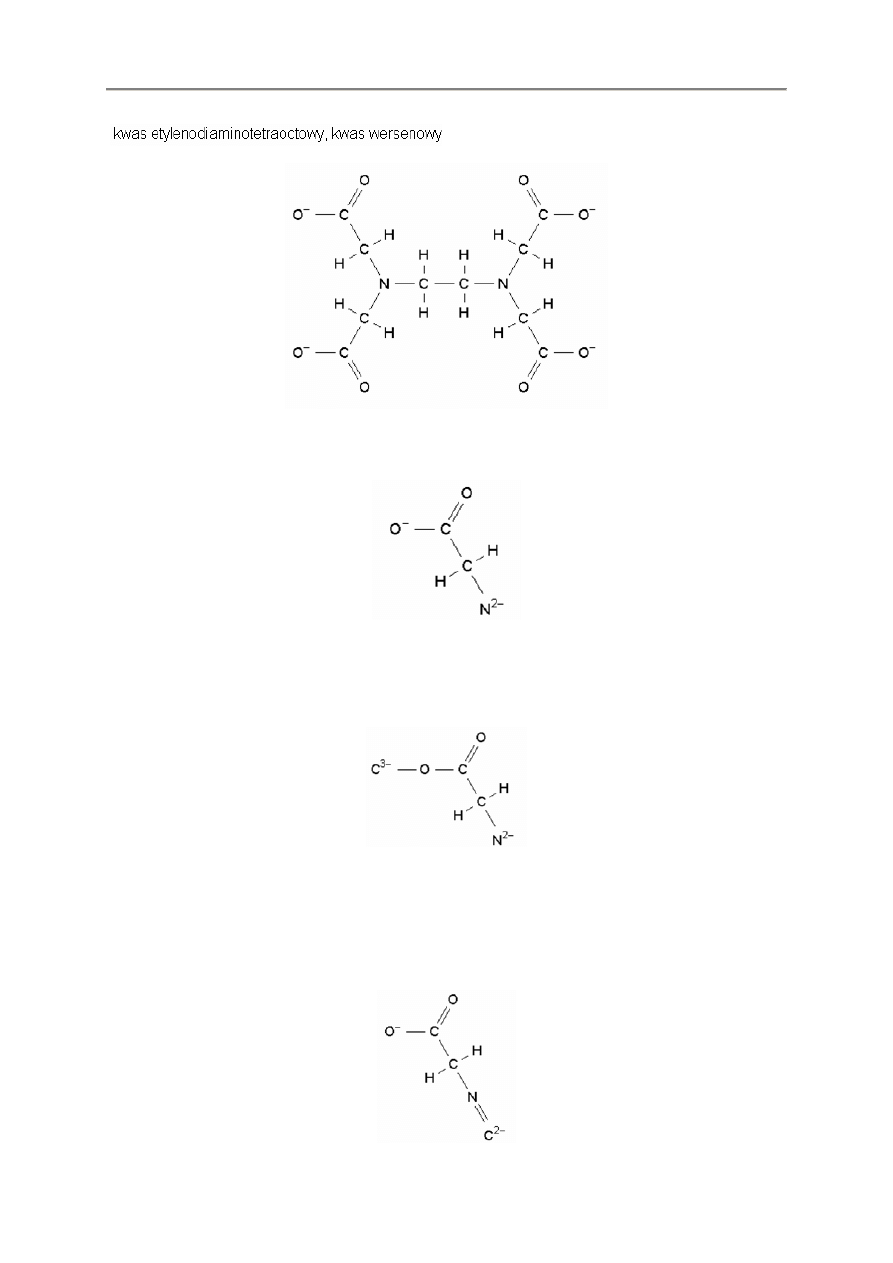
- 15 -
EDTA
Wyobra
ź
my sobie,
ż
e chcemy wprowadzi
ć
t
ą
struktur
ę
do FX ChemStruct. Mo
ż
emy zacz
ąć
od
jednego z azotów i przesun
ąć
jedno z jego ramion.
*
Jak dot
ą
d wszytko jest w porz
ą
dku. Teraz próbujemy zacz
ąć
od innego ramienia – dopisanie C
ko
ń
czy si
ę
niepowodzeniem.
nch2cooc
Program FX ChemStruct nie ma sposobu rozpoznania czy sko
ń
czyli
ś
my pierwsze rami
ę
i doł
ą
czy
ć
C
do pozostałych wi
ą
za
ń
. Potrzebujemy sposobu przekazania programowi aby si
ę
tu zatrzymał.
Mo
ż
emy to wykona
ć
korzystaj
ą
c z klawisza *.
nch2coo*c

- 16 -
Klawisz * chroni C przed wi
ą
zaniem z O, a wi
ę
c program wi
ąż
e C z nast
ę
pnym N.
W celu wprowadzenia EDTA mo
ż
emy wpisa
ć
:
nch2ch2nch2coo*ch2coo*ch2coo*ch2coo* lub nch2ch2n(ch2coo*)4
Uwaga:
Istnieje jeszcze inne sposoby wprowadzania EDTA, ale ka
ż
dy z nich wymaga stosowania klawisza *.
2.2.20. Stopnie
Mo
ż
na wprowadza
ć
znak stopni (np. 24
°
) korzystaj
ą
c z klawisza `. Jest to mały indeks górny i mo
ż
na
go zwykle znale
źć
w lewym górnym rogu klawiatury obok klawisza 1 i pod znakiem tyldy ~. Znak ten
jest automatycznie zast
ę
powany symbolem stopnia
°°°°
.
Ta sama konwencja jest stosowana w programie FX PhysEquate.
2.3. Opcje wy
ś
wietlania
Program FX ChemStruct ma kilka opcji wy
ś
wietlania, które pozwalaj
ą
na konstruowanie struktur
dokładnie tak jak chcemy je ogl
ą
da
ć
.
Opcje te s
ą
dost
ę
pne w pasku narz
ę
dzi wy
ś
wietlania.
2.3.1. Ukrywanie wi
ą
za
ń
?-H
Pierwsze dwa przyciski w pasku narz
ę
dzi wy
ś
wietlania pozwalaj
ą
na wybór czy wy
ś
wietla
ć
, czy
ukrywa
ć
wi
ą
zania ?-H. Przykładowo, gdy wpiszemy:
ch3chch2ch3ch2ch3
uzyskamy:
2.3.2. Wy
ś
wietlanie ładunków
Program FX ChemStruct oblicza ładunek ka
ż
dej wprowadzonej struktury i wy
ś
wietla go w jednym
z trzech sposobów – bez ładunku, z ładunkiem na ka
ż
dym atomie lub ł
ą
czny ładunek jonu.
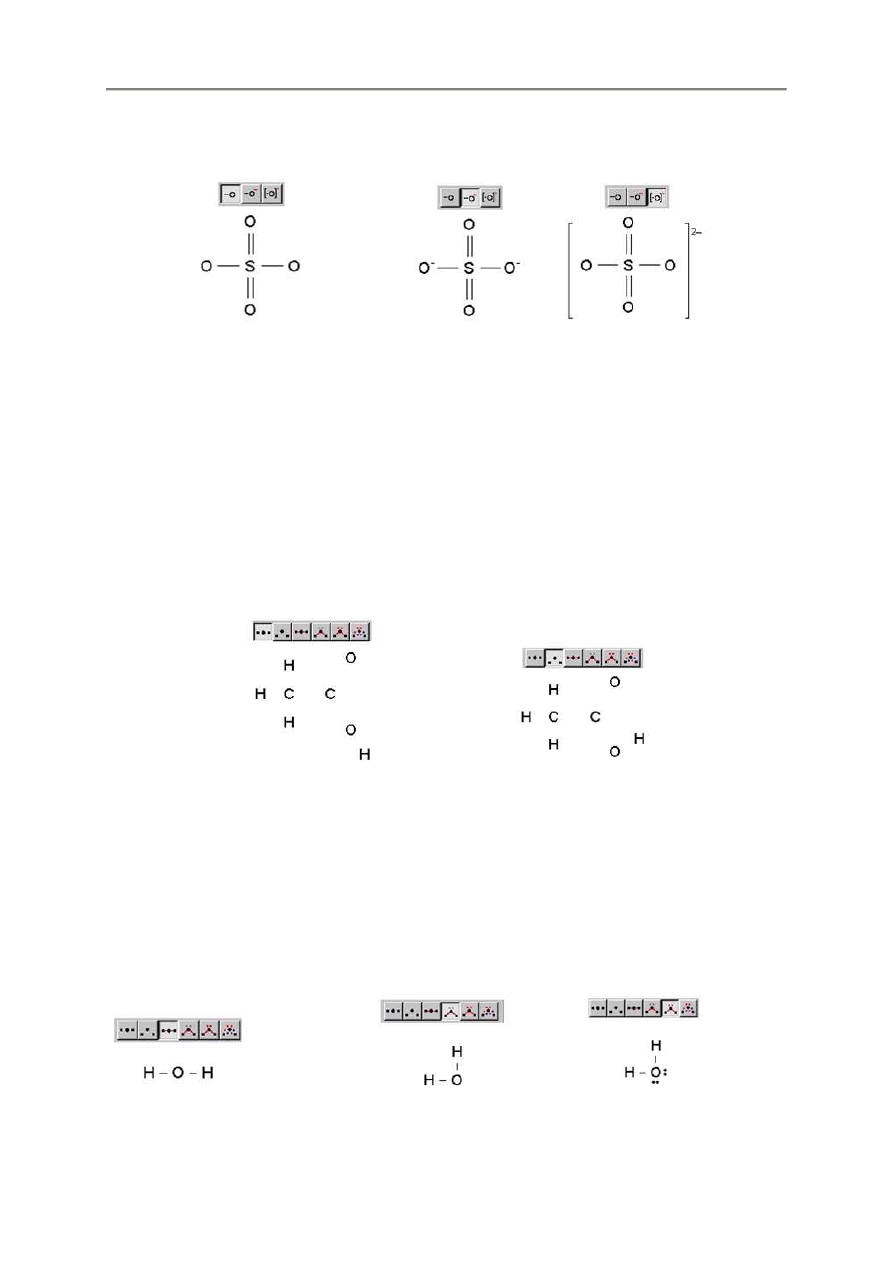
- 17 -
Przykładowo:
so4
Podkre
ś
la si
ę
,
ż
e gdy nadpisujemy ładunek i wprowadzamy własny, wówczas FX ChemStruct
wy
ś
wietla ł
ą
czny ładunek bez wzgl
ę
du na inne ustawienia.
2.3.3. Zwi
ą
zki bez wi
ą
za
ń
Czasem zachodzi potrzeba wy
ś
wietlania zwi
ą
zków, jonów lub struktur bez wy
ś
wietlania
ż
adnych
wi
ą
za
ń
lub elektronów. W ten sposób mo
ż
na konstruowa
ć
zadania dla studentów.
Istniej
ą
dwa sposoby wy
ś
wietlania zwi
ą
zków bez wi
ą
za
ń
– zezwolenie na wy
ś
wietlania niezwi
ą
zanych
pary elektronów lub zakaz wy
ś
wietlania niezwi
ą
zanych par elektronów, np.
ch3cooh
`
Zezwolenie
Zakaz
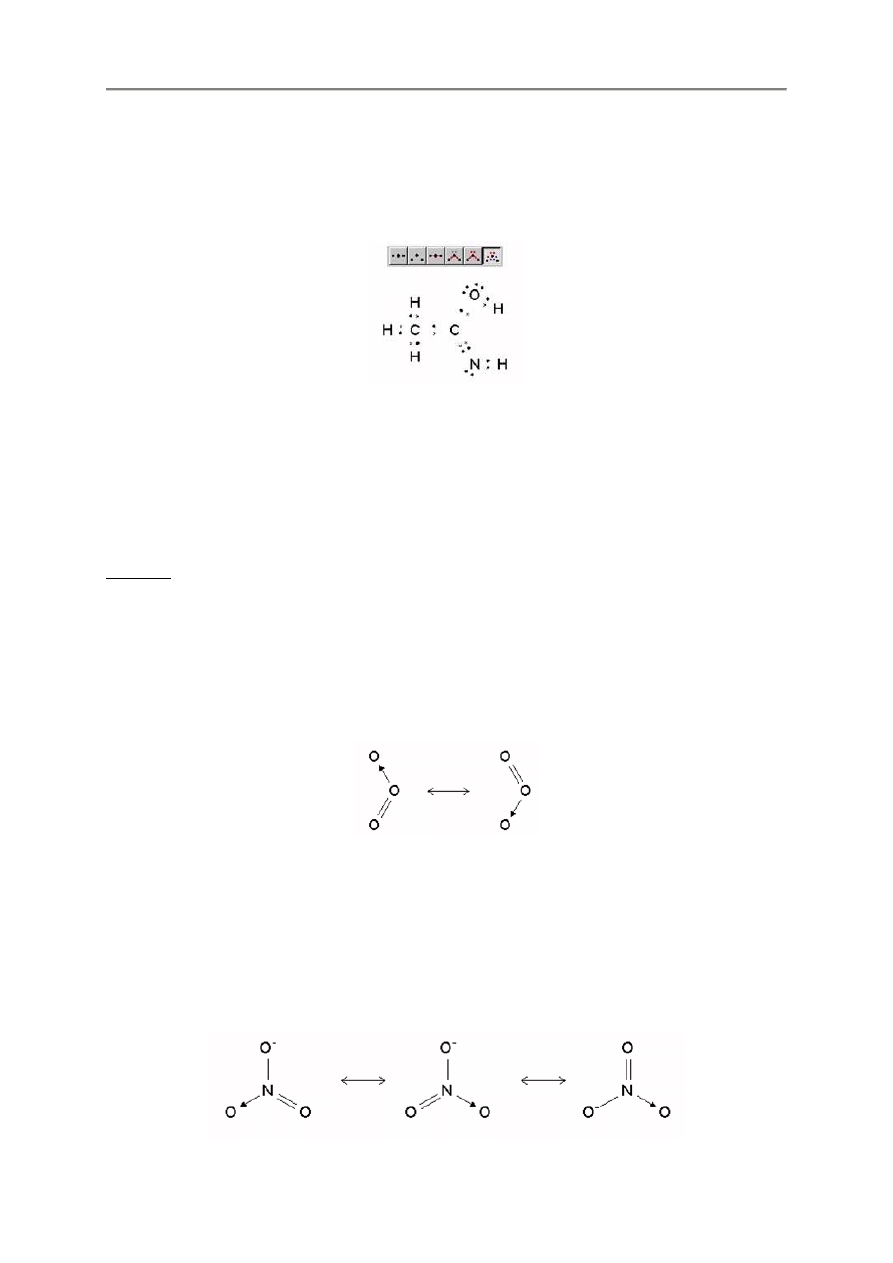
2.3.4. Zezwolenie na pary elektronowe
Program FX ChemStruct zezwala na wy
ś
wietlanie niezwi
ą
zanych par elektronów w pokazywanej
strukturze.
Mo
ż
na wybra
ć
czy lub nie chcemy wy
ś
wietla
ć
niezwi
ą
zany pary, np.
h2o
Zezwolenie
Zezwolenie ale
Zezwolenie
bez wy
ś
wietlania
z wy
ś
wietlaniem
- 18 -
2.3.5. Wy
ś
wietlanie wszystkich elektronów
Program FX ChemStruct mo
ż
4e wy
ś
wietla
ć
wszystkie elektrony w strukturze. Wystarczy w tym celu
klikn
ąć
ostatni przycisk w pasku narz
ę
dzi, bp.
ch3cohnh
2.4. Przykłady
2.4.1. Struktury rezonansowe
Niezb
ę
dne struktury rezonansowe trzeba wprowadza
ć
dwukrotnie (lub raz dla ka
ż
dego mo
ż
liwego
rozmieszczenia), a nast
ę
pnie struktury mo
ż
na odbija
ć
lub klika
ć
w celu uzyskania wymaganych
struktur rezonansowych.
Przykłady:
Ozon
Wpisujemy:
o3 <> o3
Nast
ę
pnie klikamy centralny atom tlenu drugiej cz
ą
steczki ozonu.
Jon azotanowy
Wpisujemy:
no3 <> no3 <> no3
Nast
ę
pnie klikamy centralny atom drugiego jonu i obracamy trzeci jon
- 19 -
2.4.2. W
ę
glowodany
Wi
ę
kszo
ść
w
ę
glowodorów wymaga klikania na atomie centralnym.
Przykłady:
Glukoza
ch2ohchohchohchohchohcoh
Trzeba klikn
ąć
zielony C na poni
ż
szym diagramie.
Fruktoza
ch2ohchohchohchohcoch2oh
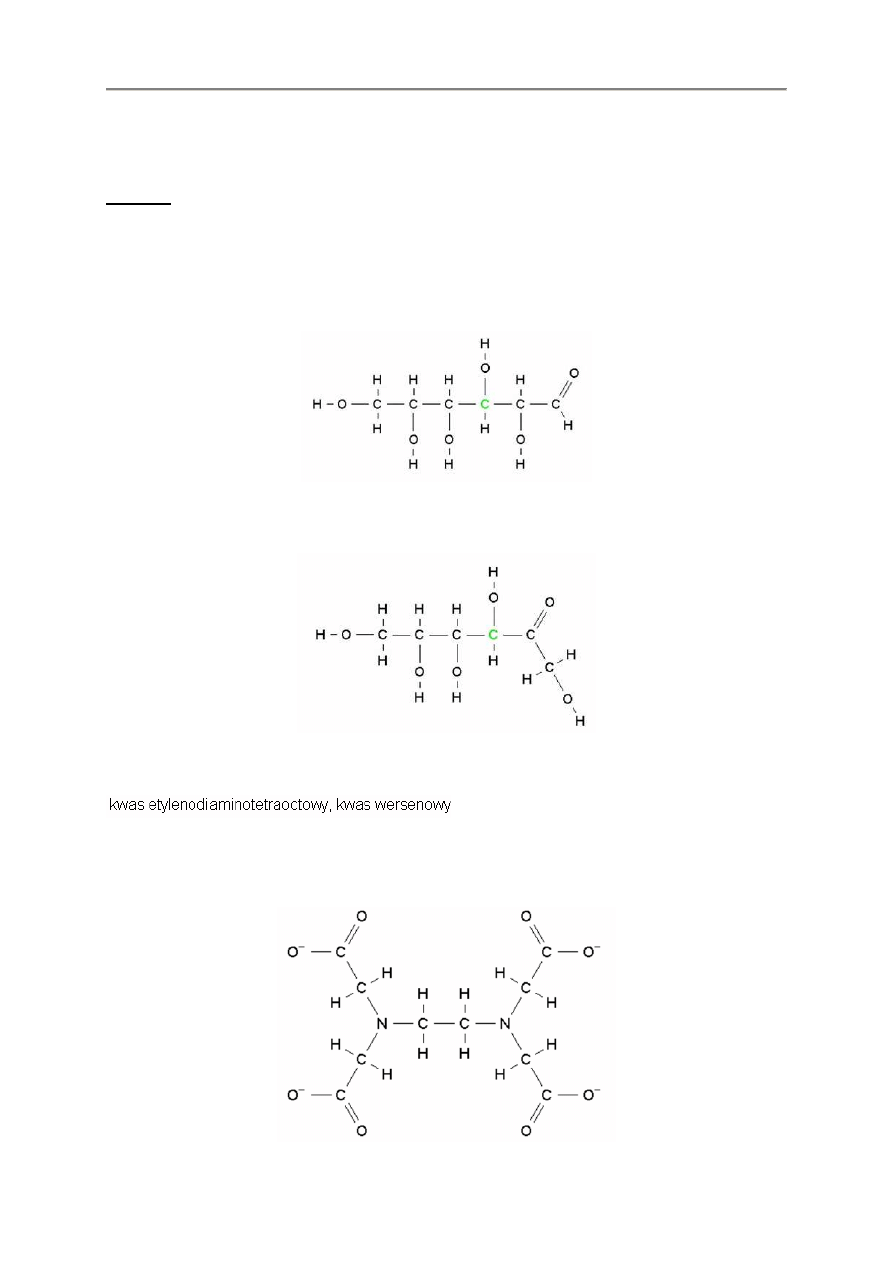
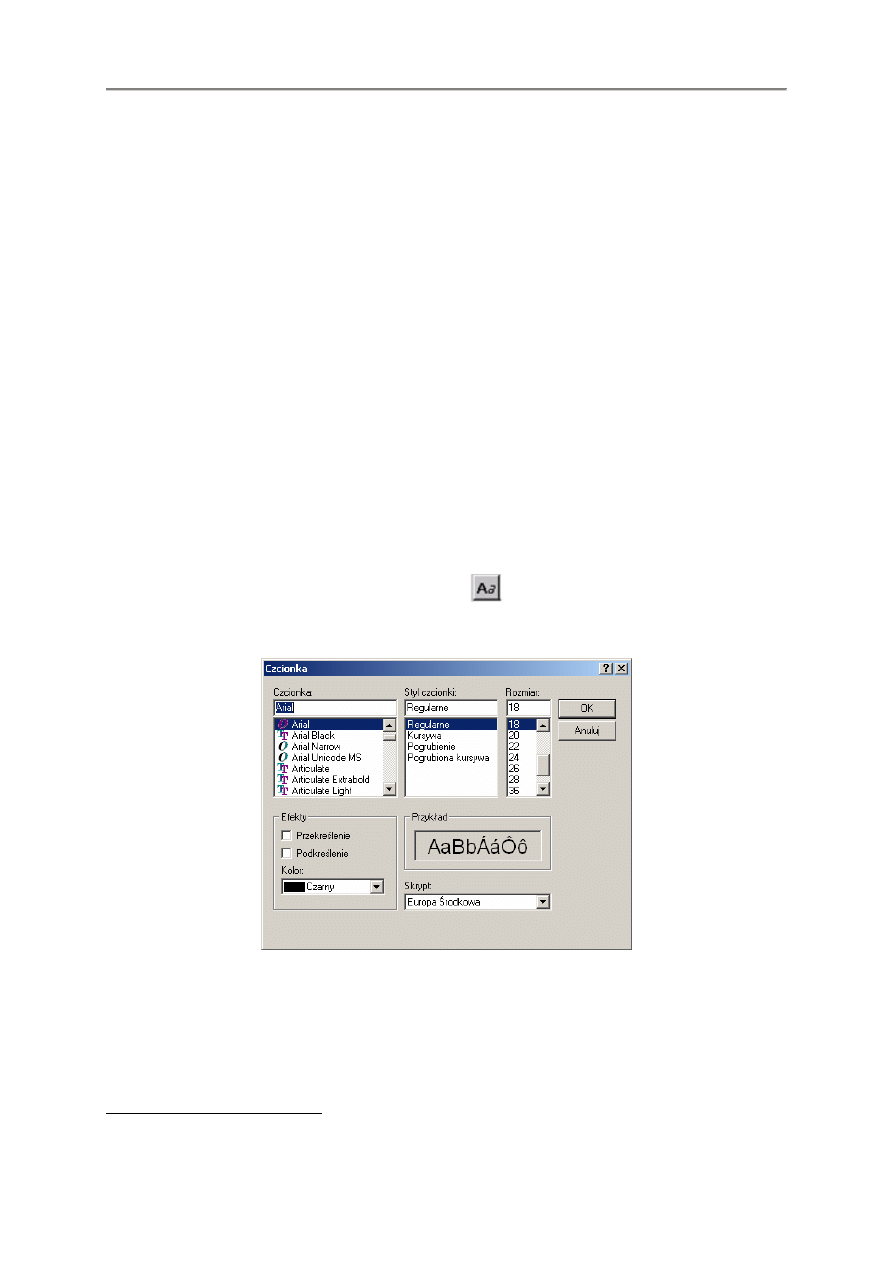
2.4.3. EDTA
EDTA wymaga skorzystania z klawisza * w celu zapobiegania wi
ą
zaniu.
nch2ch2nch2coo*ch2coo*ch2coo*ch2coo*
- 20 -
2.5. Edycja struktur
Gdy chcemy pó
ź
niej edytowa
ć
równanie wstawione do procesora tekstu (np. Word), wystarczy je
podwójnie klikn
ąć
, co powoduje powrót do FX ChemStruct. Tu mo
ż
na wprowadzi
ć
niezb
ę
dne zmiany
po czym klikn
ąć
poza równaniem gdy uznamy te zmiany za zadowalaj
ą
ce
1
.
2.6. Zmiana Co(NO3)2 na CO(NO3)2
Jedn
ą
z najbardziej przydatnych mo
ż
liwo
ś
ci programu FX ChemStruct jest jego zdolno
ść
do
kapitalizowania pierwiastków. Zaoszcz
ę
dza to wiele czasu przy wprowadzaniu równa
ń
. Niektóre
pierwiastki stwarzaj
ą
jednak problemy. Przykładowo, gdy wpiszemy co, wówczas program ZAWSZE
interpretuje to jako tlenek w
ę
gla i zmienia na CO. Jest to oczywiste i taka interpretacja jest zawsze
poprawna. Jednak je
ś
li chcemy wprowadzi
ć
kobalt Co, wówczas trzeba to wpisa
ć
poprawnie jako Co,
a program pozostawi to bez zmiany. Inaczej mówi
ą
c, je
ś
li nasz zwi
ą
zek zawiera TYLKO małe litery,
wówczas FX ChemStruct b
ę
dzie je kapitalizował wybieraj
ą
c najlepszy wariant. Jednak gdy program
znajdzie du
ż
e litery w zwi
ą
zku, wówczas pozostawi je jak zostały wpisane. W ten sposób zawsze
uzyskamy wymagany wynik.
2.7 Zmiana struktur
2.7.1. Narz
ę
dzia / Czcionki
Gdy tworzymy struktur
ę
, mo
ż
emy klikn
ąć
przycisk
w pasku narz
ę
dzi lub przej
ść
do menu
Tools | Font aby otworzy
ć
poni
ż
sze okienko dialogowe:
Mo
ż
na w nim zmieni
ć
krój czcionki, styl, rozmiar i kolor czcionki u
ż
ywanej do rysowania struktur.
Rozmiar czcionki determinuje wielko
ść
pierwiastków w strukturze.
Wszystkie inne rozmiary s
ą
do niej proporcjonalne.
1
Możliwość ta jest dostępna tylko wtedy, gdy podczas instalacji FX ChemStruct zezwolimy na integrację
programu z MS Word lub innymi aplikacjami MS Office.

- 21 -
2.7.2. Narz
ę
dzia / Opcje / Równania
Przy tworzeniu równania, mo
ż
emy klikn
ąć
przycisk
w pasku narz
ę
dzi lub przej
ść
do menu
Tools | Options | Equations aby otworzy
ć
poni
ż
sze okienko dialogowe:
Korzystaj
ą
c z niego mo
ż
na zmienia
ć
poziom i wzgl
ę
dny rozmiar ro
ż
nych składników równa
ń
.
Dzi
ę
ki temu mamy pełn
ą
kontrol
ę
nad tym co wykonujemy.
Warto wypróbowa
ć
przesuwanie suwaków i sprawdza
ć
optycznie przykładowe równanie na górze
w polu podgl
ą
du. Pokazuje ono bie
żą
ce zmiany ustawie
ń
Sekcja Equilibrium Arrow Type pozwala na wybieranie mi
ę
dzy czterema ró
ż
nymi sposobami
rysowania strzał równowagi. Mo
ż
na tu wybra
ć
najlepszy sposób dostosowany do lokalnej konwencji.
Przykładowe równanie na górze w polu podgl
ą
du odzwierciedla wygl
ą
d wybranej opcji.
Strzałki reakcji s
ą
automatycznie skalowane w dostosowaniu do tekstu znajduj
ą
cego si
ę
nad i pod
strzałk
ą
.
Gdy nie ma tekstu, strzałki przybieraj
ą
minimalny rozmiar.
Mo
ż
na ustawia
ć
domy
ś
ln
ą
wielko
ść
tego minimum za pomoc
ą
suwaka Default Arrow Width.
Zaznaczenie pola Italicize Phase determinuje czy FX ChemStruct b
ę
dzie zmieniał czcionki na
kursyw
ę
w informacji fazowej.
Przycisk Reset od
ś
wie
ż
a wszystkie opcje przywracaj
ą
c je do oryginalnych ustawie
ń
fabrycznych.
Przycisk Set as Default zapisuje aktualne ustawienia jako domy
ś
lne do korzystania z nich
w nast
ę
pnych strukturach.
2.7.3. Narz
ę
dzia / Opcje / Struktury
Przy tworzeniu struktury, mo
ż
emy klikn
ąć
przycisk
w pasku narz
ę
dzi lub przej
ść
do menu
Tools | Options | Strucrures aby otworzy
ć
poni
ż
sze okienko dialogowe:
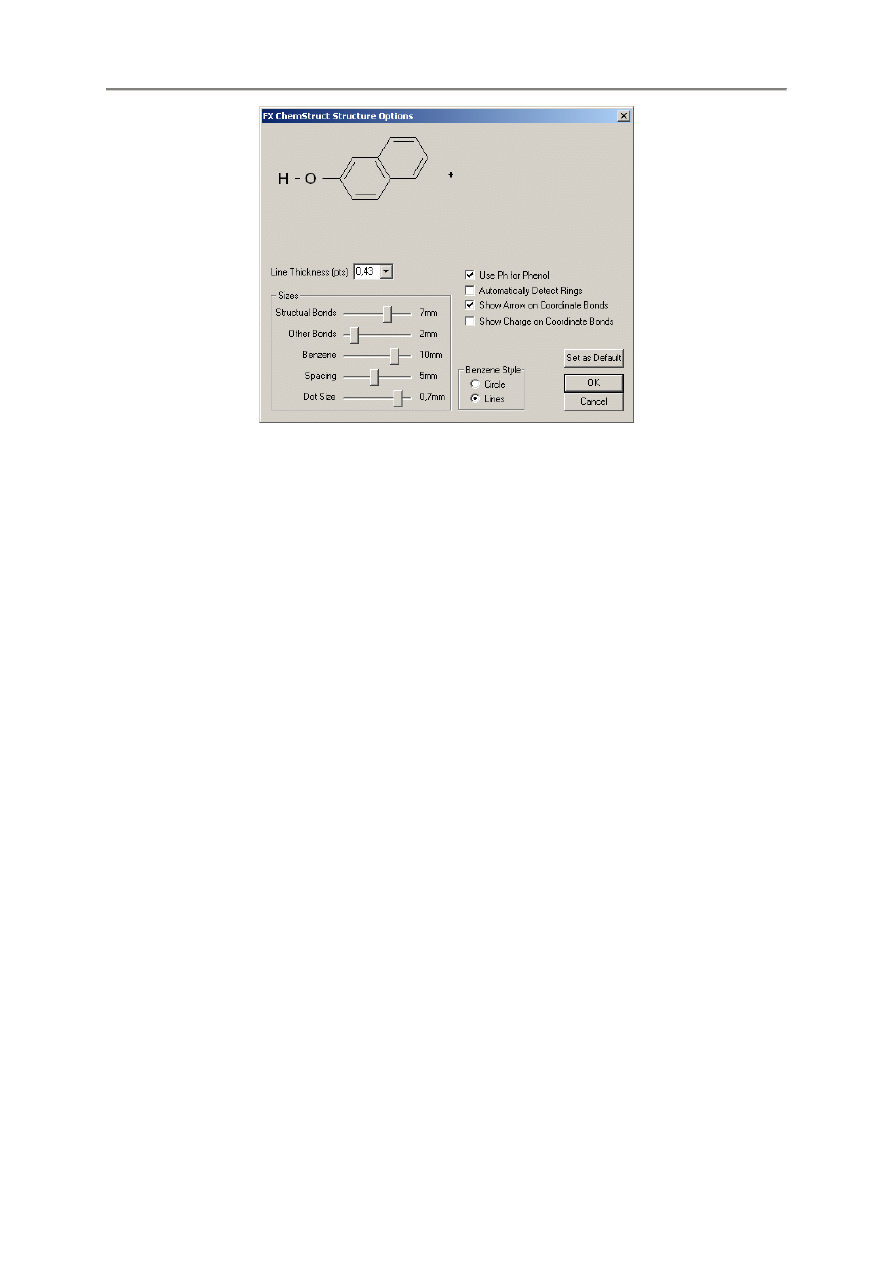
- 22 -
Powy
ż
sze okienko dialogowe pozwala sterowanie na w jaki sposób FX ChemStruct b
ę
dzie rysował
struktury.
Line Thickness – Ta rozwijalna lista ustawia grubo
ść
linii stosowanej do rysowania wi
ą
za
ń
. Rozmiar
ten wyra
ż
any jest w punktach (1 punkt = 1/72 cala = 0,35277755 mm).
Sizes – Sekcja ta pozwala na sterowanie rozmiarem ró
ż
nych składników struktury.
•
Structural Bonds / Other Bonds – Wi
ę
kszo
ść
wi
ą
za
ń
za wyj
ą
tkiem wi
ą
za
ń
?-H. Skrócenie
długo
ś
ci wi
ą
za
ń
wodorowych tworzy lepszy wynik cało
ś
ci. W tym przykładzie. Wi
ę
kszo
ść
wi
ą
za
ń
wynosi 7 mm, a wi
ą
zania wodorowe wynosz
ą
2 mm. Warto zwróci
ć
uwag
ę
,
ż
e ró
ż
nica
tych długo
ś
ci nie ma chemicznego znaczenia, jest to tylko ustawienie wy
ś
wietlania.
•
Benzene – Determinuje długo
ść
boku pier
ś
cienia benzenowego
•
Spacing – Steruje dodatkowym odst
ę
pem dodawanym automatycznie mi
ę
dzy strukturami
i innymi składnikami (w tym przykładzie jest to znak +). Mo
ż
na zawsze dodawa
ć
dalsze spacje
korzystaj
ą
c z tego suwaka.
•
Dot Size – Determinuje rozmiar kropki elektronu.
Use ph fo Phenol – Domy
ś
lnie, program FX ChemStruct pozwala na korzystanie ze skrótu ph
(do wprowadzania fenolu lub benzenu), ale mo
ż
e to by
ć
mylone z fosforem (P) i wodorem (H).
Mo
ż
na usun
ą
c zaznaczenia tego pola je
ś
li tego zechcemy.
Automatically Setect Rings – Korzystaj
ą
c z tego pola mo
ż
na wymusi
ć
aby FX ChemStruct
automatycznie wykrywał struktury pier
ś
cieniowe lub nie zaznacza
ć
go i samodzielnie deklarowa
ć
pier
ś
cienie za pomoc
ą
nawiasów klamrowych.
Show Arrow Chrage on Coordinate Bonds – Wybór wy
ś
wietlania lub ukrywania strzałek i/lub
ładunków wi
ą
za
ń
współrz
ę
dnych.
Benzene Style – Wybór wy
ś
wietlania pier
ś
cieni benzenowych z kołem aromatycznym lub liniami
podwójnych wi
ą
za
ń
.
Set as Default – Przycisk ten zapisuje aktualne ustawienia jako domy
ś
lne do korzystania z nich
w nast
ę
pnych strukturach.

- 23 -
2.8. Moje struktury nie s
ą
poprawnie formatowane !
Program FX ChemStruct próbuje jak najlepiej interpretowa
ć
w logiczny sposób to co u
ż
ytkownik
wprowadza w polu edycji. W 99 % jest to poprawne. Pozostały 1 % mo
ż
e mie
ć
inne znaczenie.
Chemia jest nauk
ą
wyj
ą
tków i nasza struktura mo
ż
e zawiera
ć
co
ś
, czego FX ChemStruct jeszcze nie
zna. Gdy poinformujecie nas o strukturze jak
ą
FX ChemStruct nie przetwarza poprawnie, podejmiemy
starania aby program j
ą
rozpoznawał.
Wyszukiwarka
Podobne podstrony:
Polski opis programu fx Calc
Polski opis programu EST
Polski opis programów pakietu winPenPack Flash 2Gb
Polski opis programu QJot Portable, Opisy programów FREE
Polski opis programu Cleanse Uninstaller Pro, Opisy programów FREE
Polski opis programu CurveFitter 2, Opisy programów FREE
Polski opis programu Autoruns
Polski opis programu Chemistry Problems
Polski opis programu RealWorld Paint, Opisy programów FREE
Polski opis programu BUSINESS CARD DESIGNER PRO
Polski opis programu InterReg 3 2 2
Opis polski FX ChemStruct, Opisy programów FREE
Polski opis FX ChemStruct 1, Opisy programów FREE
Polski opis Eigenmath, Opisy programów FREE
więcej podobnych podstron